

Abstract
The current methods for detecting circulating tumor cells (CTCs) suffer from several drawbacks. We report a novel method that is based on a chimeric virus probe and can detect CTCs with extremely high specificity and sensitivity. Moreover, it exclusively detects live CTCs, and its detection efficacy is not impacted by the variation of epithelial cell adhesion molecule (EpCAM) expression. The chimeric virus probe is composed of a capsid from human papillomavirus that provides the detection with high specificity and an SV40-based genome that can amplify extensively inside CTCs and, hence, endows the detection with high sensitivity. Furthermore, different marker genes can be incorporated into the probe to provide detection with versatility. These unique capabilities will likely improve the validity and utility of this CTC detection in several clinical applications, which is one of the drawbacks suffered by many of the current CTC detection methods.
Keywords: circulating tumor cells, chimeric viral vector, human papillomavirus, SV40, probe, GFP, luciferase, amplification, detection
Graphical abstract
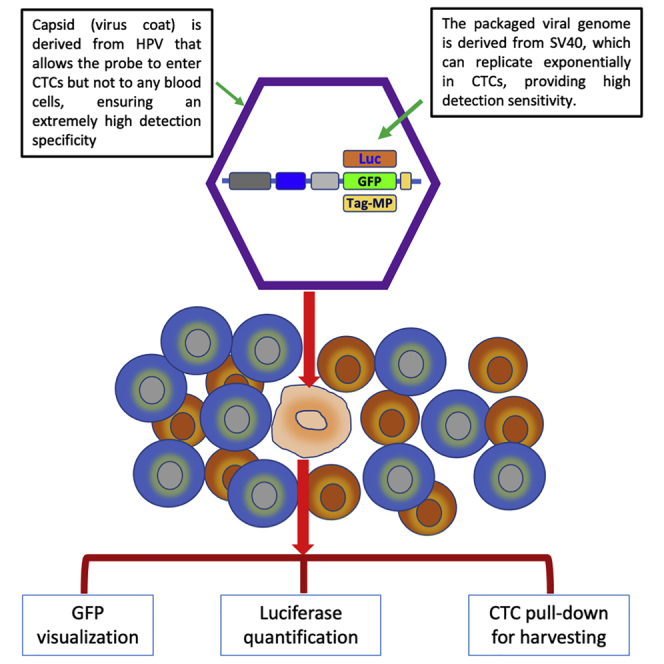
A hybrid virus probe is designed that incorporates components from both human papillomavirus (HPV) and SV40, with the former providing epithelium-specific infection for a highly selective entry of the probe into circulating tumor cells (CTCs) and the latter providing amplification for sensitive detection. Most importantly, such a detection method ensures that all the detected CTCs are alive.
Introduction
Circulating tumor cells (CTCs) are rare malignant cells that have managed to detach from local tumors and subsequently enter the bloodstream. Although the primary importance of CTCs is their ability to seed metastatic tumors, their constant presence in the blood and the easiness of access through liquid biopsy have made their detection an attractive alternative to the traditional biopsy for clinical applications such as cancer screening, therapy evaluation, and disease prognosis. However, their exceeding rareness, only a few CTCs among millions of white blood cells (WBCs) and billions of red blood cells per milliliter of blood, makes their detection extremely challenging and demands that any detection method be highly sensitive. Additionally, CTCs, like the tumors they are originally detached from, lack unique and well-defined universal biomarkers. These issues highlight the challenges of detection specificity.
Current methods for CTC detection are primarily based on two major principles. The first, and also the more common, principle is an immuno-based detection that relies on immunological recognition of a series of biomarkers that are predominantly epithelial lineage-related proteins such as epithelial cell adhesion molecule (EpCAM) and cytokeratins. An example of this approach is CELLSEARCH, which is the only CTC detection method to have received FDA approval.1 The first step of this method is to enrich CTCs from whole blood by binding the cells to the anti-EpCAM-antibody-conjugated iron nanoparticles, followed by magnetic capture. The enriched CTCs are further characterized by 4′,6-diamidino-2-phenylindole (DAPI) staining to identify nucleated cells and more antibody staining for epithelial structural cytokeratins (CK8, CK18, and CK19). Finally, anti-CD45 antibody staining is used to differentiate CTCs from circulating WBCs. The whole process is relatively cumbersome, and, because of the heterogeneous nature of tumor cell biomarkers it relies on for detection, this method may only detect a fraction of the total CTCs in the blood. For example, it is known that CTCs usually undergo epithelial to mesenchymal transition (EMT), during which epithelial markers such as EpCAM are frequently downregulated.2
The second detection principle exploits the biophysical properties of the CTCs, such as their relatively large size, distinct density, deformability, and electric charge relative to WBCs. Several methods based on this principle have been reported, such as a filter-based membrane with a specific pore size,3,4 microfluidic devices depending on both size and the ability of the CTCs to deform in their enrichment strategies,5 and devices combining density centrifugation with size-based filtration.6 One of the main drawbacks of biophysical property-based detection is that many blood cells such as monocytes/macrophages have a similar or even larger size than CTCs. As such, they tend to be inseparable from CTCs after the isolation procedure. Thus, CTCs isolated from biophysical property-based methods typically need to be further characterized by either immuno-staining or RT-PCR analysis.
Current CTC detection methods, based on either immune-affinity or biophysical property, are not designed to specifically detect live CTCs. Methods such as CELLSEARCH require CTCs to be fixed for examination. Although CTCs obtained from some of the microfluid captures are live cells, the need for further downstream characterization/confirmation by antibody staining and/or other procedures will result in the killing of these cells. A method that can unambiguously detect live CTCs is desirable to improve the relatively poor clinical correlation of the current CTC detection methods,7 which will be particularly beneficial for certain clinical applications such as evaluating therapeutic responses in real time, monitoring tumor recurrence, and/or determining patient prognosis.
