

Abstract
Misfolding and aggregation of amyloid-β peptide and hyper-phosphorylated tau are molecular markers of Alzheimer disease (AD), and although the 3D structures of these aberrantly folded proteins have been visualized in exquisite detail, no method has been able to survey protein folding across the proteome in AD. Here, we present Covalent Protein Painting (CPP), a mass spectrometry-based protein footprinting approach to quantify the accessibility of lysine ε-amines for covalent modification at the surface of natively folded proteins. We used CPP to survey the reactivity of 2,645 lysine residues and therewith the structural proteome of HEK293T cells and found that reactivity increased upon mild heat shock. CPP revealed that the accessibility of lysine residues for covalent modification in Tubulin-β (TUBB), in Succinate dehydrogenase (SHDB) and in amyloid-β peptide (Aβ) is altered in human postmortem brain samples of patients with neurodegenerative diseases. The structural alterations of TUBB and SHDB in patients with AD, Dementia with Lewy bodies (DLB) or both point to broader perturbations of the 3D proteome beyond Aβ and hyper-phosphorylated tau.
Keywords: Protein surface mapping, neurodegenerative diseases, bottom-up proteomics, MudPIT, isobaric isotopologue, chemical footprinting, quantitative mass spectrometry, structural proteomics, diffuse Lewy body disease, conformational diagnostics, molecular diagnostics
Graphical Abstract
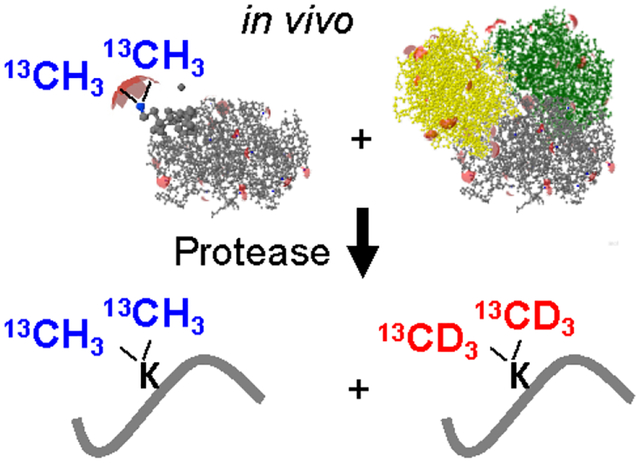
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disorder marked by progressive loss of cognition and other important mental functions1. Disease defining hallmarks of AD include the deposition of neurofibrillary tangles and senile plaques that precede neuronal cell death2. Tangles consist of macromolecular aggregates of misfolded tau protein whereas plaques mainly contain misfolded amyloid-β peptide. Misfolded proteins are targeted and removed by proteins of the proteostasis network which keeps the proteome in homeostasis3. The proteostasis netowrk recognizes incorrectly folded proteins and chaperone proteins attempt re-folding or remove misfolded proteins through proteasomal degradation. If the proteostasis network fails, conformationally altered proteins accumulate and can cause cell death. Therefore, it is tempting to link the onset of neurodegenerative diseases with a failure of the proteostasis network to re-fold or remove misfolded proteins. However, the large number of protein conformations and interactions present in cells makes it experimentally challenging to trace and pinpoint where, when, and why specific proteins misfold and persist in a misfolded state.
Monitoring protein conformer homeostasis with footprinting approaches has a long history4. Here, we attempted to measure the degree of protein misfolding in living cells using Covalent Protein Painting (CPP), a structural proteomics approach we developed to quantify changes in protein fold or altered protein-protein interaction. CPP directly determines the relative number of molecules or fraction of a chemical functionality that is accessible for covalent modification on the surface of proteins. Here, we dimethylate lysine ε-amines that are sufficiently exposed to be accessible for covalent modification, or accessible, in short. These solvent exposed primary amines on intact proteins are rapidly (within seconds) dimethylated with very high yields in this foot-printing protocol because the addition of each methyl group is a two-step reaction that is only rate limited by the initial formation of the hydroxymethylamine5. Dimethylation is performed in living cells, displays advantageous reaction kinetics, and often leaves a smaller footprint than alternative reagents used for in vitro labeling of lysine and additional residues in highly purified protein complexes such as succimidylanhydride6, diethylpyrocarbonate7, N-acetylimidazole8, or “Tandem Mass Tags” (TMT)9. Alternative labeling strategies in living cells and organisms include hydroxyl radical footprinting10 which is not restricted to a single reactive site in proteins.
After dimethylation, the labeling reaction is quenched, labeling reagents are removed, and proteins are denatured and proteolytically digested with an endoprotease that is insensitive to lysine. Proteolysis exposes previously non-accessible lysine residues, which are labeled in a second labeling step with a set of isotopically different dimethyl groups like previously explored with cysteine residue labeling approaches in vitro11. When measured with mass spectrometry, the difference in isotope composition allows CPP to directly determine the fraction of protein molecules that was accessible for covalent modification at a specific lysine site. The covalent attachment of the label sets CPP apart from other approaches, such as Protein Painting which non-covalently “coats” the protein’s surface with small molecules in order to limit endoproteolytic cleavage12, or limited proteolysis which takes advantage of a partial inaccessibility of amino acid sequences in the 3D folds of proteins during a digestion with non-specific proteases13. Thus, CPP enables an unbiased discovery of structural changes caused by misfolding and altered protein-protein interactions in living cells and tissue samples using a 3D protein structure-dependent, differential attachment of a covalent label.
Using CPP we show that mild heat shock of HEK293T cells increases accessibility of lysine sites for covalent modification. Furthermore, we compared the 3D proteome in postmortem collected brain tissue samples of patients with AD and/or LBD and controls. CPP differentiated patients with neurodegenerative disease from controls based on alterations in lysine accessibilities in Tubulin-β, Succinate dehydrogenase, and amyloid-β peptide.
Materials and Methods
Dimethylation of GAPDH
Instead of crosslinking proteins at lysine residues with formaldehyde14, the initial Schiff base of formaldehyde at the primary amine of lysine was reduced with cyanoborohydride yielding a Michael addition of a first and second methyl group. Dimethylation can label lysine residues with different combinations of carbon 12C, hydrogen H, carbon 13C and Deuterium D. The CPP protocol takes advantage of two successive and independent dimethylation reactions. The first reaction labels native proteins and the second reaction labels peptides following an endoproteolytic digest of the proteins. Each labeling reaction adds distinct, isotope-defined dimethyl groups. The specific isotope combinations for the first and the second dimethylation steps in each of the experiments are listed in Table S1.
Dimethylation of GAPDH was performed with recombinantly expressed, highly purified human GAPDH (LifeTechnologies) dissolved in 2 mM HEPES (pH 7.4). In this first step, ε-amines of lysine residues were labeled with isotope-defined reagents (H, 12C, “light”) on native proteins. Formaldehyde was used in a 10-fold molar excess over lysine residues present in the reaction mixture. Specifically, 1.7 μl H2O, 2.0 μl HEPES buffer pH 7.0 (1 M), 5 μl of GAPDH protein (1 μg/μl), 1.7 μl formaldehyde (2 % v:v, Sigma), and 0.6 μl NaBH3CN (160 mM, Sigma) were mixed (10 μl final) in a small reaction vial, and dimethylation was allowed to proceed for 5 min on ice. Following incubation, the reaction was quenched by the addition of ammonium bicarbonate in molar excess (0.5 μL of 0.3 M NH4HCO3).
Native Gel Electrophoresis
Following initial dimethylation, loading buffer (4 × NativePage Sample Buffer, Thermo Fisher Scientific) was added to samples and 1 μg of the GAPDH protein was loaded per lane on a native 4 % to 16 % Bis-Tris gel (NATIVE-PAGE, Thermo). Protein complexes were separated at 15 V/cm in a buffer cooled electrophoresis chamber (Thermo). Gels were fixed twice in 40 % MeOH/10 % acetic acid, microwaved for 45 s and agitated for 30 min at 24 °C. Gels were subsequently stained (0.02 % Coomassie Blue in 30 % MeOH/10 % acetic acid, BioRad) for 30 min and at room temperature (24 °C) and washed with 8 % acetic acid (30 min, 24 °C). Bovine serum albumin (BSA, Sigma) was used as a standard to monitor electrophoretic separation.
