

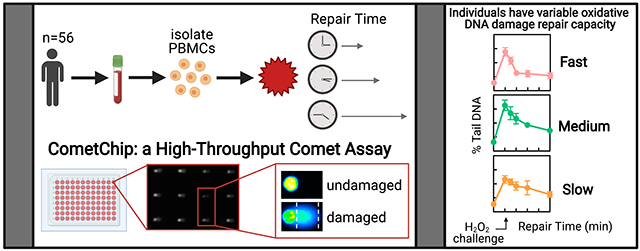
Abstract
Although DNA repair is known to impact susceptibility to cancer and other diseases, relatively few population studies have been performed to evaluate DNA repair kinetics in people due to the difficulty of assessing DNA repair in a high throughput manner. Here we use the CometChip, a high throughput comet assay, to explore inter-individual variation in repair of oxidative damage to DNA, a known risk factor for aging, cancer and other diseases. DNA repair capacity after H2O2-induced DNA oxidation damage was quantified in peripheral blood mononuclear cells (PBMCs). For 10 individuals, blood was drawn at several times over the course of 4-6 weeks. In addition, blood was drawn once from each of 56 individuals. DNA damage levels were quantified prior to exposure to H2O2 and at 0, 15, 30, 60, and 120-minutes post exposure. We found that there is significant variability in DNA repair efficiency among individuals. When subdivided into quartiles by DNA repair efficiency, we found that the average t1/2 is 81 minutes for the slowest group and 24 minutes for the fastest group. This work shows that the CometChip can be used to uncover significant differences in repair kinetics among people, pointing to its utility in future epidemiological and clinical studies.
Keywords: CometChip, DNA repair, DNA damage, repair kinetics
Graphical Abstract
INTRODUCTION
DNA damage can lead to mutations and cancer. The health effects of exposure to DNA damaging agents vary greatly among individuals, in part due to differences in their genetics. For example, among heavy smokers who are exposed to many genotoxic agents, not everyone develops lung cancer, partly due to gene-environment interactions (Wu et al. 2004; Deng et al. 2009; Yang et al. 2016; Leong et al. 2019). Furthermore, genetic differences among individuals contribute to divergent responses to DNA damaging agents used for cancer treatment with regard to both efficacy of treatment and toxic side effects (e.g., (Pollard and Gatti 2009; Cortesi et al. 2021)). Despite the relevance of DNA repair capacity as a driver of differences in susceptibility to DNA damage, population studies investigating this have been limited due to technical and throughput limitations of existing repair assays. Having a way to identify people with reduced DNA repair capacity opens doors to precision prevention (Collins and Varmus 2015; Nagel et al. 2017), as well as to personalized medicine for cancer treatment (e.g., to maximize toxicity to the tumor, while minimizing toxicity to the patient). Here, we describe the application of a higher throughput approach for monitoring DNA repair capacity in human lymphocytes.
The Base Excision Repair (BER) pathway is responsible for repair of ~30,000 lesions per cell per day (Wallace 2014; Marsden et al. 2017). Human cells have a suite of DNA glycosylases that initiate BER by recognizing and removing damaged bases from the genome, giving rise to an abasic, or AP site (Svilar et al. 2011; Kumar et al. 2020). For a key subpathway of BER, bifunctional glycosylases both remove the offending lesion and cleave the phosphodiester backbone. AP endonuclease 1 (APE1) can then hydrolyze the phosphodiester bond 5’ to the AP site, DNA polymerase β can perform repair synthesis and the remaining single strand break (SSB) can then be sealed by a DNA ligase (note that BER has multiple subpathways; for more information, see (Meira et al. 2005; Svilar et al. 2011; Krokan and Bjoras 2013; Wallace 2014)). Important connections have been drawn between BER and disease. One approach is to measure enzyme activity for a single component of a pathway, as has been done for the OGG1 DNA glycosylase. Indeed, it has been shown that there is an association between OGG1 activity and susceptibility to cancer (Paz-Elizur et al. 2006; Leitner-Dagan et al. 2014). However, BER has multiple steps and thus analysis of one step in the pathway does not necessarily reveal efficiency of the entire pathway. For example, although having high levels of a repair enzyme may seem advantageous, we now know that increased initiation of a repair pathway may have detrimental effects if downstream enzymes are rate limiting, leading to an accumulation of toxic BER intermediates (Zverina et al. 1991; Glassner et al. 1998; Calvo et al. 2016; Allocca et al. 2019). Furthermore, many studies of DNA repair have been performed for a single time point, making it difficult to discern repair kinetics. Despite the importance of repair kinetics for resolving genomic DNA damage, DNA repair kinetics are rarely measured in population studies. This gap in the literature is primarily due to the lack of assays amenable to measurements of repair pathway completion in live cells over the course of multiple time points.
In addition to activity assays for specific repair components, there are also assays that measure responses to DNA damage (e.g., phosphorylation of H2AX [γH2AX] at sites of double strand breaks [DSBs] and replication stress). These assays have proven to be highly useful, however, they are not a direct measure of DNA repair kinetics. Indeed, we found that while physical DSBs were very rapidly cleared within the first hour, γH2AX levels remained above background for more than 6 hours afterwards (Weingeist et al. 2013). Thus, to learn more about the ability of cells to repair damaged DNA, we turned our attention to cell-based assays, where pathway kinetics can be directly assessed for DNA damage present in the genomes of cells.
Recently, two traditional cell-based DNA repair assays have been reengineered for use in population studies, namely the host cell reactivation assay (HCR), and the comet assay. The traditional HCR assay involved damaging plasmids and querying restoration of the DNA structure via expression of a reporter gene. To make the assay amenable to population studies, a multiplexed fluorescence-based flow cytometric host cell reactivation assay (FM-HCR) was developed to enable higher throughput analysis of DNA repair (Nagel et al. 2014; Chaim et al. 2017). In addition to HCR, the traditional comet assay is also an effective approach for evaluating DNA damage (Ostling and Johanson 1984; Singh et al. 1988; Hartmann et al. 2003; Collins 2004; Olive and Banath 2006). The underlying principle of the comet assay is that damaged DNA migrates more readily than undamaged DNA when electrophoresed through agarose. In particular, SSBs release superhelical tension, resulting in loops that pull away from the nucleus during electrophoresis, creating a plume with a comet-like shape. Although effective for measuring DNA damage, the conventional comet assay suffers from being low-throughput and from being highly variable from experiment to experiment. To overcome these limitations, we previously created the CometChip (Wood et al. 2010; Weingeist et al. 2013; Ge et al. 2015).
The basis for the CometChip is the ability to create microarrays of mammalian cells. By arraying cells ∼250 μm apart in microwells of an agarose chip, the resulting comets are on the same focal plane. Since people generally analyze 100 comets per sample (via imaging them one-by-one), being able to capture 100 comets in just one or two images reduces labor significantly. The format is a 96-well plate, which can be imaged in ∼15 minutes using automated systems, and images are then processed using in-house software. As a result, the CometChip is easier, more robust, and at least two orders of magnitude faster than the conventional comet assay, paving the way toward large-scale population studies of DNA repair kinetics.
