

Summary
Inflammation-dependent base deaminases promote therapeutic resistance in many malignancies. However, their roles in human pre-leukemia stem cell (pre-LSC) evolution to acute myeloid leukemia stem cells (LSCs) had not been elucidated. Comparative whole genome and whole transcriptome sequencing analyses of FACS-purified pre-LSC from myeloproliferative neoplasm (MPN) patients reveals APOBEC3C upregulation, an increased C-to-T mutational burden, and hematopoietic stem and progenitor cell (HSPC) proliferation during progression, which can be recapitulated by lentiviral APOBEC3C overexpression. In pre-LSC, inflammatory splice isoform overexpression coincides with APOBEC3C upregulation and ADAR1p150-induced A-to-I RNA hyper-editing. Pre-LSC evolution to LSC is marked by STAT3 editing, STAT3β isoform switching, elevated phospho-STAT3, and increased ADAR1p150 expression, which can be prevented by JAK2/STAT3 inhibition with ruxolitinib or fedratinib or lentiviral ADAR1 shRNA knockdown. Conversely, lentiviral ADAR1p150 expression enhances pre-LSC replating and STAT3 splice isoform switching. Thus, pre-LSC evolution to LSC is fueled by primate-specific APOBEC3C induced pre-LSC proliferation and ADAR1-mediated splicing deregulation.
In Brief
Jiang et al. identify dual APOBEC3C and ADAR1 base deaminase deregulation as an inflammation-responsive driver of myeloproliferative neoplasm stem cell evolution to self-renewing leukemia stem cells that fuel secondary acute myeloid leukemia transformation.
Graphical Abstract
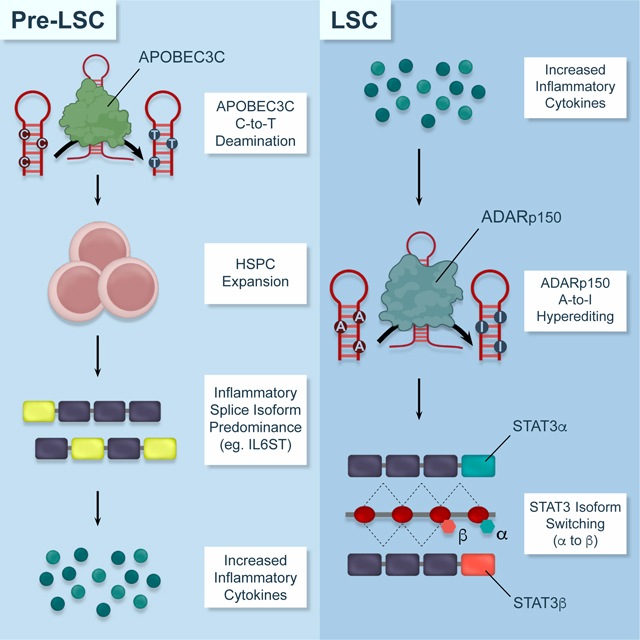
INTRODUCTION
Pro-inflammatory cytokine responsive APOBEC3 (apolipoprotein B mRNA editing enzyme, catalytic polypeptide like type 3) and ADAR1 (adenosine deaminase acting on RNA 1) base deaminases restrict viral replication (Di Giorgio et al., 2020) and LINE element retrotransposition (Mannion et al., 2014; Tan et al., 2017). However, base deaminase deregulation has been linked to both genomic and epitranscriptomic (post-transcriptional modification) instability (Alexandrov et al., 2013a; Burns et al., 2013b; Han et al., 2015; Jiang et al., 2017; Jiang et al., 2019; Lazzari et al., 2017; Peng et al., 2018; Petljak et al., 2019; Zhang and Slack, 2016; Zhou et al., 2019). In primates, APOBEC3 genes (APOBEC3A, APOBEC3B, APOBE3C, APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H) contribute to maintenance of genomic integrity. Conversely, deregulation of APOBEC3 induces genomic instability and distinctive DNA mutational spectra in many malignancies (Alexandrov et al., 2020; Alexandrov et al., 2013a; Alexandrov et al., 2013b; Burns et al., 2013a) by deaminating cytidines to thymidines (C-to-T) (Buisson et al., 2019). Although APOBEC enzymes have been fused to Cas9 nuclease-defective variants to induce targeted C-to-T transitions as a stem cell gene therapy strategy (Zafra et al., 2018), recent data suggest that APOBEC3 deaminases drive cancer-related hotspot mutagenesis (Alexandrov et al., 2020; Buisson et al., 2019). Because primate-specific APOBEC3 deaminases are activated by pro-inflammatory cytokines, such as IFNα and β, TNF-α, IL-1β and IL-6, the effects of enzymatic C-to-T deamination on the genomic landscape of cancer are inherently episodic, microenvironmentally dependent, and difficult to model (Petljak et al., 2019).
