

Abstract
Background
The opening of mitochondrial permeability transition pore and inflammation cooperatively progress myocardial ischemia‐reperfusion (IR) injury, which hampers therapeutic effects of primary reperfusion therapy for acute myocardial infarction. We examined the therapeutic effects of nanoparticle‐mediated medicine that simultaneously targets mitochondrial permeability transition pore and inflammation during IR injury.
Methods and Results
We used mice lacking cyclophilin D (CypD, a key molecule for mitochondrial permeability transition pore opening) and C‐C chemokine receptor 2 and found that CypD contributes to the progression of myocardial IR injury at early time point (30–45 minutes) after reperfusion, whereas C‐C chemokine receptor 2 contributes to IR injury at later time point (45–60 minutes) after reperfusion. Double deficiency of CypD and C‐C chemokine receptor 2 enhanced cardioprotection compared with single deficiency regardless of the durations of ischemia. Deletion of C‐C chemokine receptor 2, but not deletion of CypD, decreased the recruitment of Ly‐6Chigh monocytes after myocardial IR injury. In CypD‐knockout mice, administration of interleukin‐1β blocking antibody reduced the recruitment of these monocytes. Combined administration of polymeric nanoparticles composed of poly‐lactic/glycolic acid and encapsulating nanoparticles containing cyclosporine A or pitavastatin, which inhibit mitochondrial permeability transition pore opening and monocyte‐mediated inflammation, respectively, augmented the cardioprotection as compared with single administration of nanoparticles containing cyclosporine A or pitavastatin after myocardial IR injury.
Conclusions
Nanoparticle‐mediated simultaneous targeting of mitochondrial injury and inflammation could be a novel therapeutic strategy for the treatment of myocardial IR injury.
Keywords: cardioprotection, drug delivery system, ischemia‐reperfusion injury, nanotechnology
Subject Categories: Inflammation, Ischemia, Translational Studies
Nonstandard Abbreviations and Acronyms
- AAR
area at risk
- BMDM
bone marrow‐derived macrophages
- CCR2
C‐C chemokine receptor 2
- CsA‐NP
nanoparticles containing cyclosporine A
- CypD
cyclophilin D
- DDS
drug delivery system
- FITC‐NP
nanoparticles containing fluorescein‐isothiocyanate
- FMT
fluorescence molecular tomography
- IR
ischemia‐reperfusion
- MCP‐1/CCL2
monocyte chemoattractant protein‐1/C‐C motif chemokine 2
- mPTP
mitochondrial permeability transition pore
- Pitava‐NP
nanoparticles containing pitavastatin
- PLGA
poly‐lactic/glycolic acid
- ROS
reactive oxygen species
- TTC
2,3,5‐triphenyltetrazolium chloride
Clinical Perspective
What Is New?
Cyclophilin D‐dependent mitochondrial permeability transition pore opening significantly contributes to the infarct size in ischemia‐reperfusion injury with shorter durations of ischemia, whereas C‐C chemokine receptor 2‐mediated inflammation enlarges the infarct size in ischemia‐reperfusion injury with longer durations of ischemia.
Nanoparticle‐mediated simultaneous targeting of mitochondrial injury and monocyte‐mediated inflammation shows superior infarct‐sparing effects after myocardial ischemia‐reperfusion injury regardless of the duration of ischemia.
What Are the Clinical Implications?
Nanoparticle‐mediated delivery of cyclosporine A and statin may be a feasible and effective strategy to protect the heart from ischemia‐reperfusion injury in patients with acute myocardial infarction and undergone revascularization.
Timely restoration of coronary blood flow is currently the most effective therapy against acute myocardial infarction (MI).1 Recent heart failure pandemic, however, suggests that it is insufficient to just recanalize occluded arteries immediately after the onset of myocardial infarction.2 Restoration of blood flow to ischemic myocardium induces cell deaths and an enlargement of infarct area, which is known as ischemia‐reperfusion (IR) injury. Although hundreds of drugs have been investigated in animal experiments for their potential to reduce IR injury, clinical application of these drugs has not succeeded yet.3, 4, 5 The failure of clinical trial might be attributable to the complex pathophysiology of IR injury in which various factors are involved,1, 6 and, therefore, targeting only one mechanism of IR injury may be suboptimal. Reactive oxygen species (ROS), calcium overloads and rapid pH correction induce the opening of mitochondrial permeability transition pore (mPTP) within the first few minutes after reperfusion.6 The importance of mPTP is evident from previous reports showing that deletion of cyclophilin D (CypD), a key regulator of mPTP opening, decreases infarct size after myocardial IR injury in mice.7, 8 On the other hand, mitochondrial ROS, mitochondrial DNA, and phospholipid cardiolipin released from damaged mitochondria in necrotic tissues directly activate nucleotide‐binding oligomerization domain, leucine rich repeat, and pyrin domain containing 3 (NLRP3) inflammasome, and thus, interleukin‐1 beta (IL‐1β) expression is induced.9, 10, 11, 12 Recently, it is reported that IL‐1β enhances hematopoietic stem cell proliferation by direct actions on hematopoietic cells as well as modulation of hematopoietic microenvironment in bone marrow.13 Monocyte chemoattractant protein‐1/C‐C motif chemokine 2 induces chemotaxis of monocytes to the injured heart tissues via C‐C chemokine receptor 2 (CCR2) in the next 24 hours after MI.14 Excessive recruitments of Ly6Chigh activated monocytes are known to be harmful, and therapeutic targeting of CCR2 confers cardioprotection against IR injury.15, 16 To fulfil unmet clinical needs, we have developed novel therapeutics simultaneously targeting mitochondrial injury and inflammation, which may enhance cardioprotection against myocardial IR injury.
In terms of clinical application, one bottleneck is that the patient population is heterogeneous including durations of ischemia.17 Even aggressive interventions salvage only a part of endangered myocardial tissues after a long time of myocardial ischemia. Anti‐inflammatory therapy might be well suitable for such patients because enlargement of necrotic tissues exacerbates inflammation. In this study we, therefore, evaluated the feasibility of this strategy using an in vivo murine myocardial IR injury model with various durations of ischemia. Another possible barrier is insufficient local drug concentrations at the target cells or organelle when it is administered at the time of reperfusion. From a clinical perspective, it is essential to deliver drugs to target tissues, for example, by using a drug delivery system (DDS). We have recently developed a nanoparticle‐mediated DDS that uses bioabsorbable poly‐lactic/glycolic acid (PLGA) nanoparticles.18, 19, 20 Nano‐sized materials accumulate to the injured tissues, including the IR‐injured myocardium, where vascular permeability is enhanced.21, 22 We reported that intravenous administration of PLGA nanoparticles containing cyclosporine (CsA‐NP), a potent inhibitor of CypD, at the time of reperfusion increased the concentrations of cyclosporine in mitochondrial fraction of cardiomyocytes and protected the heart from myocardial IR injury.23 PLGA nanoparticles were also taken up by circulating monocytes after intravenous administration. We have reported that PLGA nanoparticles containing pitavastatin (Pitava‐NP) inhibited recruitment of Ly6Chigh activated monocytes by inhibiting monocyte chemoattractant protein‐1/C‐C motif chemokine 2‒mediated pathway and conferred cardioprotection against IR injury.24, 25 Therefore nanoparticle‐mediated DDS can be a promising therapeutic modality to simultaneously target mitochondria and monocytes in the IR‐injured heart.
Thus, the goals of this study were (1) to determine the contributions of mitochondrial injury (CypD) and inflammation (CCR2) in the myocardial IR injury; (2) to examine whether nanoparticle‐mediated simultaneous targeting of both mitochondrial injury and inflammation confer additive cardioprotection against myocardial IR injury.
