

To the Editor:
Frontotemporal lobar degeneration (FTLD) is a heterogeneous group of pathologies that present clinically with progressive changes in behavior, personality, and executive function with variable amounts of motor impairment (1, 2). The majority of FTLD is accounted for by TDP-43 or tau-positive inclusions (3); however, 5%–10% of FTLD cases have pathological cytoplasmic accumulation of fused in sarcoma (FUS), an RNA-binding protein that has regulatory roles in the nucleus (4). FTLD-FUS, also now known as FTLD-FET, is characterized by pathologic inclusions of the FET proteins, including FUS, Ewing sarcoma, and TATA-binding protein-associated factor 2N (TAF15) (5). Within FTLD-FET, the majority are classified as atypical FTLD with ubiquitin-positive inclusions (aFTLD-U). Patients with aFTLD-U pathology generally have a characteristic clinical presentation, with onset in the third to fifth decade, severe behavioral variant frontotemporal dementia with aggression, and lack of significant language or motor deficits (6–9). Here, we describe a case of aFTLD-U with features of corticobasal syndrome (CBS), prominent language abnormalities, and a later age of onset. Pathologically, this case showed more subtle pathology than classic aFTLD-U cases, with limited cortical involvement.
A 56-year-old man presented for evaluation of progressive language and behavioral problems. Over the past year and a half, his family had noticed a gradual decline in speech production, with the use of short phrases or single word answers to communicate. Language comprehension and knowledge of items were intact. He exhibited uncharacteristic impulsivity, risk taking, and socially inappropriate behavior without aggression. He developed a sweet-tooth and needed prompting to bathe and perform other activities aside from watching television. His initial neurological exam was notable for nonfluent aphasia with intact comprehension, telegraphic speech, echolalia, and perseveration. He had masked facies with decreased blink rate, rigidity of his right upper extremity, and a parkinsonian shuffling gait. There were no upper or lower motor neuron signs. At that time, he was clinically diagnosed with frontotemporal dementia. There were no family members with frontotemporal dementia, amyotrophic lateral sclerosis, or Alzheimer disease, but his father had been diagnosed with Parkinson’s disease in his sixties. MRI of the brain showed mild global atrophy with asymmetric left frontal and temporal lobe atrophy (Fig. 1A). FDG-PET scan showed symmetric cortical FDG uptake but markedly decreased uptake in the bilateral caudate and putamen, consistent with a parkinsonian disorder (Fig. 1B). Neuropsychological evaluation showed a Mattis Dementia Rating Scale-2 score of 111/144 (T < 20) and impairments in attention (Wechsler Adult Intelligence Scale-Fourth Edition symbol search: T = 23), executive functioning (Wisconsin Card Sorting Test: ≤1st percentile), visuospatial functioning (Benton Visual Form Discrimination Test: (24/32)), language (Boston Naming Test: T = 44, Letter fluency: T = 21, Category fluency: T < 20), and memory (California Verbal Learning Test-II overall learning score: T = 36). There was severe apathy (Frontal Systems Behavior Scale: T = 91) without anxiety or depression (Beck Depression and Anxiety Inventories: 0 and 1, respectively).
FIGURE 1.
