

Abstract
Understanding patterns of gene flow and population structure is vital for managing threatened and endangered species. The reticulated flatwoods salamander (Ambystoma bishopi) is an endangered species with a fragmented range; therefore, assessing connectivity and genetic population structure can inform future conservation. Samples collected from breeding sites (n = 5) were used to calculate structure and gene flow using three marker types: single nucleotide polymorphisms isolated from potential immune genes (SNPs), nuclear data from the major histocompatibility complex (MHC), and the mitochondrial control region. At a broad geographical scale, nuclear data (SNP and MHC) supported gene flow and little structure (F ST = 0.00–0.09) while mitochondrial structure was high (ΦST = 0.15–0.36) and gene flow was low. Mitochondrial markers also exhibited isolation by distance (IBD) between sites (p = 0.01) and within one site (p = 0.04) while nuclear markers did not show IBD between or within sites (p = 0.17 and p = 0.66). Due to the discordant results between nuclear and mitochondrial markers, our results suggest male‐biased dispersal. Overall, salamander populations showed little genetic differentiation and structure with some gene flow, at least historically, among sampling sites. Given historic gene flow and a lack of population structure, carefully considered reintroductions could begin to expand the limited range of this salamander to ensure its long‐term resilience.
Keywords: Ambystoma bishopi, male‐biased dispersal, migration, population structure, reintroductions, reticulated flatwoods salamander, translocations
1. INTRODUCTION
Amphibian populations have experienced severe declines worldwide, with up to one third of amphibian species currently facing extinction (McCallum, 2007; O’Donnell et al., 2017). Drivers of this decline include pollution, climate change, exposure to novel diseases, and habitat loss or fragmentation (Grant et al., 2016; McCallum, 2007). Increased habitat loss and fragmentation have reduced population sizes and inhibited gene flow among amphibian populations, gene flow that may prevent extirpation of populations and extinction of species (Harper et al., 2008; Whiteley et al., 2014). Re‐establishing or maintaining historical levels of gene flow by reconnecting populations should be a key component of management and a goal of habitat restoration for species with historically connected populations (Dool et al., 2016; Semlitsch et al., 2017). However, modern anthropogenic landscape changes have made it impossible for some species to naturally recolonize extirpated areas of their historic range and translocations may be necessary to reintroduce species with low vagility.
Together with population structure, estimates of contemporary and historic migration rates are important for estimating connectivity among breeding sites. Historically connected sites with little genetic structure are straightforward candidates for translocations and reintroductions. For species with population structure, moving animals to places where the species has been extirpated or where populations have been severely reduced becomes a more complex challenge. The International Union for Conservation of Nature (IUCN) defines translocations as the human‐mediated movement of living organisms from one area, with release in another. It defines reintroductions as the intentional movement and release of an organism inside its indigenous range from which it has disappeared (IUCN, 2013). Several studies have tested the effectiveness of translocations and reintroductions in order to conserve endangered amphibian species. Eastern hellbenders (Cryptobranchus alleganiensis) have survived several years post‐release in sites where they were translocated to supplement existing populations (Kraus et al., 2017; McCallen et al., 2018) and spotted salamanders (Ambystoma maculatum) have been reintroduced by moving egg masses into artificial ponds to accelerate restoration without compromising larval survival (Sacerdote, 2009). Overall, promoting dispersal has been a tool in amphibian conservation and may be used to bolster and expand populations of imperiled species.
The reticulated flatwoods salamander (RFS, Ambystoma bishopi) is a federally endangered species with a highly fragmented range. Historically, RFS occurred throughout the southeastern United States in fire‐maintained longleaf pine (Pinus palustris) ecosystems (Palis, 1996; Petranka, 2010). Over the last several decades, this species has declined for many reasons including climate change, drought, continued loss of flatwoods habitat, and a change in fire regime from summer fires when breeding habitat is dry to prescribed fires in winter when habitat is wet (Bishop & Haas, 2005; Gorman et al., 2013). Winter fires consume less vegetation and leave undesirable plants in breeding habitats, which reduces recruitment and ultimately causes localized extinctions and increased fragmentation. The RFS now only occurs in a fraction of its former range with just six known breeding sites (Farmer et al., 2016; O’Donnell et al., 2017; Semlitsch et al., 2017).
The RFS breeds in ephemeral, fishless ponds dominated by grasses and forbs (Palis, 1996, 1997). These ponds dry during the summer months but fill with rain in the winter (Chandler et al., 2016). During rain events in October and November, adults migrate to dry pond basins and lay their eggs in anticipation of the seasonal inundation (Brooks et al., 2019b; Palis, 1996, 1997). Once the basins fill and the eggs are inundated, the larvae hatch, and eventually metamorphose when the ponds begin to dry in the spring (Palis, 1996). This breeding strategy is highly dependent on predictable seasonal rainfall, and if the ponds do not fill or do not stay full all winter, the entire larval cohort can be lost (Chandler et al., 2016; Palis, 1996).
Breeding sites consist of several separate ponds that are occupied each year and are connected by suitable habitat. Individual salamanders appear to disperse among ponds within a breeding site but do not currently disperse between breeding sites, as estimated from occupancy‐based metapopulation models (Brooks, Smith, Frimpong, et al., 2019) and the lack of suitable connective habitat between breeding sites. Estimates of dispersal in relation to geographical distance have led the United States Fish and Wildlife Service (USFWS) to define any occupied pond (and a 460‐m radius around it) as a breeding population, and any grouping of ponds within 3.2 km (2 miles) of each other as a breeding site (USFWS, 2015). Populations of RFS are typically managed by USFWS at the breeding site level.
Genetic population structure, effective number of breeders, and gene flow have been previously estimated for RFS with microsatellite data, suggesting that there is some structure among ponds within breeding sites (F ST, 0.004–0.112), limited migration, and a low mean number of breeders per pond (N b: 12.5–30.1, Wendt et al., 2021). Low NE has also been observed in another endangered amphibian, the California tiger salamander (Ambystoma californiense), where NE estimates ranged from 11 to 64 per pond with an average of 30 (Wang et al., 2011). To effectively estimate population structure at small scales, a larger number of genetic markers may be needed since subtle patterns can be obscured when few makers are used (Rittmeyer & Austin, 2015). For example, several hundred single nucleotide polymorphisms (SNPs) were better able to detect fine‐scale structure in tiger salamanders (Ambystoma tigrinum) than 12 microsatellites (McCartney‐Melstad et al., 2018; Titus et al., 2014). Furthermore, mitochondrial (mtDNA) data can be included in analyses of population structure and gene flow to examine sex‐biased dispersal. Mitochondrial DNA is inherited via the matrilineal line, and by comparing estimates obtained for mtDNA and nuclear data, it may be possible to elucidate patterns of migration for males and females.
Understanding patterns of gene flow and population structure at the landscape and local scale is necessary to determine the suitability of translocations for the RFS. The aims of this research were to estimate genetic structure, effective population size, genetic variation, and historic and contemporary gene flow among five extant breeding sites of RFS. We generated potential immune gene SNP data, which were complemented by sequence data generated for two major histocompatibility (MHC) exons and one mitochondrial marker in Williams et al. (2020). Our goal was to leverage these three marker types, at the local and landscape scale, to make estimates between and within breeding sites, and to ultimately explore a more complete image of RFS population structure, genetic diversity, gene flow, and sex‐biased dispersal. Immune genes were targeted because pathogens are a major threat to amphibians globally (McCallum, 2007), and variation at these genes is associated with broad resistance to pathogens (Sommer, 2005). These genes may show whether RFS populations have responded to past infections in a way that would create population structure or indicate (through low variation) populations that may be especially vulnerable to disease. Assessing connectivity and genetic differentiation among the remaining breeding sites can inform potential reintroductions and translocations of RFS in order to begin re‐establishing this species across its former range and thus reduce its risk of extinction (Semlitsch et al., 2017).
2. METHODS
2.1. Sample collection
Tissue samples were collected from five RFS breeding sites found on public lands (Figure 1) throughout the southeast. Briefly, RFS were collected with dip nets and funnel traps at six ponds on Eglin Air Force base (AFB), Florida, between 2011 and 2017 (Table 1). These six ponds included two separate breeding sites, one in the east (ponds 4, 5, 53, and 212) and one in the west (ponds 15 and 32). In 2018 and 2019, additional RFS samples were collected at five ponds on Escribano Point Wildlife Management Area (WMA), Florida; at one pond at Garcon Point Water Management Area, Florida; and at one pond on Mayhaw WMA, Georgia. Sample sizes at some breeding sites were limited by RFS population size and permission to access sites. For example, only five samples were available from Mayhaw, GA, because this population was only discovered in 2015 and despite surveying, only a few individuals have ever been found at that location. Furthermore, the population at Garcon Point was small at the time of sampling in 2018 and is now thought to be extirpated (pers. comm., Laura Jones and Pierson Hill). More details on the sampling protocol can be found in Williams et al. (2020).