METHODS
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Reagent
Evans Blue and 2,3,5‐triphenyltetrazolium chloride (TTC), collagenase type I, collagenase type XI, DNase I, hyaluronidase, Mito‐TEMPO, Adenosine 5'‐triphosphate (ATP) (Sigma Aldrich, St. Louis, MO), AnnexinVIvo750, ProSense680 (PerkinElmer, MA), VersaLyse Lysing solution (Beckman Coulter, Brea, CA), Fc receptor with anti‐CD16/32 monoclonal antibodies, Rat anti‐mouse CD90‐PE (53‐2.1), Rat anti‐mouse B220‐PE (RA3‐6B2), Rat anti‐mouse CD49b‐PE (DX5), Mouse anti‐mouse NK1.1‐PE (PK136), Rat anti‐mouse Ly6G‐PE (1A8), Rat anti‐CD11b‐APC (M1/70), Rat anti‐Ly6C‐FITC (AL‐21) (BD Biosciences, CA), non‐immune immunoglobulin G (mabg1‐ctrlm), neutralizing monoclonal mouse antibody against mouse IL‐1β (mabg1‐mil1b), lipopolysaccharide from Escherichia coli serotype 0111: B4 (InvivoGen, San Diego, CA), RPMI 1640, UltraGlutamine (Lonza, Basel, Switzerland), Fetal bovine serum, Macrophage colony‐stimulating factor (R&D Systems, MN) were purchased commercially.
Nanoparticle Preparation
PLGA, an average molecular weight of 20 000 and a copolymer ratio 75:25 lactide to glycolide (Wako Pure Chemical Industries Ltd., Osaka, Japan), was used as a matrix for the nanoparticles; polyvinyl alcohol (PVA‐403; Kuraray, Osaka, Japan) was used as a dispersing agent. PLGA nanoparticles incorporating a fluorescent marker fluorescein‐isothiocyanate (FITC; Dojin Chemical, Tokyo, Japan) (FITC‐NP), cyclosporine (Sigma Aldrich) (CsA‐NP) or pitavastatin (Kowa Pharmaceutical Co Ltd, Tokyo, Japan) (Pitava‐NP) were prepared by an emulsion solvent diffusion method as previously described.18, 19, 20 The FITC‐NP contained 4.06% (wt/vol) FITC, the CsA‐NP contained 2.67% (wt/vol) cyclosporine and the Pitava‐NP contained 12.0% (wt/vol) pitavastatin. The diameters of FITC‐NP, CsA‐NP, and Pitava‐NP were 231 nm, 175 nm, and 159 nm, respectively. The surface charges (zeta potential) were also analyzed using a Zetasizer Nano (Sysmex, Hyogo, Japan) and were −20.3 mV (FITC‐NP), −20.2 mV (CsA‐NP), and −4.0 mV (Pitava‐NP), respectively.
Mouse Myocardial IR Model
The study protocol was reviewed and approved by the committee on the Ethics of Animal Experiments, Kyushu University Faculty of Medicine and was conducted in accordance with the American Physiological Society guidelines and National Institutes of Health guidelines. Male wild‐type (WT) mice (C57Bl/6J background) were purchased from CLEA Japan, Inc. (Tokyo, Japan), and CypD‐knockout (CypD‐KO) mice were purchased from Jackson Laboratories (Stock#: 009071, Bar Harbor, ME). CCR2‐knockout (CCR2‐KO) mice (C57Bl/6J and 129/svJae hybrids)26 backcrossed with C57Bl/6J mice at least 10 times were used for experiments. The animals were maintained on a 12‐hour light‐dark cycle with free access to normal chow and water. The murine model of myocardial IR injury was prepared by previously described methods.27 Briefly, adult male mice (age: 10–12 weeks, body weight: 20–30 g) were anesthetized by an intraperitoneal injection of pentobarbital sodium (60 mg/kg) and maintained using 1% isoflurane with a ventilator after intubation. The heart was exposed after a left thoracotomy on a heated board. Transient myocardial ischemia was induced by a ligation of the anterior descending branch of the left anterior descending coronary artery using an 8‐0 silk suture with a silicon tube placed alongside the left anterior descending coronary artery. Regional ischemia was confirmed by ECG changes (ST elevation). A masked surgeon injected therapeutic factors at the time of reperfusion. The doses of CsA‐NP (PLGA that contained 1.0 mg/kg cyclosporine in 5.0 mL/kg saline) or Pitava‐NP (PLGA that contained 1.0 mg/kg pitavastatin in 5.0 mL/kg saline) were determined according to our previous reports.23, 24 The chest was closed and animals were allowed to recover from the surgery.
Determination of MI Size
At 24 hours after reperfusion, the animals were anesthetized and intubated, and the chest was opened. The left anterior descending coronary artery was re‐ligated with the same suture that had been left at the site of the ligation. To demarcate the ischemic area at risk (AAR), 2% Evans blue dye (Sigma Aldrich) was injected via the inferior vena cava. The heart was then excised and sliced into sequential five 1.0‐mm‐thick cross sections. The sections were incubated with 2% 2,3,5‐triphenyltetrazolium chloride (TTC, Sigma Aldrich) in saline for 10 minutes at 37°C and were photographed with a stereomicroscope (HC‐2500, Nikon). The MI area (TTC‐negative, white), non‐MI area within the AAR (TTC‐positive/Evans blue‐negative, red), non‐ischemic area (TTC‐positive/Evans blue‐positive, purple), and AAR (Evans blue‐negative) were analyzed using ImageJ software (version 1.44, National Institutes of Health, http://rsb.info.nih.gov/ij/).
Experimental Protocols
Experimental Protocol 1
WT, CypD‐KO, CCR2‐KO, or double knockout (double‐KO) mice (n=32/strain) were divided into 4 groups and subjected to 30, 45, 60, or 90 minutes of ischemia followed by reperfusion. At 24 hours after reperfusion, animals were euthanized to examine the infarct size.
Experimental Protocol 2
WT, CypD‐KO, CCR2‐KO, or double‐KO mice were subjected to 45 minutes ischemia followed by reperfusion. Fluorescence molecular tomography (FMT) imaging (n=8/strain) or flow cytometric analysis (n=4/strain) were performed at 24 hours after reperfusion.
Experimental Protocol 3
WT mice were subjected to 30‐ or 60‐minutes ischemia followed by reperfusion. The mice were divided into 5 groups (n=8–9/group); each group received an intravenous injection of the following drugs at the time of reperfusion: (1) vehicle (5.0 mL/kg saline), (2) FITC‐NP (1.4 mg PLGA in 5.0 mL/kg saline), (3) CsA‐NP (PLGA that contained 1.0 mg/kg cyclosporine in 5.0 mL/kg saline), (4) Pitava‐NP (PLGA that contained 1.0 mg/kg pitavastatin in 5.0 mL/kg saline), or cocktail containing CsA/Pitava‐NP (PLGA that contained 1.0 mg/kg CsA and PLGA that contained 1.0 mg/kg pitavastatin in 5.0 mL/kg saline). The animals were euthanized 24 hours after reperfusion to examine the infarct size.
Experimental Protocol 4
CypD‐KO or CCR2‐KO (n=24/strain) mice were subjected to 45 minutes of ischemia followed by reperfusion. These mice were divided into 3 groups and injected following agents at the time of reperfusion: (1) vehicle (5.0 mL/kg saline), (2) CsA‐NP (PLGA that contained 1.0 mg/kg cyclosporine in 5.0 mL/kg saline), or (3) Pitava‐NP (PLGA that contained 1.0 mg/kg pitavastatin in 5.0 mL/kg saline). The animals were euthanized 24 hours later to quantify the infarct size.
