

Abstract
To determine whether arterial responsiveness is impaired among patients with gout, and whether arterial responsiveness inversely correlates with serum urate and inflammatory measures. This is a cross-sectional study of untreated gout subjects (n = 34) and non-gout healthy controls (n = 64). High-resolution dynamic ultrasound-measured flow-mediated dilation (FMD) and nitroglycerin-mediated dilation (NMD) assessed endothelium-dependent and endothelium-independent arterial responsiveness respectively. Serum urate (sUA) and high-sensitivity C-reactive protein (hsCRP) were measured in the gout group, and correlated with FMD and NMD responses. Both FMD (2.20 ± 0.53 vs 3.56 ± 0.31, p = 0.021) and NMD (16.69 ± 1.54 vs 24.51 ± 0.90, p = 0.00002) were impaired in the gout versus control group. Stratification for individual comorbidities suggested that no single risk factor accounted for impaired FMD/NMD in the gout subjects. However, the degree of association between gout and FMD, but not NMD impairment, was dampened after multivariable adjustment (FMD unadjusted beta = − 1.36 (SE 0.58), p = 0.02; adjusted beta = − 1.16 (SE 0.78), p = 0.14 and NMD unadjusted beta = − 7.68 (SE 1.78), p < 0.0001; adjusted beta = − 5.33 (SE 2.46), p = 0.03). Within the gout group, there was an inverse correlation between FMD and sUA (R = − 0.5, p = 0.003), and between FMD and hsCRP (R = − 0.42, p = 0.017), but not between NMD and sUA or hsCRP. Compared with healthy controls, subjects with gout have reduced arterial function. Individual comorbidities are insufficient to account for differences between gout and control groups, but multiple comorbidities may collectively contribute to impairment in endothelium-dependent arterial responsiveness. Endothelial impairment is also related to sUA and hsCRP, markers of gout severity and inflammation respectively. Studies to determine whether gout therapy may improve arterial responsiveness are warranted.
Keywords: Arterial function, Endothelium, Flow-mediated dilation, Gout, Hyperuricemia, Inflammation
Introduction
Gout is the most common inflammatory arthritis [1] and is associated with multiple comorbidities, including hypertension, chronic kidney disease (CKD), type 2 diabetes mellitus (DM), and hyperlipidemia (HL) [2, 3]. Gout is also an independent risk factor for coronary artery disease (CAD), and both gout, and its biochemical precursor hyperuricemia, are independently associated with increased CAD-associated morbidity and mortality [4–6]. Despite mounting epidemiologic evidence of the association between gout and CAD, only limited evidence is available regarding potential contributory mechanisms through which gout may adversely affect the vasculature.
Arterial function is essential to vascular health, and endothelial dysfunction is a common correlate of arterial disease. Endothelial cells regulate leukocyte and platelet adhesion and generate vasoactive substances, including nitric oxide (NO) that act on smooth muscle to promote arterial vasodilation [7]. Arterial function can be measured in the peripheral circulation through physiologic exposure of the endothelium to laminar flow, which promotes release of vasoactive substances and subsequent smooth muscle responses [7]. Flow-mediated dilation (FMD) of the brachial artery [8] is a noninvasive, accurate, reproducible [9], and commonly used technique to measure endothelial-dependent arterial responses. Subsequent administration of sublingual nitroglycerin bypasses the endothelium and permits direct assessment of endothelial-independent smooth muscle function (nitrate-mediated dilation, NMD). Endothelial dysfunction identified by FMD has been associated with an increased 5-year incidence of atherosclerosis and cardiovascular events [10].
The potential effects of gout on vascular health are multiple. In vitro, endothelial cells exposed to urate experience suppressed NO release [11], whereas smooth muscle cells respond to urate with increased migration and proliferation, possibly promoting a state of “muscle-bound” vasculature [12]. Consistent with the in vitro effects of urate, patients with hyperuricemia plus various comorbidities (e.g., chronic kidney disease [13, 14], chronic heart failure [15], and ischemic stroke [16]), as well as patients with asymptomatic hyperuricemia alone [17, 18], display worse arterial function as measured by FMD. In addition to possible direct effects of hyperuricemia, increased CAD risk in gout patients could derive from acute and chronic inflammation [19]. Atherosclerosis is an inflammatory process [20]; elevated C-reactive protein (CRP) and other inflammatory markers are associated with increased adverse events across the spectrum of CAD, and CRP reduction is independently associated with reduced coronary events [21]. Moreover, multiple rheumatic diseases characterized by systemic inflammation are associated with increased rates of CAD independent of traditional risk factors [22, 23]. CRP is elevated both acutely and chronically in gout [19, 24–26], but the impact of gouty inflammation on CAD is not well-defined.
In contrast to hyperuricemia per se, studies into the relationship between gout and cardiovascular disease have largely focused on CAD clinical outcomes, with only limited evidence available directly relevant to arterial function. To address the paucity of available data on arterial function in gout, we measured FMD and NMD in untreated gout subjects, and compared the results to a group of non-gout controls. We additionally examined the relationship between serum urate (sUA), high-sensitivity CRP (hsCRP), and arterial function in the subjects with gout.
Materials and methods
Subjects and controls
The Institutional Review Boards of the New York Harbor Health Care System of the Department of Veterans Affairs and New York University (NYU) School of Medicine approved the current study. Written informed consent was obtained from all subjects. Male subjects at least 18 years of age, who fulfilled the 2015 American College of Rheumatology classification criteria for gout [27]), and met indications for urate-lowering therapy according to the 2012 American College of Rheumatology Guidelines for Management of Gout [28], but had not received any urate-lowering therapy (allopurinol, febuxostat, or probenecid) or colchicine for at least 30 days, were enrolled in the gout group. Patients were excluded if they were actively taking glucocorticoids. Previous non-steroidal anti-inflammatory drug (NSAID) use was permitted in the prior 30 days on an as-needed but not chronic basis, and not at the time of assessment. Patients were additionally excluded if they had stage 4 or 5 CKD. Subjects were recruited through their rheumatologists or primary care physicians at the New York Campus of the New York Harbor Health Care System of the United States Department of Veterans Affairs, or through rheumatologists at the NYU Center for Musculoskeletal Care and the NYU Hospital for Joint Diseases Arthritis Clinic.
