

Abstract
Two-component signaling is a primary method by which microorganisms interact with their environments. A kinase detects stimuli and modulates autophosphorylation activity. The signal propagates by phosphotransfer from the kinase to a response regulator, eliciting a response. Response regulators operate over a range of timescales, corresponding to their related biological processes. Response regulator active site chemistry is highly conserved, but certain variable residues can influence phosphorylation kinetics. An Ala-to-Pro substitution (K+4, residue 113) in the Escherichia coli response regulator CheY triggers a constitutively active phenotype; however, the A113P substitution is too far from the active site to directly affect phosphochemistry. To better understand the activating mechanism(s) of the substitution, we analyzed receiver domain sequences to characterize the evolutionary role of the K+4 position. Although most featured Pro/Leu/Ile/Val residues, chemotaxis-related proteins exhibited atypical Ala/Gly/Asp/Glu residues at K+4. Structural and in silico analyses revealed that CheY A113P adopted a partially active configuration. Biochemical data showed that A113P shifted CheY towards a more activated state, enhancing autophosphorylation. By characterizing CheY variants, we determined that this functionality was transmitted through a hydrophobic network bounded by the β5α5 loop and the α1 helix of CheY. This region also interacts with the phosphodonor CheAP1, suggesting that binding generates an activating perturbation similar to the A113P substitution. Atypical residues like Ala at the K+4 position likely serve two purposes. First, restricting autophosphorylation may minimize background noise generated by intracellular phosphodonors such as acetyl phosphate. Second, optimizing interactions with upstream partners may help prime the receiver domain for phosphorylation.
Graphical Abstract
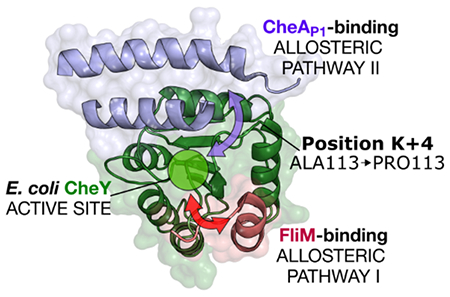
Introduction
Two-component signaling (TCS) is one of the primary means by which the microbial world senses and reacts to the environment. Microorganisms utilize TCS systems to respond to a wide range of external signals. In a canonical TCS system (reviewed in 1), the extracellular portion of a membrane-bound sensor histidine kinase (HK) functions as a detector for environmental stimuli. Upon detection, the signal is internalized across the cell membrane, modulating the cytoplasmic portion of the kinase. The HK binds adenosine triphosphate (ATP) and uses the γ-phosphoryl group to autophosphorylate on a conserved His residue. The signal is transmitted by the shuttling of the phosphoryl group to a conserved Asp residue on the receiver (rec) domain of a downstream response regulator. The phosphorylation state of the rec domain modulates the activity of the protein, ultimately eliciting a cellular response to the original stimulus. This response commonly involves some form of transcriptional regulation or additional protein-protein binding.
Rec domains self-catalyze their phosphorylation and dephosphorylation reactions.2–4 Both reactions use a catalytic pentad of highly conserved active site residues including a pair of acidic residues required for metal binding (abbreviated as DD), a conserved Thr/Ser residue (T), a conserved Lys residue (K) and a phosphorylatable Asp residue (D) (see Figure S1; reviewed in 5). Phosphorylation occurs via nucleophilic attack at the phosphorus atom center of a phosphoryl group that is usually attached through a phosphoramidate bond to a histidine residue. The reaction is assumed to proceed through an activated, negatively charged, pentavalent transition state that is stabilized by a bound metal cation (typically Mg2+ or Mn2+) and the side chains of the conserved Lys (K) and Thr/Ser (T) residues. Dephosphorylation occurs through an analogous reaction involving a nucleophilic water molecule.6
Despite exhibiting significant sequence diversity (pairwise sequence identities are typically around 20-30%), all rec domains adopt a highly conserved (βα)5 Rossmannoid fold topology, deviating from classical Rossmann fold architecture.6 Because of this common tertiary structure, rec domains share universal active site chemistry. However, these commonalities are in contrast to the highly variable kinetic properties of the corresponding response regulators that have been observed in nature. TCS systems regulate many biological processes and operate over a wide range of timescales. Responses involving phenomena such as chemotaxis or phototaxis have brief lifetimes, on the order of seconds or minutes.7–9 Other processes, such as sporulation, can last hours or even days.10, 11 The relative kinetics of the phosphorylation and dephosphorylation reactions for rec domains (and TCSs in general) must be sufficiently rapid to mediate the corresponding cellular process(es) appropriately. Available data on rec domain phosphorylation kinetics focus mainly on the water-mediated (and more easily observed) autodephosphorylation reaction, with reported rate constants spanning nearly a 106-fold range.12, 13 These dephosphorylation rates correlate well with their related biological pathways and processes.14, 15 The most notable comprehensive studies on rec domain autophosphorylation kinetics focus solely on the Escherichia coli proteins CheY and PhoB, and even this limited set of rate constants spans an approximately 103-fold range.16–20
To explain their diverse kinetic properties, studies dating back to the 1990’s have sought to characterize universal “activator” residues in rec domains, positions at which appropriate substitutions are sufficient to trigger functional activity, even in the absence of traditional environmental stimuli (summarized in 21). In this context, terms like “activator” and “activating” refer to an in vivo phenotypic state resembling one elicited as a result of rec domain phosphorylation. Using E. coli CheY (UniProtKB P0AE67) as a model, previous work identified several substitutions that triggered CheY activation, based on in vivo chemotactic ability and/or flagellar behavior.21–29 Many of the identified positions were near the sites of catalysis or partner binding; however, a few were too far away to operate through any obvious biophysical means. Contemporary efforts have used bioinformatics-based approaches to identify additional residue positions that modulate rec domain phosphorylation and/or dephosphorylation kinetics.12, 19, 20, 30 The distribution of wild-type amino acids found at variable positions that affect phosphorylation and dephosphorylation kinetics varies substantially between different types of response regulators, strongly suggesting evolutionary fine-tuning of reaction kinetics to match function.12, 19 Decades ago, a specific Ala-to-Pro substitution was identified in CheY four positions C-terminal to the active site lysine residue (K+4), which elicited significant active-like (CW) flagellar behavior in vivo. Interestingly, the substitution (referred to as CheY A113P) produced this phenotype even in the absence of the partner upstream HK, CheA.21, 23, 29 Using small molecule phosphodonors such as phosphoramidate (PAM), in vitro studies of CheY A113P revealed that the variant possessed an enhanced autophosphorylation rate (approximate 7-fold increase), a relatively unchanged autodephosphorylation rate, and a significantly increased affinity (approximate 3-fold increase) for its downstream flagellar binding partner, FliM, when compared to wild-type CheY.21 Other recent work focused on an extensive series of substitutions at another nearby pair of positions in CheY, K+1 (P110) and K+2 (F111), and found that they triggered an even more substantial enhancement of the in vitro autophosphorylation rate of CheY when replaced with appropriate residues types.19 Similar to the A113P variant, K+1 and K+2 substitutions left the autodephosphorylation reaction mostly untouched.19 All three positions are located too far from the phosphorylatable Asp residue (D57) to directly affect the chemistry of the phosphotransfer reaction in CheY (>8 Å between closest atoms; see Figure S1), suggesting that they affect reaction kinetics through an indirect mechanism. One possible explanation relates to the concept of a pre-existing conformational equilibrium. Allosteric enzymes, such as rec domains, are thought to exist as a heterogeneous population in solution, sampling a spectrum of conformations ranging from “active-like” to “inactive-like” extremes.31 In this specific context, “active” and “inactive” refer to conformational states resembling phosphorylated and unphosphorylated forms, respectively. Energetically, this equilibrium favors the unphosphorylated state in the absence of a phosphodonor. However, a small fraction of molecules is able to randomly access more active-like (higher-energy) conformations and/or characteristics.19, 32–37 Activated molecules exhibit catalytic sites that are chemically and structurally optimized for preferential reaction with a phosphodonor. Phosphorylation likely stabilizes these higher-energy, transient conformations.17 Perturbations that affect the equilibrium (such as substitutions at key positions or partner binding) can dramatically affect the population average kinetics of allosteric enzymes. Previous work has suggested that more significant changes are observed in the phosphorylation reaction, rather than dephosphorylation, presumably due to the larger field of conformational possibilities sampled by unphosphorylated rec domain molecules.19 Our findings regarding the K+1/K+2/K+4 positions imply that the region C-terminal to the active site Lys residue (K109; referred to as the β5α5 loop region in rec domains) can indirectly tune the average reaction kinetics of the E. coli CheY population, likely by altering this conformational equilibrium rather than by directly affecting phosphodonor binding or the phosphorylation reaction itself. However, it is likely that the conformational equilibrium utilized by rec domains is more complex than a simple two-state switch model, and that the transition to an activated state occurs in a disjointed or segmental fashion.17, 34 The existence of structural data featuring intermediate conformations supports the idea that functional residues can act with some independence, and a given rec domain molecule may exhibit a mixture of both active and inactive characteristics.33, 37–40
To better understand this phenomenon on a molecular and mechanistic level, we characterized the K+4 activating substitution A113P in CheY using computational, biophysical, and biochemical techniques. Sequence analysis revealed that rec domains related to the functionally short-lived chemotaxis response regulators exhibited significant diversity at the K+4 position, featuring several uncommon amino acid types. Structural and biochemical analyses indicated that the A113P substitution in E. coli CheY triggered a subtle but significant conformational shift. The change was sufficient for the A113P variant to adopt several partially active characteristics in the absence of any corresponding signal, essentially priming the rec domain for phosphorylation. These data explain the activated in vivo phenotype, the increased autophosphorylation rate, and the enhanced affinity of the CheY A113P variant for its downstream flagellar binding partner, FliM. Molecular dynamics (MD) simulations of CheY wild-type and A113P systems provided valuable insight that corroborated our previous findings and revealed a complex web of hydrophobic side chain interactions stretching from the K+4 position to the center of the active site. Using site-directed mutagenesis, we probed the extent of the predicted hydrophobic network and traced the pathway of allosteric control triggered by the A113P substitution. The results of our work are three-fold: (1) we have unraveled a decades-old mystery by demonstrating how a single Ala-to-Pro substitution at the distal K+4 position in CheY can trigger a strong active-like phenotype in E. coli; (2) we have proposed a mechanism by which other known activating substitution(s) in the β5α5 loop of CheY likely function; and (3) we have obtained additional insight into an important and relatively understudied method of kinetic tuning employed by response regulators involving manipulations of their conformational equilibria.
Materials and Methods
Sequence analysis
A previously reported, non-redundant database of bacterial response regulator receiver domain sequences was used to calculate the amino acid residue frequencies at the K+4 position that are observed in nature.12 The data structure allowed for the extraction of any given residue position relative to the critical catalytic residues found in every true receiver domain (the metal-binding acidic residue pair, the phosphorylatable Asp, the “switch” Thr/Ser, and the essential Lys). Sequences were further divided into response regulator groups based on the presence of attached output domains.12 For a higher-resolution perspective, each sequence was also scanned and assigned to a putative functional family (FunFam) using data from the CATH database (v.4.2.0).41 Due to the large number of sequences being assigned, this was performed manually using the cath-tools-genomescan tool (obtained in Aug. 2020) and HMMER3 (v.3.3.1).42, 43 FunFams are closely related groups of protein domains within CATH superfamilies that are predicted to exhibit similar functional characteristics.41 Fifty-five rec domains with known kinetic properties were included in the analysis as “reference” sequences to provide context for the resulting functional classifications. As an example, members of FunFam 151841 (into which E. coli CheY is assigned) are predicted to be involved in chemotaxis and to share rapid kinetic properties. FunFams possessing fewer than thirty detected members or lacking an assigned reference rec domain were excluded from further analysis. The amino acid residue frequencies at the K+4 position for each response regulator group and FunFam were also calculated. Physicochemically similar residue types were combined, and distributions were visualized as row-scaled (z-score) heatmaps to highlight intra-family differences.
Mutagenesis and protein purification
CheY variants that were analyzed using the fluorescence-based assay were generated by the QuikChange site-directed mutagenesis protocol (Agilent) in a pET28a expression vector using the pKC1 plasmid as a template, as previously described.16 Constructs incorporated a thrombin-cleavable His6-tag fusion at the N-terminus of CheY. Plasmids were transformed into E. coli BL21(DE3) cells for overexpression and purification.
Variants were purified as previously described with minor alterations.16 Briefly, 1 L flasks of LB media were inoculated with 10 mL overnight cultures of plasmid-containing bacteria (+30 μg/mL kanamycin) for each variant. Cells were grown at 37 °C on an orbital shaker to an OD600 of approximately 0.7. Cultures were then transferred to a room temperature shaker, induced with 1 mM isopropyl 1-thio-β-D-galactopyranoside, and agitated for approximately 12 hours. Cells were centrifuged and resuspended in lysis buffer (20 mM Tris, pH 7.5, 300 mM NaCl, 10 mM imidazole). The resuspended cells were then lysed by homogenization (Emulsiflex-C3, Avestin, Inc.) and clarified using ultracentrifugation (45 min, 4 °C, 37,000 x g). The supernatant was applied to a Nickel-NTA agarose chromatography column and rinsed with approximately 15 column volumes of wash buffer (20 mM Tris pH 8.4, 150 mM NaCl, 20 mM imidazole). Proteins were eluted with approximately 5 column volumes of elution buffer (300 mM NaCl, 20 mM Tris pH 7.5, 250 mM imidazole). Protein-containing fractions were pooled and dialyzed into 2 L of TMG buffer (25 mM Tris pH 7.5, 5 mM MgCl2 10% [v/v] glycerol) at 4 °C overnight. Simultaneously, each sample was incubated with human α-thrombin (5 units/mL sample). Samples were further purified by size exclusion chromatography (Superdex 75 16/60 column equilibrated in TMG buffer, GE Healthcare) to remove the thrombin and cleaved His6-tag.
For crystallographic studies, CheY A113P was purified as previously described.44 Briefly, the E. coli strain K0641recA (ΔcheY ΔrecA) containing pRS3A113P was grown in LB media (+100 μg/mL ampicillin) to an OD600 of approximately 1.0 at 37 °C. Cultures were then induced with 100 μg/mL β-indole acrylic acid and grown overnight. Cells were centrifuged and resuspended in TMG buffer. The resuspended cells were lysed using sonication and applied to an Affi-Gel Blue chromatography column (Bio-Rad). Cells were washed and eluted in TMG buffer + 1.0 M NaCl. Protein-containing fractions were dialyzed overnight into 2 L of TMG at 4 °C. Samples were then applied to a DE-52 ion-exchange column (equilibrated in 100 mM Tris, pH 7.5) followed by size exclusion chromatography (Superdex 75 16/60 column equilibrated in TMG buffer, GE Healthcare).
Crystallization and data collection
CheY A113P was captured in three distinct states using the hanging drop vapor diffusion method under separate room temperature crystallization conditions. Crystals of CheY A113P bound with Mg2+ in the absence of sulfate (PDB ID: 3OO1) were grown at a 1:1 drop ratio of protein:TMG buffer (18 mg/mL) in the following reservoir conditions: 28% PEG 8000, 80 mM calcium acetate, 100 mM sodium cacodylate, pH 6.0. Crystals of CheY A113P bound with Mn2+ in the presence of sulfate (PDB ID: 3OO0) were grown using a separate stock of protein (10 mg/mL) incubated with 20 mM MnCl2 at a 1:1 drop ratio of protein:TMG buffer in the following reservoir conditions: 2.2 M ammonium sulfate, 50 mM Tris-HCl, pH 8.25. Crystals of activated CheY A113P in complex with Mn2+ and BeF3− (PDB ID: 3MYY) were obtained by incubating 20 mM MnCl2, 1 mM BeCl2 and 10 mM NaF with CheY A113P (3.3 mg/mL) prior to crystallization. Diffraction-quality crystals were observed in the following reservoir conditions: 1.8-2.4 M ammonium sulfate, 0-12.5% (v/v) glycerol, 100 mM Tris, pH 7.5-8.5 at a drop ratio of 1:1.
Crystals were cryo-protected through gradually increasing glycerol levels (5-15% v/v) mixed with reservoir solution and flash-cooled in liquid N2 in preparation for data collection. X-ray diffraction data were collected using an R-Axis IV++ detector with a Ru-H3R generator (UNC home source) or a MarMosaic 300 CCD on the SERCAT 22-ID synchrotron-radiation beam line (Argonne National Laboratory). Diffraction data were reduced and scaled using the HKL-2000 suite.45
Structure determination and refinement
Structures for each CheY A113P crystal form were determined by the molecular replacement method using existing structures of CheY in its apo (PDB ID: 3CHY)46 and BeF3− bound states (PDB ID: 1FQW)47 as search models. Initial models were improved manually using Coot, followed by structural refinement with phenix.refine in the PHENIX software suite (see Table S2 for version numbers).48, 49 For the structure of CheY A113P in complex with Mn2+ grown in the presence of sulfate, TLS parameters were applied for terminal model refinement runs, with chain A as group 1 and chain B as group 2 (both protomers were CheY A113P). Final models were validated with MolProbity prior to PDB deposition.50
Small molecule phosphodonors
Because CheY exhibits autophosphorylation and autodephosphorylation activity, two small molecule phosphodonors, phosphoramidate (PAM) and acetyl phosphate (AcP), were used in the fluorescence-based assays.18 The potassium salt form of PAM was synthesized as previously described.51 The lithium potassium salt form of AcP was purchased from Sigma-Aldrich (MilliporeSigma).
