

Abstract
Background
Activating mutations in the Fms-like tyrosine kinase 3 (FLT3) are among the most prevalent oncogenic mutations in acute myeloid leukaemia. Inhibitors selectively targeting FLT3 kinase have shown promising clinical activity; their success in the clinic, however, has been limited due to the emergence of acquired resistance.
Methods
CCT245718 was identified and characterised as a dual Aurora A/FLT3 inhibitor through cell-based and biochemical assays. The ability of CCT245718 to overcome TKD-mediated resistance was evaluated in a cell line-based model of drug resistance to FLT3 inhibitors.
Results
CCT245718 exhibits potent antiproliferative activity towards FLT3-ITD + AML cell lines and strongly binds to FLT3-ITD and TKD (D835Y) mutants in vitro. Activities of both FLT3-ITD and Aurora A are also inhibited in cells. Inhibition of FLT3 results in reduced phosphorylation of STAT5, downregulation of survivin and induction of apoptotic cell death. Moreover, CCT245718 overcomes TKD-mediated resistance in a MOLM-13-derived cell line containing FLT3 with both ITD and D835Y mutations. It also inhibits FLT3 signalling in both parental and resistant cell lines compared to FLT3-specific inhibitor MLN518, which is only active in the parental cell line.
Conclusions
Our results demonstrate that CCT245718 is a potent dual FLT3/Aurora A inhibitor that can overcome TKD-mediated acquired resistance.
Subject terms: Mechanism of action, Small molecules, Drug discovery and development, Kinases
Introduction
Acute myeloid leukaemia (AML), the most common acute leukaemia in adults, is a clinically heterogeneous disease with a relatively poor prognosis. It is characterised by chromosomal abnormalities and mutations in genes regulating hematopoietic proliferation and differentiation, such as FLT3, KIT and RAS [1]. Fms-like tyrosine kinase 3 (FLT3) is a receptor tyrosine kinase that regulates the proliferation and differentiation of haematopoietic cells [2]. Constitutive activation of the FLT3 kinase is associated with one-third of the AML cases and confers poor prognosis [3]. The two most common types of activating mutations in FLT3 are internal tandem duplications (ITDs) in the juxtamembrane domain and point mutations in the tyrosine kinase domain (TKD). Activated FLT3, in turn, stimulates the downstream signalling pathways (JAK/STAT, ERK/MAPK and PI3K/AKT) that enhance survival and proliferation of immature myeloid blasts [4]. Therefore, FLT3 has been pursued as a potential therapeutic target for AML, and various FLT3 inhibitors have been discovered and evaluated in the clinic. These include relatively non-specific, multi-kinase, first-generation inhibitors [sunitinib, midostaurin, tandutinib (1; Fig. 1) and lestaurtinib] and the more potent and selective second-generation inhibitors [quizartinib (2), crenolanib and gilteritinib] [5]. Two of these inhibitors, midostaurin and gilteritinib, have consequently been approved for the treatment of FLT3-ITD-positive AML patients [6, 7]. However, the clinical success of these inhibitors as single agents has remained limited due to incomplete and transient responses in the case of the first-generation inhibitors and the acquisition of drug resistance for the potent, second-generation inhibitors [5, 8]. Nonetheless, the development of resistant mutations in the FLT3 tyrosine kinase domain in FLT3-ITD-positive patients exposed to quizartinib confirms FLT3-ITD as the driver mutation and affirms its critical importance as a target in AML [9]. Approaches to overcome resistance due to such mutations include the discovery of inhibitors with activity against FLT3 TKD mutants and combinations with inhibitors targeting additional proteins required for proliferation and survival of AML cells [5, 9]. Discovery of FLT3 inhibitors that target both FLT3 and an additional kinase such as CDK4 (AMG 925) [10], MERTK (MRX-2843) [11], MEK (E6201) [12] and Aurora kinases [CCT137690 (3)] [13] has also been used as a promising new approach to tackle resistance due to FLT3 mutations. Dual inhibition of FLT3 and microtubule polymerisation can also overcome TKD-mediated resistance to FLT3 inhibitors [14].
Fig. 1. Small molecule inhibitors of FLT3 and Aurora kinases.

Structure of some of the FLT3 (Tandutinib, Quizartinib), Aurora (Alisertib and Barasertib) and dual FLT3/Aurora inhibitors (CCT137690 and CCT245718).
Aurora kinases are a family of Ser/Thr kinases (Aurora A, B and C) that play critical roles in mitotic entry, progression and exit [15]. While the expression of Aurora C is restricted to the testis, Aurora A and B are ubiquitously expressed, distinctly localised and play non-overlapping roles during different phases of mitosis. Aurora A regulates centrosome maturation and spindle assembly, while Aurora B is a part of the chromosomal passenger complex (CPC) that is involved in chromosomal condensation, chromosomal attachment, spindle assembly checkpoint and cytokinesis [16]. Owing to their critical role in mitosis and overexpression in many types of cancers [17], several inhibitors of Aurora kinases have been discovered and evaluated in the clinic over the past decade [18]. The clinical responses to these inhibitors, however, have been limited for solid tumours, probably due to their mechanism of action that requires long exposure to drugs [19]. Haematological malignancies, on the other hand, show promising clinical responses to Aurora inhibitors such as Alisertib [MLN8237 (4), an Aurora A-selective inhibitor] [20] and Barasertib [AZD1152 (5), an Aurora B-selective inhibitor] [21, 22]. This difference in efficacy has been attributed to the higher proliferation rate in AML and inhibition of secondary targets by some of these inhibitors [19]. In addition to inhibiting Aurora B, AZD1152 can also inhibit FLT3 as a secondary target and is more potent in FLT3-ITD-positive cell lines and patient samples as compared to those with WT FLT3 [23]. Similarly, dual FLT3/pan-Aurora inhibitors have been described to inhibit Aurora kinases and clinically relevant mutants of FLT3 in preclinical studies with efficacies in FLT3-ITD-TKD human xenograft models [13, 24]. We have previously reported structure-based design and synthesis of two isoform-selective inhibitors of Aurora kinase A [25]. Here, we characterise one of these Aurora A-selective inhibitors, CCT245718 (6; compound 40f with Aurora A and B IC50 values of 0.015 and 3.045 µM, respectively [25]) as a dual FLT3/Aurora A inhibitor that can overcome resistance to FLT3 inhibitors. We show that CCT245718 also targets FLT3-ITD and FLT3 D835Y and kills FLT3-ITD AML cells that have acquired resistance due to a secondary D835Y mutation.
