

SUMMARY
KRAS G12C, the most common RAS mutation found in non-small-cell lung cancer, has been the subject of multiple recent covalent small-molecule inhibitor campaigns including efforts directed at the guanine nucleotide pocket and separate work focused on an inducible pocket adjacent to the switch motifs. Multiple conformations of switch II have been observed, suggesting that switch II pocket (SIIP) binders may be capable of engaging a range of KRAS conformations. Here we report the use of hydrogen/deuterium-exchange mass spectrometry (HDX MS) to discriminate between conformations of switch II induced by two chemical classes of SIIP binders. We investigated the structural basis for differences in HDX MS using X-ray crystallography and discovered a new SIIP configuration in response to binding of a quinazoline chemotype. These results have implications for structure-guided drug design targeting the RAS SIIP.
Graphical Abstract
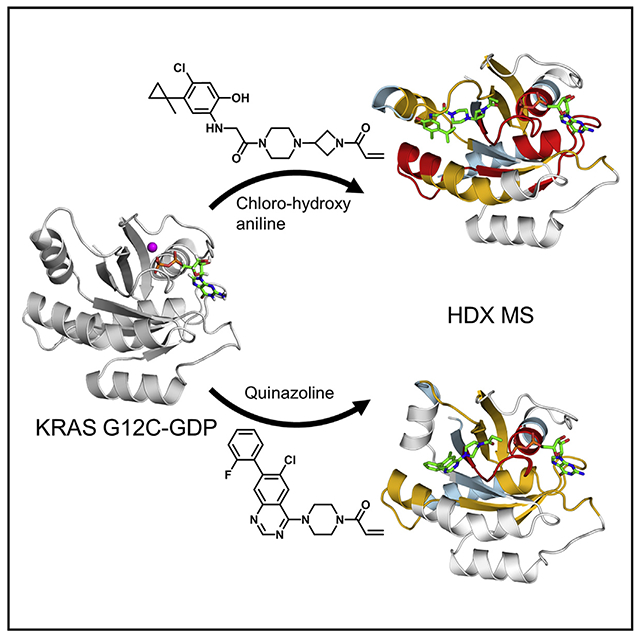
In Brief
Covalent inhibitors are promising for addressing cancers driven by KRAS G12C, the most common KRAS mutation in lung cancer. Lu et al. characterize a quinazoline switch II pocket (SIIP) inhibitor and demonstrate the utility of HDX MS for characterizing SIIP compounds.
INTRODUCTION
RAS mutations were some of the first identified acquired genetic causes of cancer and are common in many forms of malignancy, particularly gastrointestinal, skin, and lung cancers (Prior et al., 2012). Oncogenic RAS mutations result in a dysregulated excess of cellular GTP-bound RAS, the activated form for RAS, which transduces signals resulting in processes necessary for cancer such as cell growth and proliferation (Pylayeva-Gupta et al., 2011), invasiveness (Campbell and Der, 2004; Lundy et al., 1986), and evasion of immune surveillance (Seliger et al., 1998; Weijzen et al., 1999). RAS transduces signals by direct protein-protein interactions raising the possibility that specific mutations may impact RAS function within the context of cancer in unique ways. This hypothesis is supported not only by biochemical (Hunter et al., 2015) and laboratory models (Fasano et al., 1984; Feig and Cooper, 1988; Kim et al., 2016), but also by a stratification in the clinical behavior of tumors bearing certain RAS mutations (Montalvo et al., 2016). Accordingly, RAS mutation-specific strategies are under development.
The interface for most RAS-RAS effector interactions centers around two dynamic structural elements in RAS called switch I and switch II. Consistent with the role of guanosine nucleotides in controlling the signaling state of RAS, the switches also form a large portion of the guanosine nucleotide binding pocket. Accordingly, the conformations of switches I and II are responsive to the identity of the bound nucleotide, with the GTP-bound form being more compact by virtue of interactions between switch II and the terminal gamma phosphate of GTP that draw switch II toward the protein center (Figure 1A). In the absence of that interaction, switch II is less constrained and has been observed in multiple conformations in X-ray crystal and nuclear magnetic resonance structures. This mobility in switch II has enabled unanticipated strategies to target KRAS G12C directly.
Figure 1. Switch II Configuration Changes in Response to SIIP Inhibitors.

(A) GTP-bound HRAS (PDB: 4L9W) shows that the helical portion of switch II (α2) engages the gamma phosphate of GTP, resulting in a closed conformation. Switch II in green, switch I in yellow, GTP in spheres.
(B) ARS-853-bound KRAS G12C (PDB: 5F2E) shows an open switch II conformation. In addition to α2, ARS-853 (cyan spheres) also interacts with α3.
(C and D) Chemical structures of ARS-853 and 1.
