

Abstract
Many chromatin modifying enzymes require metabolic cofactors to support their catalytic activities, providing a direct path for fluctuations in metabolite availability to regulate the epigenome. Over the past decade, our knowledge of this link has grown significantly. What began with studies showing cofactor availability drives global abundances of chromatin modifications has transitioned to discoveries highlighting metabolic enzymes as loci-specific regulators of gene expression. Here, we cover our current understanding of mechanisms that facilitate the dynamic and complex relationship between metabolism and the epigenome, focusing on the roles of essential metabolic and chromatin associated enzymes. We discuss physiological conditions where availability of these ‘epi-metabolites’ are dynamically altered, highlighting known links to the epigenome and proposing other plausible connections.
Keywords: chromatin, acetylation, acylation, methylation, circadian cycles, dietary challenges
Metabolic Links to the Epigenome
In eukaryotic organisms, the storage and accessibility of genomic DNA is regulated at the level of the nucleosome (see Glossary) core particle. Along with DNA, each nucleosome particle contains an octamer of histone proteins that are small, positively charged, and globular with flexible N-terminal “tails” that protrude from the larger structure. Due to their accessibility and amino acid composition, the protruding “tails” act as a platform for reversible, chemical post-translational modifications (PTMs) that then alter chromatin structure and function. This post-translational regulation of chromatin occurs through two major mechanisms: 1) PTMs change the intrinsic chemical properties of the amino acids (e.g. acetylation-dependent charge neutralization) and can induce structural changes to chromatin, and/or 2) PTMs are recognized by “reader” proteins that function to recruit additional chromatin effectors. Similar to histones, DNA can also be chemically modified to regulate gene expression. Together, chemical modification of histones and DNA comprise the epigenome.
Metabolism is believed to be a principal regulator of the epigenome as nearly all chromatin modifying enzymes require central metabolites (Figure 1, Key Figure and Table 1) as cofactors to support their catalytic activities. However, this metabolism-epigenome link appears to be more interesting and complex than simply chromatin-modifying enzymes siphoning off choice metabolites from canonical metabolic pathways. Recently, metabolic enzymes have also been shown to moonlight as context-dependent regulators of the epigenome, introducing an emerging aspect to this dynamic relationship.
Figure 1: Central metabolites are essential cofactors for chromatin modifying enzymes.
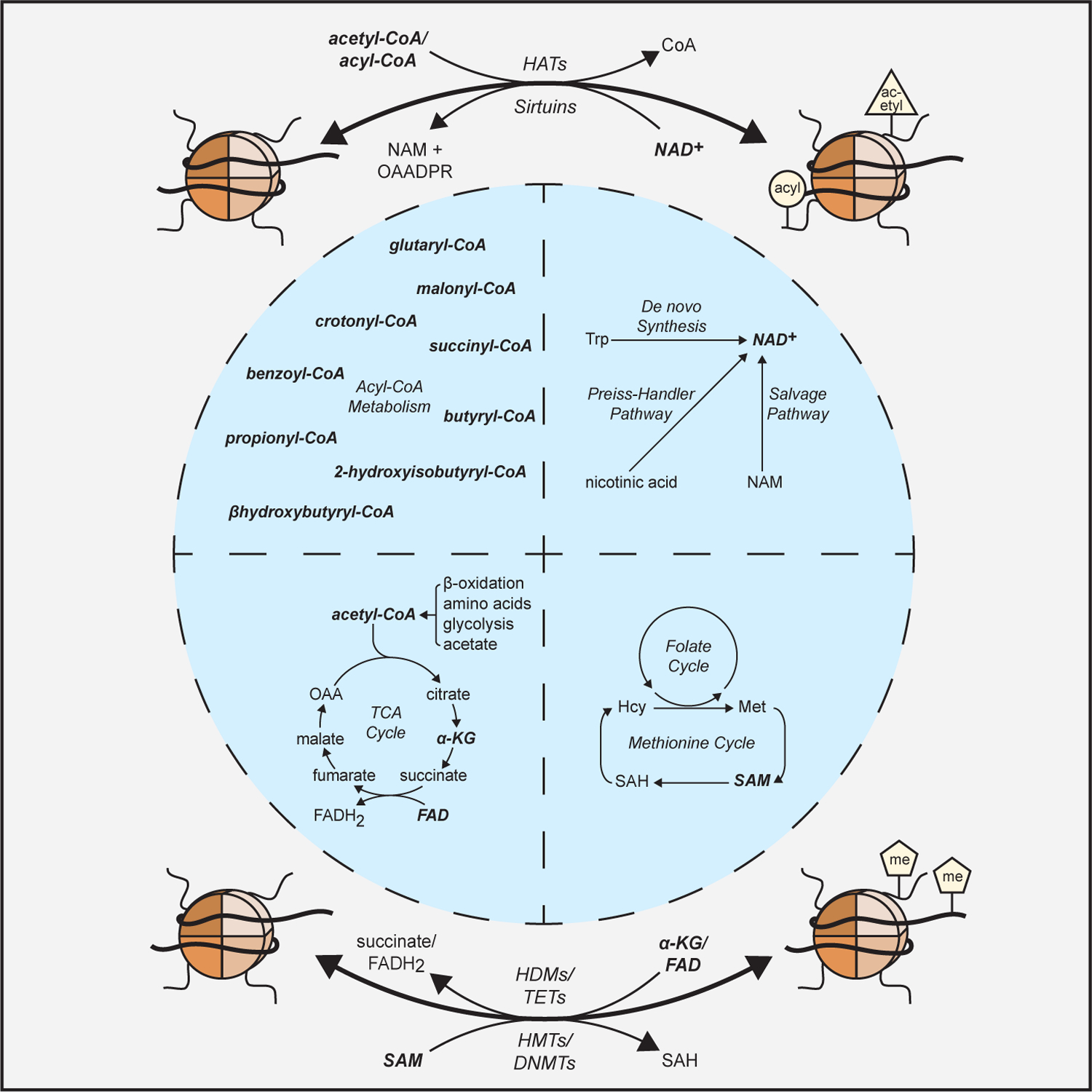
Central metabolites from diverse metabolic pathways are essential cofactors for the chromatin modifying enzymes (in bold) that regulate the epigenome. Enzyme abbreviations: HMT (histone methyltransferase); HDM (histone demethylase); DNMT (DNA methyltransferase); TET (DNA demethylase); HAT (histone acetyltransferase).
Table 1:
Essential Cofactors for Chromatin Modifying Enzymes
| Cofactor | Structure | Associated Metabolic Pathways | Supported Chromatin Modifying Reaction(s) | Common “Reader” Domains of Effectors | Citations |
|---|---|---|---|---|---|
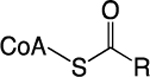
| Acetyl-CoA |
|
Glycolysis; TCA Cycle; FA Oxidation; Amino Acid Catabolism; Acetate Metabolism | Histone Acetylation | Bromo; YEATS; DPF | 1,9,11 |
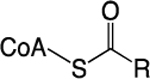
| Acyl-CoA |
|
FA Oxidation; Ketogenesis; Amino Acid Catabolism | Histone Acylation | YEATs; DPF | 1,8 |
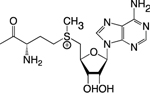
| SAM |
|
1C Metabolism; (Methionine and Folate Cycles); Phospholipid Synthesis | Histone Methylation; DNA Methylation | Chromo; PHD; Tudor, PWWP; TTD; MBD | 56,64,68 |
| NAD+ |
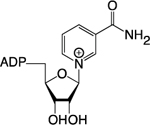
|
Amino Acid Catabolism; Preiss-Handler Pathway; NAM Salvage | Histone Deacetylation; Histone Deacylation; Histone ADP-Ribosylation | Bromo; YEATS; DPF; PBZ; WWE; Macro- | 34,36,112 |
| Alpha-KG |
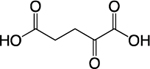
|
TCA Cycle; Amino Acid Metabolism | Histone Demethylation; DNA Demethylation | Chromo; PHD; Tudor, PWWP; TTD; MBD | 81,83,86 |
| FAD |
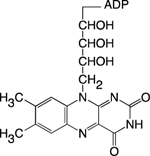
|
TCA Cycle; Oxidative Phosphorylation | Histone Demethylation | Chromo; PHD | 81–83 |
In this review, we will cover our current understanding of how essential metabolic cofactors and enzymes are able to dynamically regulate the acetyl-, acyl-, and methyl-states of the epigenome. We will highlight the chromatin effector proteins that mediate and sense these changes to the epigenome with a focus on the transcriptional consequences. Finally, we will cover physiological states (e.g. circadian cycles, aging, dietary perturbations, and diseases) associated with altered epigenetic cofactor metabolism, discussing known and potential links with the epigenome.
Metabolite-Dependent Chromatin Modifying Reactions
Histone Acetylation
Acetylation of the histone lysine ε-amino group (Kacetyl) is catalyzed by histone acetyltransferases (HATs) (Figure 2A and 2B). Three primary families of HATs have been identified (GNAT, MYST, and p300/CBP), all requiring acetyl-CoA as the acetyl-donating cofactor [1]. Histone Kacetyl supports accessible, transcriptionally active chromatin environments through two distinct yet complementary mechanisms: 1) neutralization of electrostatic interactions that contribute to higher-order chromatin structure and 2) by acting as a ligand for Kacetyl “readers.” For example, H4 Lys16 acetyl (H4K16ac) can disrupt electrostatic interactions in vitro between the H4 tail on one nucleosome particle and an acidic patch on the H2A:H2B dimer of the neighboring particle [2]. Disruption of these interactions is thought to support X-chromosome dosage compensation in Drosophila and lost transcriptional silencing in S. cerevisiae [3–5]. In addition, Kacetyl influences chromatin structure and function through its recognition by bromodomain-, YEATS domain-, and double PHD finger (DPF) domain-containing proteins [1]. By interacting with an acetyl-lysine residue, these epigenetic “readers” can recruit effectors (e.g. super elongation complex, transcription factors, ATP-dependent chromatin remodelers) that support transcriptional activation at specific genomic loci [6–8].