Materials and methods
Cell culture
Cell lines MOLM-13, MOLM-13-Res, MV-4-11, THP1 and IMR-32 were cultured in Roswell Park Memorial Institute (RPMI) medium whereas Kelly, HCT116 and SHEP were cultured in Dulbecco’s Modified Eagle Medium (DMEM). Both the culture media were supplemented with 10% foetal bovine serum (FBS) and 1× antibiotic–antimycotic. Cells in their respective culture media were grown at 37 °C in humidified incubators with 5% CO2. All the cell lines used were passaged for less than 6 months, after which cells were replaced from earlier frozen stocks.
Proliferation assays
For adherent cell lines, 1500–4000 cells per well were seeded in 96-well plates and allowed to adhere by overnight incubation at 37 °C with 5% CO2. Cells were then treated with different concentrations of CCT245718 (nine threefold dilutions starting with 50 µM) in triplicates for 72 h. At the end of incubation, cells were fixed with TCA (3% final concentration) and stained with sulforhodamine B (SRB). SRB from the stained cells was dissolved in 10 mM Tris pH 10.5, and O.D. was measured at 490 nm using a microplate reader (MicroTek). For non-adherent cells, 15,000–20,000 cells per well were split into 96-well plates and incubated with different concentrations of inhibitors. Cells were incubated with the inhibitors for 72 h at 37 °C with 5% CO2. MTS reagent (CellTiter 96 Aqueous; Promega) was then added to the cells, and they were further incubated for 2–3 h. This was followed by a measurement of absorbance at 490 nm using a microplate reader. GI50 values from both assays were calculated using GraphPad Prism. For antiproliferative activity against normal hematopoietic cells, peripheral blood mononuclear cells (PBMCs) were isolated from blood taken from consenting healthy volunteers after approval from Institutional Review Board.
Immunoblotting
Samples for immunoblotting were prepared either using SDS sample buffer or lysis buffer, depending on the time of incubation with CCT245718. For incubation time of 2 h, samples were prepared in SDS sample buffer with 200 mM DTT followed by sonication for 10 sec (at 15-micron amplitude) and boiling for 10 min. For longer periods of incubations with the inhibitors, samples were prepared using lysis buffer (120 mM NaCl, 50 mM Tris HCl pH 7.4, 1% NP40, 1%) supplemented with 10 mM NaF, protease inhibitors (Complete Mini; Roche) and phosphatase inhibitor cocktails B and C (Santa Cruz Biotechnology, Santa Cruz, CA). Soluble protein fractions were isolated by centrifugation, and protein concentrations were measured using Bradford assay (Bio-Rad). SDS sample loading buffer was added to equal amounts of proteins from all the samples, followed by boiling for 10 min. Proteins from both types of samples were resolved using 10% SDS PAGE, transferred onto a nitrocellulose membrane, followed by immunoblotting with specific antibodies. Antibodies for cleaved PARP, cleaved-caspase 3, survivin, phospho-TACC3, phospho-STAT5, STAT5, phospho-Aurora A, Aurora A and phospho-Erk were purchased from Cell Signaling. Antibodies for TACC3, and Erk were obtained from Santa Cruz.
Cell cycle analysis
For the cell cycle analysis, cells were seeded in six-well plates followed by the addition of 0.2% DMSO or increasing concentrations of CCT245718, MLN8237 or MLN518. Following incubations for indicated times, cells were washed with PBS, fixed with ice-cold 70% ethanol. Fixed cells were washed with PBS, incubated with PBS (containing 1% FBS), 0.04% propidium iodide and RNase A (0.5%) for 30 min at 37 °C followed by analysis using BD FACSCalibur.
Annexin V/PI assay for apoptosis
Apoptosis was determined through Annexin V/PI assay using Annexin V-apoptosis kit (Santa Cruz Biotechnology). Cells in six-well plates were treated with indicated concentrations of CCT245718, MLN518 or DMSO control for 48 h. Following treatment, cells were harvested, washed with PBS and incubated in 100 µL aliquot of 1× Assay buffer with 5 µL Annexin V and 2 µL of propidium iodide for 1 h. After incubation, samples were analysed using BD FACSCalibur.
Immunoprecipitation
Cells (1.5 million/mL; 5 mL total) were treated with DMSO (control) or CCT245718 at indicated concentrations and times, followed by collection in falcon tubes. After washing with PBS, cells were lysed in lysis buffer (20 mM Tris pH 7.5, 1% Triton X100, 50 mM NaCl, 1 mM EDTA and 50 mM NaF supplemented with protease and phosphatase inhibitors) for 10 min on ice. Cell lysates were cleared by centrifugation and 0.5–1 mg of proteins were incubated with 1.2 µg of anti-FLT3 antibody (S-18, Santa Cruz) overnight with rotation at 4 °C. The next day, protein/antibody complexes were added with a mixture of protein A and protein G beads and incubated for 1 h with rotation at 4 °C. After incubation, beads were washed three times with lysis buffer, boiled in 2× Laemmle buffer and analysed by immunoblotting using anti-phosphotyrosine (4G10) and Anti-FLT3 antibodies.
KD determination
The binding constant (KD) of CCT245718 for FLT3-ITD and FLT3 D835Y was determined by DiscoverX KINOMEscan (KdELECT) Profiling Service that utilises a competition binding assay.
Molecular docking studies
In preparation for the molecular docking studies of CCT245718 in Aurora A and FLT3 kinase domains, the compound structure was constructed using the “Build” tool implemented in Maestro 2011 (Schrödinger Inc.). The three-dimensional conformations of the sketched molecule were then generated using the LigPrep module [26]. Next, the protein crystal structures of kinase domains of Aurora A (4B0G) [25] and FLT3 (4XUF) [27] proteins were retrieved from the protein databank (www.rcsb.org). All water and other co-crystallised reagents were removed from these structures before the addition of the hydrogen atoms. Steric clashes among the atoms of protein structures were relieved through brief minimisation using an OPLS_2005 force field. A maximum deviation of 0.3 Å root-mean-square for each structure was allowed from its native structure. For docking studies, grid boxes around co-crystalized ligand in each kinase domain were generated with 10 Å length in X, Y and Z dimensions from the centre of the ligand. Finally, with the default settings, Glide docking tool4 was used to dock CCT245718 compound in both crystal structures, and the best binding pose was selected based on Glide [28] scoring function (G-score) and the visual inspection of binding modes of the docked CCT245718 compound was performed.
