

Abstract
Neuropathic pain is a major symptom of multiple sclerosis (MS) with up to 92% of patients reporting bodily pain, and 85% reporting pain severe enough to cause functional disability. None of the available therapeutics target MS pain. Toll-like receptors 2 and 4 (TLR2/TLR4) have emerged as targets for treating a wide array of autoimmune disorders, including MS, as well as having demonstrated success at suppressing pain in diverse animal models. The current series of studies tested systemic TLR2/TLR4 antagonists in males and females in a low-dose myelin oligodendrocyte glycoprotein (MOG) experimental autoimmune encephalitis (EAE) model, with reduced motor dysfunction to allow unconfounded testing of allodynia through 50+ days post-MOG. The data demonstrated that blocking TLR2/TLR4 suppressed EAE-related pain, equally in males and females; upregulation of dorsal spinal cord proinflammatory gene expression for TLR2, TLR4, NLRP3, interleukin-1β, IkBα, TNF-α and interleukin-17; and upregulation of dorsal spinal cord expression of glial immunoreactivity markers. In support of these results, intrathecal interleukin-1 receptor antagonist reversed EAE-induced allodynia, both early and late after EAE induction. In contrast, blocking TLR2/TLR4 did not suppress EAE-induced motor disturbances induced by a higher MOG dose. These data suggest that blocking TLR2/TLR4 prevents the production of proinflammatory factors involved in low dose EAE pathology. Moreover, in this EAE model, TLR antagonists were highly effective in reducing pain, whereas motor impairment, as seen in high dose MOG EAE, is not affected.
Keywords: Dark Agouti rats, C57Bl6/J mice, sex differences, myelin oligodendrocyte glycoprotein (MOG), allodynia, motor disturbances
Introduction
Multiple Sclerosis (MS) is a debilitating and costly disease of the central nervous system (CNS), affecting approximately 400,000 people in the United States (Medical Surveillance Monthly Report, 2000; Tullman, 2013). In addition to the motor and sensory disturbances, MS patients demonstrate significant other symptoms, including anxiety, depression, reduced social interaction, cognitive dysfunction, and neuropathic pain (Filippi and Rocca, 2013; Hirsh et al., 2009; Svendsen et al., 2005; Wallin et al., 2006). All of these symptoms have been shown to be related to ongoing CNS inflammation (Berger, 2016; Ceruti, 2018; Phillips et al., 2014; Seixas et al., 2016). Specifically, as a result of inflammation and demyelination, MS is associated with elevated CNS levels of pro-inflammatory cytokines, including nod-like receptor protein 3 (NLRP3) inflammasome-dependent release of interleukin-1β (IL-1β) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB)-dependent release of tumor necrosis factor-α (TNF-α). In addition, there are elevated CNS levels of pro-inflammatory T cell-dependent cytokines such as interferon-γ (IFN-γ) and IL-17 (Mallucci et al., 2015; Vincenzi et al., 2013). Together, such changes have been causally linked to expression of MS-induced motor dysfunction as well as MS-related neuropathic pain (Berger, 2016; Ceruti, 2018; Compston and Coles, 2008; Mallucci et al., 2015; Phillips et al., 2014; Seixas et al., 2016).
Many pharmacotherapies for MS have been developed, such as fingolimod (FTY720) and dimethyl fumarate. However, these therapies, despite being targeted to classical symptoms of MS, are only modestly effective for these outcomes (Graetz et al., 2018) and are limited by severe side effects for many patients, including increased pain (Wicks et al., 2016). Most importantly, none of these therapies have been demonstrated clinically to treat neuropathic pain or other symptoms associated with MS in patients.
Neuropathic pain is a particularly devastating symptom of MS with up to 92% of MS patients reporting bodily pain, and 85% of those patients reporting that this pain is severe enough to cause functional disability (Hirsh et al., 2009). Moreover, although both male and female MS patients report neuropathic pain, pain is more prevalent in female MS patients (Solaro et al., 2018), thus warranting a need to study sex differences associated with pain in MS. Current therapies for neuropathic pain in MS are nearly identical to therapies associated with other types of neuropathic pain, including drugs such as antidepressants, anticonvulsants, cannabinoids, and opioids (Nielsen et al., 2018; Solaro et al., 2018). Unfortunately, all are limited in their efficacy while also causing significant side effects, as is also the case for treatment of other types of neuropathic pain (Nielsen et al., 2018; Solaro et al., 2018).
We have previously shown that spinally-targeted, intrathecal immunomodulatory treatments such as anti-inflammatory interleukin-10 (IL-10) gene therapy and adenosine A2A receptor agonists are effective at reversing symptoms associated with experimental autoimmune encephalitis (EAE), a rodent model of MS (Grace et al., 2017; Loram et al., 2015; Sloane et al., 2009b; van Strien et al., 2010). These approaches are also effective in reversing neuropathic pain arising as a consequence of peripheral nerve injury (Dengler et al., 2014; Ledeboer et al., 2007; Loram et al., 2013, 2009; Milligan et al., 2006, 2005a, 2005b; Sloane et al., 2009a; Soderquist et al., 2010b, 2010a) and centrally-derived neuropathic pain (Kwilasz et al., 2018a). The mechanism of action is considered to be blockage pro-inflammatory cytokine production through inhibition of NF-κB activation (Loram et al., 2013; Vincenzi et al., 2013) and production of the anti-inflammatory cytokine, IL-10, shifting the balance of cytokine release from pro-inflammatory to anti-inflammatory (Koscsó et al., 2013; Loram et al., 2009). Although these treatment strategies may prove to be successful in human MS patients, their clinical utility is primarily limited by their need for an intrathecal route of administration.
As an alternative strategy to suppress pro-inflammatory cytokine production, Toll-like receptors 2 and 4 (TLR2/TLR4) have emerged as an important target for the treatment of a wide array of autoimmune disorders, including MS (Liu et al., 2014). Specifically, TLR2/TLR4 expression is upregulated in both MS and EAE, ligands for these TLRs are upregulated in MS and EAE demyelinating lesions, and TLR2 activation has been found to suppress oligodendrocyte precursor cell maturation and remyelination as well as enhance pro-inflammatory Th17 responses while suppressing anti-inflammatory T regulatory cells (Andersson et al., 2008; Hossain et al., 2018; Liu et al., 2014; Nyirenda et al., 2015; Sloane et al., 2010). While spinal TLR2/TLR4 involvement in EAE-related pain has not previously been examined, these TLRs are well documented to mediate other rodent peripheral and central neuropathic pain states (Ellis et al., 2016, 2014; Grace et al., 2016; Hutchinson et al., 2008), suggestive that these TLRs may have a role in central neuropathic pain from EAE as well.
TLR2/TLR4 are receptors primarily found on immune and glial cells and are responsible for detecting endogenous and exogenous signals associated with danger and pathogens. In MS, molecular factors known as danger associated molecular patterns (DAMPs) are present such as breakdown products and released mediators from myelin and other damaged/inflamed cells due to ongoing inflammation and demyelination (Cieślak et al., 2011; Piccinini and Midwood, 2010; Varhaug et al., 2017). Upon activation of TLR2/TLR4 via DAMPS, the NLRP3 inflammasome complex can be formed, leading to increased release of pro-inflammatory cytokines including IL-1β (Broz and Dixit, 2016; Haneklaus and O’Neill, 2015). IL-1β in particular has been shown to be necessary for the symptoms of MS and EAE (Acharjee et al., 2013; Burm et al., 2016; Gao et al., 2016; Gentile et al., 2016; Lalor et al., 2011; Lévesque et al., 2016; Paré et al., 2017; Prins et al., 2013), including EAE-related pain (Rodrigues et al., 2016), although other molecules such as TNF-α (Dutra et al., 2013; Rodrigues et al., 2016) and IL-17 (Hu et al., 2018) have been implicated in EAE-related pain, as well. Thus, studying systemic administration of blood-brain barrier permeable therapeutics that can block the actions of IL-1β and other pro-inflammatory cytokines may prove to be beneficial for the treatment MS.
(+)-Naltrexone [(+)-NTX], and (+)-naloxone [(+)-NLX] are (+)-isomers of the classical opioid receptor antagonists, (−)-NTX and (−)-NLX. As classical opioid receptors are strictly stereoselective for (−)-isomers (Hutchinson et al., 2011), (+)-NTX and (+)-NLX do not block opioid receptors but rather have recently been shown to act as TLR2/TLR4 antagonists in various behavioral and cellular models (Kwilasz et al., 2020, 2018b). XT-203 is a recently developed (+)-isomer derived based on the chemistry of (+)-NTX and (+)-NLX (Hutchinson et al., 2008; Selfridge et al., 2015; Theberge et al., 2013). Importantly, all of these compounds are blood-brain barrier permeable and thus active upon systemic administration. This circumvents the need for complicated routes of administration associated with other immunomodulatory approaches such as A2A receptor agonists or IL-10 gene therapy, which require intrathecal administration. We have previously shown that blockade of TLR2/TLR4 via systemic (+)-NTX and/or (+)-NLX administration reverses mechanical allodynia in several rodent models of neuropathic pain, including pain of both peripheral (Hutchinson et al., 2008; Lewis et al., 2012) and central origin (Ellis et al., 2014; Wieseler et al., 2017). In the present study, we explore whether antagonism by TLR2/TLR4 antagonists can treat both EAE-related pain and classical motor symptoms associated with EAE in male and female Dark Agouti (DA) rats and female C57BL6/J mice.
Materials and Methods
Subjects
Subjects were male and female Dark Agouti rats (225–250g; Envigo) and female C57BL6/J mice (19–22 g; Jackson) between 10–12 weeks old. Rats were housed two per cage and mice five per cage on a 12-hour light/dark cycle (lights on at 0700 hour). Experiments were conducted between 0800 and 1600 hours. All procedures were conducted in accordance with protocols approved by the University of Colorado Boulder Institutional Animal Care and Use Committee.
MOG administration
Upon arrival, the rats were randomly assigned to either the myelin oligodendrocyte glycoprotein (MOG) or saline group. For Experiments 1–8, the MOG assigned rats received a 4 μg injection of recombinant rat MOG1–125 (VU University Medical Center, Netherlands, gifted by Dr. Anne-Marie Van Dam) in a vehicle consisting of sodium acetate (pH = 3) and incomplete Freund’s adjuvant (Sigma; St. Louis, MO; (Ledeboer et al., 2003). This dose of MOG is a lower dose than that which we have found induces typical EAE motor symptoms, including full hindlimb paralysis/partial upper limb paralysis (i.e. 16 µg, the dose employed for Experiment 9). This low-dose was specifically chosen for use in Experiments 1–8 so to avoid hind limb motor/sensory dysfunction that could confound behavioral assessment of mechanical allodynia by the von Frey test.
In experiments 1 (male rats) and 2 (female rats), rats were assessed for mechanical allodynia on the same days post-MOG administration. Due to higher than expected subject attrition in experiment 2 caused primarily by cerebellar deficits which required euthanasia, a second cohort of female rats was added to experiment 2 to achieve appropriate group sizes in a follow up experiment. In this second cohort of rats, the day 19 timepoint was omitted as there was no significant difference between this timepoint and the other timepoints that were nearest to it (i.e. days 17 and 22) in the first cohort of female rats. The data from these 2 cohorts of female rats were then pooled for experiment 2, thus the day 19 timepoint is omitted in this experiment for statistical analysis, however all other timepoints between Experiment 1 and 2 are the same. The timecourse of testing for Experiments 1 and 2 were chosen specifically to test every few days in an effort to fully characterize the effect of (+)-NTX on mechanical allodynia in both males and females. In the subsequent experiments in male rats (i.e. Experiments 3–6), as timepoints between days 14 and 28 were not statistically significant than one another in the vehicle-treated rats with EAE, the timepoints were chosen to answer the specific questions regarding those experiments. Experiment 3 tested later timepoints post MOG administration, Experiment 4 tested timepoints both after (+)-NTX dosing and before (+)-NTX, Experiment 5 tested XT-203, another TLR4 antagonist, for efficacy against EAE-induced mechanical allodynia, and lastly, Experiment 6 tested whether EAE-induced mechanical allodynia was reversed by intrathecal IL-1ra at day 15 and day 29 post MOG administration, to demonstrate that spinal IL-1 is necessary for low-dose EAE-induced mechanical allodynia. The higher dose (16 µg) was used in Experiment 9 as this one study focused on motor disturbances rather than allodynia. In all cases, the injection was given intradermally at the base of the tail and the syringe left in place for 3 minutes to avoid leakage from the injection site. The rats in the saline group received saline injections following the same procedure. Also in Experiment 9, female C57Bl6/J mice were induced with EAE by subcutaneous injection of 200 µg MOG35–55 (Hooke Labs, Lawrence, MA) (100 µg injected on each flank in 100 µl complete Freund’s adjuvant vehicle), followed 1 and 24 h later by intraperitoneal administration of 400 ng pertussis toxin. Only female and not male C57BL6/J mice were used, as this model of EAE is commercially available, well-characterized, and easily reproducible in female mice for focus on motor disturbances (Hooke Labs, Lawrence, MA). All animals were between 10–12 weeks old upon receiving the injections.
Drugs
The antagonists tested were (+)-NTX (generously gifted by Dr. Kenner Rice, National Institutes of Health), XT-203 (generously gifted by Dr. Raymond Chavez, Xalud Therapeutics), recombinant human IL-1ra (Amgen; Thousand Oaks, CA), and FTY720 (Cayman Chemical; Ann Arbor, MI). Doses of (+)-NTX, IL-1ra, and FTY720 were selected based on previous studies. Additionally, the doses of subcutaneous XT-203 and osmotic minipump continuous infusions with (+)-NLX were also based on estimated in vivo potency differences between (+)-NTX and XT-203 based on in vitro studies (R. Chavez, Unpub. data).
Matching to the expression of mechanical allodynia (Experiments 1–8) or motor scores (Experiment 9), rats were equally divided across drug and vehicle treatment groups post-MOG. For Experiments 1, 2, 4–9, this occurred at two weeks post-MOG. For Experiment 3, which also included an examination of the effect of antagonists on well-established allodynia, matching occurred 5 weeks post-MOG. For experiment 6, this occurred at 2 and 4 weeks post-MOG. Upon matching, drug treatments began and were administered subcutaneously (SC) by acute injections (3 doses/day in saline; Experiments 1–5,7–9) or by osmotic minipump (in 100% DMSO; Experiment 9). The one exception to systemic dosing was Experiment 6, which examined the effect of a single intrathecal challenge with interleukin-1 receptor antagonist (IL-1ra) versus saline, administered either 2 or 4 weeks post MOG.
Systemic dosing by repeated acute injections
In Experiments 1–3, 5, 7–9, the animals received SC (+)-NTX (6 mg/kg) or XT-203 (15 mg/kg) or equivolume saline injections three times daily (0900, 1200, and 1500 hour) for approximately 2 weeks during behavioral testing for a total of 18 mg/kg/day or 45 mg/kg/day, respectively. The volume of each injection administered was 1 ml/kg. In Experiment 4, the same (+)-NTX dosing regimen was used with the exception that on days 17, 23 and 28 post-MOG, a 12 mg/kg dose was delivered at 1200 h following allodynia testing so to allow testing of allodynia prior to the first (+)-NTX dose of the day. The final dose of 6 mg/kg (+)-NTX was then administered at 1500 h, as in the other studies.
Systemic dosing by subcutaneously implanted osmotic minipumps
In Experiment 9, which examined a higher (16 µg) MOG dose to test for antagonist effects on motor paralysis, (+)-NTX and XT-203 were continuously infused SC using an osmotic minipump (Model 2001, Alzet, Cupertino, CA) for 7–14 days. Osmotic minipumps were implanted SC under brief 3% isoflurane anesthesia and became active at the time of implantation. Pumps either delivered DMSO vehicle, 18 mg/kg/day (+)-NTX, or 1 mg/kg FTY720 for rats or DMSO vehicle and 30 mg/kg/day XT-203 for mice. We chose to only use osmotic minipumps in these studies and not the other low-dose MOG studies, as we found that several of the rats/mice participated in self-injurious behavior that we deemed unacceptable for future studies. The rats/mice that participated self-injurious behavior were excluded from the studies presented here.
Acute intrathecal injections
In Experiment 6, intrathecal injections were administered to rats under brief 3% isoflurane anesthesia. The lumbar region was shaved and aseptically cleaned. An 18-gauge guide needle, with the hub removed, was inserted into the L5/L6 intervertebral space. A PE-10 catheter was inserted via the guide needle, pre-marked such that the proximal end of the PE-10 tubing rested over the L4-L6 lumbar spinal cord. Intrathecal injections occurred over 20 seconds (100 µg of IL-1ra in 1 µl followed by 2 µl of sterile saline flush) with a 30 s delay before removing the catheter and guide needle. Each rat was anesthetized for a maximum of 5 min, and none incurred observable neurological damage from the procedure.
Behavioral tests
Motor Scoring
All testing was conducted blind with respect to group assignment. Motor behavior was scored daily in all rats to assess the severity of their EAE symptoms. The motor score quantified physical paralysis and scoring was based on the following designations: 0 = no signs of paralysis, 1 = partial tail paralysis, 2 = full tail paralysis, 3 = hind limb weakness, 4 = partial hind limb paralysis, 5 = full hind limb paralysis, 6 = partial upper limb paralysis. Rats or mice that reached a score of 6 were euthanized if paralysis exceeded one day. Euthanized rats received a score of 7. Animals with a motor score of 3 or higher were excluded from von Frey mechanical allodynia testing to avoid hind limb motor/sensory dysfunction that could confound behavioral testing; however, importantly, all data analyses and figures presenting motor scores throughout this report include subjects that scored 3 or higher in order to properly assess the effects of TLR4 antagonists on this aspect of EAE.
