

Abstract
Forkhead box class O (FOXO) transcription factors play a pivotal role in regulating a variety of biological processes, including organismal development, cell signalling, cell metabolism and tumorigenesis. Therefore, we hypothesize that genetic variants in FOXO pathway genes are associated with breast cancer (BC) risk. To test this hypothesis, we conducted a large meta-analysis using 14 published GWAS datasets in the Discovery, Biology, and Risk of Inherited Variants in Breast Cancer (DRIVE) study. We assessed associations between 5,214 (365 genotyped in DRIVE and 4,849 imputed) common single-nucleotide polymorphisms (SNPs) in 55 FOXO pathway genes and BC risk. After multiple comparison correction by the Bayesian false-discovery probability method, we found five SNPs to be significantly associated with BC risk. In stepwise multivariate logistic regression analysis with adjustment for age, principal components and previously published SNPs in the same dataset, three independent SNPs (i.e., FBXO32 rs10093411 A>G, FOXO6 rs61229336 C>T and FBXO32 rs62521280 C>T) remained to be significantly associated BC risk (P = 0.0008, 0.0011 and 0.0017, respectively). Additional expression quantitative trait loci analysis revealed that the FBXO32 rs62521280 T allele was associated with decreased mRNA expression levels in breast tissue, while the FOXO6 rs61229336 T allele was found to be associated with decreased mRNA expression levels in the whole blood cells. Once replicated by other investigators, these genetic variants may serve as new biomarkers for BC risk.
Keywords: breast cancer susceptibility, expression quantitative trait loci analysis, FOXO pathway, single nucleotide polymorphism
Introduction
Worldwide data show a continuing increase in the incidence of breast cancer (BC)1 that has become the second leading cause of cancer deaths among women in the United States2. In 2020, about 279,100 cases were diagnosed with and 42,690 died from BC in the United States3. Therefore, additional biomarkers are needed to identify individuals who are at a greater risk of BC for early detection and prevention to reduce the incidence of BC.
Several risk factors (e.g. physical activities, unhealthy lifestyle and reproductive factors) are known to contribute to BC risk, and gene mutations only explain approximately 9% ~ 13% of the heritability of BC4,5. It is well recognized that single nucleotide polymorphisms (SNPs), the most common form of genetic variation, are also contributed to BC risk, suggesting their importance as a molecular biological mechanism of carcinogenesis. In the post genome-wide association study (GWAS) era, it is possible to take more sophisticated analyses to identify cancer risk-associated functional SNPs in a biological pathway manner. With such a targeted pathway-based and hypothesis-driven approach, investigators may identify cancer risk-associated SNPs from previously published GWAS datasets by using available genotyping data with further evaluation of their biological functions.
It is known that forkhead box class O (FOXO) transcription factors are important regulators of gene expression and play a pivotal role in regulating a variety of biological processes, including organismal development, cell signalling, cell metabolism and tumorigenesis6–12. FOXOs are involved in the regulation of the upstream signalling pathway of PD-L1 by transcriptional or post-translational manner. For example, FOXOs may inhibit PD-L1 expression in cancer cells and indirectly upregulate T cell response13. FOXOs are considered putative tumor suppressors, because the activation of FOXOs inhibits cell cycle and induces apoptosis in a variety types of tumor cells14–17. However, the role of FOXOs in carcinogenesis remains to be determined, because only few studies have investigated the effect of genetic variation in FOXO pathway genes on BC risk, such as an association of AKT1 gene mutation with breast cancer risk in the high-altitude Ecuadorian mestizo population18 and the relationship between SIRT1 gene polymorphisms and breast cancer in Egyptians19.
Considering the importance of the FOXO pathway in the biology of carcinogenesis, it is very likely that genetic variants in FOXO pathway genes are associated with BC risk. To test this hypothesis, we conducted a targeted pathway-based and hypothesis-driven approach to identify SNPs of FOXO pathway-related genes and examined their associations with BC risk by using available genotyping data from 14 previously published GWAS datasets of 53,107 BC case-control study participants in the DRIVE study.
Materials and Methods
Study participants
We performed a case-control meta-analysis of the participants from 14 out of the 17 previously published BC GWASs from the DRIVE study (phs001265.v1.p1), which is different from the DRIVE-Genome-Wide Association meta-analysis (phs001263.v1.p1) previously used by other researchers, and the differences between the two datasets have been described in detail elsewhere20. In brief, the major differences are as follows: 1) different sources of studies and 2) different data types: The DRIVE study we used has genotyping data with detailed SNP information, but the DIRVE study previously used by other researchers had the summary data only good for meta-analyses. The DRIVE study used in the present study was one of five projects funded in 2010 by the National Cancer Institute-supported Genetic Associations and Mechanisms in Oncology (GAME-ON). Among the 17 studies, “Women of African Ancestry Breast Cancer Study (WAABCS)” was an African ancestry study and “The Sister Study (SISTER)” and “the Two Sister Study (2 SISTER)” had different research designs from others and used cases’ sisters as controls; therefore, our meta-analysis excluded these three studies. As a result, a total of 28,758 BC cases and 24,349 controls in 14 GWAS studies of European ancestry participants were included in the final analysis, whose characteristics are summarized in Table S1.
These 14 DRIVE GWASs included: Breast Oncology Galicia Network (BREOGAN); Copenhagen General Population Study (CGPS); Cancer Prevention Study-II Nutrition Cohort (CPSII); European Prospective Investigation Into Cancer and Nutrition (EPIC); Melbourne Collaborative Cohort Study (MCCS); Multiethnic Cohort (MEC); Nashville Breast Health Study (NBHS); Nurses’ Health Study (NHS); Nurses’ Health Study 2 (NHS2); NCI Polish Breast Cancer Study (PBCS); The Prostate, Lung, Colorectal and Ovarian Cancer Screening Trial (PLCO); Study of Epidemiology and Risk factors in Cancer Heredity (SEARCH); Swedish Mammography Cohort (SMC); and Women’s Health Initiative (WHI). Illumina Infinium OncoArray-500k BeadChip genotyping platforms were used for all of the GWAS datasets, and only sex and age at interview for all the participants were available to us, while three other variables including age at diagnosis, estrogen receptor status, and tumor histology type were available to the cases; for age variables, we adopted the age at interview for the controls and the age at diagnosis for the cases. Each of the original studies approved by the Institutional Review Boards of the Participating institutions received written informed consent from the participants.