FIGURE 1.
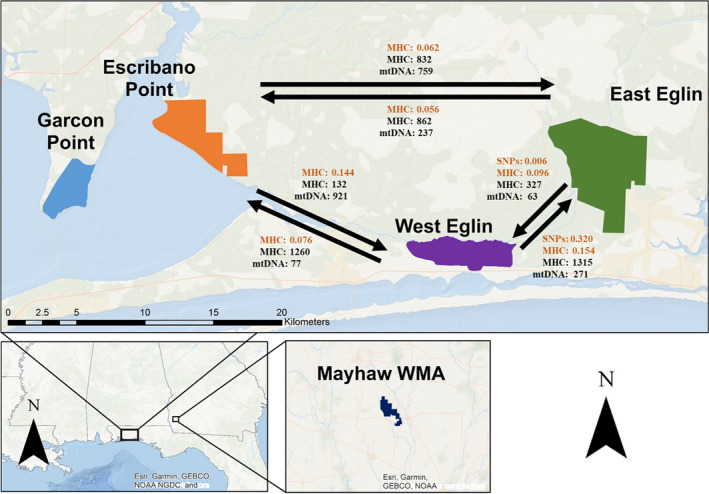
Map of sampled RFS breeding sites, arrows indicate migration rates. Black font: Estimates of mutation scaled migration rate per generation using Migrate‐N. Orange font: BayesAss estimates of migrants per generation. RFS, reticulated flatwoods salamander
TABLE 1.
Sample sizes for each genetic marker by breeding site
| Site | MHC | mtDNA | SNP |
|---|---|---|---|
| Eglin East | 29 | 143 | 73 |
| Pond 004 | 6 | 51 | 24 |
| Pond 005 | 8 | 54 | 24 |
| Pond 053 | 10 | 24 | 19 |
| Pond 212 | 5 | 14 | 6 |
| Eglin West | 18 | 30 | 23 |
| Pond 015 | 12 | 24 | 19 |
| Pond 032 | 6 | 6 | 4 |
| Escribano | 28 | 46 | 0 |
| Borrow | 5 | 10 | 0 |
| Cluster | 3 | 11 | 0 |
| Honey | 5 | 7 | 0 |
| Ghost | 6 | 9 | 0 |
| Torpedo | 7 | 8 | 0 |
| Garcon | 4 | 3 | 0 |
| Mayhaw | 5 | 5 | 0 |
| Total | 84 | 227 | 96 |
Numbers given for MHC data are the numbers of individuals sequenced at both MHC class Iα and IIβ.
2.2. SNP sequencing
DNA was extracted from tissues using a DNEasy Blood and Tissue Kit (Qiagen). Hundreds of RFS immune genes were sequenced in a target enrichment experiment using 19,339 RNA‐biotinylated baits designed to target RFS immune genes. To create custom RNA baits, the axolotl (Ambystoma mexicanum) transcriptome (Bryant et al., 2017) was filtered by the gene ontology term “immune response” using the GO2TR 1.0.8 pipeline (Gene Ontology to Target Region, Elbers & Taylor, 2015). This pipeline retained all exons related to immune response (Ortutay & Vihinen, 2006). Using these exons, Arbor Biosciences designed custom 120‐bp baits with 2x tiling to capture RFS immune genes (hereafter, baits).
In 2017, 96 samples from Eglin AFB (east = 73, west = 23) were selected for the target enrichment experiment. Due to sampling constraints, population sizes, and permit access, samples from other sites were not available for the target enrichment experiment. Libraries were prepared using 10 ng of DNA and a KAPA HyperPlus Illumina prep kit in combination with dual‐indexed 8‐bp Illumina adapters (KAPA Biosystems). Libraries were combined to create 16 equimolar pools containing six libraries each. These pooled libraries then underwent sequence capture following the MyBaits version 3.0 protocol (Arbor Biosciences). First, each pool was combined with baits and incubated in solution for 21 h at 65°C. Next pools were washed to retain only “captured” baits and amplified with 14 PCR cycles. Pools were then cleaned with 1× concentration of KAPA pure beads (Indianapolis, IN, USA) to remove excess PCR product and unincorporated baits. Before sequencing, samples were combined into final pools of equimolar concentrations. To achieve adequate coverage, this final pool of 96 enriched libraries was sequenced twice on mid‐output flow cells of an Illumina NextSeq 500 system at Pennington Biomedical Research Center using 75‐bp paired‐end reads.
Raw sequence reads were demultiplexed using the Illumina BaseSpace platform permitting zero mismatches in the barcodes. Adapter quality trimming was performed on demultiplexed paired‐end reads using all adapters sequences supplied with BBDuk 38.16 (https://sourceforge.net/projects/bbmap/) and the following options: ktrim = right, which trims kmers from 5′ to 3′ direction of a read, k = 23 which sets the kmer length to 23 bp, mink = 11 which sets the minimum kmer matching length to 11, hdist = 1 that uses a hammering distance of one to increase the number of kmers stored, tpe which trims both reads to the same length, tbo which trims adapters based on pair overlap, qtrim = rl for quality trimming right and left, and trimq = 15 which sets the quality trim to 15 using the Phred algorithm. Next, quality and adapter trimmed paired‐end reads were mapped to the axolotl 3.0.0 reference genome (https://axolotl‐omics.org/dl/AmexG_v3.0.0.fa.gz) using BBMap 38.16 (https://sourceforge.net/projects/bbmap/) and the vslow and usejni options. SAM files were converted to BAM files with SAMtools 1.9 (Li et al., 2009). Picard 2.18.10 (http://broadinstitute.github.io/picard) was used to clean, sort, add read groups, and mark duplicates. The program CallVariants 38.16 (https://sourceforge.net/projects/bbmap/) was used to call SNPs by ignoring duplicates and keeping SNPs with quality scores greater than or equal to 27. Finally, this dataset was reduced to only di‐allelic SNPs with genotype missingness of 5% or less were retained using VCFtools 0.1.15 (Danecek et al., 2011).
Retained SNPs were tested for linkage disequilibrium and Hardy–Weinberg equilibrium (HWE) using VCFtools, geno‐chisq, and hardy tests. The p.adjust function in R‐4.0.2 (R Core Team, 2018) was then used to correct for multiple tests using the false discovery rate (FDR, Benjamini & Hochberg, 1995). Gene duplication in the RFS, a pattern observed in other amphibians (McCartney‐Melstad et al., 2018; Waples, 2015), can lead to heterozygote excess causing H O >> H E. This possible gene duplication prompted filtering of the SNP data using stringent conditions. Any SNP that had a FDR less than 0.05 in one or more ponds was discarded from the dataset. Next, BayeScan 2.1 (Foll & Gaggiotti, 2008) was used to predict if SNPs were putatively under selection. As no SNPs were identified as being under selection, SNPs were used to estimate population structure and gene flow under neutral expectations. Finally, blastn v. 2.2.31+ (Altschul et al., 1990) was used to identify bait locations in the axolotl genome, and custom gene annotations were used to predict the genes each SNP might occur in (Sergej Nowoshilow, pers. comm.).
Allelic richness (AR), observed (H O), expected heterozygosity (H E), and pairwise F ST (Weir & Cockerham, 1984) were estimated at the pond and breeding site level for SNP data using hierfstat v. 0.04–22 (Goudet, 2005). Heirfstat was also used to conduct principal component analysis (PCA) for SNP data. GenePop v4.6 (Raymond & Rousset, 1995; Rousset, 2008) was used to calculate the inbreeding coefficient for each breeding site (FIS, Weir & Cockerham, 1984) along with an isolation by distance analysis (IBD). IBD was conducted using the decimal degree location for the center of each pond, 10,000 Mantel test permutations, and a minimum distance of 0.001 decimal degrees between samples. Effective population size was calculated for each breeding site using the linkage disequilibrium method in program NeEstimator v2.1 (Do et al., 2014.). Recent patterns of gene flow were estimated with BayesAss 3.0 (Wilson & Rannala, 2003). BayesAss estimates migration rate over the last three generations by calculating the probability of migrant ancestry for all individuals, assigning them as either a non‐migrant, 1st generation, or migrants that are 2nd generation or greater. For all analysis, 3 × 106 iterations were run starting with a burn‐in of 2 × 106 followed by 1 × 106 iterations with a sample taken every 100 steps for a posterior dataset of 1 × 104 samples. These analyses were repeated 10 times using different seeds to ensure convergence of models. STRUCTURE v2.3.4 (Pritchard et al., 2000) and Admixture 1.3.1 (Alexander et al., 2009) were run to assess population admixture using ancestry models to infer the number of populations (K) from the data. In program Admixture, the lowest cross‐validation value was used to select K between K = 1 to K = 7. For STRUCTURE, the model that maximizes marginal likelihood was used and results were confirmed by comparing distruct plots (Rosenberg, 2003).