Similarly, pro-inflammatory cytokines activate ADAR1p150-mediated adenosine to inosine (A-to-I) deamination of double-stranded RNA (dsRNA), particularly in the context of primate-specific Alu sequences (Chua et al., 2020). As a dynamic regulator of mRNA and miRNA stability(Jiang et al., 2013b; Lazzari et al., 2017; Tan et al., 2017), ADAR1 plays a pivotal role in embryonic development and stem cell maintenance as evidenced by murine embryonic lethality and reduced hematopoietic stem cell (HSC) multi-lineage reconstitution potential following ADAR1 deletion(Hartner et al., 2009; Jiang et al., 2017; Jiang et al., 2019; Zipeto et al., 2016). Moreover, a recent study showed that ADAR1 loss reduces induced pluripotent stem cell (iPSC) reprogramming efficiency by inducing ER stress (Guallar et al., 2020). Additionally, alternatively spliced regions frequently harbor A-to-I editing sites whereby ADAR1 deficiency impairs alternative splicing in mouse tissues (Kapoor et al., 2020). Deregulation of ADAR1-mediated A-to-I editing alters stem cell survival and self-renewal regulatory mRNA and miRNA stability (Chen et al., 2013; Han et al., 2015; Jiang et al., 2017; Jiang et al., 2019; Lazzari et al., 2017; Zipeto et al., 2016). Although deaminase deregulation has been linked to therapeutic resistance in many tumor types(Han et al., 2015; Lazzari et al., 2017), the combinatorial capacity of APOBEC3-induced DNA mutagenesis and ADAR1-mediated splicing disruption and epitranscriptomic instability to fuel pre-LSC transformation into LSC has not been elucidated.
As an important paradigm for understanding molecular drivers of progression to secondary acute myeloid leukemia (sAML), myeloproliferative neoplasms (MPNs), including polycythemia vera (PV), essential thrombocythemia (ET), myelofibrosis (MF) and chronic myeloid leukemia (CML), were the first malignancies shown to harbor somatic stem cell mutations(Eide and Druker, 2017; Jiang et al., 2017; Rossi et al., 2008). Recent reports suggest that AML transformation is not solely predicted by the baseline driver mutations (i.e., JAK2-V617F, CALR, MPL) or additional somatic mutations (i.e., ASXL1, EZH2)(Tefferi et al., 2018), but has been associated with leukocytosis, constitutional symptoms, and pathologically increased cytokines, such as IL8 (Tefferi et al., 2011). Increases in pathologically induced cytokines have been found in MPNs (Pardanani et al., 2013), and decreases in these cytokines by JAK inhibition (Verstovsek et al., 2012) may well be responsible for observed improvements in survival and decreases in risk of leukemic transformation (Verstovsek et al., 2017).
Pre-leukemia stem cells (pre-LSCs) in MPNs arise from clonally mutated hematopoietic stem and progenitor cells (HSPCs) that vary in their capacity to become dormant, resist therapy (Gishizky et al., 1993; Jamieson et al., 2004; Kleppe et al., 2018; Rossi et al., 2008), and contribute to the generation of LSC that drive sAML transformation (Mesa et al., 2017; Shlush et al., 2014). While the propensity of MPNs to transform to sAML has been difficult to ascertain based on standard prognostic guidelines (Mesa et al., 2017; Shlush et al., 2014), seminal studies demonstrate that MPN initiation is driven by heritable risks and that MPNs harbor distinctive mutational profiles that predict progression as well as overall survival (Bao et al., 2020; Grinfeld et al., 2018; Miles et al., 2020). Moreover, in response to microbial signals, IL-6 production has been shown to promote pre-leukemic myeloproliferation in Tet methylcytosine dioxygenase 2 (Tet-2)-deficient mice (Meisel et al., 2018) thereby underscoring the importance of episodic, proinflammatory cytokine induction of MPN progression. However, the primate-specific impact of cytokine-induced enzymatic mutagenesis had not been addressed. Thus, we investigated the combinatorial capacity of inflammation-dependent, primate-specific APOBEC3 and ADAR1 deaminases to drive human pre-LSC evolution to LSCs with the ultimate aim of informing the development of effective strategies that predict and prevent transformation to rapidly fatal sAML.
