

Abstract
Cyclotriazadisulfonamide (CADA) inhibits the cotranslational translocation of type I integral membrane protein human CD4 (huCD4) across the endoplasmic reticulum in a signal peptide (SP)–dependent way. Previously, sortilin was identified as a secondary substrate for CADA but showed reduced CADA sensitivity as compared with huCD4. Here, we performed a quantitative proteomic study on the crude membrane fraction of human T-cells to analyze how many proteins are sensitive to CADA. To screen for these proteins, we employed stable isotope labeling by amino acids in cell culture technique in combination with quantitative MS on CADA-treated human T-lymphoid SUP-T1 cells expressing high levels of huCD4. In line with our previous reports, our current proteomic analysis (data available via ProteomeXchange with identifier PXD027712) demonstrated that only a very small subset of proteins is depleted by CADA. Our data also confirmed that cellular expression of both huCD4 and sortilin are affected by CADA treatment of SUP-T1 cells. Furthermore, three additional targets for CADA are identified, namely, endoplasmic reticulum lectin 1 (ERLEC1), inactive tyrosine-protein kinase 7 (PTK7), and DnaJ homolog subfamily C member 3 (DNAJC3). Western blot and flow cytometry analysis of ERLEC1, PTK7, and DNAJC3 protein expression validated susceptibility of these substrates to CADA, although with varying degrees of sensitivity. Additional cell-free in vitro translation/translocation data demonstrated that the new substrates for CADA carry cleavable SPs that are targets for the cotranslational translocation inhibition exerted by CADA. Thus, our quantitative proteomic analysis demonstrates that ERLEC1, PTK7, and DNAJC3 are validated additional substrates of CADA; however, huCD4 remains the most sensitive integral membrane protein for the endoplasmic reticulum translocation inhibitor CADA. Furthermore, to our knowledge, CADA is the first compound that specifically interferes with only a very small subset of SPs and does not affect signal anchor sequences.
Keywords: CD4, cyclotriazadisulfonamide, ER cotranslational translocation, signal peptide, SILAC, small-molecule inhibitor
Abbreviations: BFP, blue fluorescent protein; CADA, cyclotriazadisulfonamide; DMSO, dimethyl sulfoxide; DNAJC3, DnaJ homolog subfamily C member 3; eGFP, enhanced GFP; ER, endoplasmic reticulum; ERAD, endoplasmic reticulum–associated degradation; ERLEC1, endoplasmic reticulum lectin 1; HEK293T, human embryonic kidney 293T; huCD4, human CD4; PL, prolactin; PTK7, inactive tyrosine-protein kinase 7; SAS, signal anchor sequence; SILAC, stable isotope labeling by amino acids in cell culture; SORT, sortilin; SP, signal peptide; TMD, transmembrane domain; tGFP, turbo GFP; V5, virus 5
Graphical Abstract
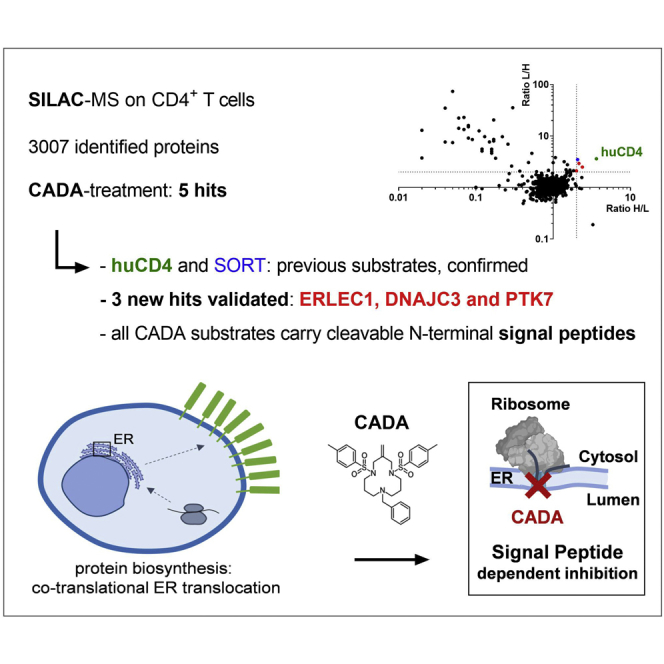
Highlights
-
•
About 3007 proteins quantified in SILAC/MS study on CD4+ T-cells treated with CADA.
-
•
Three new targets for CADA were identified: ERLEC1, PTK7, and DNAJC3.
-
•
All CADA substrates carry cleavable signal peptides for translocation into ER.
-
•
huCD4 remains the most sensitive substrate for the ER translocation inhibitor CADA.
In Brief
In mammalian cells, one-third of all polypeptides enter the secretory pathway via the ER, driven by specific targeting signals in their sequences, such as N-terminal signal peptides. The synthetic macrocycle CADA was identified as a selective ER translocation inhibitor for huCD4 and SORT. Our current SILAC/MS proteomic survey identified and validated only three additional targets for CADA, thus, confirming the high substrate selectivity of CADA, with the strongest effect on huCD4. Therefore, CADA holds great potential as an immunosuppressive drug.
In mammalian cells, one-third of all polypeptides enter the secretory pathway in order to reach their target destination, that is, the extracellular environment or plasma membrane (1, 2). Driven by specific targeting signals in their sequences, either transmembrane helices or cleavable amino-terminal signal peptides (SPs), these proteins enter the secretory pathway in order to reach their final destination (3, 4, 5, 6, 7, 8). The central component of the secretory pathway is the heterotrimeric Sec61 translocon composed of Sec61 α, β, and γ monomers that forms an aqueous polypeptide-conducting channel in the endoplasmic reticulum (ER) membrane (9, 10, 11). Cotranslational translocation, the most common form of translocation seen in higher eukaryotes, couples ribosomal protein synthesis directly to protein translocation through the Sec61 translocon (4, 12, 13). In this multistep process, the signal recognition particle recognizes the SP or transmembrane helices of nascent precursor polypeptides emerging from the translating ribosomes in the cytosol (14, 15, 16, 17). Subsequently, the ribosome-nascent chain complex is targeted to the ER membrane through binding of signal recognition particle to its receptor (18). The precursor polypeptides are then inserted into the Sec61 translocon in order to translocate the preprotein from the cytosol to the ER lumen. A lateral gate in the Sec61 translocon facilitates the integration of hydrophobic transmembrane protein segments into the lipid bilayer for the accommodation of integral membrane proteins. Finally, the signal peptidase complex and oligosaccharyl transferase allow further maturation of the translocated preprotein in the ER lumen, such as SP cleavage and glycosylation, respectively (12, 13, 19, 20, 21).
SPs are short hydrophobic sequences located at the N-terminal end of secretory or type I integral membrane proteins (3, 4). In general, SPs are highly variable in sequence and primary structure and are unique for each protein but share some common features. SPs have a typical length of 15 to 30 residues and comprise a central hydrophobic core (h-region) flanked by a positively charged “N-region” and a polar “C-region” that contains the cleavage site for the signal peptidase (5, 22). As SPs are the targeting and translocation signals for proteins to enter the secretory pathway, we can consider SPs as potential drug targets for pharmaceutical intervention of protein expression (23). It is therefore no surprise that research has been dedicated to identify inhibitors of cotranslational translocation over the ER that block the biogenesis of secretory and integral membrane proteins (24, 25).
Cotransins, a class of cyclic peptides derived from the fungal substance HUN-7293 (26, 27), have been described as compounds that potently inhibit cotranslational translocation of a number of secretory and type I transmembrane proteins in a SP-discriminatory manner by directly interacting with Sec61α (28, 29, 30, 31, 32). The cotransin variant CT08 has been reported not only to be substrate selective but also to prevent the membrane integration of a single-pass type II membrane protein with a noncleavable signal anchor sequence (SAS) (33). Other known examples of compounds that interact with the Sec61 translocon but in a substrate nonselective manner are mycolactone (34, 35), aprotoxin A (36, 37), decatransin (38), coibamide A (39, 40), and ipomoeassin F (41). In contrast, the synthetic macrocycle cyclotriazadisulfonamide (CADA; Fig. 1A) is a small-molecule ER translocation inhibitor that acts in a specific SP-dependent manner (42). Previous work from our laboratory has shown that CADA directly interacts with the SP of the human CD4 (huCD4) preprotein, preventing the receptor's cotranslational translocation process, ultimately resulting in reduced cell surface huCD4 expression levels. Moreover, specific glutamine and proline residues in the huCD4 SP are critical for full susceptibility to CADA (43).
Fig. 1.

Strategy of the SILAC study.A, chemical structure of cyclotriazadisulfonamide (CADA). B, schematic representation of the SILAC approach. Proteins of SUP-T1 cells were labeled with either 12C6l-lysine and 12C614N4l-arginine (“light” sample) or 13C6l-lysine and 13C615N4l-arginine (“heavy” sample). Cells were treated for 17 h with CADA (10 μM) or DMSO control, and pooled cell lysates were separated using an SDS gradient gel (4–12%). The protein bands were cut, proteins were in-gel digested using trypsin, extracted, and finally analyzed using LC–MS/MS. The forward experiment is shown. For the reverse experiment, isotopic coding was inverted. C, SILAC results for integral membrane proteins, which were identified and quantified by LC–MS/MS analysis (see also supplemental Table S1 for a list of the individual proteins). The ratios of protein expression following DMSO or CADA treatment of the forward (heavy/light sample = H/L) and reverse experiment (light/heavy sample = L/H) are plotted against each other. Proteins were considered CADA sensitive if the ratio was determined to be >2.0 (indicated by the dotted line) in both experiments, thus, located in the upper right quadrant of the plot. HuCD4 is marked in green, whereas the four other CADA-sensitive proteins are marked as red dots. DMSO, dimethyl sulfoxide; SILAC, stable isotope labeling by amino acids in cell culture.