2.3. MHC and mtDNA sequencing
To include other breeding sites and multiple marker types for population structure analyses, we used sequence data generated in Williams et al. (2020) for major histocompatibility complex (MHC) class Iα exon 3 and class IIβ exon 2 and the mitochondrial control region (Williams et al., 2020). An analysis of molecular variance (AMOVA) was conducted for all sampling locations with MHC and mtDNA sequence data using Arlequin 3.5.2.2 (Excoffier et al., 2005). Samples from Mayhaw and Garcon were removed from migration, STRUCTURE, and IBD analyses because of small sample sizes.
For MHC data, pairwise F ST (Weir & Cockerham, 1984) was calculated in hierfstat v.0.04–22, where MHC sequences were coded as alleles (i.e., each distinct sequence at a locus was given a number, e.g., allele 01). MHC data were only used at the site level because sample sizes at individual ponds were low (≤12) and the SNP data provided a more robust analysis for nuclear data at the smaller spatial scale. For mtDNA, ΦST was calculated in Arlequin 3.5 using mitochondrial haplotype frequencies. Unlike the MHC data, this analysis was conducted at both the pond and breeding site level.
Principal component analysis, effective population size, and program STRUCTURE v2.3.4 (Pritchard et al., 2000) were run for MHC markers as described for SNP data, but these analyses were not implemented for mitochondrial DNA because they require diploid data. GenePop v4.6 was used to estimate the inbreeding coefficient and isolation by distance. Recent migration rates between breeding sites (Eglin East, Eglin West, and Escribano) were estimated using MHC and mitochondria haplotypes in the program BayesAss 3.0 using the conditions described above for SNPs. Historic migration rates (>5 generations) were estimated by Bayesian inference in Migrate v3.7.2 (hereafter Migrate‐N, Beerli, & Felsenstein, 2001). This program estimates two metrics, the mutation scaled population size (ϴ), which is calculated as ϴ = 4N e μ where N e is effective population size and μ is mutation rate, and; a mutation scaled migration rate measured as the number of migrants per generation calculated as m/μ where m is the migration rate and μ is the mutation rate. Here, a full migration matrix model was used, which assumes direct gene flow between all populations. After testing multiple parameters, three long chains were run with a burn‐in of 5 × 106 iterations and, to avoid autocorrelation, after the burn‐in samples were taken every 50 steps for 5 × 104 iterations for a posterior dataset of 1 × 104 sampling iterations.
3. RESULTS
3.1. SNP sequencing
After quality control filtering, 263,127,494 reads were assignable to all individuals with 2,740,911 ± 2,328,524 (mean ± SD) reads assigned per individual (range = 477,444–14,876,290). Nine percent of the original reads were removed as PCR or optical duplicates, and 95% of the remaining reads were successfully aligned to the axolotl genome (2.27 × 108 reads). After alignment and comparison to the axolotl genome, 183 SNPs were identified, but after filtering, only 90 were polymorphic, in HWE, and in linkage equilibrium. All further analyses were conducted using these 90 SNPs.
Many sequencing reads were off‐target: on average, only 1.39% (min–max; 0.32–2.26%) of reads were on‐target. For this analysis, on‐target and off‐target reads were included when coverage permitted. Numerous RFS reads mapped to regions of the axolotl genome that are not predicted to represent immune genes while others could not be mapped at all. This issue may be caused by the phylogenetic distance between RFS and axolotls. Although they are members of the same genus, these two species likely diverged about 20 million years ago (Hime et al., 2021; Williams et al., 2013). Consequently, we consider the SNPs generated to be anonymous.
3.2. Genetic variation and effective population size
In the SNP dataset, allelic richness was 1.38 at East Eglin and 1.44 at West Eglin (Table 2). At East Eglin and West Eglin, H O was 15% and 18%, respectively, while H E was 12% and 15%, respectively (Table 2). Previous work shows that genetic variation was low at both MHC exons (Table 2, Williams et al., 2020). For class Iα, only three alleles were found with a nucleotide diversity of 0.001, H E of 19.9%, and a H O of 16.8% (Williams et al., 2020). MHC class IIβ had five alleles with a nucleotide diversity of 0.004, H E of 53.7%, and H O of 34.4% (Williams et al., 2020). Higher levels of H E and a H O at MHC versus SNP loci is likely a function of the marker type, as SNP markers represent a single base pair in the genome (with two possible alleles) whereas MHC exons represent several hundred base pairs that have the possibility of generating many types of alleles and heterozygotes.
TABLE 2.
Allelic richness (AR), observed heterozygosity (H O), expected heterozygosity (H E), and inbreeding coefficient (F IS) at Eglin Air Force Base using SNP and MHC data (MHC data originally presented in Williams et al., 2020)
| Marker | Site | n | AR | H O | H E | F IS |
|---|---|---|---|---|---|---|
|
SNP |
Pond 004 (East) | 24 | 1.36 | 0.13 | 0.11 | −0.22 |
| Pond 005 (East) | 24 | 1.41 | 0.16 | 0.14 | −0.22 | |
| Pond 053 (East) | 19 | 1.37 | 0.14 | 0.12 | −0.24 | |
| Pond 212 (East) | 6 | 1.4 | 0.17 | 0.14 | −0.29 | |
| Pond 015 (West) | 19 | 1.43 | 0.17 | 0.14 | −0.23 | |
| Pond 032 (West) | 4 | 1.66 | 0.19 | 0.16 | −0.22 | |
| East Eglin | 73 | 1.38 | 0.15 | 0.12 | −0.18 | |
| West Eglin | 23 | 1.44 | 0.18 | 0.15 | −0.17 | |
|
MHC Iα |
East Eglin | 105 | 1.39 | 0.11 | 0.12 | 0.11 |
| West Eglin | 37 | 1.65 | 0.24 | 0.22 | −0.12 | |
| Escribano | 39 | 2.84 | 0.59 | 0.64 | 0.08 | |
| Mayhaw | 5 | 2.00 | 0.60 | 0.56 | −0.09 | |
| Garcon Point | 4 | 1.00 | 0.00 | 0.00 | — | |
|
MHC IIβ |
East Eglin | 29 | 2.25 | 0.38 | 0.54 | 0.30 |
| West Eglin | 26 | 2.51 | 0.35 | 0.52 | 0.34 | |
| Escribano | 29 | 2.28 | 0.28 | 0.53 | 0.48 | |
| Mayhaw | 5 | 2.00 | 0.60 | 0.47 | −0.33 | |
| Garcon Point | 4 | 2.00 | 0.25 | 0.25 | 0.00 |
At East Eglin, West Eglin, and Escribano, F IS for MHC class IIβ was 0.30, 0.34, and 0.48 respectively (Table 2). Contrastingly, F IS for MHC class Iα was lower at these three sites with values of 0.106, −0.120, and 0.084. Nine mitochondrial haplotypes were observed, two of which had previously been described by Pauley et al. (2007; GenBank accession numbers H2: EU517607.1 and H3: EU517606.1) while the other seven were previously undescribed. Of the nine haplotypes, one occurred at all breeding sites (H3) while two others (H2, H9) were found at multiple breeding sites (Figure 1, Williams et al., 2020).
Effective population size could not be reliably estimated with MHC data, as values were either infinite or the 95% confidence intervals (CI) overlapped with 0 (Table S1). Infinite values indicate no genetic variation caused by drift and can be explained by sampling error or limited genetic variation (Waples & Do, 2010). Effective population size could be calculated at the site level with SNP data using a lowest allele frequency of <0.010. For East Eglin, NE was 206.3 (CI: 164.2–273.5) while NE for West Eglin was smaller at 120.2 (CI: 74.2–290.2, Table S1).