CheY partner peptides
A construct encoding the CheY-binding region of CheA (residues 124-257; P2 domain) was synthesized by GeneWiz, Inc. and incorporated into the same pET28a expression vector that was used for the CheY variants. This construct was used to express and purify the CheA124-257 P2 fragment using a similar protocol to that described above. A FliM peptide fragment corresponding to the first 16 residues on the amino terminus (MGDSILSQAEIDALLN; FliM1-16) was synthesized by GenScript Corp. and obtained in a lyophilized form (87% pure). Stock solutions of FliM1-16 were prepared at various concentrations in 100 mM HEPES, pH 7.0. For phosphorylation experiments, CheY samples were pre-incubated with either 1-2 mM FliM1-16 or 20 μM CheA124-257 P2 domain, chosen to ensure ~88% and ~90% bound to CheY, respectively (n=2 for both CheY wild-type and A113P).17, 21
Phosphorylation assays
Fluorescence measurements were made using a LS-50B spectrofluorimeter (Perkin-Elmer; minimum response time/data interval = 20 ms) using FL WINLAB (v.1.1; Perkin-Elmer). Intrinsic tryptophan fluorescence of each CheY variant was monitored at approximately 24.8 °C using excitation and emission wavelengths of 295 and 346 nm, respectively. Slit widths were optimized based on the concentrations of protein used for each assay (typically 6 nm). Proteins were prepared in an initial volume of 700 μL containing the following: 100 mM HEPES, pH 7.0, 10 mM MgCl2, 5 μM CheY variant, and 100 mM KCl. A constant ionic strength of approximately 230 mM was maintained for each experiment by altering the KCl concentration. Increasing volumes of donor solution were titrated into each sample. For experiments involving PAM, the donor solution contained the following: 100 mM HEPES, 100 mM PAM, and 10 mM MgCl2 (~230 mM total ionic strength). For experiments involving AcP, the donor solution contained the following: 100 mM HEPES, 33.3 mM AcP, and 10 mM MgCl2 (~230 mM total ionic strength). Phosphorylation of CheY resulted in a loss of tryptophan fluorescence. Time courses were recorded after each titration as the solution approached equilibrium between the phosphorylation and dephosphorylation reactions. Raw fluorescence readings were corrected for dilution and converted to changes in fluorescence signal as a function of donor concentration. Resulting data were analyzed using GraphPad Prism (v.8). Curves were fitted using non-linear regression to a one-site binding hyperbolic model to obtain apparent K1/2 values, representing the concentration of the corresponding phosphodonor required to phosphorylate 50% of the protein population (n=3 for each variant).19
Dephosphorylation assays
Autodephosphorylation assays were performed on the same instrumentation and software as described above using an RX2000 rapid mixing stopped-flow accessory (Applied Photophysics; dead time = 8 ms). Pre-steady state kinetics were determined using the pH-jump method.52 CheY variants (5-20 μM, based on the strength of the intrinsic fluorescence signal) were phosphorylated with saturating levels of PAM (typically 4 x estimated K1/2). Phosphorylated solutions were rapidly mixed with 200 mM sodium carbonate, pH 10.2, inhibiting further autophosphorylation by PAM and allowing for the direct monitoring of the autodephosphorylation reaction through the restoration of tryptophan fluorescence. The fluorescence signal change as a function of time was fitted to a first-order exponential decay model to determine the autodephosphorylation rate constant, kdephos, for each variant (n=3-5, with 12-20 replicate curves for each variant).19
Ligand binding assays
Fluorescence measurements of the binding behavior between the phosphoryl group mimic BeF3− and CheY (wild-type and A113P) were performed as previously described, with minor alterations, including a higher overall ionic strength.17 A total of 1 μM of CheY was prepared in a solution of 100 mM HEPES, pH 7.0, 10 mM MgCl2,10 mM NaF, and 90 mM KCl (230 mM total ionic strength). Previous work indicates that under these conditions, BeF3− will be the predominantly formed species upon titration with BeSO4 or BeCl2.53 A constant ionic strength was maintained for each ligand binding experiment. Increasing amounts of BeSO4-containing solution (100 mM HEPES, pH 7.0, 10 mM MgCl2, 1 mM BeSO4, and 96 mM KCl, ~230 mM total ionic strength) were titrated into the protein solution. Time courses were recorded after each injection as the solution approached equilibrium. Raw fluorescence readings were corrected for dilution and converted to changes in fluorescence signal as a function of BeSO4 concentration. Resulting data were analyzed in GraphPad Prism (v.8). Observed dissociation constants were determined by fitting the converted data using a quadratic binding equation to account for ligand depletion as previously described (n=3 for both variants).17
A similar protocol was used to estimate the binding affinity between Mg2+ and CheY (wild-type and A113P). Separate CheY aliquots were incubated with EDTA so that Mg2+ and EDTA concentrations were equimolar. Prior to use, EDTA-containing samples were buffer exchanged multiple times to remove any residual chelating agent. A total of 5 μM of metal-free CheY was then prepared in a solution of 100 mM HEPES, pH 7.0, and 130 mM KCl (~230 mM total ionic strength). Increasing amounts of MgCl2-containing solution (25 mM MgCl2, 100 mM HEPES, pH 7.0, and 55 mM KCl, ~230 mM total ionic strength) were titrated into the protein solution. Time courses were recorded after each injection as the solution approached equilibrium. Raw fluorescence readings were corrected for dilution and converted to changes in fluorescence signal as a function of MgCl2 concentration. Resulting data were analyzed in GraphPad Prism (v.8). Because [CheY] << anticipated Kd, curves were fitted using non-linear regression to a one-site binding hyperbolic model to obtain apparent dissociation constants, assuming negligible ligand depletion (n=2 for both variants).
To check for physiologically relevant levels of sulfate binding, fluorescence measurements were taken upon titration with increasing amounts of Li2SO4. A total of 5 μM of CheY was prepared in a solution of 50 mM HEPES, pH 7.0, 10 mM MgCl2, and 150 mM KCl (~230 mM total ionic strength). Increasing amounts of Li2SO4-containing solution (50 mM Li2SO4, 50 mM HEPES, pH 7.0, and 10 mM MgCl2, ~230 mM total ionic strength) were titrated into the protein solution. Time courses were recorded after each injection as the solution approached equilibrium. Raw fluorescence readings were corrected for dilution and converted to changes in fluorescent signal as a function of Li2SO4 concentration. Resulting data were analyzed in GraphPad Prism (v.8). Li2SO4 binding curves closely resembled a buffer control titration curve, suggesting that there was no interpretable sulfate binding under these conditions for both CheY wild-type and A113P (n=2 for both variants).
Molecular dynamics simulations
Systems for wild-type CheY and CheY A113P were prepared in VMD using the existing crystal structure in its apo state (PDB ID: 3CHY).54 A proline residue was manually modeled at position 113 using the wild-type 3CHY structure as a template to allow for direct comparison during simulation. Proteins were solvated using the TIP3P water model with 10 Å padding, then neutralized and ionized to a final concentration of 0.1 M KCl.55 Simulations were performed using NAMD (v.2.9) with the CHARMM36 force field release.56, 57 A total of 12 independent replicates were run for each variant. Energy minimization and equilibration were performed under a constant pressure using gradually decreasing atomic restraints over approximately 700 ps. Systems were then run unrestrained for approximately 120 ns using an integration time step of 2 fs. Snapshots were recorded every 10 ps. The first 30 ns of each replicate production trajectory were discarded to ensure proper equilibration for each system. The last 90 ns of each trajectory were retained for study, generating a cumulative production ensemble of approximately 1.08 μs for each variant. Trajectories were stripped of solvent and counterions prior to analysis, unless otherwise described.
Structural analysis
Crystallographic structural alignments were generated in PyMOL using structure-based superpositioning. Models were analyzed in both PyMOL and Chimera.58, 59 Structural analyses of molecular dynamics simulations were primarily performed in VMD and the R package Bio3D (v.2.3-4).54, 63, 64 For analysis and visualization purposes, production frames were aligned using the most invariant core region(s) estimated over the entire ensemble (Figure S4). To compare conformational distributions between the wild-type and CheY A113P variants during the MD simulations, we used Kullback-Leibler (KL) divergence analysis.65 KL divergence (also called relative entropy) is an information theoretic measurement of the free energy difference between two distributions. By comparing the torsion angle distributions, the KL divergence can reveal conformational and entropic changes triggered by subtle allosteric perturbations in a protein system, such as those triggered by a point mutation or ligand binding.65–71 For our analysis, we considered the wild-type CheY ensemble as the reference set, and the CheY A113P ensemble as the perturbed set. The local KL divergence (KLl) for a given torsion angle is described by equation 1:
| (1) |
where p is the probability that a specific torsion angle in the perturbed data set (CheY A113P ensemble) falls within a certain interval of dihedral space (a bin), and q is the probability that the analogous torsion angle in the reference data set (wild-type CheY ensemble) falls within the same bin. The sum of the KL divergence of the individual torsion angles (Φ, Ψ, and χ angles) for each position provides a KL score for every residue in the system (KLres), as shown in equation 2:
| (2) |
These equations can be used to identify “population shifts” in local residue dynamics. Trajectory ensembles for each variant were split into twelve equal blocks for the analysis. The gmx chi utility within GROMACS (v.2018.3) was used to extract per-residue φ, ψ and χ dihedral angles distributions using a bin width of 15º for all frames in each replicate.72 These torsion angle data were used as inputs for the KL divergence module within the MutInf analysis package (v.2.1) using the default α threshold of 0.1 to filter out statistically insignificant differences between the two ensembles.65, 66. Positions failing to meet this threshold were assigned scores of zero. KL scores were mapped to the wild-type E. coli CheY structure (Figure 10; PDB ID: 3CHY).
Figure 10. Comparison of internal torsion angle dynamics between wild-type and CheY A113P.
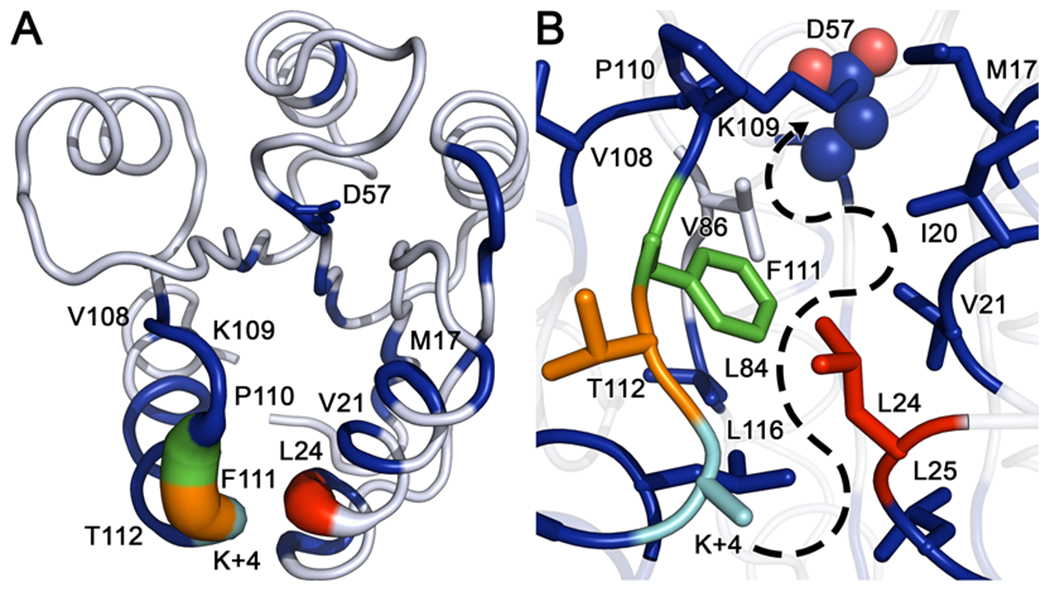
(A) Kullback-Leibler divergence between the two CheY variant ensembles mapped to the wild-type CheY crystal structure (PDB ID: 3CHY). Tube thickness and color warmth (blue to red) scale proportionally to amount of perturbation observed in dihedral angle distributions. Residues colored white failed to deviate above the thresholds set for statistical significance. (B) Closeup of the area between the β5α5 and α1 regions, showing densely packed hydrophobic cluster and putative path of signal transference from position K+4 to the active site (phosphorylatable D57 is shown as spheres). Significantly perturbed residues such as V21, L24, L84, F111 and L116 feature large hydrophobic side chains inserted into the cluster, while residues such as M17, V108, P110 and T112 are oriented away from the hydrophobic core. Residue color-coding matches panel A.
Cross-correlation network creation and community analysis
Network analysis of correlated dynamics was performed on the trajectory ensembles using the Bio3D package. A weighted graph approach was used to construct protein networks for both CheY variants, with each node representing the Cα atom of a residue. The residue-wise Cα linear mutual information (LMI; 73, 74) was calculated for each replicate as previously described using equation 3:
| (3) |
where Ci is the covariance matrix describing the Cα displacement of ith residue and Cij is the covariance matrix for residues i and j. The cross-correlation values (Cij) indicate the strength of the coupled dynamics observed between residues i and j. Individual data sets from each replicate were used to generate an average consensus matrix for each CheY variant. These were further filtered using the following protocol: correlation measurements were retained as reliably strong by meeting a Cij cutoff threshold (Cij ≥ 0.3) in 100% of replicates; more transient values still meeting the Cij cutoff threshold (Cij ≥ 0.3) in at least one replicate were trimmed if the respective Cα atoms were separate by > 10 Å in at least 60% of replicates. Positions that failed to meet the described thresholds were set to zero. As pointed out in previous work, the use of this hybrid approach allows for the retention of strongly correlated interactions, regardless of residue location, while filtering out weaker or spurious correlation using the consensus contact map.73, 75 The Bio3D package was used to construct a coarse-grained network graph with the filtered consensus LMI matrices generated above. Each node describes a single Cα atom connected by edges weighted using the formula −log(|Cij|) for a given residue pair. Residue communities are highly intraconnected groups of correlated positions with weaker outgroup correlation. Community boundaries were determined using the Girvan-Newman betweenness clustering algorithm.76 Several Cij cutoffs (0.3 – 0.6) and network modularity scores were explored during network generation to identify an optimal topology (data not shown). A common Cij cutoff of 0.3 was ultimately chosen because it produced reasonable community boundaries and increased detail during cross-correlation analysis. Residue centrality was quantified using node betweenness. Betweenness scores were normalized using equation 4 (from the igraph R package):
| (4) |
where Bnorm is the normalized betweenness score, B is the raw betweenness score, and n is the number of vertices in the residue network graph.77
Results
Chemotaxis-related receiver domains exhibit atypical residue types at position K+4
To determine the natural abundances of residues found at position K+4, we analyzed a previously described non-redundant database of over 33,000 prokaryotic rec domain sequences.12 This allowed us to determine the potential evolutionary and functional significance of amino acids with small side chains (Ala/Gly) in the K+4 position of rec domains, such as in wild-type E. coli CheY. Traditionally, rec sequences are grouped based on the presence/type of an attached output domain, which provides a reasonably accurate overview of the evolution and function of corresponding response regulators.78 We have referred to these assignments as rec domain subfamilies in previous work.12 However, this approach provides little information about the ultimate biological function or the related kinetic properties of the rec domains within a subfamily. For a more detailed analysis, we assigned each rec domain sequence in the database to a putative functional family (FunFam) using the FunFHMMER method.41, 43 This technique scans sequences against a library of known CATH database FunFam Hidden Markov Models and provides putative functional classifications for each entry. We added specific “reference” rec domain sequences known to possess a wide range of biochemically characterized kinetic properties to the non-redundant sequence library for our analysis. The reference sequences provided surrogate kinetic classifications for each FunFam in lieu of traditional biochemical data for each group. FunFams lacking an assigned reference rec domain or with <30 members were excluded. Full information on the reference sequences is included in the Supporting Information (Text S1 and Table S1). Of the >33,000 rec domain sequences within the non-redundant database, approximately 80% were successfully assigned to one of the 20 FunFams listed in Figure 1. Figures 1A and 1B show the overall size of each FunFam and rec domain subfamily, and Figures 1C and 1D show the relative distribution of amino acid types within each group at residue position K+4. The most abundant residue groups at the K+4 position among wild-type rec domains were Pro and Ile/Leu/Val (22% and 24% of all rec domain sequences in the database, respectively). However, partitioning by FunFam suggested that groups presumably related to chemotaxis (i.e., containing chemotaxis-related reference response regulators: FunFams 151438, 151841, 151695, and 151577) exhibited increased diversity, with noticeable enrichment of smaller amino acid types such as Ala and Gly (8% and 3% of all rec domain sequences, respectively) and charged residues such as Asp and Glu (5% and 4% of all rec domain sequences, respectively). Based on our current knowledge of dephosphorylation kinetics, the chemotaxis FunFams included many of the response regulators with the briefest phosphorylated lifetimes. This deviation from the common K+4 residue type (Ile/Leu/Val/Pro) strongly suggested that atypical K+4 amino acid groups possessed a functional significance. To further explore the apparent correlation between function and amino acid at position K+4, we experimentally characterized the consequences of featuring an Ala or a Pro residue at K+4 in the model response regulator CheY.
Figure 1. Sequence group sizes and amino acid distributions for residue position K+4 in a non-redundant receiver domain database.
