

Abstract
Background
Pulmonary hypertension (PH) is a deadly disease characterized by vascular stiffness and altered cellular metabolism. Current treatments focus on vasodilation and not other root causes of pathogenesis. Previously, it was demonstrated that glutamine metabolism, as catalyzed by GLS1 (glutaminase 1) activity, is mechanoactivated by matrix stiffening and the transcriptional coactivators YAP1 (yes‐associated protein 1) and transcriptional coactivator with PDZ‐binding motif (TAZ), resulting in pulmonary vascular proliferation and PH. Pharmacologic inhibition of YAP1 (by verteporfin) or glutaminase (by CB‐839) improved PH in vivo. However, systemic delivery of these agents, particularly YAP1 inhibitors, may have adverse chronic effects. Furthermore, simultaneous use of pharmacologic blockers may offer additive or synergistic benefits. Therefore, a strategy that delivers these drugs in combination to local lung tissue, thus avoiding systemic toxicity and driving more robust improvement, was investigated.
Methods and Results
We used poly(lactic‐co‐glycolic) acid polymer‐based microparticles for delivery of verteporfin and CB‐839 simultaneously to the lungs of rats suffering from monocrotaline‐induced PH. Microparticles released these drugs in a sustained fashion and delivered their payload in the lungs for 7 days. When given orotracheally to the rats weekly for 3 weeks, microparticles carrying this drug combination improved hemodynamic (right ventricular systolic pressure and right ventricle/left ventricle+septum mass ratio), histologic (vascular remodeling), and molecular markers (vascular proliferation and stiffening) of PH. Importantly, only the combination of drug delivery, but neither verteporfin nor CB‐839 alone, displayed significant improvement across all indexes of PH.
Conclusions
Simultaneous, lung‐specific, and controlled release of drugs targeting YAP1 and GLS1 improved PH in rats, addressing unmet needs for the treatment of this deadly disease.
Keywords: mechanotransduction, metabolism, nanoparticle, pulmonary hypertension, therapy
Subject Categories: Pulmonary Biology, Vascular Disease
Nonstandard Abbreviations and Acronyms
- GLS1
glutaminase 1
- PH
pulmonary hypertension
- PLGA
poly(lactic‐co‐glycolic) acid
- RVSP
right ventricular systolic pressure
- TAZ
transcriptional coactivator with PDZ‐binding motif
- YAP1
yes‐associated protein 1
Clinical Perspective
What Is New?
Simultaneous, lung‐specific, and controlled release of drugs targeting YAP1 (yes‐associated protein 1) and GLS1 (glutaminase 1) and encapsulated by poly(lactic‐co‐glycolic) acid polymer‐based microparticles act together to improve pulmonary hypertension in rats.
Such targeted combinatorial therapy performs better across multiple indexes of disease than delivery of a poly(lactic‐co‐glycolic) acid polymer‐based single drug and at much lower doses than used for systemic administration.
What Are the Clinical Implications?
These results carry broad implications regarding the development of specific, next‐generation drug combinations for pulmonary vascular disease and perhaps for pulmonary conditions beyond pulmonary hypertension that affect both normal health and disease.
Pulmonary hypertension (PH) is a poorly understood vascular disease with increasing prevalence worldwide but with inadequate treatment options.1 There exist more than a dozen approved vasodilator drugs for treatment of this disease; nonetheless, mortality rates with current therapies remain high. At the cellular and molecular levels in the diseased pulmonary vasculature, PH is characterized by metabolic dysregulation, pro‐proliferative states, and adverse pulmonary vascular remodeling and stiffness. As such, there have been recent efforts to develop novel pharmacologic approaches that target the molecular origins of PH and thus could represent disease‐modifying opportunities. Recently, we reported a key molecular connection between vessel stiffness and metabolic dysregulation that promotes PH. Namely, we found that vessel stiffness mechanoactivates the YAP1 (yes‐associated protein 1)/TAZ (transcriptional coactivator with PDZ‐binding motif) cotranscription factors to induce glutaminolysis via the induction of GLS1 (glutaminase 1), thus sustaining the metabolic needs of proliferating pulmonary vascular cells and driving PH in vivo.2
These molecular insights advanced the paradigm of vascular stiffness beyond merely a consequence of long‐standing vascular dysfunction and defined specific therapeutic targets in PH that could disrupt the mechanical and metabolic causes of vascular cell proliferation. Importantly, we demonstrated substantial reversal of PH in a monocrotaline rat model of PH by pharmacologic inhibitors of YAP1 (verteporfin) and glutaminase or GLS1 (CB‐839). When delivered systemically, these drugs improved the hemodynamic and histopathologic manifestations of PH by decreasing the hyperproliferative phenotypes of diseased vascular cells.2 Additional findings have been independently reported that emphasize the direct importance of YAP1 signaling and glutamine metabolism3 in the pathogenesis of PH.4, 5, 6 Specifically, the YAP1 inhibitor verteporfin is a drug already approved by the US Food and Drug Administration for use in age‐related macular degeneration.7 CB‐839 is a GLS1 inhibitor that is in a clinical trial for kidney cancer (Clinical Trial NCT02071862). Thus, verteporfin and CB‐839 are promising candidates for repurposing for treatment of PH in humans. Their combination as potential additive or synergistic agents is particularly appealing for PH. As such, the identification of the mechanoactivation of glutaminolysis in PH directly sets the stage for applied endeavors to develop novel clinical treatment strategies in this devastating disease.
However, because YAP1 and GLS1 are already known to be ubiquitous and active in controlling cell growth and organ size throughout the body8 as well as glutamine metabolism,2 designing an effective chronic therapy for YAP1 and GLS1 inhibition in PH while minimizing adverse effects will necessitate local rather than systemic delivery. Local lung delivery via inhalation of verteporfin and CB‐839 could achieve that goal. To do so, we endeavored to generate a poly(lactic‐co‐glycolytic) acid (PLGA) drug delivery system for application as an inhaled and controlled‐release form of verteporfin and CB‐839, singly or in combination, to target the pulmonary vascular compartment (Figure 1).
Figure 1. Local inhibition of YAP1/TAZ and glutaminase pathways for effective amelioration of pulmonary hypertension.
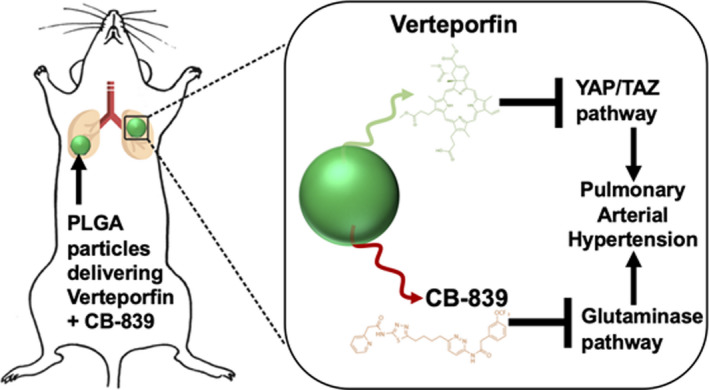
PLGA indicates poly(lactic‐co‐glycolic) acid; TAZ, transcriptional coactivator with PDZ‐binding motif; and YAP1, yes‐associated protein 1.
METHODS
The data that support the findings of this study are available from the corresponding author upon reasonable request.