Cell culture and heat shock
HEK293T cells were grown under standard conditions (37 °C, 5 % CO2) in Dulbecco’s modified Eagle’s medium containing 25 mM Glucose and supplemented with 1 mM Sodium pyruvate, 2 mM Glutamax, 10 % FBS and, 1 % Penicillin/Streptomycin (GIBCO). Following heat shock (15 min, 42 °C, 5 % CO2) cells were immediately labeled with isotope defined reagents (2 % formaldehyde, 0.3 M sodium cyanoborohydride, in 1 × Dulbecco’s phosphate buffered saline, pH 7.3) for 15 min at 0 °C. Addition of ammonium bicarbonate (1% final w:v) quenched the dimethylation reaction (15 min, 0 °C), then cells and cell fragments were collected and sonicated for 3 min in a water bath sonicator. Proteins were separated from the initial labeling reagents using a methanol-chloroform precipitation15. Precipitated proteins were resolubilized by sonication (1 h) in 1 % Rapigest (Waters), 0.1 M 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, Gibco), pH 7.5 and heat denatured (95 °C, 10 min). Disulfide bonds were reduced with 5 mM tris-(2-carboxyethyl)phosphine hydrochloride (TCEP, 20 min, 37 °C) and sulfhydryl groups were alkylated in 10 mM chloroacetamide (30 min, 24 C).
Human postmortem brain tissues
100 mg of fresh frozen human postmortem frontal cortex from neuropathologically confirmed AD and cognitively normal control cases was obtained from the Neuropathology/Brain Bank of the Shiley-Marcos Alzheimer’s Disease Research Center of the University of California, San Diego.
Purification of the insoluble brain fraction
Purification of the insoluble fraction in brain tissue samples was performed as previously described16. In brief, tissue was homogenized in 1 ml tissue lysis buffer (10 % (w:v) sucrose, 10 mM HEPES pH 7.0, 800 mM NaCl, 5 mM Ethylenediaminetetraacetic acid (EDTA), 1 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N’,N’-tetraacetic acid (EGTA), 1 × protease inhibitors Complete EDTA-free (Roche), 1 × phosphatase inhibitors (Pierce)) with a small pistil, vigorously mixed (30 s), sonicated (30 s), and tissue debris removed by centrifugation (18,000 × g, 30 min, 4 °C). The cleared tissue lysate supernatant was brought to 1 % N-lauroylsarcosine (v:v), vigorously mixed (30 min, 24 °C), centrifuged (18,000 × g, 30 min, 4 °C). Protein aggregates were precipitated from the second supernatant by ultracentrifugation (100,000 × g, 1 h, 4 °C) and the protein pellet was isotope labeled and re-solubilized in one step (2 % formaldehyde, 0.3 M sodium cyanoborohydride, in 100 mM Hepes pH 7.0, 10 μl final volume, vigorously mixing, 15 min, 24 °C). The dimethylation reaction was quenched with ammonium bicarbonate (1% final w:v, 5 min, 24 °C). Proteins were denatured (8 M guanidinium chloride, 10 mM TCEP) for 1 h at 37 °C and free sulfhydryl groups alkylated (20 mM iodoacetamide, 30 min, 24 °C).
Enzymatic digestion and second labeling step
Only brain samples were diluted to 1 M guanidinium chloride in 0.1 mM HEPES, pH 8.0, 0.02 % Rapigest. All samples were heat denatured (5 min, 95 °C), and proteins were digested with the endoprotease Chymotrypsin at either 5 μg/ml (w:v, brain samples, 16 h, 37 °C) or at a 1: 100 ratio of protease : protein (w:w, GAPDH and HEK293T samples, 16 h, 30 °C).
Rapigest was inactivated by acidification (1 % formic acid (v:v), 37 °C, 1 h) and insoluble precipitate removed by centrifugation (18,000 × g, 15 min, 4 °C) in brain-derived or GAPDH samples. Peptides were desalted by C18 reversed phase purification (C18-tips, Thermo Fisher Scientific) to remove residual reagents from the first labeling step. While still bound to the resin, newly exposed primary amines on peptides were dimethylated with isotope-defined reagents (2 % formaldehyde, 0.3 M sodium cyanoborohydride, in 100 mM HEPES, pH 7.0, occasional mixing, 15 min, 24 °C) as previously described17. Peptides were eluted with 80 % acetonitrile, 0.01 % trifluoroacetic acid. The eluted samples were evaporated almost to dryness by centrifugation under vacuum, and peptides were resolubilized in liquid chromatography solution A (5 % acetonitrile, 0.1 % formic acid).
For samples that were methanol-chloroform precipitated prior to digestion, peptides were directly labeled with isotope defined reagents (2 % formaldehyde, 0.3 M sodium cyanoborohydride, in 100 mM HEPES, pH 7.0, occasional mixing, 1 h, 24 °C). Rapigest was inactivated by acidification (1 % formic acid (v:v), 37 °C, 1 h), and samples reduced to near dryness in vacuo as described above, and finally resolubilized in liquid chromatography solution A (5 % acetonitrile, 0.1 % formic acid).
Mass spectrometry
In all experiments, peptides were ionized by electrospray at a nano-spray tip of ~0.1 μm i.d. at 1.5 kV. Full scan (400 to 1800 m/z) spectra were acquired on an Orbitrap mass spectrometer (Thermo Fisher Scientific) at a mass resolving power of R = 60,000 at m/z 400. Fragment ion spectra of > 1000 ion counts were acquired in data dependent mode for the top 20 ions with highest intensity (z = 2 or higher) using collision-induced dissociation (CID) at 35% collisional energy. To avoid sampling only the most abundant peaks, dynamic exclusion with an exclusion list of 500, repeat time of 60 s and asymmetric exclusion window of −0.51 Da and +1.50 Da was used throughout all experiments.
In each experiment samples were chromatographically separated using different methods and mass spectra acquired at different Orbitrap mass spectrometers. In the first experiment, 250 ng of GAPDH peptides were loaded onto a 300 mm reversed phase chromatographic column with 100 μm inner diameter packed with 100 Å reversed phase resin (Aqua 3, 10 Å pore size, Phenomenex). A linear chromatographic gradient of 100 % solution A (5% Acetonitrile, 0.1 % formic acid) to 60 % solution B (80 % acetonitrile, 0.1 % formic acid) was applied over 1.5 h to elute peptides. Mass spectra were acquired with an Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific).
For the heat shock experiment, 50 μg of the CPP labeled HEK293T proteome was loaded onto a MudPIT column18 and analyzed by nano-ESI LC/LC-MS/MS on a VelosPro Orbitrap mass spectrometer. The MudPIT column was placed in line with a quaternary Agilent 1200 high pressure liquid chromatography HPLC pump and peptides were separated by reversed phase liquid chromatography in 10 sequential steps, each following an initial elution of peptides from the strong cation exchange column with buffer C (500 mM ammonium acetate, 5 % acetonitrile, 0.1 % formic acid) in solution A in incrementally progressive concentrations (0 %, 10 %, 20 %, 30 %, 40 %, 50 %, 60 %, 70 %, 80 %, and 90 %) as previously described18,19.
For patient samples, 2 μg of brain sample derived peptides were loaded onto evotip C18 tips (EVOSEP, Denmark). Peptides were eluted with an EVOSEP HPLC system and separated by reversed phase chromatography on a 15 cm ReproSil C18 column (3 μm, 120 Å, id 100 μm, PepSep, Denmark) with a 45 min gradient of increasing acetonitrile concentration with 0.1% formic acid according to manufacturer’s recommendations (EVOSEP, Denmark). Following chromatographic separation, peptides were transferred into an Orbitrap Lumos mass spectrometer by electrospray ionization (nanoEasy, Thermo Fisher Scientific). The top 25 precursor peaks were picked for collision-induced fragmentation.