Figure 2: Reversible histone acetylation/acylation reaction diagrams and acetyl-CoA metabolism schematic.
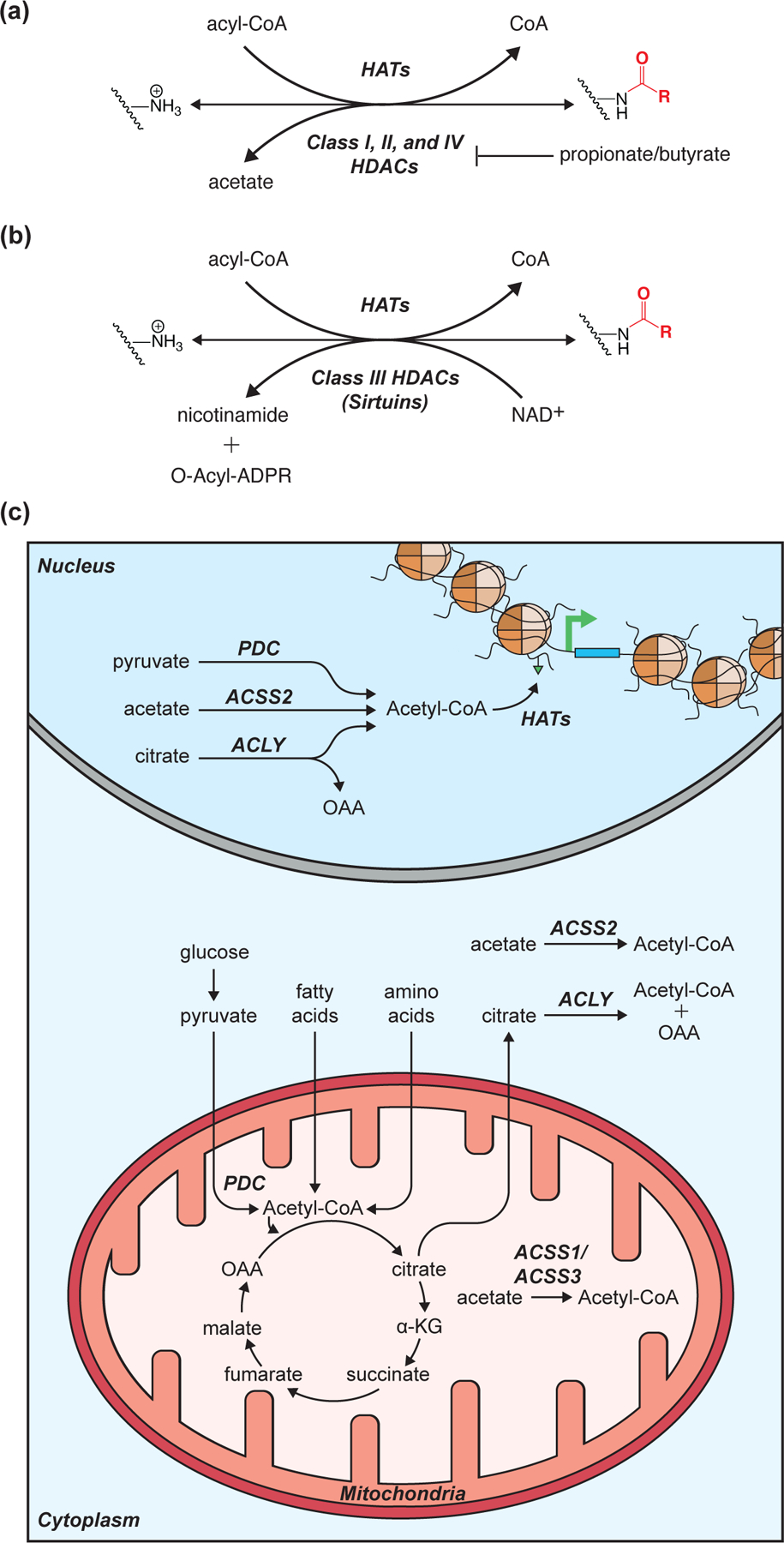
(A-B) Enzyme reaction diagrams illustrating the reversibility of histone lysine acetylation/acylation for Class I, II, and IV histone deacetylases (HDACs) (A) as well as Class III HDACs (B). (C) Schematic detailing acetyl-CoA metabolism and enzymes implicated in regulating global and/or loci specific histone acetyltransferase (HAT) reactions. Enzyme abbreviations: ACLY (ATP-citrate synthase); ACSS1–3 (acyl-CoA synthetase short-chain family member 1–3); PDC (pyruvate dehydrogenase complex).
Several enzymes are capable of generating the acetyl-CoA needed to support HAT reactions (Figure 2C). In mitochondria, acetyl-CoA can be generated by the pyruvate dehydrogenase complex (PDC), as a product of fatty acid oxidation, via amino acid degradation, or synthesized from free acetate by acyl-CoA synthetase short-chain family members 1 and 3 (ACSS1/ACSS3) [9]. However, as there is no mitochondrial acetyl-CoA exporter, acetyl-CoA must first be combined with oxaloacetate (OAA) to form citrate, which escapes mitochondria via the citrate shuttle. Once in the cytoplasm and/or nucleus, ATP-citrate synthase (ACLY) can convert the citrate back to acetyl-CoA and OAA [10]. In addition to ACLY, acyl-CoA synthetase short-chain family member 2 (ACSS2) and PDC have been implicated as nuclear acetyl-CoA synthesizing enzymes [11–13].
The catalytic activity of all three nuclear acetyl-CoA synthesizing enzymes (PDC, ACLY, and ACSS2) can maintain global histone Kacetyl levels [12–14]. However, ACLY and ACSS2 appear capable of supporting loci-specific histone Kacetyl under unique contexts. For example, ACLY-dependent Kacetyl drives the expression of key glycolytic genes during the differentiation of preadipocytes into mature adipocytes while ACSS2-dependent Kacetyl is a critical regulator of memory-related neuronal genes in the murine hippocampus [13–15]. These independent chromatin regulatory functions for ACLY and ACSS2 may be dictated, in part, by the production and/or flux of their respective substrates, citrate and acetate (Figure 2C). Such a mechanism would allow disparate metabolic states that affect acetyl-CoA precursor availability (e.g. hypoglycemia, ketosis, alcohol consumption, etc.) to initiate distinct chromatin responses.
Histone Acylation
Non-acetyl histone acylations are an emerging classification of PTM and will be referred to as acylations for the remainder of this review. Currently, nine unique lysine acylations (Kacyl) have been identified on histones, many of which are added enzymatically by HATs (Figure 2A and 2B and Figure 3A) [16–23]. Lysine malonylation and succinylation may occur in significant quantities through non-enzymatic mechanisms, given that the corresponding acyl-CoA precursors are more prone to intramolecular catalysis and anhydride formation relative to shorter-chain acyl-CoA molecules [24,25]. Acylation of the lysine ε-amino group can be recognized by proteins containing YEATs and DPF domains. These Kacyl “reader” domains possess substrate binding pockets with higher affinities for Kacyl over Kacetyl, although this is accomplished through disparate mechanisms [26–30]. YEATs domains preferentially accommodate Kacyl PTMs through aromatic residue π-stacking (Figure 3B). DPF domains, like bromodomains, rely on polar contacts with the substrate but are more structurally accommodating to acyl chains and can also utilize π-stacking to improve specificity for Kacyl residues (Figure 3C and 3D). These specific binding capabilities facilitate the unique functions for Kacyl relative to Kacetyl PTMs in regulating gene expression. For example, in S. cerevisiae, the metabolic switch from oxidative to fatty acid metabolism correlates with decreased H3K9ac and a corresponding increase in H3 Lys9 crotonyl (H3K9cro) abundance [31]. H3K9cro is then recognized by the YEATs domain-containing protein Taf14, mediating reduced expression of pro-growth genes [31].
Figure 3: Lysine acylations are chemically diverse and recognized by unique domains.