Here, we report a chimeric virus-based probe, called the CTC-UniPro, that can detect live CTCs with extremely high specificity and sensitivity. Additionally, it offers both versatility and simplicity of detection. The CTC-UniPro contains two key components. The first component is a virus-like particle (VLP) formed from the L1 and L2 proteins of human papillomavirus 16 (HPV-16). The VLP enables the probe to selectively enter CTCs via the HPV-mediated infection process, which is strictly epithelial cell specific.8 This provides the probe with extremely high specificity for detecting CTCs. The second component is a modified SV40 virus genome that contains a marker gene and is packaged into the VLP. This provides the probe with the ability to amplify the detection signal extensively via both the SV40 genome amplification and the marker gene expression. As a result, the probe has a high sensitivity at detecting CTCs. Most importantly, the detected signals (the expressed marker gene products) are strictly produced by live CTCs. The choice of different marker genes, such as the green fluorescent protein (GFP) for visual detection and numeration, the luciferase gene for luminescence detection and quantification, and a synthetic membrane protein for CTC capture, provides a high degree of versatility and simplicity. Our data show that CTC-UniPro can indeed detect CTCs with the anticipated specificity and sensitivity, and it represents a unique and probably the only method for unambiguously detecting live CTCs.
Results
The design and construction of a chimeric virus as a probe for detecting live CTCs
To our knowledge, HPV is probably the only human virus that can selectively infect cells of human epithelial origin.9 This makes it an ideal candidate to assemble a probe for CTC detection. As depicted in Figure 1A, the chimeric virus probe is composed of two parts. The outside capsid shell is from the L1 and L2 proteins of HPV-16, which can self-assemble into VLPs that possess infectivity similar to wild-type HPVs with a strong tropism for epithelial tissues. Additionally, during the VLP assembly, plasmid-like circular DNA within a certain size limit can be packaged inside the VLPs. For this reason and the fact that HPV genome replicates relatively slowly (once every 3 weeks),10 we decided to package a fast-replicating viral genome into the VLP to generate a chimeric virus that can selectively enter and rapidly replicate in CTCs. Therefore, the SV40 genome was chosen for this purpose, in addition to the following reasons. First, SV40 is known to be able to replicate in many cell types, including epithelial cells. Second, its genome replication is relatively fast, capable of producing hundreds of copies within 24 h.11 Third, its genome size is within the limits of HPV packaging capacity. Specifically, the genome of SV40 (5.2 kb) is ∼3 kb shorter than that of HPV, which allows for the insertion of a marker gene without compromising the packaging efficiency. For a maximal amplification of the probe within CTCs, we introduced a series of modifications to the SV40 genome. First, we replaced the genes encoding SV40 capsid proteins (VP 1–3) with one of the chosen marker genes (GFP, luciferase, or a tagged membrane protein) for easy, convenient, and versatile CTC detection (Figure 1B). Second, we introduced a series of modifications into the late promoter region that have been reported in the literature to increase the promoter strength.12, 13, 14 These include a series of substitutions (298C>T, 299C>T, and 304C>T) and an addition of 4 extra copies of the 72-bp repeat (Figure 1B). Third, we made an additional substitution (335A>C) at the downstream region of the late promoter to destroy the start codon in that location. This would allow the marker gene to be efficiently expressed without interference from this endogenous start codon. The exemplary construct containing the GFP marker gene is designated pSV-GFP.
Figure 1.

Probe design and construction
(A) Schematic diagram of the chimeric virus probe. The outside capsid shell is derived from HPV (formed from the L1 and L2 capsid proteins). The inside contains the modified SV genome with the insertion of one of the chosen marker genes. (B) The SV40-based self-amplifying probe. The wild-type (w.t.) SV40 genome is shown on the top. Several key modifications were introduced to the pSV40 genome for the construction of the probe (pSV-GFP). These include (1) deletion of VP1–3 capsid genes and insertion to the locus of one of the marker genes (GFP for pSV-GFP probe, luciferase for pSV-Luc probe, and Tag-MP for pSV-Tag-MP probe), (2) substitutions of 298C>T, 299C>T, and 304C>T, (3) addition of 4 extra copies of the 72-bp repeats to the downstream of the natural 21 repeats (labeled as Rs), and (4) substitution of 335A>C. (C) Construction of the control plasmid, pcDNA-GFP. (D) Construction of the packaging plasmid, pHPVL1/2, which contains both the L1 and L2 capsid genes of HPV that are separated by a copy of the 2A peptide. CMVP, CMV promoter; P-Ori-P, the SV40 early promoter-replication origin-later promoter sequences; VP1–3, SV40 capsid proteins VP 1, 2, 3; Early P, SV40 early promoter; Rs, the SV40 21-bp and 72-bp repeats; M-late P, modified SV40 late promoter. The direction of the promoter for transcription is indicated by the arrow.
As a control, we constructed a similar plasmid but without the SV40-mediated amplification mechanism. As shown in Figure 1C, pcDNA-GFP contains the same GFP gene (driven by the strong cytomegalovirus [CMV] immediate promoter) and the same SV40 replication origin (Ori) and the late promoter region. Although this plasmid can undergo amplification in 293FT cells as they constitutively express the large T antigen (LT) of the SV40, it would not be able to self-replicate in CTCs because of the lack of SV40 T antigens. Another plasmid, pHPVL1/2, was constructed by cloning both the L1 and L2 genes of the HPV16 into it (separated by a copy of the 2A peptide). This plasmid will provide the L1 and L2 HPV capsid proteins for virus particle assembly and SV-GFP probe packaging. All the constructed plasmids have been sequenced to confirm the correctness of the incorporated sequences. The gene expression from each of the plasmids was confirmed by western blot analyses (data not shown).