Identification of FOXO pathway genes and their SNP extraction
We selected candidate genes in the FOXO pathway according to the databases of KEGG, BIOCARTA, REACTOME, Canonical pathways and Gene Ontology (GO) in the “Molecular Signatures Database v7.0 (MsigDB)” (http://software.broadinstitute.org/gsea/msigdb/search.jsp) used by the keyword “FOXO”. In total, we identified 55 candidate genes after excluding 16 duplicate genes (Table S2).
We performed quality control before imputation to avoid poor quality markers to be included with the following stringent criteria: (1) the minor allelic frequency (MAF) ≥ 1%, (2) missing rate ≤ 10%, (3) genotyping success rate ≥ 95%, and (4) Hardy-Weinberg equilibrium (HWE) P ≥ 1×10−6. SNPs located in the aforementioned 55 candidate genes and their ± 500 kb flanking regions were extracted by using IMPUTE2 software with the reference panel from the 1000 Genomes Project data (phase 3)21. After quality control, imputed SNPs within 2-kb up- and down-stream of genes in the FOXO pathway were extracted for further analysis. SNPs for the final meta-analysis were selected with the following quality control criteria: (1) a genotyping call rate ≥ 95%; (2) Imputed SNPs with an information score ≥ 0.80; (3) MAF ≥ 5%; and (4) HWE P ≥ 10–6.
Statistical analysis
Principal components (PCs) were calculated for each of the GWASs, and their combined dataset was evaluated by using the Genome-wide Complex Trait Analysis22. The associations between the top 20 PCs and BC risk were evaluated by using univariate logistic regression analysis. Significant PCs together with age as covariates in the final model were adjusted for in further SNP association analysis. We calculated odds ratios (ORs) and 95% confidence intervals (CIs) for each SNP by unconditional logistic regression with adjustment for covariates (age and significant PCs). The four previously published SNPs from the same DRIVE study20 were also adjusted for in a logistic regression model to identify additional significant SNPs. A meta-analysis was further performed with the inverse variance method by combining the results of a log-additive model of the 14 studies. If the Cochran’s Q test P-value ≤ 0.1 or I2 > 50%, a random-effects model was used; otherwise a fixed-effects model was employed. The results were first corrected by false discovery rate for multiple testing correction. Because many SNPs under investigation were a high linkage disequilibrium (LD) as a result of imputation, the Bayesian false-discovery probability (BFDP) approach was also used for multiple test correction in substitution for the false discovery rate, with a cut-off value of 0.8 as recommended23. We used a prior probability of 0.01 to detect an upper bound of 3.0 for an association with variant genotypes or minor alleles of the SNPs. The number of risk genotypes (NRGs) of the independent SNPs was counted as a genetic score and subsequently used to evaluate combined effects of the SNPs. Additionally, Manhattan and LD plots were constructed by Haploview v4.224, and regional association plots for independent SNPs were produced by LocusZoom25. Other statistical analyses were performed with SAS software Version 9.4 (SAS Institute, Cary, NC), R (version 3.5.0) and PLINK (version 1.90), if not specified otherwise.
Functional analysis
Finally, we predicted potential functions of the identified independent SNPs in online functional prediction website: RegulomeDB (http://www.regulomedb.org/) and HaploReg26 (http://archive.broadinstitute.org/mammals/haploreg/haploreg.php). In addition, we performed an expression quantitative trait locus (eQTL) analysis to assess the associations between the SNPs and mRNA expression levels of their corresponding genes by using the genotyping and expression data from the lymphoblastic cell lines of 373 European descendants available in the 1000 Genomes Project27 and the Genotype-Tissue Expression (GTEx) project v8.p2 database (https://gtexportal.org/home/)28, which included genomic data from both whole blood and breast tissues. The samples used for the eQTL analysis are publically available and not from the DRIVE study.
Results
Single locus analysis
The flowchart for the analysis is shown in Figure 1. The results of the top 20 PCs of the datasets are shown in Table S3. Because of the differences in genotyping platforms used by the 14 studies, there were a range between 6,163 and 6,429 SNPs in each individual study for further analysis. In total, the final meta-analysis of the 14 studies included 5,214 SNPs that passed the quality control, including 365 genotyped SNPs and 4,849 imputed SNPs. The distribution of information score for those selected SNPs in each study is shown in Figure S1. The meta-analysis showed that 338 SNPs were significantly associated with BC risk in an additive genetic model (P < 0.05), and after multiple testing corrections by BFDP, five SNPs were still statistically noteworthy with BFDP < 0.8 (Figure 2). To further identify SNPs as independent predictors of BC risk, stepwise logistic regression analyses were performed to evaluate the independent effects of the five significant SNPs on BC risk with adjustment for age, significant PCs and another four previously published risk-associated SNPs in the same DRIVE study20. As a result, three independent SNPs (i.e., FBXO32 rs10093411 A>G, FOXO6 rs61229336 C>T and FBXO32 rs62521280 C>T) remained statistically significant in association with BC risk (P = 0.0008, 0.0011 and 0.0017, respectively), which were then used for further analyses (Table 1).
Figure 1. The workflow of the present study.
Figure 2. Manhattan plot of the 5,214 SNPs of FOXO pathway genes in the DRIVE study.