Mechanical Allodynia Testing
All testing was conducted blind with respect to group assignment. Rats received at least three 60-minute habituations to the test environment prior to behavioral testing. The von Frey test was performed as previously described in detail (Chacur et al., 2001; Chaplan et al., 1994; Grace et al., 2010; Milligan et al., 2001a). Assessments were typically made prior to MOG administration, the day after initiation of systemic drugs, and then at multi-day intervals across a timecourse. Von Frey tests for allodynia were undertaken approximately 1 h after the second of three doses of (+)-NTX or vehicle for the day. Assessments were also conducted at 1, 2, and 3 h after intrathecal (IT) administration of interleukin-1 receptor antagonist (IL-1ra). A logarithmic series of 10 calibrated Semmes-Weinstein monofilaments (von Frey hairs; Stoelting, Wood Dale, IL) were applied randomly to the left and right paws to define the threshold stimulus intensity required to elicit a paw withdrawal response. Log stiffness of the hairs ranged from manufacturer-designated 3.61 (0.40 g) to 5.18 (15.14 g) filaments. The behavioral responses were used to calculate absolute threshold (the 50% probability of response) by fitting a Gaussian integral psychometric function using a maximum-likelihood fitting method (Harvey, 1986; Treutwein and Strasburger, 1999), as described previously (Milligan et al., 2000, 2001a). This fitting method allowed parametric analyses to be undertaken (Harvey, 1986; Treutwein and Strasburger, 1999).
Real-time polymerase chain reaction (PCR)
RNA Extraction
Four weeks after MOG administration and after 2 weeks continuous (+)-NTX administration, rats were deeply anesthetized with sodium pentobarbital followed by transcardial perfusion with saline (pH 7.4). A naïve group of DA rats was added as controls for these experiments as previous studies (Giatti et al., 2013) as our own unpublished studies of IFA-treated rats showed no difference in the measured parameters between IFA-treated and naïve rats. Dorsal lumbar spinal cord tissues then dissected, snap-frozen in liquid nitrogen, and then stored at −80°C until time of assay. Dorsal lumbar spinal cord was analyzed as this tissue is directly relevant to hindpaw mechanical allodynia. At the time of assay, dorsal lumbar spinal cord tissues were homogenized on ice with 800 μl of Trizol (Thermo Fischer Scientific; Waltham, MA) and allowed to sit at room temperature for 5 minutes. To separate the organic and aqueous layers, 160 μl of chloroform (Sigma; St. Louis, MO) was added to the samples before vortexing for 15 sec and then incubating at room temperature for 3 minutes. The samples were spun in a 5810R Centrifuge (Eppendorf; Hamburg, Germany) at 12,000 g at 4°C for 15 minutes. The aqueous phase was separated from the organic phase and added in a 1:1 ratio to 100% 2-propanol where the samples incubated at room temperature for 10 minutes. The samples were then centrifuged again at the same temperature and speed for 10 minutes. The supernatant was decanted from each sample, 1 ml of 75% ethanol was added and the samples were spun in the centrifuge at 7,500 g for 5 minutes. The samples were decanted for a second time and the prior step was repeated. After the two cycles, the supernatant was decanted once more and the pellet was allowed to air dry. The pellet was then resuspended in 40 μl of nuclease-free water (Bio-Rad; Hercules, CA). The concentration and purity of each RNA sample was measured using a NanoDrop One (Thermo Fischer Scientific; Waltham, MA).
cDNA Synthesis
The volume of a sample containing 3 μg of RNA was added to polymerase chain reaction (PCR) grade water for a total of 10 μl as calculated from the previously determined RNA concentrations (260/280: 1.8–2.1; 260/230: > 1.8). The samples were then incubated in an iCycler (Bio-Rad; Hercules, CA) at 65°C for 5 minutes in Mastermix 1 consisting of 50% random primers (Thermo Fischer Scientific; Waltham, MA) and 50% dNTP mix (Thermo Fischer Scientific; Waltham, MA). The samples received a quick chill on ice before a second incubation at 25°C for 2 min in Mastermix 2. Mastermix 2 consisted of 66% 5X first strand buffer (Thermo Fischer Scientific; Waltham, MA) and 33% 0.1M DTT (Thermo Fischer Scientific; Waltham, MA). Immediately after the incubation, 1 μl of Superscript II Reverse Transcriptase (Thermo Fischer Scientific; Waltham, MA) was added to each sample. The samples were then incubated at 25°C for 10 min, 42°C for 50 min, 70°C for 15 min, and then maintained at 4°C.
RT-PCR
Samples were analyzed in duplicate on 96-well PCR plates (Bio-Rad; Hercules, CA) in a Real Time System Thermocycler C100Touch (Bio-Rad; Hercules, CA). Each well contained 13 μl SYBR Green mix (Qiagen; Hilden, Germany), 1 μl forward primer, 1 μl reverse primer, 10 μl nuclease free water, and 1 μl sample cDNA. Primer sequences were designed using GenBank, National Center for Biotechnology Information (www.ncbi.nlm.nih.gov) to span exon/exon boundaries and thus exclude amplification of genomic DNA, and they were obtained from Invitrogen. Primers included GAPDH (forward: GGAGAAACCTGCCAAGTATG; reverse: GTCATTGAGAGCAATGCCAG); IL-1β (forward: CCTTGTGCAAGTGTCTGAAG; reverse: GGGCTTGGAAGCAATCCTTA); TLR2 (forward: TGGAGGTCTCCAGGTCAAATC; reverse: ACAGAGATGCCTGGGCAGAAT); TLR4 (forward: TCCCTGCATAGAGGTACTTC; reverse: CACACCTGGATAAATCCAGC); NLRP3 (forward: AGAAGCTGGGGTTGGTGAATT; reverse: GTTGTCTAACTCCAGCATCTG); IL-17 (forward: TCCATCCATGTGCCTGATGC; reverse: ACTCTGAGCCGCAATGAGGA). PCR cycling conditions consisted of a hot start activation of Hot Start Taq DNA polymerase (94°C, 15 min) and 40 cycles of denaturation (95°C, 15 sec), annealing (57°C, 30 sec), and extension (72°C, 30 sec). PCR product was denatured (95°C, 1 min) and annealed (55°C, 1 min) before melt curve analysis was conducted. mRNA levels of IL-1β, TLR2, TLR4, NLRP3, and IL-17 were semi-quantified using the ΔCT method relative to the housekeeping gene GAPDH. There were no group differences in GAPDH expression levels.
Immunohistochemistry.
Four weeks after MOG administration and after 2 weeks continuous (+)-NTX administration, rats were deeply anesthetized with sodium pentobarbital followed by transcardial perfusion with saline (pH 7.4), followed immediately by transcardial perfusion for 5 min with 4% paraformaldehyde (pH 7.4). As in the RT-PCR experiments above, a naïve group of DA rats was added as controls for these experiments as previous studies (Giatti et al., 2013) as our own unpublished studies of IFA-treated rats showed no difference in immunohistochemcial parameters between IFA-treated and naïve rats. Lumbar spinal cords were post-fixed in 4% paraformaldehyde/0.2 M phosphate buffer (pH 7.4) for 24 h and then cryo-protected in increasing concentrations (15, 20, 30%) of sucrose in phosphate buffer (PB, pH 7.4) (Sigma-Aldrich, St. Louis, MO). Spinal cords were blocked in OCT (Fisher Scientific, Waltham, MA), frozen at −80°C, and sectioned at 16 µm on a cryostat (Leica CM1850). Sections were thaw-mounted and stored at −20°C until staining.
For staining, slides were rinsed 3x with phosphate buffered saline (PBS), permeabilized with 0.3% hydrogen peroxide, rinsed 3x in PBS, blocked for 1 h with 10% normal goat serum (NGS), 0.3% Triton-X in PBS, and incubated overnight at 4°C for 24 h in 2% normal goat serum together with primary antibodies at the following dilution ratios: rabbit ionized calcium-binding adapter molecule 1 (Iba1); 1:1000 (Abcam, Cambridge, MA) and rabbit glial fibrillary acidic protein (GFAP); 1:100 (Dako, Carpinteria, CA). Slides were then rinsed 3x with PBS and incubated for 2 h in the secondary antibody at the following dilution ratio: goat anti-rabbit; 1:200, (Jackson Immuno Research, West Grove, PA). Slides were rinsed 3x in PBS, incubated in avidin-biotin complex (ABC) solution at 1:250 dilution (Vector Laboratories, Burlingame, CA) for 2 h, rinsed 3x in PBS, and incubated in inactive 3,3’-diaminobenzidine (DAB) (Sigma-Aldrich, St. Louis, MO) for 10 min. DAB was activated with B-D glucose (10 mg/ml) and slides were incubated for 8 min, rinsed 3x with PBS, and dried overnight. Slides were dehydrated in increasing concentrations of ethanol (50, 70, 95 and 100%), cleared in Citrisolv (Fisher Scientific, Waltham, MA), dried and covered with DPX mountant (Sigma-Aldrich, St. Louis, MO).
Black and white images were acquired using an Olympus BX61 microscope (Olympus America, Center Valley, PA) with Olympus Suite CellSens Dimension software. All images of comparison were taken using the same exposure and other acquisition settings. Images were captured at 20x magnification. To conduct densitometry, images were converted to 32-bit, background subtracted, and then adjusted for threshold while blinded to the treatment conditions in NIH Image J software. Data were expressed as total area positive for staining within the region of interest (i.e. dorsal lumbar spinal cord, 0.75 mm x 0.75 mm). 2–4 images per animal in the region of interest were taken and analyzed individually. Dorsal lumbar spinal cord was analyzed as this tissue is directly relevant to hindpaw mechanical allodynia.
Statistical Analysis
Statistical analyses were conducted using GraphPad Prism v.8.30 software. Behavioral data were analyzed with paired-t tests, area under the curve, one-way ANOVA, and two-way repeated measure ANOVA, as appropriate. Immunohistochemistry data were analyzed by two-way ANOVA and PCR data were analyzed with one-way ANOVA. A significant ANOVA was followed by Tukey’s post hoc test to assess differences between specific experimental groups. For all tests, statistical significance was set to p < 0.05.
Results
Experiment 1. Repeated systemic (+)-naltrexone reverses EAE allodynia in male Dark Agouti rats
To define whether blocking TLR2/TLR4 can suppress EAE allodynia, (+)-NTX was administered systemically across a 2 week period after mechanical allodynia was established (n=5–6/group). Von Frey tests for allodynia were undertaken approximately 1 h after the second of three doses of (+)-NTX or vehicle for the day. Intradermal low-dose (4 µg) MOG-induced robust bilateral mechanical allodynia by day 11 (Figure 1A,B; paired t-test comparing baseline [BL] to day 11: p<0.005 for all groups). Allodynia remained stably expressed in saline treated rats through the end of the timecourse examined (day 28 post-MOG). In contrast, 6 and 12 mg/kg (+)-NTX (3x/day, SC beginning after behavioral testing day 15) reversed bilateral allodynia (main effect of drug: left paw: F(2,13)=41.11 p<0.0001; right paw: F(2,13)=36.88 p<0.001) and did so to a comparable degree (6 and 12 mg/kg (+)-NTX each different from saline (p<0.0001) but not different from each other).
Figure 1. Repeated systemic administration of the TLR2/TLR4 antagonist (+)-Naltrexone [(+)-NTX], beginning 15 days after intradermal myelin oligodendrocyte glycoprotein (MOG), reverses EAE-related pain in male Dark Agouti rats; no effect on motor scores.
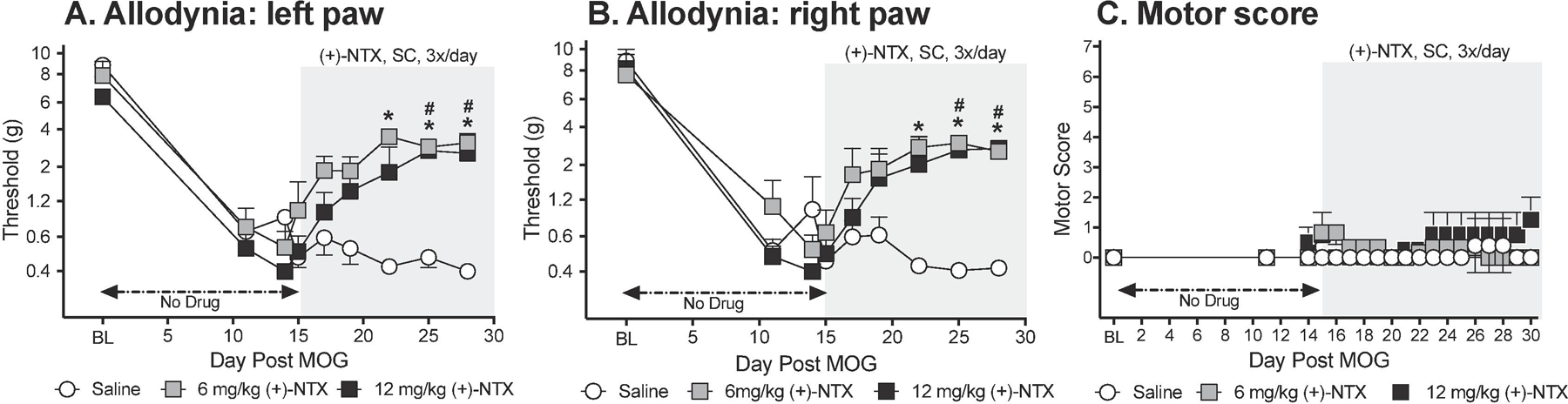
Male Dark Agouti rats were baselined (BL) for motor scores and for low-threshold mechanical withdrawal thresholds via the von Frey test, followed by intra-dermal low-dose (4 µg) myelin oligodendrocyte glycoprotein (MOG). Allodynia and motor scores were assessed thereafter across the timecourse shown. (+)-NTX (6 or 12 mg/kg subcutaneously [SC] 3x/day) vs. saline was initiated on Day 15 post-MOG, continuing through day 30 (see gray rectangle indicating span of drug delivery). Panels A and B: Both doses of (+)-NTX reversed allodynia, relative to saline control, and did so to a comparable degree. Main effect of drug: left paw: F(2,13) =41.11 p<0.0001; right paw: F(2,13) =36.88 p<0.001; posthocs: 6 and 12 mg/kg (+)-NTX each different from saline (p<0.0001) but not different from each other. Panel C: Neither (+)-NTX dose reliably increased motor scores. N=5–6/group.
This low MOG dose created, at most, mild motor effects by day 15 (i.e., prior to (+)-NTX dosing), with at maximum partial-to-full tail paralysis with no hindlimb involvement (Figure 1C). No statistically reliable between group differences in motor scores were observed across the 2 week dosing period.
Experiment 2. Repeated systemic (+)-naltrexone reverses EAE allodynia in female Dark Agouti rats
To define whether the effects observed in male rats (above) would extend to females, the effects of systemic TLR2/TLR4 inhibition were again tested on MOG-induced allodynia and motor scores (n=6/group). Von Frey tests for allodynia were undertaken approximately 1 h after the second of three doses of (+)-NTX or vehicle for the day. As was observed in male rats (above), intradermal low-dose (4 ug) MOG-induced robust bilateral mechanical allodynia in female rats by day 14 (Figure 2A,B; paired t-test comparing baseline [BL] to day 11: p<0.0005, each group). Allodynia remained stably expressed in saline treated female rats through the end of the timecourse examined (day 28 post-MOG). In contrast, 6 mg/kg (+)-NTX (3x/day, SC beginning after behavioral testing day 15) reversed bilateral allodynia (main effect of drug: left paw: F(1,10)=56.23 p<0.0001; right paw: F(1,10)=55.49 p<0.0001).
Figure 2. Repeated systemic administration of the TLR2/TLR4 antagonist (+)-Naltrexone [(+)-NTX], beginning 15 days after intradermal myelin oligodendrocyte glycoprotein (MOG), reverses EAE-related pain in female Dark Agouti rats; no effect on motor scores.
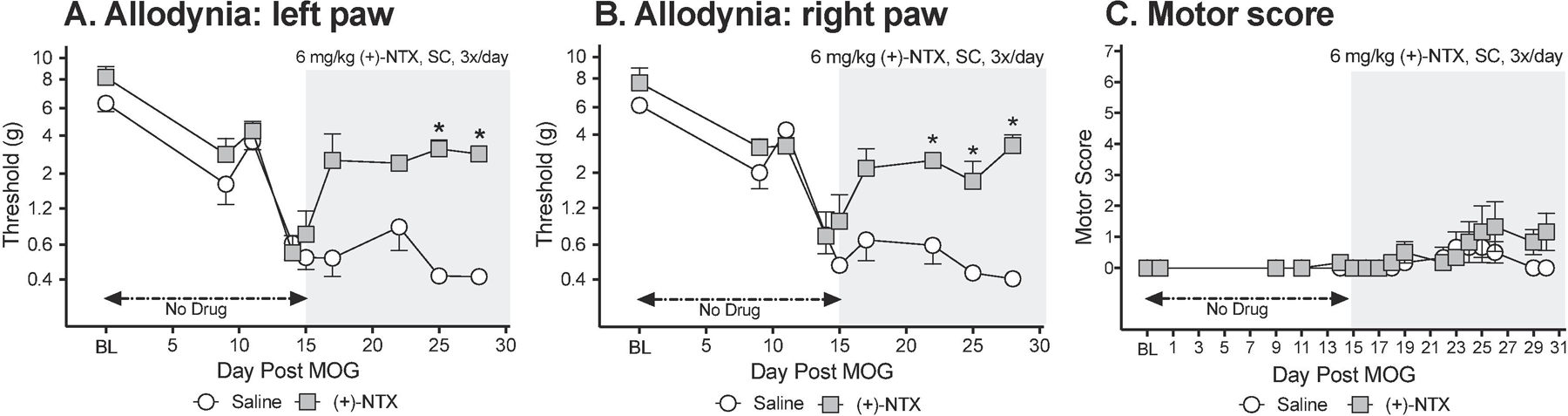
Female Dark Agouti rats were baselined (BL) for motor scores and for low-threshold mechanical withdrawal thresholds via the von Frey test, followed by intra-dermal low-dose (4 µg) myelin oligodendrocyte glycoprotein (MOG). Allodynia and motor scores were assessed thereafter across the timecourse shown. (+)-NTX (6 mg/kg subcutaneously [SC] 3x/day) vs. saline was initiated on Day 15 post-MOG, continuing through day 30 (see gray rectangle indicating span of drug delivery). Panels A and B: (+)-NTX reversed allodynia, relative to saline control. Main effect of drug: (main effect of drug: left paw: F(1,10) =56.23 p<0.0001; right paw: F(1,10) =55.49 p<0.0001. Panel C: (+)-NTX did not reliably increase motor scores. N=6/group.