PLGA Microparticle Fabrication
PLGA microparticles were fabricated using a single emulsion‐evaporation technique. For all microparticles, PLGA (50:50 lactide:glycolide, ester terminated; molecular weight (MW) 38 000–54 000; viscosity 0.45–0.6 dL/g; Millipore Sigma, St Louis, MO) was used. Specifically, 50 mg of PLGA was dissolved in 4 mL of dichloromethane (Sigma Aldrich). For single drug encapsulation, 4 mg of CB‐839 or verteporfin were directly dissolved in dichloromethane containing PLGA. In case of combinatorial drug encapsulation, 4 mg each of CB‐839 and verteporfin were added to dichloromethane containing PLGA. In case of IR780 microparticles, 5 mg of IR780 was added to the dichloromethane solution containing PLGA. In case of rhodamine microparticles, 1 mg of rhodamine 6G (Millipore Sigma, St. Louis, MO) was added to the dichloromethane solution containing PLGA. In case of blank particle generation, PLGA dissolved in dichloromethane was used as is. Next, this solution was then added to 60 mL of 2% polyvinyl‐alcohol (MW ~25 000, 98% hydrolyzed; PolySciences) and homogenized (L4RT‐A, Silverson, procured through Thermo Fisher Scientific, Pittsburgh, PA) at 10 000 rpm for 3 minutes. The homogenized mixtures were then added to 40 mL of 1% polyvinyl‐alcohol on a stir plate and left for 2 hours for the dichloromethane to evaporate. After 2 hours, the microparticles were centrifuged (2000g, 5 minutes, 4°C), washed 5 times with deionized water, and lyophilized for 48 hours (Virtis Benchtop K freeze dryer, Gardiner, NY).
Characterization of Microparticles and Assessment of Encapsulation and Release Kinetics
The morphology of the microparticles was characterized using JEOL JSM6510 scanning electron microscopy (SEM), and the average size of the blank microparticles was determined using dynamic light scattering (Malvern, Worcestershire, UK). The release kinetics of the drugs from PLGA microparticles was determined by incubating 1 mg of microparticles with or without drugs in 1 mL of 0.2% tween 80 (Thermo Fisher Scientific) in centrifuge tubes on end‐over‐end rotator at 37°C. Every day for 10 days, the tubes were centrifuged at 2000g for 5 minutes, 0.8 mL of the supernatant was retrieved and frozen at −20°C, and 0.8 mL of fresh 0.2% tween 80 was replaced in the tubes. These tubes were then returned to the incubator at 37°C.
To assess the concentration of verteporfin, an ultraviolet–visible spectroscopy plate reader (SpectraMax, Molecular Devices, Sunnyvale, CA) was used. An absorbance spectrum suggested that the maximum peak absorption of verteporfin is at 440 nm. Using this wavelength, a standard curve was plotted, and the concentration of the released verteporfin from microparticles was determined. The cumulative amount of verteporfin released from the microparticles was quantified and used to determine the percentage encapsulation efficiency and percentage loading.
To assess the concentration of CB‐839, a high‐performance liquid chromatography (HPLC–Ultimate 3000, Thermo Fisher Scientific) protocol was developed. Specifically, 18C column, 5 μm, 4.6×150 mm were used with the mobile phase of 80:20 water:methanol at 1 mL/min flow rate for 10 minutes and the absorbance was recorded at 210 nm. A standard curve of CB‐839 in 0.2% tween 80 was generated and used to quantify the concentration of the drug released over time. The cumulative amount of CB‐839 released from the microparticles was used to determine the percentage encapsulation efficiency and percentage loading.
Cell Culture
Primary human pulmonary arterial endothelial cells (Lonza, Thermo Fisher Scientific, Waltham, MA) were grown in Endothelial Growth Medium ‐ 2 (EGM‐2) cell culture media (Lonza, Thermo Fisher Scientific), and experiments were performed at passages 3–6.
Reverse Transcriptase–Quantitative Polymerase Chain Reaction
RNA extraction and reverse transcriptase–quantitative polymerase chain reaction were performed as we previously described.2 Quantitative polymerase chain reaction was performed on an Applied Biosystems (Foster City, CA) Quantstudio 6 Flex Fast Real Time polymerase chain reaction device. Fold change of RNA species was calculated using the formula 2^(−ΔΔCt), normalized to β‐actin expression. Taqman quantitative polymerase chain reaction primers for rat CTGF (connective tissue growth factor), CYR61 (cysteine‐rich angiogenic inducer 61), ANKRD1 (ankyrin repeat domain 1), and β‐actin were purchased from Thermo Fisher Scientific.
Glutaminase Activity Assay
As previously described2 and according to the manufacturer instructions (Glutaminase Microplate Assay Kit, Cohesion Biosciences, London, UK), flash frozen rat lung tissue (0.1 g/sample) was homogenized in 1 mL of assay buffer on ice and centrifuged at 8000g and 4°C for 10 minutes. Protein concentration was determined by Bradford assay. Samples, normalized to total protein (100 µg), were incubated with kit reagents for 1 hour at 37°C, and absorbances were measured at 420 nm.
Animals
Rats
To test PLGA‐encapsulated payload delivery in the lung, male Sprague‐Dawley rats (10–14 weeks old, Taconic, Germantown, NY) were administered intratracheal aerosol of PLGA microparticles with or without rhodamine 6G dye (1 mg of microparticles per dose in 0.25 mL of saline and encapsulating 5 μg dye). Alternatively, to model PH, as we previously described,4, 9 male Sprague‐Dawley rats (10–14 weeks old, Taconic) were injected with 60‐mg/kg monocrotaline at time 0; at 0 to 4 weeks postexposure, right heart catheterization was performed as described previously,2 followed by harvest of lung tissue for RNA extraction or optimal cutting temperature embedding as described in the Tissue Harvest of Rat Lungs section. At days 3, 7, 14, intratracheal aerosol administration of saline versus PLGA microparticles (1 mg of microparticles per dose in 0.25 mL of saline, which encapsulated 0.09 mg of verteporfin and 0.007 mg of CB‐839) was performed in isoflurane‐anesthetized rats at room temperature. Because of known sex differences among monocrotaline pulmonary arterial hypertension (PAH) rats,10 the assessment of PLGA‐encapsulated drug administration in female rats will be addressed in a separate study.
Randomization of the animals assigned to different experimental groups was achieved. Briefly, populations of animals sharing the same sex, same genotype, and similar body weight were generated and placed in 1 container. Then each animal was picked randomly and assigned in a logical fashion to different groups. For example, the first is assigned to group A, the second to group B, the third to group A, the fourth to group B, and so forth. For blinded analysis of right ventricular systolic pressure (RVSP) after right heart catheterization, animal subjects were only included if waveforms were found to be appropriately consistent across >30 seconds of recording. No animals were otherwise excluded from analyses.
Tissue Harvest of Rat Lungs
After physiological measurements, by direct right ventricular puncture, the pulmonary vessels were gently flushed with 1 cc of saline to remove the majority of blood cells before harvesting cardiopulmonary tissue. The heart was removed, followed by dissection and weighing of the right ventricle and the left ventricle+septum. Organs were then harvested for histological preparation or flash frozen in liquid N2 for subsequent homogenization and extraction of RNA and/or protein.
Cryostaining and Confocal Immunofluorescence of Lung Sections
To process lung tissue specifically for fluorescence microscopy, before excision, lungs were flushed with phosphate‐buffered saline (PBS) at constant low pressure (~10 mm Hg) via right ventricular cannulation, followed by tracheal inflation of the left lung in optimal cutting temperature medium (Tissue‐Tek, Sakura, Torrance, CA) at a pressure of ~20 cm H2O, followed by cryopreservation in ethanol/dry ice. Tissues were then further embedded in optimal cutting temperature, frozen solid in cryomolds, sectioned on a Microm HM 550 at 10 μm, and stored at −80°C. Cryosections were then air dried for 10 minutes at room temperature and rehydrated in 1× PBS for 15 minutes at room temperature. Sections were washed in 1× PBS, blocked in 1× PBS/BSA 3% supplemented with 10% heat‐inactivated goat serum for 1 hour at room temperature and then probed with appropriate primary antibody overnight at 4°C. After incubation, slides were washed 3 times with 1× PBS, blocked for 1 hour at room temperature, and probed with appropriate secondary antibodies in a dark room for 1 hour at room temperature. After washing, slides were mounted in mounting medium containing DAPI (Vectashield, Vector Labs, Burlingame, CA) and sealed with nail polish.