Results
CCT245718 has potent antiproliferative activity in cancer cell lines
CCT245718 is an imidazo[4,5-b]pyridine-based Aurora A kinase inhibitor that selectively inhibits Aurora A in both biochemical and cellular assays. In the HCT116 colon cancer cell line, CCT245718 potently inhibits the activation loop phosphorylation (at T288) of ectopically expressed Aurora A (IC50 = 36 nM), while inhibition of histone H3 at S10 (Aurora B substrate) is seen only at very high concentrations (25 µM and 50 µM) [25]. In order to determine the effect of Aurora A inhibition by CCT245718 on cell viability, we performed proliferation assays in cancer cell lines of different origins, including colorectal, leukaemia and neuroblastoma. As shown in Table 1, cell lines displayed a varying degree of sensitivity to CCT245718 with MV-4-11 and MOLM-13 (AML, cell lines) being the most sensitive cell lines with GI50 values of 0.191 ± 0.053 µM and 0.195 ± 0.048 µM, respectively (Table 1). Both these cell lines have constitutively active FLT3 signalling due to internal tandem duplication (ITD) in the juxtamembrane domain of FLT3. THP1, an AML cell line with wild-type FLT3 displayed significantly lower sensitivity to CCT245718, indicating that FLT3 signalling may be inhibited by this compound in addition to Aurora A. CCT245718 had no antiproliferative/cytotoxic activity in normal hematopoietic peripheral blood mononuclear cells (PBMCs) at the highest used concentration of 10 µM.
Table 1.
GI50 values of CCT245718 in cancer cell lines of different origins in 3-day proliferation assays.
| Cell line | CCT245718 GI50 µM |
|---|---|
| MOLM-13 | 0.195 ± 0.048 (n = 4) |
| MV-4-11 | 0.191 ± 0.053 (n = 4) |
| THP1 | 6.11 ± 3.58 (n = 2) |
| HCT116 | 2.99 ± 1.33 (n = 3) |
| Kelly | 4.32 ± 0.96 (n = 3) |
| IMR-32 | 2.16 ± 0.59 (n = 3) |
| SHEP WT | 3.5 ± 1.09 (n = 3) |
CCT245718 inhibits Aurora A and FLT3 signalling in cells
Guided by the potency of CCT245718 in AML cell lines (MV-4-11 and MOLM-13) with activated FLT3 (Table 1), we determined the inhibition of Aurora A and FLT3 in these cells. As shown in Fig. 2a, treatment of cells with CCT245718 for 2 h efficiently inhibited the phosphorylation of Aurora A (in the activation loop at T288) and its substrate TACC3 (at T558) [29]. A robust inhibition of FLT3 signalling by CCT245718 was also seen in both these cell lines. Phosphorylation of both Erk and STAT5 was potently inhibited by treatment with CCT245718 for 2 h in a dose-dependent manner (Fig. 2b).
Fig. 2. Inhibition of Aurora A and FLT3 in AML cell lines.
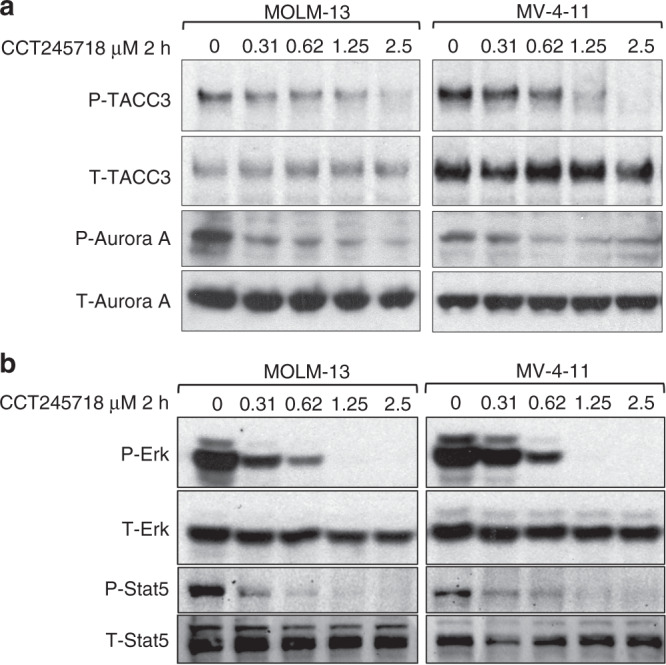
a Inhibition of phospho-TACC3 (S558) and phospho-Aurora A (T288) by different concentrations of CCT245718 in MV-4-11 and MOLM-13 cell lines following two-hour treatment. Total TACC3 and total Aurora A are shown as loading controls. b Inhibition of phospho-ERK and phospho-STAT5 by different concentrations of CCT245718 in both MV-4-11 and MOLM-13 cell lines following 2-h treatment.
CCT245718 directly binds and inhibits FLT3 activity
We next determined whether CCT245718 could directly inhibit FLT3 in cells by performing immunoprecipitation in MV-4-11 cells. As shown in Fig. 3a, treatment of cells with 1.25 µM and 2.5 µM CCT245718 for 2 h resulted in reduced tyrosine phosphorylation of FLT3. We subsequently investigated whether CCT245718 directly bound to FLT3 and thereby inhibited its activity by determining its binding constant (KD) with FLT3-ITD and FLT3 D835Y, the two most common FLT3 mutants associated with AML. Strong binding of CCT245718 with both FLT3 D835Y and FLT3-ITD was observed with KD values of 0.039 µM and 0.170 µM, respectively (Fig. 3b). These results confirmed CCT245718 as a dual Aurora A and FLT3 inhibitor in biochemical assays and in cells. In the absence of a crystal structure of CCT245718 bound to FLT3, the binding of CCT245718 to wild-type FLT3 was investigated through molecular docking studies using a crystal structure of FLT3 in an inactive conformation [27]. These docking studies indicated a binding mode similar to that of AC220, which is a selective and potent FLT3 inhibitor that binds to the kinase in a “DGF Out”, inactive conformation. The imidazopyridine ring of CCT245718 makes hydrogen bond interaction with the hinge region residue, CYS-694, similar to AC220 (Fig. 3c). Moreover, the best binding pose also generates an extra hydrogen bond interaction between the amide group of CCT245718 and ASP-698 residue present in the FLT3 active site. In the crystal structure of AC220 bound to FLT3, the inhibitor does not make direct contact with D835, and it has been suggested that mutation of D835 to a lipophilic residue such as phenylalanine and tyrosine may stabilise the activation loop in an active conformation [27]. CCT245718 has a distinctively different structure than AC220 and may bind to an active conformation that is stabilised by the D835Y mutation inhibiting its activity. We were unable to dock CCT245718 into FLT3 in an active conformation since, to the best of our knowledge, there is no reported crystal structure of an inhibitor bound to FLT3 in a DFG-in conformation. Docking of MLN518, however, predicts a different mode of binding where the inhibitor lies away from the hinge region and instead resides in the hydrophobic pocket of FLT3 (Supplementary Fig. S1). Furthermore, docking studies of this compound in the active site of Aurora A kinase indicated a binding mode similar to those of co-crystallised Aurora kinase ligands containing imidazopyridine ring moiety [25]. The overlapped binding pose with co-crystallised ligand in Fig. 3d shows that hydrogen bonding interactions with hinge region residue ALA-213 for both compounds are identical and thereby suggests that CCT245718 may bind with Aurora A kinase in this conformation experimentally.