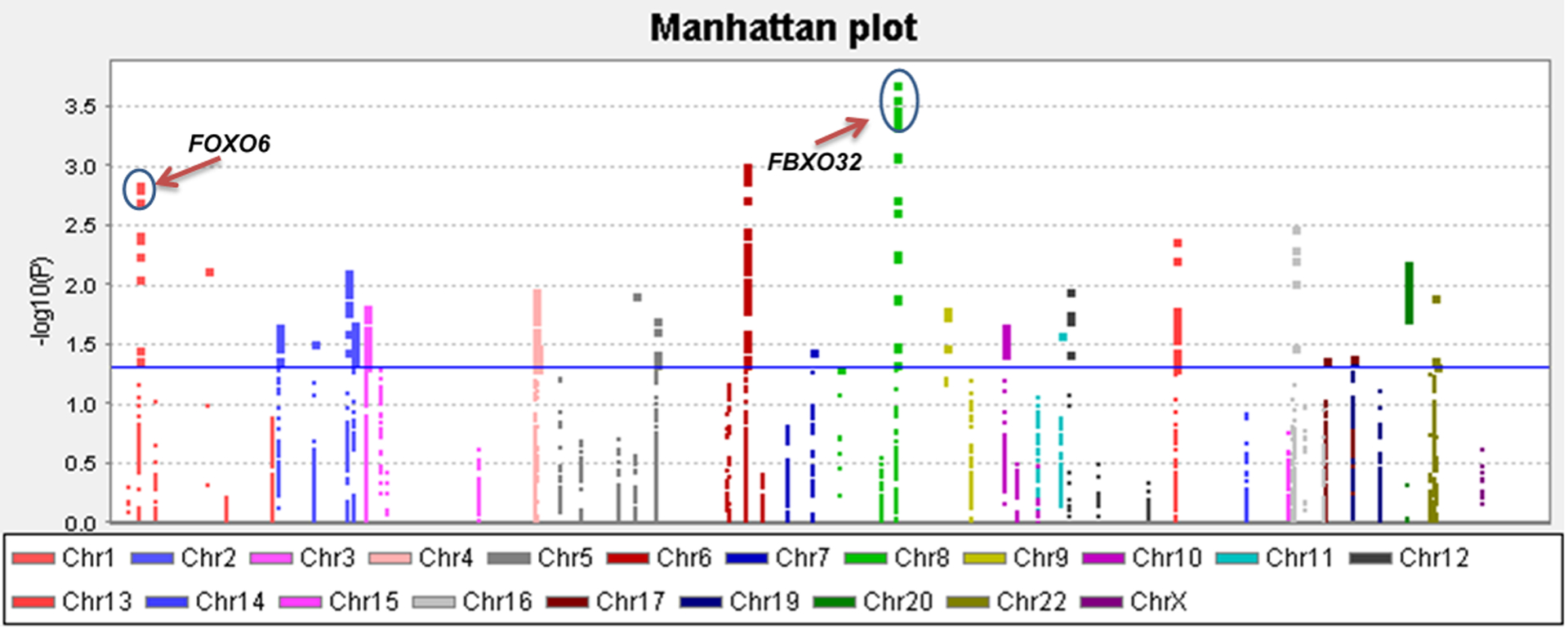
The x-axis represents each chromosome. The y-axis represents the P values for associations with breast cancer risk. The blue horizontal line indicates P value equal to 0.05. Abbreviations: DRIVE, Discovery, Biology, and Risk of Inherited Variants in Breast Cancer; FBXO32, F-box protein 32; FOXO, Forkhead box O; FOXO6, Forkhead box O6.
Table 1.
Three genetic variants as independent BC risk predictors obtained from stepwise logistic regression analysis of selected variables in the DRIVE study
| Variables* | Location | MAF | Category† | Frequency | OR (95% CI)* | P * |
|---|---|---|---|---|---|---|
| FBXO32 rs10093411_G | 8q24.13 | 0.25 | AA/AG/GG | 29736/19817/3449 | 1.05 (1.02–1.08) | 0.0008 |
| FOXO6 rs61229336_T | 1p34.2 | 0.11 | CC/CT/TT | 23192/23734/6076 | 0.96 (0.93–0.98) | 0.0011 |
| FBXO32 rs62521280_T | 8q24.13 | 0.34 | CC/CT/TT | 41990/10336/676 | 1.06 (1.02–1.11) | 0.0017 |
Stepwise multivariate logistic regression analysis included age, PC1, PC3, PC4, PC5, PC6, PC8, PC10, PC11, PC14, PC16, four previously published risk-associated SNPs (rs1323697, rs1264308, rs141308737 and rs1469412) in the same study (see ref 20) and additional five SNPs (rs10093411, rs61229336, rs62521280, rs10089663 and rs10092779) with P < 0.05 and BFDP < 0.8 of present study.
The most left-hand side “category” was used as the reference.
Note: there were 20 PCs in the combined datasets as listed in Table S3, of which 10 remained significant and were adjusted in the final stepwise multivariate logistic regression analysis.
Abbreviations: BC: breast cancer; CI: confidence interval; DRIVE: Discovery, Biology, and Risk of Inherited Variants in Breast Cancer; MAF: minor allele frequency; FBXO32, F-box protein 32; FOXO6, Forkhead box O6; OR: odds ratio.
The types of the three SNPs and their associations with BC risk are presented in Table S4, of which SNP rs10093411 was genotyped, and the other two were imputed. There was no heterogeneity existed among the 14 GWASs for the effects of these independent SNPs. The forest plots of the three SNPs by the meta-analysis are summarized in Figure S2. The results showed that two SNPs in the same gene were associated with a significantly increased risk of BC (FBXO32 rs10093411 A>G: OR = 1.06, 95% CI = 1.03–1.09, and P = 1.55 × 10–4; FBXO32 rs62521280 C>T: OR = 1.07, 95% CI = 1.03–1.11, and P = 8.35 × 10–4), while the other SNP was associated with a significantly decreased BC risk (FOXO6 rs61229336 C>T: OR = 0.96, 95% CI = 0.94–0.99, and P = 5.53 × 10–3). Regional association plots of these three independent SNPs in the 200 kb up- and down-stream regions are presented in Figure S3. As shown in Table 2, the effects of the FBXO32 rs10093411 G, FOXO6 rs61229336 T and FBXO32 rs62521280 T alleles on BC risk were statistically significant (the trend test in multivariate analysis: P = 0.0002, 0.001 and 0.0005, respectively).
Table 2.