To serve as a healthy control population, male subjects without gout were included from a control cohort participating in a prospective study of the impact of iron stores on vascular endothelial function (manuscript submitted). These individuals were voluntary blood donors who had been deferred from active donation for at least 2 years due to extended residence in Europe, or for other administrative reasons. They were otherwise in generally good health, including absence of cancer, hepatitis, HIV/AIDS, organ failure, and recreational drug use by injection. Additional exclusion criteria for the healthy control group included diabetes mellitus (DM) (defined as history of DM, fasting glucose > 100 mg/dL, or taking DM medications), current tobacco use, and surface electrocardiographic evidence consistent with CAD.
Covariates of interest
Data on patient demographics, medical history, and current medication use were obtained by direct interview and from chart review at the time of enrollment. All data were entered and maintained in a secure web-based REDCap database [29].
Assessment of vascular endothelial function and vascular smooth muscle cell responsiveness
FMD was assessed as brachial artery responsiveness after transient distal arterial occlusion, as previously described [30, 31]. Briefly, subjects and controls underwent FMD using a high-resolution duplex ultrasound imaging system (SonoSite, Inc., Bothell, WA, USA) connected to an 11-MHz linear array transducer with an axial resolution of < 0.1 mm and a computer-assisted edge detection system (AMS software) [32]. All procedures were performed by a single trained technician (ID) who was aware of the study the subject was enrolled in but otherwise blinded to the subject’s clinical condition. After allowing the subject to rest quietly for several minutes, the brachial artery diameter (trailing edge of the anterior intima-lumen interface to leading edge of the posterior lumen-intima interface) was measured at end-diastole before and after transient arterial occlusion (forearm blood pressure cuff inflation to 50 mmHg above systolic blood pressure for 5 min) [33]. FMD was determined as the percent increase in brachial diameter from baseline to 1 min after occlusion release. Subsequently, subjects were administered a single 0.4-mg dose of sublingual nitroglycerin. NMD was determined as the percent increase in brachial artery diameter from baseline (pre-blood pressure cuff inflation) to 5 min after nitroglycerin administration.
All vascular studies were performed in a quiet, temperature-controlled environment. With a small number of exceptions for patient safety or ability, studies performed in a fasting state, and subjects were requested to hold any morning dose of beta-blockers or calcium channel blockers until the completion of the procedure.
Laboratory studies
Blood was collected immediately after the FMD/NMD studies in the gout subjects, and sUA and hsCRP levels were assessed using the hospital clinical laboratory.
Statistical methods
Continuous variables were summarized using mean and standard deviation. Categorical variables were presented as proportions. Two-sample t test was used in the comparison of the mean difference between groups for continuous variables. Chi square/Fisher’s Exact test was used in the comparison of proportions between groups for categorical variables. Multivariable linear regression analysis modeled the outcome FMD and NMD over case (yes for gout and no for non-gout control) with adjustments to estimate whether gout was associated with lower values of outcome (FMD/NMD) after controlling the risk factors including age, BMI race, hyperlipidemia (HL), and hypertension (HTN). These associations were expressed as percent difference (β) and standard error (SE). Correlations between FMD/NMD and sUA and hsCRP levels among gout subjects were assessed using the Pearson correlation coefficient.
Results
Subject demographics
Ninety-eight subjects were included in the study: 34 had a diagnosis of gout and 64 were non-gout controls. Table 1 summarizes the baseline characteristics. Gout subjects had a non-significant trend for older age, and (consistent with prior studies) a significantly higher mean BMI than the control subjects. Compared to the controls, a lower proportion of the gout subjects were white. As per enrollment criteria, some gout subjects, but no controls, had CAD or DM or were current smokers. Gout subjects also had a higher prevalence of CKD, hypertension, and hyperlipidemia. Seventy-four percent of the gout subjects (n = 25) had never used urate-lowering therapy (not shown). Among the nine subjects who had used urate-lowering therapy, the mean time since prior use was 4.3 years (range, 3 months to 20 years). Twelve patients reported using NSAIDs on an as-needed basis (ibuprofen, 6 patients; naproxen, 3 patients; indomethacin, 2 patients; etodolac and meloxicam, 1 patient each); none were actively taking NSAIDs at the time of study evaluation.
Table 1.
Patient demographic data and comorbidities
| Characteristic | Gout subjects (n = 34) | Non-gout controls (n = 64) | p value |
|---|---|---|---|
| Age in years (mean ± SE) | 57.91 ± 2.32 | 53.23 ± 0.95 | 0.07 |
| BMI (mean ± SE) | 30.45 ± 0.1 | 28.51 ± 0.68 | 0.04 |
| Male (%) | 100.00 | 100.00 | |
| Race | < 0.0001 | ||
| White (%) | 44.12 | 92.19 | |
| African American (%) | 44.12 | 7.81 | |
| Other (%) | 11.76 | 0.00 | |
| Coronary artery disease (%) | 20.59 | 0.00 | 0.0004 |
| Chronic kidney disease (%) | 76.47 | 0.00 | < 0.0001 |
| Diabetes mellitus (%) | 5.88 | 0.00 | 0.12 |
| Hyperlipidemia (%) | 50.00 | 20.34 | 0.003 |
| Hypertension (%) | 70.59 | 23.73 | < 0.0001 |
| Smoking, current (%) | 29.41 | 0.00 | < 0.0001 |
Endothelial and smooth muscle arterial response in gout subjects versus controls
Both FMD and NMD were significantly impaired in the gout subjects compared to those in the non-gout controls (Fig. 1). To examine the impact of comorbid conditions on arterial impairment, we compared gout and control subgroups lacking specific comorbid risk factors. Despite a reduced number of subjects available for these sub-analyses, FMD and NMD remained numerically lower in the gout subjects versus controls. Because African American race and smoking have been reported to be associated with diminished arterial function [34, 35], we also compared FMD and NMD among whites only, and among non-smokers only, between the gout and control populations. In both of these sub-analyses, we observed persistent near-significant (FMD) and significant (NMD) differences between the gout and control groups (Table 2).
Fig. 1.