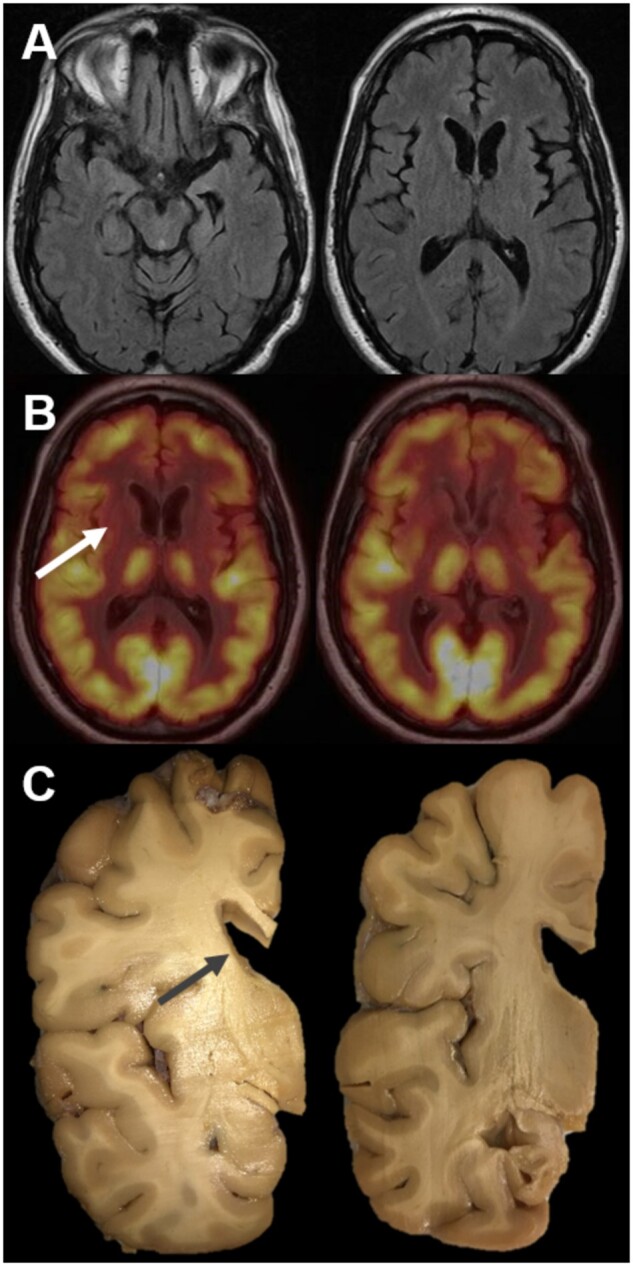
Neuroimaging and gross findings. Magnetic resonance imaging (A). Representative axial T2 Fluid Attenuated Inversion Recovery images showing mild global atrophy and also mild left temporal and left frontal segmental atrophy in this 56-year-old man with a 4-year disease history. Fluorodeoxyglucose positron emission tomography (FDG-PET) (B). Representative images showing normal symmetric cortical FDG avidity but with markedly decreased FDG avidity in the caudate and putamen bilaterally with only minimal avidity retained in the left posterior putamen. Gross examination (C). Coronal sections revealed dilation of the lateral ventricles with severe neostriatal atrophy, most prominent in the caudate (arrow).
The patient was seen 3 months later. During this time his language dysfunction worsened and he was falling regularly. His right hand was apractic and had become contracted with dystonic posturing. His postural reflexes were impaired. His working diagnosis was revised to corticobasal syndrome. The patient was admitted to an assisted living facility later that year and died approximately 2 years after presentation. At the time of his death at age 58, he was anarthric and wheelchair-bound due to balance impairments.
At autopsy, the whole fixed brain weighed 1088 g. Gross examination of the left hemi-brain revealed mild to moderate atrophy of the frontal and temporal cortex. Sectioning revealed severe atrophy of the caudate and putamen, and a severely atrophic hippocampus (Fig. 1C). Microscopically, sections showed mild thinning of the frontal and temporal cortex, but minimal neuronal drop out and astrogliosis. Sections of hippocampus showed severe hippocampal sclerosis, most prominent in CA1 (Fig. 2A, B). The basal ganglia showed severe neuron loss and reactive gliosis (Fig. 2E).
FIGURE 2.