The aim of this work was to investigate whether additional substrates for CADA, besides the earlier reported huCD4 and sortilin (SORT) (44), could be identified. We employed the stable isotope labeling by amino acids in cell culture (SILAC) technique in combination with quantitative MS on CADA-treated human T-lymphoid SUP-T1 cells expressing high levels of CD4. This proteomic screening of the crude membrane fraction of T-cells indicates only three of 3007 quantified proteins as additional substrates for CADA.
Experimental Procedures
Compounds and Antibodies
CADA hydrochloride was a gift from Dr Thomas W. Bell (Reno, Nevada). It was synthesized as described previously (45). CADA was dissolved in dimethyl sulfoxide (DMSO) and stored as a 10 mM stock at room temperature. FITC-labeled or allophycocyanin-labeled anti-huCD4 (clone SK3) (BioLegend; catalog no. 344604 and 344613) and phycoerythrin-labeled antihuman inactive tyrosine-protein kinase 7 (PTK7) (clone 188B) (MACS Miltenyi; catalog no. 130-122-923) monoclonal antibodies were used for flow cytometry. Western blot antibodies were purchased from (i) Genscript: anti-V5 (catalog no. A01724); (ii) BD transduction laboratories: anticlathrin (catalog no. 610500); (iii) Dako: horseradish peroxidase-labeled goat antimouse immunoglobulins (catalog no. P0447); and (iv) Invitrogen: anti-β-actin (catalog no. MA1-140).
Plasmids and Mutagenesis
The endoplasmic reticulum lectin 1 (ERLEC1) expression vector (pGEM-T backbone) was purchased from Sinobiological, the PTK7 expression vector (pDONR223 backbone) from Addgene, and the DNAJ homolog subfamily C member 3 (DNAJC3) expression vector (pDONR223 backbone) from DNASU plasmid repository. The pEGFP-P2A-RFP expression vector (pEGFP-N1 Clontech backbone) was a kind gift from Dr Ramanujan Hegde (46). The sequence for enhanced GFP (eGFP) was replaced by that of turbo GFP (tGFP), and wildtype huCD4 was fused to the N terminus of tGFP. In addition, the sequence for mCherry (red fluorescent protein) was replaced by that of mTag-BFP2 (blue fluorescent protein [BFP]). Constructs for Western blot were designed, which included the simian virus 5 (V5) epitope (GKPIPNPLLGLD) at the C terminus of the protein of interest. Site-directed mutagenesis of all constructs was performed with the QuikChange II kit (Stratagene), Q5 site-directed mutagenesis kit (New England Biolabs), or NEBuilder HiFi DNA assembly kit (New England Biolabs), following the manufacturer's instructions. Plasmid DNA was isolated using the Nucleospin Plasmid Transfection grade system (Machery-Nagel), supplemented with an endotoxin removal wash. The concentration of all constructs was determined with a NanoDrop 1000 spectrophotometer, and sequences were confirmed by automated capillary Sanger sequencing (Macrogen Europe).
Transient Transfection and Flow Cytometry
SUP-T1 cells were seeded at 1.5 × 105 cells/ml in Corning Costar 24-well plates, supplemented with the indicated amount of CADA or 0.1% DMSO as a control, and were incubated for 24 h. Cells were then washed in PBS and stained. Samples were stored in PBS containing 1% formaldehyde before acquisition on either a BD FACS Canto II or a BD FACS Fortessa flow cytometer (Beckton Dickinson) with BD FACSDiva 8.0.1 software. All data were analyzed in FlowJo X (FlowJo Software), version 10.
Human embryonic kidney 293T (HEK293T) cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% (v/v) fetal bovine serum. For transfection, cells were seeded at a cell density of 4 × 105 cells/ml and allowed to adhere overnight. Lipofectamine LTX (Invitrogen) was used for the transfection of plasmid DNA, according to the manufacturer's instructions. CADA and/or MG132 (Sigma) was added 6 h after transfection, and cells were collected for analysis 24 h or 48 h after transfection (as indicated in the legend to the figures).
To compare the downmodulating activity of CADA, IC50 values were calculated with GraphPad Prism 8 software (GraphPad Software, Inc) on a four parameter concentration response curve fitted to data from at least three replicate experiments with flow cytometry. The absolute IC50 value represents the concentration that produces 50% reduction in protein level.
Immunoblotting
To detect PTK7, DNAJC3, and ERLEC1, HEK293T cells were collected 24 h after transfection and lysed in ice-cold Nonidet P-40 buffer (50 mM Tris–HCl, pH 8.0, 150 mM NaCl, and 1% Nonidet P-40) supplemented with protease inhibitor cocktail (Roche) and centrifuged at 17,000g for 10 min at 4 °C to pellet nuclei and debris. The supernatant was then used for further Western blot analysis. Following cell lysis, lysate was digested overnight with endoglycosidase H (Promega) at room temperature. For SDS gel electrophoresis, samples were boiled in reducing sample buffer (120 mM Tris–HCl, pH 6.8, 4% SDS, 20% glycerol, 100 mM dithiothreitol, and 0.02% bromophenol blue). Equal protein amounts were separated on 4 to 12% Criterion XT Bis–Tris gels (Bio-Rad) using 2-(N-morpholino)ethanesulfonic acid buffer (Bio-Rad). After electrophoresis, gels were blotted onto polyvinylidene fluoride membranes using the Trans-Blot Turbo transfer system (Bio-Rad). Membranes were blocked for 1 h with 5% nonfat dried milk in 20 mM Tris–HCl, pH 7.6, 137 mM NaCl, and 0.05% Tween-20. After 1 h incubation of the primary antibodies at room temperature, membranes were washed and incubated with the secondary antibody. SuperSignal West Pico and Femto chemiluminescence reagent (Thermoscientific) was used for detection in conjunction with a ChemiDoc MP system (Bio-Rad), and signal intensities were quantified with Image Lab software, version 5.0 (Bio-Rad). Differences in protein concentration between each lane were compensated by normalization to the clathrin heavy chain or β-actin signal.
Metabolic Labeling
For the pulse labeling of the CADA substrates, CHO-K1 cells were seeded at a cell density of 2 × 105 cells/ml in Ham's F-12 medium (Gibco) supplemented with 10% fetal bovine serum and allowed to adhere overnight. The next day, cells were transfected with plasmid DNA using Lipofectamine LTX (Invitrogen) according to the manufacturer’s protocol and incubated overnight at 37 °C. Cells were pretreated with 10 μM CADA and/or 200 nM MG132 (Sigma) for 1 h prior to a 45-min starvation for methionine and cysteine. Next, cells were pulsed for 30 min with L-35S-methionine/cysteine (PerkinElmer) and chased for 45 min at 37 °C. All steps were performed under constant CADA (10 μM) and MG132 (200 nM) pressure. Chased cells were then washed with ice-cold medium and lysed with NP-40 buffer as described previously. Radiolabeled proteins were pulled down using V5-trap agarose beads (Chromotek) as described in the manufacturer's protocol. The pulled proteins were next separated with SDS-PAGE on 4 to 12% Criterion XT Bis–Tris gel (Bio-Rad) using 2-(N-morpholino)ethanesulfonic acid buffer, detected by phosphor imaging and quantified (Cyclone Plus storage phosphor system; PerkinElmer).
Cell-Free in Vitro Translation and Translocation
The Qiagen EasyXpress linear template kit was used to generate full-length complementary DNAs using PCR. The SP and first part of the mature hit protein was fused upstream of prolactin (PL) with PCR. PCR products were purified and transcribed in vitro using T7 RNA polymerase (RiboMAX system; Promega). All transcripts were translated in rabbit reticulocyte lysate (Promega) in the presence of L-35S-methionine (PerkinElmer). Translations were performed at 30 °C in the presence or the absence of ovine pancreatic microsomes and CADA as described elsewhere (47). Samples were washed with low-salt buffer (80 mM KOAc, 2 mM Mg(OAc)2, 50 mM Hepes, pH 7.6), and radiolabeled proteins were isolated by centrifugation for 10 min at 21,382g and 4 °C (Hettich 200R centrifuge with 2424B rotor). The proteins were then separated with SDS-PAGE and detected by phosphor imaging (Cyclone Plus storage phosphor system; PerkinElmer).
SILAC Methodology
SILAC experiments were carried out according to the supplier's recommendations outlined in the Pierce SILAC protein quantification kits. To obtain 90% labeling, SUP-T1 cells were grown on 60-mm diameter dishes for 2 weeks in Dulbecco's modified Eagle's medium containing either 12C6 l-lysine (1 mM) and 12C6 14N4 l-arginine (0.48 mM) (“light” sample) or 13C6 l-lysine (0.96 mM), and 13C6 15N4 l-arginine (0.45 mM) (“heavy” sample). Cells of the light sample were treated for 17 h with 10 μM of CADA, whereas cells of the heavy sample served as a DMSO-treated control. For the isolation of the total integral membrane proteins, cells were washed twice with cold PBS containing 0.5 mM phenylmethylsulfonyl fluoride (PMSF), 3.2 μg/μl soybean trypsin inhibitor, 0.5 mM benzamidine, 1.4 μg/μl trasylol; pH 7.4. Cells were disrupted in 100 μl cold fractionation buffer (250 mM sucrose, 20 mM 4(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 0.5 mM phenylmethylsulfonyl fluoride, 3.2 μg/μl soybean trypsin inhibitor, 0.5 mM benzamidine, 1.4 μg/μl trasylol; and pH 7.4) using a potter homogenizer. Lysates of the light and heavy samples were combined, and cell nuclei were removed by centrifugation (4 °C, 720g, 5 min). The crude membrane fraction was collected (4 °C, 100,000g, 1 h), resuspended in fractionation buffer, and centrifuged for a second time as described previously. The pellet containing the integral membrane proteins was resuspended in 50 μl Rotiload buffer supplemented with 15 μl Tris–HCl (100 mM, pH 8.5). Proteins were reduced using DTT (5 mM, 30 min, and 55 °C) and alkylated using iodoacetamide (15 mM, 30 min, room temperature) in the dark. Separation of the proteins by SDS-PAGE was carried out as described previously (32).