Flow Cytometry
The peripheral blood was drawn by cardiac puncture, and erythrocytes were lysed using VersaLyse Lysing solution (Beckman Coulter, Brea, CA) for 10 minutes at room temperature. The hearts were removed and digested using a cocktail composed of 450 U/mL collagenase type I, 125 U/mL collagenase type XI, 60 U/mL DNase I and 60 U/mL hyaluronidase (all enzymes were from Sigma‐Aldrich) in phosphate‐buffered saline that contained 20 mM 4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid at 37°C for 1 hour. The cell suspension was centrifuged at 300×g and 4°C for 5 minutes. After blocking the Fc receptor with anti‐CD16/32 monoclonal antibodies (BD Biosciences, CA) for 5 minutes at 4°C, the cell suspensions were incubated with a cocktail composed of monoclonal antibodies against T cells (CD90‐PE, 53‐2.1), B cells (B220‐PE, RA3‐6B2), NK cells (CD49b‐PE, DX5, and NK1.1‐PE, PK136), granulocytes (Ly6G‐PE, 1A8), myeloid cells (CD11b‐APC, M1/70), and monocyte subsets (Ly6C‐FITC, AL‐21) (BD Biosciences) for 1 hour at 4°C. All leukocytes were then analyzed using FACS Gallios (BD Biosciences). The leukocytes were also incubated with the appropriate isotype control (BD Biosciences). The monocytes/macrophages were identified as CD11bhigh (CD90/B220/CD49b/NK1.1/Ly6G)low Ly6Chigh/low, the neutrophils were identified as CD11bhigh (CD90/B220/CD49b/NK1.1/Ly6G)high, and the lymphocytes were identified as CD11blow (CD90/B220/CD49b/NK1.1/Ly6G)high as previously described.28
Fluorescence Molecular Tomography
Five nanomoles of pan‐cathepsin protease sensor, ProSence 680 (Ex/Em=680/700 nm, PerkinElmer, MA) and 2 nmol of cellular death sensor, Annexin‐Vivo 750 (Ex/Em=745/800 nm) (PerkinElmer) were intravenously administered at the time of reperfusion and 22 hours after reperfusion, respectively. Twenty‐four hours after reperfusion, the animals were scanned using an FMT‐2000 system (PerkinElmer). Mice were anesthetized by inhalation of 1.0%–1.5% isoflurane and placed in the supine position. The FMT imaging chamber was maintained at 37℃ and fluorescent signals were detected by a charge‐coupled device (CCD) camera. The collected FMT data were reconstructed by FMT‐2000 system software. Three‐dimensional region of interest for the heart was positioned based on a previous report.29 The total amount of fluorescence in the heart was calculated by FMT‐2000 system software.
Measurement of IL‐1β Protein in the Myocardial Tissue
WT, CypD‐KO, CCR2‐KO, or double‐KO mice were subjected to 30‐ or 60‐minutes ischemia followed by reperfusion. Myocardial tissue samples were collected at 24 hours after reperfusion. IL‐1β protein levels in myocardial tissue lysates were measured by ELISA (R&D systems, NE) according to the manufacturer's instruction.
Administration of Neutralizing Antibody Against IL‐1β
For systemic neutralization of IL‐1β in CypD‐knockout mice, we conducted intraperitoneal administration of non‐immune immunoglobulin G (2 mg/kg; mabg1‐ctrlm; InvivoGen, San Diego, CA) or neutralizing monoclonal mouse antibody against mouse IL‐1β (2 mg/kg; mabg1‐mil1b; InvivoGen) 2 hours before induction of myocardial ischemia. Then mice were subjected to 60 minutes of myocardial ischemia followed by reperfusion. Flow cytometry was performed 24 hours after reperfusion and then, infarct size was quantified.
In Vitro Experiments
Bone marrow‐derived macrophages (BMDM) were obtained by culturing bone marrow cells isolated from 8‐week‐old C57BL/6J mice in RPMI 1640 (Lonza, Basel, Switzerland) supplemented with 20% fetal bovine serum, 40 ng/mL macrophage colony‐stimulating factor (R&D Systems, MN) and 2 mM UltraGlutamine (Lonza) at 37°C in a 5% CO2 environment. Six days later, the BMDMs were collected and used for experiments. BMDMs from WT or CypD‐KO mice were primed with 50 ng/mL lipopolysaccharide from Escherichia coli serotype 0111: B4 (InvivoGen) for 4 hours before stimulation. BMDM were pre‐treated with the antioxidant Mito‐TEMPO (500 mM) for 15 minutes before stimulation with adenosine triphosphate (ATP) (2 mM). Culture supernatants were collected 1 hour later to measure IL‐1β, IL‐18 and tumor necrosis factor‐α protein levels by ELISA.
Statistical Analysis
The data are expressed as the mean±SD. Differences between 2 groups were analyzed by unpaired t‐tests; the differences among 3 groups or more were analyzed by an ANOVA and post‐hoc Bonferroni's multiple comparison tests with Prism Software version 4.0 (Graph Pad Software, CA). P values <0.05 were considered statistically significant.
RESULTS
Double Deficiency of CypD and CCR2 Enhanced Cardioprotection Compared With Single Deficiency Against Myocardial IR Injury
We examined infarct size in mice subjected to 30, 45, 60 or 90 minutes of coronary occlusion followed by 24 hours of reperfusion. Compared with WT mice, CypD‐KO mice showed significantly smaller infarct size after IR injury with 30 or 45 minutes of ischemia (Figure 1A and 1B), whereas no difference was observed after IR injury with 60 or 90 minutes of ischemia (Figure 1C and 1D). CCR2‐KO mice showed significant reduction in infarct size after IR injury with 45 or 60 minutes of ischemia. These findings suggested that the contributions of CypD‐dependent mPTP opening (the infarct‐sparing effects of CypD deficiency) to the infarct size are more significant in IR injury with shorter durations of ischemia, whereas those of CCR2‐mediated inflammation are larger in IR injury with longer durations of ischemia (Figure 1E). To examine whether double deficiency of CypD and CCR2 confer greater cardioprotection, we generated mice deficient with both CypD and CCR2 (double‐KO). In these double‐KO mice, a significant reduction in infarct size was consistently observed across IR injury models with different ischemia times, as compared with WT mice (Figure 1E). Notably, double deficiency conferred the greatest cardioprotection against IR injury with 45 minutes of ischemia. The percentage of AAR in the left ventricle was comparable between all study groups (Figure S1A). These data indicated that simultaneous targeting of 2 mechanisms (i.e., CypD‐dependent mPTP opening and CCR2‐mediated inflammation) could be more effective regardless of the durations of ischemia.
Figure 1. Double deficiency of cyclophilin D and CCR2 showed additive cardioprotection over single deficiency against ischemia‐reperfusion injury.
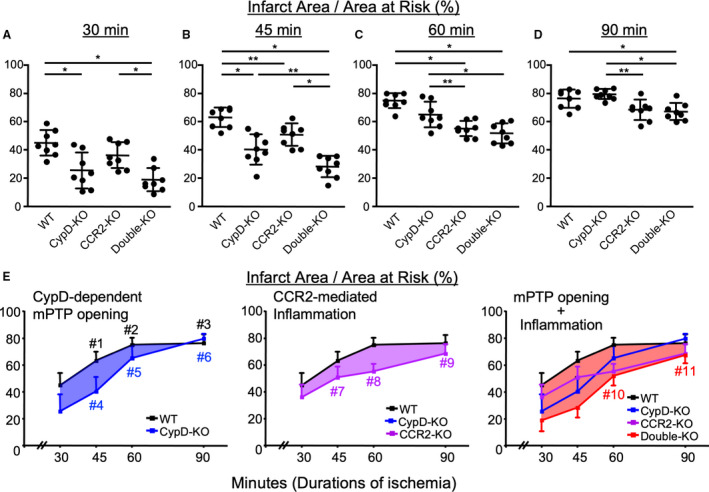
A‐D, Ratio of infarct size/area at risk in mice subjected to 30, 45, 60, or 90 minutes of myocardial ischemia followed by 24 hours of reperfusion. The horizontal lines represent mean±SD (n=8 per group). *P<0.01, **P < 0.05, 1‐way ANOVA. E, Relationship between the duration of ischemia and infarct size. Redrawn from the data in Figure 1A through 1D. Error bars indicate SD. #1, P<0.0001 vs 30 minutes, #2; P<0.0001 vs 30 minutes, P<0.05 vs 45 minutes; #3, P<0.0001 vs 30 minutes, P<0.001 vs 45 minutes; #4 P<0.01 vs 30 minutes; #5, P<0.0001 vs 30 minutes, P<0.0001 vs 45 minutes; #6, P<0.0001 vs 30 minutes, P<0.0001 vs 45 minutes, P<0.001 vs 60 minutes; #7, P<0.01 vs 30 minutes; #8, P<0.0001 vs 30 minutes; #9, P<0.0001 vs 30 minutes, P<0.001 vs 45 minutes, P<0.01 vs 60 minutes; #10, P<0.0001 vs 30 minutes, P<0.0001 vs 45 minutes; #11, P<0.0001 vs 30 minutes, P<0.0001 vs 45 minutes, P<0.001 vs 60 minutes, 2‐way ANOVA. CCR2‐KO indicates C‐C chemokine receptor 2‐knockout mice; CypD‐KO, cyclophilin D‐knockout mice; Double‐KO, cyclophilin‐D/C‐C chemokine receptor 2‐double knockout mice; mPTP, mitochondrial permeability transition pore; and WT indicates wild‐type mice.