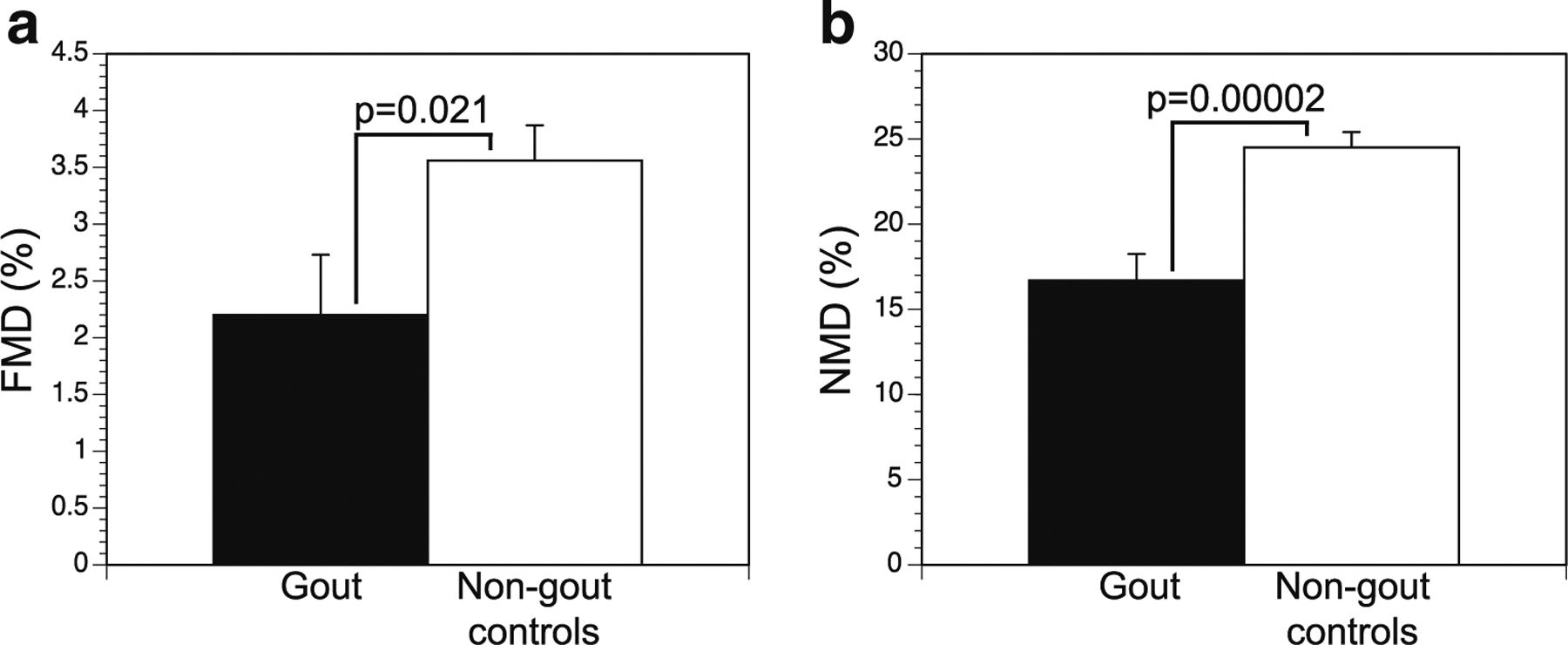
Comparison of flow-mediated dilation (FMD) (a) and nitrate-mediated dilation (NMD) (b) among gout subjects compared with non-gout controls. Data shown are mean ± SE for each condition
Table 2.
Comparison of flow-mediated dilation (FMD) and nitrate-mediated dilation (NMD) in gout subjects versus non-gout controls: stratification by demographic and comorbid risk factors
| FMD | NMD | |||||
|---|---|---|---|---|---|---|
| Stratification | Gout | Non-gout controls | p value | Gout | Non-gout controls | p value |
| None (all subjects) (34,64) | 2.20 ± 0.53 | 3.56 ± 0.31 | 0.02 | 16.69 ± 1.54 | 24.51 ± 0.90 | < 0.0001 |
| Whites only (15,59) | 2.26 ± 0.81 | 3.67 ± 0.33 | 0.09 | 19.48 ± 2.45 | 24.57 ± 0.96 | 0.04 |
| Non-smokers only (24,64) | 2.33 ± 0.87 | 3.56 ± 0.31 | 0.06 | 15.99 ± 1.62 | 24.51 ± 0.90 | < 0.0001 |
| No CAD only (27,64) | 2.07 ± 0.51 | 3.56 ± 0.31 | 0.01 | 15.56 ± 1.89 | 24.51 ± 0.90 | < 0.001 |
| No CKD only (8,64) | 2.28 ± 1.15 | 3.56 ± 0.31 | 0.20 | 18.31 ± 2.76 | 24.51 ± 0.90 | < 0.0001 |
| No HTN only (10,45) | 1.72 ± 0.90 | 3.79 ± 0.36 | 0.03 | 20.98 ± 3.41 | 24.95 ± 0.89 | 0.14 |
| No HL only (17,44) | 1.20 ± 0.63 | 3.70 ± 0.37 | 0.001 | 17.41 ± 2.28 | 24.60 ± 1.07 | 0.003 |
Numbers in parentheses after each stratification category correspond to numbers of patients in the gout and control groups, respectively
CAD coronary artery disease, CKD chronic kidney disease, HTN hypertension, HL hyperlipidemia
To explore these observations further, we modeled FMD and NMD over case (yes for gout and no for non-gout control) after controlling for multiple risk factors including age, BMI, race, hyperlipidemia, and hypertension. In these analyses, FMD values were again significantly lower for gout subjects compared with non-gout controls on univariate assessment, but did not achieve significance after multivariable adjustment. In contrast, the presence of gout associated with a lower value of NMD both in univariate analysis, and after multivariable adjustment (Table 3).
Table 3.
Regression analyses for impact of potential risk factors on flow-mediated dilation (FMD) and nitrate-mediated dilation (NMD) among gout subjects versus non-gout controls
| Outcome | Analysis | Beta (SE) for case variable | p value |
|---|---|---|---|
| FMD | Univariate | − 1.36 (0.58) | 0.02 |
| Multivariate | − 1.16 (0.78) | 0.14 | |
| NMD | Univariate | − 7.68 (1.78) | < 0.0001 |
| Multivariate | − 5.33 (2.46) | 0.03 |
Adjusted for age, BMI, race, hyperlipidemia, and hypertension
FMD and NMD in gout subjects according to comorbidities
To further understand the potential role of comorbidities on decreased arterial function in gout subjects, FMD and NMD were compared among gout subjects with versus without specific risk factors for decreased arterial responsiveness (Table 4). We observed no significant differences in FMD or NMD in gout subjects with versus without various individual cardiovascular risk factors, suggesting that the presence of any of these comorbidities individually may be insufficient to account for the diminished arterial function observed in the gout group overall. In contrast, non-gout controls with hypertension had impaired FMD compared to non-gout controls without hypertension, confirming a role for hypertension independent of gout (FMD in control subjects with vs without hypertension, 2.72 ± 0.5 vs 3.81 ± 0.4%, p = 0.05). Similarly, gout patients with hypertension had significantly impaired FMD when compared with the non-hypertensive healthy controls (2.39 ± 0.64 vs 3.81 ± 0.04%, p = 0.04). In contrast, gout patients with hypertension had non-significant impairment of FMD when compared with hypertensive healthy controls (2.39 ± 0.64 vs 2.72 ± 0.52%, p = 0.3), again underlining that both traditional risk factors and gout per se appear to contribute to endothelial dysfunction.