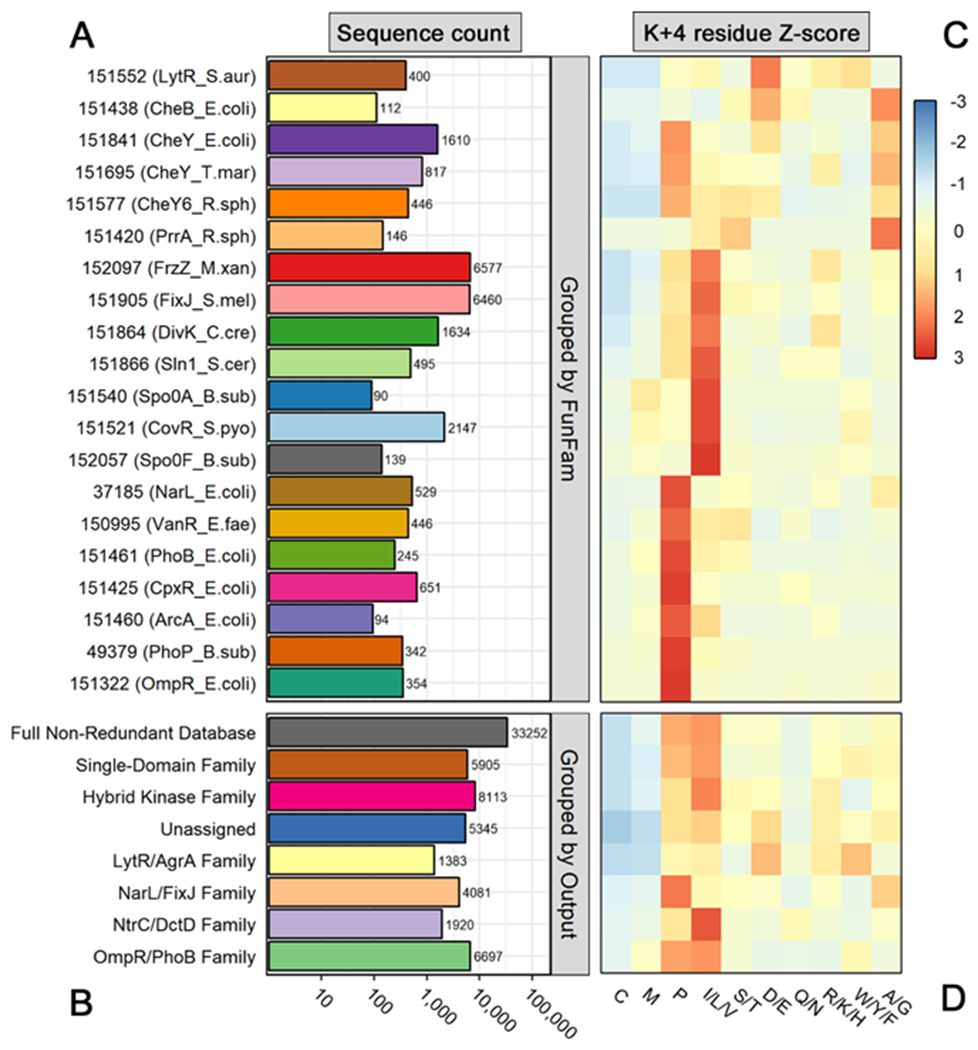
(A) Total number of sequences in each Functional Family (FunFam). Row numbers indicate CATH database FunFam codes (v.4.2.0). Relevant reference rec domain names and organisms are included in parentheses. (B) Total number of sequences in each rec domain subfamily, based on attached output domain. Note that A and B share a common logarithmic scale. (C) Heatmap showing distribution of amino acids at position K+4 of rec domains within each FunFam. (D) Heatmap showing distribution of amino acids at position K+4 of rec domains within each subfamily, based on attached output domain. Residues with similar physiological properties were combined for visualization purposes. Distributions were scaled by row (z-score) to highlight intra-family differences. Residue types with enrichment are colored red, while those with depletion are colored blue. Residue types capturing the row average are colored yellow.
CheY A113P adopts specific active-like structural characteristics in the absence of phosphorylation
To seek a mechanistic explanation for the properties of CheY A113P, we first determined high-resolution crystallographic structures of the CheY A113P variant in three distinct states: a metal-bound complex in the absence of sulfate; a metal-bound complex in the presence of sulfate (a potential phosphoryl group mimic); an activated, BeF3−-bound complex (Table S2). Given the active-like phenotype of CheY A113P in vivo (even in the absence of its own kinase), along with previous evidence suggesting that substitutions at the K+1/K+2 positions may increase the autophosphorylation ability of CheY through conformational selection, we hypothesized that the A113P variant would exhibit active-like structural characteristics in the absence of phosphorylation.19, 21, 23 To determine the structural effects of the A113P substitution on CheY conformation, we compared each CheY A113P crystallographic protomer to an inactive wild-type CheY reference model (PDB ID: 3CHY). Figure 2 shows cartoon depictions of these alignments to qualitatively compare the backbone conformations of the CheY states. Figure 2A shows an alignment of the wild-type active/phosphoryl mimic-bound CheY (CheY•BeF3−•Mn2+) and the reference CheY (inactive wild-type CheY). The relatively subtle changes associated with rec domain activation (typically < 2-4 Å shifts) makes studying their related conformational transitions challenging. However, the regions indicated by the colored discs in Figure 2A are considered hallmarks of rec domain activation (summarized in 6) and are functionally important to E. coli CheY; they are involved in upstream/downstream partner-binding and optimization of the active site geometry for phosphotransfer. Figure 2B shows an alignment between the active/phosphoryl mimic-bound CheY A113P•BeF3−•Mn2+ and the inactive wild-type CheY. Deviations appeared nearly identical to those in Figure 2A, suggesting that the active conformation of CheY A113P closely matched that of wild-type CheY. Figure 2C shows an alignment between the Mn2+-bound CheY A113P in the presence of sulfate and the inactive wild-type CheY. The differences between the experimental and reference structures were similar for both comparisons in Figures 2A and 2C, suggesting that the overall backbone changes induced by the A113P substitution were reminiscent of activation. However, we found ambiguous density for multiple conformations of the β4α4 loop (data not shown) in the first crystallographic protomer of the 3OO0 structure, suggesting heightened flexibility in that region. The second protomer of the 3OO0 structure did not exhibit this ambiguity. Figure 2D shows an alignment between two different protomers of the Mg2+-bound CheY A113P in the absence of sulfate and the inactive wild-type CheY. These protomers lacked the extensive remodeling of the β4α4 loop and more closely resembled the inactive CheY state. However, they still exhibited moderate displacements of the α1 and α5 helices, hinting at partial activation.
Figure 2. Comparisons of backbone conformations of E. coli CheY in various states reveals activated features of CheY A113P.
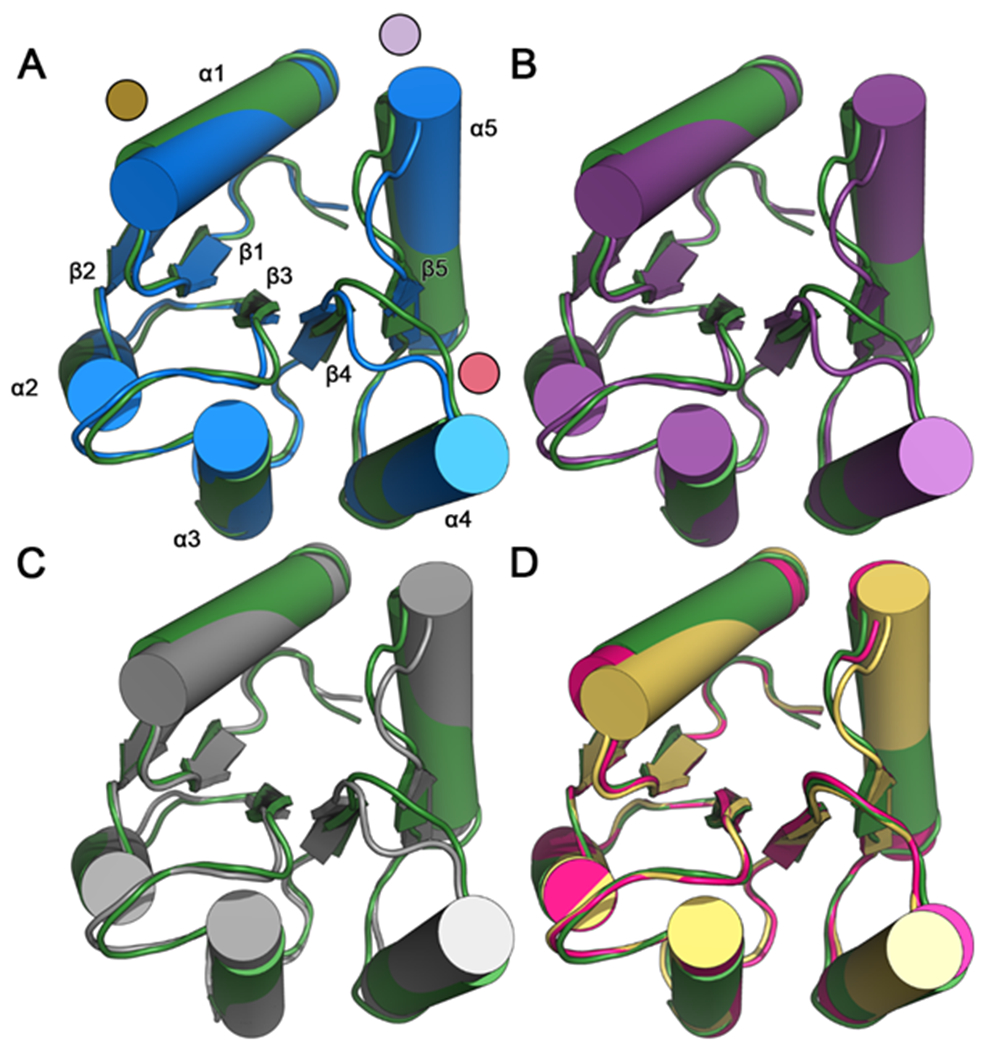
Structure-based alignment of inactive wild-type CheY (PDB ID: 3CHY; green) as a reference with: (A) active wild-type CheY•BeF3−•Mn2+ (PDB ID: 1FQW; blue; due to nearly identical backbone conformations, only one protomer is shown). Areas of the highest deviation are denoted with colored discs (gold: α1 helix; red: β4α4 loop; light purple: β5α5 loop and N-terminus of α5 helix). Each of the (βα)5 secondary structures are labeled; (B) active CheY A113P•BeF3−•Mn2+ (PDB ID: 3MYY; purple; due to nearly identical backbone conformations, only one protomer is included); (C) CheY A113P•Mn2+ in the presence of sulfate (PDB ID: 3OO0; grey; due to nearly identical backbone conformations, only one protomer is included); (D) two separate protomers of CheY A113P•Mg2+ in the absence of sulfate (PDB ID: 3OO1; pink and yellow). Note that structures 3CHY, 1FQW, and 3MMY all were determined from crystals in the P212121 space group, whereas 3OO0 was from P3221 and 3OO1 was from P21.
Next, we calculated the average per-residue Cα Root Mean Square Deviation (RMSD) in the structure-based alignments for a quantitative comparison (Figure 3; using the inactive wild-type CheY structure, 3CHY, as a reference). The largest displacements were clearly visible in the aforementioned regions (α1 and α5 helices, β4α4 and β5α5 loops; shaded in grey). The CheY A113P structure lacking sulfate (3OO1; Figure 3D) exhibited smaller changes from the inactive conformation while still matching the general pattern of deviation seen in the other comparisons. Taken together with Figure 2D, this is likely indicative of a less activated state, which would be consistent with the most dramatic changes being induced upon activation by the phosphoryl mimic, BeF3−. Collectively, the crystal structures suggested that the A113P substitution increased the probability of CheY sampling a higher-energy conformation that more closely resembled a phosphorylated state. It is uncertain to what extent the presence of sulfate influenced the activation state of the A113P variant. Several rec domain structures include a sulfate molecule in the active site partially resembling a bound phosphoryl group.79–81 However, additional biochemical binding data strongly suggested that the bound sulfate molecule was likely related to the high concentration of ammonium sulfate used in the crystallization conditions (2.2 M ammonium sulfate; see Materials and Methods) and the conformational preferences of CheY A113P (Text S2).
Figure 3. Per-residue structural differences between E. coli CheY models and the inactive state.
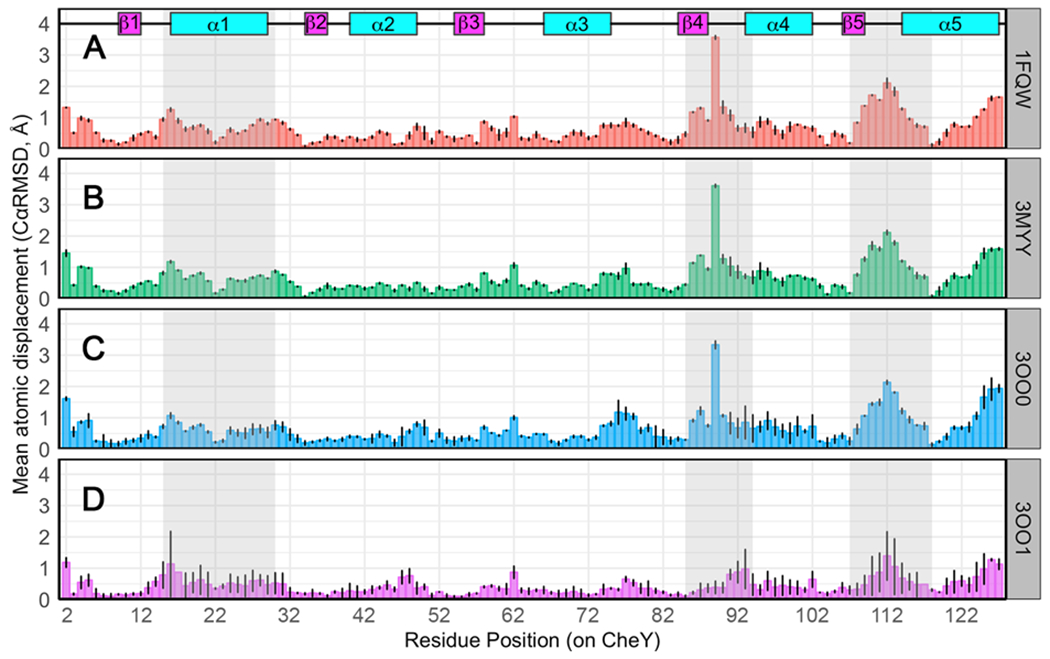
Cα deviations (Å) for each residue pair in the corresponding structural comparison. Deviations averaged over all protomers in each structure are shown. Black lines indicate ranges observed. Secondary structures are annotated at the top. Grey shaded regions indicate functional areas known to be dependent on phosphorylation state. Comparison of inactive wild-type CheY (PDB ID: 3CHY; green) as a reference with: (A) active wild-type CheY•BeF3−•Mn2+ (PDB ID: 1FQW); (B) active CheY A113P•BeF3−•Mn2+ (PDB ID: 3MYY); (C) CheY A113P•Mn2+ in the presence of sulfate (PDB ID: 3OO0); (D) two protomers of CheY A113P•Mg2+ in the absence of sulfate (PDB ID: 3OO1).
To accommodate the proposed segmental nature of rec domain activation, we performed a more comprehensive analysis on various crystal structures of E. coli CheY to quantify individual features known to be diagnostic for the activation state in rec domains. For context, we included all of the previously described crystallographic structures, along with active/phosphoryl mimic-bound, CheY•BeF3−•Mn2+ (PDB ID: 1FQW) and downstream partner-bound, CheY•FliM1-16 (PDB ID: 1F4V). Table 1 contains a detailed breakdown of these measurements. Comparisons with the activated CheY structures (incorporating BeF3− with or without the downstream binding partner, FliM) revealed that the CheY A113P variant likely falls somewhere between the inactive and active end-points of the conformational spectrum (dubbed “Partially Activated”). However, crystallographic structures provide only conformational snapshots of a protein and lack dynamic information, which is critical to the investigation of the allosteric mechanism of the A113P substitution.
Table 1.
Individual structural characteristics related to activation state in X-ray crystal structures of E. coli CheY.
| Structural description | PDB ID (chain) | State | Cα RMSD a |
β4α4 angle b | W58 χ2 | M85 χ1 | T87-D57 (Oγ1-Cγ) c | V86 Ψ | V86 Φ | Y106 χ1 | β5α5 angle d | K109-D12 (Nζ-Cγ) e | α1 to β5α5 (Cα-Cα) f |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CheY | 3CHY(A) | Inactive | - | 16.2 | −107.2 | −169.3 (−67.7) | 6.6 | 135.5 | −111.1 | 88.4 (179.9) | 160.3 | 5.0 | 5.3 |
| CheY A113P•Mg2+ | 3OO1(A) | Partially Active | 0.3 | 19.5 | −99.5 | −55.8 | 7.2 | 137.9 | −117.1 | 179.6 | 168.9 | 4.1 | 5.7 |
| CheY A113P•Mg2+ | 3OO1(B) | Partially Active | 0.4 | 7.9 h | −93.7 | −57.7 | 7.4 | 134.2 | −117.7 | 78.4 | 169.0 | 5.2 | 6.4 |
| CheY A113P•SO42−•Mn2+ | 3OO0(A) g | Partially Active | 0.6 | 110.3 (0.0; −45.1) i | −52.3 (−93.8) | −54.3 (−60.6) | 7.8 (5.3) j | 106.8 (129.2; 95.8) | −135.7 (−112.6; −117.5) | −163.8 (71.8) | 172.0 | 3.8 (5.2) | 6.4 |
| CheY A113P•SO42−Mn2+ | 3OO0(B) g | Partially Active | 0.5 | 109.3 | −49.5 | −55.7 | 5.4 | 104.0 | −134.9 | −159.5 | 171.7 | 3.8 | 6.5 |
| CheY A113P•BeF3−Mn2+ | 3MYY(A) | Active | 0.5 | 111.1 | −40.3 | −54.5 | 3.5 | 105.0 | −136.3 | −165.8 | 167.5 | 3.7 | 6.5 |
| CheY A113P•BeF3−Mn2+ | 3MYY(B) | Active | 0.5 | 111.8 | −36.3 | −55.8 | 3.4 | 109.0 | −132.0 | −157.3 | 168.2 | 3.7 | 6.5 |
| CheY•BeF3−Mn2+ | 1FQW(A) | Active | 0.5 | 109.3 | −45.2 | −57.6 | 3.6 | 100.4 | −137.4 | −164.2 | 173.6 | 3.8 | 6.7 |
| CheY•BeF3−Mn2+ | 1FQW(B) | Active | 0.5 | 110.5 | −40.3 | −55.5 | 3.4 | 100.2 | −138.4 | −160.1 | 169.1 | 3.8 | 6.5 |
| CheY•BeF3−Mg2+FliM1-16 | 1F4V(A) | Active | 0.5 | 112.8 | −50.1 | −54.0 | 3.5 | 106.4 | −133.9 | −156.4 | 169.8 | 3.7 | 6.8 |
| CheY•BeF3−Mg2+FliM1-16 | 1F4V(B) | Active | 0.5 | 113.1 | −57.1 | −56.0 | 3.4 | 94.2 | −143.3 | −155.3 | 168.5 | 3.6 | 6.6 |
| CheY•BeF3−Mg2+FliM1-16 | 1F4V(C) | Active | 0.8 | 102.9 | −53.8 | −52.4 | 3.8 | 102.2 | −130.0 | −156.2 | 162.0 | 4.0 | 6.6 |
Characteristics of inactive CheY are in orange and of active CheY are in purple.