Deletion of CCR2 Inhibits Recruitment of Ly6Chigh Activated Monocytes, While Deletion of CypD Shows Paradoxical Inflammation After IR Injury
To evaluate cardiomyocyte deaths and inflammation, we have used AnnexinVivo750 that detects the phosphatidylserine exposed in the outer leaflet of the plasma membrane, and ProSense680 that detects protease activities in inflammatory cells. Dual channel FMT imaging visualized near‐infrared radiation emitted by these probes in the IR‐injured heart (Figure 2A). Cell death determined by AnnexinVivo750 was decreased by deletion of CypD and/or CCR2, whereas inflammation determined by ProSense680 was decreased only by deletion of CCR2. Flow cytometry of myocardial tissues confirmed that CypD deficiency significantly increased the recruitment of monocytes as compared with WT (Figure 2B). In contrast, CCR2‐KO or double‐KO showed significantly decreased Ly6Chigh monocytes. It has been reported that activated monocytes/macrophages secrete IL‐1β that maintains the persistent recruitment of monocytes into injured tissues by enhancing proliferation of hematopoietic stem cells.13 We found that IL‐1β protein levels in the IR‐injured heart in CypD‐KO mice were significantly increased after 60 minutes of ischemia compared with WT mice. CCR2‐KO or double‐KO mice showed marked reduction in IL‐1β protein levels (Figure 2C). These data suggest that IL‐1β production is paradoxically enhanced in CypD‐KO mice, which worsen the recruitment of Ly6Chigh monocytes into the heart and exacerbate IR injury. These data suggest that simultaneous targeting of CypD and CCR2 complementary confers cardioprotection after myocardial IR injury.
Figure 2. Inflammation was attenuated in CCR2‐KO or double‐KO, while CypD‐KO showed residual inflammation.
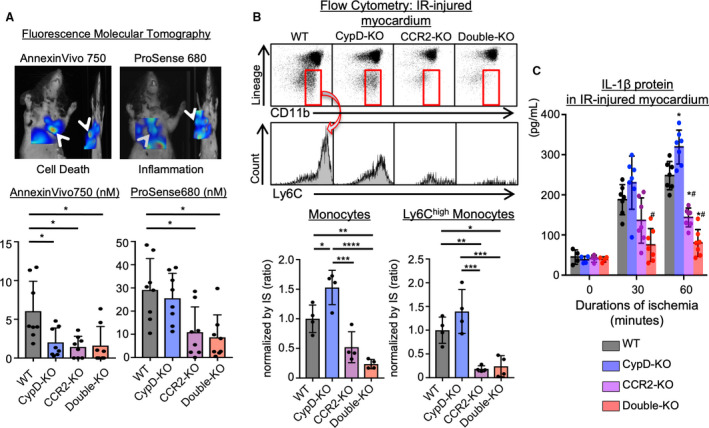
A, Dual channel fluorescence molecular tomography demonstrating cell death (AnnexinVivo750) and protease activity (ProSense680) after ischemia‐reperfusion injury. Bar graphs show quantitative data represented as mean±SD (n=8 per group). *P<0.01, 1‐way ANOVA. B, Ischemia‐reperfusion‒injured myocardium was subjected to flow cytometric analysis. The numbers of monocytes were normalized by IS. Ly6Chigh activated monocytes were decreased in CCR2‐KO or double‐KO mice, whereas CypD‐KO showed larger numbers of total monocytes. The data are mean±SD (n=4 per group). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, 1‐way ANOVA. *P<0.05 vs wild‐type, #P<0.05 vs CypD‐knockout, 1‐way ANOVA. C, IL‐1β protein levels in the ischemia‐reperfusion‒injured myocardium measured by ELISA. The data are mean±SD (n=4–7 per group). CCR2‐KO indicates C‐C chemokine receptor 2‐knockout mice; CypD‐KO, cyclophilin D‐knockout mice; Double‐KO, cyclophilin‐D/C‐C chemokine receptor 2‐double knockout mice; IL‐1β, interleukin‐1β; IR, ischemia‐reperfusion; IS, infarct size; and WT indicates wild‐type mice.
NLRP3 Inflammasome Is More Sensitive to Activation in CypD‐Knockout Macrophages
To examine the mechanism by which CypD deficiency increased IL‐1β expression, we used BMDMs. As previously reported,30 treatments with ATP induced IL‐1β and IL‐18 secretion through activation of NLRP3 inflammasome in lipopolysaccharide‐primed BMDMs from WT mice (Figure 3A and 3B). Of note, BMDMs from CypD‐KO mice showed excessive secretion of IL‐1β and IL18. In contrast, there were no significant differences in tumor necrosis factor‐α that is released by NLRP3‐independent mechanisms (Figure 3C). Since mitochondria‐derived ROS is involved in the activation of NLRP3 inflammasome, we pre‐treated BMDMs with mitochondria‐targeted antioxidant Mito‐TEMPO. As previously reported,12 Mito‐TEMPO inhibited ATP‐mediated IL‐1β and IL‐18 secretion in BMDMs from WT mice (Figure 3A and 3B). In contrast, Mito‐TEMPO failed to inhibit IL‐1β and IL‐18 secretion in BMDM from CypD‐KO. These data suggested that NLRP3 inflammasome is more sensitive to activation in CypD‐KO macrophages.
Figure 3. Deletion of cyclophilin D augmented releases of IL‐1β and IL‐18 in bone marrow‐derived macrophages.
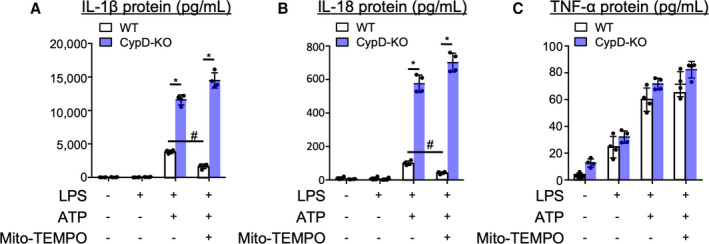
A through C, Bone marrow‐derived macrophages were pre‐treated with the antioxidant Mito‐TEMPO (500 mM) for 15 minutes before stimulation with ATP (2 mM). Culture supernatants were collected 1 hour later and IL‐1β, IL‐18 and TNF‐α protein levels were measured by ELISA. The data are mean±SD (n=4 per group). CypD‐KO indicates cyclophilin D‐knockout mice; IL‐1β, interleukin‐1β; IL‐18, interleukin‐18; TNF‐α, tumor necrosis factor‐α; and WT; wild‐type mice. *P<0.001, #P<0.001, 2‐way ANOVA.
Neutralizing IL‐1β Dampens the Recruitment of Ly6Chigh Monocytes to the IR‐Injured Heart and Reduces the Infarct Size in CypD‐KO Mice
To confirm the role of IL‐1β in the paradoxical inflammation and the progression of IR injury in CypD‐KO mice, we next examined the effects of IL‐1β neutralization on myocardial inflammation and the infarct size in CypD‐KO mice with 60 minutes of ischemia followed by 24 hours of reperfusion. Ly6Chigh monocytes in the heart after IR injury were significantly decreased by IL‐1β neutralizing antibody compared with control immunoglobulin G (Figure 4A). Antibody treatment significantly reduced the infarct size in WT‐ or CypD‐KO mice with long duration (60 minutes) of ischemia, whereas IL‐1β neutralizing antibody had no effects in CCR2‐KO mice (Figure 4B). These data suggest that the effect of IL‐1β was exerted by CCR2+ monocyte‐mediated mechanisms, and that CypD deficiency increased IL‐1β expression, which worsened inflammation and enlarged the infarct size in mice with long durations of myocardial ischemia.
Figure 4. The effects of IL‐1β neutralizing antibody on myocardial inflammation and infarct size.