Fig. 3. CCT245718 directly binds to and inhibits FLT3 activation.

a Inhibition of FLT3 by CCT245718. FLT3 was immunoprecipitated from MV-4-11 cells treated with 1.25 and 2.5 µM of CCT245718 for 2 h. Phosphorylation of FLT3 was determined through western blotting with phosphotyrosine antibodies. b CCT245718 binds to FLT3 mutants. Binding constants (KD values) of CCT245718 with FLT3 (D835Y) and FLT3 (ITD) were determined using a competition binding assay. c Superimposition of best-docked pose (green) of CCT245718 on the co-crystalized ligand, ACC220 (cyan) in the active site of FLT3 kinase domain (grey cartoon). Hydrogen bonding is shown with the dotted yellow lines. d Superimposition of best-docked pose (green) of CCT245718 on co-crystal ligand (cyan) in the active site of Aurora A kinase domain (grey cartoon). Hydrogen bonding is shown with the dotted yellow line.
CCT245718 induces FLT3 cell cycle phenotype in FLT3-ITD cells
Selective inhibition of Aurora A results in mitotic arrest due to defective spindle formation and centrosome separation [30]. Treatment of THP1 cells (containing WT FLT3) with CCT245718 for 24 h resulted in the mitotic arrest in a dose-dependent manner, similar to the effect of treatment with MLN8237, a selective Aurora A inhibitor (Fig. 4a). MLN518, a selective FLT3 inhibitor, on the other hand, only induced a small increase in G1 cells (THP1). Treatment of MV-4-11 and MOLM-13 cells with MLN518 or CCT245718 for 24 h caused G1 arrest (Fig. 4b, c, respectively), a phenotype previously reported for FLT3 inhibitors in FLT3-mutated cells [13, 31]. This contrasts with MLN8237, which led to mitotic arrest in both cell lines. The cell cycle phenotype for CCT245718, therefore, changes from that of Aurora A inhibition in THP1 cells (similar to MLN8237) to FLT3 inhibition in MV-4-11 and MOLM-13 cells (similar to MLN518).
Fig. 4. Effect of CCT245718 on cell cycle profile in THP1, MV-4-11 and MOLM-13 cells.

Cell cycle profile of THP1 (a), MV-4-11 (b) and MOLM-13 (c) cells following treatment with different inhibitors. Cells were treated with different concentrations of CCT245718, MLN518 and MLN8237 for 24 h, stained with propidium iodide and analysed by FACS.
CCT245718 induces apoptotic cell death in AML cell lines with activated FLT3 signalling
We next evaluated the ability of CCT245718 to induce apoptotic cell death in MOLM-13 and MV-4-11 cells lines. Treatment of both types of cells with different concentrations of CCT245718 for 48 h induced cleavage of caspase 3 and its substrate, Poly (ADP-ribose) polymerase (PARP), in a dose-dependent manner (Fig. 5a). Treatment with CCT245718 also caused downregulation of survivin, an inhibitor-of-apoptosis protein (IAP) that is upregulated in hematopoietic malignancies (Fig. 5a). Cleavage of caspase 3 and PARP, as well as downregulation of survivin by CCT245718, was comparable to those induced by MLN518. The induction of apoptotic cell death by CCT245718 in MV-4-11 and MOLM-13 cells was further confirmed through FACS analysis of Annexin V/PI stained cells (Fig. 5b, c, respectively). In both these cell lines, there was a dose-dependent increase in cells undergoing apoptosis following treatment with different concentrations of CCT245718, validating the results in Fig. 5a.
Fig. 5. CCT245718 induces apoptosis in AML cell lines.

a CCT245718 induces activation of caspase 3, cleavage of PARP and downregulation of survivin. MOLM-13 and MV-4-11 cells were treated with different concentrations of CCT245718 and MLN518 for 48 h. Levels of cleaved PARP, cleaved-caspase 3 and survivin were determined by western blotting. GAPDH was used as a loading control. b, c CCT245718 treatment enhances Annexin V/PI staining. MV-4-11 and MOLM-13 cells were treated with different concentrations of CCT245718 and MLN518 for 48 h. Cells were stained with FITC-labelled Annexin V/PI and analysed through FACS.
CCT245718 overcomes resistance due to TKD mutation in FLT3
The biochemical assay in Fig. 3b showed that CCT245718 potently bound to D835Y mutant of FLT3 with a KD value of 0.039 µM. In order to determine whether CCT245718 could inhibit FLT3 D835Y in cells, we used a MOLM-13-Res cell line that was generated through the incubation of MOLM-13 cells with progressively increasing concentrations of FLT3 inhibitor MLN518 [13]. We found that MOLM-13-Res cells were 11.3-fold resistant to MLN518 when compared with parental MOLM-13 cells in a 3-day proliferation assay with GI50 values of 1.162 ± 0.081 µM (n = 4) and 0.103 ± 0.072 µM (n = 4), respectively (Table 2).
Table 2.
CCT245718 overcomes TKD resistance in AML cell line.
| Cell Line | GI50 µM | |
|---|---|---|
| MLN518 | CCT245718 | |
| MOLM-13 | 0.103 ± 0.072 (n = 4) | 0.195 ± 0.048 (n = 4) |
| MOLM-13-Res | 1.162 ± 0.081 (n = 4) | 0.277 ± 0.078 (n = 5) |
| Fold difference | 11.3 | 1.42 |
GI50 values of MLN518 and CCT245718 in MOLM-13 and MOLM-13-Res cell lines in a 3-day MTS proliferation assay.