CαRMSD representing atomic displacement between the reference structure (PDB ID: 3CHY) and the mobile structure.
Pseudo-dihedral angles were measured using Cα atoms for residues T87:A88:E89:A90 (β4α4 loop conformation). Measurements for alternate conformers are included in parentheses.
Interatomic distances were calculated from the Oγ1 atom of the T87 residue to Cγ of the D57 phosphorylation site. In structures with BeF3−, the Cγ atom was replaced with the Be atom for measurement.
Pseudo-dihedral angles were measured using Cα atoms for residues K109:P110:F111:T112 (β5α5 loop conformation). Measurements for alternate conformers are included in parentheses.
Interatomic distances were calculated from the terminal Nζ atom of K109 to Cγ of D12.
Interatomic distance between Cα atoms of residue 113 (β5α5 loop) and L24 (α1).
Structures include sulfate molecule(s) bound within the active site.
No density was observed for E89 side chain, suggesting high flexibility.
Residues T87:A88:E89:A90 are highly ambiguous in the 3OO0 protomer A. E89 accesses positions equivalent to those found in both the apo and BeF3− bound CheY structures.
A third conformer was observed for T87 in the 3OO0 protomer A, nearly identical to the primary conformer.
CheY A113P exhibits a mix of both active and inactive features in silico
For additional insight into the dynamic properties affected by the A113P substitution, we performed extensive molecular dynamics simulations of the wild-type and mutant CheY proteins starting from an inactive conformation. A dozen replicates were prepared and executed independently for each system. Production simulations were performed over approximately 120 ns. We discarded the first 30 ns of each replicate to encourage sufficient equilibration and convergence (Figure S2 shows the evolution of CαRMSD for each data set), combining the final 90 ns replicates into production ensembles for analysis. In this way, we generated comprehensive data sets for both wild-type CheY and CheY A113P with over 1 μs worth of conformational data for each system.
First, we analyzed the trajectory data to examine the same structural features included in Table 1 (Figures 4, 5), along with several additional properties (Figures S3). For comparison, the gold and black dotted lines in Figures 4 and 5 respectively indicate the active and inactive values of each parameter as derived from crystal structures. Similar to the crystallographic structures of CheY A113P, the in silico A113P population exhibited certain active-like features, but failed to consistently achieve a fully realized active conformation when compared to the active wild-type CheY structure. The density distribution in Figure 4A shows that the β4α4 loop in much of the CheY A113P population exhibited the conformational remodeling characteristic of rec domain activation, though the shift was only partial, with the majority still overlapping with the wild-type CheY variant data. In rec domains, the repositioning of this β4α4 loop allows for interaction between the switch Thr/Ser residue (T87 in CheY) and the site of phosphorylation (summarized in 6). The side chain oxygen atom of the switch residue is believed to help stabilize the negatively charged phosphoryl group (or a mimic, such as BeF3−), facilitating the phosphotransfer reaction. In contrast, the distributions in Figures 4B and 4C reveal that the side chain torsion angles for W58 χ2 and M85 χ1 were virtually indistinguishable between the simulated wild-type and A113P variants (Table 1). There is evidence that the orientations of W58 and M85 are correlated with the phosphorylation state CheY, and previous studies have implicated their involvement in the allosteric network related to activation.34 The in silico findings supported our previous observations that CheY A113P resembled a partially active configuration, rather than a fully realized active state. Figure 4D illustrates one of the most dramatic differences seen between the simulated populations of wild-type and CheY A113P. One of the hallmarks of rec domain activation is the interaction between the highly conserved Thr/Ser switch residue and the phosphorylated Asp residue, thought to be facilitated by a shift in the β4α4 loop. The distribution in Figure 4D quantifies this interaction by showing the distance between the side chain Oγ1 atom of the switch residue (T87) and the side chain Cγ atom of the Asp (D57; chosen due to the equivalency of the carboxylate oxygen atoms). We observed a large shift in the CheY A113P distribution when compared to the wild-type CheY population, suggesting that the substitution elicited a substantial transition towards a more active-like state (yellow dashed line). Figure 4E shows the density distribution for the backbone torsion angle (Ψ) of V86, a metric believed to be correlated with the degree of activated character of the β4α4 loop.37 We found that the A113P population shifted slightly from the wild-type CheY ensemble distribution, but only rarely reached a fully active-like state, again supporting a partially active classification for A113P. Figure 5A shows a density distribution for the other backbone torsion angle (Φ) of V86, which also exhibited a partial shift in the A113P population towards the assumed active-like state. Figure 5B shows that the conserved aromatic switch residue (Y106) sampled roughly equal instances of buried and exposed conformations in the A113P ensemble, though the distribution appeared to shift slightly towards the active-like state. The aromatic switch residue is often considered an indicator of activation state and is thought to correlate with the rearrangement of the other conserved switch Thr/Ser residue in certain rec domains. In E. coli CheY, this has been referred to as a Y-T coupling mechanism.82, 83 However, this model is not necessarily applicable to all rec domains, and rotamerization of Y106 in CheY is at least somewhat independent of the interaction between T87 and the phosphoryl group.5, 40, 82, 84 The existence of multiple side chain rotamers for Y106, even in the inactive crystal structure of CheY (PDB ID: 3CHY), indicates that the observed distributions in simulations are within the expected range.46
Figure 4. Distribution of values for key structural features related to activation state in the simulations of CheY variants.
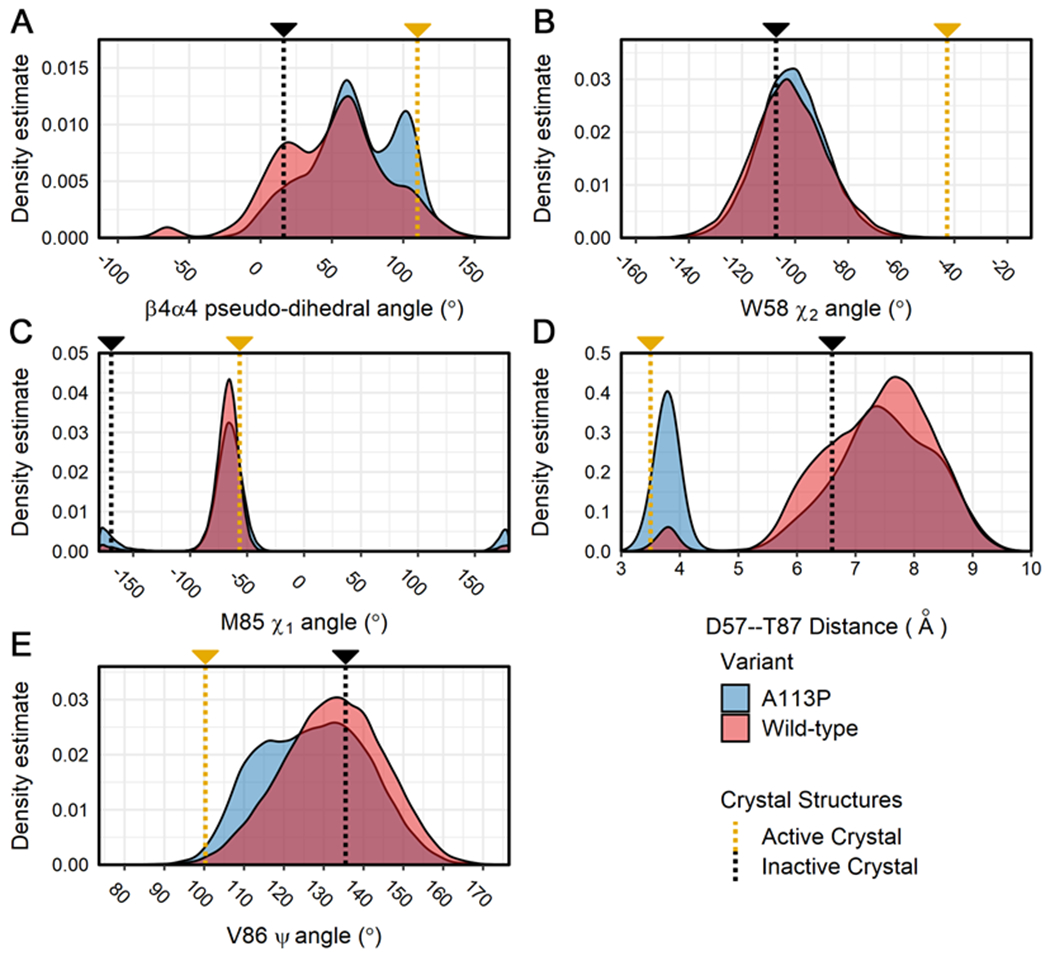
Calculations were performed on all replicates for the wild-type and A113P simulations. Density distributions were generated from each separate ensemble data set (blue = CheY A113P, red = wild-type CheY) for the following structural features: (A) the pseudo-dihedral angle of the β4α4 loop (Cα atoms of residues T87:A88:E89:A90), (B) the χ2 angle for the indole side chain of W58, (C) the χ1 angle for the hydrophobic side chain of M85, (D) the interatomic distance between the side chains of D57 (Cγ; replaced with Be in the active crystal structure) and T87 (Oγ1), (E) the Ψ backbone torsion angle of V86. Measurements for existing high-resolution crystal structures are shown on each plot for reference (gold = active CheY, PDB ID: 1FQW; black = inactive CheY, PDB ID: 3CHY).
Figure 5. Distributions for additional structural features related to activation state in the simulations of CheY variants.
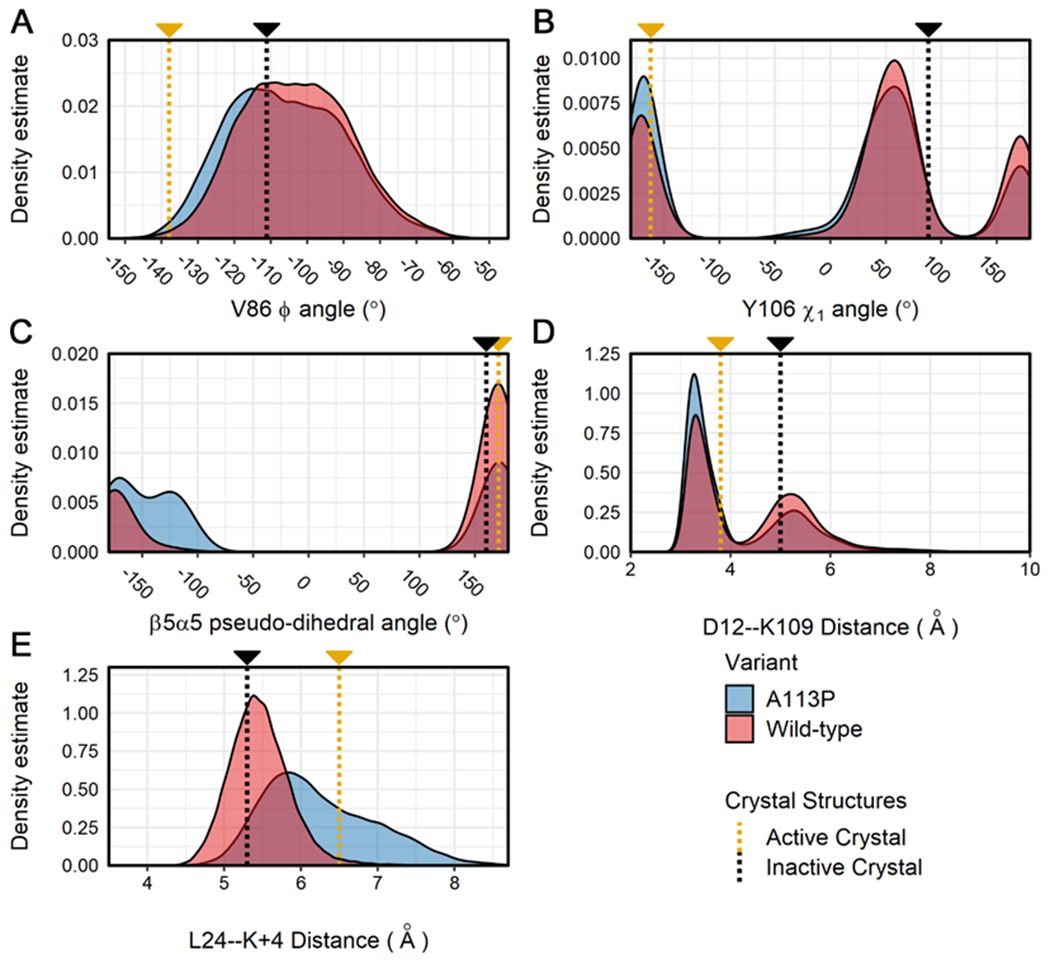
Calculations were performed on all replicates for the wild-type and A113P simulations. Density distributions are displayed for each ensemble data set (blue = CheY A113P, red = wild-type CheY) for the following structural features: (A) the Φ backbone torsion angle of V86, (B) the χ1 angle for the aromatic side chain of Y106, (C) the pseudo-dihedral angle of the β5α5 loop (Cα atoms of residues K109:P110:F111:T112), (D) the interatomic distance between the side chains of metal-binding D12 (Cγ) and K109 (terminal Nζ), (E) the interatomic distance between the β5α5 loop (Cα of position 113) and the α1 helix (Cα of L24). Measurements for existing high-resolution crystal structures are shown on each plot for reference (gold = active CheY, PDB ID: 1FQW; black = inactive CheY, PDB ID: 3CHY).
The distribution in Figure 5C reveals a small but unique feature of the CheY A113P variant. Phosphorylation in E. coli CheY causes the β5α5 loop to reposition relative to the α1 helix, but it retains largely the same conformation. This can be seen in the similar pseudo-dihedral angles of the loop (calculated using the Cα atoms of residues K109:P110:F111:T112) in the active and inactive crystal structures (Figure 5C, gold and black lines). The A113P population samples an additional orientation not seen in either crystal structure, though this is to be expected considering that the substitution introduces a conformationally-restricted Pro residue into the C-terminus of a polypeptide loop. Another small but significant change in the CheY A113P population is shown in Figure 5D, where the interatomic distance between the metal-binding D12 and the essential K109 residues was calculated. The chemistry of phosphotransfer requires a highly specific active site geometry, centered around the charged phosphoryl group and a bound divalent metal cation (often Mg2+). Previous crystal structures of wild-type CheY revealed that the adoption of this geometry involves a reorientation of the side chains of K109 and D12, forming a salt bridge. While nearly imperceptible to qualitative examination, this shift can be seen by comparing the density distribution with the annotations (yellow and black) as seen in Figure 5D, denoting the distances calculated between the side chains in the crystal structures. While both the wild-type and the A113P variant populations sampled the same two distances, the A113P ensemble tended towards a shorter bond geometry. Finally, Figure 5E shows the interatomic distance between the L24 residue on the α1 helix and the K+4 position in the simulated data sets (A113 in the wild-type CheY ensemble, P113 in the mutant ensemble). We observed a clear increase in the relative distance between position K+4 and L24 in both the active crystal structure and the A113P population, suggesting an opening motion between the β5α5 loop and α1 helix regions. Distributions for additional metrics related to activation in CheY and/or other rec domains are included in Figures S3. Of particular interest are the comparisons of the relative solvent accessible surface areas (RASAs) for the K+4 position (Figure S3D), which revealed a substantial shift in the CheY A113P population. Curiously, in several of the parameters the wild-type CheY population deviates from the expected value found in the inactive crystal structure, specifically in Figures 4C (χ1 angle of M85) and 5D (interatomic distance between side chains of conserved residues D12 and K109). Here, the wild-type CheY ensemble appears to more frequently adopt the orientation seen in the activated CheY crystal structure. We speculate that this may simply be a result of the limitations of protein crystallography, which provides only a “snapshot” of any given protein under very specific crystallization conditions.