Table 4.
Comparison of flow-mediated dilation (FMD) and nitrate-mediated dilation (NMD) in gout subjects with versus without specific demographic and comorbid risk factors
| FMD | NMD | |||||
|---|---|---|---|---|---|---|
| Condition | Condition absent | Condition present | p value | Condition absent | Condition present | p value |
| African American (15,15) | 2.36 ± 0.81 | 1.32 ± 0.19 | 0.35 | 19.48 ± 2.45 | 12.22 ± 0.63 | 0.04 |
| Smoking (24,10) | 2.33 ± 0.87 | 1.86 ± 1.02 | 0.70 | 15.99 ± 1.62 | 18.78 ± 4.6 | 0.49 |
| CAD (27,7) | 2.07 ± 0.51 | 2.70 ± 1.59 | 0.64 | 15.56 ± 1.89 | 21.89 ± 2.31 | 0.16 |
| CKD (8,26) | 2.28 ± 1.15 | 2.17 ± 0.59 | 0.93 | 18.31 ± 2.76 | 16.04 ± 2.05 | 0.57 |
| HTN (10,24) | 1.72 ± 0.9 | 2.40 ± 0.64 | 0.57 | 20.98 ± 3.41 | 15.00 ± 0.11 | 0.11 |
| HL (17,17) | 1.20 ± 0.63 | 3.19 ± 0.77 | 0.06 | 17.41 ± 2.28 | 15.00 ± 0.64 | 0.66 |
Numbers in parentheses after each stratification category correspond to numbers of patients in the gout and control groups, respectively
CAD coronary artery disease, CKD chronic kidney disease, HTN hypertension, HL hyperlipidemia
Although FMD was numerically reduced in both the African American and current smoker gout cohorts, these differences did not achieve significance. In contrast, NMD was significantly reduced among African Americans, but not among current smokers compared with non-smokers with gout. When compared with non-smoking healthy controls, smoking gout patients did show a lower mean FMD, but the difference did not achieve statistical significance, likely because the number of smokers was small (smoking gout patients vs non-smoking controls, FMD = 2.57 ± 1.4 vs 3.56 ± 0.31%, p = 0.25). Because smokers were excluded from the control group, we were not able to compare gout smokers with a smoking control group.
FMD and NMD in gout subjects: correlation with markers of gout disease severity
Hyperuricemia is a prerequisite for the development of gout, and patients with higher levels of hyperuricemia are more prone to incident gout as well as more frequent and more severe gout attacks [36]. Similarly, recent reports suggest that gout patients experience chronic as well as acute inflammation, which may be more strongly associated with duration and/or severity of disease [19]. We therefore assessed for correlations between measures of arterial responsiveness, and levels of sUA and hsCRP, within the gout group. We observed significant inverse correlations between sUA and FMD (Fig. 2a), and between hsCRP and FMD (Fig. 2b), but no significant correlations between sUA or hsCRP and NMD (Fig. 2c, d). These data suggest that gout severity, as indicated by untreated sUA and hsCRP levels, may be associated with decreased endothelial function. In contrast, smooth muscle function (NMD) appears to be diminished in gout patients in a manner independent of disease severity according to these markers.
Fig. 2.
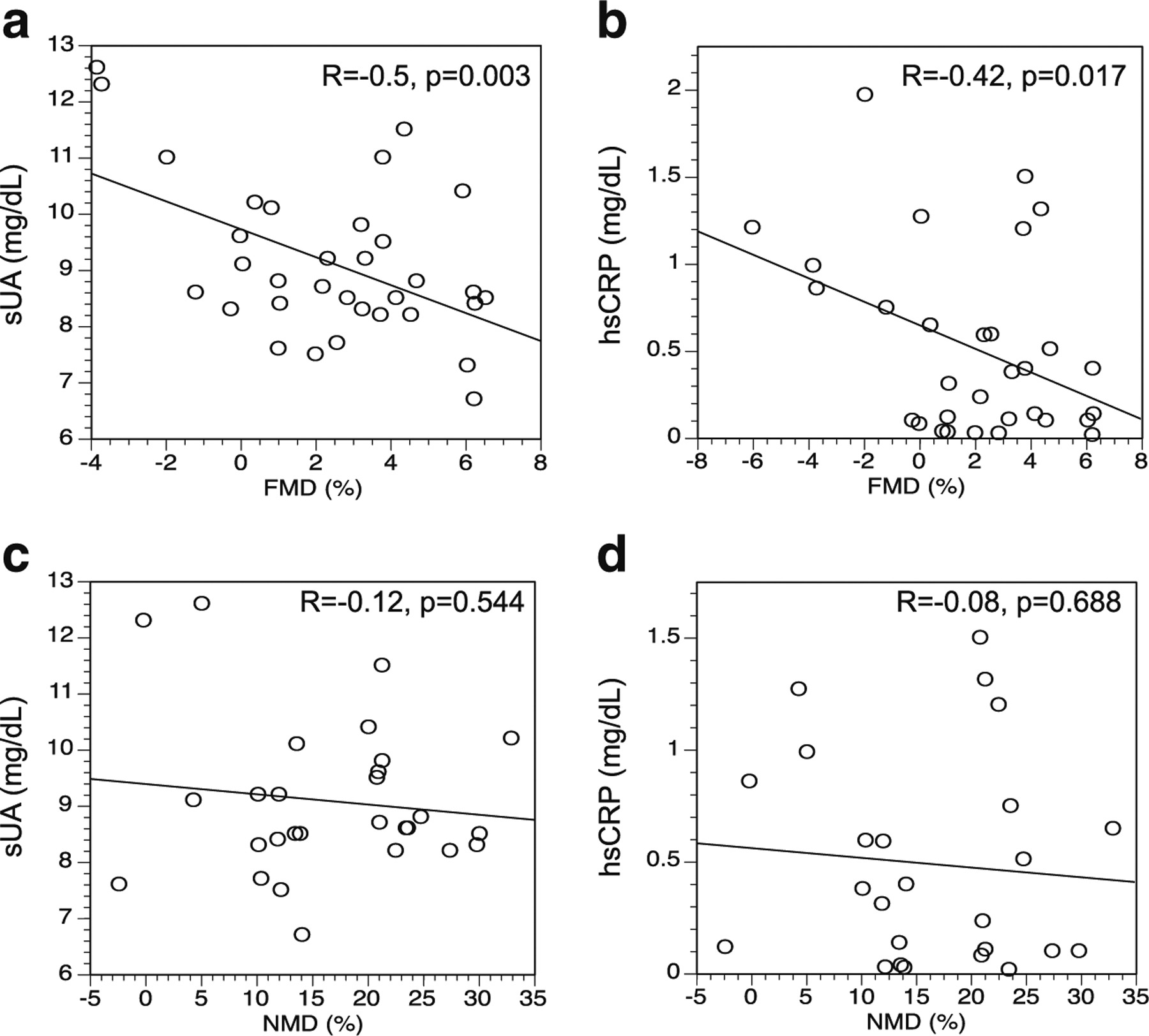
Relationships between measured arterial function and serum concentrations of urate (sUA) and high-sensitivity C-reactive protein (hsCRP). a Flow-mediated dilation (FMD) and sUA. b FMD and hsCRP. c Nitrate-mediated dilation (NMD) and sUA. d NMD and hsCRP
The presence of tophi and radiographic erosions are additional markers of gout disease severity [28]. In this study, gout subjects with both tophi and erosions had markedly reduced FMD responses compared to the non-tophaceous gout population (0.83 ± 1.78 vs 1.98 ± 0.70). However, the number of gout subjects with both tophi and erosions was small (n = 3), and the difference did not achieve statistical significance (p = 0.58). Subjects with tophi or erosions alone did not demonstrate reduced FMD responses (not shown).
Discussion
This study is the first to report that, compared with healthy controls, gout patients experience reduced arterial endothelial and smooth muscle function. While an independent association between gout and CVD has been established in multiple studies [2–4], the potential mechanisms for this association are incompletely understood. Our observation adds to the current literature and suggests an impact of gout on CVD via direct or indirect effects on arterial function.