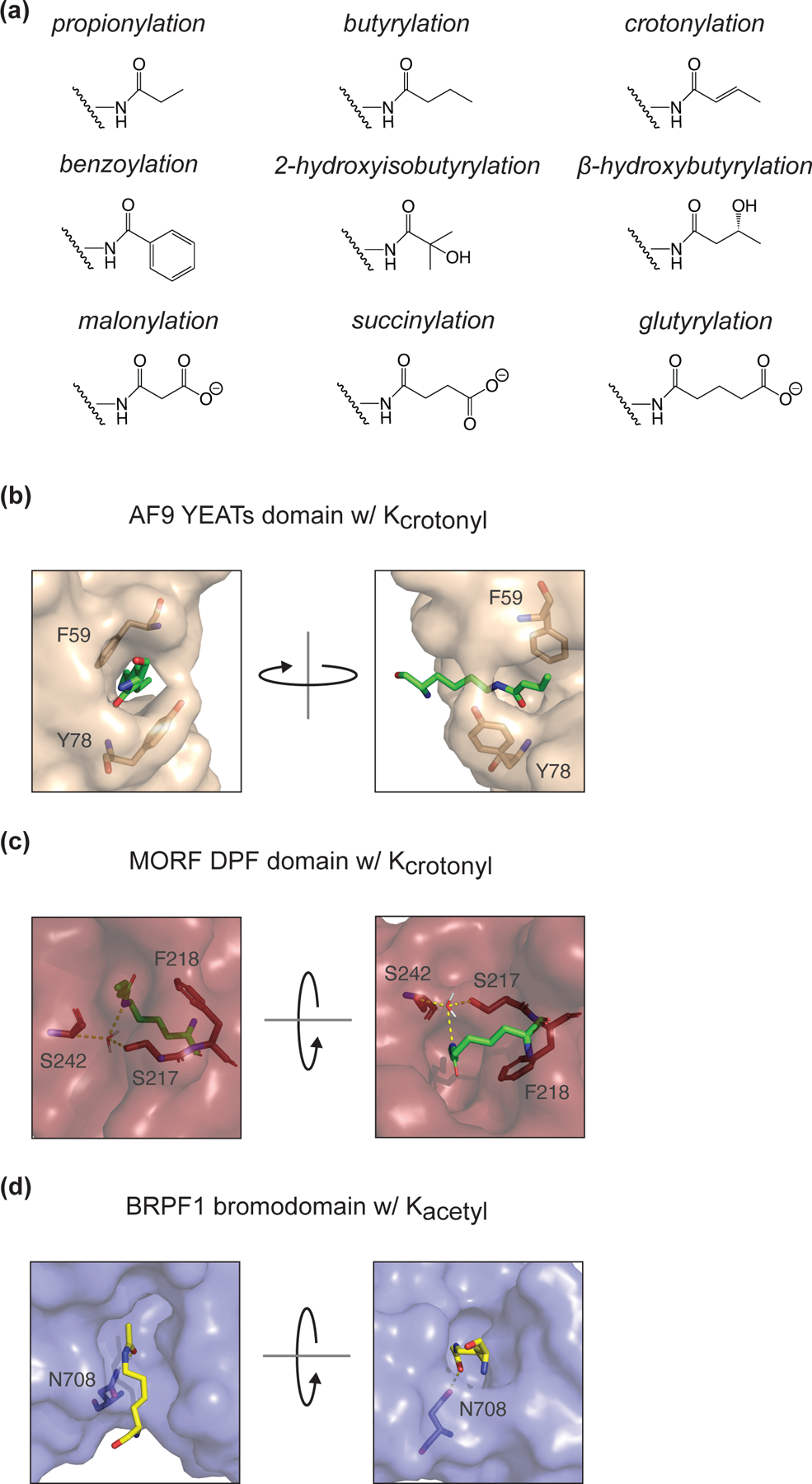
(A) Chemical structures for all identified histone lysine acylations. (B) Crystal structure diagram of Kcrotonyl in the YEATs domain binding pocket of H. sapiens AF9 (PDB: 5HJB). π-stacking is facilitated by residues F59 and Y78. (C) Crystal structure diagram of Kcrotonyl in the DPF domain binding pocket of H. sapiens MORF (PDB: 6OIE). S217 and S242 make polar contacts (mediated by a water molecule) with the crotonylated lysine. F218 improves specificity for Kcro over Kacetyl through π-stacking. (D) Crystal structure diagram of Kacetyl in the bromodomain binding pocket of H. sapiens BRPF1 (PDB: 5FFV). N708, a conserved residue in bromodomains, makes the sole polar contact with the acetylated lysine.
Unlike acetyl-CoA, the metabolic enzymes responsible for generating acyl-CoAs for chromatin acylation are not as well understood. Some acyl-CoAs are known to be generated within mitochondria (e.g. crotonyl-, malonyl-, succinyl-, and glutaryl-CoA), while others are present primarily as short chain fatty acids (SCFAs) or ketone bodies, requiring CoA-conjugation to be used as a cofactor. Tracing experiments have shown cells possess the ability to incorporate such metabolites directly onto chromatin, which may be mediated by promiscuous activity of ACSS enzymes [1,20,32]. In addition to enzymes that synthesize acyl-CoA molecules, catabolic enzymes also influence acyl-CoA abundance. Propionyl-CoA carboxylase alpha subunit knockout mice (PCAA−/−) accumulate propionyl-CoA, leading to increased global H3 Lys14 proprionyl (H3K14pro) [33]. Knockout of the butyryl-CoA catabolizing enzyme acyl-CoA dehydrogenase short-chain (ACADS−/−) also stimulates liver butyryl-CoA accumulation, but unexpectedly, has no effect on H3 Lys14 butyryl (H3K14bu) abundance [33]. Together, these findings suggest increased acyl-CoA concentrations do not always drive increased de novo histone acylation. Such observations implicate acyl-CoA sub-cellular compartmentalization and/or HAT activity as a critical regulator of Kacyl patterns.
Histone Deacetylation/Deacylation
Four main classes of histone deacetylases (HDACs) have been defined in mammalian cells, Class I, II, III, and IV, although only Class III HDACs require a metabolic cofactor to support catalytic activity (Figure 2A and 2B). These class III HDACs, also known as sirtuins, utilize a nicotinamide adenine dinucleotide (NAD+) molecule during catalysis. The reaction involves cleavage of the nicotinamide glycosidic bond and transfer of the acetyl-group from substrate to ADP-ribose, yielding nicotinamide (NAM) and O-acetyl-ADP-ribose (OAADPR) as additional products (Figure 2B) [34]. The seven mammalian sirtuins regulate diverse aspects of cell biology both in and outside the nucleus. Sirtuins 3–5 reside primarily within mitochondria, acting as protein deacetyl/deacylases, while SIRT1, SIRT2, SIRT6, and SIRT7 can target histones for nuclear deacetylation [34,35]. Given this family of enzymes’ dependence on a single cofactor, adequate NAD+ levels are critical for supporting sirtuin activity.
NAD+ synthesis occurs through three primary mechanisms within the cell: 1) de novo synthesis from tryptophan, 2) the Preiss-Handler pathway, and 3) the NAM salvage pathway [36]. The de novo NAD+ synthesis and Preiss-Handler pathways are relatively minor generators of NAD+ while the NAM salvage pathway is the major NAD+ contributor [36,37]. NAD+ molecules are the preferred cofactor for two-electron transfer redox reactions, resulting in reversible NADH formation. Unlike redox enzymes, sirtuin activity is not regulated by NAD+:NADH ratios, but is instead thought to be regulated by NAD+ abundance [38–41].
In addition to functioning as lysine deacetylases, sirtuins are capable of more general lysine deacylation. These distinct specificities are best described with sirtuins (SIRT3–5) found in mitochondria, where diverse acyl-modifications appear more prevalent. However, early evidence that sirtuins were capable of more than just deacetylation came from enzymology studies with nuclear/cytoplasmic enzymes SIRT1 and SIRT2 [42,43], paving the way for a systematic biochemical investigation of the deacylation capabilities of SIRT1–6 [44]. Surprisingly, long-chain deacylase activity was a common activity among sirtuins [44]. However, unlike SIRT1 and SIRT2, SIRT6 deacetylation and short chain (≤ crotonyl) deacylation activity were almost non-detectable [44]. In that study, Feldman et al demonstrated that free fatty acids (FFAs) could stimulate the catalytic activity for deacetylation. Following reports indicated that non-fatty acid small molecules are also able to significantly improve SIRT6 deacetylation and deacylation kinetics [44–46]. Small molecule activators with varying degrees of in vivo efficacy have been identified for SIRT1 as well, reviewed in depth by Hubbard and Sinclair [47]. Together, these findings propose a mechanism by which metabolites distinct from NAD+ can stimulate sirtuin-dependent deacetylation/deacylation to regulate gene expression. A recent study provided in situ evidence in support of this model, suggesting perilipin 5 acts as a carrier protein to deliver endogenous monounsaturated fatty acids (MUFAs) from lipid droplets to the nucleus [48]. Once in the nucleus, these MUFAs can stimulate SIRT1-dependent deacetylation and activation of the transcriptional co-regulator PGC-1α [48].
Although sirtuins are the only HDACs requiring metabolic cofactors to support catalytic activity, the SCFA metabolites butyrate and propionate are known catalytic inhibitors of Class I and II HDACs (Figure 2A). These SCFAs possess low- to mid-micromolar HDAC inhibitory IC50 values, falling in range of natural circulating concentrations [49–51]. Butyrate, having a lower IC50 value than propionate, is also the better studied in vivo HDAC inhibitor. For example, it has been shown that butyrate accumulation in cancerous colonocytes drives expression of pro-apoptotic genes through increased histone acetylation levels [52]. Interestingly, although these metabolites are known to be primarily generated via microbiota-dependent fermentation of non-digestible carbohydrates in the gut, they have also been shown to influence the epigenetic states of proximal host tissues [53,54]. While this is an exciting discovery that suggests a general mechanism by which microbiota are capable of influencing systemic host physiology, more studies are needed to determine the functional consequences of this microbial metabolism:host-chromatin axis.
Histone Methylation
Histone methylation reactions are catalyzed in a site-specific manner by histone methyltransferases (HMTs), most commonly on the ε-amino group of lysine residues (Figure 4A). Histone arginine methylation also occurs in the nucleus; however, here we focus on the metabolic influences over lysine methylation (Kme). For more information on arginine methylation, please see the review by Guccione and Richard [55].
Figure 4: Reversible chromatin methylation reaction diagrams and SAM metabolism schematic.