Probe production and purification
For producing the chimeric virus probe, pHPVL1/2 and one of the probes containing plasmids (pSV-GFP, pSV-Luc, or pSV-Tag-MP) were co-transfected into 293TT cells. Cells were collected either 48 or 72 h later and lysed either with lyse buffer or through sonication to release the packaged probe. The probe was collected by harvesting the supernatant after spinning down the cell debris. The pcDNA-GFP was prepared the same way. Since 293TT cells express high levels of both large T and small T antigens of SV40,15 pcDNA-GFP gets amplified similarly as the other ones, as it also contains a copy of the SV40 replication origin. Therefore, all the plasmids should have equivalent packaging efficiency.
The collected supernatants containing the packaged probe-containing plasmids or the control plasmid were purified by first passing through the GE Capto Core 700 column to remove protein, DNA, and RNA impurities. This was followed by passing through a PD-10 column to remove the remaining chemicals. The titer of the produced chimeric virus probes for pSV-GFP and the control pcDNA-GFP was titrated by incubating the serially diluted probes with 293TT cells for 2–3 days, followed by flow cytometry analysis. The pSV-Tag-MP probe was similarly titrated after staining the cells with an anti-Myc-tag immunoglobulin G (IgG). The titer of pSV-Luc was estimated by measuring the luciferase activity after the incubation period. The yield of the probes was usually >1 × 109 infection units (IUs) per 10-cm dish of transfection.
The self-amplification property of the CTC-UniPro can increase the marker gene signal by over 80-fold
The CTC-UniPro was designed such that the SV40-based genome can replicate in CTCs after chimeric virus-mediated entry. To test how much this self-amplification could contribute to the detection sensitivity, we incubated two lung cancer cell lines with either pSV-GFP probe or pcDNA-GFP control and constantly monitored the GFP expression. Although the GFP intensity showed a steady increase in the cells incubated with pSV-GFP, the GFP in the cells incubated with pcDNA-GFP remained at the background level of the 24-h incubation. Shown in Figure 2A is a typical field from cells incubated with these two vectors for either 24-h or 72-h incubation. To quantitate the GFP shown in Figure 2A, we lysed the cells and the released GFP was measured with a fluorescent illuminator. The result is shown in Figure 2B. It indicates that the self-amplification property in pSV-GFP contributes >80-fold signal amplification when tested in these two tumor cells.
Figure 2.

The ability of CTC-UniPro to self-amplify contributes to the increased detection sensitivity
(A) A549 and 5838 cells (human lung cancer cell lines) were incubated with either pcDNA-GFP or pSV-GFP at 2 IU per cell, and GFP expression was monitored regularly. Shown are typical micrographic fields taken at the indicated time during the incubation. Original magnification: 20×. (B) The ratio of GFP quantification at 72 h over 24 h. Hp < 0.05 compared to pcDNA-GFP.
The pSV-GFP probe shows extremely high specificity and sensitivity at detecting tumor cells versus normal blood cells
For any diagnostic method, both specificity and sensitivity are vital for detection accuracy. For determining the detection specificity of CTC-UniPro, we incubated either normal peripheral blood mononuclear cells (PBMCs) or two lung cancer cell lines (H1944 and H358) with pSV-GFP at the dose of 2 IU per cell. Cells with medium only served as a negative control. Again, GFP expression was monitored continuously. At 72 h during incubation, when usually GFP expression from the probe reaches its peak, not a single GFP-positive cell could be detected in the well with PBMCs, whereas there were extensive and strong GFP-expressing cells in the wells where lung cancer cells were seeded (Figure 3A). Next, we incubated PBMCs with increasing dosages of pSV-GFP, from 1 IU/cell up to 50 IU/cell. No single GFP-positive cell was detected even at the highest IU of the probe at 72-h incubation time (Figure 3B). It has been reported that certain oncolytic viruses, such as the one based on herpes simplex virus (HSV), because of their selective replication in tumor cells, might be used for CTC detection.16 We therefore tested two of the oncolytic HSVs constructed in our own lab, Baco-1 (derived from HSV-1) and FusOn-H2 (derived from HSV-2), both of which carry the GFP gene. PBMCs were incubated with these two viruses at 1 plaque-forming unit (PFU) per cell. Even at 24 h after infection, a significant number of GFP-positive cells could be detected in PBMCs incubated with both Baco-1 and FusOn-H2, indicating that they did not possess the same high specificity for CTCs as the pSV-GFP probe.
Figure 3.

CTC-UniPro shows high detection specificity
(A) PBMCs isolated from healthy donors or human lung cancer cells (H1944 and H358) were incubated either with medium only or in the presence of pSV-GFP at 2 IU/cell. Shown is a typical micrograph from each well taken at 72 h incubation time. (B) PBMCs were incubated with increasing doses of pSV-GFP up to 50 IU/cell. The micrographs were taken at 72 h incubation time. (C) PBMCs were incubated with 1 PFU/cell of Baco-1 or FusOn-H2. The micrographs were taken at 24 h incubation time.
To determine the detection sensitivity of CTC-UniPro, we spiked 5 million human PBMCs with different numbers of tumor cells, starting from 10 to up to 1,000. We then incubated the cell mixtures with 2 IU/cell of pSV-GFP. GFP-positive cells were counted at 72 h of incubation time. We were able to readily detect the limited number of the spiked tumor cells (10) from 5 million PBMCs by visualization in the case of pSV-GFP, as shown in Figure 4A. This detection sensitivity is similar to other CTC detection methods such as CELLSEARCH that can detect one CTC per 1 mL of a blood sample (equivalent to ∼1 CTC in ∼7 million PBMCs),17,18 Herringbone Chip (detecting up to 12 CTCs/mL of blood),19 and nanotube-CTC-chip (capable of capturing a single cell when it was spiked to 10 μL of mouse blood).20
Figure 4.