Alexa 488–conjugated, Cy3‐conjugated, and Cy5‐conjugated secondary antibodies (Thermo Fisher Scientific) were used for immunofluorescence. DAPI was obtained from Sigma Aldrich. Primary antibodies against α‐SMA (α–smooth muscle actin; ab32575; 1/1000 and ab21027; 1/300) were purchased from Abcam (Cambridge, UK). A primary antibody against proliferating cell nuclear antigen (PCNA, 13‐3900, 1/100) was purchased from Thermo Fisher Scientific. Pictures were obtained using a Nikon A1 confocal microscope. Small pulmonary vessels (<100 μm diameter) present in a given tissue section (>10 vessels/section) that were not associated with bronchial airways were selected for analysis (N>5 animals/group). Intensity of staining was quantified using ImageJ software (National Institutes of Health, Bethesda, MD). Vessel thickness was calculated as we previously described.9, 10 All measurements were performed blinded to condition.
Generation and Immunostaining of Precision Cut Tissue Slices
Precision cut tissue slices were generated for lung slices based on previous protocols11, 12 and adapted for kidney tissue.13 In brief, rat lungs were perfused with saline and subsequently inflated using a syringe pump with warm, 2% low‐melting‐point agarose in sterile DMEM/F12 media supplemented with 100 U/mL penicillin, 10 μG/mL streptomycin, and 2.5 μG/ML of amphotericin B (Sigma‐Aldrich). Lungs were kept on ice for 20 to 30 minutes to allow agarose to solidify. Kidneys were excised and kept on ice until further used. The 300‐μM thick precision cut tissue slices from kidneys and lung, respectively, were prepared using a vibratome (Ci 7000smz, Campden Instruments Ltd, Loughborough, Leicestershire, UK) at a speed of 10 to 12 μM/second with a frequency of 80 Hz and an amplitude of 1 mm. The slices were collected in fresh sterile media and immediately fixed for further processing and imaging.
For confocal immunofluorescence imaging, precision cut tissue slices were fixed with 4% FBS in PBS pH 7.4 for 30 minutes at room temperature (RT). Washed 3× with PBS and then incubated for 30 minutes in blocking buffer (2% BSA, 1% Triton‐X, 1% tween 20 in PBS pH 7.4; Sigma‐Aldrich). A fluorescein isothiocyanate (FITC)‐conjugated primary antibody against anti‐α‐SMA (1:500, F3777, Sigma‐Aldrich) was incubated overnight at 4°C. The next day, slices were washed 3×10 minutes each with PBS and mounted on glass slides with Fluormount G. Images were obtained using a Nikon A1 confocal microscope.
Picrosirius Red Stain and Quantification
Picrosirius red stain was achieved through the use of 5‐μm sections stained with 0.1% picrosirius red (Direct Red80, Sigma‐Aldrich) and counterstained with Weigert's hematoxylin to reveal fibrillar collagen. The sections were then serially imaged with an analyzer and polarizer oriented parallel and orthogonal to each other. Microscope conditions (lamp brightness, condenser opening, objective, zoom, exposure time, and gain parameters) were maintained constant throughout the imaging of all samples. A minimal threshold was set on appropriate control sections for each experiment in which only the light passing through the orthogonally oriented polarizers representing fibrous structures (ie, excluding residual light from the black background) was included. The threshold was maintained for all images across all conditions within each experiment. The area of the transferred regions that was covered by the thresholded light was calculated and at least 10 sections/vessel per condition were averaged together (ImageJ software).
Whole‐Lung Fluorescence Imaging
A total of 1 mg of PLGA microparticles encapsulating IR780 dye or blank microparticles were orotracheally administered to rats under isoflurane anaesthesia. The rats were returned to their cages for 7 days. After 7 days, another set of rats was orotracheally administered with 1 mg of PLGA microparticles encapsulating IR780 dye. All rats were euthanized, and lungs were harvested. The fluorescence in the lungs was determined using IVIS 200 (Perkin Elmer, Waltham, MA) using indocyanine green excitation and emission filters.
Statistical Analysis
In vitro experiments were performed at least 3 times and at least in triplicate for each replicate. The number of animals in each group was calculated based on the ability to detect a minimum effect size of 25 mm Hg difference between the mean RVSP after verteporfin+CB‐839 combination treatment versus blank microparticles groups (with a pooled SD=10) with a power of 80%. The estimated effect size (Cohen's d for between subjects' design with modification for small sample size) was 2.49.14 In situ expression/histologic analyses of rodent tissue and pulmonary vascular hemodynamics in rats were performed in a blinded fashion. Numerical quantifications for in vitro experiments represent mean±SD. Numerical quantifications for physiologic experiments using rodents or human reagents represent mean±SEM. To better visualize error bars on graphs for in vitro experiments, standard deviation was used, whereas for in vivo experiments, standard error was measured. Micrographs are representative of experiments in each relevant cohort. Normality of data distribution was determined by Shapiro–Wilk testing. Paired samples were compared by a 2‐tailed Student t test for normally distributed data, whereas Mann–Whitney U nonparametric testing was used for non‐normally distributed data. For comparisons among groups, 1‐way ANOVA and post hoc Bonferroni testing were performed. A P value <0.05 was considered significant.
Study Approval
All animal experiments were approved by the University of Pittsburgh School of Medicine Division of Laboratory Animal Resources.
RESULTS
PLGA Microparticles Encapsulate and Release Verteporfin and CB‐839 Simultaneously
To develop a controlled‐release formulation that could release verteporfin and CB‐839 and block YAP1/TAZ and GLS1 simultaneously, PLGA‐based microparticles were generated. Specifically, oil in water emulsions were used, where verteporfin alone, CB‐839 alone, or verteporfin with CB‐839 together were directly dissolved in the oil phase to generate the microparticles. The size of the microparticles was optimized to be in the 1 to 5 μm range (as observed using scanning electron microscope and dynamic light scattering; Figure 2A and 2B) for optimal deposition in the lungs. In the combinatorial delivery microparticle, the percentage encapsulation efficiency (%±SD) and loading (mg/mg±SD) of verteporfin were determined to be 46.5±5% and 0.09±0.01 mg/mg, respectively, and percentage encapsulation efficiency and loading of CB‐839 were determined to be 22±4% and 0.04±0.007 mg/mg, respectively. For single drug formulation, percentage encapsulation efficiency and loading of CB‐839 was observed to be 46.9±5% and 0.1±0.01 mg/mg, respectively, and percentage encapsulation efficiency and loading of verteporfin were determined to be 85±9% and 0.16±0.02 mg/mg, respectively. Moreover, the release kinetics of verteporfin and CB‐839 from different formulations suggested that verteporfin was released in a sustained manner for 6 days, and CB‐839 was released in a sustained manner for 10 days (Figure 2C and 2D; please note that the errors in Figure 2D are small and not visible).
Figure 2. PLGA microparticles are within a size range for inhalation and release verteporfin and CB‐839 in a sustained manner.