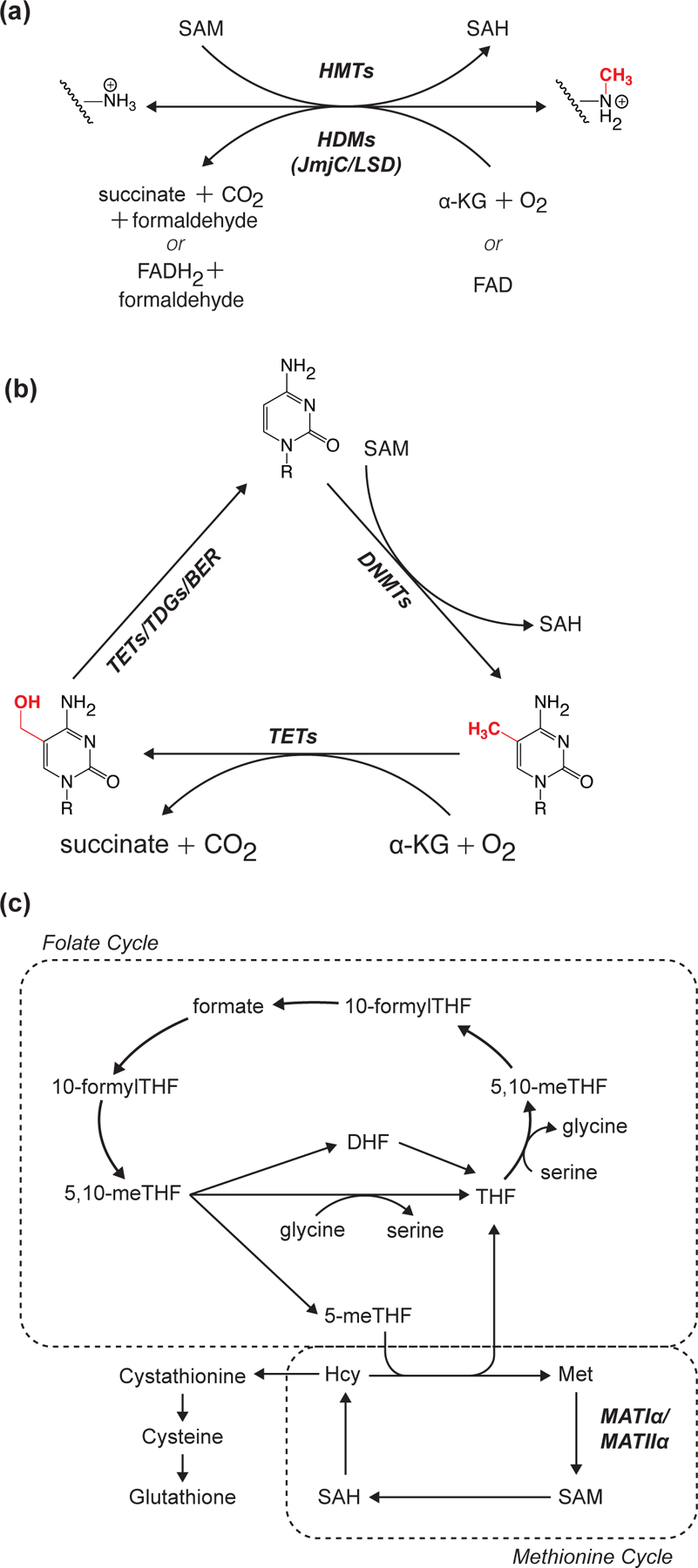
(A-B) Enzyme reaction diagrams illustrating reversibility of histone (A) and DNA (B) methylation. (C) Schematic highlighting position of SAM-synthesizing reactions at the crossroads of the folate and methionine cycles. Enzyme abbreviations: HMT (histone methyltransferase); HDM (histone demethylase); DNMT (DNA methyltransferase); TET (DNA demethylase); TDG (thymine DNA glycosylase); base excision repair (BER); MATI/IIα (methionine adenosyltransferase I/IIα).
Kme provides the most chemical diversity to the epigenome. Although only eight residues (e.g. H3K4/9/18/23/27/36/79 and H4K20) are sites of significant Kme, each lysine can support three unique methylation states (i.e. mono-, di-, or tri-methylation), resulting in 4.3x109 possible permutations of Kme for a given nucleosome. Kme is capable of supporting transcriptionally active or repressive chromatin environments, depending on the context [56]. This diverse functionality is facilitated by unique “reader” proteins that require specific methylation states and neighboring amino acids to interact with a given residue, enabling similar Kme-states to have drastically different effects on the chromatin environment. For example, H3K4me3 can be “read” by some PHD-containing transcription factors to facilitate chromatin remodeling and preinitiation complex formation at transcription start sites [57–59]. Opposingly, H3K9me2/3 can be “read” by the chromodomain-containing heterochromatin protein 1a which acts as a scaffold to recruit effector proteins that promote transcriptional silencing through the initiation and maintenance of constitutive heterochromatin [60].
All HMTs require S-adenosylmethionine (SAM) as the methyl-donor cofactor. In mammals, intracellular SAM is generated as a product of ATP-dependent methionine adenosyltransferase (MAT) reactions [61]. MAT reactions are catalyzed by only two known enzymes, MATIα and MATIIα. MATIα is expressed exclusively in the liver while MATIIα is ubiquitously expressed across tissues [61]. A third MAT protein, MATIIβ, possesses no inherent catalytic activity. Instead, MATIIβ functions as a regulatory binding partner of MATIIα, decreasing the enzyme’s Km, MET while also increasing MATIIα’s susceptibility to product inhibition [62,63]. These MAT-dependent SAM synthesis reactions are found at the core of one carbon metabolism, where the folate and methionine cycles converge, providing numerous avenues by which metabolic perturbations can influence SAM synthesis (Figure 4C) [64].
Limiting SAM availability by disrupting the metabolism and availability of its precursors can directly influence histone Kme abundance. In mouse embryonic stem cells (mESCs), disrupting the catabolism of threonine to glycine, an upstream SAM precursor, significantly reduces SAM availability as well as H3K4me2/3 abundance [65]. Subsequent studies corroborated this seminal study, highlighting the susceptibility of higher state (i.e. di- and tri-) histone Kme to decreased SAM availability [64]. Altered SAM abundance as a consequence of impaired or enhanced phospholipid metabolism has also been shown to directly influence histone methylation abundance [66,67]. Together, these direct correlations between global SAM availability and histone Kme highlight histone methylation as one of the most metabolically sensitive PTMs.
DNA Methylation
In addition to histones, individual DNA bases can be methylated. Methylation of cytosine at carbon 5 (5mC) is the most common and well-studied form of DNA methylation, typically occurring at CpG dinucleotides (Figure 4B). In mammals, these reactions are primarily catalyzed by DNA methyltransferases 3A and 3B (DNMT3A/B) at gene promoters, in the bodies of actively transcribed genes, and at repetitive DNA elements [68]. This review will focus on the links between metabolism and 5mC in gene promoters and regulatory elements. For more information on 5mC in gene bodies and at repetitive regions, please see the review by Greenberg and Bourc’his [68].
In promoters, 5mC is commonly found as part of broader CpG islands, which refer to DNA regions where CpG dinucleotides occur with a higher frequency relative to the rest of the genome [69]. Methylation of promoter CpG islands is normally associated with transcriptional repression. This gene-repressive function for 5mC can prevent transcription factor binding and/or recruitment of other chromatin modifying enzymes that support heterochromatic environments (e.g. H3K9 HMTs and HDACs) [70–76]. Contrary to common perception, 5mC can also facilitate transcriptional activation. However, unlike other epigenetic modifications associated with transcriptionally active genes (e.g. H3K9ac, H3K4me3, etc.), 5mC can act as a de-repressor through displacement of the transcriptional silencing complex PRC2 [77].
Like HMTs, DNMTs require SAM as an essential methyl-donor co-substrate, opening similar avenues for metabolic perturbations to influence 5mC profiles (Figure 4C). While mechanistic studies investigating direct links between SAM abundance and 5mC are limited, associations have been made under broader metabolic contexts [78,79]. For example, increased methionine cycle flux and glycine N-methyltransferase (GNMT) activity in the long-lived Ames-Dwarf mouse correlates with suppressed age-associated methylation at regulatory transcriptional elements [80]. Cole et al hypothesize these effects may be driven by decreased SAM availability, which if correct, would provide a direct link between perturbed methyl-donor metabolism and 5mC profiles.
Histone Demethylation
Histone demethylases (HDMs) are responsible for the active removal of Kme from histones. Two classes of HDMs have been defined based on catalytic mechanisms: 1) lysine-specific demethylase (LSD) and 2) jumonji C (JmjC) domain-containing demethylases (Figure 4A) [81]. LSDs utilize FAD-dependent oxidation of the methylated lysine ε-amine to facilitate methyl-group removal, producing FADH2 [82,83]. As this reaction requires the lysine ε-amine to have a free electron pair, LSDs may only catalyze the removal of mono- or di-methylation. JmjC demethylases initiate lysine and arginine demethylation using an Fe(II) and α-ketoglutarate (α-KG)-dependent dioxygenase mechanism, enabling this class of enzymes to target all forms of Kme for demethylation [83].