Data analysis
Following data acquisition, raw data was pre-processed and converted into ASCII file format with RawConverter20 set to monoisotopic peak detection. Converted files were uploaded in IP2 (Integrated Proteomics) and searched with ProLuCID21 for the presence of spectra that matched a theoretical peptide fragment ion spectrum based on amino acid sequences listed in the UniProt database for the human proteome release v 2016.4. Amino acid sequences in the database were either digested in silico assuming either no endoproteolytic enzyme specificity (HEK293T cells) or minimally requiring that either the N- or C-terminus of the peptide was generated by chymotryptic cleavage (GAPDH and brain samples). A 50 ppm precursor mass tolerance window was set for peptide candidate selection, carboxyamidomethylation (m = 57.021464 Da) of cysteine, and dimethylation (m = 28.0313 Da) of N-termini. Peptides including lysine residues labeled “light” or “heavy” (+8.0442 Da) were searched separately as static modifications. Results were filtered with DTASelect v 2.1.4 to a spectrum false discovery rate (FDR) of 0.1 % or less and requiring at least one Chymotrypsin specific cleavage of either peptide N- or C-terminus and a precursor mass tolerance of Δ ≤ ±10 ppm. Subsequently, relative peptide abundances were quantified based on peptide elution profiles deduced from MS survey spectra with Census22 in IP2 (Integrated Proteomics) or based on fragment ion counting when isotopically labeled peptides were isobaric23. Ratio values for each lysine residue were calculated with the SoPaX algorithm that is part of ProteinClusterQuant24 (PCQ, https://github.com/proteomicsyates/ProteinClusterQuant,). The ratio value for a lysine site is the average ratio value determined from all peptides that include the lysine site in the peptide sequence. Data presentations were assembled in Excel (Microsoft) or in Prism (GraphPad) to determine the FDR of lysine sites in two sample comparisons according to the modified statistical approach originally proposed by Benjamini and Hochberg25. Panther26 was used to determine the Gene ontology enrichment of protein groups.
Crystal structures of proteins were downloaded from the RCSB Protein Data Bank PDB (https://www.rcsb.org/pdb/home/home.do) and visualized in Jmol (v 14.19.1). All non-protein molecules and hydrogen atoms were removed. Based on the van der Waals spheres of individual atoms, the command isosurface in Jmol determined the solvent accessible surface area (SASA) at the reactive ε-amine of each lysine residue with standard parameter settings (probe radius 1.2 Å and 2 points per Å resolution). Euclidian distances of atoms were determined with the function distance dependent contacts of one residue with polar residues that is available in the WHAT IF web interface (http://swift.cmbi.ru.nl/servers/html/index.html) or with the ProteinAssessibilityCalculator (PAC, https://github.com/proteomicsyates/ProteinAccessibilityCalculator).
Results
3D proteome with CPP
We used 13C isotope-defined formaldehyde and sodium cyanoborohydride to dimethylate solvent exposed lysine ε-amines in living HEK293T cells (Figure 1A and S1, Materials and Methods). Labeled proteins were denatured and digested with the lysine-insensitive endoprotease Chymotrypsin. After digestion newly exposed primary amines in peptides were dimethylated with CDH2 methyl groups using formaldehyde and sodium cyanoborodeuteride. Following reversed-phase chromatography of peptides, tandem mass spectra of peptide fragment ions were acquired in high mass resolving power (R = 120.000 at m/z 400) on an Orbitrap Fusion mass spectrometer in order to differentiate 13CH3 from CDH2 labeled peptide fragment ions23. CPP surveyed 385 lysine residues in 246 different proteins with 2,297 individual measurements from a total of six replicate experiments which included alternative isotope combinations between the first versus second labeling step (first : second label, CDH2 : 13CH3, 13CH3 : CDH2, 13CHD2 : CD3, and CD3 : 13CHD2). The difference in the intensities of peptide ions (area under the chromatographic peak) or peptide fragment ions (signal intensity in the MS/MS mass spectrum) in the first and second isobaric label steps yield a relative abundance ratio R23. In CPP, the R value represents the proportion of protein or proteoform molecules in which a specific lysine site is accessible for covalent modification which is independent of overall protein amount in the sample assuming lysine sites are the only limiting reagent during initial labeling (% accessibility = 100 * R / (1 + R)). In 337 of 385 lysine sites (87.5 %) > 95 % of molecules were labeled in the initial dimethylation step, which was expected as lysine is the most solvent accessible amino acid in proteins. In contrast, only lysine sites that were modified during the first labeling step were identified in a heat denatured control proteome. The remaining 47 lysine sites were either less than 95% accessible in protein molecules (34 sites) or completely inaccessible (13 sites, Table S1). For example, lysine sites GAPDH#K27, GAPDH#K55, and GAPDH#K139 of the metabolic enzyme Glyceraldehyde 3-phosphate dehydrogenase GAPDH (P04406) were completely accessible for covalent modification in all molecules, whereas GAPDH#K309 was accessible in < 75 % of molecules (Figure 1B). Lysine ε-amines in crystal structures of GAPDH show that GAPDH#K309 participates in the protein-protein interface of two homo-dimers within the GAPDH homo-tetramer27 which turns the ε-amine inaccessible for covalent modification (Figure 1C and S2). Here, we found partial accessibility of GAPDH#K309 with CPP which indicated that different quaternary structures (mono-, di-, and tetramers) of GPADH may coexist in HEK293T cells.
Figure 1. Covalent Protein Painting (CPP) determines whether the ε-amino group of lysine is accessible for covalent modification.
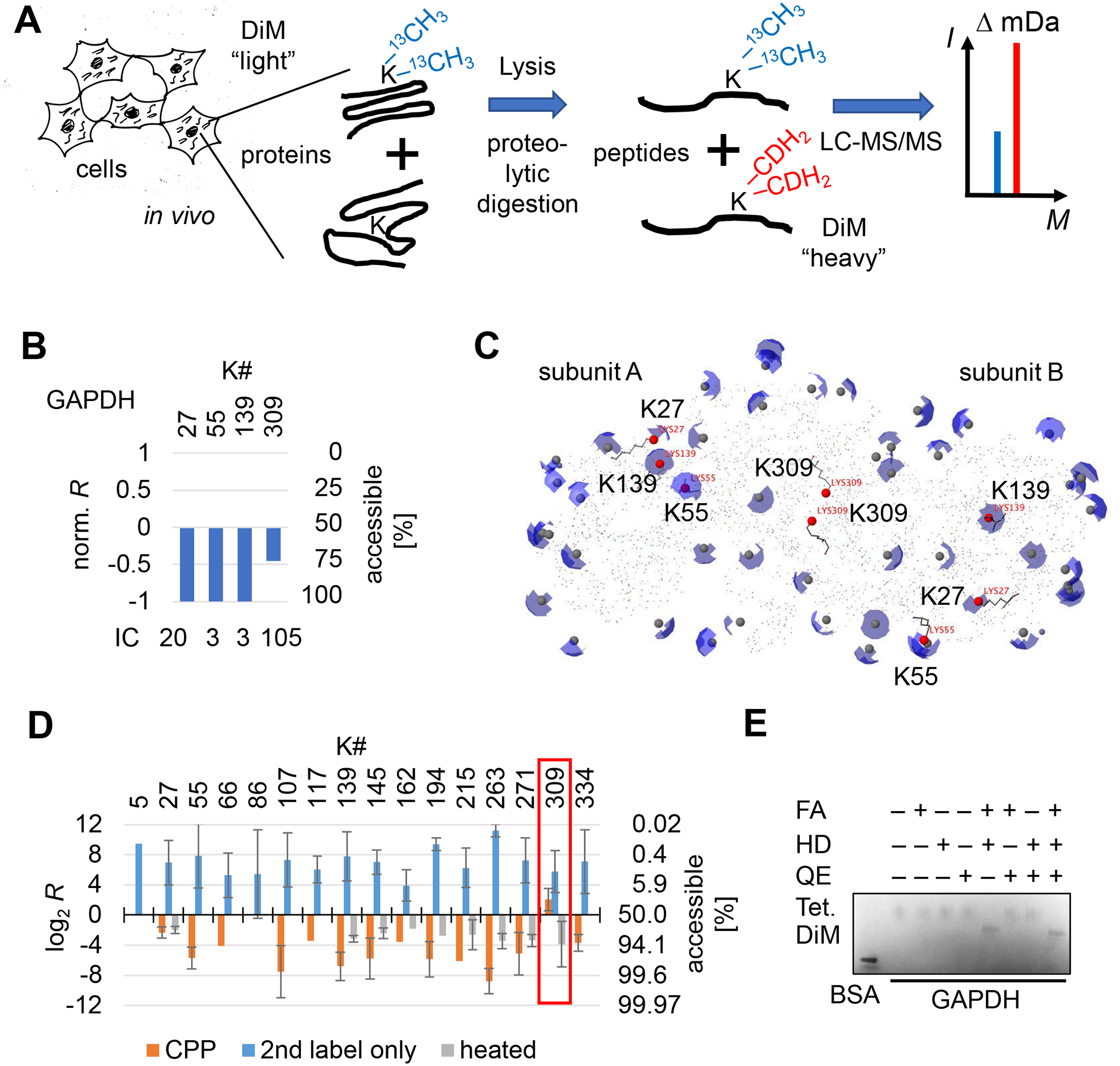