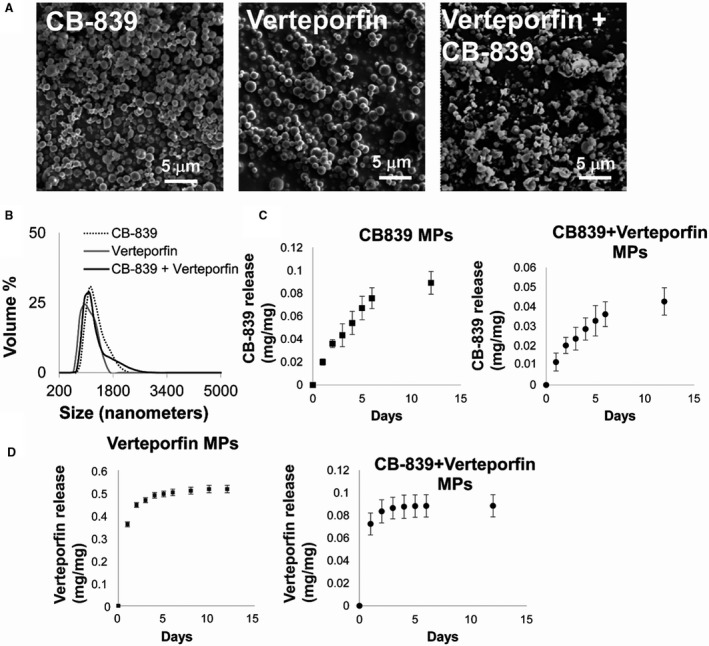
(A) Scanning electron microscope images of CB‐839 alone encapsulated, verteporfin alone encapsulated, and CB‐839 with verteporfin encapsulated microparticles displayed smooth surface morphology. (B) Size distribution of the microparticles obtained from dynamic light scattering experiments demonstrated that the average microparticle size for all the microparticles was approximately 1 μm. (C) Release kinetics of CB‐839 from PLGA microparticles encapsulating CB‐839‐verteporfin or encapsulating CB‐839 only. (D) Release kinetics of verteporfin from PLGA microparticles encapsulating CB‐839‐verteporfin or encapsulating verteporfin only. Error bars represent SD. PLGA indicates poly(lactic‐co‐glycolic) acid.
PLGA Microparticles Deposit Their Drug Payloads in the Lungs of Rats for 7 Days
To ensure extended efficacy of drug via controlled release, release of encapsulates were observed in lung tissue samples during the course of treatment. To do so, PLGA microparticles encapsulating IR780, a near infrared sensor and a substitute for the drugs used, were generated. The microparticles encapsulating IR780 dye or blank microparticles were administered to rats via a single orotracheal administration. These rats were then euthanized on day 0 or day 7 after particle delivery, and the lung and heart tissues were harvested and imaged for the presence of the dye. Microparticles deposited their payloads in the lungs of rats, and this payload was retained in the lungs for 7 days (Figure 3A). To define more precisely the histologic distribution of encapsulated payloads in lung tissue, confocal microscopy was performed 3 days after intratracheal administration with PLGA microparticles encapsulating rhodamine 6G dye versus no dye. Given the concern that rhodamine may redistribute or leak out of traditional histologic sections and staining, precision cut lung slices were analyzed. In these sections, we found that PLGA encapsulation effectively delivered rhodamine dye to muscularized pulmonary arterioles identified by anti‐α‐SMA staining (green; Figure 3B). Importantly, no rhodamine was detectable in precision cut kidney slices of rats after similar intratracheal administration, supporting the localized rather than systemic delivery of encapsulated payloads (Figure 3C).
Figure 3. PLGA microparticles deliver payload into the lungs of rats.

(A) Fluorescence image of the lungs of rats after intratracheal administration with PLGA microparticles encapsulating near infrared dye IR780 vs no dye, imaged on day 0 and day 7 after administration. (B) Confocal microscopy was performed on precision cut lung slices of rats after intratracheal administration with PLGA microparticles encapsulating rhodamine 6G dye vs no dye imaged on day 3 after administration. Lung slices were stained with antibody against α‐SMA (green) to identify muscularized pulmonary arterioles. Representative matched images are shown from each of N=3 rats/group to visualize α‐SMA (green) and rhodamine 6G dye (Rh; red; bars=50 μm). (C) Representative matched images are shown (as in B) to visualize α‐SMA (arterioles, green) and rhodamine 6G dye (Rh; red) in precision cut kidney slices of rats after intratracheal administration with PLGA microparticles encapsulating rhodamine 6G dye vs no dye imaged on day 3 after administration (bars=50 μm). α‐SMA indicates α‐smooth muscle actin; and PLGA, poly(lactic‐co‐glycolic) acid.
PLGA Microparticles Delivering Verteporfin and CB‐839 Ameliorate Multiple Indexes of PH in Monocrotaline‐Exposed Rats In Vivo
PLGA microparticles carrying verteporfin and CB‐839, singly or in combination, were tested in vivo to determine their ability to prevent PH in a rodent model of disease. Specifically, PH was induced in rats using monocrotaline injections at day 0 and studied in various groups; blank microparticles, microparticles encapsulating verteporfin alone, microparticles encapsulating CB‐839 alone, or microparticles encapsulating both verteporfin+CB‐839 were delivered intratracheally to rats weekly for 3 weeks starting at day 3 (Figure 4A). At the end of the third week, hemodynamic (RVSP, which is a surrogate of pulmonary arterial pressures as well as the right ventricle and the left ventricle+septum mass ratio or Fulton index, which is a measure of right ventricular hypertrophy), histologic (vascular remodeling as quantified by α‐SMA thickness of small pulmonary arterioles and vascular matrix remodeling as quantified by picrosirius red staining), and molecular markers (PCNA, a proliferation marker) of PH were quantified among the various comparator groups.
Figure 4. Simultaneous pharmacologic inhibition of GLS1 and YAP1/TAZ in monocrotaline‐exposed rats modulates YAP1‐specific gene expression and glutaminase activity in the lungs.