Similar to the effects observed in male rats (above), the low MOG dose created, at most, mild motor effects by day 15 (i.e., prior to (+)-NTX dosing), with at maximum partial to full tail paralysis with no hindlimb involvement (Figure 2C). No statistically reliable between group differences in motor scores were observed across the 2 week dosing period.
Experiment 3. Repeated systemic (+)-naltrexone reverses well-established EAE allodynia in male Dark Agouti rats
Here, (+)-NTX dosing was delayed until 36 days after MOG (n=6/group) in order to define whether a systemically administered TLR2/TLR4 antagonist could still reverse allodynia when initiation of drug dosing was delayed until long after allodynia was established, Von Frey tests for allodynia were undertaken approximately 1 h after the second of three doses of (+)-NTX or vehicle for the day. Intradermal low-dose (4 ug) MOG-induced robust bilateral mechanical allodynia by day 8 (Figure 3A,B; paired t-test comparing baseline [BL] to day 8: p<0.001, for each group). Allodynia remained stably expressed throughout the timecourse prior to initiation of (+)-NTX dosing on Day 36. In saline treated rats, allodynia remained stably expressed through the end of the timecourse examined (day 50 post-MOG). In contrast, 6 mg/kg (+)-NTX (3x/day, SC beginning after behavioral testing day 36) reversed bilateral allodynia (main effect of drug: left paw: F(1,10) =1452, p<0.0001; right paw: F(1,10) =3956, p<0.0001).
Figure 3. Systemic administration of the TLR2/TLR4 antagonist (+)-Naltrexone [(+)-NTX] beginning 36 days after intradermal myelin oligodendrocyte glycoprotein (MOG), reverses EAE-related pain in male Dark Agouti rats; no effect on motor scores.

Male Dark Agouti rats were baselined (BL) for motor scores and for low-threshold mechanical withdrawal thresholds via the von Frey test, followed by intra-dermal low-dose (4-µg) myelin oligodendrocyte glycoprotein (MOG). Allodynia and motor scores were assessed thereafter across the timecourse shown. (+)-NTX (6 mg/kg subcutaneously [SC] 3x/day) vs. saline was initiated on Day 36 post-MOG, continuing through day 50 (see gray rectangle indicating span of drug delivery). Panels A and B: (+)-NTX reversed allodynia, relative to saline control. Main effect of drug: left paw: F(1,10) =1452, p<0.0001; right paw: F(1,10) =3956, p<0.0001. Panel C: (+)-NTX did not reliably increase motor scores. N=6/group.
This low MOG dose created, at most, mild motor effects by day 36 (i.e., prior to (+)-NTX dosing), with at maximum partial to full tail paralysis with no hindlimb involvement (Figure 3C). Little-to-no motor effects were observed in either the saline or (+)-NTX group through the 4 week dosing period, with no reliable between group differences.
Experiment 4. Repeated systemic (+)-naltrexone provides overnight resolution of EAE allodynia in male Dark Agouti rats
One of the challenges for adequate pain relief is whether a therapeutic can provide overnight pain control without additional dosing. Given that Experiments 1–3 tested allodynia approximately 1 h after the second (+)-NTX dose of the day, the data above do not clarify whether resolution of allodynia would extend overnight. Given that the half-life of (+)-NTX and (+)-NLX 1.2 and 1.5 h in blood after SC administration, respectively (Lewis et al., 2012), duration of pain resolution is an open issue. Hence, in the present study, low-dose MOG was again administered (n=4/group), inducing robust bilateral allodynia by day 14 (Figure 4A,B; paired t-test comparing baseline [BL] to day 14: p<0.01, for each group). (+)-NTX (6 mg/kg) vs. saline was then initiated SC, with 3 doses per day, as in prior studies. Allodynia was stably maintained throughout the 2 week dosing period in the group receiving saline. In response to (+)-NTX, bilateral allodynia was suppressed across the 2 week regimen (main effect of drug: left paw: F(1,6) =431.5, p<0.0001; right paw: F(1,6) =173.4, p<0.0001). Notably, von Frey thresholds measured in the (+)-NTX group prior to the first drug dose of the day (17-pre, 23-pre, 28-pre) were no different than those measured approximately 1 h after the second (+)-NTX of the same day (Figure 4A,B). Hence, overnight suppression of allodynia consistently and robustly occurred with (+)-NTX administration.
Figure 4. Systemic administration of the TLR2/TLR4 antagonist (+)-Naltrexone [(+)-NTX] beginning 14 days after intradermal myelin oligodendrocyte glycoprotein (MOG), reverses EAE-related pain and provides overnight pain relief in male Dark Agouti rats.
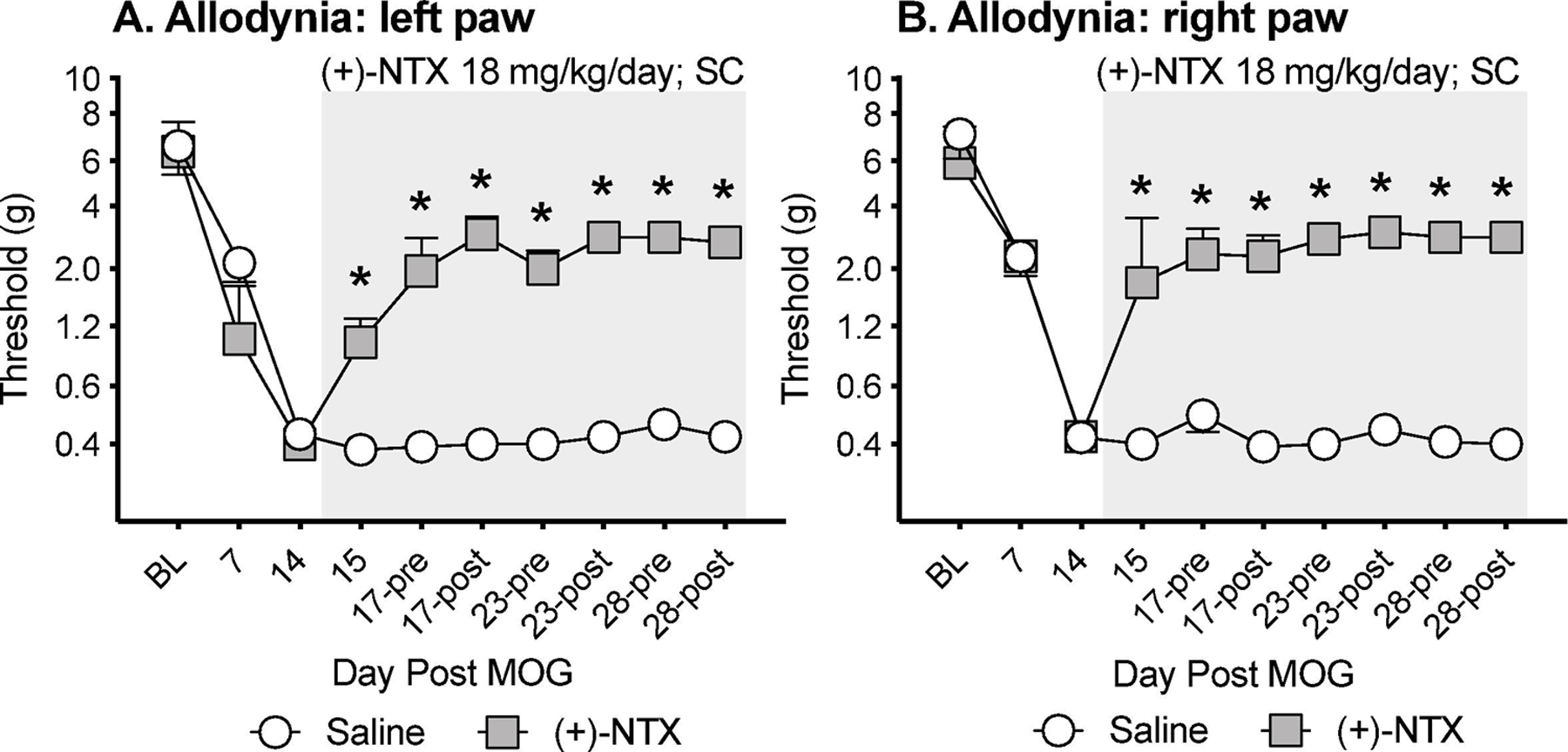
Male Dark Agouti rats were baselined (BL) for low-threshold mechanical withdrawal thresholds via the von Frey test, followed by intra-dermal low-dose (4-µg) myelin oligodendrocyte glycoprotein (MOG). Allodynia was assessed thereafter across the timecourse shown. (+)-NTX (6 mg/kg subcutaneously [SC] 3x/day) vs. saline was initiated on Day 14 post-MOG, continuing through day 28 (see gray rectangle indicating span of drug delivery). Panels A and B: (+)-NTX reversed allodynia, relative to saline control (main effect of drug: left paw: F(1,6) =431.5, p<0.0001; right paw: F(1,6) =173.4, p<0.0001). Notably, withdrawal thresholds measured prior to each day’s initiation of (+)-NTX dosing on days 17, 23 and 28 (17-pre, 23-pre, 28-pre) were comparable to withdrawal measures post-(+)-NTX on those same days supportive of overnight pain suppression.
Experiment 5. Repeated systemic XT-203 reverses EAE allodynia in male Dark Agouti rats
XT-203 is a potent, blood-brain permeable TLR4 antagonist, derived from the (+)-NTX parent compound. However, whether XT-203 also inhibits TLR2 is unknown. To test whether a second TLR4 antagonist could resolve EAE-induced allodynia, the design of Experiment 1 was repeated, now substituting 15 mg/kg XT-203 3x/day SC for (+)-NTX (n=5/group). This dose was derived from independent efficacy testing in mouse pain models (data not published/shown). As before, low-dose MOG-induced robust bilateral allodynia prior to initiation of XT-203 dosing (Figure 5A,B; paired t-test comparing baseline [BL] to day 14: p<0.0005, for each group). While allodynia remained stable in the saline treated group throughout the 2 week dosing period, bilateral allodynia resolved in response to XT-203 dosing (main effect of drug: left paw: F(1,8) =11.39, p<0.01; right paw: F(1,8) =75.8, p<0.0001).
Figure 5. Systemic administration of the TLR4 antagonist XT-203 beginning 15 days after intradermal myelin oligodendrocyte glycoprotein (MOG), reverses EAE-related pain in male Dark Agouti rats.
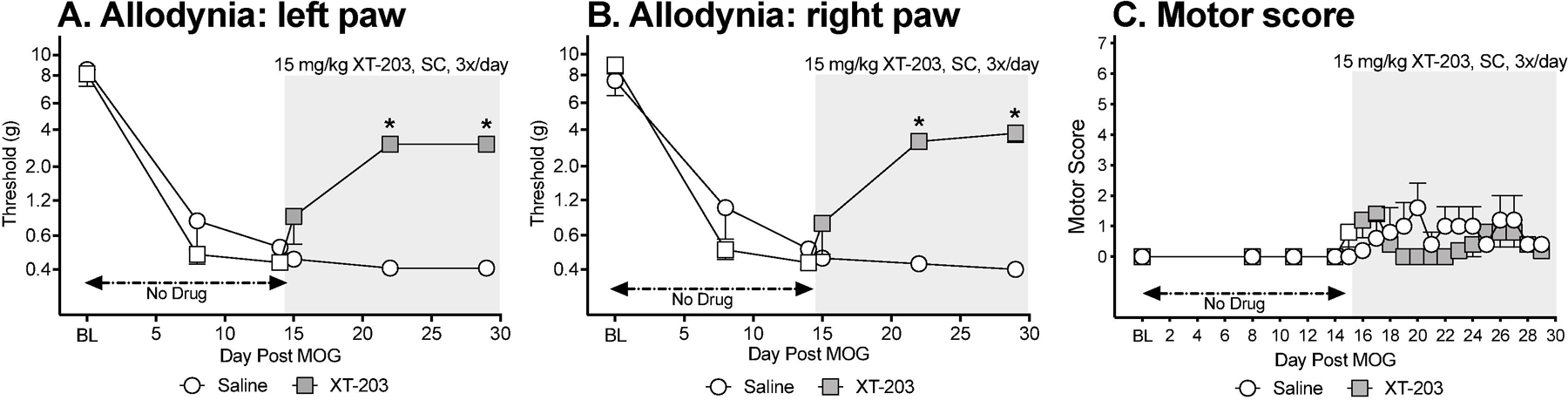
Male Dark Agouti rats were baselined (BL) for motor scores and for low-threshold mechanical withdrawal thresholds via the von Frey test, followed by intra-dermal low-dose (4 µg) myelin oligodendrocyte glycoprotein (MOG). Allodynia and motor scores were assessed thereafter across the timecourse shown. XT-203 (15 mg/kg subcutaneously [SC] 3x/day) vs. saline was initiated on Day 15 post-MOG, continuing through day 30 (see gray rectangle indicating span of drug delivery). Panels A and B: XT-203 reversed allodynia, relative to saline control. Main effect of drug: left paw: F(1,8) =11.39, p<0.01; right paw: F(1,8) =75.8, p<0.0001. Panel C: (+)-NTX did not reliably increase motor scores. N=5/group.
This low MOG dose created, at most, mild motor effects by day 15 (i.e., prior to XT-203 dosing; Figure 5C). No between group differences in motor scores in response to XT-203 vs. saline were observed, with motor scores remaining very mild across time.
Experiment 6. Early and late EAE allodynia are both mediated by spinal interleukin-1β in male Dark Agouti rats
Given that activation of either TLR2 or TLR4 can lead to the production and release of interleukin-1β (IL-1β) (Bowie and O’Neill, 2000) and that spinal IL-1β can induce allodynia (Milligan et al., 2003, 2001b), the question arises as to whether suppression of allodynia by TLR2/TLR4 antagonists may predict that EAE allodynia is mediated, at least in part, by IL-1β at the level of the spinal cord. If so, might IL-1β may be involved in allodynia both early after allodynia induction as well as in well-established pain. To test this, low-dose MOG was administered (n=7–8/group), which induced robust bilateral allodynia prior to antagonist challenge in separate groups of rats on day 15 or 29 (Figure 6A,B: paired t-test comparing baseline [BL] to day 15: p<0.0001, for each group; Figure 6C,D: paired t-test comparing BL to day 29: p<0.0001, for each group). Rats were then challenged with a single intrathecal dose of IL-1 receptor antagonist (IL-1ra). Allodynia was assessed 1, 2, and 3 h after. This single IL-1ra dose reliably suppressed both early (main effect of drug: left paw: F(1,13) =72.96, p<0.0001; right paw: F(1,13) =12.21, p<0.005) and well-established allodynia (main effect of drug: left paw: F(1,12) =148, p<0.0001; right paw: F(1,12) =50.38, p<0.0001), supportive that EAE allodynia is persistently mediated by the release of spinal IL-1β. These results support the potential that systemic TLR2/TLR4 antagonists act, at least in part, by suppressing IL-1β release at spinal levels.
Figure 6. Intrathecal (IT) interleukin-1 receptor antagonist (IL-1ra) administered either 15 days (Panels A,B) or 29 days (Panels C,D) after intradermal myelin oligodendrocyte glycoprotein (MOG), reverses EAE-related pain in male Dark Agouti rats.