Here, we leveraged the CometChip to measure repair kinetics for DNA oxidation damage in human peripheral blood mononuclear cells (PBMCs). Others had previously shown that the comet assay is compatible with analysis of oxidation damage to DNA starting with frozen leukocytes (Akor-Dewu et al. 2014; Bohn et al. 2019). In addition, we have previously shown that the CometChip is able to detect repair of DNA oxidation damage in live PBMCs (Sykora et al. 2018). Here, we have extended upon our prior work to study repair in dozens of individuals. Our rationale for selecting PBMCs reflects our interest in epidemiological studies. Specifically, there are many cryopreserved lymphocyte samples that have been banked as part of retrospective studies that can be used to study the relationship between DNA repair kinetics and disease. In addition, we have focused on oxidative stress because it is considered a critical driver of cancer, neurological diseases, heart disease and many other conditions (Cooke et al. 2003). As such, we have focused on reactive oxygen species (ROS) by challenging cells with H2O2, which induces a spectrum of damage similar to that produced endogenously during inflammation or following exposure to certain exogenous chemicals and radiation (Evans et al. 2004). Damage to DNA includes directly formed SSBs, oxidized bases, and the downstream base excision repair intermediates formed during repair of oxidized bases (Massie et al. 1972; Lesko et al. 1980; Demple and Linn 1982; Kennedy et al. 1997; Valverde et al. 2018), all of which are detectable using the CometChip. Key advances described here include the use of the CometChip for analysis of over 1,500 samples (equal to over 150,000 comets), which would be exceedingly difficult using the traditional comet assay. Taken together, this work demonstrates the utility of the CometChip for population studies and adds to comet literature pointing to significant differences in DNA repair capacity among individuals (Pool-Zobel et al. 1998; Jenkinson et al. 1999; Collins et al. 2001a; Collins et al. 2001b; Collins et al. 2003; Trzeciak et al. 2008a; Trzeciak et al. 2008b; Gaivao et al. 2009; Hoelzl et al. 2009; Chang et al. 2010; Nair-Shalliker et al. 2012; Trzeciak et al. 2012; Garm et al. 2013; Slyskova et al. 2014; Czarny et al. 2020; Kazmierczak-Baranska et al. 2020; Moller et al. 2020; Niu et al. 2020).
MATERIALS AND METHODS
CometChip fabrication
Materials.
Sylgar™ 184 silicone elastomer kit (102092-312) and bottomless 96-well plates (82050-714) were purchased from VWR, Radnor, PA. GelBond® Film (53761) was obtained from Lonza, Portsmouth, NH. UltraPure™ agarose (16500100) and UltraPure™ low melting point agarose (16520100) were purchased from ThermoFisher Scientific, Waltham, MA.
Procedure.
The microwells were fabricated as described previously (Wood et al. 2010; Ge et al. 2013; Weingeist et al. 2013; Ge et al. 2014; Ge et al. 2015). Briefly, 1% w/v agarose solution in Dulbecco’s phosphate-buffered saline (DPBS) was prepared. A polydimethylsiloxane (PDMS) stamp with an array of micropegs was fabricated using the Sylgar™ 184 kit, as described previously (Wood et al. 2010). The stamp was pressed into the molten agarose solution on top of the hydrophilic side of a sheet of GelBond® Film. The agarose was allowed to gelate at room temperature (RT) for ∼15 minutes. The stamp was removed to reveal an array of microwells with ∼40-50 μm in both diameter and depth. The microwells were spaced 240 μm apart. A bottomless 96-well plate was pressed on top of the agarose chip to form 96 macrowells. The bottom of each macrowell was an array of ∼300 microwells.
To load cells into microwells by gravity, ∼2,000 or more cells in suspension were placed into each macrowell, and the chip was incubated at 37°C in the presence of 5% CO2 for 15 minutes. Excess cells were then washed off with DPBS by shear force. The chip was covered with an agarose overlay (1% w/v low-melting point agarose solution in DPBS, kept molten at 43°C until use). For complete gelation of the overlay, the chip was kept at RT for 2 minutes followed by 2 minutes at 4°C.
Alkaline CometChip assay
Chemicals.
Sodium chloride (NaCl, 7581), disodium EDTA (Na2EDTA, 4931), and sodium hydroxide pellets (NaOH, 7708) were purchased from VWR, Radnor, PA. Trizma® base (T1503), Trizma® HCl (T5941), and Triton X-100 (X-100) were obtained from MilliporeSigma, St. Louis, MO. 10,000X SYBR™ Gold nucleic acid gel stain was obtained from ThermoFisher Scientific, Waltham, MA.
Buffers.
The lysis buffer (pH ∼ 10) is a solution of 2.5 M NaCl, 100 mM Na2EDTA, 10 mM Trizma® base, and 1% v/v Triton X-100. The alkaline unwinding buffer is 0.3 M NaOH and 1 mM Na2EDTA at pH 13.5. The neutralization buffer is 0.4 M Trizma® HCl at pH ∼7.5.
Procedure.
Cells encapsulated in the CometChip were incubated in lysis buffer overnight at 4°C. The nuclei were unwound in the alkaline unwinding buffer for 40 minutes at 4°C prior to electrophoresis in the same buffer and at the same temperature for 30 minutes at 1 V/cm and ∼300 mA. The CometChip was then washed three times in the neutralization buffer by submerging for five minutes each time at RT. The DNA was then stained via incubation in 1X SYBR™ Gold diluted in PBS for 15 minutes, protected from light. Fluorescent images of the comets were captured at 40X magnification using an epifluorescence microscope (Nikon Eclipse 80i, Nikon Instruments, Inc., Melville, NY) with a 480 nm excitation filter. Image acquisition was achieved by automatic scanning using a motorized XY stage. Comet images were automatically analyzed using Guicometanalyzer, a custom software developed in MATLAB (The MathWorks Inc., Natick, MA) as previously described (Wood et al. 2010). Outputs from Guicometanalyzer were processed and imported to a spreadsheet (Microsoft Excel, Microsoft Office Suite 2016) using Comet2Excel, an in-house software developed in Python (Python Software Foundation, Python version 2.7.10). Software is available upon request.
Culture of cell lines
Reagents.
RPMI-1640 and Pen-Strep were purchased from ThermoFisher Scientific, Waltham, MA. Fetal bovine serum (FBS) was obtained from Atlanta Biologicals, Inc., Flowery Branch, GA.
Cell lines.
TK6 cells (Skopek et al. 1978; Liber and Thilly 1982), a human B-lymphoblastoid cell line, were cultured in RPMI-1640 with GlutaMAX™ supplemented with 100 U/mL Pen-Strep. TK6 cell suspension was directly obtained from the suspension culture. Cell viability and cell number were analyzed using an automated Trypan Blue exclusion system (Vi-CELL™ cell counter [Beckman Coulter Life Sciences, Brea, CA]). TK6 cells were cryopreserved in multiple vials at the same passage to be used as an internal control for the assays.
Peripheral blood mononuclear cells (PBMCs)
Reagents.
Ficoll-Paque PLUS solution was purchased from GE Healthcare Bio-Sciences. Heat-inactivated fetal bovine serum (HI-FBS, 100-106) was obtained from Gemini Bio-Products, West Sacramento, CA. Phytohemagglutinin-L (PHA-L, L4144) and 100% dimethyl sulfoxide (DMSO, D8418) were obtained from MilliporeSigma, St. Louis, MO. D-Glucose (dextrose, BDH9230) was purchased from VWR, Radnor, PA.
Human whole blood.
For assay optimization and internal controls, fresh whole blood from one anonymous apparently healthy donor collected in sodium heparin Vacutainer collection tubes was purchased from Research Blood Components, Brighton, MA. For our population study, human subjects protocols were approved by the Committee on the Use of Humans as Experimental Subjects at MIT and informed consent was obtained from volunteers. Fresh whole blood from 56 healthy volunteers was collected in sodium heparin Vacutainer collection tubes by a clinical professional at the Clinical Research Center at Massachusetts Institute of Technology, Cambridge, MA. Ten of the 56 volunteers had multiple blood draws (up to five visits) over a period of 4-6 weeks while the remaining 46 volunteers had one single blood draw per person.