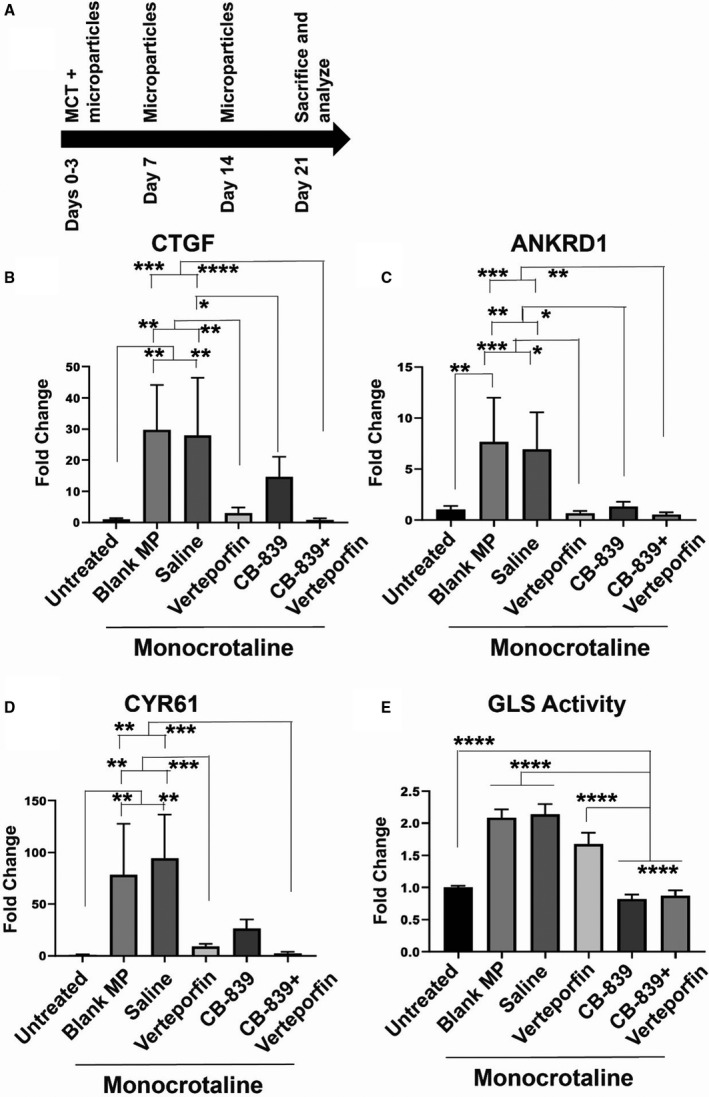
(A) Study design is shown for the induction of PH using monocrotaline via intraperitoneal injection followed by administration of microparticles orotracheally for treatments. (B–D) As assessed by reverse transcriptase–quantitative polymerase chain reaction of rat lung, YAP1‐dependent gene transcripts for CTGF (B), ANKRD1 (C), and CYR61 (D) were downregulated modestly by PLGA‐encapsulated CB‐839 and more robustly by inhaled PLGA‐encapsulated verteporfin. (E) As assessed by glutaminase activity assay of rat lung, glutaminase activity was robustly downregulated by inhaled PLGA‐encapsulated CB‐839 but not verteporfin alone. In all images, mean expression in untreated groups was assigned a fold change of 1, to which relevant samples were compared. In each group, n=3 (untreated), n=5 (blank microparticles), n=3 (saline), n=5 (verteporfin), n=5 (CB‐839), and n=6 (CB839+verteporfin). Error bars represent mean±SEM. By 1‐way ANOVA and post hoc Bonferroni testing, significantly different values are represented by *<0.05, *<0.01, ***<0.001, and ****<0.00001. ANKRD1 indicates ankyrin repeat domain 1; CTGF, connective tissue growth factor; CYR61, cysteine‐rich angiogenic inducer 61; GLS1, glutaminase 1; MCT, monocrotaline; PLGA, poly(lactic‐co‐glycolic) acid; TAZ, transcriptional coactivator with PDZ‐binding motif; and YAP1, yes‐associated protein 1.
In monocrotaline PH rats, reverse transcriptase–quantitative polymerase chain reaction demonstrated that PLGA‐based delivery of drugs potently modulated YAP1‐specific gene expression, including CTGF, ANKRD1, and CYR61 (Figure 4B through 4D). Notably, PLGA‐encapsulated CB‐839 drove modest downregulation of these transcripts, consistent with the positive feedback loop between glutaminase activity, vascular stiffness, and YAP1 activation.2 Importantly, PLGA‐encapsulated verteporfin more robustly repressed these transcripts, and the combination of both verteporfin and CB‐839 led to trends of even lower expression. In parallel, as assessed by the glutaminase activity assay of rat lung, glutaminase activity was robustly downregulated by orotracheally delivered PLGA‐encapsulated CB‐839 alone or verteporfin+CB839 together (Figure 4E). A trend toward downregulated glutaminase activity was also seen by PLGA‐encapsulated verteporfin alone.
In the context of such successful drug delivery, we found that simultaneous drug delivery led to significant and substantial decreases of RVSP and Fulton index compared with blank microparticles (Figure 5A and 5B). Consistent with the efficacy of single drugs alone delivered systemically via serial intraperitoneal administration,2 verteporfin alone also promoted significant hemodynamic amelioration (Figure 5B), and CB‐839 demonstrated nonsignificant trends toward similar improvement (Figure 5A and 5B). By confocal in situ staining of lung tissue and quantification of smooth muscle arteriolar (<100 μm diameter) thickness via α‐SMA staining (Figure 6A), histopathologic pulmonary vascular remodeling of monocrotaline PH rats with saline or blank microparticles was reduced most robustly by simultaneous PLGA delivery of both drugs (Figure 6C). In comparison, verteporfin delivery alone also decreased remodeling but to a lesser degree (Figure 6C) compared with the drug combination; CB‐839 delivery alone exhibited a slight but nonsignificant trend toward improvement of remodeling. By in situ staining of pulmonary arterioles for the proliferation marker PCNA, only the verteporfin+CB‐839 combination displayed a significant decrease of the elevated vascular PCNA levels in saline or blank particle controls (Figure 6B); either verteporfin or CB‐839 alone displayed a modest but nonsignificant decrease of vascular PCNA expression. Finally, by in situ picrosirius red staining to quantify the level of pulmonary vascular matrix remodeling, only the verteporfin+CB‐839 combination displayed a significant decrease of both pulmonary arteriolar collagen deposition (nonpolarized light) and collagen cross‐linking (polarized light) compared with saline or blank particle controls (Figure 7). Thus, all indexes demonstrated significant and substantial improvement with combination drug delivery. For some indexes, either verteporfin or CB‐839 alone demonstrated improvement. However, only the combination of drugs—but neither verteporfin nor CB‐839 alone—displayed significant improvement across all indexes of PH.
Figure 5. Delivery of verteporfin and CB‐839 simultaneously in vivo improves hemodynamic manifestations of PH in monocrotaline‐exposed rats.

(A) Poly(lactic‐co‐glycolic) acid MP delivering verteporfin and CB‐839 and poly(lactic‐co‐glycolic) acid MP delivering verteporfin alone significantly decreased the Fulton index (RV/LV+S mass) compared with blank MP. (B) PLGA MP delivering verteporfin and CB‐839 significantly decreased RVSP compared with the control of blank MP, and this RVSP was not significantly different than the untreated rats (n=3–6; error bars represent mean±SEM. By 1‐way ANOVA and post hoc Bonferroni testing, significantly different values are represented by *<0.05, *<0.01, ***<0.001, and ****<0.00001. LV+S indicates left ventricle+septum; MP, microparticles; RV, right ventricle; and RVSP, right ventricular systolic pressure.
Figure 6. Simultaneous pharmacologic inhibition of GLS1 and YAP1/TAZ in monocrotaline‐exposed rats decreases pulmonary vascular cell proliferation and pulmonary vascular remodeling.