Protein Identification by MS
Each gel lane was cut by hand into 14 slices, and in-gel tryptic digestion was performed as described (48). Tryptic peptides were analyzed by a reversed-phase capillary LC system (Ultimate 3000 nanoLC system; Thermo Scientific) connected to an Orbitrap Elite mass spectrometer (Thermo Scientific). LC separations were performed on a capillary column (Acclaim PepMap100, C18, 3 μm, 100 Å, 250 mm × 75 μm i.d.; Thermo Scientific) at an eluent flow rate of 300 nl/min using a linear gradient of 3 to 35% B in 60 min. Mobile phase A contained 0.1% formic acid in water, and mobile phase B contained 0.1% formic acid in acetonitrile. Mass spectra were acquired in a data-dependent mode with one MS survey scan with a resolution of 60,000 (Orbitrap Elite) and MS/MS scans of the 15 most intense precursor ions in the linear trap quadrupole.
Identification and quantification of proteins were performed using MaxQuant software (version 1.5.2.8). Data were searched against the Uniprot protein database (July 2015; 131,108 human sequences with 46,761,785 residues). Trypsin cleavage C-terminal region to arginine or lysine residues was used (except where these residues are directly followed by a proline residue). A maximum of two missed cleavages was allowed. The initial maximum mass deviation of the precursor ions was set at 20 ppm, and the maximum mass deviation of the fragment ions was set at 0.35 Da. Methionine oxidation and carbamidomethylation of cysteine were used as variable modifications. Protein quantification was based on calculations of isotope intensity ratios of at least two tryptic peptides. False discovery rates were <1% based on matches to reversed sequences in the concatenated target-decoy database.
Experimental Design and Statistical Rationale
All SILAC experiments were repeated with switched isotopic coding (forward and reverse experiments) and analyzed separately. Proteins were considered if at least two sequenced peptides were identified (at least one unique peptide and at least two razor + unique peptides). The criterion for quantification was a ratio count of at least two. In our experiments, we considered proteins as CADA sensitive if the ratio of protein expression following DMSO or CADA treatment (DMSO/CADA) was higher than 2.0 in both experiments. For the flow cytometry and Western blotting validation experiments, at least three independent experiments were performed using a different passage of cultured cells. For the in vitro translation/translocation experiments, at least two independent experiments were performed using a fresh aliquot of rabbit reticulocyte.
Results
CADA Sensitivity of Integral Membrane Proteins of SUP-T1 Cells
The SILAC approach used is outlined in Figure 1B. For our study, human T-lymphoid SUP-T1 cells were selected because of their high endogenous huCD4 expression, the main target of CADA (42). The IC50 values for CADA-mediated CD4 downmodulation in T-cell lines and huCD4-transfected cells were reported to be in a range of 0.25 to 1 μM (42, 43, 49). Taking these IC50 values into account, we used a CADA concentration of 10 μM for our study, a saturating concentration for huCD4 downmodulation (43). It was previously demonstrated that CADA does not affect transcription (42, 50) and does not cause cytotoxicity in SUP-T1 cells at a concentration of 75 μM (49).
Cells were grown in medium either containing 12C6 l-lysine and 12C6 14N4 l-arginine (“light” sample) or 13C6 l-lysine and 13C6 15N4 l-arginine (“heavy” sample) (Fig. 1B). Cells of the light sample were treated with CADA, whereas cells of the heavy sample served as a DMSO-treated control. For the analysis of the integral membrane, lumenal ER, and Golgi proteins, total cell lysates of the light and heavy samples were mixed, and cell fractionations were performed. Proteins of crude membrane preparations were separated by SDS-PAGE, in-gel digested with trypsin, and the resulting peptides were subjected to LC–MS/MS analysis. The experiment was repeated in a crossover mode, in which cells of the light sample were treated with DMSO and the heavy sample exposed to CADA. In our experiments, we considered proteins as CADA sensitive if the ratio of protein expression following DMSO or CADA treatment (DMSO/CADA) was higher than 2.0 in both experiments. A total of 3007 proteins could be identified and quantified (Fig. 1C and supplemental Table S1), of which 500 contained a SP and/or transmembrane domain (TMD), and are routed to the secretory pathway. As expected, for the T-cell surface glycoprotein, CD4 heavy/light (and light/heavy) isotope ratios >3 were found, indicating that CADA treatment of the cells was successful. Surprisingly, only five of the 3007 identified proteins showed a significant decrease in protein expression following CADA treatment (Table 1). Thus, at this saturating concentration (10 μM), CADA does not affect the expression of many proteins additional to huCD4. Interestingly, the proteomic data also indicated SORT as a substrate for CADA, thus, confirming our previous report (44). The three additional hits for CADA were PTK7, DNAJC3, and ERLEC1.
Table 1.
Potential CADA-affected SILAC targets selected for validation
| UniProt_ID | Protein | Function | Cellular localization | Ratio H/La | Ratio L/Hb |
|---|---|---|---|---|---|
| P01730 | Human T-cell surface glycoprotein CD4 (cluster of differentiation 4) | Major histocompatibility complex class-II antigen/T-cell coreceptor | Cell surface | 3.63 | 3.61 |
| Q13308 | Human PTK7, CCK-4 (colon carcinoma kinase 4) | Inactive tyrosine kinase involved in Wnt signaling pathway, both the noncanonical (Wnt/planar cell polarity signaling) and the canonical pathway | Cell surface | 2.39 | 2.49 |
| Q13217 | Human DNAJC3, ERdj6 (endoplasmic reticulum DNA J domain–containing protein 6) | Involved in the unfolded protein response (UPR) during ER stress | ER lumen | 2.16 | 2.93 |
| Q99523 | Sortilin, NTR3 (human neurotensin receptor 3) | Sorting receptor in the Golgi compartment, required for protein transport from the Golgi apparatus. Also acts as a receptor for neurotensin | Nucleus, ER, Golgi, and cell surface | 2.07 | 3.48 |
| Q96DZ1 | Human ERLEC1, XTP3B (XTP3-transactivated gene B protein) | Probable lectin that binds selectively to improperly folded lumenal proteins. May function in ER quality control and ERAD | ER lumen | 2.00 | 2.11 |
Ratio of the heavy sample (H) over light sample (L).
Ratio of the light sample (L) over heavy sample (H).
PTK7 (also known as colon carcinoma kinase-4) is a receptor tyrosine kinase–like molecule containing an extracellular domain with seven immunoglobulin-like loops, a TMD, and an inactive kinase domain (51). PTK7 is a key regulator of multiple Wnt pathways under normal and possible pathological conditions, including cancer (52, 53). DNAJC3, previously known as P58IPK, is expressed in the ER and, as a cochaperone of BiP (immunoglobulin heavy-chain binding protein), it plays a crucial role in ER protein folding as well as in the unfolded protein response during ER stress (54, 55). ERLEC1, also known as XTP3-B, is a lectin that selectively binds to improperly folded lumenal proteins. It may function in ER quality control and endoplasmic reticulum–associated degradation (ERAD) of both nonglycosylated proteins and glycoproteins (56, 57).
Confirmation of the SILAC Hits by Expression Analysis of Potential CADA Targets With Flow Cytometry and Western Blotting
To confirm the potential CADA hits from the SILAC experiments, we evaluated the expression of the identified integral membrane proteins in CADA-treated or DMSO-treated SUP-T1 cells. First, flow cytometry was used to analyze the natural cell surface expression levels of the target protein in SUP-T1 cells that were used for the proteomics study. PTK7 is an integral type I membrane protein that is expressed on the surface of SUP-T1 cells. Consequently, PTK7 can easily be detected by surface staining with a fluorescently labeled antibody. In parallel, huCD4 was included as our positive control. As summarized in Figure 2A, CADA concentration dependently suppressed the expression of huCD4 (IC50 of 0.79 μM) and PTK7 (IC50 of 2.05 μM) in SUP-T1 cells, but the downmodulating effect of CADA on PTK7 was less pronounced (57% reduction in surface expression at 10 μM of CADA as compared with 74% reduction for huCD4). Thus, the expression of PTK7 is substantially affected by CADA, indicating that PTK7 is a validated substrate of CADA.
Fig. 2.

CADA downmodulates cell-surface huCD4 and PTK7 in lymphoid cells and the transient expression of huCD4, SORT, PTK7, ERLEC1, and DNAJC3 in transfected cells.A, concentration–response curves of CADA for huCD4 and PTK7 in SUP-T1 cells. Cells were incubated with CADA for 24 h. Tested CADA concentrations were 50, 10, 2, 0.4, 0.08, and 0.016 μM. Expression of the surface receptors was measured with flow cytometry and normalized to the DMSO control. A four parameter concentration response curve was fitted to data from at least three replicate experiments. Values are mean ± SD; n ≥ 3. B, Western blot images of cell lysates from nontransfected (NT) and huCD4-V5, SORT-V5, PTK7-V5, ERLEC1-V5, or DNAJC3-V5 transfected HEK293T cells treated for 24 h with different CADA concentrations. Protein bands were visualized using an antibody against the V5 tag, whereas for the cell loading control, an anticlathrin or anti–β-actin antibody was used. One representative experiment out of 3 to 5 is shown. C, concentration–response curves of CADA for huCD4, SORT, PTK7, ERLEC1, and DNAJC3 in transfected HEK293T cells. Samples from (B) were quantified and normalized to the clathrin (huCD4, ERLEC1, and DNAJC3) or β-actin (SORT and PTK7) internal control. A four parameter concentration response curve was fitted to the data from at least three replicate experiments. Values are mean ± SD; n ≥ 3. CADA, cyclotriazadisulfonamide; DMSO, dimethyl sulfoxide; DNAJC3, DnaJ homolog subfamily C member 3; ERLEC1, endoplasmic reticulum lectin 1; HEK293T, human embryonic kidney 293T cells; huCD4, human CD4; PTK7, inactive tyrosine-protein kinase 7; SORT, sortilin.