PBMC Isolation.
PBMCs were isolated using standard Ficoll gradient density centrifugation (Boyum 1968b; Boyum 1968a; Cheng et al. 2001). Briefly, 10 mL of fresh whole blood was diluted with 10 mL of warm RPMI-1640 solution. 8 mL of the Ficoll-Paque PLUS solution was gently injected underneath the diluted blood sample without mixing the two solutions. The whole tube was then centrifuged at 400 g for 40 minutes at 18°C with brakes off. The upper layer containing plasma and platelets was aspirated off, providing access to the PBMC layer (buffy coat). PBMCs were transferred to a new centrifuge tube. PBMCs were washed by diluting in warm RPMI-1640 solution to a final volume of 50 mL and centrifuged at 600 g for 20 minutes at 18°C. The supernatant was aspirated, and the pellet was resuspended in 20 mL of warm RPMI-1640 solution. Cells from 10 μL of the suspension were stained with crystal violet, and the number of mononuclear cells were counted with a hemocytometer. The remaining suspension was centrifuged at 400 g for 15 minutes at 18°C. The supernatant was aspirated, and the pellet was suspended in the freezing medium (40% RPMI-1640 + 50% HI-FBS + 10% DMSO). The volume for the freezing medium was one-tenth the volume of the original blood sample (i.e., 1 mL freezing medium to suspend a pellet isolated from 10 mL of fresh whole blood). The average cell density was ∼10x106 cells/mL.
PBMC cryopreservation and storage.
1 mL aliquots of PBMCs suspended in the freezing medium were transferred to separate cryovials. The vials were stored in a Styrofoam container and placed in a −80°C freezer. After 24 hours, the frozen vials were transferred to a Dewar containing liquid nitrogen.
PBMC recovery and T-lymphocyte stimulation.
Cryopreserved PBMCs were rapidly thawed by placing the vial containing the cells in a 37°C water bath for ∼1 minute. Cells were transferred to a centrifuge tube containing 9 mL warm thawing medium (40% RPMI-1640 + 50% HI-FBS + 10% D-glucose) and centrifuged at 600 g for 5 minutes at RT. The pellet was resuspended in 10 mL of stimulation medium (RMPI-1640 + 20 % HI-FBS + 100 U/mL Pen-Strep + 5 μg/mL PHA-L). 500 μL of the suspension was analyzed for cell viability using an automated Trypan Blue exclusion system (Vi-CELL™ cell counter). The recovery procedure typically yielded more than 85% viable cells. T-lymphocytes were stimulated for three days by placing the remaining suspension in a tissue-culture incubator (37°C, 5% CO2).
CometChip cell loading.
For each experiment, ∼10x105 of PHA-stimulated T-lymphocytes were harvested and suspended in 5 mL of complete medium (RPMI-1640 with 20% HI-FBS and 100 U/mL Pen-Strep). Cells were loaded into microwells by gravity by placing 50 μL of the suspension into a macrowell and incubating for 15 minutes in a tissue-culture incubator (37°C, 5% CO2).
DNA damage induction and evaluation of repair kinetics
H2O2 treatment.
Doses of H2O2 were prepared immediately before use by diluting 30% stock solution (10 M) with cold PBS. Suspension of TK6 cells was obtained directly from exponentially growing culture and loaded onto the CometChip. A new bottomless 96-well plate was placed on top of the CometChip and 100 μL of 100 μM H2O2 solution was pipetted into each well. The CometChip was then incubated at 4°C for 20 minutes (protected from light). Afterward, the H2O2 solution was aspirated, and the bottomless plate was taken off. The CometChip was rinsed by submerging in cold PBS.
DNA damage and repair.
To analyze DNA damage immediately after treatment, the CometChip was placed in cold lysis buffer and processed following the remaining steps of the alkaline comet assay. To study repair kinetics, the CometChip was cut into ∼5 cm x 5 cm pieces using a pair of sterile surgical scissors and incubated in culture medium in a tissue-culture incubator (37°C, 5% CO2) for up to two hours. At each time point, a piece of the CometChip was removed and placed in cold lysis buffer prior to standard processing, as described above.
Data analysis of repair kinetics
Multiple comparisons between individuals.
One-way ANOVA followed by Tukey-Kramer’s multiple comparison test was performed using the Real Statistics Resource Pack software (Release 5.1).
Mathematical modeling.
Background-corrected data were obtained by subtracting the baseline damage level from the DNA damage level at each repair time point. Non-linear regression was performed using GraphPad Prism version 7.01 for Windows to fit the background-corrected data to a biphasic exponential decay model:
where f(t) is the level of DNA damage at time t, kf and Ks are two rate constants corresponding to the fast and the slow phases of the repair kinetics, and f(0) = F + S is the initial DNA damage level. Therefore, if t1/2 is the time required to repair half of the initial damage (half-time), then
The value of t1/2 was approximated using the Solver add-in in Microsoft Excel (Microsoft Office Suite 2016).
RESULTS
Quantification of repair kinetics for DNA oxidation damage in PBMCs from healthy volunteers
To study inter-individual variation in repair capacity for oxidative damage to DNA, we set out to measure H2O2-induced DNA damage and its repair in PBMCs. PBMCs were isolated from blood drawn from 56 healthy volunteers including 29 females and 27 males who were between 21 and 66 years old. Isolated PBMCs from each blood sample were viably cryopreserved in four separate vials and stored in liquid nitrogen (three vials were used for subsequent studies). Note that there is evidence that repair is correlated in fresh versus cryopreserved lymphocytes (Trzeciak et al. 2008b). For each experiment, cells from one vial were thawed, and T-lymphocytes were stimulated to divide using PHA-L for three days (Nowell 1960; Chen et al. 2003) before exposure to H2O2 (Figure 1). Our rationale for using PHA is that it increases sensitivity of the assay, enabling more robust assessment of DNA repair kinetics. Three independent experiments, corresponding to three vials of PBMCs thawed on different days, were analyzed using the alkaline comet approach, which is appropriate for studies of SSBs.
Figure 1.

Experimental workflow. For each blood draw, lymphocytes (PBMCs) were purified and cryopreserved in four vials. Three vials were used for subsequent analysis. Vials were thawed and cultured with PHA for three days. Cells were then loaded in the CometChip for H2O2 treatment, lysis, electrophoresis, imaging, and analysis.
Repair kinetics for control lymphoblastoid cells and primary PBMCs
We included TK6 lymphoblastoid cells as an internal control for each experiment where we tested DNA repair kinetics in PBMCs. In preparation for the study, TK6 cells were therefore cultured, cryopreserved in multiple vials at the same passage, and stored in liquid nitrogen. In addition, to control for PBMC processing procedures (e.g., thawing and PHA-stimulation), we also included PBMCs from one individual (#00) in all of the experiments. We purchased 440 mL of whole blood from #00, isolated PBMCs, and stored multiple vials of cryopreserved cells in liquid nitrogen.
Repair of DNA oxidation damage was evaluated as previously described (Ge et al. 2014). Specifically, PHA-stimulated lymphocytes were embedded in the CometChip, exposed to 100 μM H2O2 for 20 minutes at 4°C in the dark and then submerged in medium, allowing up to two hours for repair. As shown in Figure 2A, both TK6 cells and PHA-stimulated lymphocytes from #00 repair nearly all of the H2O2-induced DNA damage within 60 minutes. While most of the damage is cleared within 30 minutes for the TK6 cells, there is still residual damage in the #00 cells, indicating a slower rate of repair. We therefore estimated the repair rate by fitting a biphasic exponential decay model to the data, as had been done previously with primary PBMCs exposed to radiation (Trzeciak et al. 2008b). We then used the resulting equations to calculate the time it takes to repair 50% of the initial damage (t1/2) for both cell types (Figure 2B). Notably, the t1/2 for TK6 cells is ∼24 minutes, which is >1.5 times faster than that of sample #00. In terms of analysis of kinetics in PBMCs, these experiments show that repair of oxidative damage to DNA is quite rapid, and that the t1/2 can be estimated via a non-linear regression model.