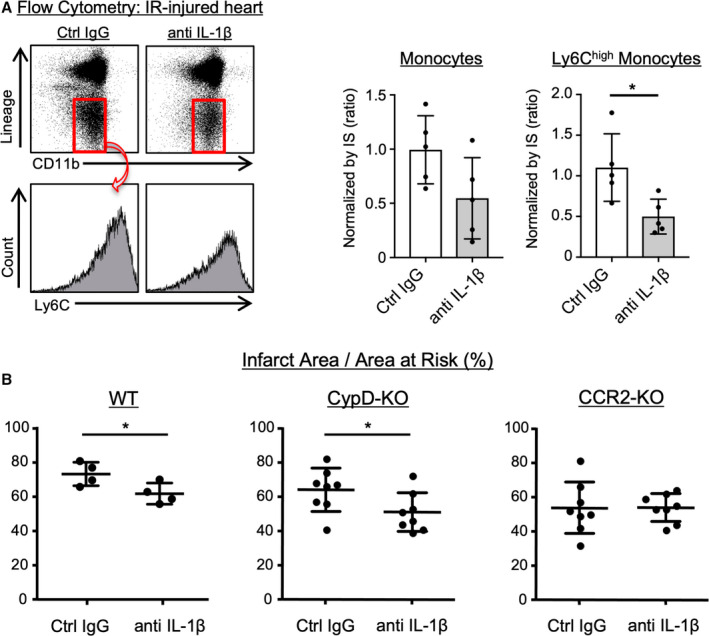
*P<0.05, unpaired t tests. A, IR‐injured hearts harvested from CypD‐KO mice were subjected to flow cytometric analysis. The number of monocytes were normalized by IS. The data are mean±SD (n=5 per group). *P<0.05, unpaired t tests. B, The therapeutic effects of IL‐1β neutralizing antibody on the infarct size in WT, CypD‐KO or CCR2‐KO mice subjected to 60 minutes of myocardial ischemia followed by 24 hours of reperfusion. The horizontal lines represent mean±SD (n=4–8 per group). CCR2‐KO indicates C‐C chemokine receptor 2‐knockout mice; Ctrl, control; CypD‐KO, cyclophilin D‐knockout mice; IgG, immunoglobulin G; IL‐1β, interleukin‐1β; IR, ischemia‐reperfusion; IS, infarct size; and WT indicates wild‐type mice.
Nanoparticle‐Mediated Targeting of Both CypD and CCR2 Confers Additive Cardioprotection Against IR Injury
We have engineered nanoparticle‐mediated DDS and reported that intravenous administration of PLGA CsA‐NP containing 1.0 mg/kg cyclosporine, a potent inhibitor of CypD, reduced myocardial IR injury.23 We also reported that Pitava‐NP containing 1.0 mg/kg pitavastatin inhibited recruitment of Ly6Chigh activated monocytes through inhibition of monocyte chemoattractant protein‐1/C‐C motif chemokine 2‒mediated pathway and conferred cardioprotection against IR injury in preclinical animal models.24, 25 Here, we have tested the efficacy of nano‐DDS which simultaneously targets mitochondrial injury and monocyte‐mediated inflammation. WT mice were subjected to short duration (30 minutes) or long duration (60 minutes) of myocardial ischemia followed by 24 hours of reperfusion. At the time of reperfusion, saline or nanoparticles containing FITC (FITC‐NP), CsA‐NP containing 1.0 mg/kg cyclosporine, Pitava‐NP containing 1.0 mg/kg pitavastatin, or cocktail of CsA‐NP and Pitava‐NP were intravenously injected. In mice with 30 minutes of ischemia, administration of CsA‐NP or Pitava‐NP significantly reduced infarct size (Figure 5A and 5B). Combined administration of CsA‐NP and Pitava‐NP further decreased the infarct size. To ensure the mechanisms by which CsA‐NP or Pitava‐NP confer cardioprotection, we examine the therapeutic effects of these nanoparticles in CypD‐KO and CCR2‐KO mice. In CypD‐KO mice, CsA‐NP did not show therapeutic effects, whereas Pitava‐NP reduced the infarct size (Figure 5C). In contrast, therapeutic effects of Pitava‐NP were abolished in CCR2‐KO mice, whereas CsA‐NP reduced the infarct size. These data suggest that cardioprotective effects of CsA‐NP depend on mPTP opening regulated by CypD, whereas those of Pitava‐NP depend on CCR2‐mediated recruitment of activated monocytes. The percentage of AAR in the left ventricle was comparable between all study groups (Figure S1B and 1C).
Figure 5. Nanoparticle‐mediated simultaneous targeting of CypD and C‐C chemokine receptor 2 confer superior cardioprotection against IR injury.
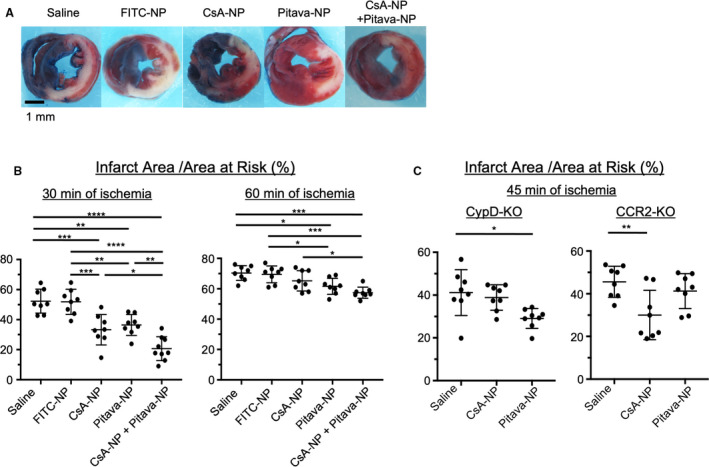
*P<0.05, **P<0.01, 1‐way ANOVA. A, Gross appearance of left ventricle myocardial sections after Evans blue and triphenyltetrazolium chloride staining. B, The therapeutic effects of CsA‐NP, Pitava‐NP or CsA/Pitava‐NP on the infarct size in wild‐type mice with 30 or 60 minutes of ischemia followed by 24 hours of reperfusion. The data represent the mean±SD (n=8–9 per group). *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, 1‐way ANOVA. C, The infarct‐sparing effects of CsA‐NP or Pitava‐NP in CypD‐knockout or CCR2‐knockout with 45 minutes of ischemia followed by 24 hours of reperfusion. The horizontal lines represent mean±SD (n=8 per group). CCR2‐KO indicates C‐C chemokine receptor 2‐knockout mice; CypD‐KO, cyclophilin D‐knockout mice; CsA‐NP, nanoparticles containing Cyclosporine A; FITC‐NP, nanoparticles containing fluorescein‐isothiocyanate; IR, ischemia‐reperfusion; Pitava‐NP, nanoparticles containing pitavastatin.
Combined Administration of CsA‐NP and Pitava‐NP Reduces Cell Death and Inflammation
We next examined the effects of nanoparticles on inflammation in the IR‐injured heart. Flow cytometric analysis revealed that combined administration of CsA‐NP and Pitava‐NP decreased recruitment of Ly‐6Chigh monocytes to the heart after IR injury as compared with control group (Figure 6A). At protein level, administration of CsA‐NP did not decrease IL‐1β concentration measured at 24 hours after reperfusion in the heart exposed to 45 minutes ischemia. In contrast, combined administration of CsA‐NP and Pitava‐NP significantly decreased IL‐1β protein (Figure 6B). Dual channel FMT demonstrated that near‐infrared signals emitted by AnnexinVivo750, indicating cell deaths, were decreased in CsA‐NP treated mice (Figure 6C). Again, protease activities were comparable between saline and CsA‐NP group. Administration of Pitava‐NP or combined administration of CsA‐NP and Pitava‐NP significantly decreased both cell deaths and protease activities. Taken together, these data suggested that nanoparticle‐mediated DDS targets both mitochondrial injury and monocytes‐mediated inflammation at the same time in the IR‐injured myocardium, and simultaneous administration of CsA‐NP and Pitava‐NP complementarily confers cardioprotection against IR injury.
Figure 6. The effects of CsA‐NP or Pitava‐NP on inflammation.
