

Abstract
Dopamine transporter (DAT) blockers like cocaine and many other abused and therapeutic drugs bind and stabilize an inactive form of the transporter inhibiting reuptake of extracellular dopamine (DA). The resulting increases in DA lead to the ability of these drugs to induce psychomotor alterations and addiction, but paradoxical findings in animal models indicate that not all DAT antagonists induce cocaine-like behavioral outcomes. How this occurs is not known, but one possibility is that uptake inhibitors may bind at multiple locations or in different poses to stabilize distinct conformational transporter states associated with differential neurochemical endpoints. Understanding the molecular mechanisms governing the pharmacological inhibition of DAT is therefore key for understanding the requisite interactions for behavioral modulation and addiction. Previously, we leveraged complementary computational docking, mutagenesis, peptide mapping, and substituted cysteine accessibility strategies to identify the specific adduction site and binding pose for the crosslinkable, photoactive cocaine analog, RTI 82, which contains a photoactive azide attached at the 2β position of the tropane pharmacophore. Here, we utilize similar methodology with a different cocaine analog N-[4-(4-azido-3-I-iodophenyl)- butyl]-2-carbomethoxy-3- (4-chlorophenyl) tropane, MFZ 2–24, where the photoactive azide is attached to the tropane nitrogen. In contrast to RTI 82, which crosslinked into residue Phe319 of transmembrane domain (TM) 6, our findings show that MFZ 2–24 adducts to Leu80 in TM1 with modeling and biochemical data indicating that MFZ 2–24, like RTI 82, occupies the central S1 binding pocket with the (+)-charged tropane ring nitrogen coordinating with the (−)-charged carboxyl side chain of Asp79. The superimposition of the tropane ring in the three-dimensional binding poses of these two distinct ligands provides strong experimental evidence for cocaine binding to DAT in the S1 site and the importance of the tropane moiety in competitive mechanisms of DA uptake inhibition. These findings set a structure-function baseline for comparison of typical and atypical DAT inhibitors and how their interactions with DAT could lead to the loss of cocaine-like behaviors.
Keywords: dopamine transporter, photoaffinity labeling, addiction, cocaine, computational modeling
Graphical Abstract
1. Introduction
The dopamine transporter (DAT) is a member of the Na+/Cl−-dependent SLC6A family of solute carriers and functions to maintain dopamine (DA) homeostasis through reuptake of transmitter from the neuronal synapse [1–3]. Altered DAT function through dysregulation or mutations can impact extracellular DA levels and has been linked to several neurological diseases and disorders including attention deficit hyperactivity disorder, addiction, depression, autism spectrum disorder, bipolar disorder, Parkinson disease, schizophrenia, Tourette syndrome, and hereditary DAT deficiency syndrome [1,4–8]. DAT activity can be modulated endogenously by various interacting proteins and post-translational modifications, including phosphorylation and palmitoylation [9]. Recently, DAT movement on and off the plasma membrane was demonstrated to be rapidly responsive to changes in membrane potential [10] and there is long-standing evidence that DAT function at the membrane can be impacted by exogenous drugs of abuse like cocaine and amphetamines, which block or reverse the transport of DA by DAT and lead to large increases in extracellular DA levels [11–15]. Enigmatically, some DAT inhibitors including bupropion, methylphenidate, and the benztropines bind and block DA reuptake but fail to induce the euphoric and addictive behavioral effects seen with cocaine [16–18]. Notably, methylphenidate can exhibit addictive cocaine-like effects upon abuse at excessive doses and administered intranasally or intravenously [19]. Understanding the underlying molecular and physiological mechanisms that dictate whether or not a DAT blocker will induce psychotropic and addictive behaviors might greatly advance the development of pharmacotherapeutics that could be used to treat drug abuse and addiction as well as attention deficit and mood disorders.
Major structural insights into DAT and the other monoamine transporters for serotonin and norepinephrine (SERT and NET) have resulted from the crystal structures of substrate and inhibitor bound complexes from both prokaryotic and eukaryotic SLC6 transporters [20–24]. These proteins share a common structure of 12 transmembrane (TM) domains connected by intracellular and extracellular loops, with cytoplasmic N- and C-termini. The topology reveals an architecture of two 5-helix bundles arranged antiparallel to one another in the membrane [25–27]. Comparative modeling utilizing the leucine transporter (LeuT) and Drosophila DAT (dDAT) templates assisted in understanding the three-dimensional structural architecture of mammalian monoamine transporters as well [28,29].
Examination of the substrate and inhibitor binding pockets in the crystal structures of LeuT, dDAT, and human SERT (hSERT) demonstrated that antagonists and substrates occupy a high affinity central substrate binding pocket (S1), generated by TM domains 1, 3, 6, and 8 [20,30], as well as a secondary low-affinity binding site (S2) located in the external vestibule [21,31] that may accommodate a second molecule of substrate or antagonist. Antagonist occupancy of the S1 site would inhibit transport competitively whereas occupancy of the S2 site is thought to allosterically modulate the S1-bound inhibitor effects through reduction or enhancement of ligand dissociation from S1 [32–35].
Findings from biochemical experiments, cocaine QSAR studies, and dDAT/cocaine co-crystal structures suggest cocaine occupancy of the S1 substrate binding pocket in DAT [30,36–41]. Site-directed mutagenesis studies showing strong negative impacts of S1 residue mutations on cocaine or cocaine analog binding. However, cocaine binding is also affected, though normally weaker, by numerous other mutants in non-S1 regions that span from the cytosolic termini to extracellular loops [27,41–44]. Whether these changes are induced indirectly by long-range conformational impacts on S1 or represent possible non-S1 cocaine interaction sites on DAT remains unknown [45–49]. The biogenic amine transporters have been shown to possess allosteric antagonist binding sites that may function in regulation of various properties [50–56], however, to our knowledge cocaine-mediated allosteric properties have not been demonstrated for these transporters [35].
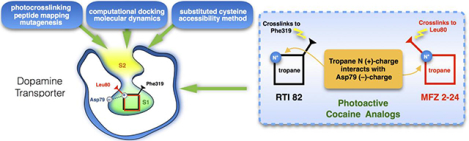
Here, we utilized the irreversible cocaine analog [125I]N-[4-(4′-azido-3′-iodophenyl)- butyl]-2-β-carbomethoxy-3β-(4-chlorophenyl) ([125I]MFZ 2–24) (Fig. 1) [57,58] in combination with comparative modeling, small molecule docking, molecular dynamics, peptide mapping, and substituted cysteine accessibility method (SCAM) [59] protection analyses to identify the adduction site corresponding to high affinity binding of the photoactivatable ligand to DAT. MFZ 2–24, cocaine, and the cocaine analog (−)-2β-carbomethoxy-3β-(4-fluorophenyl)tropane (CFT) share a common pharmacophore consisting of the tropane ring and 3β-phenyl group that results in high affinity binding [60,61]. All of these compounds bind noncovalently to DAT, but [125I]MFZ 2–24 contains a photoactivatable phenyl azido moiety that upon ultraviolet activation forms a highly reactive singlet nitrene that reacts with proximal C–H or N–H groups forming a covalent bond with the protein [62,63]. Using irreversible labeling as a strategy for determining the site of ligand-protein interaction, we previously demonstrated that [125I]MFZ 2–24 cross-links to human (h) DAT between Asp68 and Leu80, a domain corresponding to the central and extracellularly directed regions of TM1 in DAT [64]. In this report, our computational approaches predicted MFZ 2–24 adduction at Leu-80 on TM1 in DAT and this finding was validated by mutagenesis and peptide mapping of the adduction site. Furthermore, our docking models predicted a S1 placement of the tropane core of MFZ 2–24, verified by SCAM analysis, a placement that overlaps with the binding site and orientation of the tropane core of another cocaine analog, RTI 82 [65]. These findings provide the second-known example of a direct interaction of the core tropane pharmacophore of MFZ 2–24 with the central, S1 binding site, providing support for a competitive mechanism of action of cocaine for blocking DA transport and further advance the idea that the tropane pharmacophore is primarily responsible in defining affinity and position in the binding site.
Figure 1. Chemical structures of cocaine, CFT, [125I]MFZ 2–24 and [125I]RTI 82.
Structures of (A) cocaine with tropane N and 3-phenyl ester moieties indicated by stars, (B) CFT with phenyl fluoride moiety directly appended to the 3β position on the tropane pharmacophore (star), (C) [125I]MFZ 2–24 with AIP moiety appended directly to the tropane N, and (D) [125I]RTI 82 with AIP group attached to the 2β; methylester.
2. Materials and methods
2.1. Comparative DAT homology model construction
The outward-occluded and open-to-out rDAT homology models used in this study were generated previously [65] using Rosetta3.1 [66]. The top 10 scoring models (Rosetta Etotal score) for each template were compared visually to the starting structure to verify overall structural integrity before moving forward with docking studies.
2.2. Molecular dynamics analysis of DAT in complex with MFZ 2–24
The top scoring MFZ 2–24 poses, in complex with DAT, from RosettaLigand [67] and Induced Fit Docking (IFD) [68] were then subjected to molecular dynamic analysis for almost 200 ns using GROMACS [65].
2.3. Synthesis and radiolabeling of MFZ 2–24
Synthesis and radiolabeling of MFZ 2–24 was carried out as previously described [57,58].
2.4. Chemicals and materials
[3H]CFT (76 Ci/mmol) was obtained from Perkin-Elmer (Waltham, MA, USA). [3H]DA (45 Ci/mmol), Protein A Sepharose, and Hyperfilm MP were from GE Healthcare Life Sciences (Piscataway, NJ). Site directed mutagenesis QuikChange® kit was from Stratagene (La Jolla, CA). All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO) or Fisher Scientific (Pittsburgh, PA).
2.5. Cell culture and site-directed mutagenesis
Mutant constructs used for peptide mapping were stably expressed in Lewis Lung Carcinoma-Porcine Kidney (LLC-PK1) cells (ATCC) (Passages 5–15) and were characterized previously (M111L/M116L, L80M/M111L/M116L) [64] or were generated in the M111L/M116L rDAT cDNA background using the Stratagene QuikChange® kit and verified by sequencing (Northwoods DNA, Solway, MN). For ease of discussion, mutants V73M, A77M, D79M, and L80M generated in the M111L/M116L background are referred by the TM1 mutation only. For stable expression of new constructs, LLC-PK1 cells were transfected using FuGENE transfection reagent and selected with 800 μg/mL of G418. Cells were maintained in a humidified chamber with 5% CO2 at 37°C in α-minimum essential medium (AMEM with 5% fetal bovine serum, 2 mM L-glutamine, 200 μg/mL G418, and 100 μg/mL penicillin/streptomycin). Mutants for SCAM analysis were generated in the E2C background (C90A and C305A) of pcDNA3-rDAT using the Stratagene QuikChange® kit and verified by sequencing (Eurofins MWG Operon, Huntsville, AL). WT and mutant cDNAs were expressed transiently in HEK-GripTite cells (licensed from ThermoFisher) using TransIT®-LT1 (Mirus) (1 μl per 200 ng of DNA). Cells were maintained in a humidified chamber with 5% CO2 at 37 °C in Dulbecco’s modified Eagle’s medium (DMEM: 10% FBS, 600 μg/ml G418).