Figure 2.

Repair kinetics for the TK6 cell line and PBMCs from one individual (#00), whose cells served as an internal control. (A) Baseline damage is indicated as untreated (UT). DNA damage was induced with 100 μM H2O2 at time 0, and then repair was allowed to occur for 15, 30, 60, 90, and 120 minutes in warm media. (B) Background corrected graphs of repair kinetics profiles. Repair rates were estimated by fitting a biphasic exponential decay model to the data. The resulting equations were used to calculate the time it takes to repair 50% of the initial DNA damage (t1/2). Mean and standard error of the mean (SEM) are shown.
Analysis of intra-individual variation in DNA repair kinetics
Before investigating inter-individual variation in repair capacity, we first studied the variability within individuals over time (intra-individual variation). Ten volunteers from our pilot study (#01, #02, #05, #06, #09, #11, #12, #18, #19, and #23) had their blood drawn in multiple visits on different days, over the span of 4-6 weeks. PBMCs were isolated from each of 4-5 blood draws and cryopreserved prior to storage in liquid nitrogen. To measure DNA repair capacity, the cryopreserved PBMCs were thawed, stimulated with PHA-L, and then analyzed using the CometChip assay. The repair curves for PBMCs obtained from serial visits by individuals #01, #02, #05, #06, #09, #11, #12, #18, #19, and #23 are shown in Figure 3A. Visual inspection shows that repair curves for each individual are quite consistent from visit to visit.
Figure 3.

Repair kinetics for individuals who provided multiple samples. (A) Repair kinetics curves of 10 individuals who each came in for four to five repeat blood draws. (B) Repair curves for the average of all visits for each individual. Differences between individuals at each timepoint were evaluated using analysis of variance (one-way ANOVA). (C) Background corrected graphs of repeat visit individuals’ repair kinetics profiles. Calculated values of t1/2 for each individual are indicated. *statistically significant differences (p < 0.05). Mean and SEM are shown on all graphs.
Inter-individual differences in DNA repair kinetics
We next studied variability in damage levels among individuals. After averaging across visits, there is inter-individual variation in the initial levels of damage, as well the kinetics of repair. Specifically, there is a larger variability among individuals than within individuals when sampled at different times (Figure 3B). Statistically significant differences (p < 0.05, one-way ANOVA) are observed for the baseline damage level (UT) for different individuals, as well as at all repair time points (with the exception of the values immediately after exposure to H2O2) (Figure 3B). We also performed post-hoc analysis (Tukey’s honest significance test) to query whether there are statistically significant differences when the level of DNA damage for one individual is compared to the others (Table S1). Specifically, for each time point we queried whether a person’s damage levels are statistically significantly different from any of the other individuals. Only one person (#23) displays no statistically significant difference from anyone else. Interestingly, only one pair of individuals (#05 and #09) shows a significant difference at the baseline damage level, whereas six pairs differ significantly at the latest repair time point (Table S1). To learn more about differences among people, we quantified the rate of repair for each individual, based on the average value for all of the visits. Similar to TK6 and #00, we applied biphasic exponential decay equations to calculate t1/2 for each person’s PBMCs (Figure 3C).
Variability in repair kinetics of oxidative damage to DNA among 56 individuals
While serial blood draws yield information about intra-individual variability in repair kinetics, a single blood draw enables larger studies of inter-individual variation. To investigate differences in DNA repair capacity among people from a larger population, we studied PBMCs from 56 volunteers (the first blood draw from the 10 individuals whose data is displayed in Figure 3 plus an additional 46 individuals who came in for a single blood draw). Isolated PBMCs were again stimulated with PHA-L, treated with 100 μM H2O2, and analyzed with the alkaline CometChip assay. The repair kinetic curves for each individual, shown in Figure 4A, are the average results from three independent vials collected from a single blood draw. Visual inspection shows that curve shapes are highly variable among the 56 volunteers. Further analysis of the repair curves for specific individuals shows that there is variation in the shapes of the repair curves among individuals (Figure 4A). For example, #03, #04, and #38 rapidly repair most of the initial damage within the first 30 minutes, and there is no significant reduction of strand breaks after 30 minutes (Figure 4A). On the other hand, #19 and #26 display a gradual reduction in strand breaks over the entire 120 minutes of repair (Figure 4A).
Figure 4.

Repair kinetics for 56 individuals. (A) Repair kinetics curves of a single blood draw from each of 56 individuals. Note that for the 10 individuals who came in for repeat visits, only data from the first visit is included. Mean and SEM are shown. Kinetics for all individuals are shown. (B) Coefficient of variation (CV) values of technical replicates, among visits, and among individuals. (C) Rank ordered estimation of t1/2 for each individual, separating individuals into four quartiles of repair: very fast (pink), fast (blue), medium (green), and slow (orange).
Despite variation among experiments (indicated by the error bars showing the SEM for each time point), Figure S1 shows that the variability among people is significantly greater than the variability among experiments at 30 and 60 minutes (see also Tables S2 and S3) (p < 0.05, one-way ANOVA for 46 single visit individuals). The coefficient of variation (CV) was also calculated for technical replicates, among visits, and among individuals. As illustrated by Figure 4B, the CV is very similar for technical replicates and among visits at most timepoints, while the CV values are consistently higher among individuals. To quantify the repair rates, we parameterized the kinetic curves by fitting the data to a biphasic exponential decay model, as described above. Results from one blood draw from each of the 56 volunteers show a broad range of t1/2 values, with a 9.5-fold difference between the fastest repair rate (13 minutes) and the slowest repair rate (123 minutes) (Figure 4C). We then divided the values of t1/2 into quartiles, representing individuals with “very fast”, “fast”, “medium”, and “slow” repair (Figures 4A and 4C). As expected, #03, #04, and #38 (described above as showing nearly complete repair within the first 30 minutes) all belong to the “very fast” group. Additionally, both #19 and #26 belong to the “slow” repair group, with #19 being the slowest of the 56 people. The average t1/2 values for the “very fast”, “fast”, “medium”, and “slow” repair groups are 24 minutes, 35 minutes, 50 minutes, and 81 minutes, respectively. A closer examination of the “very fast” group reveals a common trend, where most of the initial DNA damage is rapidly reduced during the first 30-60 minutes (rapid phase) while any further reduction of damage afterward appears to be much slower (slow phase) (Figure 5A). The shapes of the “fast” and “medium” group show features in between the “very fast” and “slow” groups (Figure 5B and 5C). Furthermore, the “slow” group appears to have a relatively consistent rate of damage reduction over the entire 120 minutes (Figure 5D).
Figure 5.

Comparisons of repair kinetics among quartiles and individuals. Background corrected kinetics of the 56 individuals grouped by the repair kinetics groups (A) very fast, (B) fast, (C) medium, (D) slow, as indicated in Figure 4. (E) Background corrected data for cells that had the fastest and the slowest repair kinetics. (F) Representative background corrected data for the slow and medium sets. Mean and SEM are shown for all repair kinetics graphs. (G) PROAST t1/2 values of 46 single visit individuals with non-overlapping confidence intervals.