KRAS G12C is the most common KRAS mutation found in non-small-cell lung cancer and presents a fortuitous opportunity to selectively target this oncogenic KRAS mutation directly using covalent inhibitors, given that cysteine 12 extends a nucleophilic thiol side chain susceptible to electrophilic attack. The established use of cysteine as a target for rationally designed covalent warheads in small-molecule inhibitors (Liu et al., 2013) prompted several groups, including ours, to explore the KRAS G12C mutation as a handle for direct KRAS targeting in a G12C, tumor-selective manner. We demonstrated that the guanine nucleotide binding site immediately adjacent to the G12C mutation is targetable by using a covalent guanosine diphosphate (GDP)-mimetic compound, SML 8-73-1 (Lim et al., 2014). SML 8-73-1 competed efficiently with more than three times physiologic concentrations of GDP and guanosine triphosphate (GTP) to irreversibly inactivate KRAS G12C and is highly selective for KRAS G12C over other GTP-binding proteins (Hunter et al., 2014). Simultaneously, the group led by Shokat and Wells showed that a binding site on the opposite side of Cys12, beneath switch II, could also be utilized to target Cys12 (Ostrem et al., 2013). This class of compounds, termed switch II pocket (SIIP) binders, requires a substantial rearrangement of switch II to accommodate binding (Figure 1B). This class of compounds has been evolved to improve both potency and selectivity to yield lead compounds with potencies in the low micromolar range (Patricelli et al., 2016).
Multiple chemical scaffolds have been reported as SIIP binders, but structural information is available for one major class consisting of a chloro hydroxy aniline core with a reactive acrylamide warhead (Figure 1C) (Patricelli et al., 2016). However, quinazoline-containing compounds have also been reported (Figure 1D; Table S1) (Li et al., 2015), although characterized in a limited fashion. The flexibility and conformational dynamics of switch II complicates structure-guided drug design work on quinazoline scaffolds because the conformation of switch II is uncertain. Here we demonstrate that hydrogen/deuterium-exchange mass spectrometry (HDX MS), a method capable of detecting differences in switch II dynamics for native RAS family proteins (Harrison et al., 2016), can detect differences in the conformational state of switch II induced by the quinazoline versus chloro hydroxy aniline scaffolds. Further, we use X-ray crystallography to explain the differences in HDX MS signatures seen after treatment with quinazoline-containing compounds. This analysis reveals a new configuration of the SIIP and has implications for subsequent iterations of compound optimization or design.
RESULTS
Differential Effects of Classes of SIIP Binders
When the original series of chloro hydroxy aniline compounds was reported, we also noted that a series of 4-piperazino quinazoline compounds were also included in patent claims but characterization was limited (Li et al., 2015). We hypothesized that these compounds, with their divergent chemical scaffolds, had the potential to engage KRAS G12C SIIP in different ways leading to rearrangement of switch II and the binding pocket. As high level tests for differences in how these two classes of compounds interact, we analyzed these compounds in biochemical and cellular assays.
To assess the relative impact of the two different compounds on the thermodynamic stability of these two protein-ligand complexes, we conducted differential scanning fluorimetry (DSF) on compound-labeled KRAS G12C protein samples. DSF is a convenient method for measuring thermal stability that relies on a dye that becomes fluorescent when in contact with hydrophobic residues as proteins unfold upon heating, and has been used previously to characterize ARS-853 (Lito et al., 2016). KRAS G12C was overexpressed and purified as previously reported, then exposed to either ARS-632, a chloro hydroxy aniline compound similar to ARS-853 (Table S1), and 1, a chemotype based on a 4-piperazino quinazoline compound, until covalent labeling was complete as measured by mass spectrometry (MS) (Figure S1). The melting temperatures (Tm) of KRAS G12C labeled with either 1 or ARS-632 demonstrated a substantial increase in Tm of 10.5 or 13.5, respectively, compared with GDP-bound protein (Figures 2A and 2B).
Figure 2. KRAS G12C-1 and KRAS G12C-ARS-632 Both Show Enhanced Thermal Stability and Similar in Potency for Inhibition of KRAS G12C-Depenent MAPK Signaling in H358 Cells.

(A) Raw DSF measurements for KRAS G12C fully labeled with either ARS-632 or 1.
(B) First derivative of (A) shows the shifts in Tm (dashed vertical lines) associated with protein labeling.
(C) Impact of compound exposure on MAPK signaling. Levels of pERK were evaluated in the KRAS-G12C-containing cancer cell line H358 as a surrogate for RAS signaling through the MAPK pathway. Cells were treated with compound 1 or ARS-632 as indicated for 6 hr. Phosphorylation of ERK1/2 was determined by immunoblotting.
As an additional measure of compound reactivity, we performed a kinetic chemosensor assay that detects the availability of free thiol in compound-exposed KRAS G12C over time using a cysteine-reactive compound, 7-diethylamino-3-(4-maleimidophenyl)-4-methylcoumarin. Of note, we have previously used a similar assay setup to evaluate binding of covalent GDP-competitive compounds (Hunter et al., 2014). In this assay, all three compounds were statistically similar in their labeling rates, with half-lives of 10–15 min (Figure S2).
To assess the relative effects of 1 versus ARS-632 on KRAS signaling in cells, we subjected the KRAS G12C mutant cell line H358 to escalating concentrations of compounds and measured the phosphorylation of ERK, a prominent downstream member of the MAPK signaling pathway. Compounds showed roughly equivalent potencies such that 10 μM concentrations were required to reduce pERK by roughly 50% (Figure 2C). Of note, no reduction was seen in cancer cell lines containing other non-G12C mutations (A549, H441, and HCT116), indicating the selectivity of compound 1 for KRAS G12C (Figure S3). Interestingly, with escalating compound concentrations we also noted a decrease in the RAS band and simultaneous appearance of a new higher-molecular-weight band. We demonstrated that this phenomenon is due to a shift in the protein mobility after compound labeling by performing SDS-PAGE on compound-labeled recombinant KRAS G12C (Figure S4). Of note, this phenomenon is further characterized and confirmed in a separate manuscript reporting analogs of 1 (Zeng et al., 2017). Together, these results suggest that the currently available quinazoline class compounds are similar in potency to previously reported chloro hydroxy anilines. We further characterized 1 to understand if further optimization is possible for the quinazoline class of compounds.