(A) The schematic displays the workflow of CPP. Reductive alkylation labels lysine residues with isotope-defined “light” dimethyl moieties in living cells. Following digestion into peptides with a lysine-insensitive protease (Chymotrypsin), newly solvent exposed lysine residues are modified with isotope-defined “heavy” dimethyl moieties. Bottom up mass spectrometry is used to analyze the ratio of light to heavy isotope labeled peptide molecules per lysine site. (B) Lysine residue GAPDH#K309 is only partially accessible for chemical dimethylation in HEK293T cells. Proteins in HEK293T cells were covalently modified with CPP using isobaric isotopologue methyl moieties with 13CH3 for light and CDH2 for heavy, and the relative surface accessibility determined as described in (A). Numbers above the bars indicate the position of the lysine residue in GAPDH. The y-axis is the ratio of light to heavy fragment ion counts normalized to the total number of ion counts shown below each bar. A ratio of R = 1:1 (log2(1) = 0) indicates that the lysine site was accessible for chemical modification in 50 % of protein molecules. Ion counts (IC) denotes the sum of fragment ion peaks that contributed to the quantification. (C) One dimer of the GAPDH homo-tetramer is displayed (PDB: 4wnc). Partial spheres (blue) highlight solvent accessible surface area (SASA) of each individual lysine ε-amine (grey spheres). Lysine residues that were assayed with CPP in (B) are highlighted in red. GAPDH#K309 resides within the contact surface of two GAPDH monomers in the GPADH dimer and is almost completely inaccessible for covalent modification. (D) The bar graph shows accessibility of lysine sites for covalent modification in highly purified, native GAPDH tetramers (orange) or heat denatured GAPDH (grey) or when the initial labeling step was omitted (blue). CPP results obtained for GAPDH#K309 are highlighted (red box). (E) Blue-native gel® electrophoresis of purified GAPDH indicates structural stability of the GAPDH homo-tetramer following chemical dimethylation. GAPDH was pre-incubated with labeling reagents formaldehyde (FA), sodium cyanoborohydride (HD), and the quencher ammonium bicarbonate (QE). Bovine serum albumin (BSA, 66 kD) was included as molecular size indicator. Tetrameric GAPDH protein complexes migrated distinctively faster following CPP. Error bars are standard deviation (σ). Abbreviations: DiM, dimethyl moieties; Tet., homo-tetramers.
Furthermore, GAPDH#K309 was accessible in < 20 % of recombinantly expressed, highly purified human GAPDH in vitro when exposed to labeling reagents in an excess of time (Material S1) and reagents (Material S2). Monomethylation instead of dimethylation at the initial labeling step of GAPDH#K309 and additional lysine sites in GAPDH was observed only when lysine sites were labeled for a very short time period only (5 sec, time limited labeling, Material S3). 13 of the remaining 25 lysine sites were solvent exposed in 97 % of GAPDH molecules on average (σ = ±1.8 of log2R, Figure 1D and S3). Additional 12 lysine sites were either not detected or peptides harbored more than one lysine residue which precluded a site-specific quantitation based on chromatographic elution profiles22. All quantified lysine residues became accessible in > 80 % or in 87 % of GAPDH molecules on average upon heat denaturation at 95 °C for 5 min (σ = ±0.7, Figure 1D, grey bars). However, overall heat denaturation lowered the accessibility of normally solvent exposed lysine residues in native GAPDH from 97 % to 87 %. This difference suggests that random protein aggregation following heat denaturation rendered lysine residues less, rather than more, accessible. When the first labeling step is omitted, all lysine residues of endoproteolytically digested GAPDH are dimethylated with the second isotope label only (Figure 1D, blue bars). In this case, lysine sites were accessible for labeling on average in 99.3 % of GAPDH molecules (σ = ±1.9). The residual 0.7 % most likely reflected random chemical noise picked up during mass spectrometric data acquisition and quantification of elution profiles.
Native gel electrophoresis showed that dimethylation did not affect the quaternary structure of GAPDH (Figure 1E). Dimethylated GAPDH homo-tetramers (147 kD) migrated as a sharp signal slightly below non-modified GAPDH homo-tetramers, but well above bovine serum albumin (BSA, 66.5 kD). The molecular weight of BSA is close to the calculated molecular weight of GAPDH dimers (which were not observed). Dimethylation did not diminish signal intensity, suggesting that GAPDH tetramers did not disassemble. A comparison of these results with the solvent accessible surface area (SASA) of lysine ε-amines in crystal structures of GAPDH indicated that ε-amines required a SASA of > 1 Å2 in order to be covalently modified with formaldehyde (Material S4). Our labeling results matched the crystal structure data obtained for GAPDH homotetramers; therefore, we conclude that CPP measures the proportion of protein molecules in which a specific lysine residue is accessible for covalent modification.
3D proteome alterations following heat shock
A rise in temperature from 37 °C to 42 °C for 15 min in eukaryotic cells is a physical stress that leads to protein unfolding or misfolding and elicits a coordinated cellular response of the proteostasis network known as the heat shock response to limit proteome-wide damage28. We applied CPP to HEK293T cells to find out whether heat preferentially misfolds a specific subset of proteins or if it leads to wide-spread random protein unfolding. We used multidimensional protein identification technology (MudPIT)18 on an Orbitrap Velos mass spectrometer to survey the proteome in three biological replicates of HEK293T cells (Material S5) that were either untreated or exposed to heat. The experiment yielded 16,081 different peptide measurements covering a total of > 7,000 different lysine sites. 2,645 of which were quantified at least twice per condition in 979 different protein groups and proteoforms (Table S2).
In total, 2,484 out of 2,645 lysine residues were accessible for covalent modification in > 33 % of protein molecules in controls (Figure S4). Individual lysine sites were accessible for labeling on average in 94.4 % of protein molecules and accessibility normally distributed from 99.7 % to 50 % (Figure 2A). The remaining 161 lysine residues clustered in a distinct second peak in the frequency distribution plot and were accessible in ≤ 33 % of protein molecules (Figure 2A Inset). A positive control for the second labeling step in CPP represents the exogenously added endoprotease Chymotrypsin that was not subjected to the initial labeling step. Following proteolytic digestion, the autolysis products of Chymotrypsin are labeled in the second labeling step only, and thus none of its peptides should be quantified as accessible. Lysine site CTRB1#K54 of Chymotrypsinogen (P17538 and Q6GPI1) was measured as “accessible” in 0.16 % of molecules, which is most likely due to random chemical noise in mass spectra.
Figure 2. CPP quantified protein unfolding in HEK293T cells upon mild heat shock.