3.3. Population structure
Pairwise F ST was estimated with SNP and MHC data for Eglin and pairwise F ST was estimated with MHC data for Escribano. Using the SNP dataset for individual ponds within breeding sites on Eglin, all pairwise F ST were ≤0.056 while F ST between Eastern and Western Eglin was 0.004 (Table 3). No pattern of isolation by distance was detected when comparing all ponds on Eglin (p = 0.113). For MHC loci, F ST values between breeding sites ranged from −0.007 to 0.091 (Table 4). MHC F ST values between ponds within breeding sites could not be calculated due to small sample sizes at individual ponds.
TABLE 3.
FST calculated with SNP data and ΦST calculated with mtDNA data by pond on Eglin
| Pond 004 [East] | Pond 005 [East] | Pond 053 [East] | Pond 212 [East] | Pond 015 [West] | Pond 032 [West] | |
|---|---|---|---|---|---|---|
| Pond 004 [East] | — | 0.46 | 0.28 | 0.48 | 15.67 | 15.54 |
| Pond 005 [East] | 0.007 (0.033) | — | 0.36 | 0.94 | 15.45 | 15.32 |
| Pond 053 [East] | 0.002 (−0.018) | 0.012 (0.065) | — | 0.68 | 15.4 | 15.26 |
| Pond 212 [East] | 0.005 (0.068) | 0.007 (0.202) | 0.013 (0.025) | — | 15.9 | 15.77 |
| Pond 015 [West] | 0.006 (0.396) | 0.0002 (0.401) | 0.006 (0.427) | 0.001 (0.559) | — | 0.14 |
| Pond 032 [West] | 0.042 (0.549) | 0.021 (0.541) | 0.056 (0.608) | 0.031 (0.874) | 0.018 (0.057) | — |
FST and ΦST values are below the diagonal and approximate Euclidean distance (km) is given above the diagonal. F ST values calculated with SNP data are listed first and the mtDNA ΦST values are given in parentheses. Bolded values are significant at p < 0.05. Pairwise F ST between the two breeding sites was 0.004.
TABLE 4.
FST calculated with MHC data and ΦST calculated with mtDNA data by site
| Eglin East | Eglin West | Escribano | |
|---|---|---|---|
| Eglin East | — | 13.01 | 30.17 |
| Eglin West | −0.007 (0.356) | — | 19.64 |
| Escribano | 0.091 (0.152) | 0.082 (0.354) | — |
FST and ΦST values are below the diagonal and approximate Euclidean distance (km) is given above the diagonal. F ST values calculated with MHC data are listed first and the mtDNA ΦST values are given in parentheses. Bolded values are significant at p < 0.05.
The mitochondrial control region was used to estimate ΦST between individual ponds within Eglin or Escribano and between the three breeding sites. For ponds within East Eglin, ΦST values ranged from −0.169 to 0.203 while the pairwise comparison between the two West Eglin ponds was 0.057 (Table 3). On Escribano, ΦST values were between −0.063 and 0.420 (Table 5). Mitochondrial data did show a pattern of isolation by distance at some sites. When comparing ponds within East or West Eglin separately, there was no isolation by distance (IBD p = 0.173 and p = 0.663, respectively) but when comparing East and West Eglin there was isolation by distance (IBD p = 0.014). Furthermore, there was a pattern of isolation by distance (IBD p = 0.036) for ponds on Escribano. When individual ponds within breeding sites were examined more closely, the highest ΦST values (0.280–0.420) were between ponds separated by more than 1.5 km. Ponds less than 0.9 km apart had Φ values below 0.07. Finally, between the breeding sites of East Eglin, West Eglin, and Escribano, ΦST ranged from 0.152 to 0.356 (Table 4).
TABLE 5.
ΦST calculated with mtDNA data by pond on Escribano
| Borrow | Cluster | Honey | Ghost | Torpedo | |
|---|---|---|---|---|---|
| Borrow | — | 1.01 | 3.40 | 3.30 | 2.80 |
| Cluster | −0.063 | — | 2.34 | 2.28 | 2.33 |
| Honey | 0.354 | 0.400 | — | 0.130 | 0.530 |
| Ghost | 0.287 | 0.361 | −0.087 | — | 0.630 |
| Torpedo | 0.386 | 0.420 | −0.147 | −0.030 | — |
ΦST values below the diagonal and approximate Euclidean distance (km) above the diagonal. Bolded values are significant at p < 0.05.
AMOVA results with MHC data indicated little population structure: Only 9.53% of the total variation was among breeding sites (Table 6). Greater population structure was detected with mitochondrial data: 25.49% of the total variation was among breeding sites (Table 6). Program STRUCTURE cannot calculate ΔK when K = 1, so to investigate K = 1–7, we used distruct plots as well as log likelihoods to determine K. With the SNP dataset, STRUCTURE suggested a single population (K = 1) on Eglin. This result was confirmed by comparing distruct plots of K = 1–3 (Figure 2). Program Admixture also suggested a single population on Eglin based on the cross‐validation (CV) values for K = 1–7 in the SNP and MHC datasets (Table S2). For MHC data at Eglin and Escribano, program STRUCTURE suggested two populations using ΔK, but one population using log‐likelihood values. To further evaluate K = 2, distruct plots were compared (Figure 2). K = 2 showed no distinct patterns between populations and so K = 1 was accepted. These results are supported by the PCAs obtained for both SNP and MHC data (Figures S1 and S2). For the PCAs, SNP data clustered in a single group regardless of collection location or dataset, and MHC data showed no discernable pattern based on sampling site.
TABLE 6.
AMOVA as estimated with MHC and mtDNA data
| Markers | Source of variation | Degrees of freedom | Sum of squares | Percentage Variation |
|---|---|---|---|---|
| MHC Iα & IIβ | Among breeding sites | 4 | 8.42 | 9.53 |
| Among individuals within breeding sites | 85 | 46.86 | 19.53 | |
| Within individuals | 90 | 32 | 70.95 | |
| mtDNA | Among breeding sites | 4 | 14.56 | 25.49 |
| Within individuals | 233 | 67.97 | 74.51 |
All estimated values are significant.
FIGURE 2.
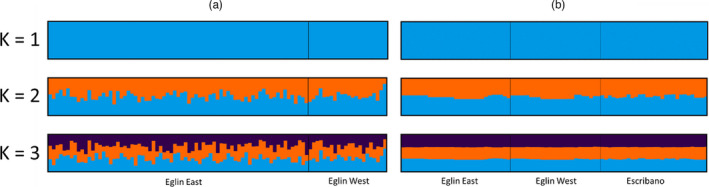
Distruct plots for SNP (a) and MHC class Iα and IIβ (b) data. For K = 2 and 3, individual membership does not align with sampling location indicating a lack of population structure and supporting K = 1. Distruct plots of SNP data broken down by pond found in Figure S3. MHC, major histocompatibility complex; SNP, single nucleotide polymorphisms
3.4. Gene flow
For program Migrate‐N, the mode migration estimate for SNP, MHC, and mitochondrial data was reported, because for SNP and MHC markers, the mean, median, and mode were almost identical (Table S3), but for mitochondrial estimates, the mode better represented the peak of the distribution on the curve (Figures S4–S6). For mitochondrial data, migration rates were not multiplied by four (to account for mtDNA NE) to equate them with migration rates for a nuclear marker; instead, they were left unadjusted to estimate migration for this haploid, maternally inherited marker. Migrate‐N estimates had wide posterior distributions for both migration and ϴ estimates. For SNP data, migration rates for East Eglin to West Eglin were 10.1 and 1.2 from West to East (Table S3 and Figure S4). Migration rates for MHC class IIβ were highest from East to West Eglin at 1315.8 with West to East migration rates of 372.5. Migration rates for West Eglin to Escribano were 1260.8, while Escribano to West Eglin were 132.5. Migration rates between East Eglin and Escribano were moderate and almost equal in both directions at 832.5 and 862.5 (Table S3 and Figure S5). For mitochondrial data, models gave wide estimates with long tails. Estimates of migration with mitochondrial data were highest from Escribano to East and West Eglin at 759.0 and 921.0, respectively, while migration out of West Eglin was the lowest at 63.0 to East Eglin and 77.0 to Escribano (Table S3 and Figure S6). Although the lowest confidence intervals for many mitochondrial migration rates were at or near zero, mean, median, and mode estimates were always more than 60 individuals. MHC class Iα estimates were uninformative, probably because range‐wide genetic variation was very low.