HDE MS
HDX MS has been used to compare the conformations of protein states, such as native versus non-native, wild-type versus mutant, and apo- versus protein- or ligand-bound (Engen, 2003; Pirrone et al., 2015). We previously used HDX MS to differentiate between guanosine monophosphate-purine nucleoside phosphorylase- and GDP-bound KRAS G12C, and showed that the protein dynamics of KRAS G12C covalently bound to GDP-mimetic SML-8-73-1 are similar to the inactive, GDP-bound protein (Lim et al., 2014). In another study, we used HDX MS to elucidate differences in switch II behavior that distinguish members of the Ras subfamily from the Rho subfamily (Harrison et al., 2016). Given the ability of HDX MS to detect changes in switch II dynamics, we hypothesized that HDX MS would be able to detect different conformations of switch II induced by the two major classes of SIIP binders, if such differences exist.
Samples containing KRAS G12C alone and covalently bound to the compounds were independently exposed to deuterium, the exchange reaction quenched, the protein digested into peptides using pepsin, and the relative deuterium level of each peptide measured using MS. To determine the effects of each compound on KRAS G12C dynamics, the relative deuterium level of protein alone was subtracted from the relative deuterium level of compound-bound protein at each labeling time point for each peptide (Figure 3A). Covalent attachment to either SIIP inhibitor significantly altered the protein dynamics of KRAS G12C, as demonstrated by the reduction of deuteration throughout many regions of KRAS G12C upon binding either ARS-632 or 1. The greatest difference in HDX of KRAS G12C when bound to these compounds occurred in switch II (Figures 3B and 3C). In switch II, the difference in deuteration was >1.0 Da at every single time point (see also Figure S5). While binding either compound impacted identical regions of KRAS G12C, the effects of ARS-632 were always larger than those of 1. The C-terminal portion of switch II incorporated 2.0–2.5 Da less deuterium between 1 and 10 min when bound to ARS than when bound to 1. The effects of ARS-632 were more than those of 1 in other regions as well, including in the region containing the DxxG motif and the region immediately N-terminal to the N/TKxD motif (containing α portion of an a helix that is proximal to the SIIP). Other parts of KRAS (the P loop (GxxGxGKS/T), switch I, the N/TKxD motif, and near the C terminus), were only subtly affected by compound binding and the differences between the HDX signatures was very small. The HDX results reveal that these compounds clearly affected the structures to different degrees and in different places.
Figure 3. HDX MS Detects Changes in Switch II dynamics Induced by SIIP Binders.

(A) Differences in HDX are represented by relative deuterium levels of inactive protein covalently bound to compound minus that of inactive GDP-bound protein alone, scale shown at top. Two-dimensional representations of KRAS G12C are given in linear fashion from N terminus (top) to C terminus (bottom), and the locations of key structural elements are shown on the left. All deuterium-labeling time points are shown, increasing from left to right.
(B and C) Crystal structures of 1 (B) or ARS-853 (C) bound to KRAS G12C with HDX MS differences at labeling time point 1 min (upper panel) and 4 hr (lower panel) annotated using the color scheme from subpanel (A).
(D) KRAS G12C bound to GDP alone (PDB: 4LDJ) provided for comparison. In (D), switch I is shown in yellow and switch II in green; magnesium is represented as a magenta sphere.
Crystal Structure of 1-KRAS G12C
To understand the structural basis for the HDX MS findings, we solved an X-ray crystal structure of KRAS G12C bound to 1. Hexagon-shaped protein crystals were readily obtained from 1.8 M sodium phosphate monobasic monohydrate, potassium phosphate dibasic, pH 6.9, and diffracted to 2.2 Å when subjected to synchrotron radiation. Analysis revealed a P3 space group with a unit cell a = 84.9, b = 84.9, and c = 130.8 Å. The structure was solved using molecular replacement with GDP-bound KRAS (PDB ID: 4OBE) as a search model (Table 1).
Table 1.
X-Ray Diffraction and Refinement Statistics
| Data Collection | G12C-1 |
|---|---|
| Space group | P3 |
| Cell dimensions | |
| a, b, c (Å) | 84.93, 84.93, 130.79 |
| α, β, γ (°) | 90.00, 90.00, 120.00 |
| Resolution (Å) | 50.00–2.23 (2.27–2.23) |
| Rsym or Rmerge | 0.183 (1.536) |
| I/sI | 17.00 (1.33) |
| Completeness (%) | 87.7 (48.0) |
| Redundancy | 7.8 (5.0) |
| Refinement | |
| Resolution (Å) | 40.39–2.23 |
| No. of reflections | 45,197 |
| Rwork/Rfree | 0.189/0.230 |
| No. of atoms | |
| Protein | 8,082 |
| Ligand/ion | 342 |
| Water | 375 |
| B factors | |
| Protein | 42.10 |
| Ligand/ion | 32.09 |
| Water | 37.19 |
| RMSD | |
| Bond lengths (Å) | 0.002 |
| Bond angles (°) | 0.596 |
RMSD, root-mean-square deviation.