RESULTS
APOBEC3C Deaminase Activation Promotes Human Pre-LSC Proliferation
To identify pre-LSC DNA mutational hierarchies and deaminase mutational signatures, whole-genome sequencing analysis (WGS) was performed on CD34+ stem cells purified from peripheral blood of 39 individuals with various MPNs, as well as 4 non-MPN controls, including 1 chronic lymphocytic leukemia (CLL) (Fig. 1a and Table S1). Somatic mutations were identified in the genomes of CD34+ stem cells from the 39 MPN patients using two complimentary approaches: (i) ensemble variant calling comparing CD34+ stems cells in peripheral blood to bulk saliva; and (ii) identification of somatic mutations, without using matched normal tissues, by employing tumor-only somatic variant filtering. These two complimentary approaches were used to mitigate the risk of identifying somatic mutations in the setting of matched-normal tissue (i.e., saliva) contamination with MPN cells (i.e., peripheral blood).
Fig. 1. MPN Pre-Leukemia Stem Cell Expansion and APOBEC3C Activation.

A. Sample Distribution in this study. Samples were distributed among Polycythemia Vera (PV, n=5), Essential Thrombocythemia (ET, n=4), Myelofibrosis (MF, n=28), Chronic Myeloid Leukemia (CML, n=3) and non-MPN control individuals (n=4, including 3 healthy volunteers and 1 CLL with CALR SNP). In parallel, whole-genome sequencing of 43 peripheral blood samples of a sample distribution of PV (n=6), ET (n=4), MF (n=26), CML (n=3) and non-MPN control individuals (n=4, including 1 CLL with CALR SNP). The somatic mutations were obtained from MPN patient samples (n=37) and non-MPN controls (healthy controls n=3 and CLL with CALR SNP n=1) with matching saliva (30X coverage) and peripheral blood (n=41, shown in solid black). Whole-transcriptomic sequencing (RNA-seq) was performed on 78 samples distributed as follows: PV (n=6), ET (n=2), MF (n=29), CML (n=5), AML (n=12), and non-MPN control individuals (n=24). These samples can further be broken down based on tissue of collection (peripheral blood or bone marrow) and cell types (stem cells and progenitor). In summary, from 54 subjects and 24 non-MPN controls, 113 samples were represented in the RNA sequencing analysis. B. Mutational burden of single point mutations (log-scaled). Each dot represents the number of substitutions per megabase in an individual MPN sample. Red lines reflect median numbers. Mutational profiles of substitutions are shown using six subtypes: C>A, C>G, C>T, T>A, T>C, T>G. Underneath each subtype are 16 bars reflecting the sequence contexts determined by the four possible bases 5’ and 3’ each mutated base. Average contributions of the two clock-like signatures across PCAWG MPN and MCCWG MPN samples are shown in different colors. C. Mutations in 69 MPN-associated genes (Grinfeld et al., 2018) in peripheral blood divided by MPN disease stage. Clinical-grade confirmation of JAK2 V617F mutation was marked as light yellow in MPN patients. MPN disease stage depicted in colored bar at the bottom of the figure. *, patient deceased since sample collection; +, patient has another malignancy; &, patient progressed after sample collection, and &&, patient progressed to AML after sample collection. D. A boxplot depicting the number of somatic mutations in peripheral blood or saliva based on transitions (Ti) or transversions (Tv). Both somatic and germline variants were included. E. A boxplot depicting the expression levels of APOBEC3 in ABM, YBM, intermediate-risk myelofibrosis (Int-MF), high-risk myelofibrosis (HR-MF) and sAML stem cell populations using normalized RNA-Seq. APOBEC3C expression was illustrated for each stem cell sample compared with ABM normal controls. (p < 0.05 =*). F. Comparison of the HSC percentage in MPN samples by flow cytometry (CML n=4, PV n=3, ET n=2, MF n=23 and AML n=3). G. A representative brightfield microscopic image of cord blood CD34+ cells lentivirally transduced with APOBEC3C compared with a lentiviral backbone control (left). H. Flow cytometry analysis of cord blood CD34+ cells 48 hours after lentiviral transduction. Error bars show SEM and significance determined by 2way ANOVA.