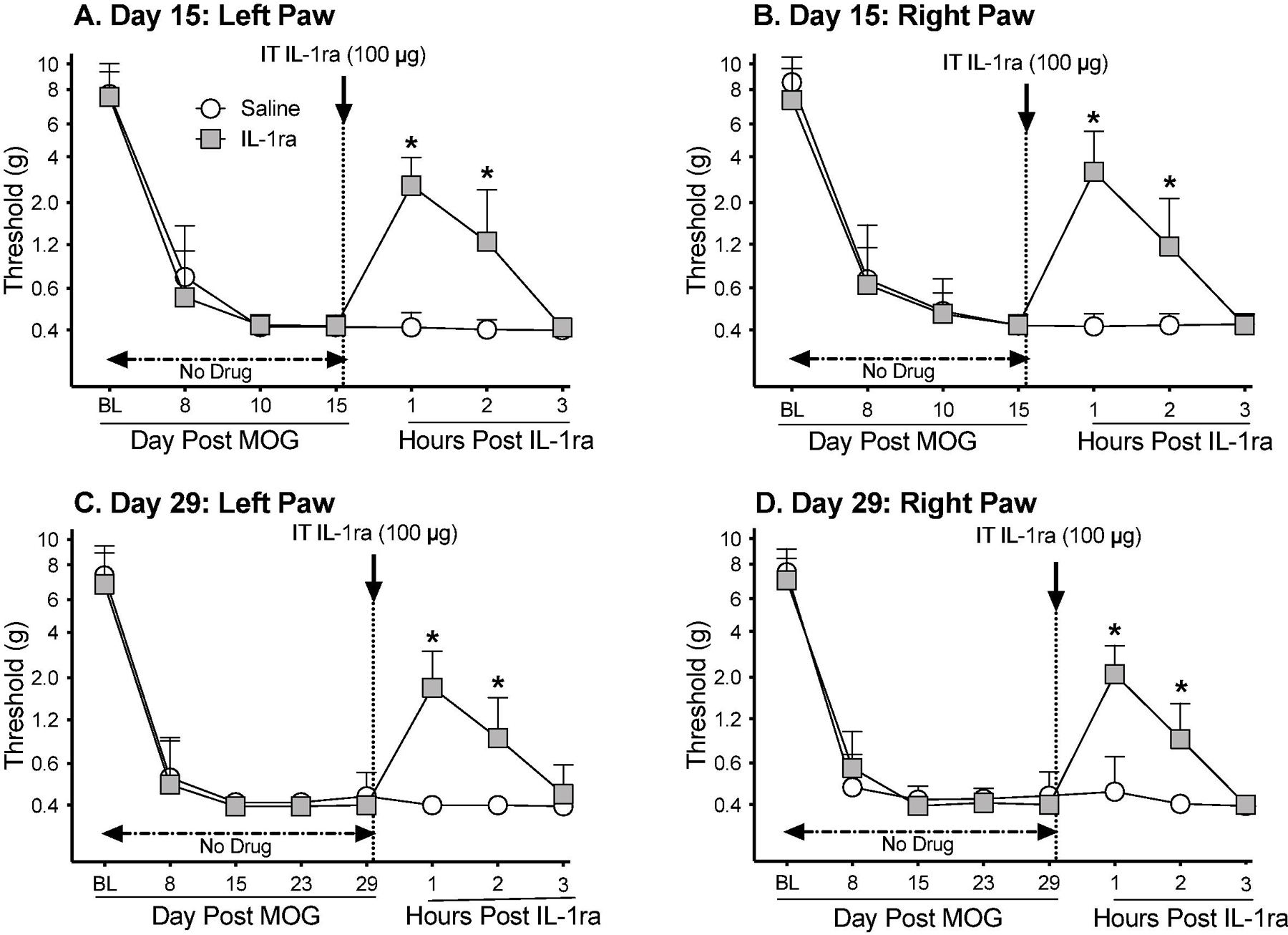
Male Dark Agouti rats were baselined (BL) for low-threshold mechanical withdrawal thresholds via the von Frey test, followed by intra-dermal low-dose (4 µg) myelin oligodendrocyte glycoprotein (MOG). Allodynia was assessed thereafter across the timecourse shown. IT IL-1ra (100 µg) vs. saline was administered either on Day 15 or Day 29 post-MOG. Panels A and B: IL-1ra reversed allodynia when administered Day 15 post-MOG, relative to saline control (main effect of drug: left paw: F(1,13) =72.96, p<0.0001; right paw: F(1,13) =12.21, p<0.005). Panels C and D: IL-1ra also reversed allodynia when administered Day 29 post-MOG, relative to saline control (main effect of drug: left paw: F(1,12) =148, p<0.0001; right paw: F(1,12) =50.38, p<0.0001).
Experiment 7. EAE allodynia increases dorsal lumbar spinal cord neuroinflammatory gene expression in male Dark Agouti rats, effects suppressed by (+)-Naltrexone
The results of Experiment 6 (above) indicate that EAE induces a proinflammatory milieu in dorsal lumbar spinal cord, leading to induction of allodynia. The present study sought to characterize the proinflammatory gene expression changes in dorsal lumbar spinal cord in response to MOG and whether these changes are suppressed by (+)-NTX, as would be anticipated were TLR2/TLR4 to drive these MOG-induced spinal gene expression changes. (+)-NTX (6 mg/kg; 3x/day) began day 16 post-MOG with tissues collected upon 10 days of (+)-NTX dosing. Dorsal lumbar spinal cord mRNAs were analyzed for a series of proinflammatory mediators: TLR2, TLR4, IL-1β. NLRP3 (inflammasome component important in TLR2/TLR4 induction of IL-1β), IκBα (reflection of NFκB activation), TNF-α and IL-17. As evident in Figure 7A-G, dorsal lumbar spinal cord harvested at a day 26 post-MOG where robust allodynia is observed exhibits significant increases in gene expression for every one of the proinflammatory markers assessed (1-way ANOVA comparing naïve to MOG+saline; IL-1β: p<0.005; NLRP3: p<0.001; TLR4: p<0.0001; TLR2: p<0.0005; IL-17: p<0.01; IκBα: p<0.0001; TNF-α: p<0.005), Notably, every one of these proinflammatory changes were suppressed by multi-day treatment with systemic (+)-NTX (1-way ANOVA comparing MOG+saline to MOG+(+)-NTX; IL-1β: p<0.05; NLRP3: p<0.05; TLR4: p<0.05; TLR2: p<0.05; IL-17: p=0.05; IκBα: p<0.001; TNF-α: p<0.05). These results support that EAE allodynia is correlated with a robust enhancement in gene expression for a family of proinflammatory mediators, each of which is suppressed by blockade by systemic delivery of a blood-brain barrier permeable TLR2/TLR4 inhibitor.
Figure 7. Repeated systemic administration of the TLR2/TLR4 antagonist (+)-Naltrexone [(+)-NTX], beginning 16 days after intradermal MOG, with tissue collection after 10 days of (+)-NTX treatment, suppresses EAE-induced neuroinflammatory markers in dorsal spinal cord of male Dark Agouti rats.
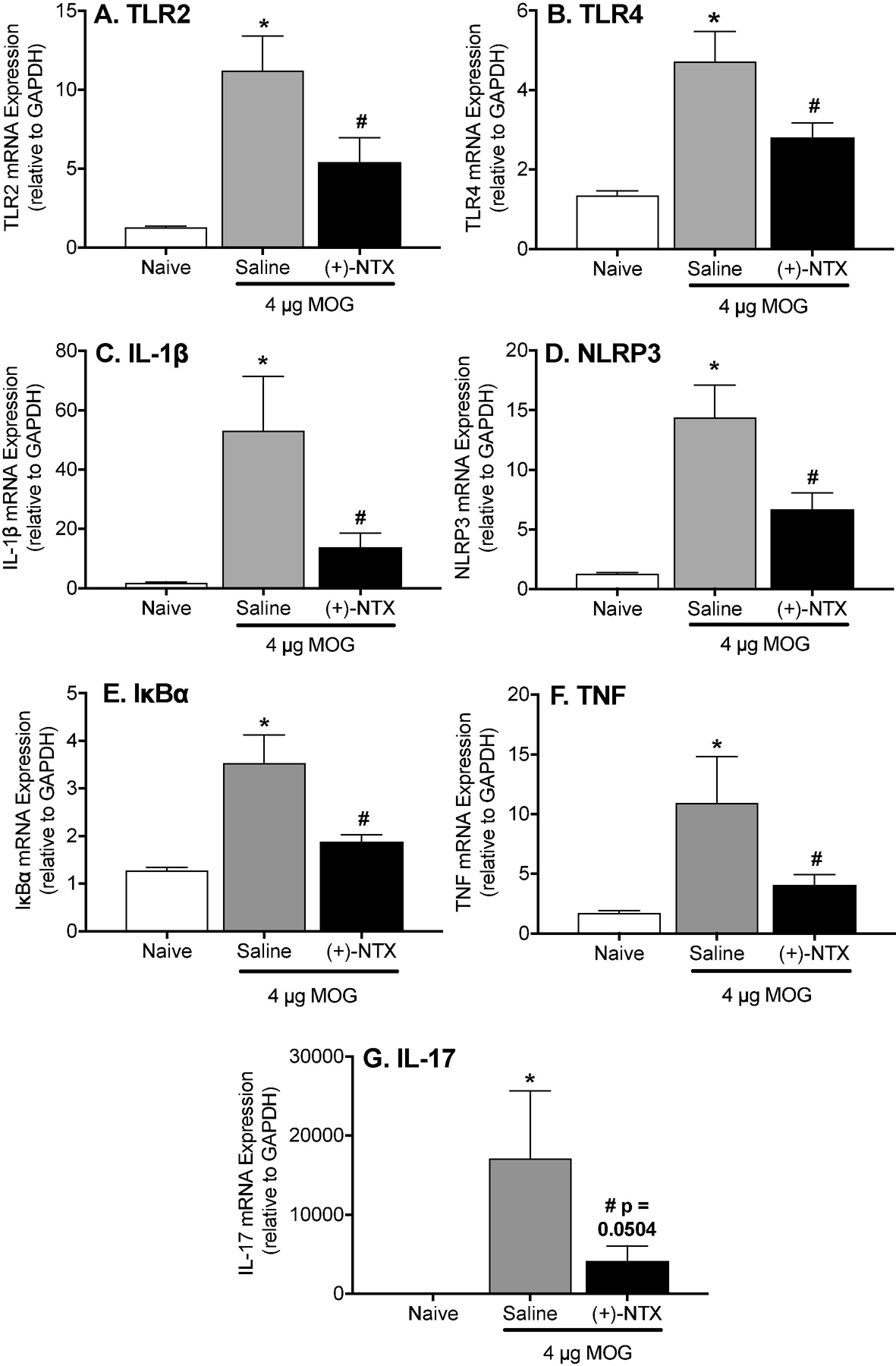
Male Dark Agouti rats received intra-dermal low-dose (4 µg) myelin oligodendrocyte glycoprotein (MOG). (+)-NTX (6 mg/kg subcutaneously [SC] 3x/day) vs. saline was initiated on Day 16 post-MOG, continuing for 10 days at which time tissues were collected for analysis. Each proinflammatory product (A: TLR2; B: TLR4; C: IL-1β; D: NLRP3; E: IκBα (reflective of NFκB activation); F: TNF-α; G: IL-17) was increased in response to EAE (1-way ANOVA comparing naïve to MOG+saline; IL-1β: p<0.005; NLRP3: p<0.001; TLR4: p<0.0001; TLR2: p<0.0005; IL-17: p<0.01; IκBα p<0.0001; TNF-α: p<0.005), and suppressed by (+)-NTX (1-way ANOVA comparing MOG+saline to MOG+(+)-NTX; IL-1β: p<0.05; NLRP3: p<0.05; TLR4: p<0.05; TLR2: p<0.05; IL-17: p=0.05; IκBα: p<0.001; TNF-α: p<0.05).
Experiment 8. EAE allodynia increases dorsal horn lumbar spinal cord glial immunoreactivity in male and female Dark Agouti rats
Experiment 8 utilized the male and female Dark Agouti rats tested for allodynia and motor scoring in Experiments 1 and 2. Tissues were collected on day 30 post-MOG after completion of behavioral testing, while continuing to receive 3x daily (+)-NTX dosing.
For male Dark Agouti rats, MOG increased expression of the microglial immunoreactivity marker (Iba1) relative to naives in the dorsal horn of the lumbar spinal cord (Figure 8A; 1-way ANOVA comparing naïve to MOG+saline: p< 0.0001). This MOG-induced enhancement of Iba1 expression in males was reduced by (+)-NTX (1-way ANOVA comparing MOG+saline to MOG+(+)-NTX: p< 0.05). There was no increase in astrocyte GFAP expression in the dorsal horn of the lumbar spinal cord in males in response to MOG, nor was expression suppressed by (+)-NTX (Figure 8C).
Figure 8. MOG increases the expression of glial immunoreactivity markers in male and female Dark Agouti rats, effects suppressed by (+)-NTX.
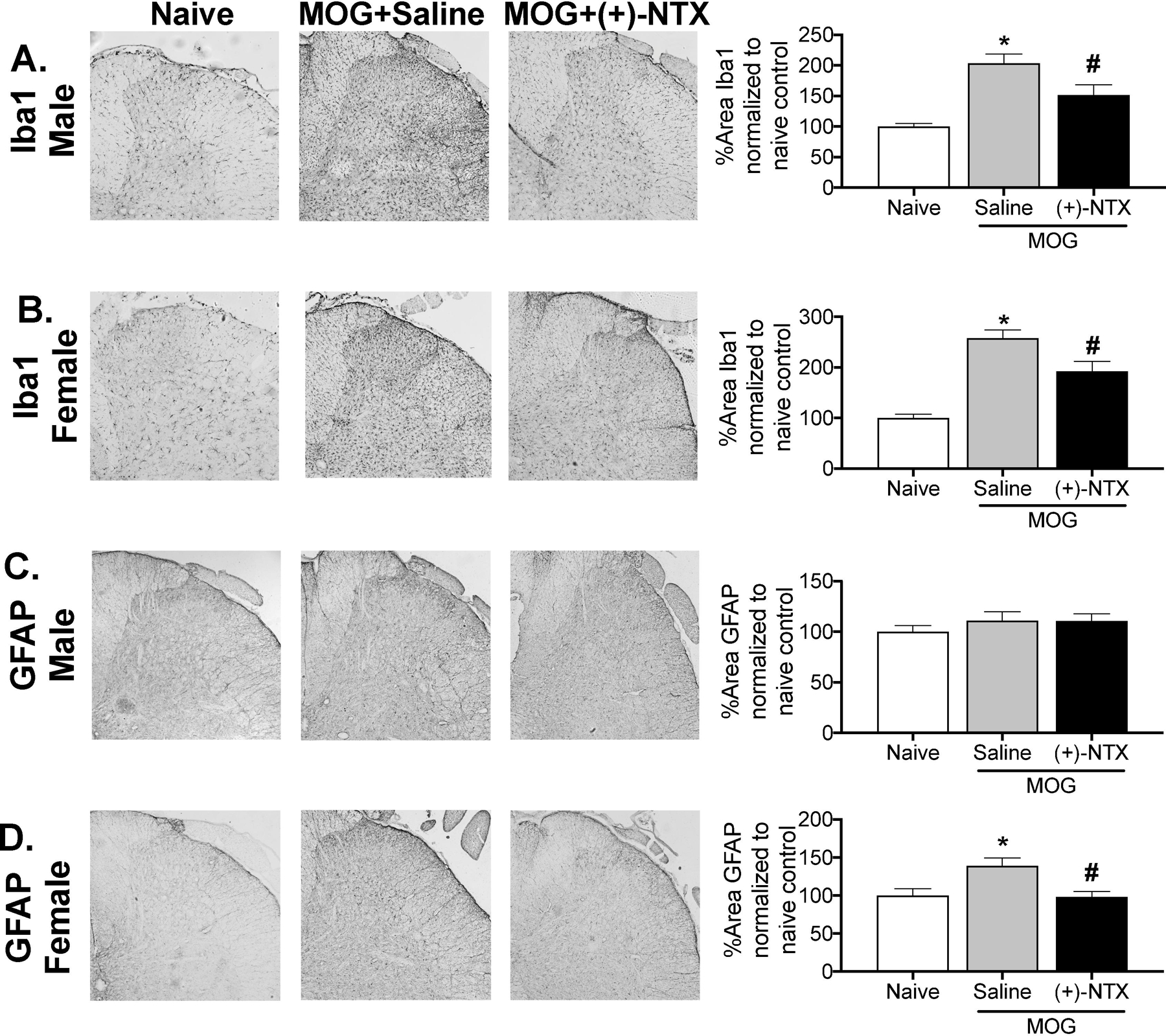
Spinal cord tissues from male and female Dark Agouti rats of Experiments 1 and 2 were collected for analysis here, upon completion of behavioral testing. For male Dark Agouti rats, MOG increased expression of the microglial immunoreactivity marker (Iba1) relative to naives in the dorsal horn of the lumbar spinal cord (Panel A; 1-way ANOVA comparing naïve to MOG+saline: p< 0.0001). This MOG-induced enhancement of Iba1 expression in males was reduced by (+)-NTX (1-way ANOVA comparing MOG+saline to MOG+(+)-NTX: p< 0.05). There was no increase in astrocyte GFAP expression in the dorsal horn of the lumbar spinal cord in males in response to MOG, nor was expression suppressed by (+)-NTX (Panel C). For female Dark Agouti rats, MOG again increased expression of the microglial immunoreactivity marker (Iba1) relative to naives in the dorsal horn of the lumbar spinal cord (Panel B; 1-way ANOVA comparing naïve to MOG+saline: p< 0.0001). As for males, this MOG-induced enhancement of Iba1 expression in females was reduced by (+)-NTX (1-way ANOVA comparing MOG+saline to MOG+(+)-NTX: p< 0.001). Unlike males, MOG increased expression of the astrocyte immunoreactivity marker GFAP relative to naives in the dorsal horn of the lumbar spinal cord (Panel D: 1-way ANOVA comparing naïve to MOG+saline: p< 0.05), an effect again reduced by (+)-NTX (1-way ANOVA comparing MOG+saline to MOG+(+)-NTX: p<0.01). For both male and female rats, n=3–9/group with 2–4 pictures analyzed/subject.
For female Dark Agouti rats, MOG again increased expression of the microglial immunoreactivity marker (Iba1) relative to naives in the dorsal horn of the lumbar spinal cord (Figure 8B; 1-way ANOVA comparing naïve to MOG+saline: p< 0.0001). As for males, this MOG-induced enhancement of Iba1 expression in females was reduced by (+)-NTX (1-way ANOVA comparing MOG+saline to MOG+(+)-NTX: p< 0.001). Unlike males, MOG increased expression of the astrocyte immunoreactivity marker GFAP in the dorsal horn of the lumbar spinal cord (Figure 8D; 1-way ANOVA comparing naïve to MOG+saline: p< 0.05), an effect again reduced by (+)-NTX (1-way ANOVA comparing MOG+saline to MOG+(+)-NTX: p<0.01). For both male and female rats, n=3–9/group with 2–4 pictures analyzed/subject.