Additional electron density within the SIIP confirmed that 1 binds between helix α2 and helix α3 in the presence of GDP, similar to previously reported SIIP binders, such as compounds 12 and ARS-853, ARS-853 being identical to ARS-632 with the exception of a methyl substituent. However, different from prior co-crystal structures, a new configuration of switch II was observed with the helix α2 (part of switch II) extended away from the main body of the protein compared with ARS- or GDP-only-bound protein (Figures 3B-3D). The extended configuration of switch II is consistent with the HDX MS findings showing rapid deuterium exchange for switch II in the sample treated with 1, based on the greater degree of switch II solvent exposure (Figures 3B and 3D). It is also consistent with the relatively more rapid exchange in helix α3 in 1-bound protein attributable to fewer interactions between helix α3, helix α2, and inhibitor, as seen in the ARS-bound structures.
The open switch II is partially driven by conformational changes to avoid a steric clash that would occur between the quinazoline ring and Glu62 if Glu62 remained in the same position as seen with ARS-853. As a result, interactions between the compound and Tyr64 and Arg68 are lost, leaving switch II less unconstrained. Met72 is rotated away from helix α3 to accommodate 1’s phenyl ring, also contributing to an outward rotation of helix α2. The piperazine ring of 1 also displaces loop 4 (part of switch II) away from the binding pocket. As a result, the entirety of switch II is shifted away from the protein main body. Otherwise, 1 binding is stabilized by hydrogen bonds to His95 and Lys16 in addition to multiple hydrophobic interactions with Tyr96, Gln99, Thr58, and Met72. In contrast, when bound to ARS-853, Met72 points toward helix α3 because the phenolic hydroxyl group and carbonyl of ARS-853 form hydrogen bonds with Asp69 and Arg68 on helix α2, respectively. A diagrammatic representation is shown in Figure 4.
Figure 4. Schematic Representation of Key Interactions between Switch II Compounds and Adjacent α Helices.

(A and B) Different interaction patterns are observed for 1 versus ARS-853 in the switch II binding pocket: (A) 1 is stabilized by pi-pi interaction with helix α3 residues His95 and Tyr96. (B) Arg68 and Asp69 in helix α2 (switch II) make hydrogen bonds to stabilize ARS-853.
DISCUSSION
Here we describe the structure of a previously unreported SIIP inhibitor-induced configuration of KRAS G12C resulting from binding to a quinazoline scaffold wherein the switch II position is dramatically altered, presenting a reconfigured SIIP that is applicable to RAS-directed drug design. These conformational changes in KRAS were readily demonstrated by HDX MS such that binding to ARS-632 resulted in a reduction in deuterium exchange in the SIIP relative to unbound protein, while exchange in the SIIP of KRAS G12C bound to 1 was accelerated relative to ARS-632 because of fewer interactions between 1 and switch II, leading to a solvent exposed switch II. All other portions of the compound-bound proteins exchanged deuterium similarly whether bound to ARS-632 or 1, consistent with our X-ray crystal structure. Both prototypical chemotypes showed similar thermal stability and efficacy for inhibition of MAPK signaling in KRAS G12C-dependent cells.
These results have implications for future efforts to rationally design new iterations of SIIP-targeted RAS therapies. Key principles related to how ligands engage the dominant protein-ligand interfaces within the SIIP and how these interactions in turn dictate the state of switch II can be discerned from these results. The binding pocket is composed of three main interfaces: negatively charged residues Glu62 and Glu63 on loop 4, the charged residues Arg68 and Asp69 on helix α2, and the hydrophobic residues His95 and Tyr96 on helix α3. The nature of interactions with these groups dictates the overall switch II conformation, particularly Arg68. ARS-632 primarily engages two interfaces on switch II while 1 primarily interacts with the hydrophobic residues on helix α3. Key among the ARS-632 interactions is hydrogen bonds between ARS-632 and Arg68 and Asp69, which constrain switch II in a closed conformation that is less solvent accessible (Figure S6). Interestingly, previous SIIP binders (Ostrem et al., 2013), such as compound 8 (Figure S7), which do not form hydrogen bonds with Arg68 but instead engage in long-range hydrophobic interactions with gamma and delta carbons in the Arg68 side chain, adopt an intermediate (half-way between that seen with ARS-632 versus 1) switch II conformation (Figure S7). Of note, a comparison of temperature factors for helix α2 between PDB: 5F2E (ARS-853) and PDB: 5V71 (current structure) show averages of 12.6 versus 11.9 for helix α2 versus total protein in PDB: 5F2E (1.4 Å structure) and 56.2 versus 42.1 for PDB: 5V71, suggesting relatively more flexibility for helix α2 in PDB: 5V71. If this flexibility enables formation of additional interactions between helix α3 and derivatives of 1, this would be predicted to improve the potency of this class of compounds. Harnessing these structural principles will likely be important for KRAS G12C-directed targeting strategies, where compound potency remains an issue given the micromolar potency of ARS-632 and 1 for inhibition of MAPK signaling (Patricelli et al., 2016). Our new structure will not only aid further exploration of the quinazoline chemotype, it also reveals a new conformation for the KRAS SIIP that may be targetable by other entirely different chemotypes discoverable through structure-guided design principles.