CTC-UniPro showed a high detection sensitivity, reaching a single-cell level
(A) 5 × 106 PBMCs were either seeded alone or mixed with 10 tumor cells (A549 and H1944, lung cancer; MDA-MB-231, breast cancer; Mpanc-96, pancreatic cancer) in the presence of 2 IU/cell of pSV-GFP. The micrographs were taken at 72 incubation time. The spiked tumor cells were readily detectable, as indicated by one of the typical fields where a tumor was identified. (B) 5 × 106 PBMCs were either seeded alone or mixed with 10 tumor cells. The cells were cultured either with medium only or in the presence of 2 IU/cell of pSV-Luc. The supernatants were collected at 72 h incubation time and were used for luciferase assay. Shown are either the total luciferase unit (PBMC with medium, PBMC with pSV-Luc) or the luciferase unit per cell by dividing the total reading with the number of cells spiked.
Next, we did the same tumor cell spiking as in Figure 4A. The cell mixtures were then incubated with pSV-Luc at 2 IU/cell. The supernatants were collected 72 h later, and the luciferase activity was determined via luminescence reading by a luminometer. The luciferase activity was converted to a per cell basis by dividing the total with the number of cells spiked. The result in Figure 4B showed that the luciferase activity per cell was well over the background, indicating that CTC-UniPro has the detection sensitivity to detect a single CTC from 5 million PBMCs.
We then moved on to test pSV-GFP on tumor cells of different tissue origins, including prostate cancer, colon cancer, ovarian cancer, head and neck cancer, skin cancer, kidney cancer, bladder cancer, and cervical cancer. The tests showed that the result presented in Figure 4A was reproducible in all the tumor cells that had been tested. Together, the data demonstrate that CTC-UniPro is a highly sensitive and specific detection method that can be used for the detection of CTCs of many different carcinomas.
The detection efficiency of CTC-UniPro is not impacted by the level of EpCAM expression on tumor cells
Many of the current methods rely on EpCAM expression on CTCs as a key biomarker for detection. It is known that the level of EpCAM expression on CTCs varies widely, and it is believed that EMT plays a major role in this fluctuation.21, 22, 23 As such, it has been reported that CTCs can escape EpCAM-based detection because of EMT.2,23 To evaluate the impact of EpCAM expression on the detection efficiency of pSV-GFP, we initially treated A549 cells with transforming growth factor β (TGF-β), which is a well-known factor that can efficiently induce EMT when added to cell culture.24 The results in Figure 5A showed that EpCAM was readily detectable in the untreated A549 cells, and TGF-β treatment led to a significant reduction of its expression. However, the EpCAM reduction induced by TGF-β had little impact on the probe signal (the intensity of GFP expression) of the pSV-GFP. Rather, the signal was slightly stronger in the TGF-β-treated cells. Part of the reason for the increased GFP signal is probably a higher copy number of the probe in the cells undergoing TGF-β-induced EMT, as it has been reported that SV40 can replicate profoundly in many mesenchymal cells.25 Regardless, these results suggest that, unlike those methods that heavily rely on the level of EpCAM detection on the surface of CTCs, CTC-UniPro is independent of this epithelial adhesion molecule. Thus, its detection is less likely to be influenced by the fluctuation of EpCAM expression on CTCs.
Figure 5.

The detection efficiency of CTC-UniPro is not affected by the EMT-induced downregulation of EpCAM
A549 cells were cultured in medium only (No treatment) or with TGF-β1 at 5 ng/mL for 4 days to induce EMT. (A) Flow cytometry detection of EpCAM expression of A549 cells with or without TGF-β1 treatment. (B) Comparison of GFP probe detection efficiency of A549 cells with or without TGF-β1 treatment.
pSV-Tag-MP probe allows live CTCs to be readily retrieved for further analysis
We constructed a pSV-Tag-MP probe, with high efficiency and specificity, for the purpose of capturing live CTCs from whole blood for further analyses such as continuous culture for drug sensitivity testing, etc. The key component of this probe is a chimeric artificial membrane protein (Tag-MP) that is composed of, in sequential order, a signal peptide (SP), a HA tag (HA), GFP, and the transmembrane domain (TMD) (Figure 6A). When the probe has entered CTCs, the Tag-MP will be expressed on the cell surface with the HA tag extruding out of the membrane, allowing easy and specific harvest with anti-HA IgG-conjugated magnetic particles (Figure 6B). Indeed, when tumor cells were incubated with the pSV-Tag-MP probe, the expressed GFP showed predominantly as the membrane form, which was in contrast to the unmodified GFP that showed an even distribution across the nucleus and the cytoplasm (Figure 6C). These cells could be efficiently captured with the anti-HA IgG-conjugated magnetic particles, whereas none of the same tumor cells incubated with pSV-GFP could be pulled down with the same procedure (Figure 6D). These results show that pSV-Tag-MP represents another versatile format of CTC-UniPro that can be used in applications such as harvesting CTCs with high efficiency and specificity.
Figure 6.

pSV-Gap-MP probe allows convenient harvest of CTCs with the same high efficiency and specificity
(A) The design of pSV-Gap-MP. The key to this probe is the modified membrane form of GFP that is tagged with the HA tag (HA), which allows the CTCs to be easily pulled down by anti-HA IgG-conjugated magnetic nanoparticles. (B) Schematic illustration of pSV-Tag-MP-enabled pull-down of CTCs, by expressing the artificial chimeric protein (Tag-MP) on the surface of CTCs. (C) Differential distribution pattern of regular GFP and Tag-MP after expression in the human ovarian cancer cells (Hey). The top images show a micrographic field, and the bottom images show a magnified cell image from each of them. The regular GFP showed even or slightly higher nuclear distribution. In contrast, Tag-MP localizes predominantly on the cell membrane. (D) Pull-down of the spiked tumor cells incubated with pSV-Tag-MP but not pSV-GFP. The pulled-down tumor cells are indicated by the red arrows on the images at the top. The bottom image shows one of the pull-down cells 24 h later in higher magnification.