Urate is the major antioxidant in human plasma and on that basis was previously considered to have possible beneficial cardiovascular effects [37]. However, multiple recent studies have reported associations between elevated sUA levels and adverse cardiovascular outcomes [4, 5, 38–40]. Consistent with these observations, we observed an inverse correlation between sUA and FMD in gout subjects. Our data thus resemble those of Mercuro et al., who reported that non-gout, high-CAD risk subjects with hyperuricemia (mean sUA 9.3 ± 1.8) had considerably impaired FMD compared to normouricemic, otherwise well-matched control subjects [41]. Our data may also be consistent with studies suggesting that elevated sUA concentrations may reduce arterial responsiveness specifically via the inhibition of endothelial function [11]. How urate may mediate endothelial function remains uncertain, but some evidence suggests that endothelial cells have transporters that may take up urate to adversely alter cellular responses, and that increased levels of urate additionally downregulate the expression of endothelial cell surface efflux transporters to synergistically promote intracellular urate elevations [42]. Alternatively, it is possible that elevated urate levels are a surrogate for xanthine oxidase activity, and therefore for the generation of oxidants which could affect vascular responsiveness [43].
It is increasingly appreciated that gout is characterized not only by excessive inflammation during acute flares, but also by chronic inflammation during intercritical periods [5–7]. The well-established observation that almost any form of chronic systemic inflammation (evidenced in part through CRP elevation) is a risk factor for arterial dysfunction and atherosclerosis [19, 20] provides a conceptual framework in which patients with gout might experience endothelial dysfunction as a consequence of their chronic inflammatory states. In support of such a model, we observed, among gout subjects, a significant inverse correlation between hsCRP and FMD, in which higher hsCRP levels were associated with poorer arterial function. Interestingly, when endothelial cells are exposed to urate in vitro, CRP production increases [44], suggesting a possible entanglement between the urate and inflammatory effects. In this context, it is interesting to note a recent report by Rongen et al., exploring the impact of cessation of anti-TNF therapy on vasodilator response in patients with rheumatoid arthritis (RA). In that study, RA subjects in remission experienced diminished vasodilator responsiveness to acetylcholine compared to patients continuing treatment, but only if they also experienced recurrent flare of their RA, suggesting that the impact of some rheumatic diseases on arterial responsiveness is mediated through the exposure to inflammation [45]. Whether the adverse effects of inflammation are driven by the presence of cytokines, or perhaps by the urate-driven intracellular activation of the NALP3 inflammasome [46], remains a matter of speculation.
In the current study, subjects with gout were also more likely to have deficits in endothelial-independent smooth muscle responsiveness, as measured by NMD. Vascular smooth muscle dysfunction is uncommon; studies of patients with rheumatoid arthritis and lupus patients, for example, report impaired FMD but normal NMD [47, 48]. Impaired NMD may therefore distinguish gout from other chronic inflammatory states, and may result in additional resistance to already-impaired endothelial signaling. Decreased NMD responses in gout would appear to be consistent with reports that in vitro exposure of smooth muscle cells to solubilized urate promotes cellular migration, hypertrophy, and inflammatory responses [12]. However, in contrast to the effects we observed on FMD, NMD failed to correlate with either sUA or CRP levels. Thus, NMD impairment in gout may be a consequence of multiple factors, some of which remain to be determined.