Migration rates as estimated with SNP data in BayesAss showed an asymmetrical pattern: 32.0% of individuals in the West were migrants from the East while 0.6% of individuals in the East were migrants from the West (Table S4). BayesAss estimates for MHC data were generally lower than estimates obtained with SNP data. West Eglin had the most individuals of migrant ancestry (14.4–15.4%) while East Eglin and Escribano had fewer individuals of migrant ancestry (5.6–8.8%; Table S4).
4. DISCUSSION
Reticulated flatwoods salamanders showed little population structure and some gene flow across the core of their remaining range as estimated with SNP data; however, due to the discordant results obtained for nuclear and mitochondrial markers, our results suggested males drive dispersal as analyses with mtDNA data demonstrated structure and isolation by distance. At a fine geographical scale among ponds on Eglin and among ponds on Escribano, nuclear markers showed gene flow and little population structure, a result that is consistent for all analyses (F ST, PCA, STRUCTURE, IBD, and Admixture). This finding is supported by the AMOVA results, which showed that the majority of genetic variation occurs within individuals and only 9.5% of variation was explained among breeding sites. This result is lower, but similar, to previous research on Eglin, which estimated structure with microsatellite data (within East Eglin mean F ST = 0.055 (range = 0.009–0.112); within West Eglin mean F ST = 0.035 (range = 0.004–0.079), between East and West Eglin = 0.104 (range = 0.061–0.163, Wendt et al., 2021). Although estimates obtained with microsatellite, SNP, and MHC markers cannot be compared directly, an examination of overall patterns is useful. F ST estimates between ponds within East Eglin or within West Eglin are low for both microsatellite and SNP data. However, microsatellite F ST values are higher between the two Eglin breeding sites than the F ST values generated with SNP or MHC data. In contrast to nuclear markers, mtDNA showed structure among some ponds on Escribano and between East and West Eglin (Tables 4 and 5).
At a broad geographical scale, the combined data from ponds within each breeding site (i.e., East Eglin, West Eglin, and Escribano) continued to support gene flow and a lack of genetic structure as estimated with SNP and MHC data. However, structure was high between all breeding sites as estimated with mitochondrial DNA (Table 4). The disparity in genetic structure and dispersal estimates for nuclear and mitochondrial DNA is indicative of male‐biased dispersal, a pattern observed in other amphibians including the red‐backed salamander (Plethodon cinereus) and the alpine salamander (Salamandra atra, Helfer et al., 2012, Liebgold et al., 2011). Females may be capable of moving long distances, but as fall breeders, they rely on cues like previous experience to select suitable egg laying habitat. As a result, they may have a stronger philopatric connection to their natal pond in contrast to males that orient based on receptive females. Thus, females do not appear to disperse as widely given the pressure to select suitable egg laying habitat (Burkhart et al., 2017; Moore & Whiteman, 2016; Peterman et al., 2015).
Although there was some mitochondrial genetic structure over short distances, analyses indicated that ΦST increased considerably when ponds were separated by more than 1.5 km (Tables 3 and 4), a value similar to the dispersal distances calculated using spatially explicit occupancy models (Brooks, Smith, Frimpong, et al., 2019). This was apparent on Eglin where IBD analysis of mitochondrial data showed no relationship between distance and structure within breeding sites, but did show a significant correlation between East and West Eglin (Tables 3 and 4). In contrast, MHC and SNP data showed no isolation by distance within or between Eglin sites indicating that males may move, or historically moved, between these ponds at a higher rate than females. Notably, although Escribano is treated as one breeding site by the USFWS, mitochondrial and IBD data showed that highly philopatric females may effectively occupy two distinct breeding sites at this location (Table 5): Borrow and Cluster ponds, which are separated by approximately 2.3 kilometers from the second group of ponds, Honey, Torpedo, and Ghost. However, it is difficult to estimate the precise relationship between dispersal and distance because distances between sampled ponds have a clumped distribution (currently occupied ponds are either 0.1–1.5 km apart, 2.5–3.5 km apart, or >10.0 km apart) and do not fall on a continuum. Mitochondrial data suggested that females typically do not disperse more than 1.5 km but that may be an underestimate because there were no sampled ponds in the 1.5–2.5 km range.
Other studies have found greater population structure in fall breeding ambystomatids, such as the RFS, than in spring breeders on the same landscape. For example, ringed salamanders (Ambystoma annulatum) and marbled salamanders (Ambystoma opacum), both fall breeders, showed more population structure on Fort Leonard Wood, MO than spotted salamanders, which breed in the spring (Burkhart et al., 2017, Peterman et al., 2015). Fall breeders may show increased philopatry to breeding ponds because they mate when ponds are dry and must deposit eggs in the dry basin. Accordingly, selecting suitable egg laying habitat is more difficult because females must anticipate inundation as compared to the spring where water is present when breeding begins. Consequently, for females, there is a strong selective pressure to choose breeding sites based on cues such as previous experience or pond‐associated vegetation (Brooks, Smith, Frimpong, et al., 2019). This pressure to select breeding sites may contribute to greater philopatry in fall breeding salamanders thus decreasing gene flow and increasing genetic structure (Burkhart et al., 2016, Peterman et al., 2015).
We attempted to isolate SNPs from immune genes, and although most were off‐target and effectively anonymous, it is possible that selection may play a role in population structure despite our BayeScan results indicating SNP neutrality. However, had there been selection on specific SNPs with different disease challenges at the various sampling sites, we would expect to find population structure rather than its absence. It would seem unlikely that 90 SNPs would all be under selection in the same way at multiple locations so as to cause a consistent lack of population structure across sampling sites. Future work with a larger number of SNP loci located throughout the genome may help to clarify population structure in RFS.
Measures of SNP genetic diversity were low and mostly uniform across East and West Eglin. MHC data showed that Escribano had the most diversity whereas East Eglin and Garcon Point had less than the other sites (Williams et al., 2020). Allelic richness, observed heterozygosity, and expected heterozygosity were all lower than a similar endangered amphibian, the California tiger salamander. Using 11 microsatellites across multiple California tiger salamander breeding ponds, all measures of genetic diversity were higher than the RFS (AR =15.6–24.0, H O = 0.70–0.92, H E = 0.75–0.89, Wang et al., 2011). Despite lower estimates of diversity, estimates of effective population size were similar between the RFS and California tiger salamander. Using SNP data, NE for the RFS was 206.3 (164.2–273.5) at East Eglin and 120.2 (74.2–290.2) at West Eglin. For the California tiger salamander, NE was 203 when summed across 10 breeding ponds (Wang et al., 2011). Although estimates obtained with microsatellites cannot be directly compared with SNP and MHC markers, it appears that diversity is low across the RFS’s sampled range, but NE may be comparable to other endangered ambystomatids.
FIS values were below zero for SNP data (Table 2) in contrast to MHC F IS values, which ranged up to 0.48 (Williams et al., 2020). The larger SNP dataset may have provided a more precise estimate of inbreeding, but additionally, selection for specific MHC alleles caused by disease history may have reduced genetic diversity and increased FIS in that region without affecting the SNP regions (Williams et al., 2020).
Data from all marker types suggest that some migration occurs, or historically occurred, between each of the East Eglin, West Eglin, and Escribano breeding sites. Notably, MHC markers had much higher estimated migration rates compared to SNP data (MHC = 132–1315, SNP = 1.2–10.1). The disparity between these estimates may be caused by similar selective pressures at MHC genes, which could inflate migration estimates. Given the considerable distance between East and West Eglin (13 km), and limited mobility for salamanders, it is likely that MHC data overestimated the amount of migration between these distant sites. Nevertheless, migration estimates generated from our datasets suggest that some migration, though perhaps limited, has occurred between East Eglin, West Eglin, and Escribano.
Both Migrate‐N (mtDNA) and BayesAss (SNP and MHC) suggest that West Eglin receives more immigrants from and contributes fewer emigrants to Escribano and East Eglin overall. Asymmetrical gene flow is most pronounced on Eglin with an approximate 3:1 migration rate from East to West Eglin (Migrate‐N) as estimated with all three marker types. This uneven gene flow could have biological implications, chiefly that West Eglin may have been a population sink in the past. Because West Eglin also has a smaller NE than East Eglin, it may have lower recruitment than East Eglin, leading to more immigration and less emigration. However, this asymmetrical migration rate could be an artifact of different demographic histories on each site. For instance, directional selection at immune genes (especially MHC) could skew allele frequencies and inflate migration estimates. Alternatively, a more severe bottleneck at one site may have removed genetic variants found at other sites, creating an uneven pattern of diversity that may be interpreted as asymmetrical migration (Wilson & Rannala, 2003). However, it is possible that asymmetrical gene flow is real and caused by topographical conditions that skew migration patterns, for example, directional water flow that may influence larval dispersal during flooding events, a possibility that requires further study.