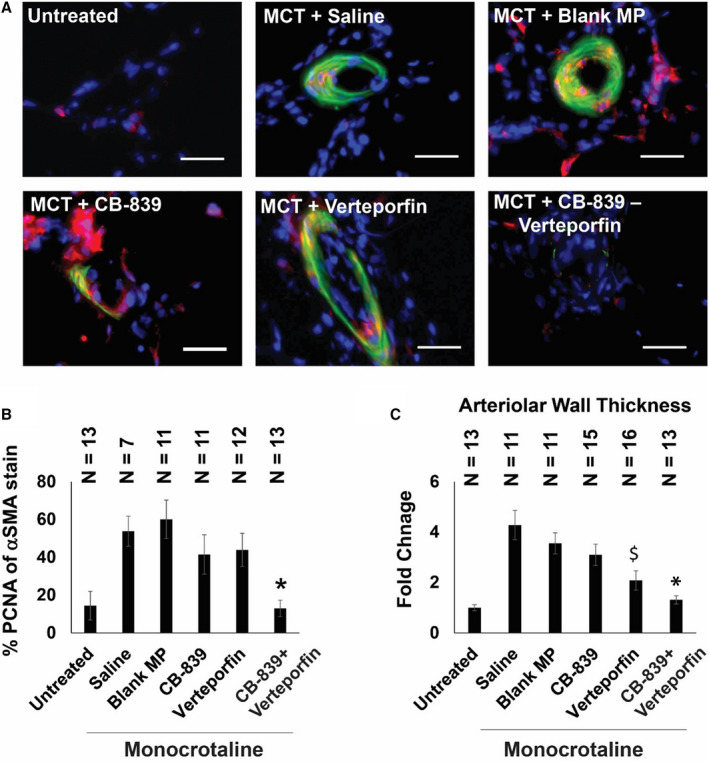
(A) Representative images of small pulmonary arterioles (<100 μm diameter) of the lungs (blue, nuclei; red, PCNA; green, α‐SMA; bar=30 μm). (B) The percentage of PCNA of α‐SMA–positive vascular cells in the CB‐839 and verteporfin combination group was significantly lower than negative controls of saline and blank MP and significantly different than single drug treatments alone (n=7–13; error bars represent mean±SEM). By 1‐way ANOVA and post hoc Bonferroni testing, significant P values were calculated as follows for CB‐839+verteporfin‐treated rodents: *P=0.0003 vs saline, P=0.0001 vs blank MP, P=0.0147 vs CB‐839, P=0.0041 vs verteporfin. Notably, P=0.8684 vs untreated. Other comparisons among monocrotaline‐PAH rat cohorts were not significant (P>0.05). (C) The wall thickness of pulmonary arterioles (diameter<100 μm) in the verteporfin+CB‐839 combination treatment group was significantly lower than either single drug treatment or negative controls of saline and blank MP; mean expression in the untreated group was assigned a fold change of 1, to which relevant samples were compared (n=11–16 vessels; error bars represent mean±SEM). By 1‐way ANOVA and post hoc Bonferroni testing, significant P values were calculated as follows for CB‐839+verteporfin‐treated rodents: *P=2.36E‐05 vs saline, P=2.40E‐05 vs blank MP, P=0.0009 vs CB‐839, P=0.0465 vs verteporfin. Notably, P=0.1241 vs untreated. For verteporfin‐treated rodents, significant P values included the following: $P=3.08E‐05 vs saline, P=0.0001 vs blank MP, P=0.017 vs CB‐839, P=0.0465 vs CB‐839+verteporfin, P=0.002 vs untreated. Other comparisons among monocrotaline‐PAH rat cohorts were not significant (P>0.05). α‐SMA indicates α‐smooth muscle actin; GLS1, glutaminase 1; MCT, monocrotaline; MP, microparticles; PCNA, proliferating cell nuclear antigen; TAZ, transcriptional coactivator with PDZ‐binding motif; and YAP1, yes‐associated protein 1.
Figure 7. Simultaneous pharmacologic inhibition of GLS1 and YAP1/TAZ in monocrotaline‐exposed rats decreases collagen deposition and collagen cross‐linking in pulmonary arterioles.
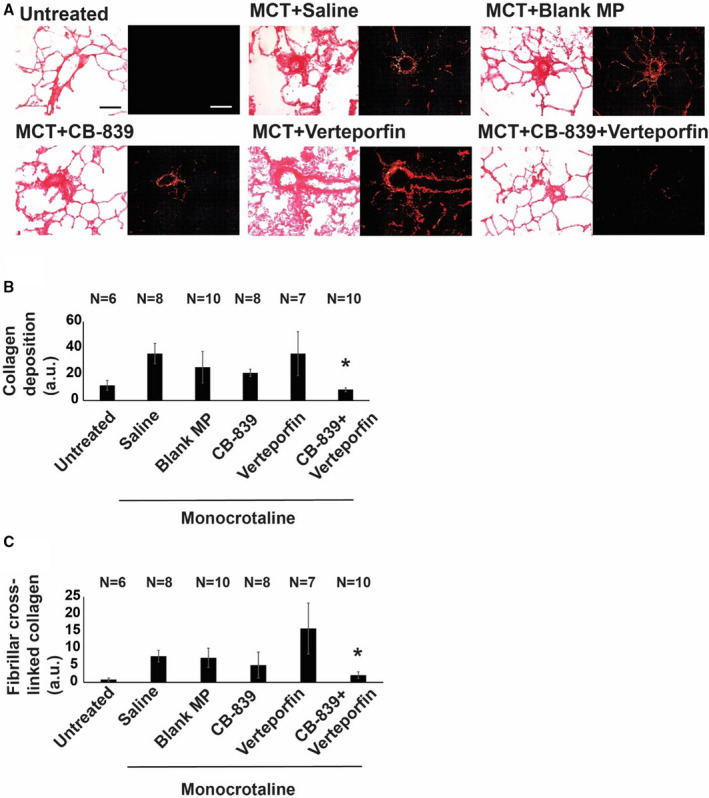
(A) Representative images of picrosirius red stain of lung tissues showing fibrillar collagen deposition (red, bright field) and cross‐linked fibrillar collagen assembly (using orthogonal polarized images, red is collagen type I and green is collagen type III [bar=50 μm]). (B) Quantification of the percent area of picrosirius red stain under nonpolarized light (represented as a.u.) showed that the CB‐839 and verteporfin combination significantly decreased pulmonary arteriolar collagen deposition compared with negative controls of saline and blank MP and was significantly different than either single drug treatment alone (n=6–10; error bars represent mean±SEM). By 1‐way ANOVA and post hoc Bonferroni testing, P values were calculated as follows for CB‐839+verteporfin‐treated rodents: *P=0.0003 vs saline, P=0.0438 vs blank MP, P=0.0036 vs CB‐839; P=0.0361 vs verteporfin. Notably, P=0.3312 vs untreated. Other comparisons among monocrotaline‐PAH rat cohorts were not significant (P>0.05). (C) Quantification of the percent area of picrosirius red stain under polarized light (represented as a.u.) demonstrated that the CB‐839 and verteporfin combination group significantly decreased pulmonary arteriolar cross‐linked collagen compared with negative controls of saline and blank MP and was significantly different than verteporfin‐alone treatment (n=6–10; error bars represent mean±SEM). By 1‐way ANOVA and post hoc Bonferroni testing, significant P values were calculated as follows for CB‐839+verteporfin‐treated rodents: *P=3.65E‐05 vs saline, P=0.0047 vs blank MP, P=0.012 vs verteporfin. Notably, P=0.1363 vs CB‐839 and P=0.095 vs untreated. Other comparisons among monocrotaline‐PAH rat cohorts were not significant (P>0.05). a.u. indicates arbitrary units; GLS1, glutaminase 1; MCT, monocrotaline; MP, microparticles; TAZ, transcriptional coactivator with PDZ‐binding motif; and YAP1, yes‐associated protein 1.
DISCUSSION
The findings of this study reveal that PLGA microparticle encapsulation is effective for controlled and sustained pulmonary vascular delivery of verteporfin and CB‐839. These data also emphasize that PLGA‐based delivery of this combination of drugs simultaneously is effective in improving PH in vivo and performs better across multiple indexes of disease than delivery of a PLGA‐based single drug alone. As such, these results carry broad implications regarding the development of specific, next‐generation drug combinations for pulmonary vascular disease and perhaps for pulmonary conditions beyond PH that affect both normal health and disease.
By coupling local delivery with combination drug therapy, this approach addresses key concerns that have emerged regarding the development of novel therapies for PH. Although prior drug development in PH has focused on compounds that target major vasodilatory pathways,15 a great majority of the next generation of drugs being tested in this disease focus on targeting the proliferative and often metabolic cancer‐like phenotypes of the diseased pulmonary vascular cells.16 In fact, the concept of repurposing chemotherapeutic drugs such as receptor tyrosine kinase inhibitors has been touted and continues to be explored.17 In parallel, a number of metabolic therapies, such as dicholoroacetate18 and bardoxolone,19 have been progressing in clinical trials designed to reverse metabolic dysfunction in PH. Nonetheless, because of the broad‐reaching effects of such antiproliferative and metabolic therapies, there is growing concern that these therapies may carry substantial risk because of unintended off‐target or systemic effects. Clinical trial data have supported that notion, demonstrating substantial adverse effects in PH with the receptor tyrosine kinase inhibitor imatinib20 despite its hemodynamic and pulmonary vascular benefits. PLGA microparticles for local tissue and pulmonary vascular delivery of such next‐generation therapies in PH may effectively address these issues not only by limiting the breadth of tissues affected but also by maximizing the local effective concentration of drug to vascular cells and thus allowing for an overall decrease of drug needed for administration.