Discussion
Because of its non-invasive, easy access and real-time monitoring functionalities, liquid biopsy can potentially become an attractive alternative to the traditional tissue biopsy in many clinical applications in cancer management. Liquid biopsy mainly detects either CTCs or circulating tumor-derived factors such as cell-free tumor DNA (ctDNA). Despite its many potential advantages, liquid biopsy has not yet become a standard clinical diagnostic method. There are several factors likely contributing to this. First, although many improvements on liquid biopsy techniques have been reported in recent years, the current methods still suffer from imperfect detection sensitivity and specificity and variability. Second, there is a lack of sufficient clinical validity and utility. In particular, a solid demonstration of its clinical correlation has not yet been unambiguously established.
The current CTC detection methods are mainly based on two principles, either immuno-based detection or biophysical property-based separation. Despite the recently reported improvements, there is still a need to further improve their detection specificity and sensitivity because of the scarcity of CTCs in the blood. However, two additional drawbacks of the current methods likely deserve more attention. First, none of the current methods has the capability to detect live CTCs exclusively and unambiguously. Possessing such a capability may be required for certain clinical applications of CTC detection, such as real-time monitoring of therapeutic efficacy during cancer treatment. Second, the immuno-based CTC detection methods are prone to the heterogeneous nature of EpCAM expression on CTCs. As such, a particular concern is that these methods may not be able to accurately detect the CTCs that have undergone EMT, which frequently results in downregulation of EpCAM expression. This intrinsic defect may compromise another important clinical application of CTC monitoring—prognosis (e.g., cancer metastasis and disease relapse)—both of which are intimately linked to EMT.26, 27, 28
The design of CTC-UniPro is unique compared to other reported CTC detection methods. It is based on a chimeric virus that incorporates the high selectivity of HPV infection for epithelial cells and efficient amplification of the SV40 plasmid genome inside CTCs. This combination results in CTC detection with high specificity and sensitivity. Most importantly, CTC-UniPro is designed for detecting live CTCs only, as the marker genes (GFP, luciferase, or the artificially assembled Tag-MP) can only be produced in a live CTC. We believe that the ability of CTC-UniPro to detect live CTCs exclusively may be particularly useful for real-time monitoring of the therapeutic effect of many cancer treatments, as the viability of CTCs is vital for accurately predicting the therapy effectiveness. This would allow oncologists to detect resistance to the chosen therapy at an early stage, so that more effective treatment could be offered before the tumor burden becomes excessive and incurable. Additionally, the patients would not need to suffer additional unwanted side effects caused by the ineffective therapy. In a recent clinical study, one of the current CTC detection methods, the FDA-approved CELLSEARCH, was found to be unable to demonstrate a clinical utility, as it failed to accurately predict late-line chemotherapy for metastatic breast cancer.29 We believe that the inability of CELLSEARCH to rigorously detect live CTCs might have contributed to this failure. We are planning to conduct a clinical trial in the near future to test CTC-UniPro for its ability to accurately predict the effectiveness of the chosen treatments for some common cancer types such as lung and breast cancer.
Our data also showed that, unlike the immuno-based detection methods, the detection sensitivity of CTC-UniPro is not affected by the reduced EpCAM expression derived from EMT. This property probably will provide CTC-UniPro with an additional advantage over other methods in clinical applications such as predicting prognosis and disease relapse. Mechanism-wise, although HPV is known for its strong tropism for epithelial cells, its entry does not depend on a single epithelial marker. Rather, it likely involves a combination of several epithelial receptors.30 This probably provides a unique advantage for this chimeric virus-based probe, allowing it to enter CTCs (in the context of blood cells) with strong specificity without being impacted by the downregulation of a single signature epithelial marker.
Materials and methods
Cell lines and viruses
Lung cancer cell lines A549, H1944, 5838, and H358; ovarian cancer cell line Hey; and African green monkey kidney (Vero) cells were obtained from American Type Culture Collection (Rockville, MD, USA). The 293TT cell line was obtained from the National Cancer Institute. Cells were cultured in DMEM containing 10% fetal bovine serum (FBS). 293TT cells were cultured in DMEM (GE Healthcare Life Sciences HyClone Laboratories, South Logan, UT, USA) supplemented with 10% FBS (Mediatech, Manassas, VA, USA), 100 units/mL penicillin and 100 μg/mL streptomycin, 1% nonessential amino acids, and 1% GlutaMAX (Thermo Fisher Scientific, Waltham, MA, USA). All cells were incubated at 37°C in a humidified atmosphere with 5% CO2. Type I HSV (HSV-1)-based oncolytic virus Baco-1 and HSV-2 based FusOn-H2 were originally constructed in our own laboratory.31,32 Both viruses contain the GFP gene, and thus their infection could be readily identifiable by the appearance of green fluorescent cells or plaques. They were grown and titrated on Vero cells as described previously.31,32
Preparation and purification of CTC-UniPro and the control vector
The details of the construction strategy for CTC-UniPro are illustrated in Figure 1. CTC-UniPro contains two key components: the inner probe-containing component (pSV-GFP, pSV-Luc, or pSV-Tag-MP) and an outside component (pHPV1/2, for packaging the inner component). The inner component was designed and based on the wild-type (WT) SV40 genome with several modifications. Initially, the entire VP 1-3 capsid genes from nucleotides (nt) 371 to 2532 of the SV40 genome were deleted and replaced with a marker gene (GFP, luciferase, or Tag-MP). To maximize the marker gene expression, we introduced the following modifications to the SV 40 later promoter region to increase the promoter strength.12, 13, 14 These include a series of substitutions (298C>T, 299C>T, and 304C>T) and the addition of 4 extra copies of the 72-bp repeat (Figure 1B). We also made an additional substitution (335A>C) at the downstream region of the late promoter to destroy the start codon in that location. This would allow the marker gene to be efficiently expressed without interference from this endogenous start codon.