Looking at repair curves for specific individuals is also informative. Figure 5E shows the results for individuals with the fastest (#05) and the slowest time to repair initial damage (#19), which clearly have different repair curves. There are also interesting differences between the repair rates of individuals in the “medium” and “slow” repair groups. In Figure 5F, a comparison between #19 and #46 (from the “slow” and “medium” groups, respectively) shows that both samples have comparable strand break levels at the earlier repair time points (0, 15, and 30 minutes), however, #46 has a repair rate ∼2 fold faster (t1/2 = 57 minutes vs 123 minutes) and thus the amount of remaining damage differs at the later time points (60 and 120 minutes).
Since there are four fitted parameters in the biphasic exponential decay model, a risk of overfitting exists. To test the robustness of the information derived from the biphasic exponential decay model, we estimated the repair half-times for the 46 single-visit individuals using an alternative analysis approach for nonlinear regression, namely PROAST (Slob 2014a; Slob 2014b). Importantly, analysis using the PROAST model is quite consistent with results using the biphasic model, described above. For example, similar to the biphasic model, the PROAST approach again shows over a seven-fold difference in t1/2 values between the fastest (18 minutes) and the slowest (130 minutes) (Figure S2). Further, most of the 23 individuals with the fastest PROAST repair rates (top half) belong to the “very fast” and “fast” groups from the biphasic model (Figure S2). The 23 slowest PROAST repair rates (bottom half) also appear to correlate well with the “medium” and “slow” groups (Figure S2).
In order to learn more about the statistical significance of the differences between the PROAST t1/2 values, we compared their 90% confidence intervals (90% CIs). Figure 5G highlights two groups where the 90% CIs do not overlap, indicating that one group has statistically significantly faster repair than the other. Importantly, most of the members of the “faster” PROAST group are also members of the “very fast” and “fast” groups from the biphasic model, while almost all of the “slower” PROAST group members belong to the biphasic “slow” group. Taken together, the results from both the biphasic model and the PROAST analysis show that there is a wide range of repair rates among PBMCs from apparently healthy individuals, and that distinct groups with significantly different repair rates can be identified.
DISCUSSION
DNA damage causes mutations that drive cancer. Nevertheless, relatively few studies have been performed to explore variation in DNA repair capacity among people. Interestingly, while there are over 10,000 studies using the comet assay (PubMed search on “comet”), to our knowledge, only one research team has measured t1/2 in over a dozen people (Trzeciak et al. 2008a; Trzeciak et al. 2008b; Trzeciak et al. 2012), since this requires quantification of DNA damage at multiple time points post exposure which is difficult to do using the traditional comet assay. Thus, despite broad appreciation of the importance of DNA repair as a modulator of cancer risk and response to cancer therapy, there has been scant use of the comet assay for population studies of DNA repair capacity. Here, we have analyzed DNA repair capacity in over 50 people, and we have revealed significant differences in repair capacity among individuals.
We had previously developed the CometChip, a higher throughput and more robust version of the well-established traditional comet assay (Wood et al. 2010), which is effective for analysis of many kinds of DNA damage, including oxidative damage to DNA, which is an important cancer susceptibility factor (Wallace 2014). Here, we have applied the CometChip to learn about variation in repair kinetics for oxidative damage to DNA. A strength of the approach is that the assay detects repair of both directly-induced SSBs as well as base lesions (revealed by the kinetics of clearance of BER intermediates). We also observed that there is even greater variability when comparing DNA repair kinetics among individuals. Looking at repair kinetics for specific individuals, we find that the dynamics of DNA repair are variable, with some individuals showing initially rapid repair followed by slower repair kinetics, while others show a steady decline in damage levels. Being able to discern such differences in the shape of the repair kinetics curve enables future studies to learn the impact of rapid versus slow initial clearance of DNA damage. Together, these studies show the efficacy of the CometChip for population studies and thus open doors to new understanding of how endogenous and environmental factors impinge on risk of cancer.
To study repair kinetics, we measured the initial levels of DNA damage, as well as the levels of DNA damage at multiple timepoints following exposure to oxidative stress. Using a biphasic model, we calculated the t1/2 for each individual’s PBMCs, and we rank-ordered individuals based on their repair kinetics. We observed significant differences in t1/2 for the fastest and the slowest repair groups. Using an alternative approach for deriving t1/2 and for calculating confidence intervals (PROAST), the fastest and the slowest repair groups show statistically significant differences in repair kinetics.
We found that inter-individual variation is higher than both technical and intra-individual variation, pointing to a possible role for genetics and/or environmental factors in influencing repair rate, as has been previously reported (Garm et al. 2013; Slyskova et al. 2014; Czarny et al. 2020; Moller et al. 2020; Niu et al. 2020). In terms of the range of repair rates, we observed a 9.5-fold variation in t1/2 of H2O2-induced damage when analyzing a single blood draw from 56 individuals. This magnitude in difference among individuals is similar to previous estimates using cell extracts (Gaivao et al. 2009). We also observed differences in repair kinetics between visits for the same individual. However, these differences were similar in magnitude to differences among technical repeats, so the extent to which DNA repair kinetics vary over time for the same person is not clear from these particular studies. Notably, previous studies using either the live cell comet assay (e.g. the conditions in this report) or using cell extracts (e.g. the “in vitro” comet assay) have shown that steady-state DNA damage levels vary over time for the same individual, and furthermore that they may be affected by diet and lifestyle (Pool-Zobel et al. 1998; Jenkinson et al. 1999; Collins et al. 2001b; Collins et al. 2003; Freese 2006; Briviba et al. 2008; Hoelzl et al. 2009; Chang et al. 2010; Fenech and Bonassi 2011; Thomas et al. 2011; Nair-Shalliker et al. 2012; Slyskova et al. 2014; Xie et al. 2015; Kazmierczak-Baranska et al. 2020). Further studies are warranted to learn more about the extent to which intra-individual variation in repair kinetics can be influenced by diet or other environmental factors.
Replication forks can break down when they encounter SSBs formed during BER (Yang et al. 2004; Ensminger et al. 2014). BRCA2 is one of the key proteins involved in the repair of the resulting double strand breaks (Scully and Livingston 2000; Nagaraju and Scully 2007). PARP inhibitors are now well established as cancer chemotherapeutics, and their mode of action is to suppress the rate of single strand break repair, which increases replication fork breakdown events. Broken forks are repaired by BRCA2-mediated homologous recombination, so PARP inhibitors are particularly toxic for cells deficient in homologous recombination, such as BRCA2 deficient tumors (Curtin and Szabo 2020; Rose et al. 2020). Remarkably, with a potent PARP inhibitor, the t1/2 for BER increases ∼8 fold, going from ∼3 minutes to ∼24 minutes (Strom et al. 2011) which is similar in magnitude to the difference between the fastest and slowest repair rates (the t1/2 ranged from 13 minutes to 123 minutes). As such, differences in repair rates among individuals may affect likelihood of spontaneous fork breakdown events, which could affect sensitivity to defects in BRCA2 (as well as other proteins involved in fork repair).
There are many valuable approaches for studying BER. For example, incubation of extracts with oligonucleotides containing site-specific DNA adducts is an excellent approach for assessing efficiency of particular steps in BER (Sauvaigo et al. 2004; Li et al. 2018; Healing et al. 2019). Another approach that more closely reflects pathway efficiency is a modified comet assay wherein cell extracts are incubated with damaged nucleoid substrates (reviewed in (Collins et al. 2001a)). Another option is the FM-HCR assay, which allows for monitoring of repair of specific DNA lesions in living cells (Nagel et al. 2014; Chaim et al. 2017; Nagel et al. 2019). While proven effective, a key difference between these approaches and the comet assay is that they do not detect repair in the context of intact chromatin. Clearly, using multiple approaches to assess DNA repair will deepen our mechanistic understanding of the relationship between BER and disease. Interestingly, despite the difficulties of performing the traditional comet assay, there have nevertheless been detailed studies of DNA repair kinetics among people following exposure to ionizing radiation (Trzeciak et al. 2008a; Trzeciak et al. 2012). These studies showed that there are biphasic repair kinetics, and that the rate of the fast component of repair is impacted by sex, age, and race. Extending upon these exciting results is now more feasible and will likely lead to a deeper understanding of the impacts of these important factors.