2.6. Photoaffinity labeling and CNBr peptide mapping
These procedures were performed as described previously [65]. Briefly, cells expressing WT or mutant DATs were washed twice with ice-cold Krebs-Ringers-HEPES (KRH) buffer (25 mM HEPES, 125 mM NaCl, 4.8 mM KCl, 1.2 mM KH2PO4, 1.3 mM CaCl2, 1.2 mM MgSO4, 5.6 mM glucose, pH 7.4) and incubated with 5 nM [125I]MFZ 2–24 in KRH for 2 h on ice in the presence or absence of 30 μM (−)-cocaine. Cells were irradiated with ultraviolet light (254 nm) for 5 min to covalently attach the ligand to DAT, washed twice with cold KRH, and lysed with radioimmunoprecipitation assay RIPA buffer (150 mM NaCl, 125 mM NaH2PO4, 2 mM EDTA, 1% Triton X-100, 0.1% SDS, pH 7.4) containing protease inhibitor (Pierce Protease Inhibitor minitablets, ThermoFisher) for 30 min on ice with shaking. The lysates were centrifuged at 20,000 × g for 12 min at 4°C and supernatants were subjected to SDS-PAGE and autoradiography on 8% Tris-glycine polyacrylamide gels. Photolabeled DAT bands were excised from the gel and protein was electroeluted and dialyzed against purified H2O. Aliquots were counted in a scintillation counter and equal amounts of radioactivity from each sample were subjected to peptide mapping. For CNBr proteolysis, samples were lyophilized to dryness, and resuspended in 70 μL of 70% formic acid with or without addition of 1 M CNBr and incubated for 24 h at 22°C in the dark. Reactions were quenched with 1 mL of purified water and samples were lyophilized to dryness, followed by removal of CNBr with four additional rounds of resuspension with water and lyophilization. The final samples were suspended in immunoprecipitation buffer and divided into aliquots. Part of the sample (25–50%) was subjected directly to SDS-PAGE/autoradiography to visualize the total CNBr digestion pattern and the remainder was immunoprecipitated with monoclonal Ab 16 (mAb 16) prior to SDS-PAGE/autoradiography. For each experiment, 2–3 mutants were photoaffinity labeled and analyzed with WT and L80M DAT exactly in parallel and all results were replicated at least 3 times. CNBr peptide masses were calculated using PeptideCutter via ExPASy.
2.7. [3H]CFT binding
LLC-PK1 cells (Passage 5–15) stably expressing WT and mutant rDATs were grown to ~80% confluency and washed with Hank’s balanced salt solution (HBSS). Cells were incubated with 10 nM [3H]CFT in KRH buffer for 2 h at 4°C. Binding was performed in triplicate with nonspecific binding determined in the presence of 10 μM mazindol. At the end of the incubation, cells were washed twice with KRH buffer, lysed with 1% Triton X-100, and samples assessed for radioactivity by liquid scintillation counting.
2.8. [3H]DA uptake
WT or mutant rDAT-LLCPK1 (Passage 5–15) cells were grown in 24-well plates to ~80% confluency in AMEM at 37°C. Cells were rinsed twice with 0.5 mL KRH buffer followed by addition of 0.5 mL warmed KRH (37°C). Uptake was performed in triplicate and initiated by addition of 10 nM [3H]DA plus 3 μM unlabeled DA, with nonspecific uptake determined in the presence of 100 μM (–)-cocaine (Sigma, St Louis, MO). Uptake was allowed to proceed for 8 min at 37°C and cells were rapidly washed three times with ice-cold KRH and solubilized in 1% Triton X-100. Radioactivity contained in lysates was assessed by liquid scintillation counting and protein content was determined using BCA colorimetric reagent.
2.9. SCAM protection analysis of S1- and S2-binding sites
GripTite cells (ThermoFisher Scientific) (Passage 2–15) were plated at a density of 150,000 cells/cm2 in 24-well culture plates, incubated for 24 h, and transfected with rDAT constructs using TransIT®-LT1 (Mirus). Following transfection (48 h), cells were processed as described previously [39,65]. Briefly, cells were incubated with 10 or 50 μM MFZ 2–24 or vehicle for 5 min in PBS/CM (137 mM NaCl, 2.7 mM KCl, 10.1 mM Na2HPO4, 1.8 mM KH2PO4, 0.1 mM CaCl2, 1.0 mM MgCl2, pH 7.4) followed by addition of 0.1 mM MTSEA-biotin (Biotium) for 10 min (Biotium, Fremont, CA). Cell lysates were obtained with RIPA solubilization buffer (10 mM Tris, pH 7.4, 150 mM NaCl, 0.1% SDS, 1% Triton X-100, 1% sodium deoxycholate) containing Halt™ Protease Inhibitor Single-Use Cocktail (ThermoFisher) and incubated with NeutrAvidin-agarose resin (ThermoFisher) to extract surface proteins labeled by MTSEA-Biotin. Following a BCA assay (ThermoFisher) to determine protein concentration, equal amounts of protein from total samples and equivalent volumes of surface protein pools were processed by SDS-PAGE and immunoblotting using anti-DAT monoclonal antibody, mAb 16 [69,70]. DAT levels were quantitated from the density of immunoblot bands from at least three separate assays using ImageJ (National Institutes of Health). Surface values were normalized to total DAT levels and are expressed as a percent of untreated samples. Data were analyzed using a paired Student’s t-test (Prism 4, Graphpad, Inc).
3. Results
3.1. Computational docking of MFZ 2–24 to rDAT homology models
To identify the site of MFZ 2–24 adduction to DAT and inform the biochemical crosslinking studies, in silico docking was performed on the open-to-out and outward-occluded rDAT comparative models (based on the LeuT crystal structures 2A65 and 3F3A) [20,22] using two independent methods, RosettaLigand (RL) and Induced Fit Docking (IFD). A recent review details how LeuT remains a useful and valid template for SLC6 modeling [71].
3.2. RosettaLigand docking
RL docking was performed and the top-scoring 5% (based on the interface_delta score, see methods) of the 25,000 RL-docked complexes (termed decoys) were filtered using three biochemically-defined molecular distance constraints. These constraints were defined as 5Å distance cutoffs between: [1] the MFZ 2–24 azido moiety and any atom in the residues Asp-79 and Leu-80, [2] the tropane N of MFZ 2–24 and the side chain of Asp-79 [65], and [3] the phenyl chloride of MFZ 2–24 and the side chain of Asn-157 [41,65]. Using both filters (1) and (2) resulted in 89 and 5 decoys from the open-to-out and outward-occluded models, respectively. The addition of filter (3) reduced the number of decoys fulfilling the constraints to 9 and 0 for the open-to-out and outward-occluded models, respectively. The top two scoring decoys had poses for MFZ 2–24 that only differed by 1.288 Å RMSD, however, evaluation of the distance between the nitrene-forming N atom on the azido group of MFZ 2–24 and the nearest C atom on the Leu-80 side chain indicated that the second best-scoring decoy (interface_delta score, −12.8) with intramolecular distance of 3.8Å (Fig. 2A) was more consistent with the molecular constraints than the top scoring decoy (interface_delta score, −15.4) which had an intramolecular distance of 5.8Å. Therefore, the second-best scoring pose was chosen and carried forward for molecular dynamic simulations.
Figure 2. MFZ 2–24 and DAT RosettaLigand docking complexes and molecular dynamics simulation.
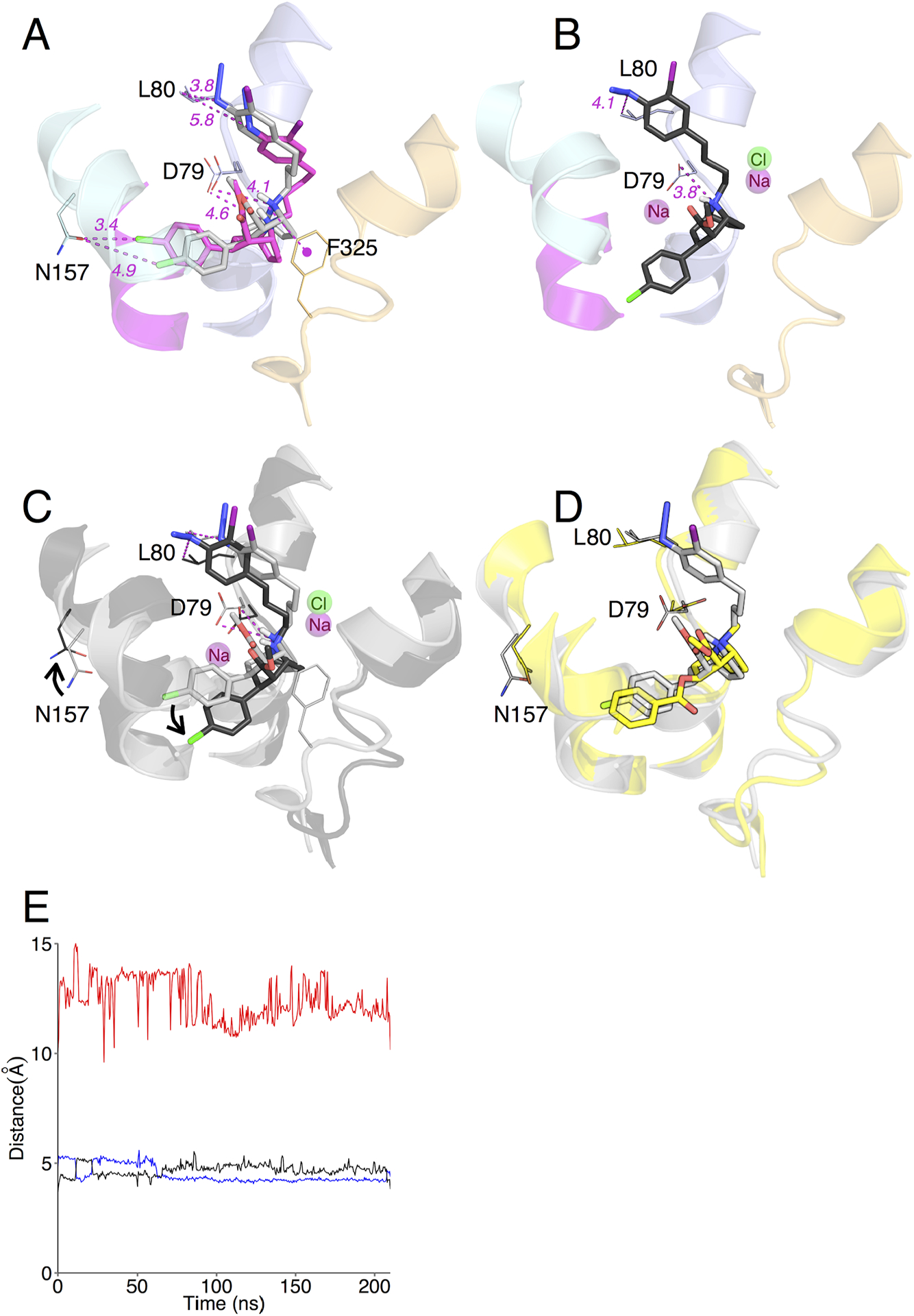
(A) Best RL rDAT/MFZ 2–24-docked complex based on energy and constraint fulfillment. Image depicts the LeuT-based rDAT homology model from the crystal structure of the open-to-out template 3F3A. (B) Molecular dynamics simulation of the RL rDAT/MFZ 2–24-docked complex. (C) Superimposition of RL-docked MFZ 2–24 before (grey) and after (black) simulation. (D) Superimposition of RL-docked MFZ 2–24 pre-simulation (grey) and cocaine (yellow) from Drosophila DAT co-crystal. (E) Time-dependent changes in distances following molecular dynamics simulation between the protonated nitrogen of MFZ 2–24 and the side chain oxygen atom of Asp79 (blue and black traces for ∂O1 and ∂O2) and the phenyl chloride of MFZ 2–24 and the amide nitrogen of Asn157 (red traces).