The packaging construct, pHPVL1/2, was constructed by cloning both the L1 and L2 genes from HPV-16 by separating them with a 2A sequence (T2A). The above-mentioned modifications were done through DNA synthesis with codon optimization for human expression, and the synthesis was done by GenScript (Piscataway, NJ, USA). The control vector was derived from pcDNA-GFP. It contains a copy of the SV40 Ori-P sequence as well as a GFP cassette in which the marker gene was driven by the CMV promoter.
For the production of CTC-UniPro and the control vector, 293TT cells were plated in a 10- or 15-cm dish 16 h prior to transfection. Plasmid pHPVL1/2 was mixed with one of the probes (pSV-GFP, pSV-Luc, and pSV-Tag-MP) or the pcDNA-GFP control plasmid, and the mixtures were then co-transfected into the 293TT cells with Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA). From these co-transfections, the L1 and L2 expressed from pHPV1/2 would form VLPs, which could then package the probe plasmid or the control plasmid (pcDNA-GFP). The transfected cells were harvested at 48 or 72 h after transfection. This prolonged time is needed for the probe to amplify to enough copies so that the GFP signal can be readily detectable. Cell pellets were washed once with phosphate-buffered saline (PBS) before they were suspended and lysed with the lysis buffer: PBS supplemented with 9.5 mM MgCl2 (Sigma, St. Louis, MO, USA), 0.5% Triton 100 (Thermo Fisher Scientific, Waltham, MA, USA), 0.1% Benzonase (Sigma), and 25 mM (NH4)2SO4 (pH 9). The lysates were incubated for 24 h at 37°C to allow the VLPs to mature. The lysates were briefly chilled on ice before they were centrifuged at 5,000 × g for 5 min at 4°C. The supernatant was transferred to a fresh siliconized tube. The pellet was re-extracted with two volumes of DPBS and centrifuged for 5 min at 5,000 × g. These clarified probes were then combined, passed through a 5-μm filter, and further purified with GE Capto Core 700 and DP-10 columns (GE Healthcare Bio-Sciences, Pittsburgh, PA, USA) according to the manufacturer’s protocols. The purified probes were quantitated by both flow cytometry (BD Biosciences, San Jose, CA, USA) for GFP expression (for pSV-GFP and pSV-Tag-MP) and quantitative real-time polymerase chain reaction.
Human PBMC preparation
Human PBMCs were prepared from buffy coats obtained from Gulf Coast Regional Blood Center (Houston, TX, USA). The buffy coats were mixed with an equal amount of PBS before they were loaded on Lymphoprep (STEMCELL Technologies, Vancouver, BC, Canada) for centrifugation for 30 min at 800 × g with the brake off at room temperature. PBMCs were collected from the layer at the plasma/Lymphoprep interface and were washed twice with 2% FBS-PBS. The cell pellets were treated with red blood cell lysing buffer (Sigma) to remove red blood cells and were washed again twice with 2% FBS-PBS. The purified PBMCs were used directly for the experiments.
Tumor cell detection by pSV-GFP probe
For testing the ability of pSV-GFP to amplify in tumor cells, 1–2 × 105 tumor cells of different tissue origins were plated in 12- or 24-well plates overnight. Cells were then incubated with either pSV-GFP or the control vector at 2 IU per cell for 24–96 h. Cells were then analyzed with the Nikon Ti-U Inverted Fluorescence Microscope.
To mimic the situation of clinical diagnosis, 10 or 100 cancer cells were mixed with 5 × 106 PBMCs in RPMI 1640 plus 10% FBS at 12-well plates. The mixed cells or PBMCs alone were incubated with 2 × 105 IU of CTC-UniPro at 37°C for 72 h. For testing the ability of oncolytic HSV to infect PBMCs, cells were infected with either Baco-1 or FusOn-H2 at 1 PFU for 72 h.
To quantitatively measure the probe (GFP) intensity in cancer cells, the indicated cells were incubated with either pSV-GFP or the control vector at 2 IU. Cells were harvested 72 h later. The quantitative measurement of GFP was performed with the GFP Quantification Kit (BioVision, Milpitas, CA, USA). Briefly, the harvested cells were lysed with assay buffer for 10 min. The supernatants were cleared by centrifugation before they were transferred to a 96-well plate. The samples were quantitated on a fluorescence microplate reader (Victor X4, PerkinElmer, Akron, OH, USA).
Tumor cell detection by pSV-Luc probe
The initial incubation step for detection with the pSV-Luc probe was the same as for pSV-GFP. After 24- to 96-h incubation, the medium was removed. 100 μL of Bright-Glo Assay Reagent (Promega, Madison, WI, USA) was added to each well and incubated for 5 min at room temperature to lyse the cells. Afterward, luciferase activity was measured with the SpectraMax 5 plate reader (Molecular Devices, San Jose, CA, USA).
Tumor cell pull-down using the pSV-Tag-MP probe
For checking the expression and location of pSV-Tag-MP in tumor cells, 2 × 105 Hey cells were transfected with either pSV-GFP or pSV-Tag-MP in 12-well plates. The cells were imaged 48 h after transfection under the Nikon Ti-U Inverted Fluorescence Microscope.