The observation that our unselected gout subjects carried many more comorbidities than a healthy non-gout control group is consistent with prior reports [2, 3], and suggests the possibility that impaired endothelial and smooth muscle responsiveness among gout subjects could be due to comorbid conditions associated with impaired vascular function. However, upon stratification of the subjects to distinguish individuals with or without specific cardiovascular risk factors or comorbidities, we generally observed persistent FMD and NMD impairment in the gout compared with the non-gout control group. Moreover, within the gout subject group itself, FMD and NMD measurements were generally similar between subjects with versus without the individual comorbidities. These data suggest that no single comorbidity was sufficient to account for the observed impairment, but do not exclude the possibility that a combination of comorbidities may have contributed to impaired endothelial function. Consistent with such an impact, the observed decreases in FMD, but not NMD, lost statistical significance upon multivariable analysis. However, the current data do not exclude the alternative possibility that the presence of multiple comorbidities may serve as an indicator of gout severity, which according to other markers (sUA and CRP) in our study was associated with decreased endothelial function. Regardless of the causal association, these observations identify gout patients as likely to have impaired endothelial and smooth muscle function. While we confirmed the previous observation that African American subjects have impaired FMD and NMD compared with white subjects [34], a stratification of only white individuals suggested that race alone was insufficient to account for the impact of gout on vascular function.
Only one prior study has examined the interaction between gout and peripheral arterial function. In contrast to our results, Brook et al. reported no impaired vascular function (including measurements of FMD and NMD) among 20 gout subjects versus 20 non-gout controls [49]. However, that study excluded gout patients with diabetes, smoking history, renal disease, or statin use—essentially eliminating many cardiovascular risk factors and ensuring that gout subjects and controls were similar in most respects other than gout itself. Thus, generalizability to typical gout patients, who on average carry approximately four comorbidities each [3], was diminished. Additionally, most gout subjects in the Brook study had been receiving urate-lowering and/or anti-inflammatory therapies, in many cases for years, and underwent treatment washout only for brief periods before assessment of sUA, hsCRP, and vascular function. Thus, in contrast to our study of currently untreated gout subjects, the Brook study compared vascular function between controls and chronically treated, non-comorbid gout patients, whose vascular disease may have been modified by prior therapy. Additional studies will be needed to clarify the discrepancies between the study by Brook et al. and our current report.
Strengths of our study include an unbiased inclusion of “typical” gout patients (who tend to have multiple comorbidities [2, 3]), the inclusion only of subjects not receiving gout therapies, and the inclusion of a racially diverse population. Our use of both ACR gout classification and treatment guidelines ensured enrollment of gout patients with a meaningful disease burden. Limitations include the use of a single tool (FMD/NMD) in the assessment of arterial function. However, this tool is the current gold standard modality for peripheral vascular assessment. Another limitation is the relatively limited number of subjects enrolled, which may have affected our ability to observe statistical significance for some sub-analyses. In part, because the majority of gout subjects were enrolled from the Veterans Affairs system, no females were enrolled. While our observations therefore require validation in females, the majority of gout sufferers in the USA are male, by a ratio of approximately 3:1 [1].
Conclusion
In conclusion, our data show for the first time that patients with gout have impaired endothelial-dependent arterial function, as well as impaired endothelial-independent smooth muscle responses, compared to healthy controls. Both elevated sUA levels, and greater systemic inflammation (elevated hsCRP), were associated with poorer endothelial-dependent function. While neither vascular endothelial nor smooth muscle dysfunction could be fully explained by any individual gout-associated comorbidity, we cannot exclude the possibility that the accrual of multiple comorbidities among gout patients can collectively impact the vascular response. Whether appropriate anti-inflammatory and/or urate-lowering therapy can improve FMD and/or NMD in these subjects remains to be determined.
Acknowledgements
The authors thank Drs. Jonathan Samuels, Cesar Fors, Stephen Bernstein, Adey Berhanu, and Sabina Sandigursky for referring their patients for the study, and Dr. Bruce Cronstein for helpful input.
Funding information
This work was supported by a New York State Empire Clinical Research Investigator Program (ECRIP) award, to S.D.K. and S.K. S.K. was also supported in part by an Investigator Award from the Rheumatology Research Foundation. B.S. was supported in part by the Biomedical Laboratory Research & Development Service of the VA Office of Research and Development (I01BX007080). M.H.P. and S.D.K. received support from a CTSA award (1UL1TR001445) to New York University from the National Center for the Advancement of Translational Science, National Institutes of Health.
Conflict of interest statement
None of the authors have any conflict of interest regarding this manuscript. For the purposes of full transparency, we acknowledge the following disclosures. S.K. has served as a consultant for Crealta, Horizon, and Ironwood. B.S. serves on the Philips Volcano Medical Education Steering Committee and has a research grant from Siemens Medical Solutions. M.H.P. serves and/or has served as a consultant for AstraZeneca, Crealta, Horizon, Ironwood, and SOBI, and has been an investigative site for a sponsored trial by Takeda.
Footnotes
Approval and consent statement The studies described in this manuscript were approved by the Institutional Review Boards of New York University School of Medicine and the New York Harbor Health Care System, United States Department of Veterans Affairs. All participating subjects gave their informed consent in writing prior to participation in the study.