3.3. IFD docking
MFZ 2–24 was also docked into the open-to-out and outward-occluded models using IFD, which allows for the incorporation of the Na+ and Cl– ions along with their energetic and density contributions during the docking analysis [20,65,72–75]. IFD docking to the open-to-out and outward-occluded models yielded 12 and 2 structures, respectively. Of the 12 open-to-out docked structures, only 1 complex fulfilled the nitrene/Leu-80 distance constraint (Fig. 3A). The nitrene/Leu-80 distance of the remaining 11 structures was >7.4Å, which eliminated them from further consideration. Analysis of the two outward-occluded IFD docked complexes revealed the distance between the nitrene group and the Leu-80 side chain was 5.3 and 5.4Å, which is just outside of the molecular cutoff. However, to determine if refinement would result in a ligand pose that met the filter requirements, the best scoring complex was carried forward for MD analysis.
Figure 3. MFZ 2–24 and DAT Induced Fit Docking complexes and molecular dynamics simulation.
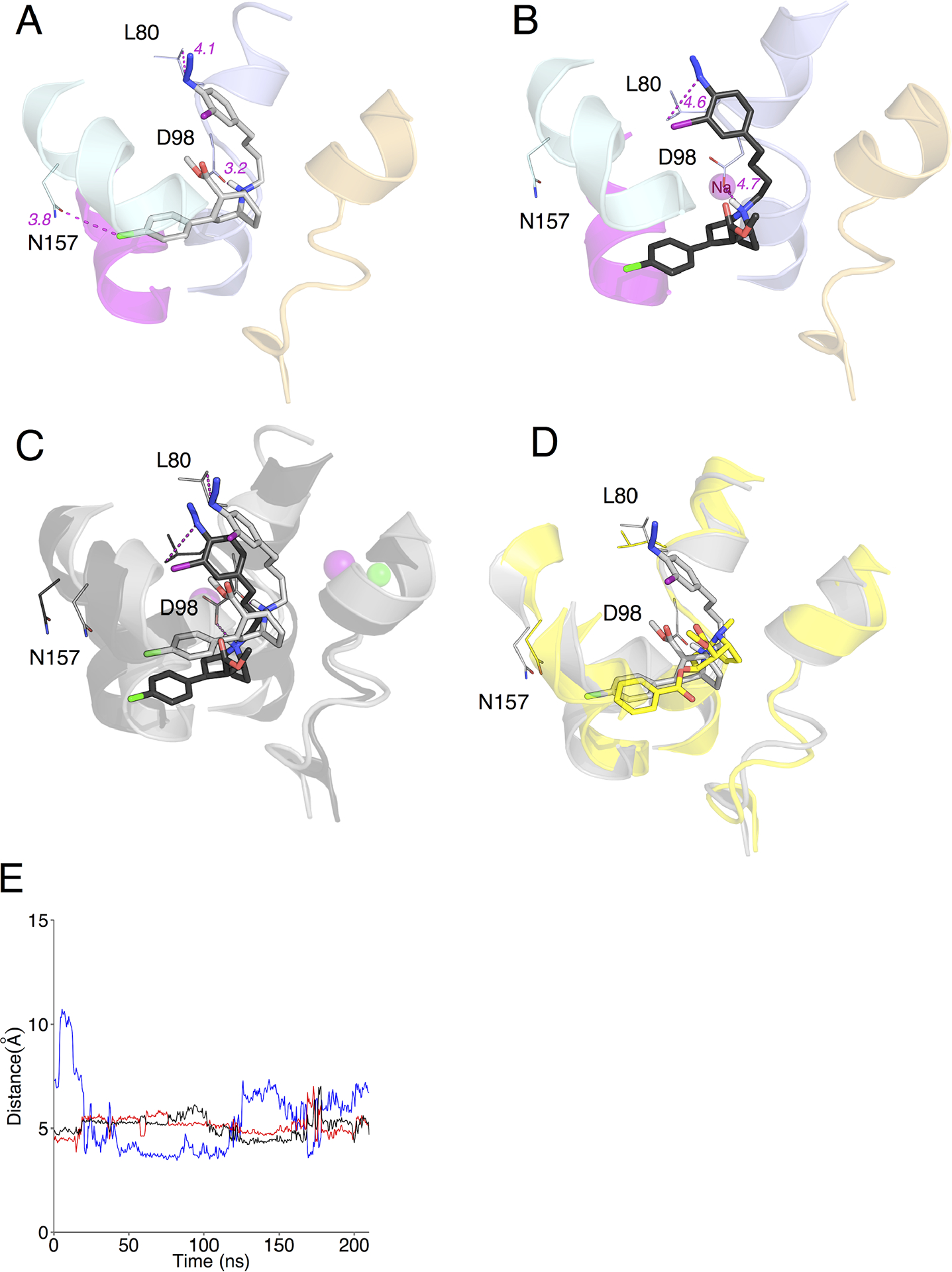
(A) Best IFD rDAT/MFZ 2–24-docked complex based on energy and constraint fulfillment. Image depicts the LeuT-based rDAT homology model from the crystal structure of the open-to-out template 3F3A. (B) Molecular dynamics simulation of the IFD rDAT/MFZ 2–24-docked complex. (C) Superimposition of IFD-docked MFZ 2–24 before (grey) and after (black) simulation. (D) Superimposition of IFD-docked MFZ 2–24 pre-simulation (grey) and cocaine (yellow) from Drosophila DAT co-crystal. (E) Time-dependent changes in distances following molecular dynamics simulation between the protonated nitrogen of MFZ 2–24 and the side chain oxygen atom of Asp79 (blue and black traces for ∂O1 and ∂O2) and the phenyl chloride of MFZ 2–24 and the amide nitrogen of Asn157 (red traces).
3.4. MD simulation of RL and IFD docked MFZ 2–24 poses
To refine the selected RL and IFD poses the ligand-transporter complexes were placed in a POPC lipid bilayer and subjected to more than 150 ns of MD simulation using GROMACS [76]. Simulation of the RL f_1217 docked structure resulted in a pivoting movement of MFZ 2–24 around the tropane N such that the phenyl Cl group moved down and away from Asn-157 to a distance of 8.5Å and the phenyl iodo azido arm moved slightly away from TM1 to a distance of 4.1Å (Fig. 2B and 2C). The ∂O atoms of the Asp-79 side chain appear to alternate in coordinating the tropane N over the duration of the simulation (Fig. 2E), which was also observed in our previous analysis with the cocaine analog RTI 82 [65]. However, the intermolecular distance between Asn-157 and the phenyl Cl increased substantially (Fig 2E). This constraint was used based on previous comparative modeling and mutagenesis work supporting interaction between the phenyl Fl of the cocaine analog CFT and Asn-157 on TM3 [41]. Similar to RL, MD analysis of the IFD pose resulted in moderate translation of MFZ 2–24 in the binding pocket. The Leu-80 and Asp-79 side chains also shifted such that they maintained a <5Å distance to the nitrene and tropane N, respectively (Fig. 3B, 3C and 3E). However, in the IFD pose, the intermolecular distance between Asn-157 and the phenyl Cl remained relatively consistent at ~ 5Å over the simulation (Fig 3E). Alignment of the pre- and post-MD RL and IFD-docked structures with the recent co-crystal of cocaine in the dDAT [30] revealed that the MFZ 2–24 poses from the pre-MD structures are closer to cocaine in the co-crystal structure than structures following refinement by MD simulations (Fig. 2D and 3D). After MD analysis, the “outward-occluded” MFZ 2–24 structures failed to meet the molecular constraints and were therefore eliminated.
3.5. Peptide mapping of photoaffinity labeled site
In previous studies we used Met substitution mutagenesis, CNBr proteolysis, and epitope specific immunoprecipitation to demonstrate that [125I]MFZ 2–24 cross-links to DAT in TM1 between Ile67 and Leu80 [64]. Here we use these procedures to further narrow the adduction site and test the cross-linking predictions obtained from computational modeling. For these experiments, endogenous residues between positions 68 and 80 are substituted one at a time with Met to generate novel CNBr cleavage sites for peptide mapping. The proteins are photolabeled, digested with CNBr, and the fragments subjected to immunoprecipitation with mAb 16 which is directed against N-terminal residues 42–59. There are no endogenous Mets in DAT between positions 11 and 106, thus if [125I]MFZ 2–24 adduction occurs N-terminal to the inserted Met, the labeled fragment will contain the epitope for mAb 16 and will immunoprecipitate, whereas if adduction occurs C-terminal to the inserted Met, CNBr digestion will separate the ligand from the antibody epitope and the photolabeled fragment will not immunoprecipitate (see Figure 4).
Figure 4. CNBr mapping of [125I]MFZ 2–24-labeled DAT mutants.