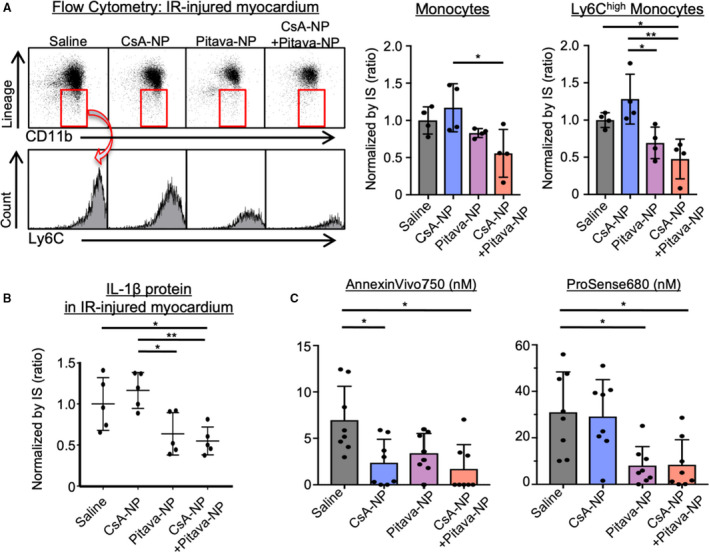
*P<0.05, 1‐way ANOVA. A, Ischemia‐reperfusion‒injured myocardium was subjected to flow cytometry. The number of monocytes were normalized by IS. Ly6Chigh activated monocytes were decreased in CsA/Pitava‐NP treated mice. The data are mean±SD (n=4 per group). *P<0.05, **P<0.01, 1‐way ANOVA. B, IL‐1β protein levels measured 24 hours after reperfusion and normalized by infarct size. The horizontal lines represent mean±SD (n=5 per group). *P<0.05, **P<0.01, 1‐way ANOVA. C, Dual channel FMT imaging revealed that CsA‐NP reduced cell death (AnnexinVivo750), whereas protease activities (ProSense680) were comparable with saline group. Treatment with Pitava‐NP or CsA/Pitava‐NP reduced both Annexin750 and ProSense680 signals. The data are mean±SD (n=7–8 per group). CsA‐NP, nanoparticles containing Cyclosporine A; FMT, fluorescence molecular tomography; IL‐1β, interleukin‐1β; IR, ischemia‐reperfusion; IS, infarct size; Pitava‐NP, nanoparticles containing pitavastatin.
DISCUSSION
The novel findings herein are as follows; (1) blockade of CypD‐mediated mPTP opening is insufficient for cardioprotection after long duration of ischemia; (2) additional blockade of CCR2 abrogated monocyte‐mediated inflammation and enhanced cardioprotection after long duration of ischemia in CypD‐knockout mice; (3) nanoparticle‐mediated simultaneous targeting of mitochondrial injury and monocyte‐mediated inflammation showed potent infarct‐sparing effects after myocardial IR injury.
The role of CypD in the mechanisms of myocardial IR injury has been already reported in experimental animals.7, 8, 31 In clinical trials, however, therapeutic efficacy of CsA has not been established. For example, in the CIRCUS trial (Cyclosporine to Improve Clinical Outcome in ST‐Elevation Myocardial Infarction Patients), in which patients referred to coronary intervention because of acute anterior MI were enrolled, intravenous administration of 2.5 mg/kg CsA (395 patients) failed to improve clinical outcomes and prevent adverse left ventricular remodeling evaluated at 1 year after the treatment.32 This negative result may be explained by heterogeneity of patients including duration of ischemia. In the CIRCUS trial, patients within 12 hours after the onset of symptoms were enrolled, and >87.5% of patients were supposed to have at least 2 hours of myocardial ischemia. We found in mice that the relative importance of mPTP opening decreases as the duration of ischemia becomes longer (>90 minutes) (Figure 1E). Therefore, additional therapies that complementarily confer cardioprotection against IR injury after long duration of ischemia are needed. Here, we have demonstrated that deletion of CCR2 decreased infarct size in CypD‐KO mice even 90 minutes after reperfusion (Figure 1E), suggesting that therapeutic strategy that simultaneously targets CypD and CCR2 is promising. Another possibility why the CIRCUS trial could not show therapeutic efficacy is insufficient CsA concentration in the heart, especially in mitochondria of cardiomyocytes. To overcome this point, we have developed nano‐DDS composed of bioabsorbable PLGA nanoparticles. In preclinical animal models, we have demonstrated that PLGA nanoparticles are delivered to myocardial tissues and inflammatory monocytes after myocardial IR injury.23, 24, 28 In addition, we have shown that intravenous administration of CsA‐NP at the time of reperfusion increases the concentrations of cyclosporine in mitochondrial fractions of cardiomyocytes and decreased infarct size after myocardial IR injury.23 We have also shown that Pitava‐NP inhibited recruitment of monocytes and conferred cardioprotection against IR injury.24 Therefore, nanoparticle‐mediated DDS could be a promising therapeutic modality which simultaneously targets mitochondria in the injured myocardium and monocytes.
We found that the protective effects of CypD inhibition were compromised in IR injury with long duration of ischemia. Myocardial tissue IL‐1β levels were significantly increased in CypD‐deficient mice after IR injury with 60 minutes ischemia (Figure 2C), suggesting that the activation of paradoxical inflammasome may counteract the infarct‐sparing effects of CypD inhibition (Figure 1). Other mitochondrial mechanisms may also compromise the beneficial effects of CypD inhibition. It is reported that mPTP opening is still observed in CypD‐deficient cells at substantially higher concentration of calcium and oxidants, suggesting that mPTP may open via CypD‐independent mechanisms.7, 33 In addition, IR injury is known to induce apoptosis via B‐cell lymphoma 2 (BCL2)‐associated X protein/BCL2 homologous antagonist/killer (BAX/BAK)‐dependent mitochondrial outer membrane permeabilization, a CypD‐independent mitochondrial process.34 BAX/BAK‐double KO mice showed smaller infarct size in mice myocardial IR injury model, suggesting the importance of mitochondrial outer membrane permeabilization‐mediated apoptosis in IR injury.35 These findings suggest that the infarct‐sparing effects of CypD inhibition can be offset by CypD‐independent mitochondrial mechanisms in IR injury with long duration of ischemia. To confirm this, we may need to examine the effects of CypD inhibition on mitochondrial phenotypes such as mPTP opening, mitochondrial function, and apoptosis in IR injury with different durations of ischemia. However, it is extremely challenging to determine the exact contribution of CypD inhibition to mitochondrial phenotypes, because various processes may affect these phenotypes during IR injury. In addition, IR‐injured myocardium is highly heterogeneous and consists of various populations of cells with different degrees of injury, which makes the in vivo assessments of the mitochondrial phenotypes difficult.
The IR‐injured myocardium releases various danger signals including mitochondria‐related mediators, which activate NLRP3 inflammasome to produce IL‐1β and IL‐18. Therefore, inhibition of mitochondrial injury might suppress inflammation by inhibiting the supply of mitochondria‐related mediators to inflammatory cells. This notion was supported by a report that mitochondrial stabilization with cyclosporine reduced IL‐1β release after the treatment with various danger signals using lipopolysaccharide‐primed macrophages.12 Unexpectedly, we found that CypD deficiency significantly increased IL‐1β in the myocardium (Figure 2). Recruitment of Ly6Chigh monocytes and residual protease activities were observed 24 hours after IR injury in CypD‐KO mice. Mitochondria‐targeted CsA‐NP treatment showed trends toward increased IL‐1β levels and the numbers of monocytes in myocardial tissues (Figure 6A and 6B). These findings suggested that inhibition of CypD‐dependent mPTP opening induces inflammatory response. We found that BMDM from CypD‐KO mice showed excessive IL‐1β and IL‐18 release in a mitochondria‐derived ROS independent manner (Figure 3). Although the precise mechanism remains to be elucidated, we consider that CypD inhibition promotes the NLRP3 inflammasome activation through accumulation of damaged mitochondria that escaped from autophagic clearance. It has been reported that Parkin, a key molecule of mitochondrial autophagy (mitophagy), translocates to mitochondria upon dissipation of the mitochondrial membrane potential induced by mPTP opening.36 Genetic or pharmacological inhibition mPTP opening in cardiomyocytes abrogate mitophagy during IR injury, resulting in the accumulation of the damaged mitochondria, which may activate the NLRP3 inflammasome.37 The significance of mitophagy during IR injury is evident from the report that deletion of dynamin‐related protein 1 (Drp1), a key molecule for mitochondrial division, suppressed mitophagy in the myocardium, and myocardial Drp1‐knockout mice showed greater infarct size after IR injury.38 In the present study, we have demonstrated that blockade of CCR2 abrogated residual inflammation and decreased the infarct size in CypD‐KO mice, suggesting the importance of simultaneous targeting of mPTP opening and inflammation.