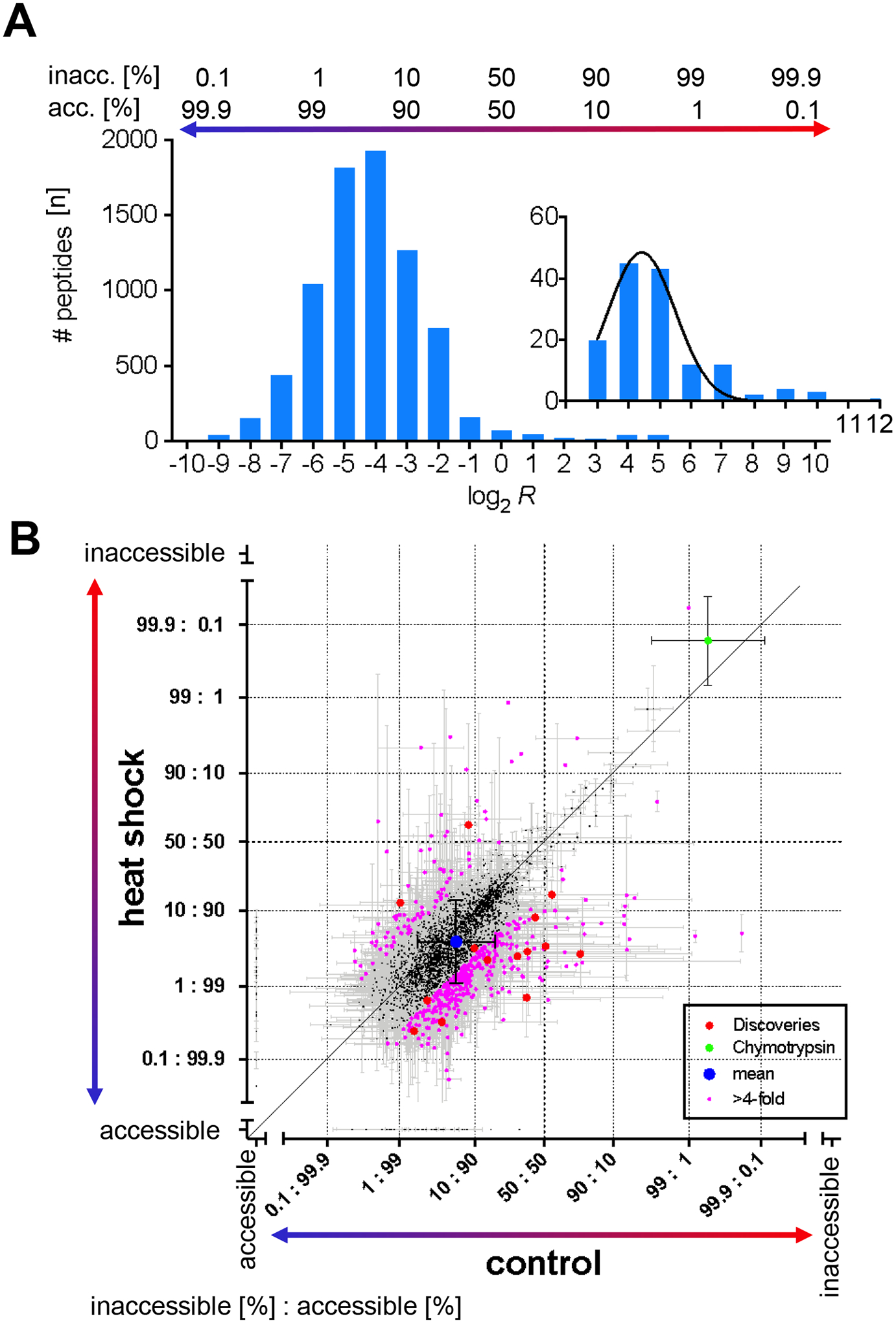
(A) The frequency plot and Gaussian fit (black) shows the number of peptides (n) in which a lysine site was accessible for covalent modification as measured by CPP (log2 R) in the proteome of control HEK293T cells. Log2 R values were binned by integer. The inset highlights the frequency distribution of lysine sites that are predominantly inaccessible for covalent modification with CPP. The relative number of protein molecules [%] in which lysine sites were accessible or conversely inaccessible to covalent modification is indicated above the bar graph. (B) The scatterplot compares the relative number of protein molecules in which a lysine residue was accessible for covalent modification in control versus heat shock treated HEK293T cells. “Accessible” and “inaccessible” on the scale bars indicate lysine sites that were measured as either completely accessible or inaccessible for covalent modification. Individual lysine residues differed by Δ > 2 in relative accessibility for covalent modification (pink). Significantly different lysine sites passed the discovery threshold of q < 0.01 (Benjamini-Hochberg corrected, red). The control lysine site CRBT1#K54 of exogenously added Chymotrypsin (green) and overall mean (blue) are shown. The diagonal denotes no change in relative covalent modification. Error bars are standard deviation (σ).
Heat shock altered relative accessibility for covalent modification by 1.8 % towards more accessible overall (p = 0.0001, Student’s t-test) relative to control, which is significantly higher than the average variation between biological replicate experiments (Δ = 0.7 ± 0.3 %) and chemical noise during mass spectrometric data acquisition as determined with the positive control (chymotrypsin: Δ = 0.4 %). Heat shock increased the relative accessibility for covalent modification by ≥ 4-fold in 461 of 2,645 lysine sites (Figure 2B). Reactivity increased at 369 lysine sites whereas it decreased at 92 lysine sites, indicating that heat shock preferentially unfolded proteins or weakened protein-protein interactions. Notably, the fractional change in the number of protein molecules with increased accessibility was < 20 % for the majority of the 369 lysine sites (Figure S5). Heat shock significantly altered accessibility for covalent modification in 14 of 2,645 lysine sites (Figure 2B) and flipped the ratio of accessible to inaccessible for 4 of the 14 lysine sites (Table S3). Three lysine sites whose accessibility was inverted with heat stress moved from predominantly inaccessible to accessible in > 50 % of protein molecules. PDCD11#K1402 was accessible in 24.0 % of programmed cell death protein 11 protein molecules (RRP5 homolog NFκB binding protein, NFBP, Q14690) and shifted to accessible in 97.3 % of molecules upon heat shock, and GSR#K501 in mitochondrial Glutathione reductase (P00390) and GLMN#K507 in Glomulin (Q92990) changed from accessible in 43.9 % and 49.9 % of molecules to accessible in 84.3 % and 96.5 % of molecules, respectively. In contrast, the fourth lysine site whose ratio of accessibility was inverted by heat shock became more inaccessible rather than accessible. PSMD3#K273 in the 26S proteasome non-ATPase regulatory subunit 3 (O43242) was 91.7 % accessible in the control and 36.8 % accessible upon heat shock. Thus, most conformational alterations are reversible except conformational alterations that inverted the accessibility of lysine sites and are therefore potentially irreversible.
Prolonged heat shock causes extensive post translational protein modifications (PTM) on lysine, including ubiquitinylation and sumoylation29, so we determined if ubiquitinylation or sumoylation occurred at the lysine sites that were quantified by CPP. Since CPP labels only primary, non-modified ε-amines, it does not quantify changes in PTM abundance at a lysine site. Naturally occurring lysine dimethylation can influence CPP results depending on the choice of isotope-defined reagents used in the design of the experiment (Material S6). Overall, 68 proteins were either ubiquitinylated or sumoylated in the control and the number of modified proteins increased upon heat shock (as determined by spectral counts (SpC)). Overall, the 68 proteins were enriched for the Gene Ontology term “protein folding”, and several ubiquitinylation sites were identified with ≥ 5 SpC across all biological replicates, including tubulin-α (13 SpC), splicing factor U2AF2 (13 SpC), heterogeneous nuclear ribonucleoprotein R HNRNPR (9 SpC), and apoptosis inhibitor 5 API5 (5 SpC). Proteins only ubiquitinylated in heat shock-exposed cells included the ubiquitin-like modifier-activating enzyme 1 (UBA1, 13 SpC), transcription intermediary factor 1β (TRIM28, 8 SpC), and heat shock 70 kD protein (HSP70, 5 SpC). Changes in accessibility for covalent modification after heat shock were < 2-fold for most of the 20 lysine sites that were ubiquitinylated or sumoylated (68 lysine sites in total) as well as quantified with CPP. However, lysine sites TBA1B#K430 in Tubulin-α1B (P68363) and TPI1#K256 in triosephosphate isomerase 1 (P60174) were sumoylated and less accessible in 3.4- and 3.8-fold more molecules, respectively. HSPA4#K84 of heat shock protein 70 kD protein 4 (P34932) was inaccessible in 3.6-fold fewer molecules while being ubiquitinylated upon heat shock. Thus, while a subset of lysine sites whose accessibility changed after heat shock were also PTM modified, changes in lysine site reactivity did not directly reflect alterations in PTM status.
Differentiating neurodegenerative diseases based on 3D alterations
Next, we used CPP as a conformational diagnostic tool to measure protein misfolding in human neurodegenerative diseases. We analyzed prefrontal cortex samples of 10 controls and 10 patients that were diagnosed with Alzheimer’s disease (AD) or Lewis body disease (LBD) or with diffuse Lewis bodies in addition to AD (AD-dLBD, Table S4). CPP was applied to whole tissue lysate, to supernatant and to pellet obtained following ultracentrifugation (UC) to enrich for protein aggregates16. Overall, the experiment quantified 559, 342, and 303 lysine sites in lysate, pellet, and supernatant, respectively.