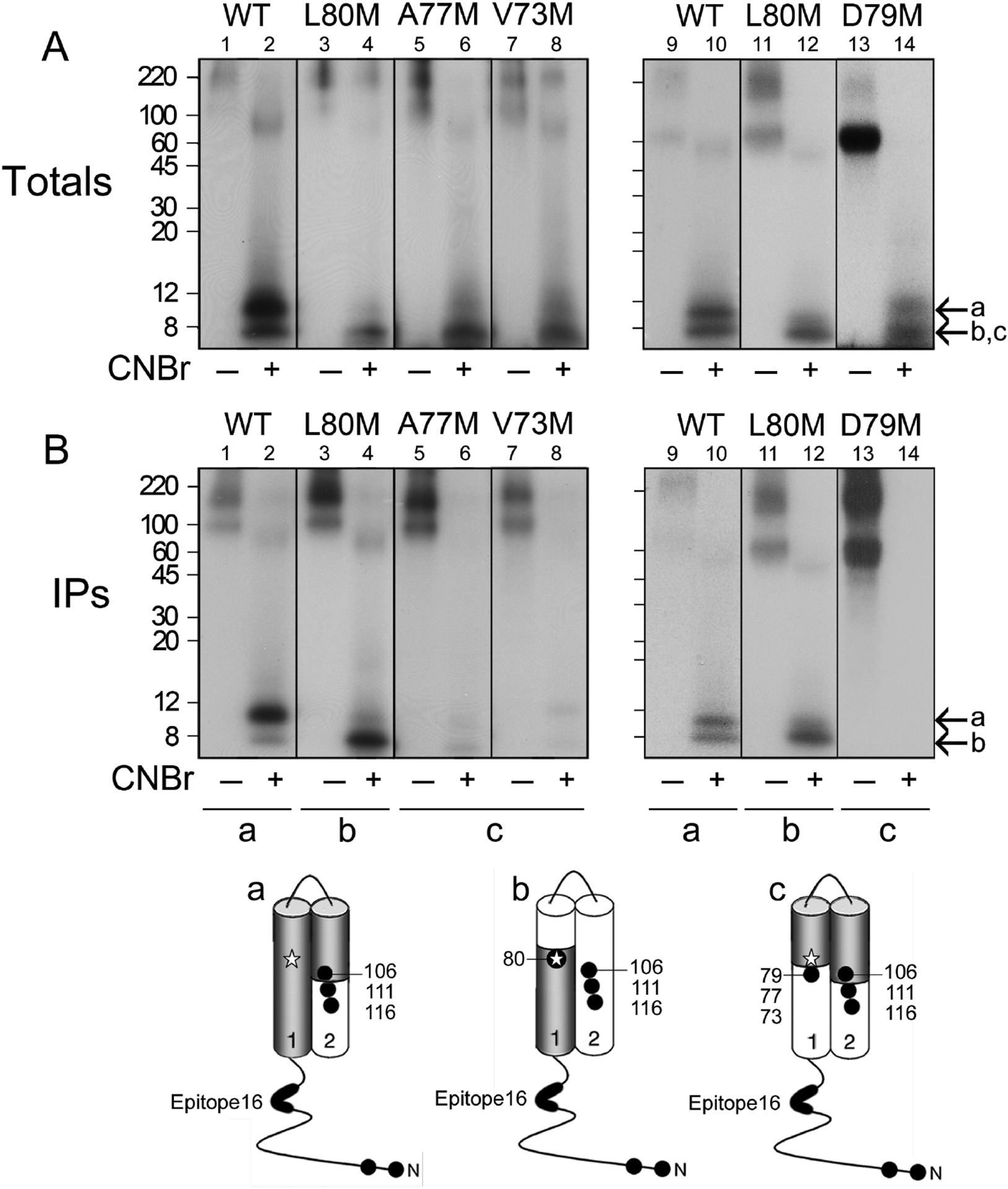
The indicated DAT forms were photolabeled with [125I]MFZ 2–24, gel purified, and equal amounts of radioactivity were treated with vehicle (odd numbered lanes) or CNBr (even numbered lanes). Aliquots of each sample were analyzed directly by SDS/PAGE-autoradiography to visualize the total pattern of CNBr fragments (A), or were immunoprecipitated with mAb 16 prior to SDS/PAGE-autoradiography (B). Left and right panels show independent experiments, and molecular mass markers shown on left gels are indicated by tic marks on right gels. Arrow a indicates migration of ~12 kDa CNBr fragment that extends from the Met-1/11 to Met106/111/116 and arrows b and c indicate ~3–8 kDa fragments produced by cleavage of exogenous TM1 Mets. Schematic diagrams show the cytosolic N-terminus, TMs 1 and 2, positions of endogenous and exogenous Met residues (filled circles), epitope for mAb 16 (bold line), adduction site of [125I]MFZ 2–24 (star), and origin of photolabeled CNBr fragments in primary sequence (grey shading).
Because molecular modeling suggested adduction of [125I]MFZ 2–24 at Asp79 or Leu80, we focused our efforts on residues close to these sites, inserting Met in place of Val73, Ala77, and Asp79. Mutants were stably expressed in LLC-PK1 cells and analyzed for expression, [3H]DA uptake, [3H]CFT binding, and [125I]MFZ 2–24 labeling. V73M and A77M DATs were expressed as full-length protein and showed robust cocaine-sensitive transport and CFT binding activity. D79M DAT expressed poorly and, as expected, showed negligible transport, but the full-length protein displayed a small amount of [125I]MFZ 2–24 labeling and was analyzed due to molecular modeling findings that indicated Asp79 as a possible adduction site.
For peptide mapping studies, the photolabeled proteins were gel purified and subjected to treatment with vehicle (formic acid) or CNBr. For all experiments, WT and L80M DATs were labeled, digested, immunoprecipitated, and electrophoresed in parallel with the new constructs to provide positive controls to validate generation and immunoprecipitation of labeled fragments. Digested samples were divided into aliquots that were directly subjected to SDS-PAGE/autoradiography to visualize the total spectrum of CNBr fragments or were immunoprecipitated with mAb 16 prior to SDS-PAGE/autoradiography. Within each experiment, samples from WT and mutant forms were adjusted to contain equal amounts of radioactivity to allow for direct comparison of fragment production and immunoprecipitation signals.
Figure 4 shows the results of these analyses with the accompanying schematic diagrams indicating the origin of the labeled fragments in the primary sequence. Panel A shows the spectrum of total CNBr fragments generated from [125I]MFZ 2–24 labeled WT and mutant DATs with left and right panels showing independent analyses of the indicated mutants. In vehicle-treated samples, full length DAT migrates at ~90 kDa (odd numbered lanes) with no low molecular weight fragments observed. Aggregates seen at ≥180 kDa are most likely induced by the formic acid treatment, as they were not seen in samples subjected directly to electrophoresis. CNBr treatment of WT rDAT produced a labeled fragment of ~12 kDa (lane 2; arrow a) that corresponds to the region between Met-1/11 and Met106/111/116 (shaded region in diagram a). The presence of doublets in some experiments is likely due to different proteolysis combinations, as the calculated mass of fragments extending from Met1 to Met116 is 12.7 kDa and the calculated mass of fragments extending from Met11 to Met106 is 10.5 kDa. CNBr treatment of [125I]MFZ 2–24 labeled L80M DAT produced a fragment of ~8 kDa, which is consistent with peptides extending from Met1 or Met11 to L80M (shaded region in schematic diagram b) with calculated masses of 7.5 and 8.7 kDa, as demonstrated previously [64]. All of the new mutants showed similar levels of CNBr fragments in the low molecular mass region of the gel indicating that the ligand became adducted to the protein in a similar manner and arguing against the possibility that the mutation induced adduction of [125I]MFZ 2–24 in a different region of the primary sequence that would generate a different fragment pattern. For V73M, A77M, and D79M DATs, a labeled fragment similar to that generated from L80M DAT would be produced if the ligand becomes adducted N-terminal to the new CNBr site whereas a labeled fragment of ~3 kDa extending from the new Met to Met106 (shown in diagram c) would be produced if the ligand adducts C-terminal to the new CNBr site. These possible fragments would not be distinguishable by SDS-PAGE and both forms are indicated in panel A with arrows b and c. Importantly, the intensity of fragment production for mutant DATs was comparable to that of the WT and L80M proteins allowing for direct comparison of immunoprecipitation signals.
Panel B shows the mAb 16 immunoprecipitation profiles of the total digests shown in the upper panels. As we previously demonstrated [64], mAb 16 immunoprecipitated the 12 kDa fragment from the WT DAT (lanes 2 and 10, arrow a) and the 8 kDa fragment from L80M DAT (lanes 4 and 12, arrow b) indicating that ligand incorporation occurs at or N-terminal to Leu80 (diagrams a and b). In striking contrast, even though labeled fragments were present at equal intensities in the total digests from V73M, A77M, or D79M DATs, these samples showed negligible immunoprecipitation by mAb 16 (lanes 6, 8, and 14). This strongly indicates that cleavage of TM1 at Mets 73, 77, and 79 separated epitope 16 from the [125I]MFZ 2–24 adduction site and thus that adduction occurred C-terminal to those Mets (diagram c). Therefore, because CNBr cleaves the peptide bond on the C-terminal side of Met residues, these data provide strong experimental evidence that [125I]MFZ 2–24 adduction occurs at Leu-80.
3.6. Analysis of MFZ 2–24 protection of S1- and S2-binding pockets from Cys-directed biotinylation
Our computational docking and peptide mapping results strongly indicate that binding of the MFZ 2–24 tropane pharmacophore occurs within the S1-binding site and positions the azido group for adduction to Leu80. In order to confirm binding of MFZ 2–24 to S1, we analyzed several residues present in the S1- or S2-binding sites in addition to several that line the transition between S1 and S2 (intermediate residues) in the DAT permeation pathway. Mutants were constructed in a methanethiosulfonate (MTS)-insensitive rDAT E2C (C90A and C305A) background and assessed for MFZ 2–24-induced protection from the SCAM reagent MTSEA-biotin.
Previously [65], we identified that surface rDAT was readily labeled with MTSEA-biotin whereas labeling of rDAT E2C is undetectable (Fig. 5A, Control), confirming it as a cysteine-reduced, MTS-insensitive background suitable for analysis of inserted Cys residues. Utilizing rDAT E2C as a template, we generated Cys mutations of the S1 residues Asp79, Ala81, Asn82, Val152, and Ser421, the S2 residues Trp84, Arg85, Ile159, and Asp475, and the intermediate residues Phe319, Ala479, and Ile483. MTSEA-biotin protection assays were performed using 10 μM MFZ 2–24, except for the mutants D79C and S421C where marked losses in potency to inhibit binding were observed, which required a higher concentration (50 μM) for protection analyses.
Figure 5. SCAM protection analysis of S1 and S2 binding pockets.