In addition to acting as histone demethylase substrates, FAD and α-KG support mitochondrial oxidative metabolism. FAD is an electron acceptor for the TCA cycle enzyme succinate dehydrogenase, producing FADH2 which is used by Complex II of the electron transport chain to drive ATP synthesis. In addition to its direct role in supporting energy metabolism, FAD availability has also been shown regulate LSD1 repression of energy expenditure target-genes in 3T3-L1 pre-adipocytes [84]. α-KG is a TCA cycle intermediate that can be generated by the oxidative decarboxylation of isocitrate or through glutamate anaplerosis. Furthermore, the JmjC reaction product succinate, as well as the structurally similar TCA cycle intermediate fumarate, can inhibit JmjC catalysis [85]. As a result, changes in mitochondrial metabolism are capable of directly influencing histone methylation profiles and subsequent gene expression.
DNA Demethylation
DNA methylation, like histone methylation, is a reversible epigenetic modification. Removal of 5mC DNA methylation can occur passively via dilution during DNA replication and/or actively by Fe(II) and α-KG-dependent dioxygenase enzymes. This family of DNA demethylases, known as TETs, catalyze several rounds of 5mC oxidation before thymine DNA glycosylase (TDG) and the base excision repair (BER) pathway replace the base with an unmodified cytosine (Figure 4B) [86]. TETs and JmjC demethylases utilize a similar catalytic mechanism and therefore possess the same metabolic dependencies and susceptibilities. For example, in mESCs, glycolysis- and glutamine-dependent intracellular α-KG supports the activities of JmjC and TET demethylases to remove repressive methyl-modifications, driving the expression of pluripotency associated genes [87,88].
Physiologic States with Altered Metabolism of Chromatin Modifying Cofactors
Due to the requirement of metabolic cofactors by many chromatin modifying enzymes (as described above), the epigenome is susceptible to perturbations in central metabolism. Such fluctuations in cofactor abundance can occur frequently over an organism’s lifespan, stemming from natural cellular processes (e.g. circadian regulation and aging) as well as external factors (e.g. changing dietary patterns and disease onset). While these fluctuations may not be solely responsible for stimulating changes to the epigenome, there are a number of cases in which some degree of causality can be illustrated. Here, we describe a subset of specific conditions where altered levels of metabolites directly affect epigenetic states.
Circadian Regulation
Circadian rhythm, or the internally regulated 24-hour oscillations of life processes that occur within organisms, can illicit significant changes in metabolite availability. The abundance of nearly 50% of mouse liver metabolites are circadian regulated, with 28% of this subset also being controlled by circadian mechanisms in human cells [89]. These cyclically regulated metabolites include cofactors for chromatin modifying enzymes (e.g. SAM, NAD+, and FAD), suggesting that the epigenome may be dynamically regulated in tune with circadian rhythms [89]. We propose that this mechanism requires a subset of epigenetic modifications to be sensitive to metabolic fluctuations, while the remaining fraction is largely unaffected (Figure 5A and 5B). This would enable the epigenome, and subsequent transcriptome, to have metabolic flexibility as needed while critical chromatin functions can continue unperturbed.
Figure 5: Known and Proposed Effects of NAD+, FAD, and SAM availability on chromatin structure and function.
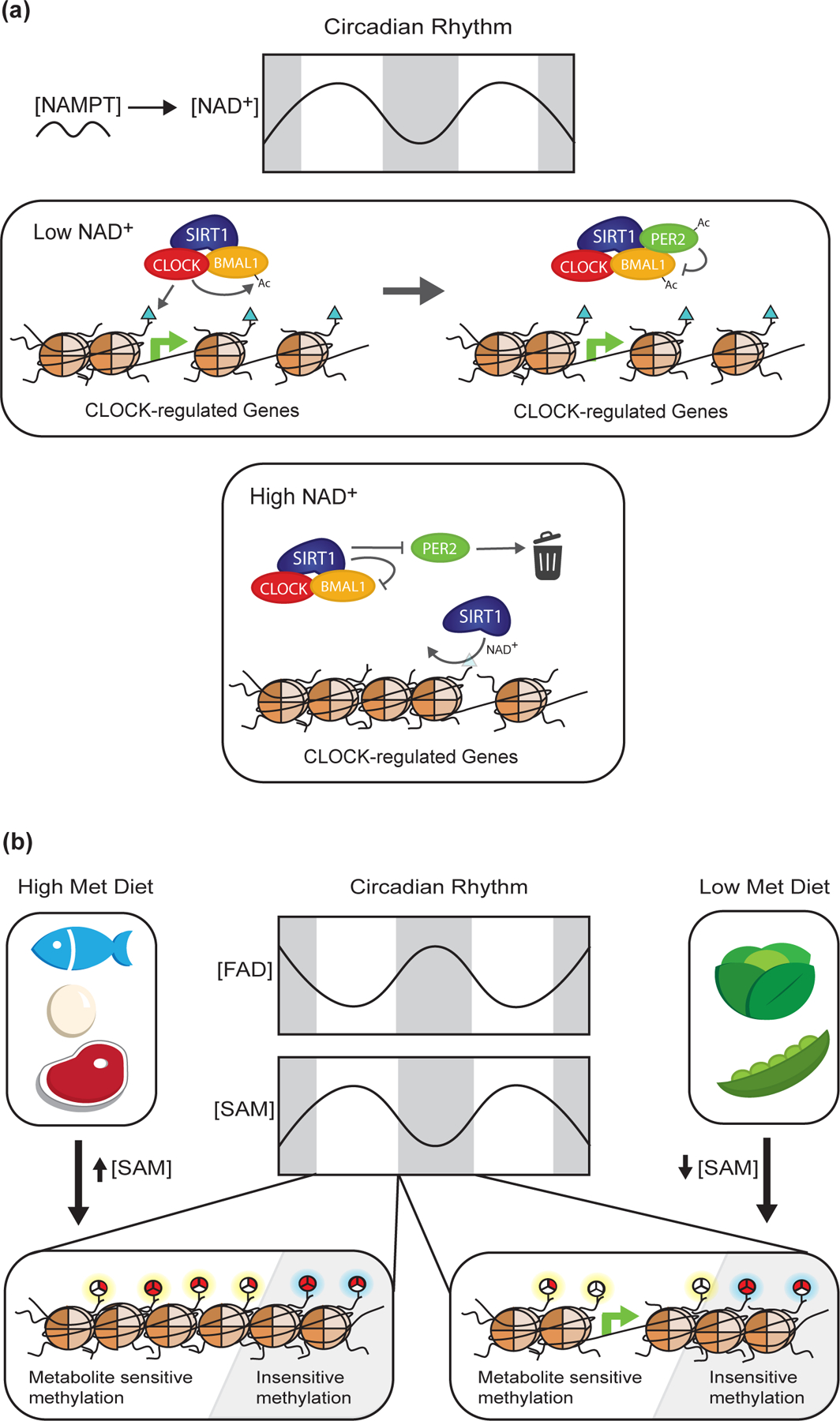
Circadian rhythm and dietary intake affect cellular availability of key metabolites that act as cofactors for chromatin modifying enzymes such as NAD+, FAD, and SAM. (A) Fluctuations in NAD+ levels enable SIRT1-dependent regulation of CLOCK target genes (e.g. NAMPT) through 2 independent yet non-exclusive mechanisms: 1) histone/BMAL1 deacetylation and 2) deacetylation of the CLOCK:BMAL1 negative regulator PER2. (B) Changes in SAM and FAD availability have been shown to directly influence chromatin methylation states, although the effect of circadian fluctuations in the abundances of these cofactors is unknown. Interestingly, the literature suggests methyl-modifications at certain loci are more sensitive (highlighted in yellow) to altered SAM and FAD availability than other loci (highlighted in blue). This mechanism may allow for dynamic flexibility in chromatin structure and function by enabling cells to adapt to various perturbations, while still supporting critical chromatin functions.
For example, oscillating regulation of NAD+ biosynthesis and availability have been shown to directly influence sensitive sites of Kacetyl targeted by the core circadian machinery complex, CLOCK:BMAL1 (Figure 5A). The NAD+ salvage pathway rate-limiting enzyme nicotinamide phosphoribosyltransferase (NAMPT) is regulated in a circadian manner at both the mRNA and protein level, creating similar fluctuations in NAD+ levels [90,91]. NAD+ is an essential cofactor for the deacetylase SIRT1, which was reported to antagonize H3K9ac and H3K14ac controlled by CLOCK:BMAL1, as well as BMAL1 acetylation [92,93]. An alternative mechanism by which fluctuating NAD+ regulates circadian transcription involves SIRT1-dependent deacetylation of PER2, a CLOCK:BMAL1 inhibitor (Figure 5A) [94]. Regardless of the mechanism, there is compelling evidence suggesting SIRT1’s dependency on intrinsic NAD+ fluctuations leads to the oscillating repression of circadian-regulated genes.
Aging
At the level of chromatin, aging is generally characterized as an increase in transcriptional noise as alterations to the patterns of epigenetic modifications leads to dysregulation of gene expression and chromatin structure [95]. While the mechanisms contributing to the loss of epigenetic information are still unknown, it is hypothesized that dysregulation of metabolic pathways may play a role in exacerbating aging phenotypes.
Acetyl-CoA levels have been shown to increase with age in Drosophila, caused by an increase in the activity of the acetyl-CoA-synthesizing enzyme, ATPCL. Rising levels of acetyl-CoA promote significant increases in H4K12ac, H3K9ac, H3K9acK14ac, and H3K23ac, provoking age-associated increases in aberrant gene transcription [96]. Additionally, age-dependent decreases in the histone deacetylase Sir2, the yeast homolog to mammalian SIRT1, and its essential cofactor NAD+, further support loss of chromatin maintenance. Diminishing HDAC activity leads to global increases in H4K16ac levels, compromising heterochromatin stability in genomic regions such as telomeres, ribosomal DNA (rDNA), and silenced mating type loci [97].