Associations between three independent SNPs in the FOXO pathway genes and BC in the DRIVE study
| Genotype | Ncontrol/Ncase | Univariate analysis | Multivariate analysis* | ||
|---|---|---|---|---|---|
| OR (95% CI) | P | OR (95% CI) | P | ||
| FBXO32 rsl0093411 A>G | |||||
| AA | 13782/15954 | 1.00 | 1.00 | ||
| AG | 8967/10850 | 1.05 (1.01–1.08) | 0.016 | 1.05 (1.01–1.09) | 0.010 |
| GG | 1500/1949 | 1.12 (1.05–1.21) | 0.002 | 1.13 (1.05–1.21) | 0.001 |
| Trend test | 0.0003 | 0.0002 | |||
| AG+GG | 10467/12799 | 1.06 (1.02–1.09) | 0.002 | 1.06 (1.02–1.10) | 0.001 |
| FOXO6 rs61229336 OT | |||||
| CC | 10434/12758 | 1.00 | 1.00 | ||
| CT | 10956/12778 | 0.95 (0.92–0.99) | 0.011 | 0.96 (0.92–0.99) | 0.015 |
| TT | 2859/3217 | 0.92 (0.87–0.97) | 0.004 | 0.92 (0.87–0.97) | 0.004 |
| Trend test | 0.0009 | 0.001 | |||
| CT+TT | 13815/15995 | 0.95 (0.92–0.98) | 0.002 | 0.95 (0.92–0.98) | 0.003 |
| FBXO32 rs62521280 C>T | |||||
| CC | 19400/22590 | 1.00 | 1.00 | ||
| CT | 4535/5801 | 1.10 (1.05–1.15) | <0.0001 | 1.10 (1.05–1.14) | <0.0001 |
| TT | 314/362 | 0.99 (0.85–1.15) | 0.898 | 0.99 (0.85–1.16) | 0.909 |
| Trend test | 0.0003 | 0.0005 | |||
| CT+TT | 4849/6163 | 1.09 (1.05–1.14) | <0.0001 | 1.09 (1.04–1.14) | 0.0001 |
| Number of combined risk genotypes † | |||||
| 0 | 6519/7378 | 1.00 | 1.00 | ||
| 1 | 10789/12517 | 1.03 (0.98–1.07) | 0.249 | 1.02 (0.98–1.07) | 0.270 |
| 2 | 5862/7371 | 1.11 (1.06–1.17) | <0.0001 | 1.11 (1.06–1.16) | <0.0001 |
| 3 | 1079/1487 | 1.22 (1.12–1.33) | <0.0001 | 1.22 (1.11–1.33) | <0.0001 |
| Trend test | <0.0001 | <0.0001 | |||
| 0–1 | 17308/19895 | 1.00 | 1.00 | ||
| 2–3 | 6941/8858 | 1.11 (1.07–1.15) | <0.0001 | 1.11 (1.07–1.15) | <0.0001 |
Abbreviations: BC: breast cancer; CI: confidence interval; DRIVE: Discovery, Biology, and Risk of Inherited Variants in Breast Cancer; FBXO32, F-box protein 32; FOXO: Forkhead box O; FOXO6, Forkhead box O6; OR: odd ratio; SNP: single nucleotide polymorphism.
Adjusted for age, PC1, PC3, PC4, PC5, PC6, PC8, PC10, PC11, PC14 and PC16.
Risk genotypes were rs10093411 AG+GG, rs61229336 CC and rs62521280 CT+TT.
Joint effect analysis
We subsequently combined risk genotypes of FBXO32 rs10093411 AG+GG, FOXO6 rs61229336 CC and FBXO32 rs62521280 CT+TT into a genetic risk score as the NRG to assess the joint effect of the genotypes of these three independent SNPs on BC risk. All the participants were divided into four groups of zero, one, two and three risk genotypes. The trend test indicated that the increased NRG was significantly associated with an increased BC risk (P < 0.0001, Table 2). According to the effect values and the frequency of each group, we further dichotomized all the participants into two groups: low-risk (0–1 NRG) and high-risk (2–3 NRG), and we found that the risk associated with NRG was more evident in the high-risk group (in multivariate analysis: OR = 1.11, 95% CI = 1.07–1.15, P < 0.0001, Table 2). Further stratified analyses by subgroups of age, ER status and invasiveness showed that risk associated with NRG was more evident in the subgroup of age ≤60 (in multivariate analysis: OR = 1.13, 95% CI = 1.07–1.19, P < 0.0001), particularly among individuals with ER+ the cases with an invasive tumor. However, no heterogeneity or interaction was observed between these strata (all P > 0.05) (Table 3).
Table 3.
Stratified analysis for associations between risk genotypes and BC in the DRIVE study
| Characteristics | NRG 0–1 | NRG 2–3 | Univariate analysis | Multivariate analysis* | P inter † | ||||
|---|---|---|---|---|---|---|---|---|---|
| Control | Case | Control | Case | OR (95% CI) | P | OR (95% CI) | P | ||
| Age | |||||||||
| ≤60 | 9090 | 8544 | 3635 | 3882 | 1.14 (1.08–1.20) | <0.0001 | 1.13 (1.07–1.19) | <0.0001 | 0.348 |
| >60 | 8218 | 11351 | 3306 | 4976 | 1.09 (1.03–1.15) | 0.001 | 1.09 (1.03–1.15) | 0.001 | |
| ER+ vs. control | |||||||||
| ≤60 | 9090 | 5274 | 3635 | 2329 | 1.10 (1.04–1.18) | 0.002 | 1.12 (1.05–1.19) | 0.0006 | 0.518 |
| >60 | 8218 | 8022 | 3306 | 3477 | 1.08 (1.02–1.14) | 0.010 | 1.08 (1.02–1.15) | 0.006 | |
| ER− vs. control | |||||||||
| ≤60 | 9090 | 1281 | 3635 | 636 | 1.24 (1.12–1.38) | <0.0001 | 1.17 (1.05–1.30) | 0.004 | 0.966 |
| >60 | 8218 | 1215 | 3306 | 574 | 1.17 (1.06–1.31) | 0.003 | 1.16 (1.04–1.30) | 0.006 | |
| Invasiveness vs. control | |||||||||
| ≤60 | 9090 | 7647 | 3635 | 3438 | 1.12 (1.06–1.19) | <0.0001 | 1.12 (1.06–1.19) | <0.0001 | 0.356 |
| >60 | 8218 | 10349 | 3306 | 4502 | 1.08 (1.02–1.14) | 0.004 | 1.08 (1.03–1.14) | 0.003 | |
| In-situ vs. control | |||||||||
| ≤60 | 9090 | 744 | 3635 | 348 | 1.17 (1.02–1.34) | 0.021 | 1.17 (1.02–1.34) | 0.022 | 0.538 |
| >60 | 8218 | 832 | 3306 | 370 | 1.11 (0.97–1.26) | 0.128 | 1.10 (0.97–1.25) | 0.155 | |
Adjusted for PC1, PC3, PC4, PC5, PC6, PC8, PC10, PC11, PC14 and PC16.