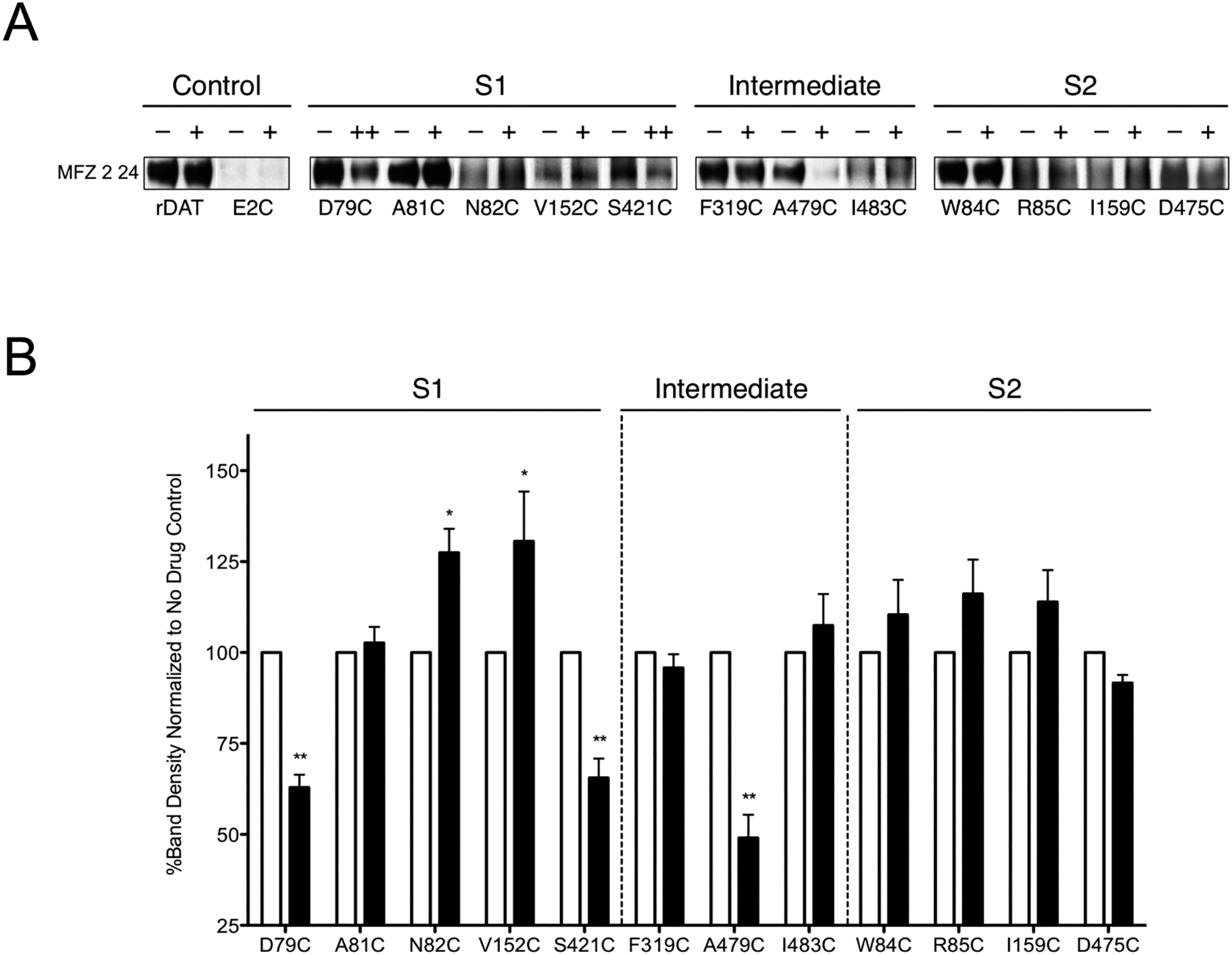
(A) Immunoblots of the surface pool of DAT Cys mutants in the rDAT E2C background purified with MTSEA-biotin in the absence (−) or presence of MFZ 2–24 (+, 10 μM; ++, 50 μM). (B) Quantification of DAT bands in A and total DAT expression (data not shown) were determined using ImageJ (National Institutes of Health). Surface samples were normalized to total DAT then expressed as a percent of MFZ 2–24-treated samples (black bars) to the respective untreated samples (white bars). These data represent three or more independent experiments. Significant differences between treated and untreated samples were determined with a paired Student’s t-test *, p < 0.05; **, p < 0.01.
Western Blot analyses revealed that treatment with MFZ 2–24 did not affect recovery of biotinylated rDAT surface protein (Fig. 5A, Control) indicating Cys90 and/or Cys305, which are located on the extracellular ends of TMs 1 and 6, respectively, remain reactive in the presence of the ligand. Recovery of the biotinylated S1 mutants D79C and S421C was significantly reduced in the presence of MFZ 2–24 (Fig. 5, S1), demonstrating protection of these residues from the MTS reagent. Mutant A81C showed no change in recovery whereas N82C and V152C exhibited increased recovery (greater MTS sensitivity), a pattern we previously observed with cocaine [65]. This difference in cocaine analog protection likely arises from the position of the phenyl azido moiety, as the RTI 82 extension is oriented toward the external vestibule whereas on MFZ 2–24 the group is bent inwards. In the S2 mutants, MFZ 2–24 did not alter recovery from W84C, R85C, I159C, and D475C (Fig. 5, S2) indicating a lack of protection of these residues from MTS attack. In combination, these data reveal binding of MFZ 2–24 to DAT in the S1, but not S2, site.
The intermediate residues Phe319, Ala479, and Ile483 that lie at the interface between the S1- and S2-binding sites provide insight into the conformation of DAT adopted upon antagonist binding. MFZ 2–24, like RTI 82 [65], only protected mutant A479C from MTS modification (Fig. 5, Intermediate) whereas cocaine protected all intermediate sites. Though the azido groups of RTI 82 and MFZ 2–24 adduct to distinct sites on DAT, each analog has the addition of a phenyl azido moiety that, through its added bulk, may prevent DAT from transitioning to an occluded conformation. Cocaine, however, lacks the phenyl azido group that may allow for transition to a more occluded structure thereby providing protection of these residues.
4. Discussion
Cocaine impacts on DAT are complex and remain incompletely understood, with binding occurring via both high and low affinity states [77,78] and uptake inhibition mediated through competitive and noncompetitive mechanisms [79,80]. Efforts to elucidate the structural bases of these processes have primarily involved loss-of-function mutagenesis approaches and co-crystal structures of bacterial and insect transporters that are thermostabilized by mutations, truncations, or antibody methods, and are often diminished in or devoid of transport activity [30,81]. These methods have provided valuable information though in some cases the biochemical data and crystallographic data for antagonist binding is contradictory [82,83]. In the dDAT crystal structure (PDBID 4PX4) [30] bound cocaine was identified in the central S1 binding site where it could directly compete with DA [41,44,65]. However, some computational and biochemical studies suggest cocaine could bind to the S2 site located in the vestibule on the extracellular side of the outer gate [84–86]. Binding of cocaine to either of these sites would likely stabilize the transporter in an inactive conformation, which would block substrate uptake, but would exert these effects by distinct mechanisms that could be subject to differential pharmacological manipulation [87]. To address these issues, we developed a series of irreversible binding cocaine analogs as a positive-function approach to directly identify their site of incorporation in functional mammalian transporters in combination with molecular modeling and biochemical validation.
Our findings demonstrate covalent attachment of the cocaine photoaffinity analog MFZ 2–24 to the TM1 residue Leu80 and, in conjunction with modeling and SCAM analysis, determined that the binding of the tropane pharmacophore occurs in the S1 substrate binding site. Similar to our previous findings regarding the cocaine photoaffinity analog RTI 82 [65], our top scoring RL and IFD models maintain a salt-bridge interaction between the positively charged tropane nitrogen of MFZ 2–24 and the negatively charged side-chain of residue Asp79, a pose recently supported by the dDAT/cocaine co-crystal [30]. These models also support an interaction between the phenyl chloride of MFZ 2–24 and Asn157, an interaction that parallels previous findings with the phenyl chloride of RTI 82 [65] and the phenyl fluoride of CFT [41]. Cocaine possesses a nonhalogenated benzoyl ester that does not interact with Asn157 [30], suggesting that interaction of a compound with Asn157 may be dependent on the presence of a halogen. Though this interaction was originally thought to contribute to the higher affinity binding of CFT, RTI 82, and MFZ 2–24 [88], recent crystal structures of the cocaine analogs β-CFT and RTI-55 in complex with DAT suggest that Asn157 does not directly participate in binding and that the halogen substituted phenyl groups do not markedly affect the architecture of the binding pocket [30]. This observation supports why, after MD simulation, the phenyl Cl group of MFZ 2–24 in our RL and IFD docked structures moves away from Asn157 to distances greater than 5Å.
Leu80, the adduction site of MFZ 2–24, coordinates substrate binding by forming a hydrophobic-aromatic interaction with Tyr156 and there is evidence that mutation of Leu80 destabilizes Tyr156 interactions resulting in loss of DA uptake though cocaine and CFT binding are unaffected [41,64]. Mutation of the homologous site in SERT also resulted in decreased substrate transport, suggestive of a conserved role for this position in substrate transport [39]. Leu80 is an S1 residue located adjacent to TM1 residue Asp79 which interacts with the tropane nitrogen of MFZ 2–24 offering strong support that MFZ 2–24 inhibits DA uptake through a competitive mechanism either by binding to S1 in an occluded or outward-facing conformation. Our models reveal that an outward-facing conformation is more likely as our docking methods were unable to identify an MFZ 2–24/DAT complex based on an occluded homology model. Furthermore, our SCAM data supports binding of MFZ 2–24 in a manner similar to RTI 82. Cocaine, in contrast to MFZ 2–24 and RTI 82, was able to protect intermediate residues from MTS inactivation suggesting cocaine binds to an occluded conformation of the transporter which would close off the intermediate residues due to interhelical side chain packing whereas the added bulk of the phenyl azido moiety on MFZ 2–24 and RTI 82 likely prevents closure of the extracellular gate and, therefore, stabilizes an outward-facing conformation leaving the intermediate residues accessible to MTS.
Therefore, this study identifies the TM1 S1 residue Leu80 as the single amino acid site of attachment for the photoactive cocaine analog MFZ 2–24 providing additional independent support that tropane-based, cocaine-like compounds bind to S1 and competitively inhibit DA binding. These findings support previous biochemical and crystallographic studies and provide further evidence for the importance of the tropane pharmacophore in driving the interaction of these cocaine-analogs despite addition of the phenyl-azido crosslinking moiety arm to the molecule core.
The parameters of our study do present some limitations. We are restricted by the concentration of radiolabeled probe that can be used and therefore only report high-affinity binding interactions which precludes measurements of binding and adduction at the low-affinity S2 site. Clearly, studies elucidating the full role of S2 are essential and worth pursuing to understand antagonist interactions with DAT. Additionally, the cocaine photoaffinity analogs do have structural differences from cocaine that could result in distinct binding poses. However, the agreement we see between RTI 82 and MFZ 2–24 binding as well as the dDAT-cocaine crystal structure provides strong support for our findings that the tropane core directs the binding of these drugs.