The SIIP may have utility beyond covalent inhibitors that can only be applied to the KRAS G12C mutation. The SIIP was discovered in, and to date has primarily been considered in the context of, KRAS G12C, which presents an accessible cancer-specific cysteine as a handle for covalent binding of small molecules containing an electrophilic warhead. However, in addition to covalent inhibitors, the SIIP may also be applicable to selective reversible inhibition of other oncogenic mutants or other cancer-associated RAS superfamily members. We believe that mutation-specific selectivity is plausible because the locations of oncogenic mutation hotspots in KRAS, codons 12, 13, and 61, either form part of the SIIP or are immediately adjacent to it, raising the possibility of engineering specificity by creating interactions between specific cancer-associated KRAS mutations and substituents of SIIP-binding compounds. Indeed, it is encouraging that recent attempts to develop reversible inhibitors of KRAS G12D based on interactions with switch I and switch II have gained traction (Welsch et al., 2017). However, it should be noted that these new inhibitors do not utilize the SIIP, which may hold advantages both for potency and selectivity. Nevertheless, the key barrier to generating reversible SIIP-binding compounds will be achieving sufficient binding affinity, a property that has not been measured.
It bears mentioning that the most commonly used biochemical assay techniques for screening and evaluating the relative potencies of SIIP binders have been MS-based measures of irreversible compound binding, and these techniques cannot be used to measure affinities of reversible candidate SIIP compounds. So far, development of assays that can measure non-covalent interactions within the SIIP has been challenging. Indeed, as part of this study we attempted to measure the affinity between a non-covalent analog of compound 1 and KRAS G12C using isothermal calorimetry but could not detect binding. This could be due to limitations inherent to the assay technique or due to lack of binding activity for the experimental compound, but distinguishing between the two is difficult in the absence of a reversible SIIP-binding positive control. Lack of effective assays that accurately assess the state of switch II and the SIIP limits progress in searching for reversible SIIP-binding compounds. As shown here, HDX MS can be used to effectively compare the effects of various compounds, although HDX MS would likely not be attractive as a primary screening assay due to practical limits to throughput. Indeed, development of efficient screening methods focused on the SIIP would be greatly facilitated by the availability of intermediate- to high-potency reversible probes that could be conjugated to fluorophores or other easily detectable compounds; however, such probes are currently unavailable. Additional assay techniques will be required to develop such probes. This work, together with our prior published work, implicate HDX MS as a promising technique for identifying the next generation of SIIP-binding inhibitors given its ability to detect changes in switch II dynamics.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Kenneth Westover (Kenneth.Westover@UTSouthwestern.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
Sources of Cell Lines
Human lung cancer cell lines, including cell lines H358 (KRAS G12C), A549 (KRAS G12S), and H441 (KRAS G13V) were authenticated and obtained from Pasi A. Janne’s laboratory. Cell line HCT116 (KRAS G13D) was obtained from ATCC and was not further authenticated. All cells were grown in RPMI1640 or DMEM medium (Life Technologies), supplemented with 10% fetal bovine serum (Gibco), 50 units/mL penicillin, 50 units/mL streptomycin, and maintained in humidified 37°C/5% CO2 incubator.
Sources of Bacterial Strains
Escherichia coli BL21(DE3)
METHOD DETAILS
HDX MS
Each compound (1 and ARS, 10 mM in DMSO) was slowly diluted into sample buffer (10 mM HEPES, 25 mM NaCl, pH 8.0) until a final concentration of 1000 μM was achieved. This solution was then combined with an equal volume (10:1 molar excess, compound:protein) of KRAS G12C (100 μM in 20 mM HEPES, 25 mM NaCl, pH 8.0) and allowed to incubate for approximately 18 hours at room temperature. A compound-free protein sample was also prepared by combining KRAS G12C alone with an equal volume of 90:10 sample buffer:DMSO, and this solution was also allowed to incubate in an identical manner. Intact mass analyses were performed to confirm that nearly all KRAS G12C was labeled with compound (Figure S1), and that all protein samples would remain stable for the duration of analyses. It was also confirmed that the 5% DMSO in all protein samples used for labeling did not significantly impact HDX MS results (data not shown).
HDX MS experiments were performed in a similar manner to those described previously (Harrison et al., 2016; Lim et al., 2014). Samples containing KRAS G12C alone and bound to each compound (1 and ARS) were independently labeled with deuterium in triplicate, using identical experimental conditions so that each compound-bound form could be compared to KRAS G12C alone. HDX was initiated by diluting 4.0 μL of protein sample (50 μM in 15 mM HEPES, 25 mM NaCl, pH 8.0) 15-fold in labeling buffer (10 mM HEPES, 25 mM NaCl, pD 8.0) containing 99% deuterium oxide at room temperature. The labeling reaction was quenched at five predetermined time points (10 s, 1 m, 10 m, 1 h, 4 h) through the addition of 64.0 μL quench buffer (2.0 M guanidinium chloride, 0.8% formic acid, pH 2.1) at 0°C. Quenched samples were immediately flash frozen using dry ice and were stored at −80°C for less than one week prior to analysis. Deuterium measurement with mass spectrometry was performed as previously described (Harrison et al., 2016; Lim et al., 2014). Deuterium incorporation graphs (Figure S5) were generated using DynamX 3.0 software (Waters) by subtracting the centroid of the isotopic distribution at each labeling time point from the centroid of the isotopic distribution of the undeuterated reference species. Since the data were not corrected for back-exchange, each data point represents the relative deuterium level at each time point for each peptide (Wales and Engen, 2006). The average error of triplicate measurements at a single data point was ±0.08 Da, therefore any differences in relative deuterium level >0.50 Da between compound-bound and compound-free KRAS G12C were considered to be meaningful.