Both ERLEC1 and DNAJC3 are ER-resident proteins that possess a cleavable SP and an ER retention signal but lack a TMD. No antibodies are available for flow cytometric analysis of these proteins. We therefore obtained the coding sequence of wildtype full-length human ERLEC1 and DNAJC3 and incorporated the simian V5 epitope at the C terminus of both proteins for detection purposes. To allow for a comparative analysis, we also applied the same procedure to PTK7, and to our previously identified control proteins SORT and huCD4. HEK293T cells were transfected with the plasmids, treated with increasing concentrations of CADA, and subjected to immunoblotting. As shown in Figure 2B, in nontreated control cells, transfection with the different plasmids resulted in high expression levels of the V5-tagged proteins as visualized by the use of an anti-V5 antibody. Treatment with CADA reduced protein expression concentration dependently (Fig. 2C). The weakest effect of CADA was observed on PTK7. For ERLEC1, a complete inhibition of protein expression was achieved with 10 μM of CADA, as determined by quantification of the V5 signal. For DNAJC3, a clear concentration-dependent effect of CADA was observed. Thus, these data demonstrate that ERLEC1 and DNAJC3 are also valuable substrates of CADA.
Next, to compare the relative CADA sensitivity of the different hits side by side in a more quantitative way, we cloned the coding sequence of each target protein in the same vector backbone to obtain a GFP-fused reporter. As outlined in Figure 3A, the SP and 62 residues of the mature protein of huCD4 were exchanged by the corresponding region of each target to obtain a chimeric CD4 protein. This chimera contains the relevant preprotein sequence of each hit (i.e., N-terminal region with the SP) important for proper protein biogenesis and ER translocation (58). In addition, tGFP was fused to the C-terminal end of huCD4 (i.e., cytosolic tail). As previously determined, the addition of tGFP did not affect the sensitivity of huCD4 to CADA (42, 43). In addition, the reporter construct holds the BFP downstream of tGFP. Here, GFP and BFP are separated by the viral P2A sequence (Fig. 3A). At the P2A sequence, ribosomes skip a peptide bond without interrupting translation elongation (59, 60). This way, each polycistronic mRNA encodes two separated fluorescent proteins in equal amounts. The tGFP signal serves as measurement for target protein expression, whereas cytosolic BFP serves as an internal expression control that correlates with the number of transcripts. Here, we show that both PTK7 and DNAJC3 are concentration dependently downmodulated by CADA in an identical manner: about 70% reduction in protein expression at 10 μM CADA (Fig. 3B), whereas for SORT, only 50% reduction in protein expression was measured, which is in line with a previous report (44). Interestingly, ERLEC1 was affected the most, with 81% reduction in protein level at 10 μM CADA, and almost copied huCD4 (85% reduction) (Fig. 3B).
Fig. 3.

SILAC hits differentially respond to CADA in transfected cells.A, schematic representation of the expected mRNA and protein products of the tGFP-P2A-BFP construct. The constructs express huCD4 that is anchored in the plasma membrane via its transmembrane domain and with tGFP at the cytosolic tail. As the SP is cleaved by the ER lumenal signal peptidase during protein biogenesis, the mature huCD4 variants differ only in their N terminus (62 residues). B, four parameter concentration–response curves for CADA of huCD4, SORT, PTK7, ERLEC1, and DNAJC3 cloned in the same tGFP-P2A-BFP plasmid backbone as shown in (A). HEK293T cells were transiently transfected with the tGFP-P2A-BFP constructs and incubated with different CADA concentrations for 24 h. Protein levels in CADA-treated samples are normalized to the DMSO control (set at 1.0). Curves are fitted to data from 3 to 4 replicate experiments. Values are mean ± SD; n ≥ 3. BFP, blue fluorescent protein; CADA, cyclotriazadisulfonamide; DMSO, dimethyl sulfoxide; DNAJC3, DnaJ homolog subfamily C member 3; ER, endoplasmic reticulum; ERLEC1, endoplasmic reticulum lectin 1; HEK293T, human embryonic kidney 293T cells; huCD4, human CD4; PTK7, inactive tyrosine-protein kinase 7; SILAC, stable isotope labeling by amino acids in cell culture; SORT, sortilin; SP, signal peptide; tGFP, turbo GFP.
CADA Diverts the Nonglycosylated Preprotein for Proteasomal Degradation
Previous experiments clearly indicated that treatment with CADA reduces the cellular expression of the selected substrates. Although CADA has been reported as an inhibitor of cotranslational translocation, we next questioned if CADA has an additional binding and destabilizing effect on the mature protein part of the different substrates. First, we performed a pulse-labeling experiment with the V5-tagged huCD4 in the presence of a proteasome inhibitor to rescue the different unstable protein species that are getting degraded. Briefly, a 30-min pulse, followed by a 45-min chase and a V5-mediated pull down resulted in a clear reduction in the mature huCD4 form when CADA was administered (Fig. 4A; lane 4, open arrowhead). Interestingly, treatment of the cells with CADA resulted in a considerable fraction of a faster migrating form, presumably the nonglycosylated huCD4 precursor (Fig. 4A; lane 4, solid arrowhead and red bar in graph), which was enriched by the addition of MG132 (lane 5, solid arrowhead). However, treatment with MG132 neither resulted in the rescue of additional mature huCD4 protein or in the enhanced protein expression of the translocated species (lanes 3 and 5, open arrowhead). This indicates that CADA interacts with the precursor form (for proteasomal degradation) rather than with the mature protein and induces the proteosomal degradation of the precursor proteins.
Fig. 4.

CADA diverts the nonglycosylated preprotein for proteasomal degradation.A, autoradiogram of cell lysates from nontransfected (NT) and huCD4-V5 transfected CHO-K1 cells pulsed for 30 min with [35S]methionine/cysteine. Pulsed-labeled cells were then chased for 45 min in the presence or the absence of CADA (10 μM) and/or MG132 (200 nM). Open arrowhead represents the translocated (and glycosylated) huCD4 mature protein fraction, and solid arrowhead represents the nonglycosylated huCD4 preprotein. Graph on the right shows the quantification of the presented radioblot. For each sample, the fraction of preprotein and mature protein is calculated and given as percentage of total amount of protein. B, Western blot images of cell lysates from nontransfected (NT) and huCD4-V5, SORT-V5, and PTK7-V5 transfected HEK293T cells treated for 24 h with different CADA concentrations and 200 nM MG132. Protein bands were visualized using an antibody against the V5 tag, whereas anticlathrin (huCD4) or anti-β-actin (SORT and PTK7) antibody was used for the cell loading controls. Do note that the combination of MG132 with 10 or 2 μM CADA strongly reduces the level of mature protein, presumably by blocking the residual translocation of substrate that is normally seen with CADA. C, same as in (B) but for 10 μM CADA and/or 200 nM MG132. Lysates were treated with endo H prior to SDS-PAGE and Western blotting to remove N-linked glycosylations. Protein bands were visualized through V5. Do note that, because of the stronger denaturation conditions during the endo H treatment, V5 detection of the endo H treated samples was more efficient, giving thicker protein bands (although the same sample loading was used). PTK7 is a highly glycosylated protein (ten potential N-glycosylations) that is partially resistant to endo H treatment, resulting in only a minor fraction that is deglycosylated by endo H (solid arrowhead). CADA, cyclotriazadisulfonamide; Endo H, endoglycosidase H; HEK293T, human embryonic kidney 293T cells; huCD4, human CD4; PTK7, inactive tyrosine-protein kinase 7; SORT, sortilin.
A similar effect was also observed when cells were transiently transfected with huCD4, treated with both CADA and MG132, and when separated proteins were subsequently visualized by immunoblotting through V5 staining 24 h after transfection. Although no (unstable) preprotein could be detected with CADA-treatment only (Fig. 4B, lanes 4 and 6), again, a clear fraction of the huCD4 precursor form could be rescued when a proteasome inhibitor was administered together with CADA (Fig. 4B, lanes 5 and 7). Notably, under these conditions, the mature form was nearly completely depleted (lane 5). Additional endoglycosidase H treatment experiments confirmed that, in the presence of CADA and MG132, the rescued protein fraction represents the nonglycosylated preprotein (Fig. 4C, solid arrowhead). Furthermore, for the two other glycosylated substrates, SORT and PTK7, a similar concentration-dependent effect with CADA was observed, with preservation of the preprotein fraction when MG132 was added simultaneously with CADA (Fig. 4B, lanes 5, 7, and 9).
CADA Inhibits the ER Cotranslational Translocation of its Substrates
As CADA is known to inhibit the cotranslational translocation through the ER membrane of both huCD4 and SORT, we next determined if PTK7, DNAJC3, and ERLEC1 are substrates of CADA in the context of ER protein translocation. Analysis of the protein sequences showed that the identified substrates carry a cleavable SP, an indispensable property of secretory and type I integral membrane proteins to enter the ER-targeted secretory pathway (5, 22). As summarized in Figure 5, the SPs of the hits differ in length and protein sequence. At first sight, the primary sequence of the SPs does not display a similarity in amino acid composition. Also, no putative consensus motif that mediates CADA sensitivity could be deduced from the aligned amino acid sequences.
Fig. 5.