The physiological effects and economic burden imparted by cocaine abuse are several factors that highlight the importance of understanding the mechanism of cocaine binding and transport inhibition. The advent of the benztropine class of DAT antagonists, compounds that do not produce cocaine-like behavioral profiles [89,90], suggests that a cocaine-abuse pharmacotherapeutic is possible. GBR12909 for example was a promising pharmacotherapeutic as it decreased self-administration of cocaine following pretreatment [91–93], but due to prolonged QTc intervals in patients participating in phase II clinical trials [94] and the ability to block multiple ion channels [95], it may support treatment of abnormal heart rhythms instead [96,97]. Recently, R-modafinil has been reported to bind the DAT in a unique fashion [54,98] and has demonstrated efficacy in rodent models of nicotine abuse [99]. Moreover, novel analogues of modafinil that exhibit an atypical DAT inhibitor profile show promise for development as medications to treat psychostimulant use disorders [100–103]. A recent study of the sigma-1 receptor provides even further evidence that interactions of DAT with cytosolic protein partners may be dependent on bound antagonists and suggests these interactions are important for establishment of cocaine-associated behaviors [104]. If DAT blockade was solely required for the cocaine-induced behavioral responses, we would expect all DAT antagonists to elicit invariable outcomes at inhibitory doses. However, this singular mechanism is not supported experimentally and points to a more complex explanation. Indeed, structurally distinct DAT inhibitors are likely to confer local conformational changes that in some cases could be communicated through the protein structure and hide and/or expose regulatory domains involved in protein-protein interactions and/or impact DAT phosphorylation and palmitoylation. Such changes could alter downstream signalling events and account for differential physiological and behavioral outcomes. Hence, understanding cocaine binding in relation to typical and atypical DAT inhibitors and how these compounds can modulate DAT structure and function [87] will provide additional and critical insights to identify promising lead molecules that could be leveraged or structurally augmented to alter these downstream events and possibly lead to the development of cocaine-abuse pharmacotherapeutics.
Acknowledgements
We are grateful to Stephen Combs for providing a Rosetta-based script for distance filtering. DK and LKH designed and performed the substituted cysteine mutagenesis studies. RAD, MJT, JDF and RAV designed and performed the photoaffinity crosslinking studies. ABP, BS and LKH designed and performed the in silico docking analyses. ABP and LKH performed and evaluated the molecular dynamics analyses. MFZ, CB,AHN, and JRL were responsible for design, synthesis and purification of the photoaffinity ligand. The paper was primarily written by DK, RAD, RAV, and LKH but all authors contributed to the writing, reviewed the results, and approved the final version of the manuscript. This work was supported in whole or in part by National Institutes of Health Grants from the INBRE Program of the NCRR and the NIDA-Intramural Research Program and grants Z1A DA000389 Z1A DA000610 and (to AHN.), Grants DA027845 (to LKH and RAV), P20 RR017699 (to University of North Dakota) from the COBRE Program of the NCRR, and Grant P20 RR016741 from ND EPSCoR IIG (to RAV and JDF.). The authors declare that they do not have any conflicts of interest either financial or non-financial with the material contained or discussed in this manuscript.
Footnotes
Chemical compounds cited in this article
CFT (PubChem CID:105056); dopamine HCl (PubChem CID: 65340); RTI-82 (PubChem CID:132374); G418 (PubChem CID:123865); cocaine (PubChem CID:446220); mazindol (PubChem CID:4020)
REFERENCES
- [1].Pramod AB, Foster J, Carvelli L, Henry LK, SLC6 transporters: structure, function, regulation, disease association and therapeutics, Mol. Aspects Med 34 (2013) 197–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kristensen AS, Andersen J, Jørgensen TN, Sørensen L, Eriksen J, Loland CJ, Strømgaard K, Gether U, SLC6 neurotransmitter transporters: structure, function, and regulation, Pharmacol. Rev 63 (2011) 585–640. [DOI] [PubMed] [Google Scholar]
- [3].Bröer S, Gether U, The solute carrier 6 family of transporters, Br. J. Pharmacol 167 (2012) 256–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gainetdinov RR, Caron MG, MONOAMINE TRANSPORTERS: From Genes to Behavior, Annu. Rev. Pharmacol. Toxicol 43 (2003) 261–284. [DOI] [PubMed] [Google Scholar]
- [5].Gether U, Andersen PH, Larsson OM, Schousboe A, Neurotransmitter transporters: molecular function of important drug targets, Trends Pharmacol. Sci 27 (2006) 375–383. [DOI] [PubMed] [Google Scholar]
- [6].Torres GE, Amara SG, Glutamate and monoamine transporters: new visions of form and function, Curr. Opin. Neurobiol 17 (2007) 304–312. [DOI] [PubMed] [Google Scholar]
- [7].Hahn MK, Blakely RD, The functional impact of SLC6 transporter genetic variation, Annu. Rev. Pharmacol. Toxicol 47 (2007) 401–441. [DOI] [PubMed] [Google Scholar]
- [8].Kurian MA, Li Y, Zhen J, Meyer E, Hai N, Christen H-J, Hoffmann GF, Jardine P, von Moers A, Mordekar SR, O’Callaghan F, Wassmer E, Wraige E, Dietrich C, Lewis T, Hyland K, Heales S Jr, Sanger T, Gissen P, Assmann BE, Reith MEA, Maher ER, Clinical and molecular characterisation of hereditary dopamine transporter deficiency syndrome: an observational cohort and experimental study, Lancet Neurol 10 (2011) 54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Vaughan RA, Foster JD, Mechanisms of dopamine transporter regulation in normal and disease states, Trends Pharmacol. Sci 34 (2013) 489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Richardson BD, Saha K, Krout D, Cabrera E, Felts B, Henry LK, Swant J, Zou M-F, Newman AH, Khoshbouei H, Membrane potential shapes regulation of dopamine transporter trafficking at the plasma membrane, Nat. Commun 7 (2016) 10423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ritz MC, Lamb RJ, Goldberg SR, Kuhar MJ, Cocaine receptors on dopamine transporters are related to self-administration of cocaine, Science. 237 (1987) 1219–1223. [DOI] [PubMed] [Google Scholar]
- [12].Bergman J, Madras BK, Johnson SE, Spealman RD, Effects of cocaine and related drugs in nonhuman primates. III. Self-administration by squirrel monkeys, J. Pharmacol. Exp. Ther 251 (1989) 150–155. [PubMed] [Google Scholar]
- [13].Khoshbouei H, Wang H, Lechleiter JD, Javitch JA, Galli A, Amphetamine-induced dopamine efflux. A voltage-sensitive and intracellular Na+-dependent mechanism, J. Biol. Chem 278 (2003) 12070–12077. [DOI] [PubMed] [Google Scholar]
- [14].Wall SC, Gu H, Rudnick G, Biogenic amine flux mediated by cloned transporters stably expressed in cultured cell lines: amphetamine specificity for inhibition and efflux, Mol. Pharmacol 47 (1995) 544–550. [PubMed] [Google Scholar]
- [15].Sitte HH, Huck S, Reither H, Boehm S, Singer EA, Pifl C, Carrier-mediated release, transport rates, and charge transfer induced by amphetamine, tyramine, and dopamine in mammalian cells transfected with the human dopamine transporter, J. Neurochem 71 (1998) 1289–1297. [DOI] [PubMed] [Google Scholar]
- [16].Volkow ND, Wang GJ, Fowler JS, Gatley SJ, Ding YS, Logan J, Dewey SL, Hitzemann R, Lieberman J, Relationship between psychostimulant-induced“ high” and dopamine transporter occupancy, Proceedings of the National Academy of Sciences. 93 (1996) 10388–10392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Rothman RB, Baumann MH, Prisinzano TE, Newman AH, Dopamine transport inhibitors based on GBR12909 and benztropine as potential medications to treat cocaine addiction, Biochem. Pharmacol 75 (2008) 2–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Stoops WW, Lile JA, Glaser PEA, Hays LR, Rush CR, Influence of acute bupropion pretreatment on the effects of intranasal cocaine, Addiction. 107 (2012) 1140–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Morton WA, Stockton GG, Methylphenidate Abuse and Psychiatric Side Effects, Prim. Care Companion J. Clin. Psychiatry 2 (2000) 159–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E, Crystal structure of a bacterial homologue of Na+/Cl--dependent neurotransmitter transporters, Nature. 437 (2005) 215–223. [DOI] [PubMed] [Google Scholar]
- [21].Singh SK, Yamashita A, Gouaux E, Antidepressant binding site in a bacterial homologue of neurotransmitter transporters, Nature. 448 (2007) 952–956. [DOI] [PubMed] [Google Scholar]
- [22].Singh SK, Piscitelli CL, Yamashita A, Gouaux E, A competitive inhibitor traps LeuT in an open-to-out conformation, Science. 322 (2008) 1655–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Krishnamurthy H, Gouaux E, X-ray structures of LeuT in substrate-free outward-open and apo inward-open states, Nature. 481 (2012) 469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Piscitelli CL, Gouaux E, Insights into transport mechanism from LeuT engineered to transport tryptophan, EMBO J. 31 (2012) 228–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Shimada S, Kitayama S, Lin CL, Patel A, Nanthakumar E, Gregor P, Kuhar M, Uhl G, Cloning and expression of a cocaine-sensitive dopamine transporter complementary DNA, Science. 254 (1991) 576–578. [DOI] [PubMed] [Google Scholar]
- [26].Giros B, el Mestikawy S, Godinot N, Zheng K, Han H, Yang-Feng T, Caron MG, Cloning, pharmacological characterization, and chromosome assignment of the human dopamine transporter, Mol. Pharmacol 42 (1992) 383–390. [PubMed] [Google Scholar]