Looking ahead, there remain some challenges to be addressed. First, there is variation in the results among different vials of PBMCs collected at the same time. Future studies could be directed toward evaluating the underlying cause of such variability, which may reflect differences in sample handling among experiments. In addition, comparing the same sample among multiple experiments (for TK6 and #00) showed some evidence of a batch effect. It is noteworthy that we observed significant differences between individuals even without any sort of correction for batch effects. Nevertheless, further study is warranted as correcting for batch effects could be a way to suppress experimental noise.
Taken together, using the CometChip, we have revealed significant differences in DNA repair capacity among individuals, consistent with previous studies (Pool-Zobel et al. 1998; Jenkinson et al. 1999; Collins et al. 2001a; Collins et al. 2001b; Collins et al. 2003; Freese 2006; Briviba et al. 2008; Trzeciak et al. 2008a; Trzeciak et al. 2008b; Hoelzl et al. 2009; Chang et al. 2010; Fenech and Bonassi 2011; Thomas et al. 2011; Nair-Shalliker et al. 2012; Trzeciak et al. 2012; Slyskova et al. 2014; Xie et al. 2015; Kazmierczak-Baranska et al. 2020). Given the higher throughput of the CometChip compared to the traditional assay, this work calls attention to the utility of this approach for future population studies, with relevance to both public health and personalized medicine.
Supplementary Material
Highlights.
The comet assay can be used to measure DNA oxidation damage
CometChip is a higher throughput DNA damage assay
CometChip can be used to measure repair kinetics
Repair rates have been measured in dividing lymphocytes (PBMCs)
Repair capacity varies for PBMCs from different individuals
ACKNOWLEDGMENTS
This work was supported by the National Institute of Environmental Health Sciences (5U01ES029520, and P30ES000002 to Z.D.N.); the Superfund Research Program (P42 ES027707); and the Massachusetts Institute of Technology Center for Environmental Health Sciences (P30-ES002109). Funding sources were not involved in study design, collection, analysis, or interpretation of data, nor in the decision to submit the article for publication. Figure 1 was created using Biorender.com.
Footnotes
Declaration of competing financial interests: B.P.E. is a co-inventor on the patent for the CometChip.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Akor-Dewu MB, El Yamani N, Bilyk O, Holtung L, Tjelle TE, Blomhoff R, Collins AR. 2014. Leucocytes isolated from simply frozen whole blood can be used in human biomonitoring for DNA damage measurement with the comet assay. Cell Biochem Funct 32: 299–302. [DOI] [PubMed] [Google Scholar]
- Allocca M, Corrigan JJ, Mazumder A, Fake KR, Samson LD. 2019. Inflammation, necrosis, and the kinase RIP3 are key mediators of AAG-dependent alkylation-induced retinal degeneration. Sci Signal 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn SK, Vebraite V, Shaposhnikov S, Collins AR. 2019. Isolation of leukocytes from frozen buffy coat for comet assay analysis of DNA damage. Mutat Res 843: 18–23. [DOI] [PubMed] [Google Scholar]
- Boyum A 1968a. Isolation of mononuclear cells and granulocytes from human blood. Isolation of monuclear cells by one centrifugation, and of granulocytes by combining centrifugation and sedimentation at 1 g. Scand J Clin Lab Invest Suppl 97: 77–89. [PubMed] [Google Scholar]
- -. 1968b. Separation of leukocytes from blood and bone marrow. Introduction. Scand J Clin Lab Invest Suppl 97: 7. [PubMed] [Google Scholar]
- Briviba K, Bub A, Moseneder J, Schwerdtle T, Hartwig A, Kulling S, Watzl B. 2008. No differences in DNA damage and antioxidant capacity between intervention groups of healthy, nonsmoking men receiving 2, 5, or 8 servings/day of vegetables and fruit. Nutr Cancer 60: 164–170. [DOI] [PubMed] [Google Scholar]
- Calvo JA, Allocca M, Fake KR, Muthupalani S, Corrigan JJ, Bronson RT, Samson LD. 2016. Parp1 protects against Aag-dependent alkylation-induced nephrotoxicity in a sex-dependent manner. Oncotarget 7: 44950–44965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaim IA, Nagel ZD, Jordan JJ, Mazzucato P, Ngo LP, Samson LD. 2017. In vivo measurements of interindividual differences in DNA glycosylases and APE1 activities. Proc Natl Acad Sci U S A 114: E10379–E10388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JL, Chen G, Ulrich CM, Bigler J, King IB, Schwarz Y, Li S, Li L, Potter JD, Lampe JW. 2010. DNA damage and repair: fruit and vegetable effects in a feeding trial. Nutr Cancer 62: 329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SK, Hsieh WA, Tsai MH, Chen CC, Hong AI, Wei YH, Chang WP. 2003. Age-associated decrease of oxidative repair enzymes, human 8-oxoguanine DNA glycosylases (hOgg1), in human aging. J Radiat Res 44: 31–35. [DOI] [PubMed] [Google Scholar]
- Cheng L, Wang LE, Spitz MR, Wei Q. 2001. Cryopreserving whole blood for functional assays using viable lymphocytes in molecular epidemiology studies. Cancer Lett 166: 155–163. [DOI] [PubMed] [Google Scholar]
- Collins AR. 2004. The comet assay for DNA damage and repair: principles, applications, and limitations. Mol Biotechnol 26: 249–261. [DOI] [PubMed] [Google Scholar]
- Collins AR, Dusinska M, Horvathova E, Munro E, Savio M, Stetina R. 2001a. Inter-individual differences in repair of DNA base oxidation, measured in vitro with the comet assay. Mutagenesis 16: 297–301. [DOI] [PubMed] [Google Scholar]
- Collins AR, Harrington V, Drew J, Melvin R. 2003. Nutritional modulation of DNA repair in a human intervention study. Carcinogenesis 24: 511–515. [DOI] [PubMed] [Google Scholar]
- Collins BH, Horska A, Hotten PM, Riddoch C, Collins AR. 2001b. Kiwifruit protects against oxidative DNA damage in human cells and in vitro. Nutr Cancer 39: 148–153. [DOI] [PubMed] [Google Scholar]
- Collins FS, Varmus H. 2015. A new initiative on precision medicine. N Engl J Med 372: 793–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke MS, Evans MD, Dizdaroglu M, Lunec J. 2003. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J 17: 1195–1214. [DOI] [PubMed] [Google Scholar]
- Cortesi L, Rugo HS, Jackisch C. 2021. An Overview of PARP Inhibitors for the Treatment of Breast Cancer. Target Oncol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtin NJ, Szabo C. 2020. Poly(ADP-ribose) polymerase inhibition: past, present and future. Nat Rev Drug Discov 19: 711–736. [DOI] [PubMed] [Google Scholar]
- Czarny P, Bialek K, Ziolkowska S, Strycharz J, Sliwinski T. 2020. DNA damage and repair in neuropsychiatric disorders. What do we know and what are the future perspectives? Mutagenesis 35: 79–106. [DOI] [PubMed] [Google Scholar]