Managing all RFS populations in a metapopulation context, in which there is limited gene flow among breeding sites, is an option that may maintain genetic diversity over the long term. In theory, as different alleles drift to fixation in individual breeding sites, global heterozygosity is effectively frozen in the total population, but there is still sufficient gene flow to prevent inbreeding over the long term within breeding sites and spread advantageous alleles across the species’ range (Allendorf et al., 2013). In practice, this approach is most suitable when (1) the population size of individual breeding sites is large enough to avoid inbreeding over the short‐term; (2) there are enough breeding sites for different alleles to randomly fix without being lost in the total population; and (3) there is evidence that the species has evolved in a metapopulation context. In RFS, these conditions arguably do not exist: some populations are very small (e.g., Mayhaw WMA and Garcon Point) and may experience inbreeding over the short term, reducing survival and reproductive success in an endangered species, if gene flow among breeding sites remains low. Moreover, very few breeding sites (n ≈ 6) exist, reducing the likelihood that all alleles will be present in at least one breeding site as they drift to fixation, and increasing the risk that stochastic events could reduce the remaining diversity. Finally, the SNP, MHC, and mitochondrial results obtained here showed little population structure and, at least historically, some gene flow among Escribano, East Eglin, and West Eglin breeding sites. This suggests that RFS may not have evolved in a metapopulation scenario with little gene flow among breeding sites. For these reasons, the historic connection among breeding sites might be maintained to facilitate gene flow, thereby reducing genetic drift and inbreeding at individual breeding sites, although we acknowledge that there are other considerations such as limiting the potential spread of disease or undermining currently stable populations. This historic connection could be maintained by expanding suitable habitat or hydrologic management and must take into consideration the population size of donor and recipient populations. However, gene flow among populations may be important given the evident low diversity at immune genes and potential limited ability to combat infections in RFS (Williams et al., 2020). At this time, it is important to maintain and expand current breeding sites to improve habitat, increase population size as well as facilitate gene flow. In future, managing RFS breeding sites as metapopulations may be a better option if population sizes recover sufficiently to limit inbreeding over the short term, and enough breeding sites have been re‐established to ensure that all alleles are likely to be present in the total population as they become randomly fixed in individual breeding sites.
Overall, RFS in this area may have existed as a series of connected populations rather than several discrete metapopulations. Given historic gene flow and a lack of population structure, carefully considered reintroductions could be used as a method to help re‐establish RFS populations on historically occupied sites that are now extirpated (Palis et al., 1997). Several of these extirpated sites are being restored to ensure the survival of RFS in future with the removal of undesirable vegetation, protection of core areas from habitat loss, and a return of important summer fires (pers comm. Kelly Jones and Charlie Abeles). Recolonization of extirpated sites is unlikely to occur without human aid as they are too far from currently active sites to naturally re‐establish, especially because females appear unlikely to disperse across long distances. Even if male salamanders disperse to unoccupied sites, they are unlikely to encounter females. However, RFS reintroductions could move both sexes and begin to expand the limited range of this salamander to ensure its long‐term resilience.
These reintroductions could initially be conducted in locations far from existing RFS populations but using multiple source ponds whose hydrology is similar to the reintroduction location. This approach would minimize the risk of spreading disease and perhaps preserve any local adaptations to environmental variables such as hydroperiod (Richter‐Boix et al., 2015; Wendt et al., 2021). Sites further from the Gulf of Mexico should be prioritized for reintroduction in order to minimize the risks posed by saltwater intrusion associated with hurricanes and sea level rise.
In conclusion, at a broad geographical scale, RFS populations showed little population structure with nuclear SNP data (F ST = 0.00–0.09) but higher structure with mitochondrial DNA (ΦST = 0.15–0.36). At this same scale, mitochondrial DNA exhibited isolation by distance, whereas nuclear markers did not. These discordant results suggest that males drive dispersal whereas females are more philopatric. Using the distance between sampled sites, and sharp increases in ΦST, it is likely that females disperse less than 1.5 km from their natal pond (Tables 3, 4, 5). Finally, program Migrate‐N and BayesAss supported some migration, at least historically, between East Eglin, West Eglin, and Escribano breeding sites.
Future amphibian conservation will require both molecular and traditional conservation work to inform appropriate management strategies, especially in fragmented or shrinking habitats with increasingly less connected populations and fewer active breeding sites. Combining molecular methods with traditional conservation techniques can help identify appropriate source populations for reintroductions, identify sex differences in gene flow, and preserve potentially unique diversity and adaptive potential. Finally, time is a resource that is in short supply: the increasing frequency of strong hurricanes (e.g., Hurricane Michael in 2018 and Hurricane Sally in 2020) as well as the risk posed by global amphibian diseases and inbreeding in small populations underlines the need to begin re‐establishing populations in order to decrease the risk of extinction.
CONFLICT OF INTEREST
The authors have no conflict of interest for this article.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
We are thankful to Dr. Carola Haas, Dr. Jamie Roberts, Dr. Kim Terrell, Dr. Chris Austin, and members of the Taylor laboratory for extensive edits and stimulating discussion concerning this manuscript. Thank you to everyone who helped in sample acquisition: K. Jones, B. Rincon, V. Porter, C. Abeles, A. Farmer, P. Hill, L. Smith, H. Mitchell, S. Piccolomini, R. Bilbow, J. de Silva, K. Erwin, B. Moore, J. Sandoval, E. Browning, and A. Wendt. Portions of this research were conducted with high‐performance computational resources provided by the Louisiana Optical Network Infrastructure (http://www.loni.org). We would like to thank the Pennington Biomedical Research Center for the use of their facilities and Richard Carmouche for his genomics assistance. Collection of tissue samples and extraction of DNA from salamanders on Eglin Air Force Base was supported by Eglin Air Force Base Jackson Guard, the US Air Force Civil Engineer Center, and the US Fish and Wildlife Service. Salamander samples from Eglin Air Force Base were collected under US Fish and Wildlife Service Threatened and Endangered Species Permit #TE049502 and Virginia Tech IACUC protocols #13‐129 and #16‐100. Samples from Mayhaw WMA were collected under Georgia Department of Natural Resources scientific collection permit #029, and samples at Garcon Point and Escribano Point were collected under the section 6 agreement between USFWS and Florida Fish and Wildlife Conservation Commission. This work would not be possible without the funding provided by Audubon Center for Research of Endangered Species (ACRES) grant. This project/work used Genomics core facilities that are supported in part by COBRE (NIH8 1P30GM118430‐02) and NORC (NIH 2P30DK072476) center grants from the National Institutes of Health. This material is also based upon work that is supported by the National Institute of Food and Agriculture, U.S. Department of Agriculture, McIntire Stennis program.
Williams, S. T., Elbers, J. P., & Taylor, S. S. (2021). Population structure, gene flow, and sex‐biased dispersal in the reticulated flatwoods salamander (Ambystoma bishopi): Implications for translocations. Evolutionary Applications, 14, 2231–2243. 10.1111/eva.13287
Funding information
This work would not be possible without the funding provided by Audubon Center for Research of Endangered Species (ACRES) grant. This project/work used Genomics core facilities that are supported in part by COBRE (NIH8 1P30GM118430‐02) and NORC (NIH 2P30DK072476) center grants from the National Institutes of Health. This material is also based upon work that is supported by the National Institute of Food and Agriculture, U.S. Department of Agriculture, McIntire Stennis program.
DATA AVAILABILITY STATEMENT
MHC and mtDNA data for this study are available at the NCBI GenBank repository (MT475763‐MT475765 and MT425246‐MT425250). Next‐generation sequencing data for this study are available at the NCBI Sequence Read Archive, BioProject, PRJNA750462.