Pinter: P value for interaction analysis between age and NRG.
Abbreviations: BC: breast cancer; CI: confidence interval; DRIVE: Discovery, Biology, and Risk of Inherited Variants in Breast Cancer; ER: estrogen receptor; NRG: number of risk genotypes; OR: odds ratio.
Genotype-phenotype correlation analysis
Functional prediction by RegulomeDB showed that FBXO32 rs10093411 A>G, FOXO6 rs61229336 C>T and FBXO32 rs62521280 C>T had a RegulomeDB score of 3a, 7 and 5, respectively and that these SNPs may be located at transcription factor binding sites or DNase I regulating sites. We also searched for SNPs in high LD (r2 ≥ 0.8) with these three independent SNPs, and their functional prediction was made as well by using HaploReg. The results suggest that FBXO32 rs10093411 A>G is located in an intron and may change the motifs of MZF1; FOXO6 rs61229336 C>T is also located in an intron with the selected eQTL for three hits and may change the motifs of CACD, PU.1 and Zbtb3, which may be markers of promoter histone in THYM and enhancer histone in 9 tissues; whereas FBXO32 rs62521280 C>T is located in the potential enhancer region with histone methylation in 5 tissues (Table S4). We further assessed potential functions of these three independent SNPs by using data from the ENCODE Project, which suggests that FOXO6 rs61229336 C>T is located in DNase I hypersensitive sites and has considerable levels of H3K4Me1 acetylation (Figure S4).
We further explored correlations between genotypes of the three independent SNPs and their corresponding mRNA expression levels in the publically available RNA-seq data of lymphoblastoid cell lines generated from 373 European descendants in the 1000 Genomes Project. Unfortunately, no significant results were found (Figure S5). We further performed the correlation analysis by using data from the GTEx Project, and the results showed that rs10093411 A>G was not significantly associated with FBXO32 mRNA expression levels in breast tissue (P = 0.100, Figure 3a) and the whole blood (P = 0.260, Figure 3b); however, the rs62521280 C>T was significantly associated with decreased levels of mRNA expression of FBXO32 in breast tissues (P = 0.030, Figure 3c) but not for the whole blood (P = 0.540, Figure 3d). Although rs61229336 C>T was not significantly associated with levels of mRNA expression of FOXO6 in breast tissue (P = 0.180, Figure 3e), it was significantly associated with decreased levels of mRNA expression of FOXO6 in the whole blood (P = 8.70 × 10−4, Figure 3f).
Figure 3. The expression quantitative trait loci (eQTLs) analysis for FBXO32 rs10093411, rs62521280 and FOXO6 rs61229336 in the GTEx project.
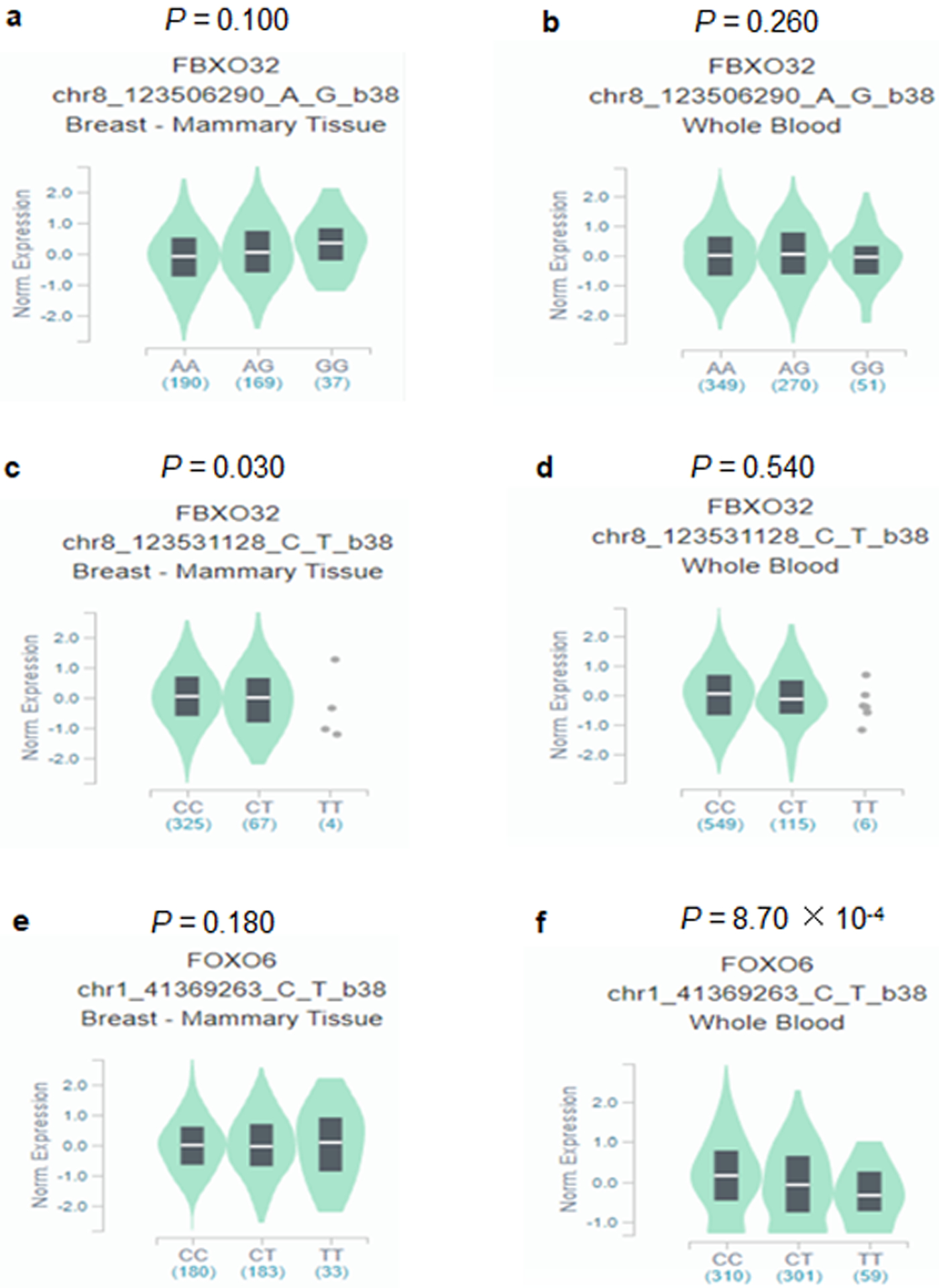
Correlation between FBXO32 mRNA expression and rs10093411 genotype (a) Breast tissue, (b) Whole blood. Correlation between FBXO32 mRNA expression and rs62521280 genotype (c) Breast tissue, (d) Whole blood. Correlation between FOXO6 mRNA expression and rs61229336 genotype (e) Breast tissue, (f) Whole blood. Abbreviations: GTEx: Genotype-Tissue Expression; FBXO32, F-box protein 32; FOXO6, Forkhead box O6.