However, CCT245718 inhibited the proliferation of both resistant and parental cell lines with similar potency (GI50 values of 0.277 ± 0.078 µM n = 5 and 0.195 ± 0.048 µM n = 4 for MOLM-13-Res and MOLM-13, respectively; Table 2). Treatment of MOLM-13 control cells with both MLN518 and CCT245718 for 48 h caused inhibition of FLT3 signalling (P-Erk) and induction of apoptotic biomarkers (cleaved-caspase 3, cleaved PARP and downregulation of survivin) as expected (Fig. 6). In MOLM-13-Res cells, however, these biomarkers for FLT3 inhibition and apoptotic cells death were only modulated by CCT245718 (Fig. 6a). Both concentrations of MLN518, which were active in MOLM-13 control cells, failed to inhibit P-Erk or induce apoptotic biomarkers. CCT245718 also inhibited FLT3 phosphorylation in MOLM-13-Res cells (Fig. 6b).
Fig. 6. CCT245718 overcomes TKD-mediated resistance.

a MOLM-13 and MOLM-13-Res (D835Y) cells were treated with different concentrations of CCT245718 and MLN518 for 48 h. Phosphorylation of Erk, cleavage of PARP and caspase 3 and expression of survivin were analysed through western blotting. Levels of total Erk and GAPDH were used as loading controls. b Immunoprecipitation of FLT3 from MOLM-13-Res cells. Cells were treated with 2.5 µM CCT245718 for 24 h. Cell lysates were immunoprecipitated with Anti-FLT3 antibodies and probed with Anti-phosphotyrosine and Anti-FLT3 antibodies.
Discussion
The response of adult AML patients to standard anthracycline/cytarabine-based chemotherapy is mainly determined by age, with complete remission in 70–80% of patients under the age of 60. Older patients, in contrast, suffer from lower remission (50%) and higher relapse (85% at 3 years) rates [32]. The better prognosis for younger patients (<60 years), however, can be adversely affected by activating mutations in FLT3 [33], the most frequent type of somatic mutations associated with AML [34]. The in-frame ITD mutations in the juxtamembrane domain and mutations in the TKD can both lead to constitutive activation of FLT3 and account for ~25% and 7% of all AML cases, respectively [35, 36]. The resultant uncontrolled proliferation and enhanced survival of AML blasts mean that the FLT3 oncogenic addiction provides an opportunity for targeting this subset of AML using small-molecule inhibitors. A number of small-molecule inhibitors with varying degrees of specificity for FLT3 have been evaluated in the clinic during the last decade resulting in approval of midostaurin and gilteritinib [6, 7]. However, the success of many FLT3 inhibitors in the clinic has been limited due to transient responses and the acquisition of drug resistance [5].
Inhibition of a target in addition to FLT3 (dual FLT3 inhibitors) has previously been used to improve efficacy and overcome TKD mutation-based resistance [10–12]. FLT3 is also a secondary target for several Aurora kinase inhibitors that consequently show more preclinical and clinical activity in FLT3-mutated AML compared to FLT3 wild-type AML [37]. Moreover, the dual FLT3/Aurora kinase inhibitors, including CCT137690, which can overcome TKD mutation-mediated resistance to FLT3 inhibitors have demonstrated efficacy in xenograft models of AML [13, 24]. Aurora inhibitors selectively targeting Aurora A, Aurora B, or both kinases have been evaluated in the clinic; however, an ideal profile in terms of selectivity towards Aurora isoforms is still unclear [19, 38, 39]. Aurora A-selective inhibitor, MLN8237, has only shown modest activity in Phase 2 clinical trials in AML patients with an overall response rate of 17% and a stable disease in 49% of patients [20]. Inhibition of FLT3 in addition to Aurora A could improve responses in a subgroup of AML patients with FLT3 mutations. Such an approach has been used for the design, synthesis and evaluation of dual FLT3-Aurora A inhibitors that exhibit marked in vitro activity in AML cell lines [40]. Here, we describe the identification and characterisation of an Aurora A-selective inhibitor, CCT245718, as a dual FTL3/Aurora A kinase inhibitor with the ability to overcome D835Y resistance in vitro. CCT245718 has >200-fold selectivity for Aurora A versus Aurora B in biochemical assays and also inhibits Aurora A selectively and potently in HCT116 cells with an IC50 value of 0.036 µM [25]. When evaluated for its antiproliferative activity, CCT245718 was most potent in AML cell lines that were FLT3-ITD positive with GI50 values of ~200 nM. Its potency in cancer cell lines derived from solid tumours or AML cell line with wild-type FLT3, however, was significantly lower (11–31-fold), indicating that its effectiveness in FLT3-mutated cells may depend on FLT3 inhibition in addition to Aurora A. CCT245718 did indeed inhibit both FLT3 and Aurora A signalling in MV-4-11 and MOLM-13 cell lines containing homozygous and heterozygous FLT3-ITD mutations, respectively (Fig. 2) [41]. Inhibition of FLT3 in cells and affinity for FLT3 in biochemical assay further confirmed CCT245718 as a dual FLT3/Aurora A inhibitor (Fig. 3a, b).
Although CCT245718 inhibited both targets potently, it induced a G1 arrest in FLT3-mutated MV-4-11 and MOLM-13 cells following 24-h treatment, a FLT3 phenotype also induced by FLT3-specific inhibitor MLN518 in these cells (Fig. 4). This was in contrast to THP1 cells (with WT FLT3) where CCT245718 induced G2/M arrest, a phenotype similar to one induced by Aurora A-selective inhibitor MLN8237. The MLN8237-induced G2/M arrest was seen in all the cell lines irrespective of FLT3 status. This suggests that the efficacy of CCT245718 in FLT3-mutated cells is predominantly due to FLT3 inhibition. The contribution of Aurora A inhibition towards its efficacy, however, cannot be ignored considering its antiproliferative activity in cells with WT FLT3 and almost no drop in activity from biochemical IC50 against FLT3-ITD (170 nM) to the GI50 value (191 nM) in these cells. Similar results have been reported for dual FLT3/pan-Aurora kinase inhibitor CCT137690 that produces a G1/S phenotype in MOLM-13 cells in contrast to an Aurora B phenotype (endoreduplication) in FLT3 wild-type KG1a cells [13]. Similarly, the dual FLT3/SYK activity of midostaurin in FLT3-ITD cells with activated SYK is superior to targeted FLT3 inhibition and can be potentiated by additional inhibition of SYK [42].