For the cell pull-down experiment, 5 × 106 PBMCs were spiked with 100 Hey cells that had been incubated with 2 IU of either pSV-GFP or pSV-Tag-MP for 72 h. Cells were incubated in 2-mL tubes with 50 μL of Pierce Anti-HA Magnetic Beads (Thermo Fisher Scientific, Waltham, MA, USA) in 1 mL of 0.1% BSA-PBS by rotation for 30 min at 4°C. Afterward, the tubes were loaded to the EasySep Magnet (STEMCELL Technologies, Cambridge, MA, USA) for 1 min to allow the tagged tumor cells to attach to the magnetic side of the tube. PBS with the unbound cells was discarded. After two washes with 1 mL of 0.1% BSA-PBS, the beads with bound cells were suspended in 100 μL of DMEM containing 10% FBS and seeded in a 96-well plate. The pull-down cells were analyzed under a Nikon Ti-U Inverted Fluorescence Microscope.
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
The data and code that support the findings of this study are contained in the supplementary files or available from the authors upon request.
Acknowledgments
This work was supported in part by National Cancer Institute Grant R01CA203852, CPRIT Grant RP200464, and a grant from the William and Ella Owens Medical Research Foundation (to X.Z.). We thank Dr. Janet S. Butel (Baylor College of Medicine) for discussion and advice and Dr. Tho Tran for the careful reading of the manuscript.
Author contributions
X.F. and X.Z. conceived and designed the experiments. X.F. and L.T. executed all the experiments in the study. X.F. and X.Z. wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
References
- 1.Riethdorf S., Fritsche H., Müller V., Rau T., Schindlbeck C., Rack B., Janni W., Coith C., Beck K., Jänicke F. Detection of circulating tumor cells in peripheral blood of patients with metastatic breast cancer: a validation study of the CellSearch system. Clin. Cancer Res. 2007;13:920–928. doi: 10.1158/1078-0432.CCR-06-1695. [DOI] [PubMed] [Google Scholar]
- 2.Gorges T.M., Tinhofer I., Drosch M., Röse L., Zollner T.M., Krahn T., von Ahsen O. Circulating tumour cells escape from EpCAM-based detection due to epithelial-to-mesenchymal transition. BMC Cancer. 2012;12:178. doi: 10.1186/1471-2407-12-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lin H.K., Zheng S., Williams A.J., Balic M., Groshen S., Scher H.I., Fleisher M., Stadler W., Datar R.H., Tai Y.C., Cote R.J. Portable filter-based microdevice for detection and characterization of circulating tumor cells. Clin. Cancer Res. 2010;16:5011–5018. doi: 10.1158/1078-0432.CCR-10-1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vona G., Sabile A., Louha M., Sitruk V., Romana S., Schütze K., Capron F., Franco D., Pazzagli M., Vekemans M. Isolation by size of epithelial tumor cells : a new method for the immunomorphological and molecular characterization of circulating tumor cells. Am. J. Pathol. 2000;156:57–63. doi: 10.1016/S0002-9440(10)64706-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hvichia G.E., Parveen Z., Wagner C., Janning M., Quidde J., Stein A., Müller V., Loges S., Neves R.P., Stoecklein N.H. A novel microfluidic platform for size and deformability based separation and the subsequent molecular characterization of viable circulating tumor cells. Int. J. Cancer. 2016;138:2894–2904. doi: 10.1002/ijc.30007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosenberg R., Gertler R., Friederichs J., Fuehrer K., Dahm M., Phelps R., Thorban S., Nekarda H., Siewert J.R. Comparison of two density gradient centrifugation systems for the enrichment of disseminated tumor cells in blood. Cytometry. 2002;49:150–158. doi: 10.1002/cyto.10161. [DOI] [PubMed] [Google Scholar]
- 7.Shishido S.N., Carlsson A., Nieva J., Bethel K., Hicks J.B., Bazhenova L., Kuhn P. Circulating tumor cells as a response monitor in stage IV non-small cell lung cancer. J. Transl. Med. 2019;17:294. doi: 10.1186/s12967-019-2035-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.zur Hausen H. Papillomaviruses causing cancer: evasion from host-cell control in early events in carcinogenesis. J. Natl. Cancer Inst. 2000;92:690–698. doi: 10.1093/jnci/92.9.690. [DOI] [PubMed] [Google Scholar]
- 9.Aksoy P., Gottschalk E.Y., Meneses P.I. HPV entry into cells. Mutat. Res. Rev. Mutat. Res. 2017;772:13–22. doi: 10.1016/j.mrrev.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kadaja M., Silla T., Ustav E., Ustav M. Papillomavirus DNA replication - from initiation to genomic instability. Virology. 2009;384:360–368. doi: 10.1016/j.virol.2008.11.032. [DOI] [PubMed] [Google Scholar]
- 11.Gluzman Y. SV40-transformed simian cells support the replication of early SV40 mutants. Cell. 1981;23:175–182. doi: 10.1016/0092-8674(81)90282-8. [DOI] [PubMed] [Google Scholar]
- 12.Brady J., Radonovich M., Thoren M., Das G., Salzman N.P. Simian virus 40 major late promoter: an upstream DNA sequence required for efficient in vitro transcription. Mol. Cell. Biol. 1984;4:133–141. doi: 10.1128/mcb.4.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wiley S.R., Kraus R.J., Zuo F., Murray E.E., Loritz K., Mertz J.E. SV40 early-to-late switch involves titration of cellular transcriptional repressors. Genes Dev. 1993;7:2206–2219. doi: 10.1101/gad.7.11.2206. [DOI] [PubMed] [Google Scholar]
- 14.Lednicky J.A., Butel J.S. Simian virus 40 regulatory region structural diversity and the association of viral archetypal regulatory regions with human brain tumors. Semin. Cancer Biol. 2001;11:39–47. doi: 10.1006/scbi.2000.0345. [DOI] [PubMed] [Google Scholar]