Another concern in developing novel pharmacologic therapies in PH that is mitigated by our approach addresses the question of potency of a given next‐generation drug intending to target only a single molecule or pathway. Given the extreme complexity of networks and overlap of mechanisms surrounding metabolic reprogramming and the hyperproliferative state in PH,21 there can be a substantial chance that targeting a single proliferative or metabolic factor may lead to compensatory responses that obviate the beneficial effects of that single drug. Systematic inhibition of multiple targets in the same pathogenic pathway as in the YAP1‐GLS1 axis2 in theory holds a much higher likelihood of achieving more substantial potency and disease modification. Indeed, our findings that the combination of verteporfin and CB‐839 performs better across multiple indexes of PH than PLGA‐based single drug delivery alone strengthen that notion. Coupling these robust effects with local delivery and decreased drug dose also mitigates the chance of systemic toxicity, facilitating this potency specifically in diseased lung and pulmonary vasculature.
Another insight derived from this work suggests a new direction for development of locally delivered therapies for PH. Although current PH therapies involving inhaled prostacyclin tend to reduce systemic side effects on peripheral vasculature,22 there has been a delay in development of long‐acting, controlled‐release prostacyclin products that could be used effectively as an inhaled therapy. In this study, we chose to pursue a solution involving PLGA‐based microparticles for a number of reasons. Specifically, PLGA microparticles have an excellent US Food and Drug Administration approval track record.23, 24 Furthermore, the drugs can be encapsulated and released from these microparticles in a sustained manner,24, 25, 26 leading to advantages for less frequent dosing and lower amounts of drug needed for each administration. Specifically, in this work, we attained robust inhibition of both YAP1‐specific and GLS1‐specific activity (Figure 4) by administering an ~600‐fold lower dose of verteporfin and an ~3000‐fold lower dose of CB‐839 in total via weekly delivery of PLGA microparticles compared with daily systemic intraperitoneal injections that we previously reported.2 The microparticles can be designed to be in different size ranges (1–5 μm in this report) for effective delivery to the lungs and potentially targeting pulmonary arterioles. The release kinetics of the encapsulated drugs can be tailored so that a sustained release of drugs for 3 to 4 weeks can be achieved.27 Moreover, these formulations are also amenable to be functionalized with different molecules to prevent macrophage‐mediated phagocytosis and clearance.28
Finally, lung delivery of PLGA‐encapsulated drugs that simultaneously target YAP1 and GLS1 may be effective in pulmonary diseases far beyond PH. For instance, in idiopathic pulmonary fibrosis independent of the development of PH, there is evidence of the pathogenic importance of increased YAP1/TAZ activity29 as well as glutaminolysis.30 To an even greater extent, YAP1/TAZ activation has emerged as a leading therapeutic candidate for multiple types of cancer, including lung cancer.31 Similarly, development and progression of specific types of lung cancer have displayed a striking dependence on glutaminolysis.32 Although our results do not test the direct effects of PLGA delivery of verteporfin and CB‐839, PLGA particle imaging suggests that orotracheal delivery can attain substantial functional coverage of lung parenchyma as well as pulmonary vasculature (Figures 3 and 4). Thus, the translation and clinical utility of this specific combination drug delivery may have broad possibilities across diverse aspects of pulmonary disease.
In conclusion, we report that pulmonary delivery of aerosolized PLGA microspheres is effective for sustained and local drug delivery to lung tissue. Using this system, delivery of a combination of drugs targeting the YAP1‐GLS1 circuit robustly improves multiple indexes of PH in vivo and performs better in aggregate than PLGA‐based single drug delivery alone. These findings establish a much‐needed foundation for further development for locally specific, sustained, and combinatorial therapies in PH and perhaps other lung diseases.
Sources of Funding
This work was supported by National Institutes of Health Grants R01 HL124021, HL122596, HL138437, UH2/UH3 TR002073 (S.Y.C.); American Heart Association Grant 18EIA33900027 (S.Y.C.); and French National Research Agency Grants ANR‐18‐CE14‐0025 and ANR‐20‐CE14‐0006‐02 (T.B.).
Disclosures
S.Y.C. has served as a consultant for Zogenix and United Therapeutics and has held grants from Pfizer and Actelion. S.Y.C. serves as a founder, director, and officer of Synhale Therapeutics. Patent applications (S.Y.C., T.B., A.P.A., S.R.L.) have been filed regarding targeting YAP1 (yes‐associated protein 1)/TAZ (transcriptional coactivator with PDZ‐binding motif), glutaminase, and glutamine metabolism in pulmonary hypertension. The remaining authors have no disclosures to report.
Acknowledgments
We thank Dr Nouraie (University of Pittsburgh School of Medicine) for statistical advice.
(J Am Heart Assoc. 2021;10:e019091. DOI: 10.1161/JAHA.120.019091.)
For Sources of Funding and Disclosures, see page 14.
References
- 1.Chan SY, Loscalzo J. Pathogenic mechanisms of pulmonary arterial hypertension. J Mol Cell Cardiol. 2008;44:14–30. DOI: 10.1016/j.yjmcc.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bertero T, Oldham WM, Cottrill KA, Pisano S, Vanderpool RR, Yu Q, Zhao J, Tai Y, Tang Y, Zhang Y‐Y, et al. Vascular stiffness mechanoactivates YAP/TAZ‐dependent glutaminolysis to drive pulmonary hypertension. J Clin Invest. 2016;126:3313–3335. DOI: 10.1172/JCI86387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dumas SJ, Bru‐Mercier G, Courboulin A, Quatredeniers M, Rücker‐Martin C, Antigny F, Nakhleh MK, Ranchoux B, Gouadon E, Vinhas M‐C, et al. NMDA‐type glutamate receptor activation promotes vascular remodeling and pulmonary arterial hypertension. Circulation. 2018;137:2371–2389. DOI: 10.1161/CIRCULATIONAHA.117.029930. [DOI] [PubMed] [Google Scholar]
- 4.Bertero T, Cottrill K, Lu YU, Haeger C, Dieffenbach P, Annis S, Hale A, Bhat B, Kaimal V, Zhang Y‐Y, et al. Matrix remodeling promotes pulmonary hypertension through feedback mechanoactivation of the YAP/TAZ‐miR‐130/301 circuit. Cell Rep. 2015;13:1016–1032. DOI: 10.1016/j.celrep.2015.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kudryashova TV, Goncharov DA, Pena A, Kelly N, Vanderpool R, Baust J, Kobir A, Shufesky W, Mora AL, Morelli AE, et al. Hippo‐integrin‐linked kinase cross‐talk controls self‐sustaining proliferation and survival in pulmonary hypertension. Am J Respir Crit Care Med. 2016;194:866–877. DOI: 10.1164/rccm.201510-2003OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dieffenbach PB, Haeger CM, Coronata AMF, Choi KM, Varelas X, Tschumperlin DJ, Fredenburgh LE. Arterial stiffness induces remodeling phenotypes in pulmonary artery smooth muscle cells via YAP/TAZ‐mediated repression of cyclooxygenase‐2. Am J Physiol Lung Cell Mol Physiol. 2017;313:L628–L647. DOI: 10.1152/ajplung.00173.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arnold JJ. Age‐related macular degeneration: anti‐vascular endothelial growth factor treatment. BMJ Clin Evid. 2016;2016:0701. [PMC free article] [PubMed] [Google Scholar]