Discussion
Some published studies have used the data from different GWAS studies to identify the associations of different pathway genes and BC risk1,4,29,30, but few have investigated the association between FOXO pathway genes and BC risk. One study found that AKT1 rs3803304 may be a predictive biomarker for BC risk in the high-altitude Ecuadorian mestizo population18. In another Egyptian population, SIRT1 rs3758391 and rs12778366 polymorphisms were found to be associated with BC risk and prognosis19. In the present study, we have investigated whether SNPs of FOXO pathway-related genes are associated with BC risk by using available genotyping data from the previously published 14 GWASs in the DRIVE study, and the study population was different from the above-mentioned two studies. We identified three independent SNPs in two genes (i.e., FBXO32 rs10093411 and rs62521280 at 8q24.13, and FOXO6 rs61229336 at 1p34.2) to be associated with BC risk. There was a significant effect of the combined genotypes of these three SNPs on BC risk. In stratified analyses, all the results with Pinter > 0.05 suggested that there was no interaction between age and NRG in all subgroups. Further eQTL analyses showed that the FBXO32 rs62521280 T allele was associated with decreased mRNA expression levels in breast tissues, while the FOXO6 rs61229336 T allele was found to be associated with decreased mRNA expression levels in the whole blood cells. These eQTL results provide some support for biological plausibility of the observed associations.
FBXO32, located on chromosome 8q24.13, encodes an F-box only protein 32 (FBXO32), an E3 ubiquitin ligase that is essential for hallmark phenotypic changes and gene expression underlying epithelial-mesenchymal transition as well as involved in the process of tumorigenesis31–34. Several studies have indicated that FBXO32 may be a functional tumor suppressor in some cancer types via promoter methylation35–38. Even with the known importance of FBXO32 in cancers, few reported studies have investigated the roles of FBXO32 in BC risk. One study found that an increased expression of FBXO32 facilitated apoptosis of multiple breast cancer cell lines39, and another study showed that FBXO32 deficiency in breast cancer cells led to the accumulation of Krüppel-like factor 4 and promoted tumorigenesis40. A more recent GWAS study identified a locus in RNTSKP155 that shares the same location of 8q24.13 as FBXO3241, but the previously reported rs58847541 SNP in RNTSKP155 was not in LD with rs10093411 and rs62521280 (Figure S6a). Interestingly, we found in the present study that the rs62521280 T allele might down-regulate the expression of FBXO32, suggesting that FBXO32 may play a protective role in BC risk, but additional experimental studies are needed to explain how FBXO32 rs62521280 C>T influences BC risk.
FOXO6, located on chromosome 1p34.2, encodes a forkhead box protein O6 (FOXO6)42, but little is known about the roles of FOXO6 in tumorigenesis, especially in BC. One study showed that FOXO6 was overexpressed in both colorectal cancer cell lines and tumor tissues and that FOXO6 knockdown inhibited the migration and invasion of colorectal cancer cells43; another study showed that overexpression of FOXO6 promoted gastric cancer cell tumorigenicity through regulation of C-myc expression44; likewise, FOXO6 was also shown to be highly overexpressed in both breast cell lines and tumor tissues45, indicating that FOXO6 may act as an oncogene in BC. Although one GWAS study identified a locus on HIVEP3 that shares the same location 1p34.2 as FOXO641, FOXO6 rs61229336 is not in LD with the previously published HIVEP3 rs79724016 (Figure S6b) without functional analysis. In the present study, we found that rs61229336 T allele might down-regulate expression of the likely oncogenic FOXO6, leading to a reduced BC risk. Nevertheless, we did not have additional experimental data that could explain how FOXO6 rs61229336 C>T influenced BC risk.
It should be pointed out that there are several limitations in the present study. First of all, the small sample sizes in some of DRIVE GWAS studies, such as WAABCS with African Americans, were not sufficient for stratification analysis for ethnic difference in genetic background and therefore excluded from the analysis. Thus, the genotype data available for the analysis consisted of non-Hispanic whites only; therefore, the results may not be generalizable to the general population. Second, several known risk factors, such as physical activities, unhealthy lifestyle and reproductive factors, prevented us from performing complete adjustment in the analyses. Finally, the publically available data for the eQTL analysis was very limited, and gene expression analysis of biological samples from the participants in the DRIVE study was not performed, because relevant data from the samples of target breast tissues were not available.
In summary, we analyzed the associations between genetic variants in 55 FOXO pathway-related genes and BC risk by using genotyping data from 14 previously published GWASs of 53,107 participants of European descendants in the DRIVE study. We identified three independent BC susceptibility loci in FOXO pathway genes (i.e., FBXO32 rs10093411 and rs62521280 at 8q24.13, and FOXO6 rs61229336 at 1p34.2), which may help with the identification of individuals at high-risk of developing BC, once these SNPs are further validated by other studies. Our findings offer some new evidence for genetic variants in the FOXO pathway as possible biomarkers that may provide additional insights into molecular biological mechanisms underlying the observed associations with BC risk.