CADA substrates differ in their signal peptide (SP) sequence. Alignment of the N-terminal amino acid sequence of the identified SILAC hits. For each protein, the amino acid composition of the SP is given, together with 20 residues of the N terminus of the mature protein. The hyphen represents the SP cleavage site. The critical residues in the huCD4 SP are indicated in bold, whereas for the other SPs, the putative residues involved in CADA sensitivity are underlined. CADA, cyclotriazadisulfonamide; SILAC, stable isotope labeling by amino acids in cell culture.
To address whether CADA also inhibits protein expression of PTK7, DNAJC3, and ERLEC1 in a SP-dependent manner, we tested the impact of CADA on the translocation of PL chimeric constructs in a cell-free in vitro translation system as described in a recent study (43). As schematically represented in Figure 6A, the PL chimeric constructs hold the SP and the first residues of the mature hit protein fused to mature PL, a CADA-resistant protein as previously determined (42). Briefly, the constructs of Figure 6A were linearized by PCR and transcribed in vitro. The transcripts of the PL chimera were translated in the rabbit reticulocyte system in the absence or the presence of ovine pancreatic microsomal membranes and exposed to different concentrations of CADA. As expected, translation and translocation of wildtype PL was not affected by CADA, whereas the translocation of the huCD4-PL chimera, as evidenced by the faster migrating SP-cleaved species (open arrowhead), was strongly inhibited with 15 μM of CADA and higher (Fig. 6B). The cotranslational translocation of the SORT, ERLEC1, DNAJC3, and PTK7 constructs was also inhibited by CADA, although in various degrees. This indicates that the SPs of the hit proteins exert differential sensitivity toward CADA, with PTK7 and SORT as the least affected substrates and the huCD4 SP as the most sensitive target.
Fig. 6.

CADA inhibits the signal peptide (SP)–dependent cotranslational translocation of SILAC hits.A, schematic representation of the chimeric construct used in radiolabeled cell-free in vitro translation reactions. For each SILAC hit, the SP and first 6 to 11 residues of the N-terminal part of the mature protein were fused to mature prolactin (pPL). B, cell-free in vitro translation and translocation in rabbit reticulocyte lysate supplemented with ovine microsomes. Representative autoradiogram of the in vitro translated and translocated prolactin variants shown in (A). In the presence of microsomes, the preprotein (black arrowhead) is translocated into the ER lumen and the SP is cleaved, resulting in a faster migrating mature protein (open arrowhead). Relative protein translocation for the 150 and 15 μM CADA-treated samples is shown in the graphs, calculated from the signal intensities of the translocated protein fraction (open arrowhead) and the preprotein fraction (black arrowhead). Bars are mean ± SE; n ≥ 2. CADA, cyclotriazadisulfonamide; ER, endoplasmic reticulum; SILAC, stable isotope labeling by amino acids in cell culture.
Discussion
Previous studies for the small-molecule CADA identified two susceptible substrates, that is, human integral membrane proteins CD4 and SORT (42, 44). Original interest in CADA came from its antiviral potency against HIV (50). As CADA significantly downmodulates surface expression of huCD4—the main cellular receptor for HIV entry—potential target cells are being protected from infection with various HIV clinical isolates (61). To systematically explore whether additional cellular receptors are downmodulated by CADA, we used SILAC-based MS to perform a proteomic survey on the cellular membrane fraction. SILAC is a potent technique for direct qualitative and quantitative comparison of proteomes (62), which has been successfully used to evidence biological pathways affected in drug-resistant cells and to investigate the mode of action of compounds (32, 48, 63, 64). Our SILAC screen in the huCD4+ T-cell lymphoma SUP-T1 identified and quantified a total of 3007 proteins, of which 500 proteins were classified for having an SP and/or a TMD, thus, belonging to the secretory pathway. Remarkably, only five hits showed a significant decrease in protein expression following CADA treatment, including the two previously confirmed targets huCD4 and SORT. By means of independent assays, we validated the newly identified hits as being valuable substrates of CADA. This implies that the used SILAC technique is very accurate as it confirms previously validated substrates and identifies new targets without selecting false positives. In an additional study on the immunosuppressive potential of CADA, CD137 has recently been identified as a new substrate of CADA (65). This type I integral membrane protein is transiently expressed after activation of immune cells, mainly cytotoxic CD8+ T cells (66). However, in SUP-T1 cells, the basal level of CD137 is nearly undetectable, which might explain that this protein was not identified in our current proteomics analysis. Even though our current proteomics study is limited to a single cell line (SUP-T1) that does not express the complete proteome, our analysis clearly demonstrates that CADA, in general, has a low impact on the cellular membranome. In addition, it would be interesting to expand this proteomic study to the secretory protein fraction in order to characterize the effect of CADA on all proteins that enter the secretory pathway. However, our attempts to analyze the supernatant of the cells did not result in successful and reliable detection of secreted proteins. Future work will therefore focus on the analysis of the CADA-treated secretome.
So far, the identified CADA substrates have various subcellular localizations and are involved in different cellular pathways and functions (Table 1), except for ERLEC1 and DNAJC3. Both proteins take part in ER quality control and ERAD but are supposed to have distinct tasks in clearance of the ER lumen (54, 55, 56, 57). Transfection experiments and the supplemental in vitro translocation data clearly demonstrate a direct inhibitory effect of CADA on each of the selected proteomics hits. However, given that ERLEC1 and DNAJC3 are involved in ER quality control, we cannot exclude that reduced levels of both proteins might affect the net outcome of protein downmodulation by CADA on the other substrates.
PTK7 is highly expressed in several cancers, such as gastric cancer, lung cancer, and colon cancer; hence, a potential role of PTK7 in the progression of multiple cancers has been proposed (67, 68, 69). Thus, pharmaceutical targeting of PTK7 might hold some clinical potential in cancer treatment, justifying the further exploration of the effect of CADA-induced PTK7 downmodulation in the field of cancer. Currently, CADA is being investigated in a breast cancer model for its suppression of SORT, as SORT inhibition has been reported to limit secretion-induced progranulin-dependent breast cancer progression and cancer stem cell expansion (70). Genetic loss of function of DNAJC3 has been linked to a monogenic and recessive form of juvenile-onset diabetes and neurodegeneration (71, 72); however, the impact of partial reduction in DNAJC3 expression therapeutically needs to be further evaluated. ERLEC1, a lectin that binds to misfolded glycosylated proteins in the ER lumen, is involved in ERAD (56, 57). Notably, knockdown of ERLEC1 by RNAi did not affect the degradation kinetics of a terminally misfolded glycoprotein (57), suggesting that these ER-resident lectins are functionally redundant in ERAD, and that downmodulation of ERLEC1 might have little impact on cell viability.
In order to get a better understanding of the mode of action of CADA, we designed a tGFP-P2A-BFP reporter assay to compare the relative susceptibility of the substrates to CADA in a same genetic background. This flow cytometric assay proved to be a very sensitive and reproducible system to measure and accurately compare susceptibilities of potential targets to CADA (43). Here, it reveals that the impact of CADA on the cellular expression of the different substrates varies roughly between 50% and 85% reduction, suggesting a substrate-specific feature. Metabolic labeling experiments and addition of the proteasome inhibitor MG132 to the cells revealed the presence of the nonglycosylated preprotein in the CADA-treated samples and not an enrichment of the mature protein. This indicates that it is very unlikely that CADA interacts with the mature part of the substrates and exerts a destabilizing effect on the mature protein. However, the rescue of the preprotein of the integral membrane proteins huCD4, SORT, and PTK7 in the MG132-treated CADA samples rather suggests the putative interaction of CADA with the SP of its substrates.
Comparison of the SP primary sequence of the different CADA substrates did not reveal a clear similarity in amino acid composition. Unfortunately, no putative consensus motif that mediates full sensitivity to CADA could be deduced from the aligned amino acid sequences. However, as recently reported (43), the presence of a glutamine and proline in the hydrophobic core of the SP of huCD4 is key for susceptibility to CADA. Even though no Gln and Pro residues could be identified at the same position relative to the SP cleavage site in the aligned SPs (Fig. 5), the N terminus of each target protein does contain at least one Gln residue in the vicinity of the cleavage site (either in the SP or in the N-terminal end of the mature protein). The same holds true for the presence of a proline (and/or the other alpha helix terminator glycine). As the drug is expected to hold the SP in a locked position in the translocon, the compound might indeed interact with Gln or Pro that are positioned more upstream or downstream the nascent chain. Also, given that the nascent polypeptide forms a loop inside the translocon, residues of both SP and N-terminal part of the mature protein may be involved in the interaction with the drug, making the absolute sequence orientation (N terminus or C terminus facing ER lumen) less determinant. In addition, the presence of positively charged residues in the SP can also contribute to higher susceptibility to CADA (43).