- 8.Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179–183. DOI: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- 9.Bertero T, Lu YU, Annis S, Hale A, Bhat B, Saggar R, Saggar R, Wallace WD, Ross DJ, Vargas SO, et al. Systems‐level regulation of microRNA networks by miR‐130/301 promotes pulmonary hypertension. J Clin Invest. 2014;124:3514–3528. DOI: 10.1172/JCI74773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tofovic SP. Estrogens and development of pulmonary hypertension: Interaction of estradiol metabolism and pulmonary vascular disease. J Cardiovasc Pharmacol. 2010;56:696–708. DOI: 10.1097/FJC.0b013e3181f9ea8d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uhl FE, Vierkotten S, Wagner DE, Burgstaller G, Costa R, Koch I, Lindner M, Meiners S, Eickelberg O, Konigshoff M. Preclinical validation and imaging of Wnt‐induced repair in human 3D lung tissue cultures. Eur Respir J. 2015;46:1150–1166. DOI: 10.1183/09031936.00183214. [DOI] [PubMed] [Google Scholar]
- 12.Alsafadi HN, Uhl FE, Pineda RH, Bailey KE, Rojas M, Wagner DE, Konigshoff M. Applications and approaches for three‐dimensional precision‐cut lung slices. Disease modeling and drug discovery. Am J Respir Cell Mol Biol. 2020;62:681–691. DOI: 10.1165/rcmb.2019-0276TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stribos EG, Hillebrands JL, Olinga P, Mutsaers HA. Renal fibrosis in precision‐cut kidney slices. Eur J Pharmacol. 2016;790:57–61. DOI: 10.1016/j.ejphar.2016.06.057. [DOI] [PubMed] [Google Scholar]
- 14.Goulet‐Pelletier J‐C, Cousineau D. A review of effect sizes and their confidence intervals, part I: the Cohen's d family. Quant Method Psychol. 2018;14:242–265. DOI: 10.20982/tqmp.14.4.p242. [DOI] [Google Scholar]
- 15.Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med. 2004;351:1425–1436. DOI: 10.1056/NEJMra040291. [DOI] [PubMed] [Google Scholar]
- 16.Pullamsetti SS, Savai R, Seeger W, Goncharova EA. Translational advances in the field of pulmonary hypertension. From cancer biology to new pulmonary arterial hypertension therapeutics. Targeting cell growth and proliferation signaling hubs. Am J Respir Crit Care Med. 2017;195:425–437. DOI: 10.1164/rccm.201606-1226PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Godinas L, Guignabert C, Seferian A, Perros F, Bergot E, Sibille Y, Humbert M, Montani D. Tyrosine kinase inhibitors in pulmonary arterial hypertension: a double‐edge sword? Semin Respir Crit Care Med. 2013;34:714–724. DOI: 10.1055/s-0033-1356494. [DOI] [PubMed] [Google Scholar]
- 18.Michelakis ED, Gurtu V, Webster L, Barnes G, Watson G, Howard L, Cupitt J, Paterson I, Thompson RB, Chow K, et al. Inhibition of pyruvate dehydrogenase kinase improves pulmonary arterial hypertension in genetically susceptible patients. Sci Transl Med. 2017;9:eaao4583. DOI: 10.1126/scitranslmed.aao4583. [DOI] [PubMed] [Google Scholar]
- 19.Hu J, Xu Q, McTiernan C, Lai YC, Osei‐Hwedieh D, Gladwin M. Novel targets of drug treatment for pulmonary hypertension. Am J Cardiovasc Drugs. 2015;15:225–234. DOI: 10.1007/s40256-015-0125-4. [DOI] [PubMed] [Google Scholar]
- 20.Hoeper MM, Barst RJ, Bourge RC, Feldman J, Frost AE, Galié N, Gómez‐Sánchez MA, Grimminger F, Grünig E, Hassoun PM, et al. Imatinib mesylate as add‐on therapy for pulmonary arterial hypertension: results of the randomized impres study. Circulation. 2013;127:1128–1138. DOI: 10.1161/CIRCULATIONAHA.112.000765. [DOI] [PubMed] [Google Scholar]
- 21.Chan SY, Rubin LJ. Metabolic dysfunction in pulmonary hypertension: from basic science to clinical practice. Eur Respir Rev. 2017;26;170094. DOI: 10.1183/16000617.0094-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zamanian RT, Levine DJ, Bourge RC, De Souza SA, Rosenzweig EB, Alnuaimat H, Burger C, Mathai SC, Leedom N, DeAngelis K, et al. An observational study of inhaled‐treprostinil respiratory‐related safety in patients with pulmonary arterial hypertension. Pulm Circ. 2016;6:329–337. DOI: 10.1086/688059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jain RA. The manufacturing techniques of various drug loaded biodegradable poly(lactide‐co‐glycolide) (PLGA) devices. Biomaterials. 2000;21:2475–2490. DOI: 10.1016/S0142-9612(00)00115-0. [DOI] [PubMed] [Google Scholar]
- 24.Acharya AP, Clare‐Salzler MJ, Keselowsky BG. A high‐throughput microparticle microarray platform for dendritic cell‐targeting vaccines. Biomaterials. 2009;30:4168–4177. DOI: 10.1016/j.biomaterials.2009.04.032. [DOI] [PubMed] [Google Scholar]
- 25.Fisher JD, Acharya AP, Little SR. Micro and nanoparticle drug delivery systems for preventing allotransplant rejection. Clin Immunol. 2015;160:24–35. DOI: 10.1016/j.clim.2015.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Acharya AP, Carstens MR, Lewis JS, Dolgova N, Xia CQ, Clare‐Salzler MJ, Keselowsky BG. A cell‐based microarray to investigate combinatorial effects of microparticle‐encapsulated adjuvants on dendritic cell activation. J Mater Chem B. 2016;4:1672–1685. DOI: 10.1039/C5TB01754H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ratay ML, Balmert SC, Acharya AP, Greene AC, Meyyappan T, Little SR. Tri microspheres prevent key signs of dry eye disease in a murine, inflammatory model. Sci Rep. 2017;7:17527. DOI: 10.1038/s41598-017-17869-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schneider CS, Xu Q, Boylan NJ, Chisholm J, Tang BC, Schuster BS, Henning A, Ensign LM, Lee E, Adstamongkonkul P, et al. Nanoparticles that do not adhere to mucus provide uniform and long‐lasting drug delivery to airways following inhalation. Sci Adv. 2017;3:e1601556. DOI: 10.1126/sciadv.1601556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu F, Lagares D, Choi KM, Stopfer L, Marinković A, Vrbanac V, Probst CK, Hiemer SE, Sisson TH, Horowitz JC, et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am J Physiol Lung Cell Mol Physiol. 2015;308:L344–L357. DOI: 10.1152/ajplung.00300.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ge J, Cui H, Xie N, Banerjee S, Guo S, Dubey S, Barnes S, Liu G. Glutaminolysis promotes collagen translation and stability via alpha‐ketoglutarate‐mediated mTOR activation and proline hydroxylation. Am J Respir Cell Mol Biol. 2018;58:378–390. DOI: 10.1165/rcmb.2017-0238OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lo Sardo F, Strano S, Blandino G. YAP and TAZ in lung cancer: oncogenic role and clinical targeting. Cancers (Basel). 2018;10:137. DOI: 10.3390/cancers10050137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Romero R, Sayin VI, Davidson SM, Bauer MR, Singh SX, LeBoeuf SE, Karakousi TR, Ellis DC, Bhutkar A, Sánchez‐Rivera FJ, et al. Keap1 loss promotes Kras‐driven lung cancer and results in dependence on glutaminolysis. Nat Med. 2017;23:1362–1368. DOI: 10.1038/nm.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]