References
- 1.Zhu Y, Pandya BJ, Choi HK (2011) Prevalence of gout and hyperuricemia in the US general population: the National Health and Nutrition Examination Survey 2007–2008. Arthritis Rheum 63(10):3136–3141 [DOI] [PubMed] [Google Scholar]
- 2.Zhu Y, Pandya BJ, Choi HK (2012) Comorbidities of gout and hyperuricemia in the US general population: NHANES 2007–2008. Am J Med 125(7):679–687 e671 [DOI] [PubMed] [Google Scholar]
- 3.Keenan RT, O’Brien WR, Lee KH, Crittenden DB, Fisher MC, Goldfarb DS, Krasnokutsky S, Oh C, Pillinger MH (2011) Prevalence of contraindications and prescription of pharmacologic therapies for gout. Am J Med 124(2):155–163 [DOI] [PubMed] [Google Scholar]
- 4.Choi HK, Curhan G (2007) Independent impact of gout on mortality and risk for coronary heart disease. Circulation 116(8):894–900 [DOI] [PubMed] [Google Scholar]
- 5.Ndrepepa G, Braun S, King L, Hadamitzky M, Haase HU, Birkmeier KA, Schomig A, Kastrati A (2012) Association of uric acid with mortality in patients with stable coronary artery disease. Metabolism 61(12):1780–1786 [DOI] [PubMed] [Google Scholar]
- 6.Krishnan E, Baker JF, Furst DE, Schumacher HR (2006) Gout and the risk of acute myocardial infarction. Arthritis Rheum 54(8): 2688–2696 [DOI] [PubMed] [Google Scholar]
- 7.Monica FZ, Bian K, Murad F (2016) The endothelium-dependent nitric oxide-cGMP pathway. Adv Pharmacol 77:1–27 [DOI] [PubMed] [Google Scholar]
- 8.Celermajer DS, Sorensen KE, Gooch VM, Spiegelhalter DJ, Miller OI, Sullivan ID, Lloyd JK, Deanfield JE (1992) Non-invasive detection of endothelial dysfunction in children and adults at risk of atherosclerosis. Lancet 340(8828):1111–1115 [DOI] [PubMed] [Google Scholar]
- 9.Sorensen KE, Celermajer DS, Spiegelhalter DJ, Georgakopoulos D, Robinson J, Thomas O, Deanfield JE (1995) Non-invasive measurement of human endothelium dependent arterial responses: accuracy and reproducibility. Br Heart J 74(3):247–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yeboah J, Folsom AR, Burke GL, Johnson C, Polak JF, Post W, Lima JA, Crouse JR, Herrington DM (2009) Predictive value of brachial flow-mediated dilation for incident cardiovascular events in a population-based study: the multi-ethnic study of atherosclerosis. Circulation 120(6):502–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi YJ, Yoon Y, Lee KY, Hien TT, Kang KW, Kim KC, Lee J, Lee MY, Lee SM, Kang DH, Lee BH (2014) Uric acid induces endothelial dysfunction by vascular insulin resistance associated with the impairment of nitric oxide synthesis. FASEB J 28(7):3197–3204 [DOI] [PubMed] [Google Scholar]
- 12.Kang DH, Han L, Ouyang X, Kahn AM, Kanellis J, Li P, Feng L, Nakagawa T, Watanabe S, Hosoyamada M, Endou H, Lipkowitz M, Abramson R, Mu W, Johnson RJ (2005) Uric acid causes vascular smooth muscle cell proliferation by entering cells via a functional urate transporter. Am J Nephrol 25(5):425–433 [DOI] [PubMed] [Google Scholar]
- 13.Kanbay M, Yilmaz MI, Sonmez A, Turgut F, Saglam M, Cakir E, Yenicesu M, Covic A, Jalal D, Johnson RJ (2011) Serum uric acid level and endothelial dysfunction in patients with nondiabetic chronic kidney disease. Am J Nephrol 33(4):298–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yelken B, Caliskan Y, Gorgulu N, Altun I, Yilmaz A, Yazici H, Oflaz H, Yildiz A (2012) Reduction of uric acid levels with allopurinol treatment improves endothelial function in patients with chronic kidney disease. Clin Nephrol 77(4):275–282 [DOI] [PubMed] [Google Scholar]
- 15.Doehner W, Schoene N, Rauchhaus M, Leyva-Leon F, Pavitt DV, Reaveley DA, Schuler G, Coats AJ, Anker SD, Hambrecht R (2002) Effects of xanthine oxidase inhibition with allopurinol on endothelial function and peripheral blood flow in hyperuricemic patients with chronic heart failure: results from 2 placebo-controlled studies. Circulation 105(22):2619–2624 [DOI] [PubMed] [Google Scholar]
- 16.Muir SW, Harrow C, Dawson J, Lees KR, Weir CJ, Sattar N, Walters MR (2008) Allopurinol use yields potentially beneficial effects on inflammatory indices in those with recent ischemic stroke: a randomized, double-blind, placebo-controlled trial. Stroke 39(12):3303–3307 [DOI] [PubMed] [Google Scholar]
- 17.Kanbay M, Huddam B, Azak A, Solak Y, Kadioglu GK, Kirbas I, Duranay M, Covic A, Johnson RJ (2011) A randomized study of allopurinol on endothelial function and estimated glomular filtration rate in asymptomatic hyperuricemic subjects with normal renal function. Clin J Am Soc Nephrol 6(8):1887–1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Melendez-Ramirez G, Perez-Mendez O, Lopez-Osorio C, Kuri-Alfaro J, Espinola-Zavaleta N (2012) Effect of the treatment with allopurinol on the endothelial function in patients with hyperuricemia. Endocr Res 37(1):1–6 [DOI] [PubMed] [Google Scholar]
- 19.Perez-Ruiz F, Becker MA (2015) Inflammation: a possible mechanism for a causative role of hyperuricemia/gout in cardiovascular disease. Curr Med Res Opin 31(Suppl 2):9–14 [DOI] [PubMed] [Google Scholar]
- 20.Hansson GK (2005) Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med 352(16):1685–1695 [DOI] [PubMed] [Google Scholar]
- 21.Abd TT, Eapen DJ, Bajpai A, Goyal A, Dollar A, Sperling L (2011) The role of C-reactive protein as a risk predictor of coronary atherosclerosis: implications from the JUPITER trial. Curr Atheroscler Rep 13(2):154–161 [DOI] [PubMed] [Google Scholar]
- 22.Schieir O, Tosevski C, Glazier RH, Hogg-Johnson S, Badley EM (2017) Incident myocardial infarction associated with major types of arthritis in the general population: a systematic review and meta-analysis. Ann Rheum Dis 76:1396–1404 [DOI] [PubMed] [Google Scholar]
- 23.Avina-Zubieta JA, To F, Vostretsova K, De Vera M, Sayre EC, Esdaile JM: Risk of myocardial infarction and stroke in newly diagnosed systemic lupus erythematosus: a general population-based study. Arthritis Care Res (Hoboken) 2017 [DOI] [PubMed] [Google Scholar]
- 24.Yeh ET, Anderson HV, Pasceri V, Willerson JT (2001) C-reactive protein: linking inflammation to cardiovascular complications. Circulation 104(9):974–975 [DOI] [PubMed] [Google Scholar]