Supplementary Material
Acknowledgement
DRIVE (dbGaP Study Accession: phs001265.v1.p1): The genotyping and phenotype data obtained from the Discovery, Biology, and Risk of Inherited Variants in Breast Cancer (DRIVE), breast-cancer case control samples was supported by U19 CA148065 and X01 HG007491 and by Cancer Research UK (C1287/A16563). Genotyping was performed by the Centre for Inherited Disease Research (CIDR), University of Cambridge, Centre for Cancer Genetic Epidemiology, and the National Cancer Institute. Germline DNA for breast cancer cases and controls were provided by the following studies: the Two Sister Study (2SISTER), Breast Oncology Galicia Network (BREOGAN), Copenhagen General Population Study (CGPS), Cancer Prevention Study 2 (CPSII), The European Prospective Investigation into Cancer and Nutrition (EPIC), Melbourne Collaborative Cohort Study (MCCS), Multi-ethnic Cohort (MEC), Nashville Breast Health Study (NBHS), Nurses’ Health Study (NHS), Nurses’ Health Study 2 (NHS2), Polish Breast Cancer Study (PBCS), Prostate Lung Colorectal and Ovarian Cancer Screening Trial (PLCO), Studies of Epidemiology and Risk Factors in Cancer Heredity (SEARCH), The Sister Study (SISTER), Swedish Mammographic Cohort (SMC), Women of African Ancestry Breast Cancer Study (WAABCS), Women’s Health Initiative (WHI).
GTEx: The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data described in this manuscript were obtained from: GTEx Analysis v8.p2 (dbGaP Accession phs000424.v8.p2).
Grant sponsor:
Duke Cancer Institute’s P30 Cancer Center Support Grant;
Grant number:
NIH CA014236.
Abbreviations:
- BC
breast cancer
- BFDP
Bayesian false-discovery probability
- BREOGAN
Breast Oncology Galicia Network
- CGPS
Copenhagen General Population Study
- CI
confidence interval
- CPSII
Cancer Prevention Study-II Nutrition Cohort
- DRIVE
Discovery, Biology, and Risk of Inherited Variants in Breast Cancer
- EPIC
European Prospective Investigation Into Cancer and Nutrition
- eQTL
expression quantitative trait loci
- FBXO32
F-box protein 32
- FOXO
Forkhead box O
- FOXO6
Forkhead box O6
- GWAS
genome-wide association study
- LD
linkage disequilibrium
- MAF
minor allele frequency
- MCCS
Melbourne Collaborative Cohort Study
- MEC
Multiethnic Cohort
- NBHS
Nashville Breast Health Study
- NHS
Nurses’ Health Study
- NHS2
Nurses’ Health Study 2
- NRG
number of risk genotype
- OR
odds ratio
- PBCS
NCI Polish Breast Cancer Study
- PCs
principal components
- PLCO
The Prostate, Lung, Colorectal and Ovarian Cancer Screening Trial
- SEARCH
Study of Epidemiology and Risk factors in Cancer Heredity
- SMC
Swedish Mammography Cohort
- SNP
single nucleotide polymorphism
- WHI
Women’s Health Initiative
Footnotes
Conflict of interest: The authors state no conflict of interest.
Data Availability Statement:
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
References
- 1.Harkness EF, Astley SM, Evans DG. Risk-based breast cancer screening strategies in women. Best Pract Res Clin Obstet Gynaecol. 2020;65:3–17. [DOI] [PubMed] [Google Scholar]
- 2.DeSantis CE, Ma J, Gaudet MM, et al. Breast cancer statistics, 2019. CA Cancer J Clin. 2019;69(6):438–451. [DOI] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30. [DOI] [PubMed] [Google Scholar]
- 4.Jiang X, Finucane HK, Schumacher FR, et al. Shared heritability and functional enrichment across six solid cancers. Nat Commun. 2019;10(1):431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zavala VA, Serrano-Gomez SJ, Dutil J, Fejerman L. Genetic Epidemiology of Breast Cancer in Latin America. Genes (Basel). 2019;10(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma J, Matkar S, He X, Hua X. FOXO family in regulating cancer and metabolism. Semin Cancer Biol. 2018;50:32–41. [DOI] [PubMed] [Google Scholar]
- 7.Myatt SS, Lam EW. The emerging roles of forkhead box (Fox) proteins in cancer. Nat Rev Cancer. 2007;7(11):847–859. [DOI] [PubMed] [Google Scholar]
- 8.Eijkelenboom A, Burgering BM. FOXOs: signalling integrators for homeostasis maintenance. Nat Rev Mol Cell Biol. 2013;14(2):83–97. [DOI] [PubMed] [Google Scholar]
- 9.Deng Y, Wang F, Hughes T, Yu J. FOXOs in cancer immunity: Knowns and unknowns. Semin Cancer Biol. 2018;50:53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tenbaum SP, Ordóñez-Morán P, Puig I, et al. β-catenin confers resistance to PI3K and AKT inhibitors and subverts FOXO3a to promote metastasis in colon cancer. Nat Med. 2012;18(6):892–901. [DOI] [PubMed] [Google Scholar]
- 11.Sykes SM, Lane SW, Bullinger L, et al. AKT/FOXO signaling enforces reversible differentiation blockade in myeloid leukemias. Cell. 2011;146(5):697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Naka K, Hoshii T, Muraguchi T, et al. TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature. 2010;463(7281):676–680. [DOI] [PubMed] [Google Scholar]
- 13.Gan B, Lim C, Chu G, et al. FoxOs enforce a progression checkpoint to constrain mTORC1-activated renal tumorigenesis. Cancer Cell. 2010;18(5):472–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paik JH, Kollipara R, Chu G, et al. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell. 2007;128(2):309–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hornsveld M, Tenhagen M, van de Ven RA, et al. Restraining FOXO3-dependent transcriptional BMF activation underpins tumour growth and metastasis of E-cadherin-negative breast cancer. Cell Death Differ. 2016;23(9):1483–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim CG, Lee H, Gupta N, et al. Role of Forkhead Box Class O proteins in cancer progression and metastasis. Semin Cancer Biol. 2018;50:142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hornsveld M, Smits LMM, Meerlo M, et al. FOXO Transcription Factors Both Suppress and Support Breast Cancer Progression. Cancer Res. 2018;78(9):2356–2369. [DOI] [PubMed] [Google Scholar]