We previously reported that CADA inhibits the cotranslational translocation over the ER membrane of both huCD4 and SORT (42, 44), and CD137 (65), a process that relies on the presence of a cleavable SP of the preprotein. Interestingly, the three additional CADA substrates of this study are also proteins with cleavable SPs and not membrane proteins with SASs. This is in contrast with cotransin, another ER translocation inhibitor with a wide substrate spectrum, including membrane proteins with an SAS for which a consensus sequence could be deduced for cotransin sensitivity (32). In our cell-free in vitro translation experiments, we could evaluate the inhibitory potential of CADA directly on the protein translocation of the three new targets. Translation of the substrates as such was not affected by CADA (not shown), whereas reduced translocation of ERLEC1, DNAJC3, and PTK7 after CADA exposure was observed. This indicates that CADA blocks the SP-dependent ER translocation process of each target protein. However, the impact of CADA on ERLEC1 translocation was rather limited and did not correlate with the Western blot data. The overall low translation and translocation efficiency of ERLEC1 seen in this (artificial) cell-free system may skew the intrinsic susceptibility of ERLEC1 to CADA in a cellular context. In contrast, the data of ERLEC1 in HEK293T cells are based on the transient overexpression of a V5-tagged protein that also not represents the physiological condition. In that context, the results from flow cytometry in SUP-T1 cells are the most relevant data where we quantify the endogenous cell surface expression level of the CADA substrates huCD4 and PTK7. The relative high residual huCD4 level in SUP-T1 after CADA exposure as compared with HEK293T cells is because of the presence of p56lck in T cells, resulting in reduced endocytosis and a low turnover of huCD4 (73). Nevertheless, the flow cytometry and the cell-free in vitro translocation data demonstrate that huCD4 is affected the most by CADA, confirming that huCD4 remains the main target of CADA. HuCD4 is known to play a crucial role in immune activation (74, 75, 76). The in vitro and in vivo immunosuppressive potential of nondepleting anti-CD4 monoclonal antibodies further emphasize a major role of huCD4 in our immune system (77, 78, 79). In that perspective, CADA might hold great potential as an immunosuppressive drug, for instance in the treatment of transplant rejection, given that CADA exerts significant immunosuppressive activity in several in vitro models of T-cell activation (65).
In conclusion, the present study suggested that CADA has a low impact on the expression of more than 3000 different proteins. It also revealed that CADA inhibits ER translocation of PTK7, DNAJC3, and ERLEC1 in a SP-dependent way with subsequent protein downmodulating effect on these substrates. However, the high selectivity of CADA for huCD4 makes it an interesting drug to further explore its potential as an alternative immunosuppressive agent.
Data Availability
The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (80, 81, 82) partner repository with the dataset identifier PXD027712. All validating data by various biological and cell-free in vitro experiments are contained within the article.
Supplemental data
This article contains supplemental data.
Conflict of interest
The authors declare that they have no conflicts of interest with the contents of this article.
Acknowledgments
We thank Anita Camps and Geert Schoofs for excellent technical assistance. Dr Thomas W. Bell (UNR, NV) is acknowledged for providing CADA compound. We also thank Prof Dr Fan Liu for her help in preparing our data for the PRIDE.
Author contributions
E. P., C. R., R. S., and K. V. methodology; E. P., C. R., B. P., J. S., D. S., E. H., E. K., H. S., R. S., and K. V. validation; E. P., C. R., E. K., and H. S. formal analysis; E. P., C. R., B. P., and J. S. investigation; D. S., E. H., and E. K. resources; E. P., C. R., E. K., R. S., and K. V. writing–original draft; E. P., C. R., B. P., J. S., D. S., E. H., E. K., H. S., R. S., and K. V. writing–review and editing.
Supplemental Data
References
- 1.Uhlen M., Fagerberg L., Hallstrom B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson A., Kampf C., Sjostedt E., Asplund A., Olsson I., Edlund K., Lundberg E., Navani S., Szigyarto C.A. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 2.Wickner W., Schekman R. Protein translocation across biological membranes. Science. 2005;310:1452–1456. doi: 10.1126/science.1113752. [DOI] [PubMed] [Google Scholar]
- 3.Blobel G., Dobberstein B. Transfer of proteins across membranes. I. Presence of proteolytically processed and unprocessed nascent immunoglobulin light chains on membrane-bound ribosomes of murine myeloma. J. Cell Biol. 1975;67:835–851. doi: 10.1083/jcb.67.3.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walter P., Gilmore R., Blobel G. Protein translocation across the endoplasmic reticulum. Cell. 1984;38:5–8. doi: 10.1016/0092-8674(84)90520-8. [DOI] [PubMed] [Google Scholar]
- 5.von Heijne G. Signal sequences. The limits of variation. J. Mol. Biol. 1985;184:99–105. doi: 10.1016/0022-2836(85)90046-4. [DOI] [PubMed] [Google Scholar]
- 6.Pfeffer S., Dudek J., Zimmermann R., Forster F. Organization of the native ribosome-translocon complex at the mammalian endoplasmic reticulum membrane. Biochim. Biophys. Acta. 2016;1860:2122–2129. doi: 10.1016/j.bbagen.2016.06.024. [DOI] [PubMed] [Google Scholar]
- 7.Blobel G., Dobberstein B. Transfer of proteins across membranes. II. Reconstitution of functional rough microsomes from heterologous components. J. Cell Biol. 1975;67:852–862. doi: 10.1083/jcb.67.3.852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.von Heijne G., Gavel Y. Topogenic signals in integral membrane proteins. Eur. J. Biochem. 1988;174:671–678. doi: 10.1111/j.1432-1033.1988.tb14150.x. [DOI] [PubMed] [Google Scholar]
- 9.van den Berg B., Clemons W.M., Jr., Collinson I., Modis Y., Hartmann E., Harrison S.C., Rapoport T.A. X-ray structure of a protein-conducting channel. Nature. 2004;427:36–44. doi: 10.1038/nature02218. [DOI] [PubMed] [Google Scholar]
- 10.Voorhees R.M., Hegde R.S. Structure of the Sec61 channel opened by a signal sequence. Science. 2016;351:88–91. doi: 10.1126/science.aad4992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simon S.M., Blobel G. A protein-conducting channel in the endoplasmic reticulum. Cell. 1991;65:371–380. doi: 10.1016/0092-8674(91)90455-8. [DOI] [PubMed] [Google Scholar]
- 12.Rapoport T.A. Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature. 2007;450:663–669. doi: 10.1038/nature06384. [DOI] [PubMed] [Google Scholar]
- 13.Hegde R.S., Kang S.W. The concept of translocational regulation. J. Cell Biol. 2008;182:225–232. doi: 10.1083/jcb.200804157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walter P., Blobel G. Translocation of proteins across the endoplasmic reticulum. II. Signal recognition protein (SRP) mediates the selective binding to microsomal membranes of in-vitro-assembled polysomes synthesizing secretory protein. J. Cell Biol. 1981;91:551–556. doi: 10.1083/jcb.91.2.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keenan R.J., Freymann D.M., Stroud R.M., Walter P. The signal recognition particle. Annu. Rev. Biochem. 2001;70:755–775. doi: 10.1146/annurev.biochem.70.1.755. [DOI] [PubMed] [Google Scholar]
- 16.Halic M., Blau M., Becker T., Mielke T., Pool M.R., Wild K., Sinning I., Beckmann R. Following the signal sequence from ribosomal tunnel exit to signal recognition particle. Nature. 2006;444:507–511. doi: 10.1038/nature05326. [DOI] [PubMed] [Google Scholar]
- 17.Nilsson I., Lara P., Hessa T., Johnson A.E., von Heijne G., Karamyshev A.L. The code for directing proteins for translocation across ER membrane: SRP cotranslationally recognizes specific features of a signal sequence. J. Mol. Biol. 2015;427:1191–1201. doi: 10.1016/j.jmb.2014.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gilmore R., Walter P., Blobel G. Protein translocation across the endoplasmic reticulum. II. Isolation and characterization of the signal recognition particle receptor. J. Cell Biol. 1982;95:470–477. doi: 10.1083/jcb.95.2.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pfeffer S., Dudek J., Gogala M., Schorr S., Linxweiler J., Lang S., Becker T., Beckmann R., Zimmermann R., Forster F. Structure of the mammalian oligosaccharyl-transferase complex in the native ER protein translocon. Nat. Commun. 2014;5:3072. doi: 10.1038/ncomms4072. [DOI] [PubMed] [Google Scholar]
- 20.Park E., Rapoport T.A. Mechanisms of Sec61/SecY-mediated protein translocation across membranes. Annu. Rev. Biophys. 2012;41:21–40. doi: 10.1146/annurev-biophys-050511-102312. [DOI] [PubMed] [Google Scholar]
- 21.Gemmer M., Forster F. A clearer picture of the ER translocon complex. J. Cell Sci. 2020;133 doi: 10.1242/jcs.231340. [DOI] [PubMed] [Google Scholar]