Experiment 9. TLR4 antagonists do not reverse motor disturbances associated with standard dose-EAE
To define whether blocking TLR2/TLR4 can suppress EAE-induced motor disturbances in standard-dose EAE models, (+)-NTX or XT-203 was administered systemically in osmotic minipump across a 2 week period beginning after motor disturbances were established (i.e. Day 14 post EAE induction; n=5–9/group). Motor scoring was undertaken each morning at approximately 9:00 AM. Intradermal standard-dose (16 µg) MOG-induced motor disturbances by day 11 in male Dark Agouti rats (Figure 9A; paired t-test comparing baseline [BL] to day 11: p<0.001 for all groups) and subcutaneous standard-dose (200 µg) MOG combined with 400 ng PTX at 1 and 24 h post-MOG-induced motor disturbances by day 10 in female C57BL6 mice (Figure 9B; paired t-test comparing baseline [BL] to day 10: p<0.01 for all groups). Motor disturbances remained stably expressed in MOG-treated animals through the end of the timecourse examined (day 28 post-MOG for rats and day 22 for mice). 18 mg/kg/day (+)-NTX was ineffective to reverse motor disturbances whereas the clinical MS drug FTY720 at 1 mg/kg/day reversed motor disturbances and served as a positive control. Repeated measures two-way ANOVA revealed significant differences between vehicle and FTY720 treatment (main effect of drug: F(2,28)=9.66 p<0.0001); days 22 (p<0.01), 25 (p<0.0001), and 27 (p<0.0001), but did not reveal any significant differences of (+)-NTX treatment. Similar to the results in the rats, 30 mg/kg/day XT-203 did not reverse motor disturbances associated with standard dose EAE in mice.
Figure 9. Standard-dose MOG induces motor disturbances in male Dark Agouti rats and female C57BL6/J mice; no suppression by TLR4 antagonists.
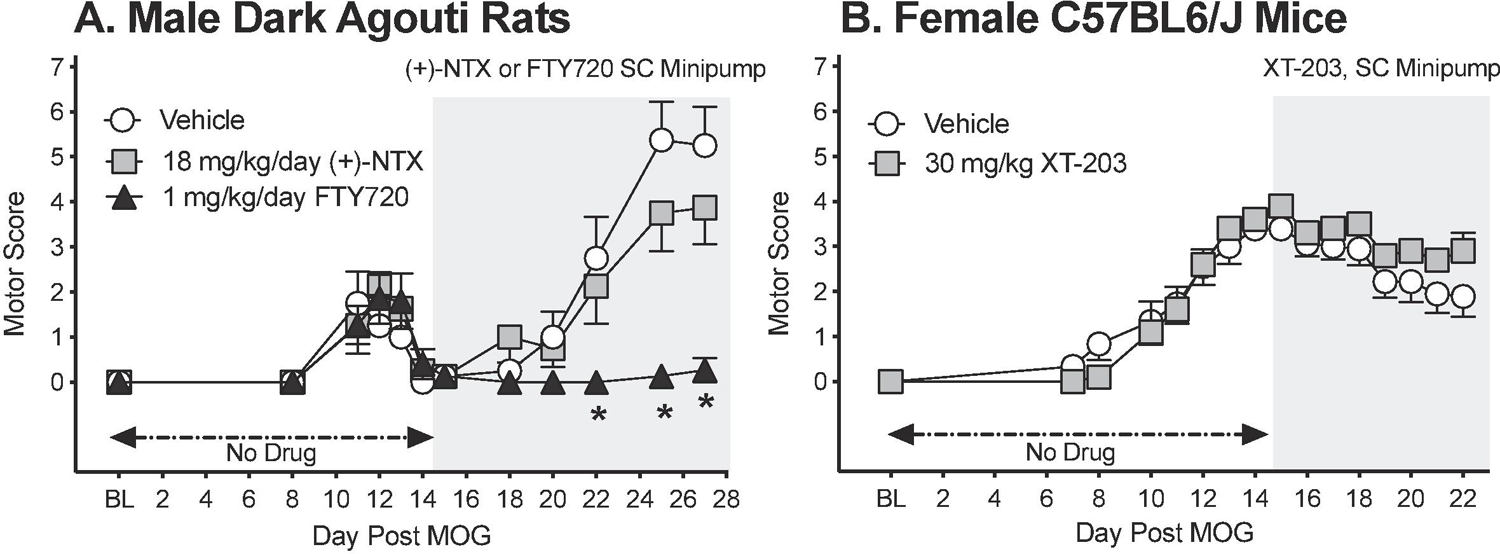
Male Dark Agouti rats and C57BL6/J mice were induced with standard EAE and motor scores were assessed thereafter across the timecourse shown. Panel A: For rats, (+)-NTX (18 mg/kg/day), FTY720 (1 mg/kg/day) or DMSO in SC osmotic minipump was initiated on Day 14 post-MOG, continuing through day 28 (see gray rectangle indicating span of drug delivery). (+)-NTX failed to reverse motor disturbances, whereas the clinically-effective MS drug and positive control FTY720 succeeded to reversed motor disturbances (main effect of drug: F(2,28)=9.66 p<0.0001). Panel B: For mice, XT-203 (30 mg/kg/day) or DMSO was initiated on day 14 post-MOG continuing until day 22 (see gray rectangle indicating span of drug delivery). Similar to the results with (+)-NTX for rats, XT-203 also failed to reverse motor disturbances in mice. N=5–9/group.
Discussion
The present series of studies aimed to address whether TLR2/TLR4 antagonists would reverse mechanical allodynia and motor disturbances associated with low-dose and standard-dose MOG-induced EAE in both male and female rodents. Our data show that 1) mechanical allodynia during low-dose MOG-induced EAE occurs independent of sex, 2) spinal IL-1β contributes to low-dose MOG-induced mechanical allodynia in males 3) antagonism of TLR2/TLR4 reduces low-dose MOG-induced mechanical allodynia but not standard-dose MOG-induced motor disturbances in both males and females, 4) (+)-NTX treatment blocks EAE-induced increases in spinal TLR2 and TLR4 expression, pro-inflammatory cytokines in males, and glial cell immunoreactivity in both males and females, and 5) spinal microglial cells appear to respond similarly, whereas spinal astrocytes respond differently to low-dose MOG-induced EAE in male versus female rats.
The low-dose MOG model employed in the present series of studies as well as in our previous studies (Grace et al., 2017) is advantageous for the study of EAE-associated pain enhancement for several reasons. First, this model recapitulates the typical symptoms of MS where pain and motor symptoms are commonly comorbid (Compston and Coles, 2008), however, motor symptoms are also not severe enough to interfere with movement of the hindpaw that could lead to difficulty detecting mechanical allodynia. Indeed, we show here as others have previously shown (Duffy et al., 2016; Segal et al., 2020; Sloane et al., 2009c) that as motor symptoms become more severe, EAE-related mechanical allodynia becomes more difficult to detect and is associated with high levels of subject attrition/exclusion due to motor scores at 3 or higher on testing day (Figure S1). Second, in our previous studies, we have also shown that this same low-dose EAE model produces memory deficits and is associated with increased serum MOG antibodies (Kwilasz et al., 2020). Collectively, these data suggest that in this low dose EAE Dark Agouti rat model, CNS myelin is under immunological attack by systemic MOG administration that produces anti-MOG antibodies which infiltrate the CNS, ultimately leading to pain, spinal glial cell activation, and motor disturbances. Importantly, the contributions of blood brain barrier breakdown, T cell activation, demyelination, and axonal damage in this model remain to be determined. However, notably, in one study, dorsal horn spinal cord glial activation and EAE-related pain preceded motor disturbances and spinal T cell infiltration, but, after motor symptom onset, pain and glial activation were correlated spinal T cell infiltration (Duffy et al., 2016), suggesting spinal T cell infiltration is likely also occurring the low-dose model presented here. Lastly, a final advantage of the low-dose EAE model presented here is the extended timecourse of EAE-related pain that allows for multi-day assessment of treatment related effects, as well as allowing for drug administration to begin at a clinically-relevant timepoint after the establishment of motor disturbances and EAE-related pain.
Understanding how EAE-related pain is mediated is important given the high percentage of MS patients suffering from such pain (Heitmann et al., 2020). Prior studies of EAE allodynia have, with rare exception (Murphy et al., 2020; Rahn et al., 2014), been restricted to the early expression of the disease state prior to progression of symptoms including hindlimb motor disturbances (Duffy et al., 2016; Rodrigues et al., 2016; Sloane et al., 2009c; Zhang et al., 2019), which make interpretation difficult given the lack of comorbidity of pain and motor symptoms that are common in MS patients (Compston and Coles, 2008). Others have also shown that facial allodynia occurs in the presence of motor disturbances in a standard-dose EAE model, detection of which avoids confounding hindpaw motor impairment (Duffy et al., 2016; Thorburn et al., 2016), but importantly only recapitulates pain from trigeminal-related pathways and not spinally-mediated pathways as is demonstrated here, both of which are common in MS-related pain (Bergamaschi et al., 1994; Compston and Coles, 2008). Recently, it was also demonstrated that by manipulating blood brain barrier permeability with pertussis toxin, a similar low-dose EAE-related pain model could also be conducted without confounding hindpaw motor impairment in mice with a similarly extended timecourse of pain (i.e. 28 days). Given the stability of both reduced motor disturbances and allodynia expression, the present study was able to extend the study of EAE-related pain for 50+ days after MOG dosing, allowing the role of TLR2/TLR4 in pain to be clearly assessed over an extended period of time. Mechanistically, our data demonstrate that spinal IL-1β is necessary for EAE-related mechanical allodynia throughout at least approximately one month post-low-dose MOG, as is indicated by the ability of intrathecal IL-1ra to reverse mechanical allodynia on days 14 and 28 post-MOG. These findings are consistent with a prior study in mice that demonstrated systemic IL-1ra can block mechanical allodynia associated with EAE (Rodrigues et al., 2016) and suggest that inhibition of spinal IL-1β may be one mechanism by which (+)-NTX exerts its therapeutic effects given the ability of (+)-NTX to also reverse spinal IL-1b mRNA at a similar timepoint approximately 1 month post MOG dosing.
The fact that blocking TLR2/TLR4 effectively reverses EAE-induced allodynia, but has no effect on motor disturbances from either low- or standard-dose MOG, may also provide insight into the underlying mechanisms. While speculative, it would seem logical that this profile would be observed if TLR2/TLR4 blockade does not prevent EAE-induced CNS myelin damage but does block downstream neuroinflammatory consequences. In support of this, we have demonstrated that, like standard higher-dose MOG models (Lalive et al., 2011), our low-dose MOG paradigm induces anti-MOG IgG antibodies (Kwilasz et al., 2020). Notably, in the present series of studies, the TLR2/TLR4 antagonists were administered long after MOG dosing, explicitly to avoid their presence during induction of the anti-MOG antibody response. This was done so to allow anti-MOG antibodies and EAE to develop normally. Further, we have confirmed that this TLR2/TLR4 antagonist regimen does not alter blood levels of anti-MOG antibodies (Kwilasz et al., 2020), suggesting that CNS myelin attack is likely still occurring with administration of these drugs (Kinzel and Weber, 2017; Westland et al., 1999). Anti-MOG antibody attack causes the release of alarmins (a specific danger associated molecular pattern [DAMP]) which are endogenous danger signals released in response to cell stress, damage and death (Thundyil and Lim, 2015). Both TLR2 and TLR4 are activated by DAMPs (Bachtell et al., 2015; Jafarzadeh et al., 2019; Kato and Svensson, 2015), and furthermore, allodynia is created by DAMPs in spinal cord (Grace et al., 2018; O’Connor et al., 2003). Thus, taken together, a straightforward interpretation of blockade of allodynia, but no blockade of motor disturbances by TLR2/TLR4 antagonists is that anti-MOG antibodies are causing damage within the spinal cord, leading to the release of DAMPs, and it is at this point that TLR2/TLR4 antagonism is preventing allodynia.
TLR2 and TLR4 recognize a wide array of DAMPs that have been implicated in EAE, including but not limited to heat shock protein 60 (HSP60), HSP70, high mobility group box 1 (HMGB1), alpha-synuclein, hyaluronic acid, fibrinogen, and biglycan (Grace et al., 2018; Jafarzadeh et al., 2019; Kato and Svensson, 2015; Kim et al., 2014; Papadopoulos et al., 2006; Uzawa et al., 2013). These TLRs are expressed within the spinal cord by intrinsic microglia, astrocytes, oligodendrocytes, and endothelial cells (Grace et al., 2014; Leitner et al., 2019; Miranda-Hernandez and Baxter, 2013), as well as dorsal root ganglion (DRG) neurons and DRG satellite glia (T.-T. Wang et al., 2020; Xing et al., 2018; Zhang et al., 2019). In addition, TLR2/TLR4 are expressed by a wide array of infiltrating inflammatory cells, including dendritic cells, lymphocytes (CD4+ T cells, CD8+ T cells, B cells), monocytes, and macrophages (Jafarzadeh et al., 2019; Kato and Svensson, 2015). Hence, which cell type(s) contribute to the resolution of EAE allodynia by TLR2/TLR4 antagonists is/are as yet unclear. However, what is clear is that the downstream effects of blocking TLR2/TLR4 extend well beyond simply preventing the induction and release of typical glial cell-related proinflammatory cytokines, as is indicated here by (+)-NTX blocking EAE-induced increases in TLR2 and TLR4 expression, glial activation, and IL-17 expression. Indeed, these findings are consistent with previous studies that have linked activation of these TLRs to a host of additional EAE- and MS-associated alterations that can contribute to pathology, including but not limited to enhancement of Th1/Th17 cell responses, downregulation of regulatory T cells, induction of IL-17+ γδ T cells, inhibition of oligodendrocyte maturation, and induction of poly ADP-ribose polymerase-1 (PARP-1)-dependent pathways in microglia, macrophages and astrocytes (Besnard et al., 2012; Ferreira et al., 2018; Jafarzadeh et al., 2019; Ni et al., 2020; Srivastava et al., 2018).
While one prior study in mice implicated TLR4-expressing dorsal root ganglion (DRG) neurons in allodynia observed early in EAE prior to motor symptom onset (Zhang et al., 2019), the present series of studies are the first to focus on the role of TLR2/TLR4 in dorsal lumbar spinal cord in mediating neuroinflammatory and EAE-related pain changes occurring in EAE. This extends the demonstrated pain modulatory role of TLR2 and/or TLR4 in diverse enhanced pain states, including but not limited to, peripheral nerve injury (Hutchinson et al., 2008; Jurga et al., 2016; Lewis et al., 2012; Stokes et al., 2013), intrathecal lipopolysaccharide (Hutchinson et al., 2009; Woller et al., 2016), subcutaneous formalin (Woller et al., 2016), bone cancer (Mao-Ying et al., 2012), supradural inflammatory mediators (Su et al., 2018; Wieseler et al., 2017), paclitaxel chemotherapy (Zhang et al., 2016) and spinal cord injury (Ellis et al., 2014).
One aspect that makes the present results with TLR2/TLR4 blockade of EAE allodynia distinct from several prior publications with other pain models is that TLR2/TLR4 blockade suppressed EAE-related pain equally in females and males. This contrasts prior reports that TLR4 mediates pain enhancement in males but not females, where pain enhancement was induced in response to intrathecal lipopolysaccharide (Sorge et al., 2011; Woller et al., 2016), spinal nerve ligation (Stokes et al., 2013), spared nerve injury (Sorge et al., 2011), and inflammation from subcutaneous complete Freund’s adjuvant (Sorge et al., 2011). On the other hand, TLR4 involvement in pain enhancement in females has been implicated in bone cancer (Kong et al., 2017; Li et al., 2013; Liu et al., 2010; Mao-Ying et al., 2012), remifentanil-induced hyperalgesia (Aguado et al., 2018), subcutaneous formalin (Woller et al., 2016), arthritis (Agalave et al., 2014; Miller et al., 2015), intrathecal HMGB1 (Miller et al., 2015), type 2 diabetic neuropathy (Zhang et al., 2013), intrathecal morphine-3-glucoronide (the TLR4 agonist morphine metabolite previously studied in males) (Due et al., 2012; Lewis et al., 2010), and intrathecal lipopolysaccharide (Clark et al., 2010). While what distinguishes models wherein TLR4 is mechanistically important in females versus not is as yet unclear, the growing number of observations of TLR4 involvement in enhanced pain states in females is striking and important.
Taken together from the data presented here, it is clear that TLR2 and/or TLR4 are key players in EAE-related pain both females and males. Whether the same TLR2/TLR4-expressing cell types are involved and/or whether the same downstream mechanisms are recruited is as yet unknown. In males, EAE induced the upregulation of spinal gene expression for TLR2, TLR4, IL-1β NLRP3, IκBα (a reflection of NFkB activation), TNF-α, and IL-17, all of which were suppressed by (+)-NTX treatment. As female tissues were not available for analysis, it remains for future to define whether this profile is mirrored in females. What was defined was that, at the time point of tissue collection (30 days post-MOG), female dorsal spinal cord exhibited upregulation of glial immunoreactivity markers for both microglia (Iba1) and astrocytes (GFAP), whereas in males only an increase in microglial immunoreactivity was observed. Notably, in all cases (microglia and astrocytes in females; microglia in males), the upregulation of immunoreactivity marker expression was suppressed by blocking TLR2/TLR4, providing further support of a key role of these TLRs in females, as well as males. The observation of upregulation of GFAP expression only in females is in contrast to a recent publication wherein increased GFAP expression in spinal cord was observed at day 31 post-MOG only in male mice (Murphy et al., 2020) and, at disease onset, robustly upregulated in both male and female mice, with greater expression in males (Catuneanu et al., 2019). Like the present results here, Iba1 expression has been shown to increase in both sexes in EAE mice, but far more robustly in males than that observed in females at day 31 post-MOG (Murphy et al., 2020).