Protein Preparation and Crystallization
Protein was expressed and purified as described previously (Hunter et al., 2014; Lim et al., 2014). Point mutations were generated using the GeneArt® site-directed mutagenesis system (Life Technologies). A construct encoding codon-optimized N-terminal His-tobacco etch virus (TEV)-G12C V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog (K-Ras) in the pJExpress vector (DNA2.0) was synthesized and used to transform BL21(DE3) cells. Cells were grown in Luria broth (LB) to OD 600 0.7 and induced with 250 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) for 16 h at 16°C. Cells were pelleted and resuspended in lysis buffer [20 mM sodium phosphate (pH 8.0), 500 mM NaCl, 10 mM imidazole, 1 mM 2-mercaptoethanol (BME), 5% (vol/vol) glycerol] containing PMSF, benzamidine, and 1 mg/mL lysozyme. Lysates were flash-frozen and stored at −80°C until use. Protein was purified over an IMAC cartridge (BioRad) following standard Ni-affinity protocols and desalted into crys-tallization buffer [20 mM Hepes (pH 8.0), 150 mM NaCl, 5 mM MgCl2, 0.5 mM DTT]. The N-terminal His tag was cleaved by overnight digestion with a 1:5 ratio of TEV protease at 4°C, and the TEV and Tag were removed by reverse purification over an IMAC cartridge. Protein was concentrated to 30–40 mg/mL in a 10-kDa cutoff Amicon filter (Millipore), aliquoted, and then flash-frozen and stored under liquid nitrogen.
1 was dissolved in 100% DMSO and incubated with KRAS G12C at a 3:1 molar ratio at 20°C for two hours and then 4°C for overnight. The mixture was analyzed by mass spectrometry to confirm 100% labeling, and was further purified by Superdex75 in buffer: 20 mM Hepes pH 8.0, 150 mM NaCl, 5 mM MgCl2 and 0.5 mM DTT. Crystals grew from sitting vapor diffusion drops with the following condition: 1.8M sodium phosphate monobasic monohydrate, potassium phosphate dibasic pH6.9. Crystals were cryoprotected in mother liquor with 20% glycerol and flash frozen in liquid nitrogen.
Crystal Structure Determination
Diffraction images were collected at the advanced photon source beamline 19-ID. Data were integrated and scaled using HKL2000/3000 packages (Otwinowski and Minor, 1997). Molecular replacement was performed with 4OBE as the search model using Phaser software. Manual and automated model building and refinement were performed using Phenix package and coot software (Adams et al., 2010; Emsley et al., 2010). Figure images were prepared using Pymol (The PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC) and ChemDraw Professional 15.0. Final model and scaled reflection data were deposited at the protein databank (5V71). Final collection and refinement statistics are presented in Table 1.
Differential Scanning Fluorimetry
KRAS G12C protein was diluted to 10 μM in analysis buffer consisting of 20mM HEPES pH 7.5, 100mM NaCl, 5% glycerol and subjected to a range of test compound concentrations at 25°C for 1 hour (diluted from 10mM DMSO stock). SYPRO® Orange (Sigma-Aldrich, Cat. S5692) was added in DSF buffer and samples analyzed in a CFX Connect™ RT-PCR Detection System (Bio-Rad). Samples were heated from 25°C to 100°C at a rate of 1.5°C/minute, taking a fluorescence reading every 0.5°C using a SYBR_FAM channel matching the excitation and emission wavelengths of SYPRO orange (λex 470 nm; λem 570 nm). Melting points for samples were determined by taking the first derivative of fluorescence readings and inspecting for peaks in the derivative signal using Bio-Rad CFX Manager and Prism GraphPad 7.01. (GraphPad Software, Inc., La Jolla, CA).
Chemosensor Assay
The chemosensor assay was performed as previously described (Hunter et al., 2014). Briefly, G12C or WT KRAS were incubated with DMSO or compounds at ambient temperature for the indicated durations. At each time point, protein sample was mixed with CPM in a black 384-well plate and fluorescence was read at 384/470 nm. Normalization was as follows: background was subtracted from each time point using the signal from WT KRAS and percent CPM was calculated using the signal derived from KRAS G12C protein alone at each time point as the maximal possible signal (100% labeling). Other readings are normalized accordingly. Prism GraphPad 7.01. was used to run the data analysis.