Due to the significant remodeling of the α4β5α5 region seen in the A113P variant simulations (Figures 4A, 4D, 5C and 5E) and the expectation that the introduction of a Pro residue near the β5α5 loop might rigidify the region, we next estimated the per-residue Cα Root Mean Square Fluctuation (RMSF) for each CheY variant ensemble. These findings are included in Figure 6, with colored shading representing the mean and standard deviation for both simulated data sets. A two-sided t-test was performed to detect statistically significant differences in the two populations (p < 0.01). Residues near the phosphorylatable D57 exhibited relatively high levels of flexibility in both of the CheY variants, but only the β5α5 region (shaded residues) demonstrated a unique increase in fluctuation as a result of the A113P substitution. This initially seemed counterintuitive, given the restrictive nature of a Pro residue in a polypeptide chain. However, we suggest that the presence of Pro at K+4 subtly shifted the internal conformation of the β5α5 loop in CheY A113P (Figure 5C), affecting the intramolecular interaction(s) between it and the rest of the protein and altering the orientation, plasticity, and dynamics of the region. As a consequence of this, the CheY A113P variant exhibited enhanced fluctuations in the β5α5 loop as a whole. Previous preliminary NMR studies examining backbone and side chain order parameter values (S2 and S2axis) also predicted a small but significant increase in flexibility throughout the CheY A113P structure compared to wild-type, including the region near Y106 and continuing through the α5 helix.85 This enhanced level of motion in a functionally relevant region may make sampling a phospho-competent active conformation more likely and/or favorable for the CheY A113P variant.
Figure 6. Average CαRMSF for CheY variants during simulation.
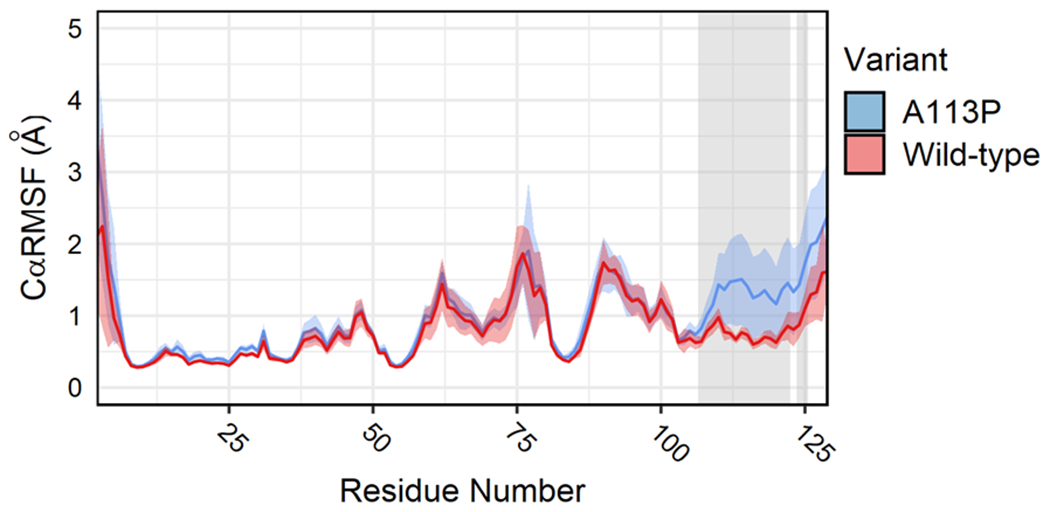
Per-residue CαRMSF was calculated across production replicates to quantify atomic fluctuations during the simulations. Solid lines indicate ensemble averages. Colored shaded regions represent standard deviations of the mean (one frame = 20 ps; blue = CheY A113P, red = wild-type CheY). Grey shaded areas indicate regions of statistically significant differences detected between the two CheY variants (two-tailed t-test, p < 0.01, minimum mean difference > 0.15 Å).
Network analysis reveals that the A113P substitution significantly alters the allosteric communication system within CheY
To search for potential changes in the interactions between the functional regions in CheY triggered by the A113P substitution, which might explain the increase in flexibility shown in Figure 6, we analyzed the dynamical motion within both the wild-type and the A113P mutant systems. We first calculated the pairwise cross-correlation between residues (Cα atoms) using linear mutual information (LMI).74 This allowed us to quantify the correlated motions between protein regions in the two ensembles and compare them directly. In the literature, atomic cross-correlations in proteins have been used to identify areas of functional or structural importance, as well as to provide insight into the flow of allosteric information within enzymes.33, 73, 86–88 Figures 7A and 7B show the consensus matrices for the wild-type and the A113P trajectories, respectively. Figure 7C shows the difference matrix between the two variants (A113P minus wild-type). The majority of the correlated movement in both systems was found along the diagonal (perfect correlation = 1; residues are perfectly correlated to themselves), attributable to the residues being in close proximity as components of common secondary structures. We focused only on regions that exhibited changes in correlation that would likely be attributable to the A113P substitution. These differences were generally subtle, with small increases and decreases occurring throughout the structure. However, changes in correlation within the following regions (outlined and labeled in black in Figure 7C) were predicted to be relevant: α1—β2α2 (A); β1α1—β3 (B); α1—β4 (C); α1—β5α5 (D); α2—α3 (E); β3α3—β4α4β5 (F); β4—β5α5 (G). Each of these areas includes two noncontiguous regions within CheY that are likely to be involved in the activation of the A113P mutant. In particular, boxes D, F, and G in Figure 7C highlight changes involving the α4β5α5 region that may provide insight into the findings shown in Figure 6.
Figure 7. Consensus cross-correlation matrices for each CheY variant ensemble.
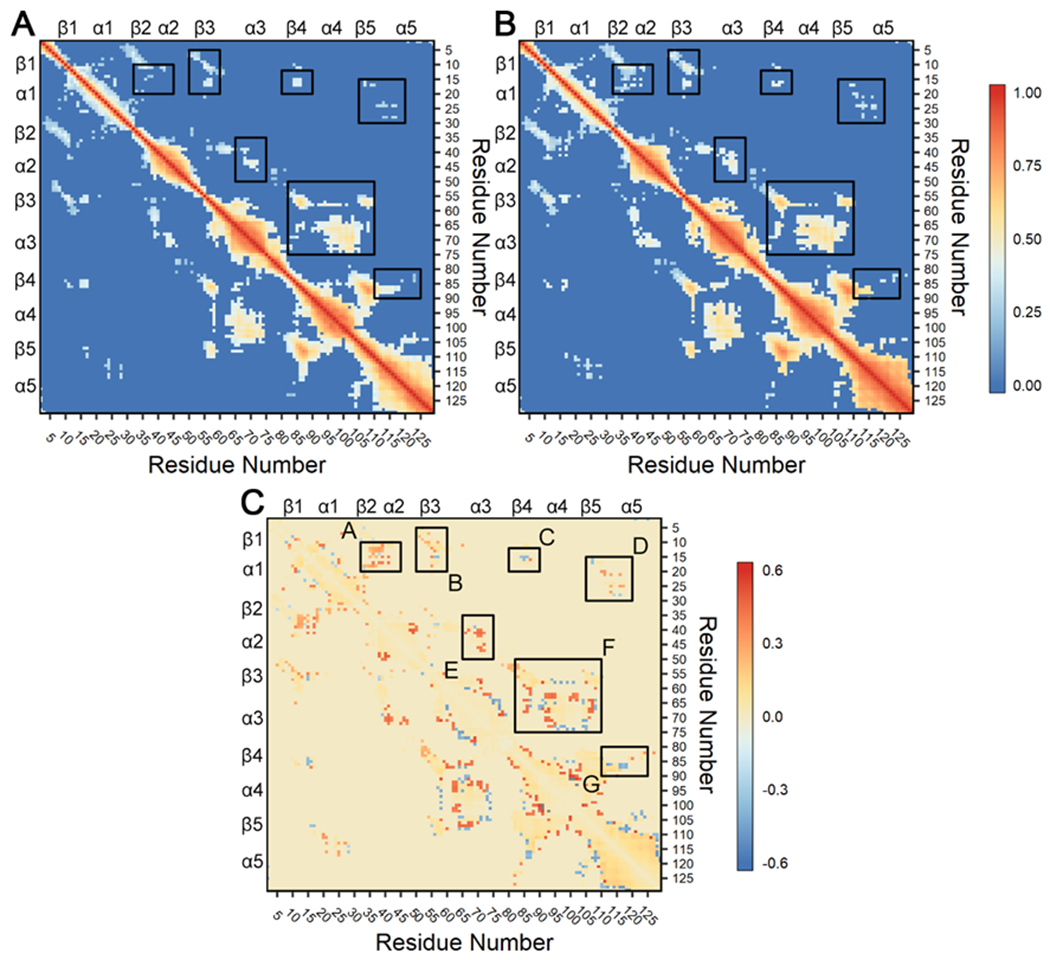
Calculations were performed on all replicates for the wild-type and A113P simulations and filtered with a consensus contact map as described in 73, 75, 89. Briefly, analysis used linear mutual information between Cα atoms to create a covariance matrix for each CheY variant.74 Correlations ≥ 0.30 in all twelve replicates were retained as reliable couplings. All other correlations ≥ 0.30 in at least one replicate were excluded if the respective Cα atoms were separated by > 10 Å in 60% of cumulative frames in each replicate trajectory. Correlations failing to meet these requirements were set to zero. Additional cutoffs were examined with similar findings. (A) Correlation observed in the wild-type CheY simulations. (B) Correlation observed in the CheY A113P simulations. In both panels, positive values (red) indicate strongly correlated regions. (C) Difference matrix (B – A) showing changes in dynamical cross-correlation caused by the A113P substitution. Positive values (red) indicate stronger correlation in the A113P variant compared to wild-type CheY, while negative values (blue) indicate weaker correlation. Black boxes highlight areas with differences in correlation between residues in distinct secondary structures, indicating potential functional significance. Note that panels A and B share a common scale (0 to 1), while panel C uses a separate scale (−0.6 to +0.6).
To gain a more comprehensive understanding of the changes triggered by the A113P substitution to the signaling pathways within CheY, correlation-based, coarse-grained residue networks were constructed for both wild-type and A113P systems using the LMI matrices described above. Each residue (Cα atom) was represented by a single network node. Nodes were connected to one another through edges weighted by corresponding correlation values. We then used community analysis to identify various clusters of highly intracorrelated blocs of residues under the assumption that these groups would possess functionally related roles within the CheY structure (Figure 8; a full list of network assignments can be found in Table S3). Visualization of these groups revealed several relevant intercommunity interactions (Figures 8C and 8F). While typically weaker than intracommunity couplings, these interactions can provide valuable insight into the allosteric network of the protein, highlighting key pathways of signal transfer between distant sites.
Figure 8. Residue community analysis for CheY wild-type and A113P variants.
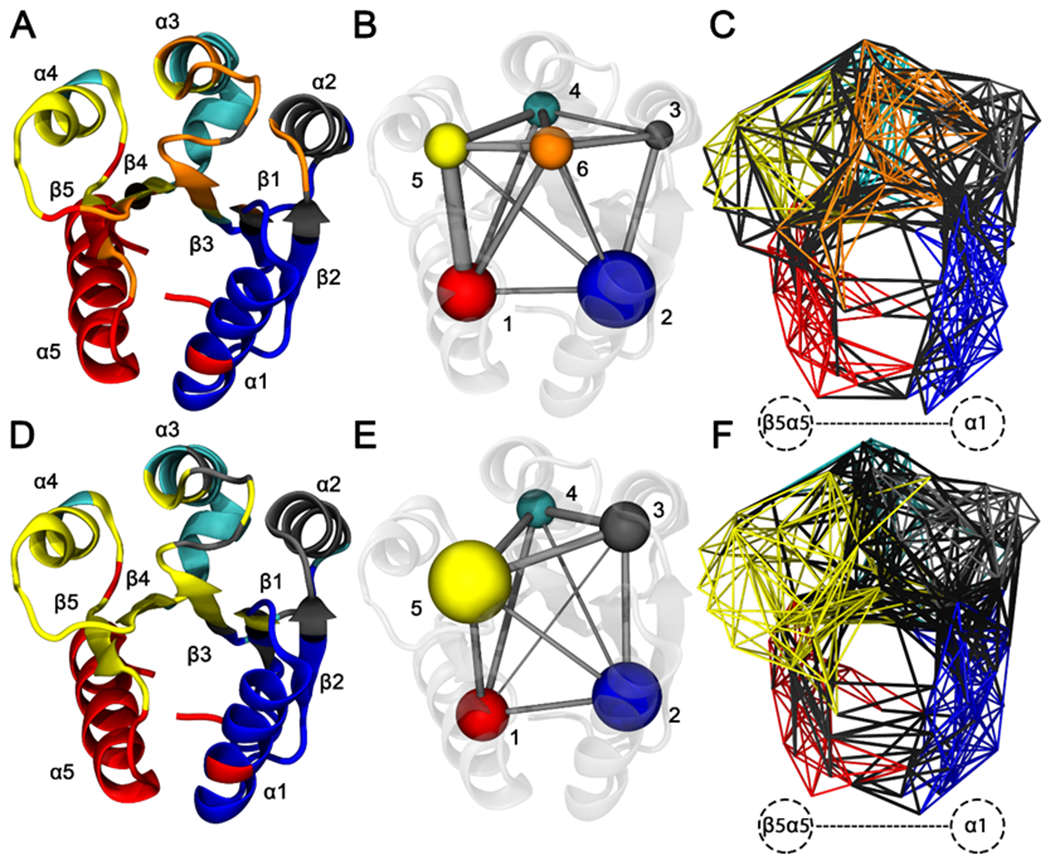
Calculations were performed using the filtered consensus LMI matrices. Residue Cα atoms were represented by network nodes. Pairs of nodes were connected by edges weighted by the correlation between the corresponding residues. Community detection was performed using the Girvan-Newman betweenness clustering algorithm. Residues were color coded based on optimal community membership. Wild-type CheY is shown in the top row, and CheY A113P is shown in the bottom row. (A and D) Cartoon diagram of wild-type CheY, with secondary structures colored based on residue community assignments. (B and E) Simplified schematics showing differences in residue network topologies. (C and F) All-residue depictions showing full network connectivity. Colored edges indicate intracommunity couplings. Black edges indicate intercommunity couplings. Labeled region in panels C and F show substantially altered interactions between the β5α5 region and the α1 helix.
Overall network architectures were highly similar between the two CheY variants, with most communities centered on the surrounding helical regions. Six clusters were identified in the wild-type CheY ensemble: one for each of the five α helices and one for the active site. Communities #1 and #2, featuring the α1 and α5 helices (Figures 8A and 8B, blue and red regions), were the largest groups and shared moderately strong intercommunity couplings (Figure 7A, box D; Figure 8C, black edges). Community #6 (Figure 8B, orange region) contained the phosphorylatable D57 and other functionally important residues, including positions T+1/T+2, D+1/D+2, D, K and K+1.12, 14, 19, 20, 25, 28, 30 Also notable in the wild-type CheY network was a distinct community formed by the α4 helix and a portion of the β4α4 loop (Figures 8A and 8B, community #5, yellow region). Unlike the wild-type network, only five communities were detected in the CheY A113P ensemble; the previously mentioned community #5 (Figure 8B, yellow region) was combined with portions of community #6 (Figure 8B, orange region) to create a single larger residue group (Figures 8D and 8E, yellow region). Additionally, certain residues previously assigned to community #6 (the “active site” bloc) were instead incorporated into community #3 in the A113P variant (Figure 8F, grey region). Figures 8C and 8F highlight the region between the β5α5 loop and the α1 helix, where a substantial increase in the number of intercommunity correlations (black edges) was detected in the A113P variant when compared to the wild-type network. These findings further highlight the change in the intramolecular interaction(s) between the β5α5 region and the rest of the system, indicating a subtle but significant remodeling of the allosteric network in the protein and corroborating the results shown in Figure 6. They also suggest that the functionally relevant residues involved in the conformational transition from an inactive to an active state interact differently in the CheY A113P network, particularly when considering the merger of the β4α4 region and the active site communities.
Using the correlation network constructed above, we calculated per-residue centrality scores based on normalized node “betweenness.” Betweenness is defined as the number of unique shortest paths crossing a given node. Residues with high centrality/betweenness are known to be significant for enzymatic function, frequently serving as active site residues or key parts of an allosteric pathway.90–92 By quantifying and comparing the centrality of each residue in the two variants, we gained insight into the allosteric changes triggered by the A113P substitution. Figure 9 shows a per-residue plot of normalized betweenness for the wild-type and A113P variants using the network structures in Figure 8. While the two centrality profiles exhibited much overlap, several positions were found to have contrasting levels of betweenness (see top labels for residues with noteworthy scores). M17, L24, L43, V86, A101, S104, and V107 all scored higher in the wild-type network than in the A113P network, while G39, D57, G65, K91, K109, F111, T112, L116, and E117 all exhibited greater centrality in the A113P mutant network than in the wild-type network. It is uncertain what roles G39, L43, G65, A101, and S104 might play in an allosteric network related to the β5α5 loop, given that they are outside the region between the K+4 position and the active site. However, the remaining residues were located either near the K+4 position or between it and the active site, making them ideal candidates for additional study.
Figure 9. Normalized residue-wise centrality for the CheY variant networks.