- [27].Itokawa M, Lin Z, Cai N-S, Wu C, Kitayama S, Wang J-B, Uhl GR, Dopamine Transporter Transmembrane Domain Polar Mutants: ΔG and ΔΔG Values Implicate Regions Important for Transporter Functions, Mol. Pharmacol 57 (2000) 1093–1103. [PubMed] [Google Scholar]
- [28].Huang X, Zhan C-G, How dopamine transporter interacts with dopamine: insights from molecular modeling and simulation, Biophys. J 93 (2007) 3627–3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Koldsø H, Christiansen AB, Sinning S, Schiøtt B, Comparative modeling of the human monoamine transporters: similarities in substrate binding, ACS Chem. Neurosci 4 (2013) 295–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wang KH, Penmatsa A, Gouaux E, Neurotransmitter and psychostimulant recognition by the dopamine transporter, Nature. 521 (2015) 322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Coleman JA, Green EM, Gouaux E, X-ray structures and mechanism of the human serotonin transporter, Nature. 532 (2016) 334–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chen F, Larsen MB, Sánchez C, Wiborg O, The S-enantiomer of R,S-citalopram, increases inhibitor binding to the human serotonin transporter by an allosteric mechanism. Comparison with other serotonin transporter inhibitors, Eur. Neuropsychopharmacol 15 (2005) 193–198. [DOI] [PubMed] [Google Scholar]
- [33].Neubauer HA, Hansen CG, Wiborg O, Dissection of an allosteric mechanism on the serotonin transporter: a cross-species study, Mol. Pharmacol 69 (2006) 1242–1250. [DOI] [PubMed] [Google Scholar]
- [34].Plenge P, Shi L, Beuming T, Te J, Newman AH, Weinstein H, Gether U, Loland CJ, Steric hindrance mutagenesis in the conserved extracellular vestibule impedes allosteric binding of antidepressants to the serotonin transporter, J. Biol. Chem 287 (2012) 39316–39326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Plenge P, Mellerup ET, Antidepressive drugs can change the affinity of [3H] imipramine and [3H] paroxetine binding to platelet and neuronal membranes, Eur. J. Pharmacol 119 (1985) 1–8. [DOI] [PubMed] [Google Scholar]
- [36].Carroll FI, Gao Y, Abraham P, Lewin AH, Lew R, Patel A, Boja JW, Kuhar MJ, Probes for the cocaine receptor. Potentially irreversible ligands for the dopamine transporter, J. Med. Chem 35 (1992) 1813–1817. [DOI] [PubMed] [Google Scholar]
- [37].Kitayama S, Shimada S, Xu H, Markham L, Donovan DM, Uhl GR, Dopamine transporter site-directed mutations differentially alter substrate transport and cocaine binding, Proc. Natl. Acad. Sci. U. S. A 89 (1992) 7782–7785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Chen JG, Sachpatzidis A, Rudnick G, The third transmembrane domain of the serotonin transporter contains residues associated with substrate and cocaine binding, J. Biol. Chem 272 (1997) 28321–28327. [DOI] [PubMed] [Google Scholar]
- [39].Henry LK, Adkins EM, Han Q, Blakely RD, Serotonin and cocaine-sensitive inactivation of human serotonin transporters by methanethiosulfonates targeted to transmembrane domain I, J. Biol. Chem 278 (2003) 37052–37063. [DOI] [PubMed] [Google Scholar]
- [40].Henry LK, Field JR, Adkins EM, Parnas ML, Vaughan RA, Zou M-F, Newman AH, Blakely RD, Tyr-95 and Ile-172 in transmembrane segments 1 and 3 of human serotonin transporters interact to establish high affinity recognition of antidepressants, J. Biol. Chem 281 (2006) 2012–2023. [DOI] [PubMed] [Google Scholar]
- [41].Beuming T, Kniazeff J, Bergmann ML, Shi L, Gracia L, Raniszewska K, Newman AH, Javitch JA, Weinstein H, Gether U, Loland CJ, The binding sites for cocaine and dopamine in the dopamine transporter overlap, Nat. Neurosci 11 (2008) 780–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wu X, Gu HH, Cocaine affinity decreased by mutations of aromatic residue phenylalanine 105 in the transmembrane domain 2 of dopamine transporter, Mol. Pharmacol 63 (2003) 653–658. [DOI] [PubMed] [Google Scholar]
- [43].Chen R, Han DD, Gu HH, A triple mutation in the second transmembrane domain of mouse dopamine transporter markedly decreases sensitivity to cocaine and methylphenidate, J. Neurochem 94 (2005) 352–359. [DOI] [PubMed] [Google Scholar]
- [44].Bisgaard H, Larsen MAB, Mazier S, Beuming T, Newman AH, Weinstein H, Shi L, Loland CJ, Gether U, The binding sites for benztropines and dopamine in the dopamine transporter overlap, Neuropharmacology. 60 (2011) 182–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Uhl GR, Lin Z, The top 20 dopamine transporter mutants: structure–function relationships and cocaine actions, Eur. J. Pharmacol 479 (2003) 71–82. [DOI] [PubMed] [Google Scholar]
- [46].Volz TJ, Schenk JO, A comprehensive atlas of the topography of functional groups of the dopamine transporter, Synapse. 58 (2005) 72–94. [DOI] [PubMed] [Google Scholar]
- [47].Surratt CK, Ukairo OT, Ramanujapuram S , Recognition of psychostimulants, antidepressants, and other inhibitors of synaptic neurotransmitter uptake by the plasma membrane monoamine transporters, AAPS J 7 (2005) E739–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Gaffaney JD, Shetty M, Felts B, Pramod A-B, Foster JD, Henry LK, Vaughan RA, Antagonist-induced conformational changes in dopamine transporter extracellular loop two involve residues in a potential salt bridge, Neurochem. Int 73 (2014) 16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Trudell ML, Izenwasser S, Dopamine transporters: chemistry, biology, and pharmacology, Wiley, Hoboken, N.J, 2008. [Google Scholar]
- [50].Plenge P, Mellerup ET, Nielsen M, Inhibitory and regulatory binding sites on the rat brain serotonin transporter: molecular weight of the [3H]paroxetine and [3H]citalopram binding proteins, Eur. J. Pharmacol 189 (1990) 129–134. [DOI] [PubMed] [Google Scholar]
- [51].Plenge P, Wiborg O, High- and low-affinity binding of S-citalopram to the human serotonin transporter mutated at 20 putatively important amino acid positions, Neurosci. Lett 383 (2005) 203–208. [DOI] [PubMed] [Google Scholar]
- [52].Plenge P, Gether U, Rasmussen SG, Allosteric effects of R- and S-citalopram on the human 5-HT transporter: evidence for distinct high- and low-affinity binding sites, Eur. J. Pharmacol 567 (2007) 1–9. [DOI] [PubMed] [Google Scholar]
- [53].Larsen MAB, Plenge P, Andersen J, Eildal JNN, Kristensen AS, Bøgesø KP, Gether U, Strømgaard K, Bang-Andersen B, Loland CJ, Structure-activity relationship studies of citalopram derivatives: examining substituents conferring selectivity for the allosteric site in the 5-HT transporter, Br. J. Pharmacol 173 (2016) 925–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Schmitt KC, Reith MEA, The atypical stimulant and nootropic modafinil interacts with the dopamine transporter in a different manner than classical cocaine-like inhibitors, PLoS One. 6 (2011) e25790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Rothman RB, Ananthan S, Partilla JS, Saini SK, Moukha-Chafiq O, Pathak V, Baumann MH, Studies of the biogenic amine transporters 15. Identification of novel allosteric dopamine transporter ligands with nanomolar potency, J. Pharmacol. Exp. Ther 353 (2015) 529–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Kortagere S, Fontana ACK, Rose DR, Mortensen OV, Identification of an allosteric modulator of the serotonin transporter with novel mechanism of action, Neuropharmacology. 72 (2013) 282–290. [DOI] [PubMed] [Google Scholar]
- [57].Zou MF, Kopajtic T, Katz JL, Wirtz S, Justice JB Jr, Newman AH, Novel tropane-based irreversible ligands for the dopamine transporter, J. Med. Chem 44 (2001) 4453–4461. [DOI] [PubMed] [Google Scholar]
- [58].Lever JR, Zou M-F, Parnas ML, Duval RA, Wirtz SE, Justice JB, Vaughan RA, Newman AH, Radioiodinated azide and isothiocyanate derivatives of cocaine for irreversible labeling of dopamine transporters: synthesis and covalent binding studies, Bioconjug. Chem 16 (2005) 644–649. [DOI] [PubMed] [Google Scholar]
- [59].Karlin A, Akabas MH, Substituted-cysteine accessibility method, Methods Enzymol. 293 (1998) 123–145. [DOI] [PubMed] [Google Scholar]
- [60].Carroll FI, Gao YG, Rahman MA, Abraham P, Parham K, Lewin AH, Boja JW, Kuhar MJ, Synthesis, ligand binding, QSAR, and CoMFA study of 3 beta-(p-substituted phenyl)tropane-2 beta-carboxylic acid methyl esters, J. Med. Chem 34 (1991) 2719–2725. [DOI] [PubMed] [Google Scholar]
- [61].Carroll FI, Mascarella SW, Kuzemko MA, Gao Y, Abraham P, Lewin AH, Boja JW, Kuhar MJ, Synthesis, ligand binding, and QSAR (CoMFA and classical) study of 3 beta-(3’-substituted phenyl)-, 3 beta-(4’-substituted phenyl)-, and 3 beta-(3’,4’-disubstituted phenyl)tropane-2 beta-carboxylic acid methyl esters, J. Med. Chem 37 (1994) 2865–2873. [DOI] [PubMed] [Google Scholar]
- [62].Kotzyba-Hibert F, Kapfer I, Goeldner M, Recent trends in photoaffinity labeling, Angewandte Chemie International Edition in English. 34 (1995) 1296–1312. [Google Scholar]
- [63].Geurink PP, Prely LM, van der Marel GA, Bischoff R, Overkleeft HS, Photoaffinity labeling in activity-based protein profiling, Top. Curr. Chem 324 (2012) 85–113. [DOI] [PubMed] [Google Scholar]
- [64].Parnas ML, Gaffaney JD, Zou MF, Lever JR, Newman AH, Vaughan RA, Labeling of dopamine transporter transmembrane domain 1 with the tropane ligand N-[4-(4-azido-3-[125I]iodophenyl)butyl]-2beta-carbomethoxy-3beta-(4-chlorophenyl)tropane implicates proximity of cocaine and substrate active sites, Mol. Pharmacol 73 (2008) 1141–1150. [DOI] [PubMed] [Google Scholar]