One well-established dietary intervention to delay onset of age-related diseases and extend longevity is caloric restriction (CR), the method of limiting calorie intake without causing malnutrition. Interestingly, recent studies have associated CR with reduced “methylation drift”, a scenario where normally hypermethylated CpG islands become hypomethylated and vise-versa [98]. This loss of specific methylation patterns is a common epigenetic phenotype of aging and correlates with alterations in gene expression [98]. Some of these age-associated changes in CpG methylation are robust enough within mammalian populations that they can be used as predictors of chronological age [99]. In rhesus monkeys, CR by 30% for 7–14 years resulted in a “methylation age” 7 years younger, on average, when compared to chronologically age-matched controls. In mice, an even more pronounced phenotype was observed in animals fed a 40% caloric restrictive diet, where on average, mice displayed a methylation age of 0.8 years compared to their actual chronological age of 2.8 years [98]. Although such alterations to DNA methylation have not been shown to be causal in slowing aging, the ability of CR to counteract aging and age-related diseases could be mediated at the epigenetic level through the alterations in epi-metabolites discussed in this review. In more general terms, it is plausible that the metabolic reprogramming of transcriptional states associated with CR is driven by chronic alterations in epi-metabolites, hormetic responses, and signaling pathways that sense and transmit metabolic restriction to chromatin.
Cancer
Reprogrammed energy metabolism is a trademark of tumor cells, highlighted by the presence of the Warburg effect. A hypothesized benefit of the Warburg effect is increased availability of glycolysis intermediates that can be shunted into various biosynthesis pathways that produce macromolecules required for proliferation and growth [100]. A secondary consequence of the Warburg effect is elevated intracellular concentrations of acetyl-CoA, providing a direct link between altered cancer metabolism and the epigenome [101].
In fact, alteration of histone acetylation patterns is a hallmark of many solid tumors. In cooperation with increased glycolytic flux, oncogenic AKT has been shown to drive increases in acetyl-CoA levels through phosphorylation and activation of ACLY. This AKT-dependent rise in acetyl-CoA is sufficient to increase global histone acetylation levels [102]. The citrate used by ACLY is supplied by increased glycolytic flux, highlighting the cooperativity of these oncogenic mechanisms [101]. More studies are needed to determine if ACLY-dependent hyperacetylation might promote cancer pathology through specific transcriptional states or through a more general mechanism involving global changes to chromatin and increased genomic instability.
In addition to histone acetylation, altered cancer metabolism is directly linked to changes in histone and DNA methylation abundance as well. Mutations in isocitrate dehydrogenase genes IDH1 and IDH2 (commonly found in gliomas, melanomas, and acute myeloid leukemias) encode a mutant enzyme that catalyzes the reduction of α-KG to 2-hydroxyglutarate (2-HG) [103]. 2-HG functions as a competitive inhibitor for JmjC and TET demethylases, binding to the same region as α-KG, causing genome wide alterations to histone and DNA methylation patterns. Loss of function mutations to succinate and fumarate dehydrogenase, TCA cycle enzymes downstream of IDH, are also associated with various cancers. Such mutations result in the accumulation of succinate and fumarate, respectively, which are capable of inhibiting JmjC and TET demethylases through a similar mechanism as 2-HG [85,104,105]. Together, these metabolite-induced changes in epigenetic methylation status are believed to block differentiation and promote tumorigenesis, emphasizing the detrimental effects that oncometabolites, produced through altered cancer metabolism, can have on the epigenome.
Dietary Alterations
Many metabolic pathways depend on dietary intake of essential amino acids (EAAs) as organisms lack the ability for endogenous synthesis. EAAs are often precursors to cofactors used by chromatin modifying enzymes (Figure 1 and Table 1), showcasing a potential regulatory link between diet and the epigenome. For instance, foods such as eggs, poultry, red-meat, and fish contain high levels of methionine, the obligatory precursor of the methyl-donor cofactor SAM. Consumption of plant-based diets that avoid such foods correlate with significantly lower plasma methionine levels, among other amino acids, which may impact an organism’s methylation capacity (Figure 5B) [106]. While the relationship between dietary methionine restriction, as well as that of other EAAs, and the epigenome is documented, the phenotypes elicited by these diets are typically positive [64,107–109]. From a chromatin perspective, this appears counterintuitive as the literature implies decreased availability of essential metabolic precursors and cofactors would result in dysregulation of the epigenome, promoting negative pathologies. Interestingly, Haws et al. recently demonstrated cells/organisms possess the ability to adaptively regulate and preserve critical chromatin regions during fluctuations in methyl-metabolite availability [67]. These findings suggest fluctuations in the abundance of other EAAs or essential metabolic cofactors may stimulate similar responses, proposing a general mechanism by which regulation of essential genomic loci can be preserved during severe metabolic perturbations.
In addition to EAA restricted diets, high energy diets (high fat and simple sugars) can also impact the epigenome. Keleher et al. showed such diets stimulate altered DNA methylation profiles in SM/J mouse liver [110]. Interestingly, type 2 diabetes mellitus (T2DM), a common pathophysiological consequence of chronic high energy diet intake, is associated with altered DNA methylation profiles in pancreatic β-cells as well. These changes include hypermethylation of promoters for genes which regulate cellular metabolism and β-cell function, correlating with glycolated hemoglobin A1c (HbA1c) plasma levels, a long-term readout of plasma glucose concentrations [111]. While the mechanisms that outline how high energy diet/T2DM alter DNA methylation require further investigation, these collections of studies highlight how metabolic perturbations are capable of affecting the epigenome.
Concluding Remarks
Susceptibility of the epigenome to altered metabolic cofactor availability provides a direct path for metabolism to regulate a cell’s and/or organism’s transcriptional environment. This allows chromatin to act as a signal integrator of diverse metabolic pathways, like those described here, to regulate both canonical (e.g. acetylation and methylation) and emerging (e.g. acylation, ADP-ribosylation, O-GlcNAcylation, and lactylation, etc.) epigenetic modifications [112–114]. Numerous studies have provided evidence in support of this general model, yet it remains largely unclear why certain regions of the epigenome are more or less susceptible to metabolic perturbations than others (see Outstanding Questions). Recent insights into the capabilities of loci-specific regulation of the epigenome by metabolic enzymes introduces potential mechanisms for more directed regulation of chromatin modifications during fluctuations in cofactor availability. However, these metabolic enzymes cannot act in isolation as the PTMs they support will require coordinated regulation of relevant chromatin effectors to facilitate a functional transcriptional response. Broad experimental approaches (i.e. biochemical, genetic, physiological, etc.) will be needed to uncover such complex mechanisms as the metabolism-epigenome relationship paradigm continues to shift, opening exciting opportunities for the field moving forward.
Outstanding Questions.
Why are PTMs at certain genomic loci more susceptible to fluctuations in epigenetic cofactor availability than others?
Do adaptive mechanisms maintain regulation of critical chromatin regions during physiologic fluctuations in cofactor availability?
How do metabolic enzymes support loci-specific regulation of gene expression?
What signal(s) determine whether metabolism-induced changes to the epigenome revert upon metabolic recovery or are instead retained permanently?
What are the functional consequences of altered epigenetic profiles during natural or induced fluctuations in epigenetic cofactor availability?
How much does tissue and/or cell-type-specific expression of chromatin modifying enzymes impact the metabolic sensitivity of a cell’s epigenome?
Highlights.
Metabolites from diverse pathways act as essential cofactors for chromatin modifying enzymes.
Distinct reader domains of chromatin effectors facilitate the downstream functions for individual post-translational modifications.
Metabolic enzymes are capable of moonlighting as loci-specific regulators of the epigenome.
Physiologic fluctuations in metabolite cofactor availability can be stimulated by natural and exogenous stimuli.
Acknowledgements
This work was supported through grants from the NIH (S.A.H. –T32 DK007665 and J.M.D. – R37 GM059785). We apologize to our colleagues whose work was unable to be cited given strict limitations in reference number.
Glossary
- Acetylation
Chemical reaction that adds an acetyl group to a protein or molecule. Histone acetylation takes place on lysine residues residing within histone tails, requires histone acetyltransferases (HATS) and the acetyl-donor acetyl-CoA, and can elicit varying functions to chromatin structure and function depending on the site of modification.
- Chromatin Effectors
Proteins that bind to chromatin and elicit specific modifications or transcriptional outcomes. Also known as chromatin modifying enzymes.
- Epigenome
In this review, refers to chemical modifications on histones and DNA, where chemical modifications allow for regulation of chromatin structure and function without altering genomic sequences.
- Euchromatin
A physically open, transcriptionally permissive structure of chromatin that allows for the binding of transcription machinery to DNA.
- Heterochromatin
A physically compact, dense structure of chromatin that supports transcriptional repression through the preclusion of transcription machinery.
- Higher-order Chromatin Structure
Refers to structural states of compacting chromatin beyond ‘beads on a string’ (extended nucleosomes on a stretch of DNA) where more compaction of nucleosomes is related to less gene expression.
- Methylation
Chemical reaction that catalyzes the addition of a methyl group to a protein or molecule. Histone methylation takes place on lysine or arginine residues on histone “tails” and requires both histone methyltransferases (HMTs) and the cofactor SAM. DNA methylation takes place at cytosine carbon 5 (5mC) typically at CpG dinucleotides and requires DNA methyltransferases (DNMTs) and SAM. Histone and DNA methylation is typically associated with gene repression.