Western Blots
Human non-small cell lung cancer KRAS G12C mutant cell line H358 were grown in RPMI1640 medium (Gibco) supplemented with 10% heat-inactivated FBS (Lonza) and incubated in humidified 37°C/5% CO2 incubator. Cells were washed once with 1x phosphate buffered saline (PBS) and then lysed in RIPA buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS) supplemented with protease and phosphatase inhibitors (Roche). Protein concentrations were determined by using the Pierce BCA protein assay kit (Life Technologies). Equal amount of protein was resolved on SDS-PAGE, and was subsequently transferred onto the nitrocellulose membrane (Bio-Rad). The membrane was blocked with 5% non-fat milk in TBS-0.1%Tween-20 and was then incubated with primary antibodies overnight at 4°C with gentle rotating. After washing, the membrane was incubated with fluorophore-conjugated secondary antibodies for 1 hour at room temperature. The membrane was then washed and scanned with an Odyssey Infrared scanner (Li-Cor Biosciences). Primary antibodies included anti-KRAS (Sigma #SAB1404011), anti-Phospho-p44/42 MAPK (Erk1/2) (Cell Signaling Technology # 9101S), and anti-alpha-Tubulin (Cell Signaling Technology # 3873S). Secondary antibodies used were IRDye700-conjugated anti-mouse IgG and IRDye800-conjugated anti-rabbit IgG (Rockland, Gilbertsville, PA).
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were produced using Prism 6 (GraphPad Software).
DATA AND SOFTWARE AVAILABILITY
The crystal structure of 1-KRAS G12C has been deposited in the RCSB Protein Data Bank under code: 5V71.
Deuterium incorporation graphs were generated using DynamX 3.0 software (Waters). Diffraction data were integrated and scaled using HKL2000/3000 packages (Otwinowski and Minor, 1997). Molecular replacement was performed with 4OBE as the search model using Phaser software. Manual and automated model building and refinement were performed using Phenix package and coot software (Adams et al., 2010; Emsley et al., 2010).
Figure images were prepared using Pymol (The PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC) and ChemDraw Professional 15.0.
The Supplemental_final.pdf file was provided as Supplemental Material.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-KRAS | Sigma-Aldrich | Cat# SAB1404011 |
| Rabbit monoclonal anti-phospho-ERK | Cell Signaling Technology | Cat# 4370S |
| Rabbit monoclonal anti-ERK | Cell Signaling Technology | Cat# 4695S |
| Mouse monoclonal anti-alpha-Tubulin | Cell Signaling Technology | Cat# 3873S |
| Chemicals, Peptides, and Recombinant Proteins | ||
| GeneArt® site-directed mutagenesis system for Human KRAS G12C | Life Technologies | Cat# A13282 |
| KRAS G12C and WT | Westover lab | N/A |
| Deposited Data | ||
| KRAS G12C/compound 1 structure | Protein databank | PDB: 5V71 |
| Experimental Models: Cell Lines | ||
| Human: H358 (Male) | Pasi Janne Lab | ATCC-CRL-5807 |
| Human: A549 (Male) | Pasi Janne Lab | ATCC-CCL-185 |
| Human: H441 (Male) | Pasi Janne Lab | ATCC-HTB-174 |
| Human: HCT116 (Male) | Pasi Janne Lab | ATCC-CCL-247 |
| Recombinant DNA | ||
| Plasmid pcDNA2.0_KRAS_WT | Westover lab | N/A |
| Plasmid pcDNA2.0_KRAS_G12C | Westover lab | N/A |
| Software and Algorithms | ||
| GraphPad Prism | Graphpad Software Inc | https://www.graphpad.com/scientific-software/prism/ |
| DynamX 3.0 | Waters | http://www.waters.com/waters/ |
| HKL2000/3000 | HKL Research, Inc. | http://www.hkl-xray.com/hkl-3000 |
| Phenix | https://www.phenix-online.org/ | |
| Coot | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ | |
| Pymol | The PyMOL Molecular Graphics System, Version 1.5.0.4 | Schrödinger, LLC |
| ChemDraw Professional 15.0 | PerkinElmer | http://www.cambridgesoft.com/ |
Highlights.
HDX mass spectrometry detects differences between classes of RAS SIIP inhibitors
X-ray crystal structures show conformational dynamics upon SIIP inhibitor binding
Quinazoline SIIP inhibitor has similar activity to chloro hydroxy aniline inhibitor
ACKNOWLEDGMENTS
Department of Defence W81XWH-16-1-0106 (K.D.W.), Jimmy V Foundation (K.D.W.), Astellas (K.D.W. and N.S.G.), NIH (R01-GM101135) (J.R.E.), and a research collaboration with the Waters Corporation (J.R.E.). X-ray crystallography was performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne is operated by UChicago Argonne, LLC, for the US Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and one table and can be found with this article online at http://dx.doi.org/10.1016/j.str.2017.07.003.