- 15.Buck C.B., Pastrana D.V., Lowy D.R., Schiller J.T. Efficient intracellular assembly of papillomaviral vectors. J. Virol. 2004;78:751–757. doi: 10.1128/JVI.78.2.751-757.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang W., Bao L., Yang S., Qian Z., Dong M., Yin L., Zhao Q., Ge K., Deng Z., Zhang J. Tumor-selective replication herpes simplex virus-based technology significantly improves clinical detection and prognostication of viable circulating tumor cells. Oncotarget. 2016;7:39768–39783. doi: 10.18632/oncotarget.9465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Swennenhuis J.F., van Dalum G., Zeune L.L., Terstappen L.W. Improving the CellSearch® system. Expert Rev. Mol. Diagn. 2016;16:1291–1305. doi: 10.1080/14737159.2016.1255144. [DOI] [PubMed] [Google Scholar]
- 18.Wang L., Balasubramanian P., Chen A.P., Kummar S., Evrard Y.A., Kinders R.J. Promise and limits of the CellSearch platform for evaluating pharmacodynamics in circulating tumor cells. Semin. Oncol. 2016;43:464–475. doi: 10.1053/j.seminoncol.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stott S.L., Hsu C.H., Tsukrov D.I., Yu M., Miyamoto D.T., Waltman B.A., Rothenberg S.M., Shah A.M., Smas M.E., Korir G.K. Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proc. Natl. Acad. Sci. USA. 2010;107:18392–18397. doi: 10.1073/pnas.1012539107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loeian M.S., Mehdi Aghaei S., Farhadi F., Rai V., Yang H.W., Johnson M.D., Aqil F., Mandadi M., Rai S.N., Panchapakesan B. Liquid biopsy using the nanotube-CTC-chip: capture of invasive CTCs with high purity using preferential adherence in breast cancer patients. Lab Chip. 2019;19:1899–1915. doi: 10.1039/c9lc00274j. [DOI] [PubMed] [Google Scholar]
- 21.Yu M., Bardia A., Wittner B.S., Stott S.L., Smas M.E., Ting D.T., Isakoff S.J., Ciciliano J.C., Wells M.N., Shah A.M. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science. 2013;339:580–584. doi: 10.1126/science.1228522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Armstrong A.J., Marengo M.S., Oltean S., Kemeny G., Bitting R.L., Turnbull J.D., Herold C.I., Marcom P.K., George D.J., Garcia-Blanco M.A. Circulating tumor cells from patients with advanced prostate and breast cancer display both epithelial and mesenchymal markers. Mol. Cancer Res. 2011;9:997–1007. doi: 10.1158/1541-7786.MCR-10-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hyun K.-A., Koo G.-B., Han H., Sohn J., Choi W., Kim S.-I., Jung H.-I., Kim Y.-S. Epithelial-to-mesenchymal transition leads to loss of EpCAM and different physical properties in circulating tumor cells from metastatic breast cancer. Oncotarget. 2016;7:24677–24687. doi: 10.18632/oncotarget.8250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu J., Lamouille S., Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009;19:156–172. doi: 10.1038/cr.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daya-Grosjean L., Azzarone B., Maunoury R., Zaech P., Elia G., Zaniratti S., Benedetto A. SV40 immortalization of adult human mesenchymal cells from neuroretina. Biological, functional and molecular characterization. Int. J. Cancer. 1984;33:319–329. doi: 10.1002/ijc.2910330308. [DOI] [PubMed] [Google Scholar]
- 26.Tiwari N., Gheldof A., Tatari M., Christofori G. EMT as the ultimate survival mechanism of cancer cells. Semin. Cancer Biol. 2012;22:194–207. doi: 10.1016/j.semcancer.2012.02.013. [DOI] [PubMed] [Google Scholar]
- 27.Shao B., Bjaanæs M.M., Helland Å., Schütte C., Conrad T. EMT network-based feature selection improves prognosis prediction in lung adenocarcinoma. PLoS ONE. 2019;14:e0204186. doi: 10.1371/journal.pone.0204186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mitra A., Mishra L., Li S. EMT, CTCs and CSCs in tumor relapse and drug-resistance. Oncotarget. 2015;6:10697–10711. doi: 10.18632/oncotarget.4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cabel L., Berger F., Cottu P., Loirat D., Rampanou A., Brain E., Cyrille S., Bourgeois H., Kiavue N., Deluche E. Clinical utility of circulating tumour cell-based monitoring of late-line chemotherapy for metastatic breast cancer: the randomised CirCe01 trial. Br. J. Cancer. 2021;124:1207–1213. doi: 10.1038/s41416-020-01227-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raff A.B., Woodham A.W., Raff L.M., Skeate J.G., Yan L., Da Silva D.M., Schelhaas M., Kast W.M. The evolving field of human papillomavirus receptor research: a review of binding and entry. J. Virol. 2013;87:6062–6072. doi: 10.1128/JVI.00330-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fu X., Tao L., Cai R., Prigge J., Zhang X. A mutant type 2 herpes simplex virus deleted for the protein kinase domain of the ICP10 gene is a potent oncolytic virus. Mol. Ther. 2006;13:882–890. doi: 10.1016/j.ymthe.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 32.Fu X., Tao L., Jin A., Vile R., Brenner M., Zhang X. Expression of a fusogenic membrane glycoprotein by an oncolytic herpes simplex virus potentiates the viral antitumor effect. Mol. Ther. 2003;7:748–754. doi: 10.1016/s1525-0016(03)00092-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data and code that support the findings of this study are contained in the supplementary files or available from the authors upon request.