One of the downstream targets of activated FLT3 is survivin, an inhibitor-of-apoptosis protein (IAP) involved in the regulation of cell division and apoptosis [38, 43]. Survivin is differentially overexpressed in cancers, including AML, and its high expression levels are associated with poor prognosis [43]. Abrogation of survivin function through RNA interference or dominant-negative mutants reduces tumour growth in different cancer models, making it a validated cancer target [43–45]. Here, we show that the inhibition of FLT3 function in both MV-4-11 and MOLM-13 cell lines through 24-h treatment with CCT245718 results in a marked reduction in survivin levels (Fig. 5). The downregulation of survivin has also been demonstrated for FLT3 and Aurora A-specific inhibitors through the transcriptional and posttranslational mechanisms, respectively [46, 47]. The decrease in survivin expression levels by CCT245718 is accompanied by an increase in apoptotic cell death as indicated by activation of caspase 3 and cleavage of its substrate PARP (Fig. 5). This is in agreement with the reported inhibition of effector caspase activity by survivin [44, 48].
TKD mutations in the activation loop and gatekeeper residues (D835 and F691, respectively) confer resistance to FLT3 inhibitors and are implicated in acquired resistance to targeted FLT3 therapy. Mutations of D835 and F691 residues were identified in relapsed patients following quizartinib monotherapy and have been demonstrated to confer resistance to various FLT3 inhibitors in cell line models [9, 13, 49, 50]. FLT3 inhibitors that can inhibit secondary TKD mutations in FLT3-ITD or potently target a kinase in addition to FLT3 can overcome secondary resistance to FLT3 inhibitors or prevent the acquisition of such resistance, respectively. The dual FLT3/Aurora A inhibitor, CCT245718, had a higher in vitro affinity for FLT3-D835Y mutant compared to the FLT3-ITD mutant (KD values of 39 and 170 nM, respectively) and was able to potently inhibit the proliferation of MOLM-13 cells (MOLM-13-Res [13]) containing both ITD and D835Y mutations on the same allele (Fig. 6). The FLT3 mutants with both ITD and TKD mutations, including D835Y (FLT3-ITD-D835Y), exhibit high relative resistance (compared to FLT3-ITD alone) towards various FLT3 inhibitors, including quizartinib, sorafenib and PLX3397 [49]. The GI50 value for CCT245718 in MOLM-13-Res cells was only 1.42-fold higher than in MOLM-13 cells in contrast to MLN518 for which the fold resistance was 11.3-fold (Table 2). Similar results were seen for inhibition of FLT3 phosphorylation and downstream signalling by CCT245718 in these cells; while CCT245718 inhibited phospho-ERK, downregulated survivin and induced apoptosis in both MOLM-13 and MOLM-13-Res cells, MLN518 was only active in MOLM-13 cells, indicating that FLT3 with ITD and TKD mutations is potently inhibited by CCT245718 and not MLN518 (Fig. 6). CCT245718 binding to FLT3 was predicted to be similar to that of ACC220, which is a Type II inhibitor that binds in a “DFG Out”, inactive confirmation [27]. TKD mutations with bulky hydrophobic substitution at D835 show high resistance to Type II inhibitors. Despite ACC20-like predicted binding mode, CCT245718 potently binds to D835Y mutant and inhibits FLT3 phosphorylation and downstream signalling in cells with D835Y mutation (Figs. 3 and 6). One of our previously reported dual FLT3/Aurora inhibitor, CCT137690, an imidazopyridine, is also predicted to bind with a similar mode despite potently inhibiting FLT3-D835Y mutant (0.0033 µM IC50; Supplementary Fig. S2) [13]. The precise mode of binding, therefore, can only be determined through co-crystallisation.
The inhibition of Aurora A by CCT245718 may also contribute to its potency in these cells as discussed earlier; its exact contribution towards induction of apoptosis in FLT3-ITD cells, however, remains to be determined. Inhibition of Aurora A in addition to FLT3 can, nonetheless, contribute to evasion of development of resistance to this inhibitor. To summarise, we have identified and characterised CCT245718 as a dual FLT3/Aurora A inhibitor that overcomes D835Y-mediated resistance to FLT3 inhibitors in AML cell lines.
Supplementary information
Acknowledgements
We thank Dr. Safee Ullah Chaudhary and Dr. Ahmad Jawad Afzal for carefully reading the manuscript and providing useful insights.
Author contributions
AF, VB and SL conceived and designed the study with MUT providing additional input. AF, MUT, MF, HP, RU and MM performed all the experiments. MUT, HP and AF wrote the manuscript. RSZS, VB and SL helped in writing and reviewing the manuscript. MF helped in the revision of the manuscript. AF supervised the whole study. All the authors read and approved the final manuscript.
Funding
This study was supported by generous grants from Lahore University of Management Sciences to Amir Faisal (Faculty Initiative Fund-253 and Startup Grant STG-064).
Ethics approval and consent to participate
Peripheral blood mononuclear cells (PBMCs) were isolated from blood taken from consenting healthy volunteers after approval from Institutional Review Board.
Consent to publish
Not applicable.
Competing interests
Some of the authors are current (VB) or former (SL and AF) employees of The Institute of Cancer Research which operates a rewards to inventors scheme applicable to all current and former employees. For all the other authors, there is no financial or commercial conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41416-021-01527-2.