- 22.Hegde R.S., Bernstein H.D. The surprising complexity of signal sequences. Trends Biochem. Sci. 2006;31:563–571. doi: 10.1016/j.tibs.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 23.Lumangtad L.A., Bell T.W. The signal peptide as a new target for drug design. Bioorg. Med. Chem. Lett. 2020;30:127115. doi: 10.1016/j.bmcl.2020.127115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kalies K.U., Romisch K. Inhibitors of protein translocation across the ER membrane. Traffic. 2015;16:1027–1038. doi: 10.1111/tra.12308. [DOI] [PubMed] [Google Scholar]
- 25.Van Puyenbroeck V., Vermeire K. Inhibitors of protein translocation across membranes of the secretory pathway: Novel antimicrobial and anticancer agents. Cell. Mol. Life Sci. 2018;75:1541–1558. doi: 10.1007/s00018-017-2743-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garrison J.L., Kunkel E.J., Hegde R.S., Taunton J. A substrate-specific inhibitor of protein translocation into the endoplasmic reticulum. Nature. 2005;436:285–289. doi: 10.1038/nature03821. [DOI] [PubMed] [Google Scholar]
- 27.Besemer J., Harant H., Wang S., Oberhauser B., Marquardt K., Foster C.A., Schreiner E.P., de Vries J.E., Dascher-Nadel C., Lindley I.J. Selective inhibition of cotranslational translocation of vascular cell adhesion molecule 1. Nature. 2005;436:290–293. doi: 10.1038/nature03670. [DOI] [PubMed] [Google Scholar]
- 28.Westendorf C., Schmidt A., Coin I., Furkert J., Ridelis I., Zampatis D., Rutz C., Wiesner B., Rosenthal W., Beyermann M., Schulein R. Inhibition of biosynthesis of human endothelin B receptor by the cyclodepsipeptide cotransin. J. Biol. Chem. 2011;286:35588–35600. doi: 10.1074/jbc.M111.239244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mackinnon A.L., Paavilainen V.O., Sharma A., Hegde R.S., Taunton J. An allosteric Sec61 inhibitor traps nascent transmembrane helices at the lateral gate. Elife. 2014;3 doi: 10.7554/eLife.01483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harant H., Lettner N., Hofer L., Oberhauser B., de Vries J.E., Lindley I.J. The translocation inhibitor CAM741 interferes with vascular cell adhesion molecule 1 signal peptide insertion at the translocon. J. Biol. Chem. 2006;281:30492–30502. doi: 10.1074/jbc.M607243200. [DOI] [PubMed] [Google Scholar]
- 31.Harant H., Wolff B., Schreiner E.P., Oberhauser B., Hofer L., Lettner N., Maier S., de Vries J.E., Lindley I.J. Inhibition of vascular endothelial growth factor cotranslational translocation by the cyclopeptolide CAM741. Mol. Pharmacol. 2007;71:1657–1665. doi: 10.1124/mol.107.034249. [DOI] [PubMed] [Google Scholar]
- 32.Klein W., Westendorf C., Schmidt A., Conill-Cortes M., Rutz C., Blohs M., Beyermann M., Protze J., Krause G., Krause E., Schulein R. Defining a conformational consensus motif in cotransin-sensitive signal sequences: A proteomic and site-directed mutagenesis study. PLoS One. 2015;10 doi: 10.1371/journal.pone.0120886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maifeld S.V., MacKinnon A.L., Garrison J.L., Sharma A., Kunkel E.J., Hegde R.S., Taunton J. Secretory protein profiling reveals TNF-alpha inactivation by selective and promiscuous Sec61 modulators. Chem. Biol. 2011;18:1082–1088. doi: 10.1016/j.chembiol.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hall B.S., Hill K., McKenna M., Ogbechi J., High S., Willis A.E., Simmonds R.E. The pathogenic mechanism of the Mycobacterium ulcerans virulence factor, mycolactone, depends on blockade of protein translocation into the ER. PLoS Pathog. 2014;10 doi: 10.1371/journal.ppat.1004061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gerard S.F., Hall B.S., Zaki A.M., Corfield K.A., Mayerhofer P.U., Costa C., Whelligan D.K., Biggin P.C., Simmonds R.E., Higgins M.K. Structure of the inhibited state of the Sec translocon. Mol. Cell. 2020;79:406–415.e407. doi: 10.1016/j.molcel.2020.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paatero A.O., Kellosalo J., Dunyak B.M., Almaliti J., Gestwicki J.E., Gerwick W.H., Taunton J., Paavilainen V.O. Apratoxin kills cells by direct blockade of the Sec61 protein translocation channel. Cell Chem. Biol. 2016;23:561–566. doi: 10.1016/j.chembiol.2016.04.008. [DOI] [PubMed] [Google Scholar]
- 37.Huang K.C., Chen Z., Jiang Y., Akare S., Kolber-Simonds D., Condon K., Agoulnik S., Tendyke K., Shen Y., Wu K.M., Mathieu S., Choi H.W., Zhu X., Shimizu H., Kotake Y. Apratoxin A shows novel pancreas-targeting activity through the binding of Sec 61. Mol. Cancer Ther. 2016;15:1208–1216. doi: 10.1158/1535-7163.MCT-15-0648. [DOI] [PubMed] [Google Scholar]
- 38.Junne T., Wong J., Studer C., Aust T., Bauer B.W., Beibel M., Bhullar B., Bruccoleri R., Eichenberger J., Estoppey D., Hartmann N., Knapp B., Krastel P., Melin N., Oakeley E.J. Decatransin, a new natural product inhibiting protein translocation at the Sec61/SecYEG translocon. J. Cell Sci. 2015;128:1217–1229. doi: 10.1242/jcs.165746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Serrill J.D., Wan X., Hau A.M., Jang H.S., Coleman D.J., Indra A.K., Alani A.W., McPhail K.L., Ishmael J.E. Coibamide A, a natural lariat depsipeptide, inhibits VEGFA/VEGFR2 expression and suppresses tumor growth in glioblastoma xenografts. Invest. New Drugs. 2016;34:24–40. doi: 10.1007/s10637-015-0303-x. [DOI] [PubMed] [Google Scholar]
- 40.Tranter D., Paatero A.O., Kawaguchi S., Kazemi S., Serrill J.D., Kellosalo J., Vogel W.K., Richter U., Mattos D.R., Wan X., Thornburg C.C., Oishi S., McPhail K.L., Ishmael J.E., Paavilainen V.O. Coibamide A targets Sec61 to prevent biogenesis of secretory and membrane proteins. ACS Chem. Biol. 2020;15:2125–2136. doi: 10.1021/acschembio.0c00325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zong G., Hu Z., O'Keefe S., Tranter D., Iannotti M.J., Baron L., Hall B., Corfield K., Paatero A.O., Henderson M.J., Roboti P., Zhou J., Sun X., Govindarajan M., Rohde J.M. Ipomoeassin F binds Sec61alpha to inhibit protein translocation. J. Am. Chem. Soc. 2019;141:8450–8461. doi: 10.1021/jacs.8b13506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vermeire K., Bell T.W., Van Puyenbroeck V., Giraut A., Noppen S., Liekens S., Schols D., Hartmann E., Kalies K.U., Marsh M. Signal peptide-binding drug as a selective inhibitor of co-translational protein translocation. PLoS Biol. 2014;12 doi: 10.1371/journal.pbio.1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van Puyenbroeck V., Pauwels E., Provinciael B., Bell T.W., Schols D., Kalies K.U., Hartmann E., Vermeire K. Preprotein signature for full susceptibility to the co-translational translocation inhibitor cyclotriazadisulfonamide. Traffic. 2020;21:250–264. doi: 10.1111/tra.12713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Van Puyenbroeck V., Claeys E., Schols D., Bell T.W., Vermeire K. A proteomic survey indicates sortilin as a secondary substrate of the ER translocation inhibitor cyclotriazadisulfonamide (CADA) Mol. Cell. Proteomics. 2017;16:157–167. doi: 10.1074/mcp.M116.061051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bell T.W., Anugu S., Bailey P., Catalano V.J., Dey K., Drew M.G., Duffy N.H., Jin Q., Samala M.F., Sodoma A., Welch W.H., Schols D., Vermeire K. Synthesis and structure-activity relationship studies of CD4 down-modulating cyclotriazadisulfonamide (CADA) analogues. J. Med. Chem. 2006;49:1291–1312. doi: 10.1021/jm0582524. [DOI] [PubMed] [Google Scholar]
- 46.Juszkiewicz S., Hegde R.S. Initiation of quality control during poly(A) translation requires site-specific ribosome ubiquitination. Mol. Cell. 2017;65:743–750. doi: 10.1016/j.molcel.2016.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vermeire K., Allan S., Provinciael B., Hartmann E., Kalies K.U. Ribonuclease-neutralized pancreatic microsomal membranes from livestock for in vitro co-translational protein translocation. Anal. Biochem. 2015;484:102–104. doi: 10.1016/j.ab.2015.05.019. [DOI] [PubMed] [Google Scholar]
- 48.Lange S., Sylvester M., Schumann M., Freund C., Krause E. Identification of phosphorylation-dependent interaction partners of the adapter protein ADAP using quantitative mass spectrometry: SILAC vs (18)O-labeling. J. Proteome Res. 2010;9:4113–4122. doi: 10.1021/pr1003054. [DOI] [PubMed] [Google Scholar]
- 49.Vermeire K., Bell T.W., Choi H.J., Jin Q., Samala M.F., Sodoma A., De Clercq E., Schols D. The anti-HIV potency of cyclotriazadisulfonamide analogs is directly correlated with their ability to down-modulate the CD4 receptor. Mol. Pharmacol. 2003;63:203–210. doi: 10.1124/mol.63.1.203. [DOI] [PubMed] [Google Scholar]
- 50.Vermeire K., Zhang Y., Princen K., Hatse S., Samala M.F., Dey K., Choi H.J., Ahn Y., Sodoma A., Snoeck R., Andrei G., De Clercq E., Bell T.W., Schols D. CADA inhibits human immunodeficiency virus and human herpesvirus 7 replication by down-modulation of the cellular CD4 receptor. Virology. 2002;302:342–353. doi: 10.1006/viro.2002.1624. [DOI] [PubMed] [Google Scholar]
- 51.Park S.K., Lee H.S., Lee S.T. Characterization of the human full-length PTK7 cDNA encoding a receptor protein tyrosine kinase-like molecule closely related to chick KLG. J. Biochem. 1996;119:235–239. doi: 10.1093/oxfordjournals.jbchem.a021228. [DOI] [PubMed] [Google Scholar]
- 52.Puppo F., Thome V., Lhoumeau A.C., Cibois M., Gangar A., Lembo F., Belotti E., Marchetto S., Lecine P., Prebet T., Sebbagh M., Shin W.S., Lee S.T., Kodjabachian L., Borg J.P. Protein tyrosine kinase 7 has a conserved role in Wnt/beta-catenin canonical signalling. EMBO Rep. 2011;12:43–49. doi: 10.1038/embor.2010.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bin-Nun N., Lichtig H., Malyarova A., Levy M., Elias S., Frank D. PTK7 modulates Wnt signaling activity via LRP6. Development. 2014;141:410–421. doi: 10.1242/dev.095984. [DOI] [PubMed] [Google Scholar]