The demonstration that EAE allodynia is robustly reversed by (+)-NTX and XT-203, including overnight and into the next day without additional dosing, as well as long-after allodynia had been established, provides initial support for considering TLR2/TLR4 as clinically relevant therapeutic targets for central neuropathic pain of MS. (+)-NTX has undergone extensive screening for cross-reactivity to 60+ neurotransmitters and neuropeptides, steroids, ion channels, second messengers, growth factors and hormones, enzymes, neurotransporter binding and activities, etc. with no off-target effects being observed (Theberge et al., 2013). Additionally, screening against a panel of TLRs supported specificity of (+)-NTX for TLR4 and, to a lesser degree, TLR2 (Kwilasz et al., 2020, 2018b). XT-203 is a (+)-isomer based on the chemical structure of, but distinct from, its parent compound, (+)-NTX, designed to improve the pharmacokinetic profile of the parent. XT-203 is in preclinical development as a systemically administered, blood brain barrier permeable TLR4 antagonist. TLR2/TLR4 appear to be logical candidates as targets for MS pain, because: (a) there is no blood brain barrier permeable TLR4 antagonist yet approved by the FDA (Y. Wang et al., 2020), (b) none of the FDA approved drugs for treating MS target TLR2 or TLR4 (Rafiee Zadeh et al., 2019; Taşkapilioğlu, 2018), (c) none of the FDA approved drugs improve resolution of MS pain, but rather many have increased pain as significant side effects (Rafiee Zadeh et al., 2019; Taşkapilioğlu, 2018), and (d) lastly as is presented here, clear evidence of EAE pain resolution by both (+)-NTX and XT-203.
Results with small clinical trials with low-dose (−)-naltrexone for the treatment of MS-related neuropathic pain also provide at least preliminary support for such a clinical approach. While the clinically available form of naltrexone (brand names: Vivitrol, Revia, Depade) is purely the (−)-isomer, we have previously documented that (−)-naltrexone is comparable to (+)-naltrexone in terms of its actions as a TLR4 antagonist (Wang et al., 2016; Zhang et al., 2018). While results of small trials are mixed, there are reports indicating improvements in quality of life and self-reported pain, with subjective benefits over placebo (Cree et al., 2010; Patten et al., 2018). Whether the mechanism of action of low-dose (−)-naltrexone in MS is via TLR4 is unknown, but has been posited (Younger et al., 2013; Younger and Mackey, 2009).
In summary, utilizing a low-dose MOG protocol, the current series of studies demonstrate that systemic administration of TLR2/TLR4 antagonists suppresses EAE-related pain equally in male and female Dark Agouti rats. Blocking TLR2/TLR4 also suppressed upregulation of dorsal lumbar spinal cord proinflammatory gene expression for TLR2, TLR4, IL-1b, NLRP3, IkBa, TNF-α, and IL-17 in male rats, as well as suppressing the upregulation of dorsal lumbar spinal cord expression of glial immunoreactivity markers in both male and female rats. In keeping with this proinflammatory profile, intrathecal IL-1 receptor antagonist reversed EAE-induced allodynia, both early and late in the EAE timecourse in male rats. As blocking TLR2/TLR4 did not suppress EAE-induced motor disturbances in response to a higher MOG dose in either male rats or female mice, TLR2/TLR4 antagonists appear selective in their actions in EAE, specifically to suppress pain rather than motor disturbances.
Supplementary Material
Figure S1. 8 µg MOG induces greater motor disturbances and subject attrition von Frey testing in male Dark Agouti rats. Male Dark Agouti rats were baselined (BL) for motor scores and for low-threshold mechanical withdrawal thresholds via the von Frey test, followed by intra-dermal moderate-dose (8 µg) myelin oligodendrocyte glycoprotein (MOG). Allodynia and motor scores were assessed thereafter across the timecourse shown. Panels A and B: Von Frey testing on left and right hindpaws, n=6/group, but 50% of subjects were excluded on final day of testing due to high motor scores/cerebellar deficits (i.e. Day 28). Panel C: Motor scores. N=6/group (one subject excluded after Day 24 due to cerebellar deficits requiring euthanasia).
Highlights.
*Low-dose MOG produces allodynia without motor disturbances in male and female rats
*TLR2/4 antagonism blocks allodynia but not motor disturbances in male and female rats
*TLR2/4 antagonism blocks pro-inflammatory signals in dorsal lumbar spinal cord
*Low-dose EAE induces astrocyte activation in female rats but not in male rats
Acknowledgements:
This work was supported in part by grants from the National Institute of Neurological Disorders and Stroke (R01NS097313); the University of Colorado Biological Sciences Initiative; and by the intramural Research programs of the National Institute on Drug Abuse and National Institute on Alcohol Abuse and Alcoholism, National Institute on Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acharjee S, Nayani N, Tsutsui M, Hill MN, Ousman SS, Pittman QJ, 2013. Altered cognitive-emotional behavior in early experimental autoimmune encephalitis--cytokine and hormonal correlates. Brain. Behav. Immun 33, 164–172. 10.1016/j.bbi.2013.07.003 [DOI] [PubMed] [Google Scholar]
- Agalave NM, Larsson M, Abdelmoaty S, Su J, Baharpoor A, Lundbäck P, Palmblad K, Andersson U, Harris H, Svensson CI, 2014. Spinal HMGB1 induces TLR4-mediated long-lasting hypersensitivity and glial activation and regulates pain-like behavior in experimental arthritis. Pain 155, 1802–1813. 10.1016/j.pain.2014.06.007 [DOI] [PubMed] [Google Scholar]
- Aguado D, Bustamante R, Gómez de Segura IA, 2018. Toll-like receptor 4 deficient mice do not develop remifentanil-induced mechanical hyperalgesia: An experimental randomised animal study. Eur. J. Anaesthesiol 35, 505–510. 10.1097/EJA.0000000000000803 [DOI] [PubMed] [Google Scholar]
- Andersson A, Covacu R, Sunnemark D, Danilov AI, Dal Bianco A, Khademi M, Wallström E, Lobell A, Brundin L, Lassmann H, Harris RA, 2008. Pivotal advance: HMGB1 expression in active lesions of human and experimental multiple sclerosis. J. Leukoc. Biol 84, 1248–1255. 10.1189/jlb.1207844 [DOI] [PubMed] [Google Scholar]
- Bachtell R, Hutchinson MR, Wang X, Rice KC, Maier SF, Watkins LR, 2015. Targeting the Toll of Drug Abuse: The Translational Potential of Toll-Like Receptor 4. CNS Neurol. Disord. Drug Targets 14, 692–699. 10.2174/1871527314666150529132503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergamaschi R, Romani A, Versino M, Callieco R, Gaspari D, Citterio A, Cosi V, 1994. Usefulness of trigeminal somatosensory evoked potentials to detect subclinical trigeminal impairment in multiple sclerosis patients. Acta Neurol. Scand 89, 412–414. 10.1111/j.1600-0404.1994.tb02658.x [DOI] [PubMed] [Google Scholar]
- Berger T, 2016. Immunological processes related to cognitive impairment in MS. Acta Neurol. Scand 134Suppl 200, 34–38. 10.1111/ane.12647 [DOI] [PubMed] [Google Scholar]
- Besnard A-G, Togbe D, Couillin I, Tan Z, Zheng SG, Erard F, Le Bert M, Quesniaux V, Ryffel B, 2012. Inflammasome-IL-1-Th17 response in allergic lung inflammation. J. Mol. Cell Biol 4, 3–10. 10.1093/jmcb/mjr042 [DOI] [PubMed] [Google Scholar]
- Bowie A, O’Neill LA, 2000. The interleukin-1 receptor/Toll-like receptor superfamily: signal generators for pro-inflammatory interleukins and microbial products. J. Leukoc. Biol 67, 508–514. 10.1002/jlb.67.4.508 [DOI] [PubMed] [Google Scholar]
- Broz P, Dixit VM, 2016. Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunol 16, 407–420. 10.1038/nri.2016.58 [DOI] [PubMed] [Google Scholar]
- Burm SM, Peferoen LAN, Zuiderwijk-Sick EA, Haanstra KG, ‘t Hart BA, van der Valk P, Amor S, Bauer J, Bajramovic JJ, 2016. Expression of IL-1β in rhesus EAE and MS lesions is mainly induced in the CNS itself. J. Neuroinflammation 13, 138. 10.1186/s12974-016-0605-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catuneanu A, Paylor JW, Winship I, Colbourne F, Kerr BJ, 2019. Sex differences in central nervous system plasticity and pain in experimental autoimmune encephalomyelitis. Pain 160, 1037–1049. 10.1097/j.pain.0000000000001483 [DOI] [PubMed] [Google Scholar]
- Ceruti S, 2018. What role does multiple sclerosis play in the development of untreatable painful conditions? Pain Manag 8, 37–44. 10.2217/pmt-2017-0038 [DOI] [PubMed] [Google Scholar]
- Chacur M, Milligan ED, Gazda LS, Armstrong C, Wang H, Tracey KJ, Maier SF, Watkins LR, 2001. A new model of sciatic inflammatory neuritis (SIN): induction of unilateral and bilateral mechanical allodynia following acute unilateral peri-sciatic immune activation in rats. Pain 94, 231–244. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL, 1994. Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 53, 55–63. [DOI] [PubMed] [Google Scholar]
- Cieślak M, Kukulski F, Komoszyński M, 2011. Emerging role of extracellular nucleotides and adenosine in multiple sclerosis. Purinergic Signal 7, 393–402. 10.1007/s11302-011-9250-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AK, Staniland AA, Marchand F, Kaan TKY, McMahon SB, Malcangio M, 2010. P2X7-Dependent Release of Interleukin-1β and Nociception in the Spinal Cord following Lipopolysaccharide. J. Neurosci 30, 573–582. 10.1523/JNEUROSCI.3295-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compston A, Coles A, 2008. Multiple sclerosis. Lancet Lond. Engl 372, 1502–1517. 10.1016/S0140-6736(08)61620-7 [DOI] [PubMed] [Google Scholar]
- Cree BAC, Kornyeyeva E, Goodin DS, 2010. Pilot trial of low-dose naltrexone and quality of life in multiple sclerosis. Ann. Neurol 68, 145–150. 10.1002/ana.22006 [DOI] [PubMed] [Google Scholar]
- Dengler EC, Alberti LA, Bowman BN, Kerwin AA, Wilkerson JL, Moezzi DR, Limanovich E, Wallace JA, Milligan ED, 2014. Improvement of spinal non-viral IL-10 gene delivery by D-mannose as a transgene adjuvant to control chronic neuropathic pain. J. Neuroinflammation 11, 92. 10.1186/1742-2094-11-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Due MR, Piekarz AD, Wilson N, Feldman P, Ripsch MS, Chavez S, Yin H, Khanna R, White FA, 2012. Neuroexcitatory effects of morphine-3-glucuronide are dependent on Toll-like receptor 4 signaling. J. Neuroinflammation 9, 200. 10.1186/1742-2094-9-200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy SS, Perera CJ, Makker PGS, Lees JG, Carrive P, Moalem-Taylor G, 2016. Peripheral and Central Neuroinflammatory Changes and Pain Behaviors in an Animal Model of Multiple Sclerosis. Front. Immunol 7, 369. 10.3389/fimmu.2016.00369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutra RC, Bento AF, Leite DFP, Manjavachi MN, Marcon R, Bicca MA, Pesquero JB, Calixto JB, 2013. The role of kinin B1 and B2 receptors in the persistent pain induced by experimental autoimmune encephalomyelitis (EAE) in mice: evidence for the involvement of astrocytes. Neurobiol. Dis 54, 82–93. 10.1016/j.nbd.2013.02.007 [DOI] [PubMed] [Google Scholar]
- Ellis A, Grace PM, Wieseler J, Favret J, Springer K, Skarda B, Ayala M, Hutchinson MR, Falci S, Rice KC, Maier SF, Watkins LR, 2016. Morphine amplifies mechanical allodynia via TLR4 in a rat model of spinal cord injury. Brain. Behav. Immun 58, 348–356. 10.1016/j.bbi.2016.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis A, Wieseler J, Favret J, Johnson KW, Rice KC, Maier SF, Falci S, Watkins LR, 2014. Systemic administration of propentofylline, ibudilast, and (+)-naltrexone each reverses mechanical allodynia in a novel rat model of central neuropathic pain. J. Pain Off. J. Am. Pain Soc 15, 407–421. 10.1016/j.jpain.2013.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira TB, Hygino J, Wing AC, Kasahara TM, Sacramento PM, Camargo S, Rueda F, Alves-Leon SV, Alvarenga R, Vasconcelos CC, Agrawal A, Gupta S, Bento CAM, 2018. Different interleukin-17-secreting Toll-like receptor+ T-cell subsets are associated with disease activity in multiple sclerosis. Immunology 154, 239–252. 10.1111/imm.12872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippi M, Rocca MA, 2013. Let’s rehabilitate cognitive rehabilitation in multiple sclerosis. Neurology 81, 2060–2061. 10.1212/01.wnl.0000437311.96160.b3 [DOI] [PubMed] [Google Scholar]
- Gao Q, Zhang Y, Han C, Hu X, Zhang H, Xu X, Tian J, Liu Y, Ding Y, Liu J, Wang C, Guo Z, Yang Y, Cao X, 2016. Blockade of CD47 ameliorates autoimmune inflammation in CNS by suppressing IL-1-triggered infiltration of pathogenic Th17 cells. J. Autoimmun 69, 74–85. 10.1016/j.jaut.2016.03.002 [DOI] [PubMed] [Google Scholar]
- Gentile A, Fresegna D, Musella A, Sepman H, Bullitta S, De Vito F, Fantozzi R, Usiello A, Maccarrone M, Mercuri NB, Lutz B, Mandolesi G, Centonze D, 2016. Interaction between interleukin-1β and type-1 cannabinoid receptor is involved in anxiety-like behavior in experimental autoimmune encephalomyelitis. J. Neuroinflammation 13, 231. 10.1186/s12974-016-0682-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giatti S, Boraso M, Abbiati F, Ballarini E, Calabrese D, Santos-Galindo M, Rigolio R, Pesaresi M, Caruso D, Viviani B, Cavaletti G, Garcia-Segura LM, Melcangi RC, 2013. Multimodal analysis in acute and chronic experimental autoimmune encephalomyelitis. J. Neuroimmune Pharmacol. Off. J. Soc. NeuroImmune Pharmacol 8, 238–250. 10.1007/s11481-012-9385-9 [DOI] [PubMed] [Google Scholar]