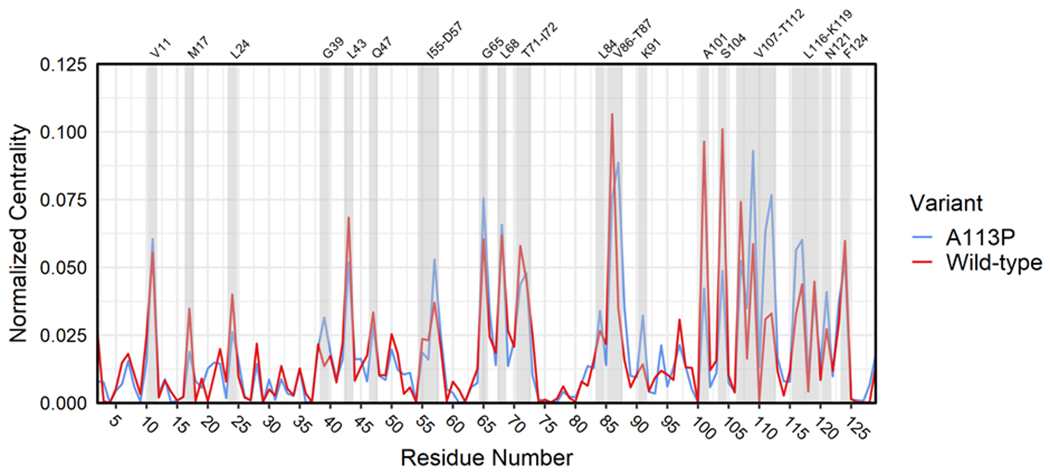
Per-residue normalized betweenness scores for wild-type and A113P networks (blue = CheY A113P, red = wild-type CheY). Grey shading indicates residues of interest with known functional roles and/or high centrality in one or both variants (labels on top).
Torsion angle dynamics reveal that the distal K+4 position affects a hydrophobic side chain network extending to critical active site residues
To further compare the conformational dynamics of the individual residues within the CheY variants, we used the Kullback-Leibler (KL) divergence to quantify changes in their conformational dynamics (see Materials and Methods). KL divergence is an information theoretic measurement of the difference between two probability distributions over the same variable.65 Comparing the per-residue KL divergence scores for the internal dihedral angles between a wild-type and a mutant ensemble can reveal dynamic structural perturbations triggered by the corresponding substitution(s). Dihedral angle measurements were extracted from the all-atom trajectories for each replicate and combined in a residue-wise manner to create two data sets of torsion angle distributions. KL divergence scores were calculated for each residue and mapped onto the wild-type CheY crystal structure (PDB ID: 3CHY) for visualization. Figure 10A shows the resulting structure as a tube representation with segment thickness and color-coding scaled proportionally to the magnitude of the detected divergence. The residues most substantially affected by the A113P substitution were near the K+4 position (Figure 10A and 10B, red, green and orange residues). Subtle but significant conformational perturbations (blue positions) were found radiating out as far as the essential catalytic residues D57 and K109. A literature search for existing phenotypic data on the full list of significantly affected residues revealed that nearly 50% of positions perturbed by the A113P substitution in CheY were involved in known activating substitutions at equivalent positions in other rec domains and/or organisms (summarized in detail in Table S4).21 When comparing normalized betweenness scores as seen in Figure 9, approximately 50% of the significantly perturbed positions also possessed noteworthy centrality within the CheY residue network(s). Both of these findings imply the existence of an underlying framework of “effector nodes” that may exist in at least some rec domains such as in CheY.
Careful examination of the residues found to be significantly affected by the A113P substitution revealed a potential path of signal transmission from the K+4 position to the active site (D57), running through a region bounded by the β5α5 loop and the innermost face of α1 helix. A close-up of this area is shown in Figure 10B, highlighting multiple hydrophobic side chains from perturbed positions found along the path: residues such as V21, L24, L25, L84, V86, F111, and L116. Other significantly affected residues (M17, V108, P110, and T112) were detected, but their involvement in the internal hydrophobic lattice was likely limited due to the location, positioning, and/or nature of their side chains. The side chains of residues V108, P110, and T112 were oriented away from the hydrophobic core, while M17 is located at the terminus of the proposed allosteric pathway. These results provided a useful guide for experimental characterization of this network and its role in the enhanced active-like phenotype of the A113P variant.
Kinetic characterization of the A113P variant combined with substitutions at several other key positions in CheY reveals the extent of the hydrophobic allosteric network
We next sought to assess the function of the hydrophobic side chain network described above using site-directed mutagenesis at strategic positions in and around the network. Based on the perturbations triggered by the A113P substitution, we predicted that disruptions to the hydrophobic lattice would produce significant non-additive effects on CheY phosphorylation kinetics when combined with the A113P substitution, while disruptions at the periphery or outside the predicted side chain network would generate more straightforward additive effects. To that end, we characterized the following substitutions in both CheY wild-type and A113P backgrounds (predicted effects and justifications are provided in parentheses): M17A (additive, on the periphery of the network; Ala was used because CheY does not tolerate polar/charged residues at this position); L24S (non-additive); V86S (additive, just outside of the predicted network; Ser was used because CheY does not tolerate large/charged residues at this position); V108T (additive, side chain orients towards the exterior of the network); F111V (non-additive, Val was used due to its known enhancement of autophosphorylation kinetics); T112A (additive, side chain orients towards the exterior of the network).19, 24 Autophosphorylation and autodephosphorylation kinetic rate constants were obtained for each of these substitutions, along with wild-type and CheY A113P replicates (Table 2).
Table 2.
Observed rate constants for E. coli CheY variants.
| Protein | kdephos (min−1) a | K1/2 (mM) b | kdephos/K1/2 (M−1s−1) c | ||
|---|---|---|---|---|---|
|
| |||||
| PAM | AcP | PAM | AcP | ||
| CheY wild-type | 4.0 ± 0.5 | 8.6 ± 2 | 7.0 ± 0.7 | 7.8 ± 2 | 9.5 ± 1 |
| CheY A113P | 4.7 ± 0.2 | 1.6 ± 0.1 | 2.2 ± 0.2 | 63 ± 5 | 36 ± 0.9 |
| CheY M17A | 1.7 ± 0.1 | 2.3 ± 0.2 | - | 12 ± 2 | - |
| CheY M17A A113P | 3.2 ± 0.1 | 0.68 ± 0.05 | - | 79 ± 7 | - |
| CheY L24S | 6.2 ± 0.1 | 2.5 ± 0.5 | - | 41 ± 5 | - |
| CheY L24S A113P | 6.4 ± 0.4 | 2.0 ± 0.1 | - | 55 ± 5 | - |
| CheY V86S | 4.6 ± 0.2 | 4.8 ± 0.6 | - | 16 ± 2 | - |
| CheY V86S A113P | 4.8 ± 0.2 | 6.9 ± 0.2 | - | 12 ± 1 | - |
| CheY V108T | 4.1 ± 0.4 | 5.6 ± 0.3 | - | 12 ± 1 | - |
| CheY V108T A113P | 5.8 ± 0.2 | 1.3 ± 0.09 | - | 74 ± 6 | - |
| CheY F111V | 7.6 ± 0.3 | 0.89 ± 0.06 | - | 140 ± 10 | - |
| CheY F111V A113P | 6.7 ± 0.08 | 0.82 ± 0.04 | - | 140 ± 8 | - |
| CheY T112A | 4.8 ± 0.2 | 4.7 ± 0.6 | - | 17 ± 2 | - |
| CheY T112A A113P | 6.2 ± 0.2 | 0.85 ± 0.1 | - | 120 ± 20 | - |
Dephosphorylation kinetics were determined using the pH-jump method (n=3-5, with 12-20 replicate curves for each variant).
K1/2 values (concentration of donor required to phosphorylate 50% of the protein population; n=3 for each variant).
kdephos/K1/2 is equivalent to the bimolecular autophosphorylation rate, kphos/Ks.19
Mean values with standard deviations of the mean are provided.
The data in Table 2 indicate that the A113P substitution accelerated autophosphorylation (estimated by the rate constant kdephos/K1/2) approximately 8-fold over wild-type, but left autodephosphorylation (estimated by the rate constant kdephos) relatively unaffected. There was no appreciable difference in divalent metal (Mg2+) binding affinities between the wild-type and A113P variants, ruling out incomplete metal saturation as an explanation for the change (Table S5). Additionally, multiple small molecule phosphodonors (acetyl phosphate or AcP, and phosphoramidate or PAM) showed kinetic enhancement (Table 2). Taken together, these findings corroborated our previous results and suggested that the A113P substitution likely affects the kinetics of autophosphorylation through conformational selection rather than by directly altering donor binding and/or the chemistry of phosphorylation.19, 21 The proposed mechanism is supported by increased affinity between CheY A113P and the phosphoryl group mimic, BeF3−, when compared to wild-type CheY (Table S5). Interestingly, data displayed in Table 2 indicate that similar to A113P, none of the other tested substitutions had a significant effect on the autodephosphorylation reaction. However, they did exhibit substantial influence on the autophosphorylation reaction, with estimated rate constants spanning a >10-fold range. Furthermore, none of the substitutions (single or double) produced a lower autophosphorylation rate constant than that of wild-type CheY. These results were reminiscent of our recent findings regarding the effect(s) of the nearby K+1/K+2 positions on the β5α5 loop on the phosphorylation kinetics of CheY.19 The full implications of each substitution (including their additive vs. non-additive natures) along with how they relate to our prior predictions are addressed in detail in the Discussion section.
Discussion
The proposed hydrophobic side chain network transmits the allosteric signal triggered by the A113P substitution
To obtain a quantitative view of the role of the proposed hydrophobic side chain network in the partial allosteric activation triggered by the A113P substitution, we compared the reaction rate constants of the chosen CheY variants shown in Table 2 using the concept of double mutant cycles. This approach is based on the proportional relationship between the activation energy (ΔG‡) of a given reaction and the logarithm of its corresponding reaction rate constant (ΔG‡ = −RTln(k)). Using the rule for the subtraction of logarithms, the difference in activation energies (ΔΔG‡) between a wild-type (k1) and a substitution variant (k2) can be described by the equation ΔΔG‡ = −RTln(k1/k2). The sum of the changes in activation energy of single substitutions made at two independent positions will equal the change in activation energy exhibited by the corresponding double substitution (ΔΔG‡1+ΔΔG‡2 = ΔΔG‡1+2), demonstrating an additive kinetic effect.14, 30, 93, 94 Positions that deviate from this rule are thought to exert their influence on the reaction collectively (either antagonistically or synergistically), exhibiting a non-additive effect. Because the A113P substitution primarily affected the autophosphorylation reaction, only the kdephos/K1/2 values were examined as approximations of the corresponding autophosphorylation rate constants (kphos; justified in 19). The logarithm of the ratios of the substitution variant to wild-type CheY kdephos/K1/2 measurements were used to calculate each ΔΔG‡ value. The final values of the double substitutions were compared to the sums of the values for the individual substitutions. Table 3 contains each of these estimates, along with the absolute deviations between the observed and predicted values.
Table 3.
Additivity of the effects of substitutions at various positions with A113P (K+4) on the autophosphorylation rate constant.
| Substitutions | Expected value (ΔΔG‡1+2) a | Observed value (ΔΔG‡) b | Absolute value of difference (|Expected – Observed|) c |
|---|---|---|---|
| CheY M17A A113P | 1.01d | 1.02 | 0.01 |
| CheY L24S A113P | 1.53 | 0.85 | 0.68 |
| CheY V86S A113P | 1.13 | 0.18 | 0.95 |
| CheY V108T A113P | 1.01 | 0.98 | 0.03 |
| CheY F111V A113P | 2.08 | 1.25 | 0.83 |
| CheY T112A A113P | 1.15 | 1.20 | 0.05 |
The expected value, assuming no interaction between the individual positions, equals log10(ratio of A113P to wild-type CheY rate constants) + log10(ratio of specified single variant to wild-type CheY rate constants).
The actual value equals log10(ratio of the specified double substitution to wild-type rate constants).
We considered a deviation from the anticipated additive effects significant if the absolute difference > 0.65, implying that one of the positions influences the contribution(s) of the other.
All table values are given in units of −RT.
Positions M17, V108, and T112 exhibited no significant differences between the observed (double substitution) and the expected (sum of single substitutions) values (Table 3). Each of these residues was predicted to have primarily additive effects on the hydrophobic lattice revealed by the in silico analysis, due to their presumably unsuitable side chain orientations and/or their location on the periphery of the putative hydrophobic network. However, positions L24, V86, and F111 showed significant deviations between the observed and expected numbers, implying stronger non-additive effects. Substitutions at each of these positions in the A113P background exhibited significantly less enhancement of the autophosphorylation rate than the sum of the individual substitutions would imply, suggesting that these substitutions were somehow antagonistic towards the effect(s) of the K+4 position. Based on the in silico results, a path of long-distance allosteric transmission can be traced from the location of the substitution to the active site (Figure 10B; K+4 to D57), traveling along the β5α5 loop and up the α1 helix. It is reasonable to assume that substitutions along this region, at appropriate residues, would produce a similar effect(s) as the A113P substitution, though not necessarily to the same degree. We propose that the substitutions L24S and F111V influence CheY autophosphorylation kinetics through a similar mechanism as A113P, i.e., altering the conformational equilibrium of the pre-phosphorylated population. Hence, the seemingly antagonistic effects of their constituent substitutions.
The conserved K109 residue acts as an electrostatic switch and is an important component for the allosteric effect triggered by substitutions in the α1α5 region
The explanation offered above does not readily account for the unexpected effect of the V86S substitution. Despite the position displaying one of the highest centrality values in both CheY variant residue networks (suggesting high functional relevance; Figure 9), the V86S substitution was not initially predicted to suppress the enhanced autophosphorylation phenotype of CheY A113P. Residue V86 consistently demonstrated only minor differences in dynamics between the wild-type and A113P CheY variants (Figures 4E, 5A and 10B). This is likely due to its role as a buried structural support with limited flexibility, as suggested by a low level of fluctuation in Figure 6. However, the antagonistic effect of V86S on A113P provided insight into the overall allosteric mechanism of the activating K+4 substitution. A hallmark of CheY (and rec domains in general) activation is the rearrangement of K109, which forms a salt bridge with the metal-binding D12 and the phosphoryl group (or phosphoryl mimic such as BeF3−).47 Metal- and FliM-binding are thought to trigger this remodeling as well.37, 95 Due to the critically conserved nature of the K109 residue in rec domains, it has long been considered a key component of an electrostatic “switch” involved in the arrangement of the rec domain active site for metal-binding/phosphorylation and the requisite conformational transition to an active-like state.23 The location of V86, deeply buried within a hydrophobic pocket directly next to K109, suggests that changes to the residue could have profound effects on the side chain of the functional lysine residue. Our findings regarding V86S implied that the allosteric effect(s) triggered by the A113P substitution (and likely by L24S and F111V) depend on the K109 residue for propagation (Figure 9 shows substantial increase in betweenness at K109 in the A113P variant). Disruption of this key position, such as perturbation of the local electrostatic environment by the V86S substitution, may disrupt allosteric signal transmission traveling through the proposed hydrophobic side chain network. What remains unclear is the exact role of V86 in the function of E. coli CheY. Previous work suggests that the in vivo activity of CheY is intolerant of large and/or charged residues at position 86, likely for the reasons described above.24 The position is also affected by both phosphorylation and partner binding in CheY.37, 96–98 Further work will be required to better understand the role of the V86 residue in CheY and related rec domains.
Residues affected by the activating A113P substitution participate in activating interactions between CheY and its binding partners.
The exact relationship(s) between rec domain donor binding, conformational transition, phosphorylation and dephosphorylation are unknown. However, the relative conformational and kinetic effects of partner binding on CheY are known, or can be surmised, based on basic biology and previous biochemical studies. E. coli CheY interacts with the upstream donor HK, CheA, through two different domains: CheAP1 and CheAP2.99 The CheAP2 domain acts to capture unphosphorylated CheY molecules, exhibiting an increased affinity for the apo form (additionally, CheAP2-bound CheY exhibits a significantly diminished autophosphorylation ability in vitro).17, 100, 101 Unphosphorylated CheY then binds the CheAP1 domain so that phosphotransfer can occur. While the conformational changes induced by an interaction with the CheAP1 domain are unclear, we can assume that CheY must adopt a primed, active-like conformation in response to, or concurrently with, binding to CheAP1 before the reaction (and the pathway) can proceed. Binding of the downstream partner, FliM, can also induce a more active-like conformation in CheY, as suggested by its increased in vitro autophosphorylation rate in the presence of the fragment FliM1-16.17 Finally, the phosphatase CheZ must bind phosphorylated CheY and maintain a sufficiently active-like conformation to assist in the hydrolytic dephosphorylation reaction. A significantly increased CheY autophosphorylation rate has also been observed in the presence of CheZ in vitro.17 A similar example of allosteric activation is found in the DesK/DesR system in Bacillus subtilis. Binding to the DesK HK is sufficient to trigger the allosteric activation of the monomeric form of the DesR response regulator, even in the absence of phosphorylation.102 The authors referred to this binding step as “preactivation”. Each of the CheY partner binding events involves key residues primarily on the α1α5 and β5α5 surfaces of the rec domain. We speculated that the activating A113P substitution perturbs a similar set of residues as those involved in CheAP1, CheZ, and/or FliM1-16 binding, eliciting an active-like phenotype by flipping a similar and likely diffuse allosteric switch.
To investigate this, we performed a comparison of residues predicted to be structurally perturbed by the A113P substitution (based on KL divergence) against every residue experimentally implicated in CheY partner binding (CheAP1, CheZ and/or FliM1-16; based on literature-sourced biochemical studies, Table S6). The comparison revealed a high degree of overlap between the two groups (nearly 60% of the predicted residues are shared, the majority being involved in CheAP1 binding; Figure 11).
Figure 11. Overlay of known CheY partner binding residues (red) and positions determined to be significantly perturbed by the A113P substitution (light blue).