REFERENCES
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell PM, and Der CJ (2004). Oncogenic Ras and its role in tumor cell invasion and metastasis. Semin. Cancer Biol 14, 105–114. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of coot. Acta Crystallogr. D Biol. Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engen JR (2003). Analysis of protein complexes with hydrogen exchange and mass spectrometry. Analyst 128, 623–628. [DOI] [PubMed] [Google Scholar]
- Fasano O, Aldrich T, Tamanoi F, Taparowsky E, Furth M, and Wigler M (1984). Analysis of the transforming potential of the human H-ras gene by random mutagenesis. Proc. Natl. Acad. Sci. USA 81, 4008–4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feig LA, and Cooper GM (1988). Relationship among guanine nucleotide exchange, GTP hydrolysis, and transforming potential of mutated ras proteins. Mol. Cell. Biol 8, 2472–2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison RA, Lu J, Carrasco M, Hunter J, Manandhar A, Gondi S, Westover KD, and Engen JR (2016). Structural dynamics in ras and related proteins upon nucleotide switching. J. Mol. Biol 428, 4723–4735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter JC, Gurbani D, Ficarro SB, Carrasco MA, Lim SM, Choi HG, Xie T, Marto JA, Chen Z, Gray NS, et al. (2014). In situ selectivity profiling and crystal structure of SML-8-73-1, an active site inhibitor of oncogenic K-Ras G12C. Proc. Natl. Acad. Sci. USA 111, 8895–8900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, and Westover KD (2015). Biochemical and structural analysis of common cancer-associated KRAS mutations. Mol. Cancer Res 13, 1325–1335. [DOI] [PubMed] [Google Scholar]
- Kim E, Ilic N, Shrestha Y, Zou L, Kamburov A, Zhu C, Yang X, Lubonja R, Tran N, and Nguyen C (2016). Systematic functional interrogation of rare cancer variants identifies oncogenic alleles. Cancer Discov. 6, 714–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Feng J, Wu T, Ren P, Liu Y, Liu Y, and Long YO. (2015). Inhibitors of kras g12c. (Google Patents; ). [Google Scholar]
- Lim SM, Westover KD, Ficarro SB, Harrison RA, Choi HG, Pacold ME, Carrasco M, Hunter J, Kim ND, Xie T, et al. (2014). Therapeutic targeting of oncogenic K-Ras by a covalent catalytic site inhibitor. Angew. Chem. Int. Ed 53, 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lito P, Solomon M, Li L-S, Hansen R, and Rosen N (2016). Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 351, 604–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Sabnis Y, Zhao Z, Zhang T, Buhrlage SJ, Jones LH, and Gray NS (2013). Developing irreversible inhibitors of the protein kinase cysteinome. Chem. Biol 20, 146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundy J, Grimson R, Mishriki Y, Chao S, Oravez S, Fromowitz F, and Viola M (1986). Elevated ras oncogene expression correlates with lymph node metastases in breast cancer patients. J. Clin. Oncol 4, 1321–1325. [DOI] [PubMed] [Google Scholar]
- Montalvo SK, Li L, and Westover KD (2016). Rationale for RAS mutation-tailored therapies. Future Oncol. 13, 263–271. [DOI] [PubMed] [Google Scholar]
- Ostrem JM, Peters U, Sos ML, Wells JA, and Shokat KM (2013). K-Ras (G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, and Minor W (1997). Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Patricelli MP, Janes MR, Li L-S, Hansen R, Peters U, Kessler LV, Chen Y, Kucharski JM, Feng J, and Ely T (2016). Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov. 6, 316–329. [DOI] [PubMed] [Google Scholar]
- Pirrone GF, Iacob RE, and Engen JR (2015). Applications of hydrogen/deuterium exchange MS from 2012 to 2014. Anal. Chem 87, 99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior IA, Lewis PD, and Mattos C (2012). A comprehensive survey of Ras mutations in cancer. Cancer Res. 72, 2457–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pylayeva-Gupta Y, Grabocka E, and Bar-Sagi D (2011). RAS oncogenes: weaving a tumorigenic web. Nat. Rev. Cancer 11, 761–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seliger B, Harders C, Lohmann S, Momburg F, Urlinger S, Tampe R, and Huber C (1998). Down-regulation of the MHC class I antigen-processing machinery after oncogenic transformation of murine fibroblasts. Eur. J. Immunol 28, 122–133. [DOI] [PubMed] [Google Scholar]
- Wales TE, and Engen JR (2006). Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom. Rev 25, 158–170. [DOI] [PubMed] [Google Scholar]
- Weijzen S, Velders M, and Kast W (1999). Modulation of the immune response and tumor growth by activated Ras. Leukemia 13, 502–513. [DOI] [PubMed] [Google Scholar]
- Welsch ME, Kaplan A, Chambers JM, Stokes ME, Bos PH, Zask A, Zhang Y, Sanchez-Martin M, Badgley MA, and Huang CS (2017). Multivalent small-molecule Pan-RAS inhibitors. Cell 168, 878–889.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng M, Lu J, Li L, Feru F, Quan C, Gero T, Ficarro S, Xiong Y, Ambrogio C, Paranal R, et al. (2017). Potent and selective covalent quinazoline inhibitors of KRAS G12C. Cell Chem Biol. 10.1016/j.chembiol.2017.06.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The crystal structure of 1-KRAS G12C has been deposited in the RCSB Protein Data Bank under code: 5V71.
Deuterium incorporation graphs were generated using DynamX 3.0 software (Waters). Diffraction data were integrated and scaled using HKL2000/3000 packages (Otwinowski and Minor, 1997). Molecular replacement was performed with 4OBE as the search model using Phaser software. Manual and automated model building and refinement were performed using Phenix package and coot software (Adams et al., 2010; Emsley et al., 2010).
Figure images were prepared using Pymol (The PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC) and ChemDraw Professional 15.0.
The Supplemental_final.pdf file was provided as Supplemental Material.