- Grace PM, Hutchinson MR, Manavis J, Somogyi AA, Rolan PE, 2010. A novel animal model of graded neuropathic pain: utility to investigate mechanisms of population heterogeneity. J. Neurosci. Methods 193, 47–53. 10.1016/j.jneumeth.2010.08.025 [DOI] [PubMed] [Google Scholar]
- Grace PM, Loram LC, Christianson JP, Strand KA, Flyer-Adams JG, Penzkover KR, Forsayeth JR, van Dam A-M, Mahoney MJ, Maier SF, Chavez RA, Watkins LR, 2017. Behavioral assessment of neuropathic pain, fatigue, and anxiety in experimental autoimmune encephalomyelitis (EAE) and attenuation by interleukin-10 gene therapy. Brain. Behav. Immun 59, 49–54. 10.1016/j.bbi.2016.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace PM, Ramos KM, Rodgers KM, Wang X, Hutchinson MR, Lewis MT, Morgan KN, Kroll JL, Taylor FR, Strand KA, Zhang Y, Berkelhammer D, Huey MG, Greene LI, Cochran TA, Yin H, Barth DS, Johnson KW, Rice KC, Maier SF, Watkins LR, 2014. Activation of adult rat CNS endothelial cells by opioid-induced toll-like receptor 4 (TLR4) signaling induces proinflammatory, biochemical, morphological, and behavioral sequelae. Neuroscience 280, 299–317. 10.1016/j.neuroscience.2014.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace PM, Strand KA, Galer EL, Rice KC, Maier SF, Watkins LR, 2018. Protraction of neuropathic pain by morphine is mediated by spinal damage associated molecular patterns in male rats. Brain. Behav. Immun 72, 45–50. 10.1016/j.bbi.2017.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace PM, Strand KA, Galer EL, Urban DJ, Wang X, Baratta MV, Fabisiak TJ, Anderson ND, Cheng K, Greene LI, Berkelhammer D, Zhang Y, Ellis AL, Yin HH, Campeau S, Rice KC, Roth BL, Maier SF, Watkins LR, 2016. Morphine paradoxically prolongs neuropathic pain in rats by amplifying spinal NLRP3 inflammasome activation. Proc. Natl. Acad. Sci. U. S. A 113, E3441–3450. 10.1073/pnas.1602070113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graetz C, Groppa S, Zipp F, Siller N, 2018. Preservation of neuronal function as measured by clinical and MRI endpoints in relapsing-remitting multiple sclerosis: how effective are current treatment strategies? Expert Rev. Neurother 18, 203–219. 10.1080/14737175.2018.1438190 [DOI] [PubMed] [Google Scholar]
- Haneklaus M, O’Neill LAJ, 2015. NLRP3 at the interface of metabolism and inflammation. Immunol. Rev 265, 53–62. 10.1111/imr.12285 [DOI] [PubMed] [Google Scholar]
- Harvey LO, 1986. Efficient estimation of sensory thresholds. Behav. Res. Methods Instrum. Comput 18, 623–632. [Google Scholar]
- Heitmann H, Haller B, Tiemann L, Mühlau M, Berthele A, Tölle TR, Salmen A, Ambrosius B, Bayas A, Asseyer S, Hartung H-P, Heesen C, Stangel M, Wildemann B, Haars S, Groppa S, Luessi F, Kümpfel T, Nischwitz S, Meuth SG, Klotz L, Linker RA, Zettl UK, Ziemann U, Tumani H, Tackenberg B, Zipp F, Wiendl H, Gold R, Hemmer B, Ploner M, 2020. Longitudinal prevalence and determinants of pain in multiple sclerosis: results from the German National Multiple Sclerosis Cohort study. Pain 161, 787–796. 10.1097/j.pain.0000000000001767 [DOI] [PubMed] [Google Scholar]
- Hirsh AT, Turner AP, Ehde DM, Haselkorn JK, 2009. Prevalence and impact of pain in multiple sclerosis: physical and psychologic contributors. Arch. Phys. Med. Rehabil 90, 646–651. 10.1016/j.apmr.2008.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain MJ, Morandi E, Tanasescu R, Frakich N, Caldano M, Onion D, Faraj TA, Erridge C, Gran B, 2018. The Soluble Form of Toll-Like Receptor 2 Is Elevated in Serum of Multiple Sclerosis Patients: A Novel Potential Disease Biomarker. Front. Immunol 9, 457. 10.3389/fimmu.2018.00457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Huang F, Wang ZJ, 2018. CaMKIIα Mediates the Effect of IL-17 To Promote Ongoing Spontaneous and Evoked Pain in Multiple Sclerosis. J. Neurosci. Off. J. Soc. Neurosci 38, 232–244. 10.1523/JNEUROSCI.2666-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Ramos KM, Loram LC, Wieseler J, Sholar PW, Kearney JJ, Lewis MT, Crysdale NY, Zhang Y, Harrison JA, Maier SF, Rice KC, Watkins LR, 2009. Evidence for a role of heat shock protein-90 in toll like receptor 4 mediated pain enhancement in rats. Neuroscience 164, 1821–1832. 10.1016/j.neuroscience.2009.09.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Shavit Y, Grace PM, Rice KC, Maier SF, Watkins LR, 2011. Exploring the neuroimmunopharmacology of opioids: an integrative review of mechanisms of central immune signaling and their implications for opioid analgesia. Pharmacol. Rev 63, 772–810. 10.1124/pr.110.004135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Zhang Y, Brown K, Coats BD, Shridhar M, Sholar PW, Patel SJ, Crysdale NY, Harrison JA, Maier SF, Rice KC, Watkins LR, 2008. Non-stereoselective reversal of neuropathic pain by naloxone and naltrexone: involvement of toll-like receptor 4 (TLR4). Eur. J. Neurosci 28, 20–29. 10.1111/j.1460-9568.2008.06321.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jafarzadeh A, Nemati M, Khorramdelazad H, Mirshafiey A, 2019. The Toll-like Receptor 2 (TLR2)-related Immunopathological Responses in the Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis. Iran. J. Allergy Asthma Immunol 18, 230–250. 10.18502/ijaai.v18i3.1117 [DOI] [PubMed] [Google Scholar]
- Jurga AM, Rojewska E, Piotrowska A, Makuch W, Pilat D, Przewlocka B, Mika J, 2016. Blockade of Toll-Like Receptors (TLR2, TLR4) Attenuates Pain and Potentiates Buprenorphine Analgesia in a Rat Neuropathic Pain Model. Neural Plast 2016. 10.1155/2016/5238730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato J, Svensson CI, 2015. Role of extracellular damage-associated molecular pattern molecules (DAMPs) as mediators of persistent pain. Prog. Mol. Biol. Transl. Sci 131, 251–279. 10.1016/bs.pmbts.2014.11.014 [DOI] [PubMed] [Google Scholar]
- Kim C, Cho E-D, Kim H-K, You S, Lee H-J, Hwang D, Lee S-J, 2014. β1-integrin-dependent migration of microglia in response to neuron-released α-synuclein. Exp. Mol. Med 46, e91. 10.1038/emm.2014.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinzel S, Weber MS, 2017. The Role of Peripheral CNS-Directed Antibodies in Promoting Inflammatory CNS Demyelination. Brain Sci 7. 10.3390/brainsci7070070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong X, Wei J, Wang D, Zhu X, Zhou Y, Wang S, Xu G-Y, Jiang G-Q, 2017. Upregulation of Spinal Voltage-Dependent Anion Channel 1 Contributes to Bone Cancer Pain Hypersensitivity in Rats. Neurosci. Bull 33, 711–721. 10.1007/s12264-017-0195-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koscsó B, Csóka B, Kókai E, Németh ZH, Pacher P, Virág L, Leibovich SJ, Haskó G, 2013. Adenosine augments IL-10-induced STAT3 signaling in M2c macrophages. J. Leukoc. Biol 94, 1309–1315. 10.1189/jlb.0113043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwilasz AJ, Ellis A, Wieseler J, Loram L, Favret J, McFadden A, Springer K, Falci S, Rieger J, Maier SF, Watkins LR, 2018a. Sustained reversal of central neuropathic pain induced by a single intrathecal injection of adenosine A2Areceptor agonists. Brain. Behav. Immun 10.1016/j.bbi.2018.01.005 [DOI] [PubMed]
- Kwilasz AJ, Todd LS, Duran-Malle J, Schrama AEW, Mitten EH, VanDam A-M, Maier SF, Rice KC, Watkins LR, Barrientos RM, 2018b. The non-opioid TLR2/4 antagonist (+)-naltrexone blocks contextual long-term memory deficits in experimental autoimmune encephalomyelitis and associated neuroinflammation in hippocampus. Proc Soc. Neurosci
- Kwilasz AJ, Todd LS, Schrama AEW, Mitten EH, Larson TA, Clements MA, Harris KM, Litwiler ST, Wang X, Van Dam A-M, Maier SF, Rice KC, Watkins LR, Barrientos RM, 2020. Experimental autoimmune encephalopathy (EAE)-induced hippocampal neuroinflammation and memory deficits are prevented with the non-opioid TLR2/TLR4 antagonist (+)-naltrexone. Behav. Brain Res In Press. [DOI] [PMC free article] [PubMed]
- Lalive PH, Molnarfi N, Benkhoucha M, Weber MS, Santiago-Raber M-L, 2011. Antibody response in MOG(35–55) induced EAE. J. Neuroimmunol 240–241, 28–33. 10.1016/j.jneuroim.2011.09.005 [DOI] [PubMed] [Google Scholar]
- Lalor SJ, Dungan LS, Sutton CE, Basdeo SA, Fletcher JM, Mills KHG, 2011. Caspase-1-processed cytokines IL-1beta and IL-18 promote IL-17 production by gammadelta and CD4 T cells that mediate autoimmunity. J. Immunol. Baltim. Md 1950 186, 5738–5748. 10.4049/jimmunol.1003597 [DOI] [PubMed] [Google Scholar]
- Ledeboer A, Jekich BM, Sloane EM, Mahoney JH, Langer SJ, Milligan ED, Martin D, Maier SF, Johnson KW, Leinwand LA, Chavez RA, Watkins LR, 2007. Intrathecal interleukin-10 gene therapy attenuates paclitaxel-induced mechanical allodynia and proinflammatory cytokine expression in dorsal root ganglia in rats. Brain. Behav. Immun 21, 686–698. 10.1016/j.bbi.2006.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledeboer A, Wierinckx A, Bol JGJM, Floris S, Renardel de Lavalette C, De Vries HE, van den Berg TK, Dijkstra CD, Tilders FJH, van dam A-M, 2003. Regional and temporal expression patterns of interleukin-10, interleukin-10 receptor and adhesion molecules in the rat spinal cord during chronic relapsing EAE. J. Neuroimmunol 136, 94–103. [DOI] [PubMed] [Google Scholar]
- Leitner GR, Wenzel TJ, Marshall N, Gates EJ, Klegeris A, 2019. Targeting toll-like receptor 4 to modulate neuroinflammation in central nervous system disorders. Expert Opin. Ther. Targets 23, 865–882. 10.1080/14728222.2019.1676416 [DOI] [PubMed] [Google Scholar]
- Lévesque SA, Paré A, Mailhot B, Bellver-Landete V, Kébir H, Lécuyer M-A, Alvarez JI, Prat A, de Rivero Vaccari JP, Keane RW, Lacroix S, 2016. Myeloid cell transmigration across the CNS vasculature triggers IL-1β-driven neuroinflammation during autoimmune encephalomyelitis in mice. J. Exp. Med 213, 929–949. 10.1084/jem.20151437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis SS, Hutchinson MR, Rezvani N, Loram LC, Zhang Y, Maier SF, Rice KC, Watkins LR, 2010. Evidence that intrathecal morphine-3-glucuronide may cause pain enhancement via toll-like receptor 4/MD-2 and interleukin-1beta. Neuroscience 165, 569–583. 10.1016/j.neuroscience.2009.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis SS, Loram LC, Hutchinson MR, Li C-M, Zhang Y, Maier SF, Huang Y, Rice KC, Watkins LR, 2012. (+)-naloxone, an opioid-inactive toll-like receptor 4 signaling inhibitor, reverses multiple models of chronic neuropathic pain in rats. J. Pain Off. J. Am. Pain Soc 13, 498–506. 10.1016/j.jpain.2012.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Wang X-W, Feng X-M, Zhou W-J, Wang Y-Q, Mao-Ying Q-L, 2013. Stage-dependent anti-allodynic effects of intrathecal Toll-like receptor 4 antagonists in a rat model of cancer induced bone pain. J. Physiol. Sci. JPS 63, 203–209. 10.1007/s12576-012-0244-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Yang J, Wang L, Jiang M, Qiu Q, Ma Z, Liu L, Li C, Ren C, Zhou J, Li W, 2010. Tibia tumor-induced cancer pain involves spinal p38 mitogen-activated protein kinase activation via TLR4-dependent mechanisms. Brain Res 1346, 213–223. 10.1016/j.brainres.2010.05.014 [DOI] [PubMed] [Google Scholar]
- Liu Y, Yin H, Zhao M, Lu Q, 2014. TLR2 and TLR4 in autoimmune diseases: a comprehensive review. Clin. Rev. Allergy Immunol 47, 136–147. 10.1007/s12016-013-8402-y [DOI] [PubMed] [Google Scholar]
- Loram LC, Harrison JA, Sloane EM, Hutchinson MR, Sholar P, Taylor FR, Berkelhammer D, Coats BD, Poole S, Milligan ED, Maier SF, Rieger J, Watkins LR, 2009. Enduring reversal of neuropathic pain by a single intrathecal injection of adenosine 2A receptor agonists: a novel therapy for neuropathic pain. J. Neurosci. Off. J. Soc. Neurosci 29, 14015–14025. 10.1523/JNEUROSCI.3447-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loram LC, Strand KA, Taylor FR, Sloane E, Van Dam A-M, Rieger J, Maier SF, Watkins LR, 2015. Adenosine 2A receptor agonism: A single intrathecal administration attenuates motor paralysis in experimental autoimmune encephalopathy in rats. Brain. Behav. Immun 46, 50–54. 10.1016/j.bbi.2015.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loram LC, Taylor FR, Strand KA, Harrison JA, Rzasalynn R, Sholar P, Rieger J, Maier SF, Watkins LR, 2013. Intrathecal injection of adenosine 2A receptor agonists reversed neuropathic allodynia through protein kinase (PK)A/PKC signaling. Brain. Behav. Immun 33, 112–122. 10.1016/j.bbi.2013.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallucci G, Peruzzotti-Jametti L, Bernstock JD, Pluchino S, 2015. The role of immune cells, glia and neurons in white and gray matter pathology in multiple sclerosis. Prog. Neurobiol 127–128, 1–22. 10.1016/j.pneurobio.2015.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao-Ying Q-L, Wang X-W, Yang C-J, Li X, Mi W-L, Wu G-C, Wang Y-Q, 2012. Robust spinal neuroinflammation mediates mechanical allodynia in Walker 256 induced bone cancer rats. Mol. Brain 5, 16. 10.1186/1756-6606-5-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medical Surveillance Monthly Report, 2000. Multiple sclerosis, active component, US Armed Forces 12–16.