Oxidative stress is a major detrimental factor in the IR injury.1 CypD may have 2 opposite effects on oxidative stress. CypD regulates mPTP opening and CypD inhibition may protect mitochondria and reduce oxidative stress via inhibition of mPTP opening.7, 8 On the other hand, CypD inhibition may hamper mitophagy and facilitates the accumulation of ROS‐generating mitochondria. In this study, CypD‐KO mice showed smaller infarct size in IR injury with shorter durations of ischemia, while the paradoxical inflammasome activation was observed and the infarct‐sparing effects of CypD‐KO was compromised in longer durations of ischemia (Figure 1 and 2C). These data suggested that CypD inhibition reduces oxidative stress in IR injury with short duration of ischemia, whereas detrimental effects of CypD‐inhibition on mitochondrial quality control may increase oxidative stress in IR injury with longer durations of ischemia. Monocytes/macrophages are primarily involved in the myocardial IR injury through monocyte chemoattractant protein‐1/C‐C motif chemokine 2 mechanism.15, 19 These leukocytes actively release ROS, resulting in increased oxidative stress in IR‐injured tissues. Consistent with this, CCR2 inhibition is reported to reduce oxidative stress in IR‐injured tissues.16
Intravenously injected PLGA nanoparticles accumulated in monocytes/macrophage and cardiomyocytes. Pitava‐NP inhibit migration of monocytes/macrophages, resulting in anti‐inflammation. On the other hand, Pitava‐NP prevented cardiomyocytes death via activation of PI3K/Akt pathway.24 Therefore, we expected that the therapeutic effects of Pitava‐NP that act through 2 different mechanisms may show superior benefit over anti‐IL‐1β therapy. We, however, found that Pitava‐NP and anti‐IL‐1β therapy showed similar therapeutic effects in IR injury with 60 minutes ischemia (61.6% versus 61.3%, Figure 4B and 5B). These results might be attributed to the difference in how the drugs were administered. In this study, we intravenously injected Pitava‐NP just after reperfusion, whereas anti‐IL‐1β antibody were intraperitoneally injected 2 hours before the induction of myocardial ischemia. Future studies of intravenous injection of Pitava‐NP versus anti‐IL‐1β antibody in large animal models would elucidate the optimal combinations of therapeutics for this strategy.
From the perspective of clinical translation, PLGA nanoparticles have several advantages. PLGA has been already used as a drug carrier with biosafety approval for human use by the US Food and Drug Administration,39, 40 the European Medicine Agency and the Japanese regulatory agency (PMDA). We have already conducted a phase I/IIa clinical trial in patients with critical limb ischemia at Kyushu University Hospital (UMIN000008011) to investigate the safety and efficacy of intramuscular injections of PLGA nanoparticles incorporated with pitavastatin.41 In addition, we have also completed a phase I clinical trial (UMIN000014940) to investigate the safety of a single and repeated intravenous injections of PLGA nanoparticles incorporated with pitavastatin in healthy volunteers.42 Based on the result of this phase I trial, we are now conducting a phase II clinical trial to test the safety and efficacy of intravenous administration of Pitava‐NP in patients with pulmonary arterial hypertension.
In conclusion, simultaneous targeting of CypD‐mediated mPTP opening and inflammation enhanced cardioprotection after myocardial IR injury. PLGA nanoparticle‐mediated delivery of CsA and pitavastatin to the heart after IR injury could be a clinical feasible and effective strategy to protect the heart from IR injury in patients with acute myocardial infarction and undergone revascularization.
Sources of Funding
This study was supported by grants from the Ministry of Education, Science, and Culture, Tokyo, Japan (JSPS KAKENHI Grant Number 17K09590 to T.M., and 25293185 to K.E.) and intractable diseases overcome research project from the Japan Agency for Medical Research and Development, AMED (to K.E.).
Disclosures
None.
Supporting information
Figure S1
Acknowledgments
We thank Kozue Nakamura, Eiko Iwata, and Satomi Abe for their technical assistance.
(J Am Heart Assoc. 2021;10:e019521. DOI: 10.1161/JAHA.120.019521.)
Supplementary Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.120.019521
For Sources of Funding and Disclosures, see page 13.
References
- 1.Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–1135. DOI: 10.1056/NEJMra071667. [DOI] [PubMed] [Google Scholar]
- 2.Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS, Falk V, González‐Juanatey JR, Harjola V‐P, Jankowska EA, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: the task force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2016;37:2129–2200. DOI: 10.1093/eurheartj/ehw128. [DOI] [PubMed] [Google Scholar]
- 3.Dirksen MT, Laarman GJ, Simoons ML, Duncker DJ. Reperfusion injury in humans: a review of clinical trials on reperfusion injury inhibitory strategies. Cardiovasc Res. 2007;74:343–355. DOI: 10.1016/j.cardiores.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 4.Gerczuk PZ, Kloner RA. An update on cardioprotection: a review of the latest adjunctive therapies to limit myocardial infarction size in clinical trials. J Am Coll Cardiol. 2012;59:969–978. DOI: 10.1016/j.jacc.2011.07.054. [DOI] [PubMed] [Google Scholar]
- 5.Miura T, Miki T. Limitation of myocardial infarct size in the clinical setting: current status and challenges in translating animal experiments into clinical therapy. Basic Res Cardiol. 2008;103:501–513. DOI: 10.1007/s00395-008-0743-y. [DOI] [PubMed] [Google Scholar]
- 6.Prasad A, Stone GW, Holmes DR, Gersh B. Reperfusion injury, microvascular dysfunction, and cardioprotection: the "dark side" of reperfusion. Circulation. 2009;120:2105–2112. DOI: 10.1161/CIRCULATIONAHA.108.814640. [DOI] [PubMed] [Google Scholar]
- 7.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. DOI: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 8.Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D‐dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–658. DOI: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 9.Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013–1022. DOI: 10.1016/j.cell.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 10.Wen H, Miao EA, Ting JP. Mechanisms of NOD‐like receptor‐associated inflammasome activation. Immunity. 2013;39:432–441. DOI: 10.1016/j.immuni.2013.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shimada K, Crother T, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan V, Wolf A, Vergnes L, Ojcius D, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36:401–414. DOI: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iyer S, He Q, Janczy J, Elliott E, Zhong Z, Olivier A, Sadler J, Knepper‐Adrian V, Han R, Qiao L, et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity. 2013;39:311–323. DOI: 10.1016/j.immuni.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sager HB, Heidt T, Hulsmans M, Dutta P, Courties G, Sebas M, Wojtkiewicz GR, Tricot B, Iwamoto Y, Sun Y, et al. Targeting interleukin‐1beta reduces leukocyte production after acute myocardial infarction. Circulation. 2015;132:1880–1890. 10.1161/CIRCULATIONAHA.115.016160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R, Pittet MJ. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037–3047. DOI: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leuschner F, Dutta P, Gorbatov R, Novobrantseva TI, Donahoe JS, Courties G, Lee KM, Kim JI, Markmann JF, Marinelli B, et al. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat Biotechnol. 2011;29:1005–1010. DOI: 10.1038/nbt.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayasaki T, Kaikita K, Okuma T, Yamamoto E, Kuziel WA, Ogawa H, Takeya M. CC chemokine receptor‐2 deficiency attenuates oxidative stress and infarct size caused by myocardial ischemia‐reperfusion in mice. Circ J. 2006;70:342–351. DOI: 10.1253/circj.70.342. [DOI] [PubMed] [Google Scholar]