References
- 1.Peterson LF, Boyapati A, Ahn E-Y, Biggs JR, Okumura AJ, Lo M-C, et al. Acute myeloid leukemia with the 8q22; 21q22 translocation: secondary mutational events and alternative t (8; 21) transcripts. Blood. J Am Soc Hematol. 2007;110:799–805. doi: 10.1182/blood-2006-11-019265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grafone T, Palmisano M, Nicci C, Storti S. An overview on the role of FLT3-tyrosine kinase receptor in acute myeloid leukemia: biology and treatment. Oncology Rev. 2012;6:64–74. [DOI] [PMC free article] [PubMed]
- 3.Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood J Am Soc Hematol. 2002;100:1532–42. doi: 10.1182/blood-2002-02-0492. [DOI] [PubMed] [Google Scholar]
- 4.Levis M. FLT3 mutations in acute myeloid leukemia: what is the best approach in 2013? Hematology. 2013;2013:220–6. doi: 10.1182/asheducation-2013.1.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Larrosa-Garcia M, Baer MR. FLT3 inhibitors in acute myeloid leukemia: current status and future directions. Mol Cancer Therapeut. 2017;16:991–1001. doi: 10.1158/1535-7163.MCT-16-0876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sidaway P. Gilteritinib improves outcomes in AML. Nat Rev Clin Oncol. 2020;17:69. [DOI] [PubMed]
- 7.Levis M. Midostaurin approved for FLT3-mutated AML. Blood. 2017;129:3403–6. doi: 10.1182/blood-2017-05-782292. [DOI] [PubMed] [Google Scholar]
- 8.Assi R, Ravandi F. FLT3 inhibitors in acute myeloid leukemia: choosing the best when the optimal does not exist. Am J Hematol. 2018;93:553–63. doi: 10.1002/ajh.25027. [DOI] [PubMed] [Google Scholar]
- 9.Smith CC, Wang Q, Chin C-S, Salerno S, Damon LE, Levis MJ, et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature. 2012;485:260–3. doi: 10.1038/nature11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li C, Liu L, Liang L, Xia Z, Li Z, Wang X, et al. AMG 925 is a dual FLT3/CDK4 inhibitor with the potential to overcome FLT3 inhibitor resistance in acute myeloid leukemia. Mol Cancer Therapeut. 2015;14:375–83. doi: 10.1158/1535-7163.MCT-14-0388. [DOI] [PubMed] [Google Scholar]
- 11.Minson KA, Smith CC, DeRyckere D, Libbrecht C, Lee-Sherick AB, Huey MG, et al. The MERTK/FLT3 inhibitor MRX-2843 overcomes resistance-conferring FLT3 mutations in acute myeloid leukemia. JCI Insight. 2016;1(3):e85630 [DOI] [PMC free article] [PubMed]
- 12.Zhang W, Borthakur G, Gao C, Chen Y, Mu H, Ruvolo VR, et al. The dual MEK/FLT3 inhibitor E6201 exerts cytotoxic activity against acute myeloid leukemia cells harboring resistance-conferring FLT3 mutations. Cancer Res. 2016;76:1528–37. doi: 10.1158/0008-5472.CAN-15-1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moore AS, Faisal A, De Castro DG, Bavetsias V, Sun C, Atrash B, et al. Selective FLT3 inhibition of FLT3-ITD+ acute myeloid leukaemia resulting in secondary D835Y mutation: a model for emerging clinical resistance patterns. Leukemia. 2012;26:1462–70. doi: 10.1038/leu.2012.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Malik HS, Bilal A, Ullah R, Iqbal M, Khan S, Ahmed I, et al. Natural and semisynthetic chalcones as dual FLT3 and microtubule polymerization inhibitors. J Nat Prod. 2020;83:3111–21. doi: 10.1021/acs.jnatprod.0c00699. [DOI] [PubMed] [Google Scholar]
- 15.Andrews PD. Aurora kinases: shining lights on the therapeutic horizon? Oncogene. 2005;24:5005–15. doi: 10.1038/sj.onc.1208752. [DOI] [PubMed] [Google Scholar]
- 16.Vader G, Lens SM. The Aurora kinase family in cell division and cancer. Biochimica et Biophysica Acta (BBA)-Rev Cancer. 2008;1786:60–72. doi: 10.1016/j.bbcan.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 17.Gautschi O, Heighway J, Mack PC, Purnell PR, Lara PN, Gandara DR. Aurora kinases as anticancer drug targets. Clin Cancer Res. 2008;14:1639–48. doi: 10.1158/1078-0432.CCR-07-2179. [DOI] [PubMed] [Google Scholar]
- 18.Pollard JR, Mortimore M. Discovery and development of aurora kinase inhibitors as anticancer agents. J Med. Chem. 2009;52:2629–51. doi: 10.1021/jm8012129. [DOI] [PubMed] [Google Scholar]
- 19.Bavetsias V, Linardopoulos S. Aurora kinase inhibitors: current status and outlook. Front Oncol. 2015;5:278. doi: 10.3389/fonc.2015.00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldberg SL, Fenaux P, Craig MD, Gyan E, Lister J, Kassis J, et al. An exploratory phase 2 study of investigational Aurora A kinase inhibitor alisertib (MLN8237) in acute myelogenous leukemia and myelodysplastic syndromes. Leuk Res Rep. 2014;3:58–61. doi: 10.1016/j.lrr.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Löwenberg B, Muus P, Ossenkoppele G, Rousselot P, Cahn J-Y, Ifrah N, et al. Phase 1/2 study to assess the safety, efficacy, and pharmacokinetics of barasertib (AZD1152) in patients with advanced acute myeloid leukemia. Blood. 2011;118:6030–6. doi: 10.1182/blood-2011-07-366930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kantarjian HM, Sekeres MA, Ribrag V, Rousselot P, Garcia-Manero G, Jabbour EJ, et al. Phase I study assessing the safety and tolerability of barasertib (AZD1152) with low-dose cytosine arabinoside in elderly patients with AML. Clin Lymphoma Myeloma Leuk. 2013;13:559–67. doi: 10.1016/j.clml.2013.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grundy M, Seedhouse C, Shang S, Richardson J, Russell N, Pallis M. The FLT3 internal tandem duplication mutation is a secondary target of the aurora B kinase inhibitor AZD1152-HQPA in acute myelogenous leukemia cells. Mol Cancer Therapeut. 2010;9:661–72. doi: 10.1158/1535-7163.MCT-09-1144. [DOI] [PubMed] [Google Scholar]
- 24.Moore AS, Faisal A, Mak GWY, Miraki-Moud F, Bavetsias V, Valenti M, et al. Quizartinib-resistant FLT3-ITD acute myeloid leukemia cells are sensitive to the FLT3-Aurora kinase inhibitor CCT241736. Blood Adv. 2020;4:1478–91. doi: 10.1182/bloodadvances.2019000986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bavetsias V, Faisal A, Crumpler S, Brown N, Kosmopoulou M, Joshi A, et al. Aurora isoform selectivity: design and synthesis of imidazo [4, 5-b] pyridine derivatives as highly selective inhibitors of Aurora-A kinase in cells. J Med Chem. 2013;56:9122–35. doi: 10.1021/jm401115g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schrodinger. Ligprep V. 2.3. Schrodinger, LLC, New York; 2009.