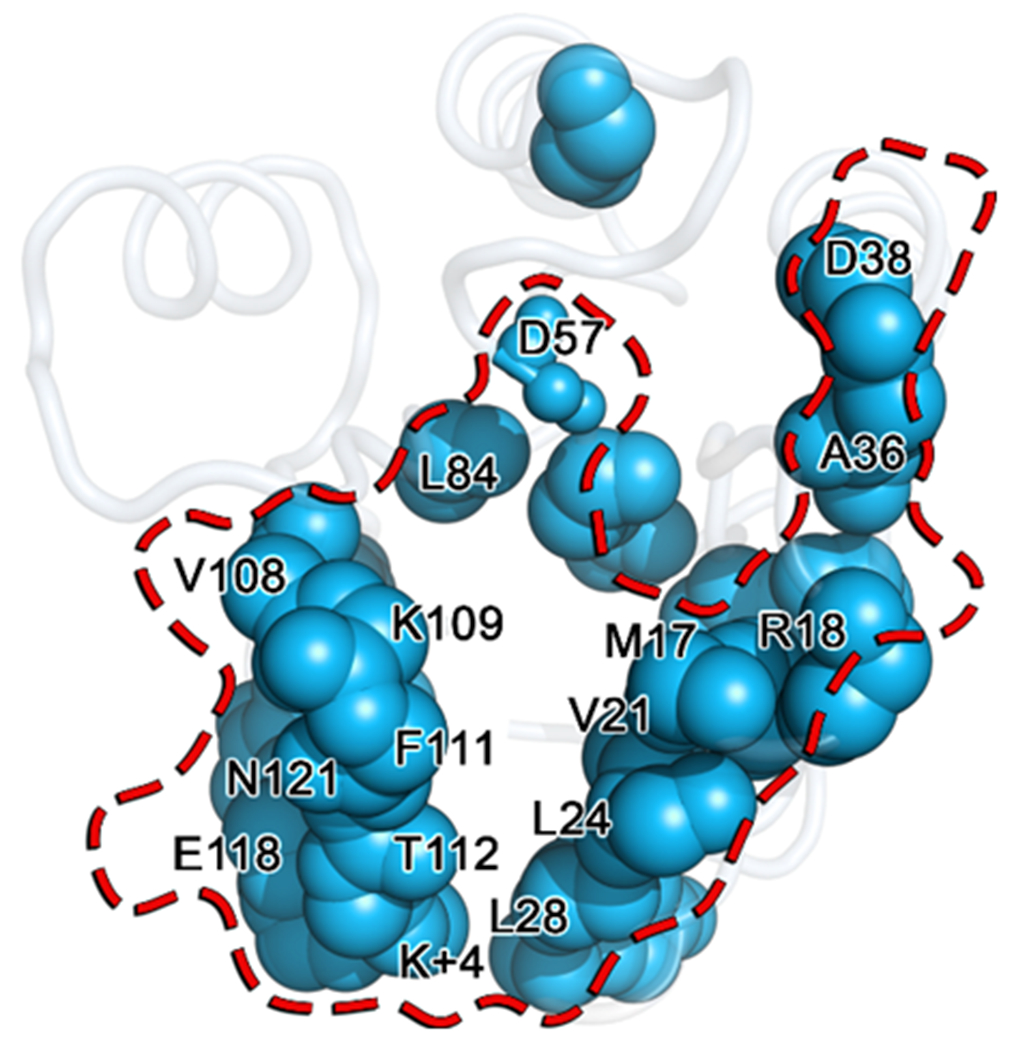
Residues identified as significantly perturbed by the A113P substitution using the KL divergence are shown as blue spheres mapped onto the wild-type CheY crystal structure (PDB ID: 3CHY). Residues known to be involved in CheY binding to its partners, CheAP1, CheZ and/or FliM1-16, are denoted with a transparent surface representation outlined in red. Residue positions shared by both groups are labeled, revealing a high degree of overlap.
Evolutionary rationale for uncommon residue types at position K+4 in receiver domains
Sequence analysis of rec domain subfamilies revealed that the most abundant amino acids in nature at position K+4 are Pro and hydrophobic branched residues such as Leu/Ile/Val (Figure 1). However, when segregated into putative FunFams, chemotaxis-related rec domains showed enrichment of uncommon residue types such as Ala/Gly and Asp/Glu. From a phosphorylation standpoint, the wild-type amino acid at the K+4 position in E. coli CheY (Ala; A113) functions less efficiently than the more common Pro seen in the majority of rec domains (see Table 2). While it is challenging to determine the exact evolutionary reason(s) that rec domain-containing proteins such as CheY might have uncommon residue types at K+4, we provide two potential explanations here.
First, the K+4 position in rec domains is likely to be strongly influenced by partner interactions. The α4β5α5 region of rec domains has long been implicated in dimerization events. CheY binds the downstream FliM and the upstream CheAP2 through this region. Additionally, as previously stated, the α1α5 region is known to bind upstream partners such as CheAP1. Residues at the interface of protein-protein interactions are typically highly specific and constrained through coevolution. This is the principle by which interfacial residues between binding partners coevolve through rounds of disruptive and compensatory changes to optimize the physicochemical properties of their binding surfaces.103 It is likely that the residue type at the K+4 position is dependent on the intermolecular protein-protein interactions in which the corresponding rec domain participates. For example, in E. coli, the estimated Kd between phosphorylated wild-type CheY and CheZ is 13-26 μM.21, 97 However, the estimated Kd between phosphorylated CheY A113P and CheZ is only 8 μM, suggesting a potential change in binding affinity of up to 2 to 3-fold as a result of the substitution.21 It is unclear what effect(s) such a change would have on the chemotaxis system in E. coli, though the significance may be greater in other rec domain examples.
Second, the K+4 position may have evolved in chemotaxis-related rec domains to minimize background noise generated by endogenous small molecule phosphodonors. While the primary phosphodonor used for this study was phosphoramidate (PAM), other compounds are known to phosphorylate rec domains. Table 2 shows that the enhanced autophosphorylation phenotype of CheY A113P is independent of phosphodonor. Unlike PAM, acetyl phosphate (AcP) is a physiologically relevant compound that can be found in vivo as an intermediate in the phosphotransacetylase-acetate kinase pathway (reviewed in 104). In certain bacteria, AcP can accumulate to relatively high levels (≥3 mM in E. coli) and is dependent on growth conditions and environmental factors.105 As shown in Table 2, the CheY A113P K1/2 value (the amount of substrate required to maintain 50% of the population in a phosphorylated state) is only 2.2 mM. This increased ability to utilize intracellular AcP as a phosphodonor could explain the constitutively active/kinase-independent/phosphorylation-dependent in vivo phenotype of the CheY A113P variant. Some rec domains, such as those related to chemotaxis, may have selected for less common and less efficient residue types at position K+4 (Ala/Gly/Asp/Glu) simply to decrease the sensitivity of the system. This would prevent off-target pathway activation and decrease the background noise generated by donors such as AcP. Some precedent exists for rec domains utilizing intracellular AcP for activation, including PmrA from S. enterica (kinase-independent, but AcP-dependent; 106), the PhoP S93N variant from S. typhimurium (constitutively active, AcP-dependent; 107), LytR from S. aureus (can utilize AcP more efficiently as a donor than its own partner kinase; 108), and DegU from B. subtilis (kinase-independent, AcP-dependent; 109).
As detailed above, it seems reasonable to suppose that increased autophosphorylation ability (allowing increased use of intracellular AcP) and increased binding affinity for the downstream flagellar motor protein FliM (regardless of phosphorylation state) allows the CheY A113P mutant to function as an in vivo activator of the chemotaxis pathway. However, we cannot fully rule out the possibility that the activating substitution operates through another phosphorylation-independent mechanism. This alternative is supported by the fact that CheY A113P exhibits two- to three-fold increased affinity for its phosphatase, CheZ21, potentially negating the increase in the population of autophosphorylated CheY A113P. Ablation of CheY A113P activity by introduction of a nonphosphorylatable D57A substitution is consistent with a mechanism dependent on phosphorylation21, but the Ala substitution might also disrupt a process that is separate from the phosphorylation reaction. Further study will be required to completely unravel the phenotype of the A113P mutant.
An independent allosteric mechanism for influencing the pre-existing conformational equilibrium through the β5α5 loop and the α1 helix
Allosteric changes induced by rec domain phosphorylation are typically observed in the β4α4 loop and α4β5α5 surface regions and can trigger downstream events such as partner binding or dimerization. It is worth noting that the NarL family of response regulators dimerizes using the α1α5 surface and thus likely employs the allosteric pathway reported here.5 In rec domains similar to CheY, allosteric signal transmission likely occurs in a bidirectional manner between the active site and these distal regions. Interaction with binding partners at the α4β5α5 surface can dramatically alter the kinetic properties of CheY (Table S7). The presence of the downstream flagellar motor protein (FliM1-16) greatly accelerated the autophosphorylation reaction (~15-fold increase for wild-type CheY), while the presence of the upstream CheY-binding domain of CheA, (CheAP2) led to a decrease in the autophosphorylation rate constant (~6-fold decrease for wild-type CheY). The reverse of this concept is true and is physiologically critical to processes such as chemotaxis. Phosphorylation of wild-type CheY increases affinity for FliM1-16 and decreases affinity for CheAP2 (by ~10-fold and ~6-fold, respectively).17 It is tempting to assume that bound FliM1-16 or CheAP2 is sufficient to force the CheY population to the conformational “endpoints” (active-like or inactive-like states, respectively), similar to the consequences of phosphorylation. However, our results suggest that the allosteric mechanism of activation achieved through the K+4 position is independent of CheAP2/FliM1-16 binding and the central face of the α4β5α5 region. This explains how the autophosphorylation rate constant of CheY A113P in the presence of FliM1-16 is increased by an amount comparable to that of the wild-type protein (10- to 15-fold increase for both CheY variants), while greatly exceeding the actual value of the wild-type rate constant. The same reasoning applies to the additive effect we observed for CheY A113P in the presence of CheAP2. Figure 11 and Table S6 show that the majority of the dynamic perturbations associated with the A113P substitution affected residues directly involved in the docking of the upstream phosphodonor domain, CheAP1. We propose that the β5α5-α1 interface serves two non-mutually exclusive functions in CheY and other related rec domains: 1) specific residue positions can influence the pre-existing conformational equilibrium, tuning the kinetics of phosphorylation, and 2) specific residue positions participate in HK binding through the formation of intermolecular interactions. It is reasonable to assume that perturbations generated by an activating substitution at a key position in a rec domain can mimic perturbations generated by an intermolecular interaction formed between the same residue and a HK.
Conclusions
Although the sequence space accessible to rec domains is likely limited, our findings advance our overall understanding of response regulator evolution and demonstrate an important facet of both rec domain function and enzymatic regulation in general. This work provides further insight into the temporal order of operations involved in rec domain phosphorylation, characterizing the roles that phosphodonor binding and related key residues play in shifting the conformational equilibrium, essentially priming the enzyme for phosphochemistry. This explains not only why phosphodonors such as CheA are far more efficient at rapidly phosphorylating their partner rec domains than small molecules, but also highlights an additional and largely understudied dimension to kinetic tuning of rec domains.
Supplementary Material
Acknowledgments
We thank Leanna R. McDonald, Andrew L. Lee, Gerald P. Guanga, and Peter J. Miller for their input and assistance with the crystallographic structures and behavior of CheY A113P, Katie M. Foster and Sarah A. Barr for their insightful input, and the anonymous reviewers for their constructive comments and thoughts.
Funding Sources
This work was funded by National Institutes of Health grant GM050860 awarded to R.B.B. Financial support for the use of beamline 22-ID at the Advanced Photon Source (Argonne National Laboratory, Argonne, IL) was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357, and the Southeast Regional Collaborative Access Team (SER-CAT).
Abbreviations
- AcP
acetyl phosphate
- ATP
adenosine triphosphate
- CheY A113P
E. coli CheY variant with Ala113 substituted with Pro at position K+4
- COM
center of mass
- CW
clockwise
- D
conserved phosphorylatable aspartate
- DD
conserved metal-binding acidic residues
- FunFam
Functional Family group
- HK
histidine kinase
- HMM
Hidden Markov Model
- K
conserved active site lysine
- K+1/K+2/K+4
position in the primary sequence that is one/two/four residues C-terminal to the active site lysine
- K1/2
amount of phosphodonor required to maintain 50% of the population in a phosphorylated state
- kdephos
autodephosphorylation rate constant
- KL
Kullback-Leibler
- kphos
autophosphorylation rate constant
- LMI
linear mutual information
- PAM
phosphoramidate
- PDB
Protein Data Bank
- RASA
relative accessible surface area
- rec
receiver domain
- RMSD
root mean square deviation
- RMSF
root mean square fluctuation
- SASA
solvent accessible surface area
- T
conserved active site threonine/serine
- TCS
two-component signaling
Footnotes
Supporting Information
The Supporting Information is included in the file Foster et al Supporting Information.docx.
Accession Codes
Coordinates for the X-ray structures used in this study are contained in the Protein Data Bank as entries 3OO1, 3OO0 and 3MYY.
The authors declare no competing financial interest. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health.