REFERENCES
- Alexander, D. H., Novembre, J., & Lange, K. (2009). Fast model‐based estimation of ancestry in unrelated individuals. Genome Research, 19(9), 1655–1664. 10.1101/gr.094052.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allendorf, F. W., Luikart, G., & Aitken, S. (2013). Conservation and the genetics of populations. Blackwell publishing. [Google Scholar]
- Altschul, S. F., Gish, W., Miller, W., Myers, E. W., & Lipman, D. J. (1990). Basic local alignment search tool. Journal of Molecular Biology, 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Beerli, P., & Felsenstein, J. (2001). Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proceedings of the National Academy of Sciences, 98(8), 4563–4568. 10.1073/pnas.081068098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini, Y., & Hochberg, Y. (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. Journal of the Royal Statistical Society. Series B, 57, 289–300. [Google Scholar]
- Bishop, D. C., & Haas, C. A. (2005). Burning trends and potential negative effects of suppressing wetland fires on flatwoods salamanders. Natural Areas Journal, 25(3), 290–294. [Google Scholar]
- Brooks, G. C., Smith, J. A., Frimpong, E. A., Gorman, T. A., Chandler, H. C., & Haas, C. A. (2019). Indirect connectivity estimates of amphibian breeding wetlands from spatially explicit occupancy models. Aquatic Conservation: Marine and Freshwater Ecosystems, 29(11), 1815–1825. 10.1002/aqc.3190 [DOI] [Google Scholar]
- Brooks, G. C., Smith, J. A., Gorman, T. A., & Haas, C. A. (2019). Discerning the environmental drivers of annual migrations in an endangered amphibian. Copeia, 107, 270–276. 10.1643/CH-18-068 [DOI] [Google Scholar]
- Bryant, D. M., Johnson, K., DiTommaso, T., Tickle, T., Couger, M. B., Payzin‐Dogru, D., Lee, T. J., Leigh, N. D., Kuo, T.‐H., Davis, F. G., Bateman, J., Bryant, S., Guzikowski, A. R., Tsai, S. L., Coyne, S., Ye, W. W., Freeman, R. M., Peshkin, L., Tabin, C. J., … Whited, J. L. (2017). A tissue‐mapped axolotl de novo transcriptome enables identification of limb regeneration factors. Cell Reports, 18(3), 762–776. 10.1016/j.celrep.2016.12.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhart, J. J., Peterman, W. E., Brocato, E. R., Romine, K. M., Willis, M. M. S., Ousterhout, B. H., Anderson, T. L., Drake, D. L., Rowland, F. E., Semlitsch, R. D., & Eggert, L. S. (2017). The influence of breeding phenology on the genetic structure of four pond‐breeding salamanders. Ecology and Evolution, 7(13), 4670–4681. 10.1002/ece3.3060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler, H. C., Rypel, A. L., Jiao, Y., Haas, C. A., & Gorman, T. A. (2016). Hindcasting historical breeding conditions for an endangered salamander in ephemeral wetlands of the southeastern USA: Implications of climate change. PLoS One, 11(2), e0150169. 10.1371/journal.pone.0150169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., Handsaker, R. E., Lunter, G., Marth, G. T., Sherry, S. T., McVean, G., Durbin, R. & 1000 Genomes Project Analysis Group . (2011). The variant call format and VCFtools. Bioinformatics, 27(15), 2156–2158. 10.1093/bioinformatics/btr330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do, C., Waples, R. S., Peel, D., Macbeth, G. M., Tillett, B. J., & Ovenden, J. R. (2014). NeEstimator V2: Re‐implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Molecular Ecology Resources, 14, 209–214. [DOI] [PubMed] [Google Scholar]
- Dool, S. E., Puechmaille, S. J., Kelleher, C., McAney, K., & Teeling, E. C. (2016). The effects of human‐mediated habitat fragmentation on a sedentary woodland‐associated species (Rhinolophus hipposideros) at its range margin. Acta Chiropterologica, 18(2), 377. 10.3161/15081109ACC2016.18.2.006 [DOI] [Google Scholar]
- Elbers, J. P., & Taylor, S. S. (2015). GO2TR: A gene ontology‐based workflow to generate target regions for target enrichment experiments. Conservation Genetics Resources, 7(4), 851–857. 10.1007/s12686-015-0487-6 [DOI] [Google Scholar]
- Excoffier, L., Laval, G., & Schneider, S. (2005). Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evolutionary Bioinformatics, 1, 117693430500100. 10.1177/117693430500100003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer, A. L., Walls, S. C., Haas, C. A., Barichivich, W. J., Gorman, T. A., Jones, K. C., Hill, P., Odonnell, K., Harris, B., Enge, K. & Mott, J. (2016). A statewide species and habitat assessment for the reticulated flatwoods salamander, frosted flatwoods salamander, and striped newt (pp. 1–29). Unpublished. 10.13140/rg.2.2.21621.22244 [DOI]
- Foll, M., & Gaggiotti, O. (2008). A genome‐scan method to identify selected loci appropriate for both dominant and codominant markers: A bayesian perspective. Genetics, 180(2), 977–993. 10.1534/genetics.108.092221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman, T. A., Haas, C. A., & Himes, J. G. (2013). Evaluating methods to restore amphibian habitat in fire‐suppressed pine flatwoods wetlands. Fire Ecology, 9(1), 96–109. 10.4996/fireecology.0901096 [DOI] [Google Scholar]
- Goudet, J. (2005). Hierfstat, a package for r to compute and test hierarchical F‐statistics. Molecular Ecology Notes, 5(1), 184–186. 10.1111/j.1471-8286.2004.00828.x [DOI] [Google Scholar]
- Grant, E. H. C., Miller, D. A. W., Schmidt, B. R., Adams, M. J., Amburgey, S. M., Chambert, T., Cruickshank, S. S., Fisher, R. N., Green, D. M., Hossack, B. R., Johnson, P. T. J., Joseph, M. B., Rittenhouse, T. A. G., Ryan, M. E., Waddle, J. H., Walls, S. C., Bailey, L. L., Fellers, G. M., Gorman, T. A., … Muths, E. (2016). Quantitative evidence for the effects of multiple drivers on continental‐scale amphibian declines. Scientific Reports, 6(1), 1–9. 10.1038/srep25625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper, E. B., Rittenhouse, T. A., & Semlitsch, R. D. (2008). Demographic consequences of terrestrial habitat loss for pool‐breeding amphibians: Predicting extinction risks associated with inadequate size of buffer zones. Conservation Biology, 22(5), 1205–1215. 10.1111/j.1523-1739.2008.01015.x [DOI] [PubMed] [Google Scholar]
- Helfer, V., Broquet, T., & Fumagalli, L. (2012). Sex‐specific estimates of dispersal show female philopatry and male dispersal in a promiscuous amphibian, the alpine salamander (Salamandra atra). Molecular Ecology, 21, 4706–4720. [DOI] [PubMed] [Google Scholar]
- Hime, P. M., Lemmon, A. R., Lemmon, E. C. M., Prendini, E., Brown, J. M., Thomson, R. C., Kratovil, J. D., Noonan, B. P., Pyron, R. A., Peloso, P. L. V., Kortyna, M. L., Keogh, J. S., Donnellan, S. C., Mueller, R. L., Raxworthy, C. J., Kunte, K., Ron, S. R., Das, S., Gaitonde, N., … Weisrock, D. W. (2021). Phylogenomics reveals ancient gene tree discordance in the Amphibian tree of life. Systematic Biology, 70(1), 49–66. 10.1093/sysbio/syaa034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- IUCN . (2013). Guidelines for reintroductions and other conservation translocations. Version 1.0. Gland, Switzerland: IUCN Species Survival Commission, viiii. [Google Scholar]
- Kraus, B. T., McCallen, E. B., & Williams, R. N. (2017). Evaluating the survival of translocated adult and captive‐reared, juvenile eastern hellbenders (Cryptobranchus alleganiensis alleganiensis). Herpetologica, 73(4), 271–276. 10.1655/Herpetologica-D-16-00009 [DOI] [Google Scholar]
- Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R., & 1000 Genome Project Data Processing Subgroup . (2009). The sequence alignment/map format and SAMtools. Bioinformatics, 25(16), 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebgold, E. B., Brodie, E. D., & Cabe, P. R. (2011). Female philopatry and male‐biased dispersal in a direct‐developing salamander, Plethodon cinereus: Sex‐biased dispersal in Plethodon cinereus . Molecular Ecology, 20(2), 249–257. 10.1111/j.1365-294X.2010.04946.x [DOI] [PubMed] [Google Scholar]