- Miller RE, Belmadani A, Ishihara S, Tran PB, Ren D, Miller RJ, Malfait A-M, 2015. Damage-Associated Molecular Patterns Generated in Osteoarthritis Directly Excite Murine Nociceptive Neurons through Toll-like Receptor 4. Arthritis Rheumatol. Hoboken NJ 67, 2933–2943. 10.1002/art.39291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan ED, Langer SJ, Sloane EM, He L, Wieseler-Frank J, O’Connor K, Martin D, Forsayeth JR, Maier SF, Johnson K, Chavez RA, Leinwand LA, Watkins LR, 2005a. Controlling pathological pain by adenovirally driven spinal production of the anti-inflammatory cytokine, interleukin-10. Eur. J. Neurosci 21, 2136–2148. 10.1111/j.1460-9568.2005.04057.x [DOI] [PubMed] [Google Scholar]
- Milligan ED, Mehmert KK, Hinde JL, Harvey LO, Martin D, Tracey KJ, Maier SF, Watkins LR, 2000. Thermal hyperalgesia and mechanical allodynia produced by intrathecal administration of the human immunodeficiency virus-1 (HIV-1) envelope glycoprotein, gp120. Brain Res 861, 105–116. [DOI] [PubMed] [Google Scholar]
- Milligan ED, O’Connor KA, Nguyen KT, Armstrong CB, Twining C, Gaykema RP, Holguin A, Martin D, Maier SF, Watkins LR, 2001a. Intrathecal HIV-1 envelope glycoprotein gp120 induces enhanced pain states mediated by spinal cord proinflammatory cytokines. J. Neurosci. Off. J. Soc. Neurosci 21, 2808–2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan ED, O’Connor KA, Nguyen KT, Armstrong CB, Twining C, Gaykema RP, Holguin A, Martin D, Maier SF, Watkins LR, 2001b. Intrathecal HIV-1 envelope glycoprotein gp120 induces enhanced pain states mediated by spinal cord proinflammatory cytokines. J. Neurosci. Off. J. Soc. Neurosci 21, 2808–2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan ED, Sloane EM, Langer SJ, Cruz PE, Chacur M, Spataro L, Wieseler-Frank J, Hammack SE, Maier SF, Flotte TR, Forsayeth JR, Leinwand LA, Chavez R, Watkins LR, 2005b. Controlling neuropathic pain by adeno-associated virus driven production of the anti-inflammatory cytokine, interleukin-10. Mol. Pain 1, 9. 10.1186/1744-8069-1-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan ED, Soderquist RG, Malone SM, Mahoney JH, Hughes TS, Langer SJ, Sloane EM, Maier SF, Leinwand LA, Watkins LR, Mahoney MJ, 2006. Intrathecal polymer-based interleukin-10 gene delivery for neuropathic pain. Neuron Glia Biol 2, 293–308. 10.1017/S1740925X07000488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan ED, Twining C, Chacur M, Biedenkapp J, O’Connor K, Poole S, Tracey K, Martin D, Maier SF, Watkins LR, 2003. Spinal glia and proinflammatory cytokines mediate mirror-image neuropathic pain in rats. J. Neurosci. Off. J. Soc. Neurosci 23, 1026–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda-Hernandez S, Baxter AG, 2013. Role of toll-like receptors in multiple sclerosis. Am. J. Clin. Exp. Immunol 2, 75–93. [PMC free article] [PubMed] [Google Scholar]
- Murphy KL, Fischer R, Swanson KA, Bhatt IJ, Oakley L, Smeyne R, Bracchi-Ricard V, Bethea JR, 2020. Synaptic alterations and immune response are sexually dimorphic in a non-pertussis toxin model of experimental autoimmune encephalomyelitis. Exp. Neurol 323, 113061. 10.1016/j.expneurol.2019.113061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni S-Y, Zhong X-L, Li Z-H, Huang D-J, Xu W-T, Zhou Y, Ou C-W, Chen M-S, 2020. Puerarin Alleviates Lipopolysaccharide-Induced Myocardial Fibrosis by Inhibiting PARP-1 to Prevent HMGB1-Mediated TLR4-NF-κB Signaling Pathway. Cardiovasc. Toxicol 10.1007/s12012-020-09571-9 [DOI] [PubMed]
- Nielsen S, Germanos R, Weier M, Pollard J, Degenhardt L, Hall W, Buckley N, Farrell M, 2018. The Use of Cannabis and Cannabinoids in Treating Symptoms of Multiple Sclerosis: a Systematic Review of Reviews. Curr. Neurol. Neurosci. Rep 18, 8. 10.1007/s11910-018-0814-x [DOI] [PubMed] [Google Scholar]
- Nyirenda MH, Morandi E, Vinkemeier U, Constantin-Teodosiu D, Drinkwater S, Mee M, King L, Podda G, Zhang G-X, Ghaemmaghami A, Constantinescu CS, Bar-Or A, Gran B, 2015. TLR2 stimulation regulates the balance between regulatory T cell and Th17 function: a novel mechanism of reduced regulatory T cell function in multiple sclerosis. J. Immunol. Baltim. Md 1950 194, 5761–5774. 10.4049/jimmunol.1400472 [DOI] [PubMed] [Google Scholar]
- O’Connor KA, Hansen MK, Rachal Pugh C, Deak MM, Biedenkapp JC, Milligan ED, Johnson JD, Wang H, Maier SF, Tracey KJ, Watkins LR, 2003. Further characterization of high mobility group box 1 (HMGB1) as a proinflammatory cytokine: central nervous system effects. Cytokine 24, 254–265. 10.1016/j.cyto.2003.08.001 [DOI] [PubMed] [Google Scholar]
- Papadopoulos D, Ewans L, Pham-Dinh D, Knott J, Reynolds R, 2006. Upregulation of alpha-synuclein in neurons and glia in inflammatory demyelinating disease. Mol. Cell. Neurosci 31, 597–612. 10.1016/j.mcn.2006.01.007 [DOI] [PubMed] [Google Scholar]
- Paré A, Mailhot B, Lévesque SA, Lacroix S, 2017. Involvement of the IL-1 system in experimental autoimmune encephalomyelitis and multiple sclerosis: Breaking the vicious cycle between IL-1β and GM-CSF. Brain. Behav. Immun 62, 1–8. 10.1016/j.bbi.2016.07.146 [DOI] [PubMed] [Google Scholar]
- Patten DK, Schultz BG, Berlau DJ, 2018. The Safety and Efficacy of Low-Dose Naltrexone in the Management of Chronic Pain and Inflammation in Multiple Sclerosis, Fibromyalgia, Crohn’s Disease, and Other Chronic Pain Disorders. Pharmacother. J. Hum. Pharmacol. Drug Ther 38, 382–389. 10.1002/phar.2086 [DOI] [PubMed] [Google Scholar]
- Phillips LH, Henry JD, Nouzova E, Cooper C, Radlak B, Summers F, 2014. Difficulties with emotion regulation in multiple sclerosis: Links to executive function, mood, and quality of life. J. Clin. Exp. Neuropsychol 36, 831–842. 10.1080/13803395.2014.946891 [DOI] [PubMed] [Google Scholar]
- Piccinini AM, Midwood KS, 2010. DAMPening inflammation by modulating TLR signalling. Mediators Inflamm 2010. 10.1155/2010/672395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins M, Eriksson C, Wierinckx A, Bol JGJM, Binnekade R, Tilders FJH, Van Dam A-M, 2013. Interleukin-1β and interleukin-1 receptor antagonist appear in grey matter additionally to white matter lesions during experimental multiple sclerosis. PloS One 8, e83835. 10.1371/journal.pone.0083835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafiee Zadeh A, Ghadimi K, Ataei A, Askari M, Sheikhinia N, Tavoosi N, Falahatian M, 2019. Mechanism and adverse effects of multiple sclerosis drugs: a review article. Part 2. Int. J. Physiol. Pathophysiol. Pharmacol 11, 105–114. [PMC free article] [PubMed] [Google Scholar]
- Rahn EJ, Iannitti T, Donahue RR, Taylor BK, 2014. Sex differences in a mouse model of multiple sclerosis: neuropathic pain behavior in females but not males and protection from neurological deficits during proestrus. Biol. Sex Differ 5, 4. 10.1186/2042-6410-5-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues DH, Leles BP, Costa VV, Miranda AS, Cisalpino D, Gomes DA, de Souza DG, Teixeira AL, 2016. IL-1β Is Involved with the Generation of Pain in Experimental Autoimmune Encephalomyelitis. Mol. Neurobiol 53, 6540–6547. 10.1007/s12035-015-9552-0 [DOI] [PubMed] [Google Scholar]
- Segal JP, Bannerman CA, Silva JR, Haird CM, Baharnoori M, Gilron I, Ghasemlou N, 2020. Chronic mechanical hypersensitivity in experimental autoimmune encephalomyelitis is regulated by disease severity and neuroinflammation. Brain. Behav. Immun 89, 314–325. 10.1016/j.bbi.2020.07.010 [DOI] [PubMed] [Google Scholar]
- Seixas D, Palace J, Tracey I, 2016. Chronic pain disrupts the reward circuitry in multiple sclerosis. Eur. J. Neurosci 44, 1928–1934. 10.1111/ejn.13272 [DOI] [PubMed] [Google Scholar]
- Selfridge BR, Wang X, Zhang Y, Yin H, Grace PM, Watkins LR, Jacobson AE, Rice KC, 2015. Structure-Activity Relationships of (+)-Naltrexone-Inspired Toll-like Receptor 4 (TLR4) Antagonists. J. Med. Chem 58, 5038–5052. 10.1021/acs.jmedchem.5b00426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloane E, Langer S, Jekich B, Mahoney J, Hughes T, Frank M, Seibert W, Huberty G, Coats B, Harrison J, Klinman D, Poole S, Maier S, Johnson K, Chavez R, Watkins LR, Leinwand L, Milligan E, 2009a. Immunological priming potentiates non-viral anti-inflammatory gene therapy treatment of neuropathic pain. Gene Ther 16, 1210–1222. 10.1038/gt.2009.79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloane E, Ledeboer A, Seibert W, Coats B, van Strien M, Maier SF, Johnson KW, Chavez R, Watkins LR, Leinwand L, Milligan ED, Van Dam AM, 2009b. Anti-inflammatory cytokine gene therapy decreases sensory and motor dysfunction in experimental Multiple Sclerosis: MOG-EAE behavioral and anatomical symptom treatment with cytokine gene therapy. Brain. Behav. Immun 23, 92–100. 10.1016/j.bbi.2008.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloane E, Ledeboer A, Seibert W, Coats B, van Strien M, Maier SF, Johnson KW, Chavez R, Watkins LR, Leinwand L, Milligan ED, Van Dam AM, 2009c. Anti-inflammatory cytokine gene therapy decreases sensory and motor dysfunction in experimental Multiple Sclerosis: MOG-EAE behavioral and anatomical symptom treatment with cytokine gene therapy. Brain. Behav. Immun 23, 92–100. 10.1016/j.bbi.2008.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloane JA, Batt C, Ma Y, Harris ZM, Trapp B, Vartanian T, 2010. Hyaluronan blocks oligodendrocyte progenitor maturation and remyelination through TLR2. Proc. Natl. Acad. Sci 107, 11555–11560. 10.1073/pnas.1006496107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderquist RG, Milligan ED, Harrison JA, Chavez RA, Johnson KW, Watkins LR, Mahoney MJ, 2010a. PEGylation of interleukin-10 for the mitigation of enhanced pain states. J. Biomed. Mater. Res. A 93, 1169–1179. 10.1002/jbm.a.32611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderquist RG, Sloane EM, Loram LC, Harrison JA, Dengler EC, Johnson SM, Amer LD, Young CS, Lewis MT, Poole S, Frank MG, Watkins LR, Milligan ED, Mahoney MJ, 2010b. Release of plasmid DNA-encoding IL-10 from PLGA microparticles facilitates long-term reversal of neuropathic pain following a single intrathecal administration. Pharm. Res 27, 841–854. 10.1007/s11095-010-0077-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solaro C, Cella M, Signori A, Martinelli V, Radaelli M, Centonze D, Sica F, Grasso MG, Clemenzi A, Bonavita S, Esposito S, Patti F, D’Amico E, Cruccu G, Truini A, Neuropathic Pain Special Interest Group of the Italian Neurological Society, 2018. Identifying neuropathic pain in patients with multiple sclerosis: a cross-sectional multicenter study using highly specific criteria. J. Neurol 10.1007/s00415-018-8758-2 [DOI] [PubMed]
- Sorge RE, LaCroix-Fralish ML, Tuttle AH, Sotocinal SG, Austin J-S, Ritchie J, Chanda ML, Graham AC, Topham L, Beggs S, Salter MW, Mogil JS, 2011. Spinal cord Toll-like receptor 4 mediates inflammatory and neuropathic hypersensitivity in male but not female mice. J. Neurosci. Off. J. Soc. Neurosci 31, 15450–15454. 10.1523/JNEUROSCI.3859-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava T, Diba P, Dean JM, Banine F, Shaver D, Hagen M, Gong X, Su W, Emery B, Marks DL, Harris EN, Baggenstoss B, Weigel PH, Sherman LS, Back SA, 2018. A TLR/AKT/FoxO3 immune tolerance-like pathway disrupts the repair capacity of oligodendrocyte progenitors. J. Clin. Invest 128, 2025–2041. 10.1172/JCI94158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes JA, Cheung J, Eddinger K, Corr M, Yaksh TL, 2013. Toll-like receptor signaling adapter proteins govern spread of neuropathic pain and recovery following nerve injury in male mice. J. Neuroinflammation 10, 148. 10.1186/1742-2094-10-148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su M, Ran Y, He Z, Zhang M, Hu G, Tang W, Zhao D, Yu S, 2018. Inhibition of toll-like receptor 4 alleviates hyperalgesia induced by acute dural inflammation in experimental migraine. Mol. Pain 14. 10.1177/1744806918754612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svendsen KB, Jensen TS, Hansen HJ, Bach FW, 2005. Sensory function and quality of life in patients with multiple sclerosis and pain. Pain 114, 473–481. 10.1016/j.pain.2005.01.015 [DOI] [PubMed] [Google Scholar]
- Taşkapilioğlu Ö, 2018. Recent Advances in the Treatment for Multiple Sclerosis; Current New Drugs Specific for Multiple Sclerosis. Arch. Neuropsychiatry 55, S15–S20. 10.29399/npa.23402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theberge FR, Li X, Kambhampati S, Pickens CL, St. Laurent R, Bossert JM, Baumann MH, Hutchinson MR, Rice KC, Watkins LR, Shaham Y, 2013. Effect of chronic delivery of the Toll-like receptor 4 antagonist (+)-Naltrexone on incubation of heroin craving. Biol. Psychiatry 73, 729–737. 10.1016/j.biopsych.2012.12.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorburn KC, Paylor JW, Webber CA, Winship IR, Kerr BJ, 2016. Facial hypersensitivity and trigeminal pathology in mice with experimental autoimmune encephalomyelitis. Pain 157, 627–642. 10.1097/j.pain.0000000000000409 [DOI] [PubMed] [Google Scholar]
- Thundyil J, Lim K-L, 2015. DAMPs and neurodegeneration. Ageing Res. Rev 24, 17–28. 10.1016/j.arr.2014.11.003 [DOI] [PubMed] [Google Scholar]
- Treutwein B, Strasburger H, 1999. Fitting the psychometric function. Percept. Psychophys 61, 87–106. [DOI] [PubMed] [Google Scholar]
- Tullman MJ, 2013. Overview of the epidemiology, diagnosis, and disease progression associated with multiple sclerosis. Am. J. Manag. Care 19, S15–20. [PubMed] [Google Scholar]
- Uzawa A, Mori M, Taniguchi J, Masuda S, Muto M, Kuwabara S, 2013. Anti-high mobility group box 1 monoclonal antibody ameliorates experimental autoimmune encephalomyelitis. Clin. Exp. Immunol 172, 37–43. 10.1111/cei.12036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Strien ME, Mercier D, Drukarch B, Brevé JJP, Poole S, Binnekade R, Bol JGJM, Blits B, Verhaagen J, van Dam A-M, 2010. Anti-inflammatory effect by lentiviral-mediated overexpression of IL-10 or IL-1 receptor antagonist in rat glial cells and macrophages. Gene Ther 17, 662–671. 10.1038/gt.2010.8 [DOI] [PubMed] [Google Scholar]
- Varhaug KN, Vedeler CA, Myhr K-M, Aarseth JH, Tzoulis C, Bindoff LA, 2017. Increased levels of cell-free mitochondrial DNA in the cerebrospinal fluid of patients with multiple sclerosis. Mitochondrion 34, 32–35. 10.1016/j.mito.2016.12.003 [DOI] [PubMed] [Google Scholar]
- Vincenzi F, Corciulo C, Targa M, Merighi S, Gessi S, Casetta I, Gentile M, Granieri E, Borea PA, Varani K, 2013. Multiple sclerosis lymphocytes upregulate A2A adenosine receptors that are antiinflammatory when stimulated. Eur. J. Immunol 43, 2206–2216. 10.1002/eji.201343314 [DOI] [PubMed] [Google Scholar]
- Wallin MT, Wilken JA, Kane R, 2006. Cognitive dysfunction in multiple sclerosis: Assessment, imaging, and risk factors. J. Rehabil. Res. Dev 43, 63–72. [DOI] [PubMed] [Google Scholar]
- Wang T-T, Xu X-Y, Lin W, Hu D-D, Shi W, Jia X, Wang H, Song N-J, Zhang Y-Q, Zhang L, 2020. Activation of Different Heterodimers of TLR2 Distinctly Mediates Pain and Itch. Neuroscience 429, 245–255. 10.1016/j.neuroscience.2020.01.010 [DOI] [PubMed] [Google Scholar]
- Wang X, Zhang Y, Peng Y, Hutchinson MR, Rice KC, Yin H, Watkins LR, 2016. Pharmacological characterization of the opioid inactive isomers (+)-naltrexone and (+)-naloxone as antagonists of toll-like receptor 4. Br. J. Pharmacol 173, 856–869. 10.1111/bph.13394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhang S, Li H, Wang H, Zhang T, Hutchinson MR, Yin H, Wang X, 2020. Small-Molecule Modulators of Toll-like Receptors. Acc. Chem. Res 10.1021/acs.accounts.9b00631 [DOI] [PubMed]
- Westland KW, Pollard JD, Sander S, Bonner JG, Linington C, McLeod JG, 1999. Activated non-neural specific T cells open the blood-brain barrier to circulating antibodies. Brain J. Neurol 122 (Pt 7), 1283–1291. 10.1093/brain/122.7.1283 [DOI] [PubMed] [Google Scholar]
- Wicks P, Rasouliyan L, Katic B, Nafees B, Flood E, Sasané R, 2016. The real-world patient experience of fingolimod and dimethyl fumarate for multiple sclerosis. BMC Res. Notes 9, 434. 10.1186/s13104-016-2243-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieseler J, Ellis A, McFadden A, Stone K, Brown K, Cady S, Bastos LF, Sprunger D, Rezvani N, Johnson K, Rice KC, Maier SF, Watkins LR, 2017. Supradural inflammatory soup in awake and freely moving rats induces facial allodynia that is blocked by putative immune modulators. Brain Res 1664, 87–94. 10.1016/j.brainres.2017.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woller SA, Ravula SB, Tucci FC, Beaton G, Corr M, Isseroff RR, Soulika AM, Chigbrow M, Eddinger KA, Yaksh TL, 2016. Systemic TAK-242 prevents intrathecal LPS evoked hyperalgesia in male, but not female mice and prevents delayed allodynia following intraplantar formalin in both male and female mice: The role of TLR4 in the evolution of a persistent pain state. Brain. Behav. Immun 56, 271–280. 10.1016/j.bbi.2016.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing F, Zhang W, Wen J, Bai L, Gu H, Li Z, Zhang J, Tao Y-X, Xu J-T, 2018. TLR4/NF-κB signaling activation in plantar tissue and dorsal root ganglion involves in the development of postoperative pain. Mol. Pain 14, 1744806918807050. 10.1177/1744806918807050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younger J, Mackey S, 2009. Fibromyalgia Symptoms Are Reduced by Low-Dose Naltrexone: A Pilot Study. Pain Med. Malden Mass 10, 663–672. 10.1111/j.1526-4637.2009.00613.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younger J, Noor N, McCue R, Mackey S, 2013. Low-dose naltrexone for the treatment of fibromyalgia: Findings of a small, randomized, double-blind, placebo-controlled, counterbalanced, crossover trial assessing daily pain levels. Arthritis Rheum 65, 529–538. 10.1002/art.37734 [DOI] [PubMed] [Google Scholar]
- Zhang H, Li Y, de Carvalho-Barbosa M, Kavelaars A, Heijnen CJ, Albrecht PJ, Dougherty PM, 2016. Dorsal root ganglion infiltration by macrophages contributes to paclitaxel chemotherapy induced peripheral neuropathy. J. Pain Off. J. Am. Pain Soc 17, 775–786. 10.1016/j.jpain.2016.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Harada Y, Hayashi Y, 2019. A TLR-CXCL1 pathway in DRG neurons induces neutrophil accumulation in the DRG and mechanical allodynia in EAE mice. Sci. Rep 9, 12003. 10.1038/s41598-019-48558-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Cui F, Chen H, Zhang T, Yang K, Wang Y, Jiang Z, Rice KC, Watkins LR, Hutchinson MR, Li Y, Peng Y, Wang X, 2018. Dissecting the Innate Immune Recognition of Opioid Inactive Isomer (+)-Naltrexone Derived Toll-like Receptor 4 (TLR4) Antagonists. J. Chem. Inf. Model 10.1021/acs.jcim.7b00717 [DOI] [PMC free article] [PubMed]
- Zhang YP, Song CY, Yuan Y, Eber A, Rodriguez Y, Levitt RC, Takacs P, Yang Z, Goldberg R, Candiotti KA, 2013. Diabetic neuropathic pain development in type 2 diabetic mouse model and the prophylactic and therapeutic effects of coenzyme Q10. Neurobiol. Dis 58, 169–178. 10.1016/j.nbd.2013.05.003 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. 8 µg MOG induces greater motor disturbances and subject attrition von Frey testing in male Dark Agouti rats. Male Dark Agouti rats were baselined (BL) for motor scores and for low-threshold mechanical withdrawal thresholds via the von Frey test, followed by intra-dermal moderate-dose (8 µg) myelin oligodendrocyte glycoprotein (MOG). Allodynia and motor scores were assessed thereafter across the timecourse shown. Panels A and B: Von Frey testing on left and right hindpaws, n=6/group, but 50% of subjects were excluded on final day of testing due to high motor scores/cerebellar deficits (i.e. Day 28). Panel C: Motor scores. N=6/group (one subject excluded after Day 24 due to cerebellar deficits requiring euthanasia).