- 25.Yeh ET (2004) CRP as a mediator of disease. Circulation 109(21 Suppl 1):II11–II14 [DOI] [PubMed] [Google Scholar]
- 26.Singh SK, Suresh MV, Voleti B, Agrawal A (2008) The connection between C-reactive protein and atherosclerosis. Ann Med 40(2): 110–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neogi T, Jansen TL, Dalbeth N, Fransen J, Schumacher HR, Berendsen D, Brown M, Choi H, Edwards NL, Janssens HJ et al. (2015) 2015 gout classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheumatol 67(10):2557–2568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khanna D, Fitzgerald JD, Khanna PP, Bae S, Singh MK, Neogi T, Pillinger MH, Merill J, Lee S, Prakash S, Kaldas M, Gogia M, Perez-Ruiz F, Taylor W, Lioté F, Choi H, Singh JA, Dalbeth N, Kaplan S, Niyyar V, Jones D, Yarows SA, Roessler B, Kerr G, King C, Levy G, Furst DE, Edwards NL, Mandell B, Schumacher HR, Robbins M, Wenger N, Terkeltaub R, American College of Rheumatology (2012) 2012 American College of Rheumatology guidelines for management of gout. Part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res (Hoboken) 64(10):1431–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG (2009) Research electronic data capture (REDCap)—a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform 42(2):377–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zheng H, Cable R, Spencer B, Votto N, Katz SD (2005) Iron stores and vascular function in voluntary blood donors. Arterioscler Thromb Vasc Biol 25(8):1577–1583 [DOI] [PubMed] [Google Scholar]
- 31.Zheng H, Patel M, Cable R, Young L, Katz SD (2007) Insulin sensitivity, vascular function, and iron stores in voluntary blood donors. Diabetes Care 30(10):2685–2689 [DOI] [PubMed] [Google Scholar]
- 32.Jelani QU, Norcliffe-Kaufmann L, Kaufmann H, Katz SD (2015) Vascular endothelial function and blood pressure regulation in afferent autonomic failure. Am J Hypertens 28(2):166–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Corretti MC, Anderson TJ, Benjamin EJ, Celermajer D, Charbonneau F, Creager MA, Deanfield J, Drexler H, Gerhard-Herman M, Herrington D, Vallance P, Vita J, Vogel R, International Brachial Artery Reactivity Task Force (2002) Guidelines for the ultrasound assessment of endothelial-dependent flow-mediated vasodilation of the brachial artery: a report of the International Brachial Artery Reactivity Task Force. J Am Coll Cardiol 39(2):257–265 [DOI] [PubMed] [Google Scholar]
- 34.Campia U, Choucair WK, Bryant MB, Waclawiw MA, Cardillo C, Panza JA (2002) Reduced endothelium-dependent and - independent dilation of conductance arteries in African Americans. J Am Coll Cardiol 40(4):754–760 [DOI] [PubMed] [Google Scholar]
- 35.Esen AM, Barutcu I, Acar M, Degirmenci B, Kaya D, Turkmen M, Melek M, Onrat E, Esen OB, Kirma C (2004) Effect of smoking on endothelial function and wall thickness of brachial artery. Circ J 68(12):1123–1126 [DOI] [PubMed] [Google Scholar]
- 36.Campion EW, Glynn RJ, DeLabry LO (1987) Asymptomatic hyperuricemia. Risks and consequences in the Normative Aging Study. Am J Med 82(3):421–426 [DOI] [PubMed] [Google Scholar]
- 37.Waring WS, McKnight JA, Webb DJ, Maxwell SR (2006) Uric acid restores endothelial function in patients with type 1 diabetes and regular smokers. Diabetes 55(11):3127–3132 [DOI] [PubMed] [Google Scholar]
- 38.Puddu P, Puddu GM, Cravero E, Vizioli L, Muscari A (2012) Relationships among hyperuricemia, endothelial dysfunction and cardiovascular disease: molecular mechanisms and clinical implications. J Cardiol 59(3):235–242 [DOI] [PubMed] [Google Scholar]
- 39.Kelkar A, Kuo A, Frishman WH (2011) Allopurinol as a cardiovascular drug. Cardiol Rev 19(6):265–271 [DOI] [PubMed] [Google Scholar]
- 40.Gagliardi AC, Miname MH, Santos RD (2009) Uric acid: a marker of increased cardiovascular risk. Atherosclerosis 202(1):11–17 [DOI] [PubMed] [Google Scholar]
- 41.Mercuro G, Vitale C, Cerquetani E, Zoncu S, Deidda M, Fini M, Rosano GM (2004) Effect of hyperuricemia upon endothelial function in patients at increased cardiovascular risk. Am J Cardiol 94(7): 932–935 [DOI] [PubMed] [Google Scholar]
- 42.Komori H, Yamada K, Tamai I (2018) Hyperuricemia enhances intracellular urate accumulation via down-regulation of cell-surface BCRP/ABCG2 expression in vascular endothelial cells. Biochim Biophys Acta 1860:973–980 [DOI] [PubMed] [Google Scholar]
- 43.Mazzali M, Kanbay M, Segal MS, Shafiu M, Jalal D, Feig DI, Johnson RJ (2010) Uric acid and hypertension: cause or effect? Curr Rheumatol Rep 12(2):108–117 [DOI] [PubMed] [Google Scholar]
- 44.Kang DH, Park SK, Lee IK, Johnson RJ (2005) Uric acid-induced C-reactive protein expression: implication on cell proliferation and nitric oxide production of human vascular cells. J Am Soc Nephrol 16(12):3553–3562 [DOI] [PubMed] [Google Scholar]
- 45.Rongen GAvI I; Kok M; Vonkeman H; Janssen M; Jansen TL: Vasodilator function worsens after cessation of tumour necrosis factor inhibitor therapy in patients with rheumatoid arthritis only if a flare occurs. Clin Rheumatol 2018, Epub ahead of print [DOI] [PubMed] [Google Scholar]
- 46.Igel TF, Krasnokutsky S, Pillinger MH (2017) Recent advances in understanding and managing gout. F1000Res 6:247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Di Minno MN, Ambrosino P, Lupoli R, Di Minno A, Tasso M, Peluso R, Tremoli E (2015) Clinical assessment of endothelial function in patients with rheumatoid arthritis: a meta-analysis of literature studies. Eur J Intern Med 26(10):835–842 [DOI] [PubMed] [Google Scholar]
- 48.Mikolajczyk TP, Osmenda G, Batko B, Wilk G, Krezelok M, Skiba D, Sliwa T, Pryjma JR, Guzik TJ (2016) Heterogeneity of peripheral blood monocytes, endothelial dysfunction and subclinical atherosclerosis in patients with systemic lupus erythematosus. Lupus 25(1):18–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brook RD, Yalavarthi S, Myles JD, Khalatbari S, Hench R, Lustig S, Marder W, Neidert A, Kaplan MJ (2011) Determinants of vascular function in patients with chronic gout. J Clin Hypertens (Greenwich) 13(3):178–188 [DOI] [PMC free article] [PubMed] [Google Scholar]