- McCallen, E. B., Kraus, B. T., Burgmeier, N. G., Fei, S., & Williams, R. N. (2018). Movement and habitat use of eastern hellbenders (Cryptobranchus alleganiensis alleganiensis) following population augmentation. Herpetologica, 74(4), 283–293. 10.1655/0018-0831.283 [DOI] [Google Scholar]
- McCallum, M. L. (2007). Amphibian decline or extinction? Current declines dwarf background extinction rate. Journal of Herpetology, 41(3), 483–491. 10.1670/0022-1511(2007)41483:ADOECD2.0.CO;2 [DOI] [Google Scholar]
- McCartney‐Melstad, E., Vu, J. K., & Shaffer, H. B. (2018). Genomic data recover previously undetectable fragmentation effects in an endangered amphibian. Molecular Ecology, 27(22), 4430–4443. 10.1111/mec.14892 [DOI] [PubMed] [Google Scholar]
- Moore, M. P., & Whiteman, H. H. (2016). Natal philopatry varies with larval condition in salamanders. Behavioral Ecology and Sociobiology, 70(8), 1247–1255. 10.1007/s00265-016-2133-z [DOI] [Google Scholar]
- O’Donnell, K. M., Messerman, A. F., Barichivich, W. J., Semlitsch, R. D., Gorman, T. A., Mitchell, H. G., Allan, N., Fenolio, D., Green, A., Johnson, F. A., Keever, A., Mandica, M., Martin, J., Mott, J., Peacock, T., Reinman, J., Romañach, S. S., Titus, G., McGowan, C. P., & Walls, S. C. (2017). Structured decision making as a conservation tool for recovery planning of two endangered salamanders. Journal for Nature Conservation, 37, 66–72. 10.1016/j.jnc.2017.02.011 [DOI] [Google Scholar]
- Ortutay, C., & Vihinen, M. (2006). Immunome: A reference set of genes and proteins for systems biology of the human immune system. Cellular Immunology, 244, 87–89. 10.1016/j.cellimm.2007.01.012 [DOI] [PubMed] [Google Scholar]
- Palis, J. (1996). Flatwoods salamander (Ambystoma cingulatum Cope). Natural Areas Journal, 16(1), 49–54. [Google Scholar]
- Palis, J. G. (1997). Breeding migration of Ambystoma cingulatum in Florida. Journal of Herpetology, 31(1), 71. 10.2307/1565331 [DOI] [Google Scholar]
- Pauly, G. B., Piskurek, O., & Shaffer, H. B. (2007). Phylogeographic concordance in the southeastern United States: The flatwoods salamander, Ambystoma cingulatum, as a test case: Flatwoods salamander phylogeography. Molecular Ecology, 16(2), 415–429. 10.1111/j.1365-294X.2006.03149.x [DOI] [PubMed] [Google Scholar]
- Peterman, W. E., Anderson, T. L., Ousterhout, B. H., Drake, D. L., Semlitsch, R. D., & Eggert, L. S. (2015). Differential dispersal shapes population structure and patterns of genetic differentiation in two sympatric pond breeding salamanders. Conservation Genetics, 16(1), 59–69. 10.1007/s10592-014-0640-x [DOI] [Google Scholar]
- Petranka, J. (2010). Salamanders of the United States and Canada. Smithsonian Institution Press. [Google Scholar]
- Pritchard, J. K., Stephens, M., & Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics, 155(2), 945–959. 10.1093/genetics/155.2.945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2018). R: A language and environment for statistical computing. R Foundation for Statistical Computing. [Google Scholar]
- Raymond, M., & Rousset, F. (1995). GENEPOP (Version 1.2): Population genetics software for exact tests and ecumenicism. Journal of Heredity, 86(3), 248–249. 10.1093/oxfordjournals.jhered.a111573 [DOI] [Google Scholar]
- Richter‐Boix, A., Katzenberger, M., Duarte, H., Quintela, M., Tejedo, M., & Laurila, A. (2015). Local divergence of thermal reaction norms among amphibian populations is affected by pond temperature variation: Thermal reaction norm divergence in frog. Evolution, 69(8), 2210–2226. 10.1111/evo.12711 [DOI] [PubMed] [Google Scholar]
- Rittmeyer, E. N., & Austin, C. C. (2015). Combined next‐generation sequencing and morphology reveal fine‐scale speciation in Crocodile Skinks (Squamata: Scincidae: Tribolonotus). Molecular Ecology, 24(2), 466–483. 10.1111/mec.13030 [DOI] [PubMed] [Google Scholar]
- Rosenberg, N. A. (2003). distruct: A program for the graphical display of population structure: PROGRAM NOTE. Molecular Ecology Notes, 4(1), 137–138. 10.1046/j.1471-8286.2003.00566.x [DOI] [Google Scholar]
- Rousset, F. (2008). genepop’007: A complete re‐implementation of the genepop software for Windows and Linux. Molecular Ecology Resources, 8(1), 103–106. 10.1111/j.1471-8286.2007.01931.x [DOI] [PubMed] [Google Scholar]
- Sacerdote, A. B. (2009). Reintroduction of extripated flatwoods amphibians into restored forested wetlands in northern Illinois: Feasibility, assessment, implementation, habitat restoration and conservation implications (PhD dissertation). Norther Illinois University, DeKalb, IL. [Google Scholar]
- Semlitsch, R. D., Walls, S. C., Barichivich, W. J., & O’Donnell, K. M. (2017). Extinction debt as a driver of amphibian declines: An example with imperiled flatwoods salamanders. Journal of Herpetology, 51(1), 12–18. 10.1670/16-090 [DOI] [Google Scholar]
- Sommer, S. (2005). The importance of immune gene variability (MHC) in evolutionary ecology and conservation. Frontiers in Zoology, 2(1), 16. 10.1186/1742-9994-2-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titus, V. R., Bell, R. C., Becker, C. G., & Zamudio, K. R. (2014). Connectivity and gene flow among eastern tiger salamander (Ambystoma tigrinum) populations in highly modified anthropogenic landscapes. Conservation Genetics, 15(6), 1447–1462. 10.1007/s10592-014-0629-5 [DOI] [Google Scholar]
- USFWS [U.S. Fish and Wildlife Service] . (2015). Reticulated flatwoods salamander (Ambystoma bishopi) 5‐year review: Summary and evaluation. Federal Register, 79, FR 56821.
- Wang, I. J., Johnson, J. R., Johnson, B. B., & Shaffer, H. B. (2011). Effective population size is strongly correlated with breeding pond size in the endangered California tiger salamander, Ambystoma californiense . Conservation Genetics, 12, 911–920. 10.1007/s10592-011-0194-0 [DOI] [Google Scholar]
- Waples, R. S. (2015). Testing for Hardy‐Weinberg proportions: Have we lost the plot? Journal of Heredity, 106(1), 1–19. 10.1093/jhered/esu062 [DOI] [PubMed] [Google Scholar]
- Waples, R. S., & Do, C. (2010). Linkage disequilibrium estimates of contemporary Ne using highly variable genetic markers: A largely untapped resource for applied conservation and evolution. Evolutionary Applications, 3(3), 244–262. 10.1111/j.1752-4571.2009.00104.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir, B. S., & Cockerham, C. C. (1984). Estimating F ‐statistics for the analysis of population structure. Evolution, 38(6), 1358–1370. 10.1111/j.1558-5646.1984.tb05657.x [DOI] [PubMed] [Google Scholar]
- Wendt, A., Haas, C. A., Gorman, T., & Roberts, J. H. (2021). Metapopulation genetics of endangered reticulated flatwoods salamanders (Ambystoma bishopi) in a dynamic and fragmented landscape. Conservation Genetics, 22(4), 551–567. 10.1007/s10592-021-01360-3 [DOI] [Google Scholar]
- Whiteley, A. R., McGarigal, K., & Schwartz, M. K. (2014). Pronounced differences in genetic structure despite overall ecological similarity for two Ambystoma salamanders in the same landscape. Conservation Genetics, 15(3), 573–591. 10.1007/s10592-014-0562-7 [DOI] [Google Scholar]
- Williams, J. S., Niedzwiecki, J. H., & Weisrock, D. W. (2013). Species tree reconstruction of a poorly resolved clade of salamanders (Ambystomatidae) using multiple nuclear loci. Molecular Phylogenetics and Evolution, 68(3), 671–682. 10.1016/j.ympev.2013.04.013. [DOI] [PubMed] [Google Scholar]
- Williams, S. T., Haas, C. A., Roberts, J. H., & Taylor, S. S. (2020). Depauperate major histocompatibility complex variation in the endangered reticulated flatwoods salamander (Ambystoma bishopi). Immunogenetics, 72(4), 263–274. 10.1007/s00251-020-01160-y [DOI] [PubMed] [Google Scholar]
- Wilson, G. A., & Rannala, B. (2003). Bayesian inference of recent migration rates using multilocus genotypes. Genetics, 163(3), 1177–1191. 10.1093/genetics/163.3.1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
MHC and mtDNA data for this study are available at the NCBI GenBank repository (MT475763‐MT475765 and MT425246‐MT425250). Next‐generation sequencing data for this study are available at the NCBI Sequence Read Archive, BioProject, PRJNA750462.