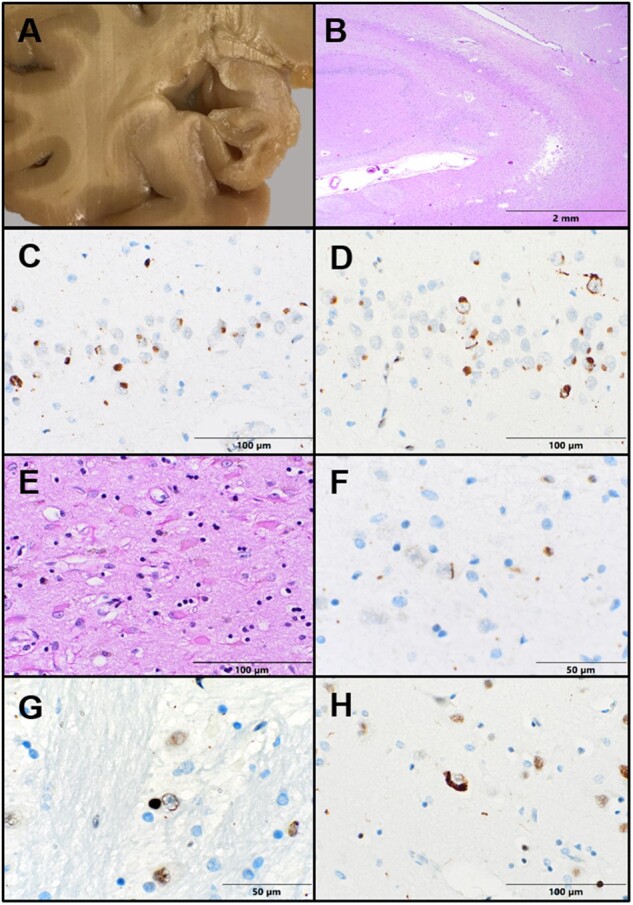
FET pathology. (A) Grossly, the hippocampus appears severely atrophic. (B) Low-power view shows severe hippocampal sclerosis with pallor and rarefaction in CA1. (C) Cytoplasmic rounded inclusions are identified in dentate gyrus granular neurons with FUS immunohistochemistry (IHC). (D) TAF15 IHC highlights cytoplasmic and intranuclear inclusions in dentate granular neurons. (E) Sections of the basal ganglia show severe neuron loss and gliosis. (F) Rare FUS-positive curvi-linear intranuclear inclusions are seen in the putamen. (G) TAF15 IHC highlights both cytoplasmic and curvilinear intranuclear inclusions in basis pontis nuclei. (H) Rare cortical neurons contain TAF15-immunoreactive cytoplasmic inclusions.
Immunohistochemistry (IHC) for tau revealed only rare tangles in the entorhinal cortex. β-amyloid, α-synuclein, and TDP-43 IHC stains were negative. Ubiquitin staining identified frequent rounded neuronal cytoplasmic inclusions (NCI) within the dentate granule cells of the hippocampus. The ubiquitin-positive inclusions in the dentate gyrus were also FUS-positive (Fig. 2C) and TAF15-positive (Fig. 2D). Many dentate granule neurons showed FUS and TAF15-positive curvilinear filamentous neuronal intranuclear inclusions (NII) (Fig. 2D). Rare FUS- and TAF15-positive NII were also seen in CA4 and the putamen (Fig. 2F). Sparse NCI and NII were identified within the pons, superior temporal gyrus, and frontal cortex with TAF15 stain but were absent in the substantia nigra and locus coeruleus (Fig. 2G, H). No neurofilament-positive inclusions were identified in the frontal cortex, hippocampus, or basal ganglia, ruling out neuronal intermediate filament inclusion disease. The morphology and distribution of NCIs and NIIs were consistent with aFLTD-U.
Clinically, this case exhibited some unusual features for aFTLD-U, including a later age of onset (10), nonfluent agrammatic aphasia, and motor features meeting diagnostic criteria for possible CBS and clinical research criteria for probable corticobasal degeneration (11). In addition, while irritability was noted, there was no history of significant aggression typical of aFTLD-U (8, 12). Although rare cases of aFTLD-U with an older age of onset (>55 years old) and language dysfunction have been reported, this is the first case of aFTLD-U clinically presenting as possible CBS with a nonfluent agrammatic aphasia (13, 14).
The pathology in this case also appears less diffuse than aFTLD-U cases described to date. Although characteristic FUS-immunoreactive NCIs and NIIs were present in the hippocampus and basal ganglia, there was notably minor cortical involvement. Minimal frontal and temporal atrophy was present microscopically, and only infrequent TAF15-positive inclusions were identified within the neocortex. This is similar to the 2 older-onset cases recently reported, which also had more subtle pathology (13) consisting of only mild to moderate cortical atrophy with numerous NCI and NII in hippocampal dentate neurons, but infrequent inclusions in the cortex. Our case further expands on this more subtle pathologic spectrum of aFTLD-U in older patients, and demonstrates the utility of TAF15 staining to highlight infrequent cortical pathology.