- 27.Zorn JA, Wang Q, Fujimura E, Barros T, Kuriyan J. Crystal structure of the FLT3 kinase domain bound to the inhibitor quizartinib (AC220) PLoS ONE. 2015;10:e0121177. doi: 10.1371/journal.pone.0121177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–49. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 29.LeRoy PJ, Hunter JJ, Hoar KM, Burke KE, Shinde V, Ruan J, et al. Localization of human TACC3 to mitotic spindles is mediated by phosphorylation on Ser558 by Aurora A: a novel pharmacodynamic method for measuring Aurora A activity. Cancer Res. 2007;67:5362–70. doi: 10.1158/0008-5472.CAN-07-0122. [DOI] [PubMed] [Google Scholar]
- 30.Manfredi MG, Ecsedy JA, Meetze KA, Balani SK, Burenkova O, Chen W, et al. Antitumor activity of MLN8054, an orally active small-molecule inhibitor of Aurora A kinase. Proc Natl Acad Sci USA. 2007;104:4106–11. doi: 10.1073/pnas.0608798104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weisberg E, Boulton C, Kelly LM, Manley P, Fabbro D, Meyer T, et al. Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412. Cancer Cell. 2002;1:433–43. doi: 10.1016/S1535-6108(02)00069-7. [DOI] [PubMed] [Google Scholar]
- 32.Burnett A, Wetzler M, Lowenberg B. Therapeutic advances in acute myeloid leukemia. J Clin Oncol. 2011;29:487–94. doi: 10.1200/JCO.2010.30.1820. [DOI] [PubMed] [Google Scholar]
- 33.Kiyoi H, Naoe T, Nakano Y, Yokota S, Minami S, Miyawaki S, et al. Prognostic implication of FLT3 and N-RAS gene mutations in acute myeloid leukemia. Blood, J Am Soc Hematol. 1999;93:3074–80. [PubMed] [Google Scholar]
- 34.Komanduri KV, Levine RL. Diagnosis and therapy of acute myeloid leukemia in the era of molecular risk stratification. Annu Rev Med. 2016;67:59–72. doi: 10.1146/annurev-med-051914-021329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thiede C, Steudel C, Mohr B, Schaich M, Schäkel U, Platzbecker U, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002;99:4326–35. doi: 10.1182/blood.V99.12.4326. [DOI] [PubMed] [Google Scholar]
- 36.Abu‐Duhier F, Goodeve A, Wilson G, Care R, Peake I, Reilly J. Identification of novel FLT‐3 Asp835 mutations in adult acute myeloid leukaemia. Br J Haematol. 2001;113:983–8. doi: 10.1046/j.1365-2141.2001.02850.x. [DOI] [PubMed] [Google Scholar]
- 37.Moore A, Blagg J, Linardopoulos S, Pearson A. Aurora kinase inhibitors: novel small molecules with promising activity in acute myeloid and Philadelphia-positive leukemias. Leukemia. 2010;24:671–8. doi: 10.1038/leu.2010.15. [DOI] [PubMed] [Google Scholar]
- 38.Fukuda S, Singh P, Moh A, Abe M, Conway EM, Boswell HS, et al. Survivin mediates aberrant hematopoietic progenitor cell proliferation and acute leukemia in mice induced by internal tandem duplication of Flt3. Blood, J Am Soc Hematol. 2009;114:394–403. doi: 10.1182/blood-2008-11-188714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheung CHA, Sarvagalla S, Lee JY-C, Huang Y-C, Coumar MS. Aurora kinase inhibitor patents and agents in clinical testing: an update (2011–2013) Expert Opin Therapeutic Pat. 2014;24:1021–38. doi: 10.1517/13543776.2014.931374. [DOI] [PubMed] [Google Scholar]
- 40.Chang Hsu Y, Ke YY, Shiao HY, Lee CC, Lin WH, Chen CH, et al. Facile identification of dual FLT3–Aurora A inhibitors: a computer‐guided drug design approach. ChemMedChem. 2014;9:953–61. doi: 10.1002/cmdc.201300571. [DOI] [PubMed] [Google Scholar]
- 41.Quentmeier H, Reinhardt J, Zaborski M, Drexler H. FLT3 mutations in acute myeloid leukemia cell lines. Leukemia. 2003;17:120–4. doi: 10.1038/sj.leu.2402740. [DOI] [PubMed] [Google Scholar]
- 42.Weisberg EL, Puissant A, Stone R, Sattler M, Buhrlage SJ, Yang J, et al. Characterization of midostaurin as a dual inhibitor of FLT3 and SYK and potentiation of FLT3 inhibition against FLT3-ITD-driven leukemia harboring activated SYK kinase. Oncotarget. 2017;8:52026–44. doi: 10.18632/oncotarget.19036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Altieri DC. Survivin, versatile modulation of cell division and apoptosis in cancer. Oncogene. 2003;22:8581–9. doi: 10.1038/sj.onc.1207113. [DOI] [PubMed] [Google Scholar]
- 44.Williams NS, Gaynor RB, Scoggin S, Verma U, Gokaslan T, Simmang C, et al. Identification and validation of genes involved in the pathogenesis of colorectal cancer using cDNA microarrays and RNA interference. Clin Cancer Res. 2003;9:931–46. [PubMed] [Google Scholar]
- 45.Grossman D, Kim PJ, Schechner JS, Altieri DC. Inhibition of melanoma tumor growth in vivo by survivin targeting. Proc Natl Acad Sci USA. 2001;98:635–40. doi: 10.1073/pnas.98.2.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yoshida A, Ookura M, Zokumasu K, Ueda T. Gö6976, a FLT3 kinase inhibitor, exerts potent cytotoxic activity against acute leukemia via inhibition of survivin and MCL-1. Biochemical Pharmacol. 2014;90:16–24. doi: 10.1016/j.bcp.2014.04.002. [DOI] [PubMed] [Google Scholar]
- 47.Kamran M, Long Z, Xu D, Lv S, Liu B, Wang C, et al. Aurora kinase A regulates survivin stability through targeting FBXL7 in gastric cancer drug resistance and prognosis. Oncogenesis. 2017;6:e298. doi: 10.1038/oncsis.2016.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shin S, Sung B-J, Cho Y-S, Kim H-J, Ha N-C, Hwang J-I, et al. An anti-apoptotic protein human survivin is a direct inhibitor of caspase-3 and-7. Biochemistry. 2001;40:1117–23. doi: 10.1021/bi001603q. [DOI] [PubMed] [Google Scholar]
- 49.Smith CC, Lin K, Stecula A, Sali A, Shah NP. FLT3 D835 mutations confer differential resistance to type II FLT3 inhibitors. Leukemia. 2015;29:2390–2. doi: 10.1038/leu.2015.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alvarado Y, Kantarjian HM, Luthra R, Ravandi F, Borthakur G, Garcia‐Manero G, et al. Treatment with FLT3 inhibitor in patients with FLT3‐mutated acute myeloid leukemia is associated with development of secondary FLT3–tyrosine kinase domain mutations. Cancer. 2014;120:2142–9. doi: 10.1002/cncr.28705. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.