The amyloid precursor protein APP (P05067) is endoproteolytically cleaved to yield amyloid-β peptide Aβ1–40 and Aβ1–42 which extends Aβ1–40 by two C-terminal amino acids. Both amyloid-β peptides misfold and form large peptide aggregates and plaques in AD patients30. The role of amyloid-β in AD has been previously studied in vitro and ex vivo with chemical footprinting techniques31–34 and with methods that provide high spatial resolution35–37 (Material S7). Inferring the most likely proteoform in the proteomic experiment24, we quantified amyloid-β peptides rather than proteolytic cleavage products of full length APP (Material S8). Both amyloid-β peptides include lysine site Aβ#K28 which is inaccessible in fibrillar Aβ aggregates38. Using CPP, we detected Aβ#K28 in AD and AD-dLBD samples whereas Aβ#K16, which is upstream of Aβ#K28 in the amyloid-β peptide, was not detected. Aβ#K28 was accessible in 65 % to 89 % of peptide molecules in patients diagnosed with AD only. It was accessible in 48 % to 75 % of peptide molecules in patients diagnosed with AD-dLBD. Thus, Aβ#K28 was on average accessible in fewer (but not significantly fewer) molecules in AD-dLBD than in AD only patients. Aβ#K28 was not detected in controls, except control B2 in which it was inaccessible in 93 % of all Aβ-peptides. We consider sample B2 to be an outlier because it was derived from an individual who did not show symptoms of neurodegeneration.
After biochemical purification of protein aggregates with ultracentrifugation (UC) amyloid-β peptide signal was detected in the pellet. Aβ#K28 was not detected in the supernatant following UC in any of the samples. Consistently more Aβ#K28 was accessible for covalent modification in the ultracentrifugation pellet than in the lysate, suggesting that the ultracentrifugation step reduced the proportion of inaccessible Aβ#K28 that was recovered. The largest relative increase was observed in AD patient sample A10 in which the percentage of peptide molecules with accessible Aβ#K28 increased from 89 % in the lysate to 99 % in the ultracentrifugation pellet. Conversely, the proportion of peptide molecules in which Aβ#K28 was inaccessible for covalent modification was reduced > 10-fold or from 11 % to 1 % (Δ = 10%). Relative accessibility remained almost unaltered in samples where Aβ#K28 was already ≥ 50 % before ultracentrifugation; for instance, in AD-dLBD patient B7 48 % and 51 % (Δ = 3%) of Aβ#K28 were accessible for covalent modification in lysate and UC pellet. The difference indicated that between 3 % to 10 % of Aβ peptide molecules with inaccessible Aβ#K28 were not recovered with UC which inversely scaled with the initial proportion of inaccessible Aβ#K28 in the lysate.
Furthermore, lysine sites SDHB#K137 in mitochondrial succinate dehydrogenase (P21912) and TUBB#K174 in tubulin-β (P07437) were significantly altered between controls and AD, AD-dLBD or DLB in the lysate (q < 0.05, Table 1). TUBB#K174 was overall accessible in 99.2 % to 97.3 % tubulin-β molecules with 0.6 % less accessibility in AD, AD-dLBD and DLB than in controls. SDHB#K137 was on average accessible for covalent modification in 21.4 % fewer protein molecules in AD, AD-dLBD, and DLB than in controls (detected in half of all controls). Conversely, > 2-fold more SDHB molecules were inaccessible at lysine site SDHB#K137 in patient-derived samples than in controls. Thus, CPP revealed structural alterations in SDHB and TUBB in addition to misfolded amyloid-β peptides in patients with AD, AD-dLBD and DLB.
Table 1. Amyloid-β lysine K28 accessibility for covalent modification with CPP in AD and LBD patient prefrontal cortex samples.
The table shows the accessibility in percent for Ab#K28 and SDHB#K137, and TUBB#K174 lysine sites in 10 patient and 10 control samples in total lysate or the precipitate following ultracentrifugation of the lysate. Abbreviations: SpC, spectrum count; %acc, percentile of peptide in which the lysine site was accessible for covalent modification.
| Ab#K28 | SDHB#K137 | TUBB#K174 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Lysate | Pellet | Lysate | Lysate | ||||||
| Diagnosis | Sample | SpC | %acc | SpC | %acc | SpC | %acc | SpC | %acc |
| AD | A10 | 2 | 89 | 2 | 99 | 0 | n.d. | 2 | 98 |
| AD | A7 | 4 | 79 | 7 | 92 | 2 | 58 | 1 | 99 |
| AD | A4 | 7 | 65 | 5 | 92 | 4 | 56 | 1 | 99 |
| AD-dLBD | A6 | 4 | 75 | 5 | 94 | 2 | 58 | 2 | 98 |
| AD-dLBD | BIO | 7 | 84 | 6 | 94 | 2 | 62 | 2 | 98 |
| AD-dLBD | B6 | 6 | 51 | 7 | 86 | 2 | 61 | 1 | 98 |
| AD-dLBD | A9 | 6 | 49 | 5 | 82 | 1 | 72 | 1 | 99 |
| AD-dLBD | B7 | 7 | 48 | 8 | 51 | 2 | 60 | 2 | 99 |
| AD-dLBD | B8 | 6 | 62 | 5 | 84 | 1 | 56 | 2 | 97 |
| LBD | B5 | 0 | n.d. | 0 | n.d. | 2 | 57 | 2 | 98 |
| Normal | A1 | 0 | n.d. | 0 | n.d. | 0 | n.d. | 2 | 99 |
| Normal | A2 | 0 | n.d. | 0 | n.d. | 0 | n.d. | 1 | 98 |
| Normal | A3 | 0 | n.d. | 0 | n.d. | 2 | 91 | 1 | 99 |
| Normal | A5 | 0 | n.d. | 0 | n.d. | 0 | n.d. | 2 | 99 |
| Normal | A8 | 0 | n.d. | 0 | n.d. | 0 | n.d. | 1 | 99 |
| Normal | B1 | 0 | n.d. | 0 | n.d. | 1 | 80 | 1 | 99 |
| Normal | B3 | 0 | n.d. | 0 | n.d. | 2 | 80 | 2 | 99 |
| Normal | B9 | 0 | n.d. | 0 | n.d. | 0 | n.d. | 1 | 99 |
| Normal | B4 | 0 | n.d. | 0 | n.d. | 2 | 79 | 2 | 99 |
| Normal | B2 | 3 | 7 | 1 | 4 | 2 | 78 | 2 | 99 |
Discussion
Here, we show that CPP can be used to quantify changes in the chemical reactivity of lysine sites in HEK293T cells. Mild heat shock of HEK293T cells preferentially unfolded or disrupted protein-protein interactions overall by 1.8 %. This very small increase in accessibility for covalent modification might be due to an increase in entropy at elevated temperatures. However, more experiments will be required to confirm a direct correlation between CPP measurements and a change in entropy in the proteome. After heat shock, predominantly inaccessible lysine sites in three proteins, PDCD11, GSR, and GLMN became accessible (> 90 %), suggesting that this change was non-random and not linear to the increase in temperature during heat shock. The molecular pathways associated with these proteins are ribosome assembly (PDCD11), oxidative stress response (GSR), and protein translation (GLMN). PDCD11 supports maturation of ribosomal subunits 40S and 80S39 and processing of 47S rRNA40 which transiently subsides during prolonged heat shock41. GLMN binds to RBX1, preventing E2 ligase recruitment and Cul1 E3 ligase-mediated ubiquitinylation of substrates42; however, GLMN#K507 does not map to the interaction surface of GLMN with RBX142.
Using CPP, we quantified misfolding and aggregation of amyloid-β peptides in samples from patients with neurodegenerative diseases based on the relative inaccessibility of Aβ#K28. In the most commonly proposed model for the amyloid-β misfold38 Aβ#K28 forms an intramolecular salt bridge with aspartate D23 which stabilizes the hairpin loop and which connects the two β-strands, so the inaccessibility of Aβ#K28 for covalent modification most likely reflects misfolded amyloid-β peptide. However, amyloid-β fibers can also associate laterally with an alternative number of neighboring fibers which then influence the solvent accessible surface area of lysine K2843. Although we cannot exclude that initial dimethylation in CPP affects protein structure, CPP quantified the relative proportion of fibrillar amyloid-β in AD, and a potentially higher proportion of fibrillar amyloid-β in AD-dLBD patient brain samples.