In summary, we report a case of aFTLD-U presenting with behavioral abnormalities, a nonfluent agrammatic aphasia, and clinical features of CBS. This case further broadens the clinical phenotype associated with aFTLD-U pathology. In addition, this case highlights the need for close examination for TAF15/FUS-positive inclusions within the hippocampus and basal ganglia, even in the absence of more severe cortical atrophy typical of most FTLDs. Finally, this unusual case expands the pathologies that can present clinically with features of CBS.
Vanessa Goodwill and David Coughlin contributed equally as co-first authors and Shauna H. Yuan and Annie Hiniker contributed equally as co-senior authors.
FUNDING
This report was supported by National Institutes of Health (NIH) P30-AG062429, Alzheimer’s Association Clinician Scientist Fellowship Grant (A.H.), NIH 1R03AG070415-01 (S.H.Y.), and ITN startup funds (S.H.Y).
COMPETING INTERESTS
The authors declare that they have no competing interests.
REFERENCES
- 1.Josephs KA, Hodges JR, Snowden JS, et al. Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol 2011;122:137–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Woollacott IOC, Rohrer JD.. The clinical spectrum of sporadic and familial forms of frontotemporal dementia. J Neurochem 2016;138:6–31 [DOI] [PubMed] [Google Scholar]
- 3.MacKenzie IRA, Neumann M, Bigio EH, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: An update. Acta Neuropathol 2010;119:1–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tan AY, Manley JL.. The TET family of proteins: Functions and roles in disease. J Mol Cell Biol 2009;1:82–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neumann M, Bentmann E, Dormann D, et al. FET proteins TAF15 and EWS are selective markers that distinguish FTLD with FUS pathology from amyotrophic lateral sclerosis with FUS mutations. Brain 2011;134:2595–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neumann M, Mackenzie IRA.. Review: Neuropathology of non-tau frontotemporal lobar degeneration. Neuropathol Appl Neurobiol 2019;45:19–40 [DOI] [PubMed] [Google Scholar]
- 7.Urwin H, Josephs KA, Rohrer JD, FReJA Consortium, et al. FUS pathology defines the majority of tau-and TDP-43-negative frontotemporal lobar degeneration. Acta Neuropathol 2010;120:33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mackenzie IRA, Foti D, Woulfe J, et al. Atypical frontotemporal lobar degeneration with ubiquitin-positive, TDP-43-negative neuronal inclusions. Brain 2008;131:1282–93 [DOI] [PubMed] [Google Scholar]
- 9.Snowden JS, Hu Q, Rollinson S, et al. The most common type of FTLD-FUS (aFTLD-U) is associated with a distinct clinical form of frontotemporal dementia but is not related to mutations in the FUS gene. Acta Neuropathol 2011;122:99–110 [DOI] [PubMed] [Google Scholar]
- 10.MacKenzie IRA, Munoz DG, Kusaka H, et al. Distinct pathological subtypes of FTLD-FUS. Acta Neuropathol 2011;121:207–18 [DOI] [PubMed] [Google Scholar]
- 11.Armstrong MJ, Litvan I, Lang AE, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology 2013;80:496–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roeber S, Mackenzie IRA, Kretzschmar HA, et al. TDP-43-negative FTLD-U is a significant new clinico-pathological subtype of FTLD. Acta Neuropathol 2008;116:147–57 [DOI] [PubMed] [Google Scholar]
- 13.Chornenka K, Hirsch-Reinshagen V, Perez-Rosendahl M, et al. Expanding the phenotype of frontotemporal lobar degeneration with FUS-positive pathology (FTLD-FUS). J Neuropathol Exp Neurol 2020;79:809–12 [DOI] [PubMed] [Google Scholar]
- 14.Bradfield NI, McLean C, Drago J, et al. Rapidly progressive Fronto-temporal dementia (FTD) associated with Frontotemporal lobar degeneration (FTLD) in the presence of Fused in Sarcoma (FUS) protein: A rare, sporadic, and aggressive form of FTD. Int Psychogeriatr 2017;29:1743–6 [DOI] [PubMed] [Google Scholar]