- Demple B, Linn S. 1982. 5,6-Saturated thymine lesions in DNA: production by ultraviolet light or hydrogen peroxide. Nucleic Acids Res 10: 3781–3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Kimmel M, Foy M, Spitz M, Wei Q, Gorlova O. 2009. Estimation of the effects of smoking and DNA repair capacity on coefficients of a carcinogenesis model for lung cancer. Int J Cancer 124: 2152–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ensminger M, Iloff L, Ebel C, Nikolova T, Kaina B, Lbrich M. 2014. DNA breaks and chromosomal aberrations arise when replication meets base excision repair. J Cell Biol 206: 29–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans MD, Dizdaroglu M, Cooke MS. 2004. Oxidative DNA damage and disease: induction, repair and significance. Mutat Res 567: 1–61. [DOI] [PubMed] [Google Scholar]
- Fenech M, Bonassi S. 2011. The effect of age, gender, diet and lifestyle on DNA damage measured using micronucleus frequency in human peripheral blood lymphocytes. Mutagenesis 26: 43–49. [DOI] [PubMed] [Google Scholar]
- Freese R 2006. Markers of oxidative DNA damage in human interventions with fruit and berries. Nutr Cancer 54: 143–147. [DOI] [PubMed] [Google Scholar]
- Gaivao I, Piasek A, Brevik A, Shaposhnikov S, Collins AR. 2009. Comet assay-based methods for measuring DNA repair in vitro; estimates of inter- and intra-individual variation. Cell Biol Toxicol 25: 45–52. [DOI] [PubMed] [Google Scholar]
- Garm C, Moreno-Villanueva M, Burkle A, Larsen LA, Bohr VA, Christensen K, Stevnsner T. 2013. Genetic and environmental influence on DNA strand break repair: a twin study. Environ Mol Mutagen 54: 414–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge J, Chow DN, Fessler JL, Weingeist DM, Wood DK, Engelward BP. 2015. Micropatterned comet assay enables high throughput and sensitive DNA damage quantification. Mutagenesis 30: 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge J, Prasongtanakij S, Wood DK, Weingeist DM, Fessler J, Navasummrit P, Ruchirawat M, Engelward BP. 2014. CometChip: a high-throughput 96-well platform for measuring DNA damage in microarrayed human cells. J Vis Exp: e50607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge J, Wood DK, Weingeist DM, Prasongtanakij S, Navasumrit P, Ruchirawat M, Engelward BP. 2013. Standard fluorescent imaging of live cells is highly genotoxic. Cytometry A 83: 552–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glassner BJ, Rasmussen LJ, Najarian MT, Posnick LM, Samson LD. 1998. Generation of a strong mutator phenotype in yeast by imbalanced base excision repair. Proc Natl Acad Sci U S A 95: 9997–10002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann A, Agurell E, Beevers C, Brendler-Schwaab S, Burlinson B, Clay P, Collins A, Smith A, Speit G, Thybaud V et al. 2003. Recommendations for conducting the in vivo alkaline Comet assay. 4th International Comet Assay Workshop. Mutagenesis 18: 45–51. [DOI] [PubMed] [Google Scholar]
- Healing E, Charlier CF, Meira LB, Elliott RM. 2019. A panel of colorimetric assays to measure enzymatic activity in the base excision DNA repair pathway. Nucleic Acids Res 47: e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoelzl C, Knasmuller S, Misik M, Collins A, Dusinska M, Nersesyan A. 2009. Use of single cell gel electrophoresis assays for the detection of DNA-protective effects of dietary factors in humans: recent results and trends. Mutat Res 681: 68–79. [DOI] [PubMed] [Google Scholar]
- Jenkinson AM, Collins AR, Duthie SJ, Wahle KW, Duthie GG. 1999. The effect of increased intakes of polyunsaturated fatty acids and vitamin E on DNA damage in human lymphocytes. FASEB J 13: 2138–2142. [DOI] [PubMed] [Google Scholar]
- Kazmierczak-Baranska J, Boguszewska K, Karwowski BT. 2020. Nutrition Can Help DNA Repair in the Case of Aging. Nutrients 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy LJ, Moore K Jr., Caulfield JL, Tannenbaum SR, Dedon PC. 1997. Quantitation of 8-oxoguanine and strand breaks produced by four oxidizing agents. Chem Res Toxicol 10: 386–392. [DOI] [PubMed] [Google Scholar]
- Krokan HE, Bjoras M. 2013. Base excision repair. Cold Spring Harb Perspect Biol 5: a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar N, Raja S, Van Houten B. 2020. The involvement of nucleotide excision repair proteins in the removal of oxidative DNA damage. Nucleic Acids Res 48: 11227–11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitner-Dagan Y, Sevilya Z, Pinchev M, Kremer R, Elinger D, Rennert HS, Schechtman E, Freedman L, Rennert G, Livneh Z et al. 2014. Enzymatic MPG DNA repair assays for two different oxidative DNA lesions reveal associations with increased lung cancer risk. Carcinogenesis 35: 2763–2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leong TL, Gayevskiy V, Steinfort DP, De Massy MR, Gonzalez-Rajal A, Marini KD, Stone E, Chin V, Havryk A, Plit M et al. 2019. Deep multi-region whole-genome sequencing reveals heterogeneity and gene-by-environment interactions in treatment-naive, metastatic lung cancer. Oncogene 38: 1661–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesko SA, Lorentzen RJ, Ts'o PO. 1980. Role of superoxide in deoxyribonucleic acid strand scission. Biochemistry 19: 3023–3028. [DOI] [PubMed] [Google Scholar]
- Li J, Svilar D, McClellan S, Kim JH, Ahn EE, Vens C, Wilson DM 3rd, Sobol RW. 2018. DNA Repair Molecular Beacon assay: a platform for real-time functional analysis of cellular DNA repair capacity. Oncotarget 9: 31719–31743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liber HL, Thilly WG. 1982. Mutation assay at the thymidine kinase locus in diploid human lymphoblasts. Mutat Res 94: 467–485. [DOI] [PubMed] [Google Scholar]
- Marsden CG, Dragon JA, Wallace SS, Sweasy JB. 2017. Base Excision Repair Variants in Cancer. Methods Enzymol 591: 119–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massie HR, Samis HV, Baird MB. 1972. The kinetics of degradation of DNA and RNA by H 2 O 2. Biochim Biophys Acta 272: 539–548. [DOI] [PubMed] [Google Scholar]
- Meira LB, Burgis NE, Samson LD. 2005. Base Excision Repair . in Genome instability in cancer development (ed. Nigg EA), pp. 125–173. Springer, New York. [Google Scholar]
- Moller P, Stopper H, Collins AR. 2020. Measurement of DNA damage with the comet assay in high-prevalence diseases: current status and future directions. Mutagenesis 35: 5–18. [DOI] [PubMed] [Google Scholar]
- Nagaraju G, Scully R. 2007. Minding the gap: the underground functions of BRCA1 and BRCA2 at stalled replication forks. DNA Repair (Amst) 6: 1018–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel ZD, Beharry AA, Mazzucato P, Kitange GJ, Sarkaria JN, Kool ET, Samson LD. 2019. Fluorescent reporter assays provide direct, accurate, quantitative measurements of MGMT status in human cells. PLoS One 14: e0208341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel ZD, Engelward BP, Brenner DJ, Begley TJ, Sobol RW, Bielas JH, Stambrook PJ, Wei Q, Hu JJ, Terry MB et al. 2017. Towards precision prevention: Technologies for identifying healthy individuals with high risk of disease. Mutat Res 800-802: 14–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel ZD, Margulies CM, Chaim IA, McRee SK, Mazzucato P, Ahmad A, Abo RP, Butty VL, Forget AL, Samson LD. 2014. Multiplexed DNA repair assays for multiple lesions and multiple doses via transcription inhibition and transcriptional mutagenesis. Proc Natl Acad Sci U S A 111: E1823–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair-Shalliker V, Armstrong BK, Fenech M. 2012. Does vitamin D protect against DNA damage? Mutat Res 733: 50–57. [DOI] [PubMed] [Google Scholar]