CPP showed that the ultracentrifugation pellet of amyloid-β aggregates had fewer molecules with inaccessible Aβ#K28 than the initial lysate, suggesting that ultracentrifugation of fibrils might alter one or several different fibrillar amyloid-β conformers or that a small proportion (3% to 10%) of amyloid-β peptides with inaccessible Aβ#K28 was not precipitated during ultracentrifugation. However, no amyloid-β peptides were detected in the supernatant, although we cannot rule out that they were below the threshold for detection. In vitro outgrowth assays of amyloid-β fibers seeded with AD patient-derived brain material recently highlighted the differences between clinical AD subtypes and the heterogeneity of amyloid-β conformers that can coexist44. Furthermore, recent cryo-EM data suggested that Aβ#K28 can be solvent accessible in distinct strains of amyloid-β fibrils45–47 which limits CPP’s ability to measure overall amyloid-β peptide aggregation based on Aβ#K28 inaccessibility. Denaturation assays also revealed up to three different states of amyloid-β aggregation in Alzheimer disease brain samples with up to 4-fold (Aβ1–40) or 20-fold (Aβ1–42) more aggregated amyloid-β than soluble amyloid-β48. Thus, the additional presence of oligomeric rather than fibrillar aggregates or the coexistence of different conformer strains might characterize a small but not independent proportion of misfolded amyloid-β peptides in AD.
In surveying > 500 proteins with CPP we found that TUBB#K147 in TUBB, and SDHB#K137 in SDHB were less accessible for covalent modification in patients with neurodegenerative diseases. The difference in CPP for TUBB#K147 was small (0.6%) between AD and AD-dLBD versus Normal. While statistically significant (Student’s t-test, P = 0.0031), any biological effect of such small change remains unclear. Mitochondrial SDHB is part of the oxidative respiration chain, a key metabolic process that fails in aging neurons. A 2-fold increase in molecules with altered accessibility to lysine site SDHB#K137 might reflect a previously unidentified alteration in SDHB protein structure or protein-protein interaction in AD. Previous work showed that the dehydrogenase activity of SDHB is blocked by amyloid-β peptide49; however, the site of physical interaction was not determined. Additional proteins are well known to undergo conformational change in sporadic AD and to aggregate in plaques and tangles. Specifically, tangles in neurons are formed by fibrillar tau, and lysine sites in fibrillar tau are at least in part solvent inaccessible according to cryo-EM studies50–52. Here, we were not able to detect tau in patient samples which suggests that either fibrillar tau needs to be enriched prior to sample digestion by immunoprecipitation or ultracentrifugation for example53, or endoproteolytic digestion conditions need to be optimized to efficiently cover lysine sites in tau.
In conclusion, CPP quantifies the proportion of protein molecules in a proteome in which a lysine site is accessible for dimethylation. With CPP, we determined the contribution of fibrillar amyloid-β with inaccessible Aβ#K28 in AD and in AD-dLBD patient brain samples and revealed that SDHB and TUBB might be conformationally altered upon neurodegeneration, suggesting that there are more conformational perturbations of the 3D proteome besides the highly aggregation-prone misfolds of amyloid-β.
Supplementary Material
Figure S1 Schematic of the CPP Method.
Figure S2 Tetrameric structure of GPADH with the ε-amine of all lysine residues highlighted with blue spheres.
Figure S3 Relative accessibility of lysine residues for covalent modification expressed as percent of molecules.
Figure S4 Protein-peptide representation of all lysine residues identified and quantified in HEK293T cells.
Figure S5 Frequency plot of differences in lysine site accessibility for covalent modification with CPP.
Material S1 Limiting Reaction Time in CPP.
Material S2 CPP of GAPDH in Reactant-limited Conditions.
Material S3 Monomethylation During Initial Labeling with CPP.
Material S4 Correlating Relative Solvent Accessibility with the Accessibility of Lysine Sites for Covalent Modification with CPP.
Material S5 Analysis of Replicate Measurements.
Material S6 CPP Analysis and PTM Status of Lysine Residues.
Material S7 Summary of Previous Structural Analysis of Amyloid-β Peptide.
Material S8 Differentiating Proteolytic Cleavage Products of Amyloid-β Peptides from the APP Precursor Protein.
Table S1 Isotope combinations selected for labeling in CPP.
Table S2 Accessibility measurements with CPP for lysine residues in HEK293T cells using isobaric methyl moieties in CPP.
Table S3 Accessibility measurements with CPP for lysine residues in heat shock and control HEK293T cells.
Table S4 Significantly different lysine sites in HEK293T cells following heat shock.
Table S5 Patient diagnosis of human brain samples.
Acknowledgements
We thank Claire Delahunty for reading the manuscript. We are thankful to Robin Park for support in mass spectrometric data analysis with IP2 (Integrated Proteomics) and Daniel McClatchy for many discussions. We thank Ivy Trinh and Jeffery Metcalf from the Rissman laboratory for technical assistance and dissecting brain tissues for this study.
Funding
Funding was provided by NIH grants R03AG047957-02 and R33CA212973-01 awarded to John R. Yates III and funding from the Shiley-Marcos ADRC Neuropathology Core (U19AG010483-26) awarded to Robert A. Rissman.
Footnotes
Competing financial interests
The authors do not declare competing financial interests.
Supporting Information
The following supporting information is available free of charge at the ACS website http://pubs.acs.org
Data Availability
Mass spectrometric raw data, search engine result files, and quantification result files can be accessed in Massive or ProteomeXchange (MassIVE MSV000083031, ProteomeXchange PXD011351).
Reference
- 1.Zlokovic BV Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci 12, 723–738, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scheltens P et al. Alzheimer’s disease. Lancet 388, 505–517, (2016). [DOI] [PubMed] [Google Scholar]
- 3.Balch WE, Morimoto RI, Dillin A & Kelly JW Adapting proteostasis for disease intervention. Science 319, 916–919, (2008). [DOI] [PubMed] [Google Scholar]
- 4.Liu XR, Zhang MM & Gross ML Mass Spectrometry-Based Protein Footprinting for Higher-Order Structure Analysis: Fundamentals and Applications. Chemical Reviews 120, 4355–4454, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kallen RG & Jencks WP Equilibria for the reaction of amines with formaldehyde and protons in aqueous solution. A re-examination of the formol titration. The Journal of biological chemistry 241, 5864–5878, (1966). [PubMed] [Google Scholar]
- 6.Kahsai AW et al. Multiple ligand-specific conformations of the beta2-adrenergic receptor. Nat Chem Biol 7, 692–700, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou Y & Vachet RW Diethylpyrocarbonate labeling for the structural analysis of proteins: label scrambling in solution and how to avoid it. Journal of the American Society for Mass Spectrometry 23, 899–907, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xiang S et al. The LC Domain of hnRNPA2 Adopts Similar Conformations in Hydrogel Polymers, Liquid-like Droplets, and Nuclei. Cell 163, 829–839, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou Y & Vachet RW Covalent labeling with isotopically encoded reagents for faster structural analysis of proteins by mass spectrometry. Analytical chemistry 85, 9664–9670, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Espino JA, Mali VS & Jones LM In Cell Footprinting Coupled with Mass Spectrometry for the Structural Analysis of Proteins in Live Cells. Analytical chemistry 87, 7971–7978, (2015). [DOI] [PubMed] [Google Scholar]
- 11.Apuy JL et al. Pulsed-Alkylation Mass Spectrometry for the Study of Protein Folding and Dynamics: Development and Application to the Study of a Folding/Unfolding Intermediate of Bacterial Luciferase. Biochemistry 40, 15153–15163, (2001). [DOI] [PubMed] [Google Scholar]
- 12.Luchini A, Espina V & Liotta LA Protein painting reveals solvent-excluded drug targets hidden within native protein-protein interfaces. Nat Commun 5, 4413, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schopper S et al. Measuring protein structural changes on a proteome-wide scale using limited proteolysis-coupled mass spectrometry. Nature protocols 12, 2391–2410, (2017). [DOI] [PubMed] [Google Scholar]
- 14.Harlow E & Lane D Using antibodies : a laboratory manual. (Cold Spring Harbor Laboratory Press, 1999). [Google Scholar]
- 15.Pankow S, Bamberger C, Calzolari D, Bamberger A & Yates JR 3rd. Deep interactome profiling of membrane proteins by co-interacting protein identification technology. Nature protocols 11, 2515–2528, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goedert M, Spillantini MG, Cairns NJ & Crowther RA Tau proteins of Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron 8, 159–168, (1992). [DOI] [PubMed] [Google Scholar]
- 17.Boersema PJ, Raijmakers R, Lemeer S, Mohammed S & Heck AJ Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nature protocols 4, 484–494, (2009). [DOI] [PubMed] [Google Scholar]