- [65].Dahal RA, Pramod AB, Sharma B, Krout D, Foster JD, Cha JH, Cao J, Newman AH, Lever JR, Vaughan RA, Henry LK, Computational and Biochemical Docking of the Irreversible Cocaine Analog RTI 82 Directly Demonstrates Ligand Positioning in the Dopamine Transporter Central Substrate-binding Site, J. Biol. Chem 289 (2014) 29712–29727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Leaver-Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, Kaufman K, Renfrew PD, Smith CA, Sheffler W, Davis IW, Cooper S, Treuille A, Mandell DJ, Richter F, Ban Y-EA, Fleishman SJ, Corn JE, Kim DE, Lyskov S, Berrondo M, Mentzer S, Popović Z, Havranek JJ, Karanicolas J, Das R, Meiler J, Kortemme T, Gray JJ, Kuhlman B, Baker D, Bradley P, ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules, Methods Enzymol. 487 (2011) 545–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Kaufmann KW, Meiler J, Using RosettaLigand for small molecule docking into comparative models, PLoS One. 7 (2012) e50769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Sherman W, Day T, Jacobson MP, Friesner RA, Farid R, Novel procedure for modeling ligand/receptor induced fit effects, J. Med. Chem 49 (2006) 534–553. [DOI] [PubMed] [Google Scholar]
- [69].Foster JD, Pananusorn B, Vaughan RA, Dopamine transporters are phosphorylated on N-terminal serines in rat striatum, J. Biol. Chem 277 (2002) 25178–25186. [DOI] [PubMed] [Google Scholar]
- [70].Gaffaney JD, Vaughan RA, Uptake inhibitors but not substrates induce protease resistance in extracellular loop two of the dopamine transporter, Mol. Pharmacol 65 (2004) 692–701. [DOI] [PubMed] [Google Scholar]
- [71].Loland CJ, The use of LeuT as a model in elucidating binding sites for substrates and inhibitors in neurotransmitter transporters, Biochim. Biophys. Acta 1850 (2015) 500–510. [DOI] [PubMed] [Google Scholar]
- [72].Wall SC, Innis RB, Rudnick G, Binding of the cocaine analog 2 beta-carbomethoxy-3 beta-(4-[125I]iodophenyl)tropane to serotonin and dopamine transporters: different ionic requirements for substrate and 2 beta-carbomethoxy-3 beta-(4-[125I]iodophenyl)tropane binding, Mol. Pharmacol 43 (1993) 264–270. [PubMed] [Google Scholar]
- [73].Barker EL, Moore KR, Rakhshan F, Blakely RD, Transmembrane domain I contributes to the permeation pathway for serotonin and ions in the serotonin transporter, J. Neurosci 19 (1999) 4705–4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Forrest LR, Tavoulari S, Zhang Y-W, Rudnick G, Honig B, Identification of a chloride ion binding site in Na+/Cl -dependent transporters, Proc. Natl. Acad. Sci. U. S. A 104 (2007) 12761–12766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Zomot E, Bendahan A, Quick M, Zhao Y, Javitch JA, Kanner BI, Mechanism of chloride interaction with neurotransmitter:sodium symporters, Nature. 449 (2007) 726–730. [DOI] [PubMed] [Google Scholar]
- [76].Hess B, Kutzner C, van der Spoel D, Lindahl E, GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation, J. Chem. Theory Comput 4 (2008) 435–447. [DOI] [PubMed] [Google Scholar]
- [77].Madras BK, Spealman RD, Fahey MA, Neumeyer JL, Saha JK, Milius RA, Cocaine receptors labeled by [3H]2 beta-carbomethoxy-3 beta-(4-fluorophenyl)tropane, Mol. Pharmacol 36 (1989) 518–524. [PubMed] [Google Scholar]
- [78].Boja JW, Carroll FI, Vaughan RA, Kopajtic T, Kuhar MJ, Multiple binding sites for [125I]RTI-121 and other cocaine analogs in rat frontal cerebral cortex, Synapse. 30 (1998) 9–17. [DOI] [PubMed] [Google Scholar]
- [79].Missale C, Castelletti L, Govoni S, Spano PF, Trabucchi M, Hanbauer I, Dopamine uptake is differentially regulated in rat striatum and nucleus accumbens, J. Neurochem 45 (1985) 51–56. [DOI] [PubMed] [Google Scholar]
- [80].McElvain JS, Schenk JO, A multisubstrate mechanism of striatal dopamine uptake and its inhibition by cocaine, Biochem. Pharmacol 43 (1992) 2189–2199. [DOI] [PubMed] [Google Scholar]
- [81].Wang H, Goehring A, Wang KH, Penmatsa A, Ressler R, Gouaux E, Structural basis for action by diverse antidepressants on biogenic amine transporters, Nature. 503 (2013) 141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Andersen J, Stuhr-Hansen N, Zachariassen LG, Koldsø H, Schiøtt B, Strømgaard K, Kristensen AS, Molecular basis for selective serotonin reuptake inhibition by the antidepressant agent fluoxetine (Prozac), Mol. Pharmacol 85 (2014) 703–714. [DOI] [PubMed] [Google Scholar]
- [83].Davis BA, Nagarajan A, Forrest LR, Singh SK, Mechanism of Paroxetine (Paxil) Inhibition of the Serotonin Transporter, Sci. Rep 6 (2016) 23789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Huang X, Gu HH, Zhan C-G, Mechanism for Cocaine Blocking the Transport of Dopamine: Insights from Molecular Modeling and Dynamics Simulations, J. Phys. Chem. B 113 (2009) 15057–15066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Merchant BA, Madura JD, Insights from molecular dynamics: The binding site of cocaine in the dopamine transporter and permeation pathways of substrates in the leucine and dopamine transporters, J. Mol. Graph. Model 38 (2012) 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Schmitt KC, Rothman RB, Reith MEA, Nonclassical pharmacology of the dopamine transporter: atypical inhibitors, allosteric modulators, and partial substrates, J. Pharmacol. Exp. Ther 346 (2013) 2–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Cheng MH, Block E, Hu F, Cobanoglu MC, Sorkin A, Bahar I, Insights into the Modulation of Dopamine Transporter Function by Amphetamine, Orphenadrine, and Cocaine Binding, Front. Neurol 6 (2015) 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Runyon SP, Carroll FI, Tropane-based dopamine transporter-uptake inhibitors, Trudell MLSI(ed) Dopamine Transporters, Chemistry, Biology, and Pharmacology. Wiley, New York. (2008) 125–170. [Google Scholar]
- [89].Agoston GE, Wu JH, Izenwasser S, George C, Katz J, Kline RH, Newman AH, Novel N-substituted 3 alpha-[bis(4’-fluorophenyl)methoxy]tropane analogues: selective ligands for the dopamine transporter, J. Med. Chem 40 (1997) 4329–4339. [DOI] [PubMed] [Google Scholar]
- [90].Katz JL, Kopajtic TA, Agoston GE, Newman AH, Effects of N-substituted analogs of benztropine: diminished cocaine-like effects in dopamine transporter ligands, J. Pharmacol. Exp. Ther 309 (2004) 650–660. [DOI] [PubMed] [Google Scholar]
- [91].Baumann MH, Char GU, De Costa BR, Rice KC, Rothman RB, GBR12909 attenuates cocaine-induced activation of mesolimbic dopamine neurons in the rat, J. Pharmacol. Exp. Ther 271 (1994) 1216–1222. [PubMed] [Google Scholar]
- [92].Glowa JR, Wojnicki FHE, Matecka D, Rice KC, Effects of dopamine reuptake inhibitors on food- and cocaine-maintained responding: II. Comparisons with other drugs and repeated administrations, Exp. Clin. Psychopharmacol 3 (1995) 232–239. [Google Scholar]
- [93].Tella SR, Effects of monoamine reuptake inhibitors on cocaine self-administration in rats, Pharmacol. Biochem. Behav 51 (1995) 687–692. [DOI] [PubMed] [Google Scholar]
- [94].Vocci FJ, Acri J, Elkashef A, Medication development for addictive disorders: the state of the science, Am. J. Psychiatry 162 (2005) 1432–1440. [DOI] [PubMed] [Google Scholar]
- [95].Lacerda AE, Kuryshev YA, Yan G-X, Waldo AL, Brown AM, Vanoxerine: cellular mechanism of a new antiarrhythmic, J. Cardiovasc. Electrophysiol 21 (2010) 301–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Obejero-Paz CA, Bruening-Wright A, Kramer J, Hawryluk P, Tatalovic M, Dittrich HC, Brown AM, Quantitative Profiling of the Effects of Vanoxerine on Human Cardiac Ion Channels and its Application to Cardiac Risk, Sci. Rep 5 (2015) 17623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Piccini JP, Simon DN, Steinberg BA, Thomas L, Allen LA, Fonarow GC, Gersh B, Hylek E, Kowey PR, Reiffel JA, Naccarelli GV, Chan PS, Spertus JA, Peterson ED, Outcomes Registry for Better Informed Treatment of Atrial Fibrillation (ORBIT-AF) Investigators and Patients, Differences in Clinical and Functional Outcomes of Atrial Fibrillation in Women and Men: Two-Year Results From the ORBIT-AF Registry, JAMA Cardiol. 1 (2016) 282–291. [DOI] [PubMed] [Google Scholar]
- [98].Loland CJ, Mereu M, Okunola OM, Cao J, Prisinzano TE, Mazier S, Kopajtic T, Shi L, Katz JL, Tanda G, Newman AH, R-modafinil (armodafinil): a unique dopamine uptake inhibitor and potential medication for psychostimulant abuse, Biol. Psychiatry 72 (2012) 405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Wang X-F, Bi G-H, He Y, Yang H-J, Gao J-T, Okunola-Bakare OM, Slack RD, Gardner EL, Xi Z-X, Newman AH, R-modafinil attenuates nicotine-taking and nicotine-seeking behavior in alcohol-preferring rats, Neuropsychopharmacology. 40 (2015) 1762–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Okunola-Bakare OM, Cao J, Kopajtic T, Katz JL, Loland CJ, Shi L, Newman AH, Elucidation of structural elements for selectivity across monoamine transporters: novel 2-[(diphenylmethyl)sulfinyl]acetamide (modafinil) analogues, J. Med. Chem 57 (2014) 1000–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Cao J, Prisinzano TE, Okunola OM, Kopajtic T, Shook M, Katz JL, Newman AH, Structure-Activity Relationships at the Monoamine Transporters for a Novel Series of Modafinil (2-[(diphenylmethyl)sulfinyl]acetamide) Analogues, ACS Med. Chem. Lett 2 (2010) 48–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Cao J, Slack RD, Bakare OM, Burzynski C, Novel and High Affinity 2-[(Diphenylmethyl) sulfinyl] acetamide (Modafinil) Analogues as Atypical Dopamine Transporter Inhibitors, Journal of Medicinal. (2016). http://pubs.acs.org/doi/abs/10.1021/acs.jmedchem.6b01373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Zhang H-Y, Bi G-H, Yang H-J, He Y, Xue G, Cao J, Tanda G, Gardner EL, Newman AH, Xi Z-X, The novel modafinil analog, JJC8–016, as a potential cocaine abuse pharmacotherapeutics, Neuropsychopharmacology. (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Hong WC, Yano H, Hiranita T, Chin FT, McCurdy CR, Su T-P, Amara SG, Katz JL, The sigma-1 receptor modulates dopamine transporter conformation and cocaine binding and may thereby potentiate cocaine self-administration in rats, J. Biol. Chem (2017). doi: 10.1074/jbc.M116.774075. [DOI] [PMC free article] [PubMed] [Google Scholar]