- Niu BY, Li WK, Li JS, Hong QH, Khodahemmati S, Gao JF, Zhou ZX. 2020. Effects of DNA Damage and Oxidative Stress in Human Bronchial Epithelial Cells Exposed to PM2.5 from Beijing, China, in Winter. Int J Environ Res Public Health 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowell PC. 1960. Phytohemagglutinin: an initiator of mitosis in cultures of normal human leukocytes. Cancer Res 20: 462–466. [PubMed] [Google Scholar]
- Olive PL, Banath JP. 2006. The comet assay: a method to measure DNA damage in individual cells. Nat Protoc 1: 23–29. [DOI] [PubMed] [Google Scholar]
- Ostling O, Johanson KJ. 1984. Microelectrophoretic study of radiation-induced DNA damages in individual mammalian cells. Biochem Biophys Res Commun 123: 291–298. [DOI] [PubMed] [Google Scholar]
- Paz-Elizur T, Ben-Yosef R, Elinger D, Vexler A, Krupsky M, Berrebi A, Shani A, Schechtman E, Freedman L, Livneh Z. 2006. Reduced repair of the oxidative 8-oxoguanine DNA damage and risk of head and neck cancer. Cancer Res 66: 11683–11689. [DOI] [PubMed] [Google Scholar]
- Pollard JM, Gatti RA. 2009. Clinical radiation sensitivity with DNA repair disorders: an overview. Int J Radiat Oncol Biol Phys 74: 1323–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pool-Zobel BL, Bub A, Liegibel UM, Treptow-van Lishaut S, Rechkemmer G. 1998. Mechanisms by which vegetable consumption reduces genetic damage in humans. Cancer Epidemiol Biomarkers Prev 7: 891–899. [PubMed] [Google Scholar]
- Rose M, Burgess JT, O'Byrne K, Richard DJ, Bolderson E. 2020. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front Cell Dev Biol 8: 564601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvaigo S, Guerniou V, Rapin D, Gasparutto D, Caillat S, Favier A. 2004. An oligonucleotide microarray for the monitoring of repair enzyme activity toward different DNA base damage. Anal Biochem 333: 182–192. [DOI] [PubMed] [Google Scholar]
- Scully R, Livingston DM. 2000. In search of the tumour-suppressor functions of BRCA1 and BRCA2. Nature 408: 429–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh NP, McCoy MT, Tice RR, Schneider EL. 1988. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res 175: 184–191. [DOI] [PubMed] [Google Scholar]
- Skopek TR, Liber HL, Penman BW, Thilly WG. 1978. Isolation of a human lymphoblastoid line heterozygous at the thymidine kinase locus: possibility for a rapid human cell mutation assay. Biochem Biophys Res Commun 84: 411–416. [DOI] [PubMed] [Google Scholar]
- Slob W 2014a. Benchmark dose and the three Rs. Part I. Getting more information from the same number of animals. Crit Rev Toxicol 44: 557–567. [DOI] [PubMed] [Google Scholar]
- -. 2014b. Benchmark dose and the three Rs. Part II. Consequences for study design and animal use. Crit Rev Toxicol 44: 568–580. [DOI] [PubMed] [Google Scholar]
- Slyskova J, Lorenzo Y, Karlsen A, Carlsen MH, Novosadova V, Blomhoff R, Vodicka P, Collins AR. 2014. Both genetic and dietary factors underlie individual differences in DNA damage levels and DNA repair capacity. DNA Repair (Amst) 16: 66–73. [DOI] [PubMed] [Google Scholar]
- Strom CE, Johansson F, Uhlen M, Szigyarto CA, Erixon K, Helleday T. 2011. Poly (ADP-ribose) polymerase (PARP) is not involved in base excision repair but PARP inhibition traps a single-strand intermediate. Nucleic Acids Res 39: 3166–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svilar D, Goellner EM, Almeida KH, Sobol RW. 2011. Base excision repair and lesion-dependent subpathways for repair of oxidative DNA damage. Antioxid Redox Signal 14: 2491–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykora P, Witt KL, Revanna P, Smith-Roe SL, Dismukes J, Lloyd DG, Engelward BP, Sobol RW. 2018. Next generation high throughput DNA damage detection platform for genotoxic compound screening. Sci Rep 8: 2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas P, Wu J, Dhillon V, Fenech M. 2011. Effect of dietary intervention on human micronucleus frequency in lymphocytes and buccal cells. Mutagenesis 26: 69–76. [DOI] [PubMed] [Google Scholar]
- Trzeciak AR, Barnes J, Ejiogu N, Foster K, Brant LJ, Zonderman AB, Evans MK. 2008a. Age, sex, and race influence single-strand break repair capacity in a human population. Free Radic Biol Med 45: 1631–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trzeciak AR, Barnes J, Evans MK. 2008b. A modified alkaline comet assay for measuring DNA repair capacity in human populations. Radiat Res 169: 110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trzeciak AR, Mohanty JG, Jacob KD, Barnes J, Ejiogu N, Lohani A, Zonderman AB, Rifkind JM, Evans MK. 2012. Oxidative damage to DNA and single strand break repair capacity: relationship to other measures of oxidative stress in a population cohort. Mutat Res 736: 93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valverde M, Lozano-Salgado J, Fortini P, Rodriguez-Sastre MA, Rojas E, Dogliotti E. 2018. Hydrogen Peroxide-Induced DNA Damage and Repair through the Differentiation of Human Adipose-Derived Mesenchymal Stem Cells. Stem Cells Int 2018: 1615497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace SS. 2014. Base excision repair: a critical player in many games. DNA Repair (Amst) 19: 14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weingeist DM, Ge J, Wood DK, Mutamba JT, Huang Q, Rowland EA, Yaffe MB, Floyd S, Engelward BP. 2013. Single-cell microarray enables high-throughput evaluation of DNA double-strand breaks and DNA repair inhibitors. Cell Cycle 12: 907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood DK, Weingeist DM, Bhatia SN, Engelward BP. 2010. Single cell trapping and DNA damage analysis using microwell arrays. Proc Natl Acad Sci U S A 107: 10008–10013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Zhao H, Suk R, Christiani DC. 2004. Genetic susceptibility to tobacco-related cancer. Oncogene 23: 6500–6523. [DOI] [PubMed] [Google Scholar]
- Xie Z, Lin H, Fang R, Shen W, Li S, Chen B. 2015. Effects of a fruit-vegetable dietary pattern on oxidative stress and genetic damage in coke oven workers: a cross-sectional study. Environ Health 14: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N, Galick H, Wallace SS. 2004. Attempted base excision repair of ionizing radiation damage in human lymphoblastoid cells produces lethal and mutagenic double strand breaks. DNA Repair (Amst) 3: 1323–1334. [DOI] [PubMed] [Google Scholar]
- Yang SY, Hsiung CN, Li YJ, Chang GC, Tsai YH, Chen KY, Huang MS, Su WC, Chen YM, Hsiung CA et al. 2016. Fanconi anemia genes in lung adenocarcinoma- a pathway-wide study on cancer susceptibility. J Biomed Sci 23: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zverina J, Zimanova J, Bartova D. 1991. [Catamnesis of a group of 84 castrated sexual offenders]. Cesk Psychiatr 87: 28–34. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.