- Nucleosome
The fundamental structural unit of chromatin, consisting of ~147bp of DNA wrapped around a histone octamer (consisting of two copies each of H2A, H2B, H3, and H4).
- Post-Translational Modifications (PTMs)
Covalent chemical modifications made to proteins that elicit specific biological functions. In the context of histones this can refer to acetylation, acylation, methylation, ADP-ribosylation, phosphorylation, ubiquitylation, sumoylation, and more.
- “Reader” Proteins
Proteins which contain reader domains that allow for the detection of epigenetic modifications and typically respond by recruiting additional proteins to carry out a specific function at genomic loci marked by a histone PTM.
- Warburg Effect
The observation that cancer cells favor aerobic glycolysis irrespective of oxygen availability.
References
- 1.Sabari BR et al. (2017) Metabolic regulation of gene expression through histone acylations. Nat. Rev. Mol. Cell Biol 18, 90–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang R et al. (2017) Histone Acetylation Regulates Chromatin Accessibility: Role of H4K16 in Inter-nucleosome Interaction. Biophys. J 112, 450–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Copur Ö et al. (2018) Sex-specific phenotypes of histone H4 point mutants establish dosage compensation as the critical function of H4K16 acetylation in Drosophila. Proc. Natl. Acad. Sci. U.S.A 115, 13336–13341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson LM (1990) Genetic evidence for an interaction between SIR3 and histone H4 in the repression of the silent mating loci in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A 87, 6286–6290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Park EC and Szostak JW (1990) Point mutations in the yeast histone H4 gene prevent silencing of the silent mating type locus HML. Mol Cell. Bio 10, 4932–4934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fujisawa T and Filippakopoulos P (2017) Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat. Rev. Mol. Cell Biol 18, 246–262 [DOI] [PubMed] [Google Scholar]
- 7.Gates LA et al. (2017) Acetylation on histone H3 lysine 9 mediates a switch from transcription initiation to elongation. J. Biol. Chem 292, 14456–14472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao D et al. (2017) YEATS Domain—A Histone Acylation Reader in Health and Disease. J. Mol. Bio 429, 1994–2002 [DOI] [PubMed] [Google Scholar]
- 9.Fan J et al. (2015) Metabolic regulation of histone post-translational modifications. ACS Chem. Biol 10, 95–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sivanand S et al. (2017) Nuclear Acetyl-CoA Production by ACLY Promotes Homologous Recombination. Mol. Cell 67, 252–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sivanand S et al. (2018) Spatiotemporal Control of Acetyl-CoA Metabolism in Chromatin Regulation. Trends Biochem. Sci 43, 61–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sutendra G et al. (2014) A Nuclear Pyruvate Dehydrogenase Complex Is Important for the Generation of Acetyl-CoA and Histone Acetylation. Cell 158, 84–97 [DOI] [PubMed] [Google Scholar]
- 13.Mews P et al. (2017) Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory. Nature 546, 381–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wellen KE et al. (2009) ATP-Citrate Lyase Links Cellular Metabolism to Histone Acetylation. Science 324, 1076–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mews P et al. (2019) Alcohol metabolism contributes to brain histone acetylation. Nature 574, 717–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang H et al. (2018) Lysine benzoylation is a histone mark regulated by SIRT2. Nat Commun DOI: 10.1038/s41467-018-05567-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simithy J et al. (2017) Characterization of histone acylations links chromatin modifications with metabolism. Nat Commun DOI: 10.1038/s41467-017-01384-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Y et al. (2007) Lysine Propionylation and Butyrylation Are Novel Post-translational Modifications in Histones. Mol. Cell. Proteomics 6, 812–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tan M et al. (2011) Identification of 67 Histone Marks and Histone Lysine Crotonylation as a New Type of Histone Modification. Cell 146, 1016–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie Z et al. (2016) Metabolic Regulation of Gene Expression by Histone Lysine β-Hydroxybutyrylation. Mol. Cel.l 62, 194–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dai L et al. (2014) Lysine 2-hydroxyisobutyrylation is a widely distributed active histone mark. Nat. Chem. Biol 10, 365–370 [DOI] [PubMed] [Google Scholar]
- 22.Xie Z et al. (2012) Lysine Succinylation and Lysine Malonylation in Histones. Mol. Cell. Proteomics 11, 100–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bao X et al. (2019) Glutarylation of Histone H4 Lysine 91 Regulates Chromatin Dynamics. Mol. Cell 76, 660–675 [DOI] [PubMed] [Google Scholar]
- 24.Wagner GR et al. (2017) A Class of Reactive Acyl-CoA Species Reveals the Non-enzymatic Origins of Protein Acylation. Cell Metab 25, 823–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trub AG and Hirschey MD (2018) Reactive Acyl-CoA Species Modify Proteins and Induce Carbon Stress. Trends Biochem. Sci 43, 369–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Y et al. (2016) Molecular Coupling of Histone Crotonylation and Active Transcription by AF9 YEATS Domain. Mol. Cel.l 62, 181–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Q et al. (2016) Structural Insights into Histone Crotonyl-Lysine Recognition by the AF9 YEATS Domain. Structure 24, 1606–1612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y et al. (2018) Identification of the YEATS domain of GAS41 as a pH-dependent reader of histone succinylation. Proc. Natl. Acad. Sci. U.S.A 115, 2365–2370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klein BJ et al. (2019) Histone H3K23-specific acetylation by MORF is coupled to H3K14 acylation. Nat. Commun DOI: 10.1038/s41467-019-12551-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andrews FH et al. (2016) The Taf14 YEATS domain is a reader of histone crotonylation. Nat. Chem. Biol 12, 396–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gowans GJ et al. (2019) Recognition of Histone Crotonylation by Taf14 Links Metabolic State to Gene Expression. Mol. Cell 76, 909–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sabari BR et al. (2017) Intracellular Crotonyl-CoA Stimulates Transcription through p300-Catalyzed Histone Crotonylation. Mol. Cell 58, 203–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kebede AF et al. (2017) Histone propionylation is a mark of active chromatin. Nat. Struct. Mol. Biol 24, 1048–1056 [DOI] [PubMed] [Google Scholar]
- 34.Feldman JL et al. (2012). Sirtuin Catalysis and Regulation. J. Biol. Chem 287, 42419–42427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jing H and Lin H (2015) Sirtuins in Epigenetic Regulation. Chem. Rev 115, 2350–2375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Verdin E (2015) NAD+ in aging, metabolism, and neurodegeneration. Science 350, 1208–1213 [DOI] [PubMed] [Google Scholar]
- 37.Revollo JR et al. (2004) The NAD Biosynthesis Pathway Mediated by Nicotinamide Phosphoribosyltransferase Regulates Sir2 Activity in Mammalian Cells. J. Biol. Chem 279, 50754–50763 [DOI] [PubMed] [Google Scholar]
- 38.Mouchiroud L et al. (2013) The NAD+/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 154, 430–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Belenky P et al. (2007) Nicotinamide Riboside Promotes Sir2 Silencing and Extends Lifespan via Nrk and Urh1/Pnp1/Meu1 Pathways to NAD+. Cell 129, 473–484 [DOI] [PubMed] [Google Scholar]
- 40.Ho C et al. (2009) SIRT1 markedly extends replicative lifespan if the NAD+ salvage pathway is enhanced. FEBS Lett 583, 3081–3085 [DOI] [PubMed] [Google Scholar]
- 41.Brown KD et al. (2014) Activation of SIRT3 by the NAD+ Precursor Nicotinamide Riboside Protects from Noise-Induced Hearing Loss. Cell Metab 20, 1059–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheng Z et al. (2009) Molecular Characterization of Propionyllysines in Non-histone Proteins. Mol. Cell. Proteomics 8, 45–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith BC and Denu JM (2007) Acetyl-lysine Analog Peptides as Mechanistic Probes of Protein Deacetylases. J. Biol. Chem 282, 37256–37265 [DOI] [PubMed] [Google Scholar]
- 44.Feldman JL et al. (2013) Activation of the Protein Deacetylase SIRT6 by Long-chain Fatty Acids and Widespread Deacylation by Mammalian Sirtuins. J. Biol. Chem 288, 31350–31356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Klein MA et al. (2020) Mechanism of activation for the sirtuin 6 protein deacylase. J. Biol. Chem 295, 1385–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rahnasto-Rilla M et al. (2018) Natural polyphenols as sirtuin 6 modulators. Sci. Rep DOI: 10.1038/s41598-018-22388-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hubbard BP and Sinclair DA (2014) Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol. Sci 35, 146–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Najt CP et al. (2020) Lipid Droplet-Derived Monounsaturated Fatty Acids Traffic via PLIN5 to Allosterically Activate SIRT1. Mol. Cell 77, 810–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Qin Y and Wade PA (2018) Crosstalk between the microbiome and epigenome: messages from bugs. J. Biochem 163, 105–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sekhavat A et al. (2007) Competitive inhibition of histone deacetylase activity by trichostatin A and butyrate. Biochem. Cell Biol 85, 751–758 [DOI] [PubMed] [Google Scholar]
- 51.Waldecker M et al. (2008) Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. J. Nutr. Biochem 19, 587–593 [DOI] [PubMed] [Google Scholar]
- 52.Donohoe DR et al. (2012) The Warburg Effect Dictates the Mechanism of Butyrate-Mediated Histone Acetylation and Cell Proliferation. Mol. Cell 48, 612–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.den Besten G et al. (2013) The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res 54, 2325–2340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Krautkramer KA et al. (2016) Diet-Microbiota Interactions Mediate Global Epigenetic Programming in Multiple Host Tissues. Mol. Cell 64, 982–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guccione E and Richard S (2019) The regulation, functions and clinical relevance of arginine methylation. Nat. Rev. Mol. Cell. Biol 20, 642–657 [DOI] [PubMed] [Google Scholar]