- 18.López-Cortés A, Leone PE, Freire-Paspuel B, et al. Mutational Analysis of Oncogenic AKT1 Gene Associated with Breast Cancer Risk in the High Altitude Ecuadorian Mestizo Population. Biomed Res Int. 2018;2018:7463832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rizk SM, Shahin NN, Shaker OG. Association between SIRT1 Gene Polymorphisms and Breast Cancer in Egyptians. PLoS One. 2016;11(3):e0151901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ge J, Liu H, Qian D, et al. Genetic variants of genes in the NER pathway associated with risk of breast cancer: A large-scale analysis of 14 published GWAS datasets in the DRIVE study. Int J Cancer. 2019;145(5):1270–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Howie B, Marchini J, Stephens M. Genotype imputation with thousands of genomes. G3 (Bethesda). 2011;1(6):457–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang J, Lee SH, Goddard ME, Visscher PM. Genome-wide complex trait analysis (GCTA): methods, data analyses, and interpretations. Methods Mol Biol. 2013;1019:215–236. [DOI] [PubMed] [Google Scholar]
- 23.Wakefield J A Bayesian measure of the probability of false discovery in genetic epidemiology studies. Am J Hum Genet. 2007;81(2):208–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21(2):263–265. [DOI] [PubMed] [Google Scholar]
- 25.Pruim RJ, Welch RP, Sanna S, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26(18):2336–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40(Database issue):D930–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lappalainen T, Sammeth M, Friedlander MR, et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013;501(7468):506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gibson G Human genetics. GTEx detects genetic effects. Science. 2015;348(6235):640–641. [DOI] [PubMed] [Google Scholar]
- 29.Aloraifi F, McDevitt T, Martiniano R, et al. Detection of novel germline mutations for breast cancer in non-BRCA1/2 families. FEBS J. 2015;282(17):3424–3437. [DOI] [PubMed] [Google Scholar]
- 30.Xu X, Li J, Zou J, et al. Association of Germline Variants in Natural Killer Cells With Tumor Immune Microenvironment Subtypes, Tumor-Infiltrating Lymphocytes, Immunotherapy Response, Clinical Outcomes, and Cancer Risk. JAMA Netw Open. 2019;2(9):e199292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sahu SK, Tiwari N, Pataskar A, et al. FBXO32 promotes microenvironment underlying epithelial-mesenchymal transition via CtBP1 during tumour metastasis and brain development. Nat Commun. 2017;8(1):1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frescas D, Pagano M. Deregulated proteolysis by the F-box proteins SKP2 and beta-TrCP: tipping the scales of cancer. Nat Rev Cancer. 2008;8(6):438–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Skaar JR, Pagan JK, Pagano M. SnapShot: F box proteins I. Cell. 2009;137(6):1160–1160 e1161. [DOI] [PubMed] [Google Scholar]
- 34.Mei Z, Zhang D, Hu B, Wang J, Shen X, Xiao W. FBXO32 Targets c-Myc for Proteasomal Degradation and Inhibits c-Myc Activity. J Biol Chem. 2015;290(26):16202–16214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo W, Zhang M, Shen S, et al. Aberrant methylation and decreased expression of the TGF-β/Smad target gene FBXO32 in esophageal squamous cell carcinoma. Cancer. 2014;120(16):2412–2423. [DOI] [PubMed] [Google Scholar]
- 36.Chou JL, Su HY, Chen LY, et al. Promoter hypermethylation of FBXO32, a novel TGF-beta/SMAD4 target gene and tumor suppressor, is associated with poor prognosis in human ovarian cancer. Lab Invest. 2010;90(3):414–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yuan X, Zhang Z, Jiang K, Wang X, Li Y. Preliminary Study of the Role F-Box Protein 32 (FBXO32) in Colorectal Neoplasms Through the Transforming Growth Factor beta (TGF-β)/Smad4 Signalling Pathway. Med Sci Monit. 2018;24:1080–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo W, Zhang M, Guo Y, Shen S, Guo X, Dong Z. FBXO32, a new TGF-β/Smad signaling pathway target gene, is epigenetically inactivated in gastric cardia adenocarcinoma. Neoplasma. 2015;62(4):646–657. [DOI] [PubMed] [Google Scholar]
- 39.Wu Z, Lee ST, Qiao Y, et al. Polycomb protein EZH2 regulates cancer cell fate decision in response to DNA damage. Cell Death Differ. 2011;18(11):1771–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou H, Liu Y, Zhu R, et al. FBXO32 suppresses breast cancer tumorigenesis through targeting KLF4 to proteasomal degradation. Oncogene. 2017;36(23):3312–3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Michailidou K, Lindstrom S, Dennis J, et al. Association analysis identifies 65 new breast cancer risk loci. Nature. 2017;551(7678):92–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jacobs FM, van der Heide LP, Wijchers PJ, Burbach JP, Hoekman MF, Smidt MP. FoxO6, a novel member of the FoxO class of transcription factors with distinct shuttling dynamics. J Biol Chem. 2003;278(38):35959–35967. [DOI] [PubMed] [Google Scholar]
- 43.Li Q, Tang H, Hu F, Qin C. Silencing of FOXO6 inhibits the proliferation, invasion, and glycolysis in colorectal cancer cells. J Cell Biochem. 2019;120(3):3853–3860. [DOI] [PubMed] [Google Scholar]
- 44.Qinyu L, Long C, Zhen-dong D, et al. FOXO6 promotes gastric cancer cell tumorigenicity via upregulation of C-myc. FEBS Lett. 2013;587(14):2105–2111. [DOI] [PubMed] [Google Scholar]
- 45.Lallemand F, Petitalot A, Vacher S, et al. Involvement of the FOXO6 transcriptional factor in breast carcinogenesis. Oncotarget. 2018;9(7):7464–7475. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.