- 17.Vander Heide RS, Steenbergen C. Cardioprotection and myocardial reperfusion: pitfalls to clinical application. Circ Res. 2013;113:464–477. DOI: 10.1161/CIRCRESAHA.113.300765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fujiwara M, Matoba T, Koga JI, Okahara A, Funamoto D, Nakano K, Tsutsui H, Egashira K. Nanoparticle incorporating Toll‐like receptor 4 inhibitor attenuates myocardial ischaemia‐reperfusion injury by inhibiting monocyte‐mediated inflammation in mice. Cardiovasc Res. 2019;115:1244–1255. DOI: 10.1093/cvr/cvz066. [DOI] [PubMed] [Google Scholar]
- 19.Tokutome M, Matoba T, Nakano Y, Okahara A, Fujiwara M, Koga JI, Nakano K, Tsutsui H, Egashira K. Peroxisome proliferator‐activated receptor‐gamma targeting nanomedicine promotes cardiac healing after acute myocardial infarction by skewing monocyte/macrophage polarization in preclinical animal models. Cardiovasc Res. 2019;115:419–431. DOI: 10.1093/cvr/cvy200. [DOI] [PubMed] [Google Scholar]
- 20.Mao Y, Koga JI, Tokutome M, Matoba T, Ikeda G, Nakano K, Egashira K. Nanoparticle‐mediated delivery of pitavastatin to monocytes/macrophages inhibits left ventricular remodeling after acute myocardial infarction by inhibiting monocyte‐mediated inflammation. Int Heart J. 2017;58:615–623. DOI: 10.1536/ihj.16-457. [DOI] [PubMed] [Google Scholar]
- 21.Dauber IM, VanBenthuysen KM, McMurtry IF, Wheeler GS, Lesnefsky EJ, Horwitz LD, Weil JV. Functional coronary microvascular injury evident as increased permeability due to brief ischemia and reperfusion. Circ Res. 1990;66:986–998. DOI: 10.1161/01.RES.66.4.986. [DOI] [PubMed] [Google Scholar]
- 22.Jin BY, Lin AJ, Golan DE, Michel T. MARCKS protein mediates hydrogen peroxide regulation of endothelial permeability. Proc Natl Acad Sci U S A. 2012;109:14864–14869. DOI: 10.1073/pnas.1204974109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ikeda G, Matoba T, Nakano Y, Nagaoka K, Ishikita A, Nakano K, Funamoto D, Sunagawa K, Egashira K. Nanoparticle‐mediated targeting of cyclosporine a enhances cardioprotection against ischemia‐reperfusion injury through inhibition of mitochondrial permeability transition pore opening. Sci Rep. 2016;6:20467. DOI: 10.1038/srep20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nagaoka K, Matoba T, Mao Y, Nakano Y, Ikeda G, Egusa S, Tokutome M, Nagahama R, Nakano K, Sunagawa K, et al. A new therapeutic modality for acute myocardial infarction: nanoparticle‐mediated delivery of pitavastatin induces cardioprotection from ischemia‐reperfusion injury via activation of PI3K/Akt pathway and anti‐inflammation in a rat model. PLoS One. 2015;10:e0132451. DOI: 10.1371/journal.pone.0132451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ichimura K, Matoba T, Nakano K, Tokutome M, Honda K, Koga J, Egashira K. A translational study of a new therapeutic approach for acute myocardial infarction: nanoparticle‐mediated delivery of pitavastatin into reperfused myocardium reduces ischemia‐reperfusion injury in a preclinical porcine model. PLoS One. 2016;11:e0162425. DOI: 10.1371/journal.pone.0162425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ishibashi M, Hiasa K‐I, Zhao Q, Inoue S, Ohtani K, Kitamoto S, Tsuchihashi M, Sugaya T, Charo IF, Kura S, et al. Critical role of monocyte chemoattractant protein‐1 receptor CCR2 on monocytes in hypertension‐induced vascular inflammation and remodeling. Circ Res. 2004;94:1203–1210. DOI: 10.1161/01.RES.0000126924.23467.A3. [DOI] [PubMed] [Google Scholar]
- 27.Hsu CP, Zhai P, Yamamoto T, Maejima Y, Matsushima S, Hariharan N, Shao D, Takagi H, Oka S, Sadoshima J. Silent information regulator 1 protects the heart from ischemia/reperfusion. Circulation. 2010;122:2170–2182. DOI: 10.1161/CIRCULATIONAHA.110.958033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Katsuki S, Matoba T, Nakashiro S, Sato K, Koga J, Nakano K, Nakano Y, Egusa S, Sunagawa K, Egashira K. Nanoparticle‐mediated delivery of pitavastatin inhibits atherosclerotic plaque destabilization/rupture in mice by regulating the recruitment of inflammatory monocytes. Circulation. 2014;129:896–906. DOI: 10.1161/CIRCULATIONAHA.113.002870. [DOI] [PubMed] [Google Scholar]
- 29.Vasquez KO, Casavant C, Peterson JD. Quantitative whole body biodistribution of fluorescent‐labeled agents by non‐invasive tomographic imaging. PLoS One. 2011;6:e20594. DOI: 10.1371/journal.pone.0020594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakahira K, Haspel JA, Rathinam VAK, Lee S‐J, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12:222–230. DOI: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Amanakis G, Sun J, Fergusson MM, McGinty S, Liu C, Molkentin JD, Murphy E. Cysteine 202 of Cyclophilin D is a site of multiple post‐translational modifications and plays a role in cardioprotection. Cardiovasc Res. 2020;117(1):212–223. DOI: 10.1093/cvr/cvaa053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cung T‐T, Morel O, Cayla G, Rioufol G, Garcia‐Dorado D, Angoulvant D, Bonnefoy‐Cudraz E, Guérin P, Elbaz M, Delarche N, et al. Cyclosporine before PCI in patients with acute myocardial infarction. N Engl J Med. 2015;373:1021–1031. DOI: 10.1056/NEJMoa1505489. [DOI] [PubMed] [Google Scholar]
- 33.Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J Biol Chem. 2005;280:18558–18561. DOI: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- 34.Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. DOI: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Whelan RS, Konstantinidis K, Wei A‐C, Chen Y, Reyna DE, Jha S, Yang Y, Calvert JW, Lindsten T, Thompson CB, et al. Bax regulates primary necrosis through mitochondrial dynamics. Proc Natl Acad Sci U S A. 2012;109:6566–6571. DOI: 10.1073/pnas.1201608109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kubli DA, Zhang X, Lee Y, Hanna RA, Quinsay MN, Nguyen CK, Jimenez R, Petrosyan S, Murphy AN, Gustafsson AB. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem. 2013;288:915–926. DOI: 10.1074/jbc.M112.411363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carreira RS, Lee Y, Ghochani M, Gustafsson ÅB, Gottlieb RA. Cyclophilin D is required for mitochondrial removal by autophagy in cardiac cells. Autophagy. 2010;6:462–472. DOI: 10.4161/auto.6.4.11553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M, et al. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res. 2015;116:264–278. DOI: 10.1161/CIRCRESAHA.116.303356. [DOI] [PubMed] [Google Scholar]
- 39.Acharya S, Sahoo SK. PLGA nanoparticles containing various anticancer agents and tumour delivery by EPR effect. Adv Drug Deliv Rev. 2011;63:170–183. DOI: 10.1016/j.addr.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 40.Vasir JK, Labhasetwar V. Biodegradable nanoparticles for cytosolic delivery of therapeutics. Adv Drug Deliv Rev. 2007;59:718–728. DOI: 10.1016/j.addr.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matsumoto T, Yamashita S, Yoshino S, Kurose S, Morisaki K, Nakano K, Koga JI, Furuyama T, Mori M, Egashira K. Therapeutic arteriogenesis/angiogenesis for peripheral arterial disease by nanoparticle‐mediated delivery of pitavastatin into vascular endothelial cells. Ann Vasc Dis. 2020;13:4–12. DOI: 10.3400/avd.ra.19-00130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakano K, Matoba T, Koga J‐I, Kashihara Y, Fukae M, Ieiri I, Shiramoto M, Irie S, Kishimoto J, Todaka K, et al. Safety, Tolerability, and Pharmacokinetics of NK‐104‐NP. Int Heart J. 2018;59:1015–1025. DOI: 10.1536/ihj.17-555. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1