- 18.Washburn MP, Wolters D & Yates JR 3rd. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nature biotechnology 19, 242–247, (2001). [DOI] [PubMed] [Google Scholar]
- 19.Pankow S et al. F508 CFTR interactome remodelling promotes rescue of cystic fibrosis. Nature 528, 510–516, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He L, Diedrich J, Chu YY & Yates JR 3rd. Extracting Accurate Precursor Information for Tandem Mass Spectra by RawConverter. Analytical chemistry 87, 11361–11367, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu T et al. ProLuCID: An improved SEQUEST-like algorithm with enhanced sensitivity and specificity. J Proteomics 129, 16–24, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park SK, Venable JD, Xu T & Yates JR 3rd. A quantitative analysis software tool for mass spectrometry-based proteomics. Nature methods 5, 319–322, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bamberger C, Pankow S, Park SK & Yates JR 3rd. Interference-free proteome quantification with MS/MS-based isobaric isotopologue detection. Journal of proteome research 13, 1494–1501, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bamberger C et al. Deducing the presence of proteins and proteoforms in quantitative proteomics. Nat Commun 9, 2320, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reiner A, Yekutieli D & Benjamini Y Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics 19, 368–375, (2003). [DOI] [PubMed] [Google Scholar]
- 26.Mi H, Muruganujan A, Casagrande JT & Thomas PD Large-scale gene function analysis with the PANTHER classification system. Nature protocols 8, 1551–1566, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.White MR et al. A dimer interface mutation in glyceraldehyde-3-phosphate dehydrogenase regulates its binding to AU-rich RNA. The Journal of biological chemistry 290, 1770–1785, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ben-Zvi A, Miller EA & Morimoto RI Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging. Proceedings of the National Academy of Sciences of the United States of America 106, 14914–14919, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Golebiowski F et al. System-wide changes to SUMO modifications in response to heat shock. Sci Signal 2, ra24, (2009). [DOI] [PubMed] [Google Scholar]
- 30.Karran E, Mercken M & De Strooper B The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov 10, 698–712, (2011). [DOI] [PubMed] [Google Scholar]
- 31.Klinger AL et al. A synchrotron-based hydroxyl radical footprinting analysis of amyloid fibrils and prefibrillar intermediates with residue-specific resolution. Biochemistry 53, 7724–7734, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li KS, Rempel DL & Gross ML Conformational-Sensitive Fast Photochemical Oxidation of Proteins and Mass Spectrometry Characterize Amyloid Beta 1–42 Aggregation. J Am Chem Soc 138, 12090–12098, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williams AD et al. Structural properties of Abeta protofibrils stabilized by a small molecule. Proceedings of the National Academy of Sciences of the United States of America 102, 7115–7120, (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramos I, Fabris D, Qi W, Fernandez EJ & Good TA Kinetic study of beta-amyloid residue accessibility using reductive alkylation and mass spectrometry. Biotechnol Bioeng 104, 181–192, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Terry RD, Gonatas NK & Weiss M Ultrastructural Studies in Alzheimer’s Presenile Dementia. Am J Pathol 44, 269–297, (1964). [PMC free article] [PubMed] [Google Scholar]
- 36.Cohen AS & Calkins E Electron microscopic observations on a fibrous component in amyloid of diverse origins. Nature 183, 1202–1203, (1959). [DOI] [PubMed] [Google Scholar]
- 37.Eanes ED & Glenner GG X-ray diffraction studies on amyloid filaments. J Histochem Cytochem 16, 673–677, (1968). [DOI] [PubMed] [Google Scholar]
- 38.Petkova AT et al. A structural model for Alzheimer’s beta -amyloid fibrils based on experimental constraints from solid state NMR. Proceedings of the National Academy of Sciences of the United States of America 99, 16742–16747, (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khoshnevis S et al. The DEAD-box Protein Rok1 Orchestrates 40S and 60S Ribosome Assembly by Promoting the Release of Rrp5 from Pre-40S Ribosomes to Allow for 60S Maturation. PLoS Biol 14, e1002480, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Young CL & Karbstein K The roles of S1 RNA-binding domains in Rrp5’s interactions with pre-rRNA. RNA 17, 512–521, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Coccia M, Rossi A, Riccio A, Trotta E & Santoro MG Human NF-kappaB repressing factor acts as a stress-regulated switch for ribosomal RNA processing and nucleolar homeostasis surveillance. Proceedings of the National Academy of Sciences of the United States of America 114, 1045–1050, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Duda DM et al. Structure of a glomulin-RBX1-CUL1 complex: inhibition of a RING E3 ligase through masking of its E2-binding surface. Molecular cell 47, 371–382, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paravastu AK, Leapman RD, Yau WM & Tycko R Molecular structural basis for polymorphism in Alzheimer’s beta-amyloid fibrils. Proceedings of the National Academy of Sciences of the United States of America 105, 18349–18354, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qiang W, Yau WM, Lu JX, Collinge J & Tycko R Structural variation in amyloid-beta fibrils from Alzheimer’s disease clinical subtypes. Nature 541, 217–221, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Walti MA et al. Atomic-resolution structure of a disease-relevant Abeta(1–42) amyloid fibril. Proceedings of the National Academy of Sciences of the United States of America 113, E4976–4984, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schmidt A, Annamalai K, Schmidt M, Grigorieff N & Fandrich M Cryo-EM reveals the steric zipper structure of a light chain-derived amyloid fibril. Proceedings of the National Academy of Sciences of the United States of America 113, 6200–6205, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gremer L et al. Fibril structure of amyloid-beta(1–42) by cryo-electron microscopy. Science 358, 116–119, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cohen ML et al. Rapidly progressive Alzheimer’s disease features distinct structures of amyloid-beta. Brain 138, 1009–1022, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kaneko I, Yamada N, Sakuraba Y, Kamenosono M & Tutumi S Suppression of mitochondrial succinate dehydrogenase, a primary target of beta-amyloid, and its derivative racemized at Ser residue. J Neurochem 65, 2585–2593, (1995). [DOI] [PubMed] [Google Scholar]
- 50.Fitzpatrick AWP et al. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547, 185–190, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Giacobini E & Gold G Alzheimer disease therapy--moving from amyloid-beta to tau. Nat Rev Neurol 9, 677–686, (2013). [DOI] [PubMed] [Google Scholar]
- 52.Alonso A, Zaidi T, Novak M, Grundke-Iqbal I & Iqbal K Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proceedings of the National Academy of Sciences of the United States of America 98, 6923–6928, (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mair W et al. FLEXITau: Quantifying Post-translational Modifications of Tau Protein in Vitro and in Human Disease. Analytical chemistry 88, 3704–3714, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Schematic of the CPP Method.
Figure S2 Tetrameric structure of GPADH with the ε-amine of all lysine residues highlighted with blue spheres.
Figure S3 Relative accessibility of lysine residues for covalent modification expressed as percent of molecules.
Figure S4 Protein-peptide representation of all lysine residues identified and quantified in HEK293T cells.
Figure S5 Frequency plot of differences in lysine site accessibility for covalent modification with CPP.
Material S1 Limiting Reaction Time in CPP.
Material S2 CPP of GAPDH in Reactant-limited Conditions.
Material S3 Monomethylation During Initial Labeling with CPP.
Material S4 Correlating Relative Solvent Accessibility with the Accessibility of Lysine Sites for Covalent Modification with CPP.
Material S5 Analysis of Replicate Measurements.
Material S6 CPP Analysis and PTM Status of Lysine Residues.
Material S7 Summary of Previous Structural Analysis of Amyloid-β Peptide.
Material S8 Differentiating Proteolytic Cleavage Products of Amyloid-β Peptides from the APP Precursor Protein.
Table S1 Isotope combinations selected for labeling in CPP.
Table S2 Accessibility measurements with CPP for lysine residues in HEK293T cells using isobaric methyl moieties in CPP.
Table S3 Accessibility measurements with CPP for lysine residues in heat shock and control HEK293T cells.
Table S4 Significantly different lysine sites in HEK293T cells following heat shock.
Table S5 Patient diagnosis of human brain samples.
Data Availability Statement
Mass spectrometric raw data, search engine result files, and quantification result files can be accessed in Massive or ProteomeXchange (MassIVE MSV000083031, ProteomeXchange PXD011351).