References
- 1.Zschiedrich CP, Keidel V, and Szurmant H (2016) Molecular mechanisms of two-component signal transduction, J. Mol. Biola 428, 3752–3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lukat GS, McCleary WR, Stock AM, and Stock JB (1992) Phosphorylation of bacterial response regulator proteins by low molecular weight phospho-donors, Proc. Natl. Acad. Sci. U. S. A 89, 718–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hess JF, Oosawa K, Matsumura P, and Simon MI (1987) Protein phosphorylation is involved in bacterial chemotaxis, Proc. Natl. Acad. Sci. U. S. A 84, 7609–7613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hess JF, Bourret RB, Oosawa K, Matsumura P, and Simon MI (1988) Protein phosphorylation and bacterial chemotaxis, Cold Spring Harbor Symposia on Quantitative Biology 53, 41–48. [DOI] [PubMed] [Google Scholar]
- 5.Gao R, Bouillet S, and Stock AM (2019) Structural basis of response regulator function, Annu. Rev. Microbiol 73, 175–197. [DOI] [PubMed] [Google Scholar]
- 6.Bourret RB (2010) Receiver domain structure and function in response regulator proteins, Curr. Opin. Microbiol 13, 142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sourjik V, and Berg HC (2002) Binding of the Escherichia coli response regulator CheY to its target measured in vivo by fluorescence resonance energy transfer, Proc. Natl. Acad. Sci. U. S. A 99, 12669–12674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alon U, Camarena L, Surette MG, Aguera y Arcas B, Liu Y, Leibler S, and Stock JB (1998) Response regulator output in bacterial chemotaxis, EMBO J. 17, 4238–4248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nutsch T, Marwan W, Oesterhelt D, and Gilles ED (2003) Signal processing and flagellar motor switching during phototaxis of Halobacterium salinarum, Genome Res. 13, 2406–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dawes IW, and Mandelstam J (1970) Sporulation of Bacillus subtilis in continuous culture, J. Bacteriol 103, 529–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zapf J, Madhusudan, Grimshaw CE, Hoch JA, Varughese KI, and Whiteley JM (1998) A source of response regulator autophosphatase activity: the critical role of a residue adjacent to the Spo0F autophosphorylation active site, Biochem. 37, 7725–7732. [DOI] [PubMed] [Google Scholar]
- 12.Page SC, Immormino RM, Miller TH, and Bourret RB (2016) Experimental analysis of functional variation within protein families: receiver domain autodephosphorylation kinetics, J. Bacteriol 198, 2483–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Porter SL, and Armitage JP (2002) Phosphotransfer in Rhodobacter sphaeroides chemotaxis, J. Mol. Biol 324, 35–45. [DOI] [PubMed] [Google Scholar]
- 14.Thomas SA, Brewster JA, and Bourret RB (2008) Two variable active site residues modulate response regulator phosphoryl group stability, Mol. Microbiol 69, 453–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pazy Y, Wollish AC, Thomas SA, Miller PJ, Collins EJ, Bourret RB, and Silversmith RE (2009) Matching biochemical reaction kinetics to the timescales of life: structural determinants that influence the autodephosphorylation rate of response regulator proteins, J. Mol. Biol 392, 1205–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Creager-Allen RL, Silversmith RE, and Bourret RB (2013) A link between dimerization and autophosphorylation of the response regulator PhoB, J. Biol. Chem 288, 21755–21769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schuster M, Silversmith RE, and Bourret RB (2001) Conformational coupling in the chemotaxis response regulator CheY, Proc. Natl. Acad. Sci. U. S. A 98, 6003–6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thomas SA, Immormino RM, Bourret RB, and Silversmith RE (2013) Nonconserved active site residues modulate CheY autophosphorylation kinetics and phosphodonor preference, Biochem. 52, 2262–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Straughn PB, Vass LR, Yuan C, Kennedy EN, Foster CA, and Bourret RB (2020) Modulation of response regulator CheY reaction kinetics by two variable residues that affect conformation, J. Bacteriol 202, e00089–00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Immormino RM, Starbird CA, Silversmith RE, and Bourret RB (2015) Probing mechanistic similarities between response regulator signaling proteins and haloacid dehalogenase phosphatases, Biochem. 54, 3514–3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith JG, Latiolais JA, Guanga GP, Pennington JD, Silversmith RE, and Bourret RB (2004) A search for amino acid substitutions that universally activate response regulators, Mol. Microbiol 51, 887–901. [DOI] [PubMed] [Google Scholar]
- 22.Bourret RB, Hess JF, and Simon MI (1990) Conserved aspartate residues and phosphorylation in signal transduction by the chemotaxis protein CheY, Proc. Natl. Acad. Sci. U. S. A 87, 41–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bourret RB, Drake SK, Chervitz SA, Simon MI, and Falke JJ (1993) Activation of the phosphosignaling protein CheY. II. Analysis of activated mutants by 19F NMR and protein engineering, J. Biol. Chem 268, 13089–13096. [PMC free article] [PubMed] [Google Scholar]
- 24.Smith JG, Latiolais JA, Guanga GP, Citineni S, Silversmith RE, and Bourret RB (2003) Investigation of the role of electrostatic charge in activation of the Escherichia coli response regulator CheY, J. Bacteriol 185, 6385–6391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Silversmith RE, Smith JG, Guanga GP, Les JT, and Bourret RB (2001) Alteration of a nonconserved active site residue in the chemotaxis response regulator CheY affects phosphorylation and interaction with CheZ, J. Biol. Chem 276, 18478–18484. [DOI] [PubMed] [Google Scholar]
- 26.Appleby JL, and Bourret RB (1998) Proposed signal transduction role for conserved CheY residue Thr87, a member of the response regulator active-site quintet, J. Bacteriol 180, 3563–3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ganguli S, Wang H, Matsumura P, and Volz K (1995) Uncoupled phosphorylation and activation in bacterial chemotaxis: the 2.1-Å structure of a threonine to isoleucine mutant at position 87 of CheY, J. Biol. Chem 270, 17386–17393. [PubMed] [Google Scholar]
- 28.Silversmith RE, Guanga GP, Betts L, Chu C, Zhao R, and Bourret RB (2003) CheZ-mediated dephosphorylation of the Escherichia coli chemotaxis response regulator CheY: role for CheY glutamate 89, J. Bacteriol 185, 1495–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Da Re S, Tolstykh T, Wolanin PM, and Stock JB (2002) Genetic analysis of response regulator activation in bacterial chemotaxis suggests an intermolecular mechanism, Protein Sci. 11, 2644–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Immormino RM, Silversmith RE, and Bourret RB (2016) A variable active site residue influences the kinetics of response regulator phosphorylation and dephosphorylation, Biochem. 55, 5595–5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Volkman BF, Lipson D, Wemmer DE, and Kern D (2001) Two-state allosteric behavior in a single-domain signaling protein, Science 291, 2429–2433. [DOI] [PubMed] [Google Scholar]
- 32.Bobay BG, Thompson RJ, Hoch JA, and Cavanagh J (2010) Long range dynamic effects of point-mutations trap a response regulator in an active conformation, FEBS Lett. 584, 4203–4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Foster CA, and West AH (2017) Use of restrained molecular dynamics to predict the conformations of phosphorylated receiver domains in two-component signaling systems, Proteins 85, 155–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McDonald LR, Boyer JA, and Lee AL (2012) Segmental motions, not a two-state concerted switch, underlie allostery in CheY, Structure 20, 1363–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pontiggia F, Pachov DV, Clarkson MW, Villali J, Hagan MF, Pande VS, and Kern D (2015) Free energy landscape of activation in a signalling protein at atomic resolution, Nat. Commun 6, 7284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stock AM, and Guhaniyogi J (2006) A new perspective on response regulator activation, J. Bacteriol 188, 7328–7330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dyer CM, and Dahlquist FW (2006) Switched or not?: the structure of unphosphorylated CheY bound to the N-terminus of FliM, J. Bacteriol 188, 7354–7363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Simonovic M, and Volz K (2001) A distinct meta-active conformation in the 1.1-A resolution structure of wild-type ApoCheY, J. Biol. Chem 276, 28637–28640. [DOI] [PubMed] [Google Scholar]
- 39.Guhaniyogi J, Robinson VL, and Stock AM (2006) Crystal structures of beryllium fluoride-free and beryllium fluoride-bound CheY in complex with the conserved C-terminal peptide of CheZ reveal dual binding modes specific to CheY conformation, J. Mol. Biol 359, 624–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma L, and Cui Q (2007) Activation mechanism of a signaling protein at atomic resolution from advanced computations, J. Am. Chem. Soc 129, 10261–10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Das S, Lee D, Sillitoe I, Dawson NL, Lees JG, and Orengo CA (2015) Functional classification of CATH superfamilies: a domain-based approach for protein function annotation, Bioinformatics 31, 3460–3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eddy SR (2011) Accelerated Profile HMM Searches, PLoS Comput. Bio 7, e1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Das S, Sillitoe I, Lee D, Lees JG, Dawson NL, Ward J, and Orengo CA (2015) CATH FunFHMMer web server: protein functional annotations using functional family assignments, Nucleic Acids Res. 43, W148–W153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fred Hess J, Bourret RB, and Simon MI (1991) Phosphorylation assays for proteins of the two-component regulatory system controlling chemotaxis in Escherichia coli, Methods Enzymol. 200, 188–204. [DOI] [PubMed] [Google Scholar]
- 45.Otwinowski Z, and Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode, Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- 46.Volz K, and Matsumura P (1991) Crystal structure of Escherichia coli CheY refined at 1.7-A resolution, J. Biol. Chem 266, 15511–15519. [DOI] [PubMed] [Google Scholar]
- 47.Lee SY, Cho HS, Pelton JG, Yan D, Berry EA, and Wemmer DE (2001) Crystal structure of activated CheY. Comparison with other activated receiver domains, J. Biol. Chem 276, 16425–16431. [DOI] [PubMed] [Google Scholar]
- 48.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, and Zwart PH (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution, Acta Crystallogr. Sect. D. Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Emsley P, and Cowtan K (2004) Coot: model-building tools for molecular graphics, Acta Crystallogr. Sect. D. Biol. Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- 50.Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010) MolProbity: all-atom structure validation for macromolecular crystallography, Acta Crystallogr. Sect. D. Biol. Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sheridan RC, McCullough JF, Wakefield ZT, Allcock HR, and Walsh EJ (1972) Phosphoramidic acid and its salts, Inorg. Synth, 23–26. [Google Scholar]
- 52.Mayover TL, Halkides CJ, and Stewart RC (1999) Kinetic characterization of CheY phosphorylation reactions: comparison of P-CheA and small-molecule phosphodonors, Biochem. 38, 2259–2271. [DOI] [PubMed] [Google Scholar]
- 53.Goldstein G (1964) Equilibrium distribution of metal-fluoride complexes, Anal. Chem 36, 243–244. [Google Scholar]
- 54.Humphrey W, Dalke A, and Schulten K (1996) VMD: visual molecular dynamics, J. Mol. Graph 14, 33–38, 27–38. [DOI] [PubMed] [Google Scholar]
- 55.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, and Klein ML (1983) Comparison of simple potential functions for simulating liquid water, J. Chem. Phys 79, 926–935. [Google Scholar]
- 56.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, and Schulten K (2005) Scalable molecular dynamics with NAMD, J. Comput. Chem 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brooks BR, Brooks CL 3rd, Mackerell AD Jr., Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, and Karplus M (2009) CHARMM: the biomolecular simulation program, J. Comput. Chem 30, 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.(2020) The PyMOL molecular graphics system, Version 2.3.3, Schrodinger, LLC. [Google Scholar]
- 59.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004) UCSF Chimera--a visualization system for exploratory research and analysis, J. Comput. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- 60.Søndergaard CR, Olsson MHM, Rostkowski M, and Jensen JH (2011) Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values, J. Chem. Theory. Comput 7, 2284–2295. [DOI] [PubMed] [Google Scholar]
- 61.Olsson MHM, Søndergaard CR, Rostkowski M, and Jensen JH (2011) PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions, J. Chem. Theory. Comput 7, 525–537. [DOI] [PubMed] [Google Scholar]
- 62.Jurrus E, Engel D, Star K, Monson K, Brandi J, Felberg LE, Brookes DH, Wilson L, Chen J, Liles K, Chun M, Li P, Gohara DW, Dolinsky T, Konecny R, Koes DR, Nielsen JE, Head-Gordon T, Geng W, Krasny R, Wei GW, Holst MJ, McCammon JA, and Baker NA (2018) Improvements to the APBS biomolecular solvation software suite, Protein Sci. 27, 112–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grant BJ, Rodrigues AP, ElSawy KM, McCammon JA, and Caves LS (2006) Bio3d: an R package for the comparative analysis of protein structures, Bioinformatics 22, 2695–2696. [DOI] [PubMed] [Google Scholar]
- 64.Team, R. C. (2018) R: a language and environment for statistical computing, R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 65.McClendon CL, Hua L, Barreiro G, and Jacobson MP (2012) Comparing conformational ensembles using the Kullback–Leibler divergence expansion, J. Chem. Theory. Comput 8, 2115–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McClendon CL, Friedland G, Mobley DL, Amirkhani H, and Jacobson MP (2009) Quantifying correlations between allosteric sites in thermodynamic ensembles, J. Chem. Theory. Comput 5, 2486–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fribourgh JL, Srivastava A, Sandate CR, Michael AK, Hsu PL, Rakers C, Nguyen LT, Torgrimson MR, Parico GCG, Tripathi S, Zheng N, Lander GC, Hirota T, Tama F, and Partch CL (2020) Dynamics at the serine loop underlie differential affinity of cryptochromes for CLOCK:BMAL1 to control circadian timing, eLife 9, e55275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moffett AS, Bender KW, Huber SC, and Shukla D (2017) Allosteric control of a plant receptor kinase through S-glutathionylation, Biophys. J 113, 2354–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Woods DC, and Wereszczynski J (2020) Elucidating the influence of linker histone variants on chromatosome dynamics and energetics, Nucleic Acids Res. 48, 3591–3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pradhan MR, Siau JW, Kannan S, Nguyen MN, Ouaray Z, Kwoh CK, Lane DP, Ghadessy F, and Verma CS (2019) Simulations of mutant p53 DNA binding domains reveal a novel druggable pocket, Nucleic Acids Res. 47, 1637–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jamieson SA, Ruan Z, Burgess AE, Curry JR, McMillan HD, Brewster JL, Dunbier AK, Axtman AD, Kannan N, and Mace PD (2018) Substrate binding allosterically relieves autoinhibition of the pseudokinase TRIB1, Sci. Signal 11, eaau0597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, and Lindahl E (2015) GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers, SoftwareX 1-2, 19–25. [Google Scholar]
- 73.Scarabelli G, and Grant BJ (2014) Kinesin-5 allosteric inhibitors uncouple the dynamics of nucleotide, microtubule, and neck-linker binding sites, Biophys. J 107, 2204–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lange OF, and Grubmüller H (2006) Generalized correlation for biomolecular dynamics, Proteins 62, 1053–1061. [DOI] [PubMed] [Google Scholar]
- 75.Yao X-Q, Malik RU, Griggs NW, Skjærven L, Traynor JR, Sivaramakrishnan S, and Grant BJ (2016) Dynamic coupling and allosteric networks in the α subunit of heterotrimeric G proteins, J. Biol. Chem 291, 4742–4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Girvan M, and Newman MEJ (2002) Community structure in social and biological networks, Proc. Natl. Acad. Sci. U. S. A 99, 7821–7826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Csardi G, and Nepusz T (2005) The igraph software package for complex network research, InterJournal Complex Systems, 1695. [Google Scholar]
- 78.Galperin MY (2010) Diversity of structure and function of response regulator output domains, Curr. Opin. Microbiol 13, 150–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Casino P, Miguel-Romero L, Huesa J, García P, García-del Portillo F, and Marina A (2017) Conformational dynamism for DNA interaction in the Salmonella RcsB response regulator, Nucleic Acids Res. 46, 456–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Casino P, Rubio V, and Marina A (2009) Structural insight into partner specificity and phosphoryl transfer in two-component signal transduction, Cell 139, 325–336. [DOI] [PubMed] [Google Scholar]
- 81.Lam KH, Ling TKW, and Au SWN (2010) Crystal structure of activated CheY1 from Helicobacter pylori, J. Bacteriol 192, 2324–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cho HS, Lee SY, Yan D, Pan X, Parkinson JS, Kustu S, Wemmer DE, and Pelton JG (2000) NMR structure of activated CheY, J. Mol. Biol 297, 543–551. [DOI] [PubMed] [Google Scholar]
- 83.Zhu X, Rebello J, Matsumura P, and Volz K (1997) Crystal structures of CheY mutants Y106W and T87I/Y106W: CheY activation correlates with movement of residue 106, J. Biol. Chem 272, 5000–5006. [DOI] [PubMed] [Google Scholar]
- 84.Villali J, Pontiggia F, Clarkson MW, Hagan MF, and Kern D (2014) Evidence against the “Y-T coupling” mechanism of activation in the response regulator NtrC, J. Mol. Biol 426, 1554–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.McDonald LR (2013) Towards understanding the mechanisms of allostery: the investigation of the relationship between dynamics, structure and function in the model allosteric protein CheY, In Biochemistry and Biophysics, University of North Carolina at Chapel Hill. [Google Scholar]
- 86.Bowerman S, and Wereszczynski J (2016) Detecting allosteric networks using molecular dynamics simulation, Methods Enzymol. 578, 429–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gasper PM, Fuglestad B, Komives EA, Markwick PRL, and McCammon JA (2012) Allosteric networks in thrombin distinguish procoagulant vs. anticoagulant activities, Proc. Natl. Acad. Sci. U. S. A 109, 21216–21222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kormos BL, Baranger AM, and Beveridge DL (2006) Do collective atomic fluctuations account for cooperative effects? Molecular dynamics studies of the U1A–RNA complex, J. Am. Chem. Soc 128, 8992–8993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ribeiro AAST, and Ortiz V (2014) Determination of signaling pathways in proteins through network theory: importance of the topology, J. Chem. Theory. Comput 10, 1762–1769. [DOI] [PubMed] [Google Scholar]
- 90.Doshi U, Holliday MJ, Eisenmesser EZ, and Hamelberg D (2016) Dynamical network of residue-residue contacts reveals coupled allosteric effects in recognition, catalysis, and mutation, Proc. Natl. Acad. Sci. U. S. A 113, 4735–4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.del Sol A, Fujihashi H, Amoros D, and Nussinov R (2006) Residue centrality, functionally important residues, and active site shape: analysis of enzyme and non-enzyme families, Protein Sci. 15, 2120–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Amitai G, Shemesh A, Sitbon E, Shklar M, Netanely D, Venger I, and Pietrokovski S (2004) Network analysis of protein structures identifies functional residues, J. Mol. Biol 344, 1135–1146. [DOI] [PubMed] [Google Scholar]
- 93.Mildvan AS (2004) Inverse thinking about double mutants of enzymes, Biochem. 43, 14517–14520. [DOI] [PubMed] [Google Scholar]
- 94.Boyer JA, Clay CJ, Luce KS, Edgell MH, and Lee AL (2010) Detection of native-state nonadditivity in double mutant cycles via hydrogen exchange, J. Am. Chem. Soc 132, 8010–8019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bellsolell L. s., Prieto J, Serrano L, and Coll M (1994) Magnesium binding to the bacterial chemotaxis protein CheY results in large conformational changes involving its functional surface, J. Mol. Biol 238, 489–495. [DOI] [PubMed] [Google Scholar]
- 96.Minato Y, Ueda T, Machiyama A, Iwaï H, and Shimada I (2017) Dynamic domain arrangement of CheA-CheY complex regulates bacterial thermotaxis, as revealed by NMR, Sci. Rep 7, 16462–16462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.McEvoy MM, Bren A, Eisenbach M, and Dahlquist FW (1999) Identification of the binding interfaces on CheY for two of its targets the phosphatase CheZ and the flagellar switch protein FliM, J. Mol. Biol 289, 1423–1433. [DOI] [PubMed] [Google Scholar]
- 98.McEvoy MM, Hausrath AC, Randolph GB, Remington SJ, and Dahlquist FW (1998) Two binding modes reveal flexibility in kinase/response regulator interactions in the bacterial chemotaxis pathway, Proc. Natl. Acad. Sci. U. S. A 95, 7333–7338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Stewart RC, Jahreis K, and Parkinson JS (2000) Rapid phosphotransfer to CheY from a CheA protein lacking the CheY-binding domain, Biochem. 39, 13157–13165. [DOI] [PubMed] [Google Scholar]
- 100.Swanson RV, Lowry DF, Matsumura P, McEvoy MM, Simon MI, and Dahlquist FW (1995) Localized perturbations in CheY structure monitored by NMR identify a CheA binding interface, Nat. Struct. Biol 2, 906–910. [DOI] [PubMed] [Google Scholar]
- 101.Li J, Swanson RV, Simon MI, and Weis RM (1995) Response regulators CheB and CheY exhibit competitive binding to the kinase CheA, Biochem. 34, 14626–14636. [DOI] [PubMed] [Google Scholar]
- 102.Trajtenberg F, Albanesi D, Ruétalo N, Botti H, Mechaly AE, Nieves M, Aguilar PS, Cybulski L, Larrieux N, de Mendoza D, and Buschiazzo A (2014) Allosteric activation of bacterial response regulators: the role of the cognate histidine kinase beyond phosphorylation, mBio 5, e02105–02114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Szurmant H, and Weigt M (2018) Inter-residue, inter-protein and inter-family coevolution: bridging the scales, Curr. Opin. Struct. Biol 50, 26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wolfe AJ (2010) Physiologically relevant small phosphodonors link metabolism to signal transduction, Curr. Opin. Microbiol 13, 204–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Klein AH, Shulla A, Reimann SA, Keating DH, and Wolfe AJ (2007) The intracellular concentration of acetyl phosphate in Escherichia coli is sufficient for direct phosphorylation of two-component response regulators, J. Bacteriol 189, 5574–5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Perez JC, and Groisman EA (2007) Acid pH activation of the PmrA/PmrB two-component regulatory system of Salmonella enterica, Mol. Microbiol 63, 283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chamnongpol S, and Groisman EA (2000) Acetyl phosphate-dependent activation of a mutant PhoP response regulator that functions independently of its cognate sensor kinase, J. Mol. Biol 300, 291–305. [DOI] [PubMed] [Google Scholar]
- 108.Patel K, and Golemi-Kotra D (2016) Signaling mechanism by the Staphylococcus aureus two-component system LytSR: role of acetyl phosphate in bypassing the cell membrane electrical potential sensor LytS, F1000Res. 4, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cairns LS, Martyn JE, Bromley K, and Stanley-Wall NR (2015) An alternate route to phosphorylating DegU of Bacillus subtilis using acetyl phosphate, BMC Microbiol. 15, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.