- 56.Hyun K et al. (2017) Writing, erasing and reading histone lysine methylations. Exp. Mol. Med DOI: 10.1038/emm.2017.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li H et al. (2006) Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature 442, 91–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wysocka J et al. (2006) A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature 442, 86–90 [DOI] [PubMed] [Google Scholar]
- 59.Lauberth SM et al. (2013) H3K4me3 Interactions with TAF3 Regulate Preinitiation Complex Assembly and Selective Gene Activation. Cell 152, 1021–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eissenberg JC and Elgin SCR (2014) HP1a: A Structural Chromosomal Protein Regulating Transcription. Trends Genet 30, 103–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Markham GD and Pajares MA (2009) Structure-function relationships in methionine adenosyltransferases. Cell. Mol. Life Sci 66, 636–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Halim AB et al. (1999) Expression and Functional Interaction of the Catalytic and Regulatory Subunits of Human Methionine Adenosyltransferase in Mammalian Cells. J. Biol. Chem 274, 29720–29725 [DOI] [PubMed] [Google Scholar]
- 63.LeGros HL et al. (2000) Cloning, Expression, and Functional Characterization of the β Regulatory Subunit of Human Methionine Adenosyltransferase (MAT II). J. Biol. Chem 275, 2359–2366 [DOI] [PubMed] [Google Scholar]
- 64.Sanderson SM et al. (2019) Methionine metabolism in health and cancer: a nexus of diet and precision medicine. Nat. Rev. Cancer 19, 625–637 [DOI] [PubMed] [Google Scholar]
- 65.Shyh-Chang N et al. (2013) Influence of Threonine Metabolism on S-Adenosylmethionine and Histone Methylation. Science 339, 222–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ye C et al. (2017) A Metabolic Function for Phospholipid and Histone Methylation. Mol. Cell 66, 180–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Haws SA et al. (2020) Methyl-Metabolite Depletion Elicits Adaptive Responses to Support Heterochromatin Stability and Epigenetic Persistence. Mol. Cell DOI: 10.1016/j.molcel.2020.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Greenberg MVC and Bourc’his D (2019) The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell. Biol 20, 590–607 [DOI] [PubMed] [Google Scholar]
- 69.Deaton AM and Bird A (2011) CpG islands and the regulation of transcription. Genes Dev 25, 1010–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yin Y et al. (2017) Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Estève PO et al. (2006) Direct interaction between DNMT1 and G9a coordinates DNA and histone methylation during replication. Genes Dev 20, 3089–3103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Myant K et al. (2011) LSH and G9a/GLP complex are required for developmentally programmed DNA methylation. Genome. Res 21, 83–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Deplus R et al. (2002) Dnmt3L is a transcriptional repressor that recruits histone deacetylase. Nucleic Acids Res 30, 3831–3838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fuks F et al. (2000) T. DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat. Genet 24, 88–91 [DOI] [PubMed] [Google Scholar]
- 75.Hendrich B and Bird A (1998) Identification and Characterization of a Family of Mammalian Methyl-CpG Binding Proteins. Mol. Cell. Biol 18, 6538–6547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Meehan RR et al. (1989) Identification of a mammalian protein that binds specifically to DNA containing methylated CpGs. Cell 58, 499–507 [DOI] [PubMed] [Google Scholar]
- 77.Holoch D and Margueron R (2017) Mechanisms Regulating PRC2 Recruitment and Enzymatic Activity. Trends Biochem. Sci 42, 531–542 [DOI] [PubMed] [Google Scholar]
- 78.Mattocks DA et al. (2017) Short term methionine restriction increases hepatic global DNA methylation in adult but not young male C57BL/6J mice. Exp. Gerontol DOI: 10.1016/j.exger.2016.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Maddocks OD et al. (2016) Serine Metabolism Supports the Methionine Cycle and DNA/RNA Methylation through De Novo ATP Synthesis in Cancer Cells. Mol. Cell 61, 210–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cole JJ et al. (2017) Diverse interventions that extend mouse lifespan suppress shared age-associated epigenetic changes at critical gene regulatory regions. Genome Biol DOI: 10.1186/s13059-017-1185-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kooistra SM and Helin K (2012) Molecular mechanisms and potential functions of histone demethylases. Nat. Rev. Mol. Cell. Biol 13, 297–311 [DOI] [PubMed] [Google Scholar]
- 82.Shi Y et al. (2004) Histone Demethylation Mediated by the Nuclear Amine Oxidase sHomolog LSD1. Cell 119, 941–953 [DOI] [PubMed] [Google Scholar]
- 83.Anand R and Marmorstein R (2007) Structure and Mechanism of Lysine-specific Demethylase Enzymes. J. Biol. Chem 282, 35425–35429 [DOI] [PubMed] [Google Scholar]
- 84.Hino S et al. (2012) FAD-dependent lysine-specific demethylase-1 regulates cellular energy expenditure. Nat. Commun DOI: 10.1038/ncomms1755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Xiao M et al. (2012) Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev 26, 1326–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wu X and Zhang Y (2017) TET-mediated active DNA demethylation: mechanism, function and beyond. Nat. Rev. Genet 18, 517–534 [DOI] [PubMed] [Google Scholar]
- 87.Carey BW et al. (2015) Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 518, 413–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hwang IY et al. (2016) Psat1-Dependent Fluctuations in α-Ketoglutarate Affect the Timing of ESC Differentiation. Cell Metab 24, 494–501 [DOI] [PubMed] [Google Scholar]
- 89.Krishnaiah SY et al. (2017) Clock Regulation of Metabolites Reveals Coupling between Transcription and Metabolism. Cell Metab 25, 961–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ramsey KM et al. (2009) Circadian Clock Feedback Cycle Through NAMPT-Mediated NAD+ Biosynthesis. Science 324, 651–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nakahata Y et al. (2009) Circadian Control of the NAD+ Salvage Pathway by CLOCK-SIRT1. Science 324, 654–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nakahata Y et al. (2008) The NAD+-Dependent Deacetylase SIRT1 Modulates CLOCK-Mediated Chromatin Remodeling and Circadian Control. Cell 134, 329–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Doi M et al. (2006) Circadian regulator CLOCK is a histone acetyltransferase. Cell 125, 497–508 [DOI] [PubMed] [Google Scholar]
- 94.Asher G et al. (2008) SIRT1 Regulates Circadian Clock Gene Expression through PER2 Deacetylation. Cell 134, 317–328 [DOI] [PubMed] [Google Scholar]
- 95.Peleg S et al. (2016) The Metabolic Impact on Histone Acetylation and Transcription in Ageing. Trends Biochem. Sci 41, 700–711 [DOI] [PubMed] [Google Scholar]
- 96.Peleg S et al. (2016) Life span extension by targeting a link between metabolism and histone acetylation in Drosophila. EMBO Rep 17, 455–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dang W et al. (2009) Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature 459, 802–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Maegawa S et al. (2017) Caloric restriction delays age-related methylation drift. Nat. Commun DOI: 10.1038/s41467-017-00607-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Weidner CI et al. (2014) Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Bio DOI: 10.1186/gb-2014-15-2-r24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hanahan D and Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 101.Liberti MV and Locasale JW (2018) The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci 41, 211–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lee JV et al. (2014) Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab 20, 306–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Xu W et al. (2011) Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 19, 17–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Letouzé E et al. (2013) SDH Mutations Establish a Hypermethylator Phenotype in Paraganglioma. Cancer Cell 23, 739–752 [DOI] [PubMed] [Google Scholar]
- 105.Cervera AM et al. (2009) Inhibition of succinate dehydrogenase dysregulates histone modification in mammalian cells. Mol. Cancer DOI: doi: 10.1186/1476-4598-8-89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schmidt JA et al. (2016) Plasma concentrations and intakes of amino acids in male meat-eaters, fish-eaters, vegetarians and vegans: a cross-sectional analysis in the EPIC-Oxford cohort. Eur. J. Clin. Nutr 70, 306–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Son SM et al. (2019) Leucine Signals to mTORC1 via Its Metabolite Acetyl-Coenzyme A. Cell Metab 29, 192–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fontana L and Partridge L (2015) Promoting Health and Longevity through Diet: From Model Organisms to Humans. Cell 161, 106–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Brown-Borg HM (2016) Reduced growth hormone signaling and methionine restriction: interventions that improve metabolic health and extend life span. Ann. N.Y. Acad. Sci 1363, 40–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Keleher MR et al. (2018) A high-fat diet alters genome-wide DNA methylation and gene expression in SM/J mice. BMC Genomics DOI: 10.1186/s12864-018-5327-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Davegårdh C et al. (2018) DNA methylation in the pathogenesis of type 2 diabetes in humans. Mol. Metabolism 14, 12–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lüscher B et al. (2018) ADP-Ribosylation, a Multifaceted Posttranslational Modification Involved in the Control of Cell Physiology in Health and Disease. Chem. Rev 118, 1092–1136 [DOI] [PubMed] [Google Scholar]
- 113.Hart GW (2019) Nutrient Regulation of Signaling and Transcription. J. Biol. Chem 294, 2211–2231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhang D et al. (2019) Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580 [DOI] [PMC free article] [PubMed] [Google Scholar]