- 54.Yan W., Frank C.L., Korth M.J., Sopher B.L., Novoa I., Ron D., Katze M.G. Control of PERK eIF2alpha kinase activity by the endoplasmic reticulum stress-induced molecular chaperone P58IPK. Proc. Natl. Acad. Sci. U. S. A. 2002;99:15920–15925. doi: 10.1073/pnas.252341799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Melville M.W., Katze M.G., Tan S.L. P58IPK, a novel cochaperone containing tetratricopeptide repeats and a J-domain with oncogenic potential. Cell. Mol. Life Sci. 2000;57:311–322. doi: 10.1007/PL00000692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cruciat C.M., Hassler C., Niehrs C. The MRH protein Erlectin is a member of the endoplasmic reticulum synexpression group and functions in N-glycan recognition. J. Biol. Chem. 2006;281:12986–12993. doi: 10.1074/jbc.M511872200. [DOI] [PubMed] [Google Scholar]
- 57.Hosokawa N., Wada I., Nagasawa K., Moriyama T., Okawa K., Nagata K. Human XTP3-B forms an endoplasmic reticulum quality control scaffold with the HRD1-SEL1L ubiquitin ligase complex and BiP. J. Biol. Chem. 2008;283:20914–20924. doi: 10.1074/jbc.M709336200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim S.J., Mitra D., Salerno J.R., Hegde R.S. Signal sequences control gating of the protein translocation channel in a substrate-specific manner. Dev. Cell. 2002;2:207–217. doi: 10.1016/s1534-5807(01)00120-4. [DOI] [PubMed] [Google Scholar]
- 59.de Felipe P., Hughes L.E., Ryan M.D., Brown J.D. Co-translational, intraribosomal cleavage of polypeptides by the foot-and-mouth disease virus 2A peptide. J. Biol. Chem. 2003;278:11441–11448. doi: 10.1074/jbc.M211644200. [DOI] [PubMed] [Google Scholar]
- 60.Lin Y.J., Huang L.H., Huang C.T. Enhancement of heterologous gene expression in Flammulina velutipes using polycistronic vectors containing a viral 2A cleavage sequence. PLoS One. 2013;8 doi: 10.1371/journal.pone.0059099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vermeire K., Princen K., Hatse S., De Clercq E., Dey K., Bell T.W., Schols D. CADA, a novel CD4-targeted HIV inhibitor, is synergistic with various anti-HIV drugs in vitro. AIDS. 2004;18:2115–2125. doi: 10.1097/00002030-200411050-00003. [DOI] [PubMed] [Google Scholar]
- 62.Ong S.E., Blagoev B., Kratchmarova I., Kristensen D.B., Steen H., Pandey A., Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 63.Chavez J.D., Hoopmann M.R., Weisbrod C.R., Takara K., Bruce J.E. Quantitative proteomic and interaction network analysis of cisplatin resistance in HeLa cells. PLoS One. 2011;6 doi: 10.1371/journal.pone.0019892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Caceres N.E., Aerts M., Marquez B., Mingeot-Leclercq M.P., Tulkens P.M., Devreese B., Van Bambeke F. Analysis of the membrane proteome of ciprofloxacin-resistant macrophages by stable isotope labeling with amino acids in cell culture (SILAC) PLoS One. 2013;8 doi: 10.1371/journal.pone.0058285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Claeys E., Pauwels E., Humblet-Baron S., Provinciael B., Schols D., Waer M., Sprangers B., Vermeire K. Small molecule cyclotriazadisulfonamide abrogates the upregulation of the human receptors CD4 and 4-1BB and suppresses in vitro activation and proliferation of T lymphocytes. Front. Immunol. 2021;12:650731. doi: 10.3389/fimmu.2021.650731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shuford W.W., Klussman K., Tritchler D.D., Loo D.T., Chalupny J., Siadak A.W., Brown T.J., Emswiler J., Raecho H., Larsen C.P., Pearson T.C., Ledbetter J.A., Aruffo A., Mittler R.S. 4-1BB costimulatory signals preferentially induce CD8+ T cell proliferation and lead to the amplification in vivo of cytotoxic T cell responses. J. Exp. Med. 1997;186:47–55. doi: 10.1084/jem.186.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen R., Khatri P., Mazur P.K., Polin M., Zheng Y., Vaka D., Hoang C.D., Shrager J., Xu Y., Vicent S., Butte A.J., Sweet-Cordero E.A. A meta-analysis of lung cancer gene expression identifies PTK7 as a survival gene in lung adenocarcinoma. Cancer Res. 2014;74:2892–2902. doi: 10.1158/0008-5472.CAN-13-2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang H., Wang A., Qi S., Cheng S., Yao B., Xu Y. Protein tyrosine kinase 7 (PTK7) as a predictor of lymph node metastases and a novel prognostic biomarker in patients with prostate cancer. Int. J. Mol. Sci. 2014;15:11665–11677. doi: 10.3390/ijms150711665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Duan F., Tang J., Kong F.L., Zou H.W., Ni B.L., Yu J.C. Identification of PTK7 as a promising therapeutic target for thyroid cancer. Eur. Rev. Med. Pharmacol. Sci. 2020;24:6809–6817. doi: 10.26355/eurrev_202006_21670. [DOI] [PubMed] [Google Scholar]
- 70.Rhost S., Hughes E., Harrison H., Rafnsdottir S., Jacobsson H., Gregersson P., Magnusson Y., Fitzpatrick P., Andersson D., Berger K., Stahlberg A., Landberg G. Sortilin inhibition limits secretion-induced progranulin-dependent breast cancer progression and cancer stem cell expansion. Breast Cancer Res. 2018;20:137. doi: 10.1186/s13058-018-1060-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Synofzik M., Haack T.B., Kopajtich R., Gorza M., Rapaport D., Greiner M., Schonfeld C., Freiberg C., Schorr S., Holl R.W., Gonzalez M.A., Fritsche A., Fallier-Becker P., Zimmermann R., Strom T.M. Absence of BiP co-chaperone DNAJC3 causes diabetes mellitus and multisystemic neurodegeneration. Am. J. Hum. Genet. 2014;95:689–697. doi: 10.1016/j.ajhg.2014.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ozon Z.A., Alikasifoglu A., Kandemir N., Aydin B., Gonc E.N., Karaosmanoglu B., Celik N.B., Eroglu-Ertugrul N.G., Taskiran E.Z., Haliloglu G., Oguz K.K., Kiper P.O., Yalnizoglu D., Utine G.E., Alikasifoglu M. Novel insights into diabetes mellitus due to DNAJC3-defect: Evolution of neurological and endocrine phenotype in the pediatric age group. Pediatr. Diabetes. 2020;21:1176–1182. doi: 10.1111/pedi.13098. [DOI] [PubMed] [Google Scholar]
- 73.Pelchen-Matthews A., Boulet I., Littman D.R., Fagard R., Marsh M. The protein tyrosine kinase p56lck inhibits CD4 endocytosis by preventing entry of CD4 into coated pits. J. Cell Biol. 1992;117:279–290. doi: 10.1083/jcb.117.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Claeys E., Vermeire K. The CD4 receptor: An indispensable protein in T cell activation and a promising target for immunosuppression. Arch. Microbiol. Immunol. 2019;3:133–150. [Google Scholar]
- 75.Janeway C.A., Jr. The role of CD4 in T-cell activation: Accessory molecule or co-receptor? Immunol. Today. 1989;10:234–238. doi: 10.1016/0167-5699(89)90260-0. [DOI] [PubMed] [Google Scholar]
- 76.Konig R., Zhou W. Signal transduction in T helper cells: CD4 coreceptors exert complex regulatory effects on T cell activation and function. Curr. Issues Mol. Biol. 2004;6:1–15. [PubMed] [Google Scholar]
- 77.Schulze-Koops H., Lipsky P.E. Anti-CD4 monoclonal antibody therapy in human autoimmune diseases. Curr. Dir. Autoimmun. 2000;2:24–49. doi: 10.1159/000060506. [DOI] [PubMed] [Google Scholar]
- 78.Mayer C.T., Huntenburg J., Nandan A., Schmitt E., Czeloth N., Sparwasser T. CD4 blockade directly inhibits mouse and human CD4+ T cell functions independent of Foxp3+ tregs. J. Autoimmun. 2013;47:73–82. doi: 10.1016/j.jaut.2013.08.008. [DOI] [PubMed] [Google Scholar]
- 79.Winsor-Hines D., Merrill C., O'Mahony M., Rao P.E., Cobbold S.P., Waldmann H., Ringler D.J., Ponath P.D. Induction of immunological tolerance/hyporesponsiveness in baboons with a nondepleting CD4 antibody. J. Immunol. 2004;173:4715–4723. doi: 10.4049/jimmunol.173.7.4715. [DOI] [PubMed] [Google Scholar]
- 80.Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D.J., Inuganti A., Griss J., Mayer G., Eisenacher M., Perez E., Uszkoreit J., Pfeuffer J., Sachsenberg T., Yilmaz S. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019;47:D442–D450. doi: 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Deutsch E.W., Bandeira N., Sharma V., Perez-Riverol Y., Carver J.J., Kundu D.J., Garcia-Seisdedos D., Jarnuczak A.F., Hewapathirana S., Pullman B.S., Wertz J., Sun Z., Kawano S., Okuda S., Watanabe Y. The ProteomeXchange consortium in 2020: Enabling 'big data' approaches in proteomics. Nucleic Acids Res. 2020;48:D1145–D1152. doi: 10.1093/nar/gkz984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Perez-Riverol Y., Xu Q.W., Wang R., Uszkoreit J., Griss J., Sanchez A., Reisinger F., Csordas A., Ternent T., Del-Toro N., Dianes J.A., Eisenacher M., Hermjakob H., Vizcaino J.A. PRIDE inspector toolsuite: Moving toward a universal visualization tool for proteomics data standard formats and quality assessment of ProteomeXchange datasets. Mol. Cell. Proteomics. 2016;15:305–317. doi: 10.1074/mcp.O115.050229. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The MS proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (80, 81, 82) partner repository with the dataset identifier PXD027712. All validating data by various biological and cell-free in vitro experiments are contained within the article.