Using this combined approach, we compared the somatic mutations derived using ensemble variant calling in our CD34+ MPN stems cells (MCCWG CD34+ MPNs) to the ones derived using ensemble variant calling by the PCAWG consortium in MPN bulk peripheral blood. Remarkably, the tumor mutational burden was lower in stem cells compared to bulk blood with a more than 4-fold depletion of single point mutations observed in CD34+ MPN cells (p<0.0012; Fig. 1b). Furthermore, focused interrogation of 69 MPN-associated genes (Grinfeld et al., 2018) provided additional confirmation of a low mutational burden in MPN stem cells compared to previously published datasets (Fig.1b-c). Overall, these results suggest that more slowly cycling pre-leukemic MPN stem cells may be less mutable than the more highly proliferative bulk blood cells. Further analysis confirmed that clock-like mutational signatures perfectly recapitulate the patterns observed in both PCAWG MPNs derived from bulk blood and MCCWG MPNs derived from CD34+ stem cells (cosine similarities: 0.97 and 0.94, respectively). Interestingly, COSMIC signature SBS1, a clock-like signature associated with cell division, was depleted in pre-leukemic MPN stem cells compared to proliferative bulk blood; while COSMIC signature SBS5, a clock-like signature putatively associated with circadian rhythm, was highly prominent in MPN CD34+ stem cells (Fig. 1b).
Subsequently, single nucleotide variant, copy number variant and structural variant analyses were performed on all samples, employing tumor-only somatic variant filtering on the peripheral blood samples and subtracting structural and copy number variants found in the non-MPN samples as common germline variants (Fig. S1a). Mutations were found in inflammation-related genes, such as IL1A (SNP) and IRF1 (duplication in 1 PV and 4 MF patients) (Fig. S1a), a known driver of APOBEC3 activation. Except for patients presenting with high-risk MF, the genes JAK2, CALR, ASXL1/3 and KMT2C were mutated in MPN CD34+ stem cell populations (Fig. 1c) at a lower frequency than bulk peripheral blood samples (Fig. 1c). Some high-risk MF patients with mutations in MPN-associated genes harbored another malignancy or progressed to AML following sample collection indicative of genomic and/or transcriptomic instability, as has been described for malignancies with APOBEC3-related mutational signatures and ADAR1 activation (Fig. 1c; Fig. S1b-f; Table S1).
The most commonly observed mutations in MPN CD34+ cells were C-to-T transitions (approximately 50%) followed by T-to-C changes (approximately 20%) (Fig. 1c-d). Frequent C-to-T mutations have been reported following activation of APOBEC3 (Alexandrov et al., 2013a; Burns et al., 2013b). Therefore, we examined the expression of APOBEC3 and other transcripts in MPN stem and progenitor populations by RNA-seq (Fig. 1e and Fig. S2a-e). Notably, APOBEC3C was upregulated in national comprehensive cancer network (NCCN) panel guideline-defined intermediate-risk (Int-MF) and high-risk myelofibrosis (HR-MF) stem cell-enriched samples, suggesting a role for APOBEC3C in pre-LSC propagation (Fig. 1e and Fig. S2e). Consistent with this hypothesis, we observed proliferation of CD34+ hematopoietic stem and progenitor cells (HSPCs) following lentiviral APOBEC3C overexpression as well as expansion of the stem cell population following MF progression to AML (Fig. 1f). Following lentivirally enforced APOBEC3C expression in CD34+ cord blood stem cells, WGS revealed a pattern of mutations similar to that of MPN CD34+ cells (cosine similarity: 0.96; Fig. 1b and Fig. S1b) suggesting that APOBEC3C contributes to MPN stem cell mutagenesis. In contrast to an editase-deficient mutant APOBEC3C, lentiviral APOBEC3C wild-type overexpression in CD34+ cord blood cells resulted in expansion of a progenitor population that lacks replating capacity and skews toward the erythroid lineage as evidenced by increased erythroid colony formation (Fig. 1g-h, Fig. S1c and S1f). Lentiviral overexpression of another inflammation-responsive deaminase, ADAR1, induced expansion of a CD19+ B cell population (Fig. S1e). These results indicate that APOBEC3C and ADAR1 play regulatory roles in hematopoietic cell fate determination. While APOBEC3 deaminases are drivers of somatic mutagenesis in many human cancers (Burns et al., 2013b), this report provides a mechanistic link between APOBEC3C and pre-LSC generation and identifies the differential roles of APOBEC3C and ADAR1 in human HSPC expansion and cell fate determination.
Inflammatory Pathway Activation in Pre-LSC
To comprehensively investigate inflammation-dependent deaminase induction of pre-LSC evolution, a comparative WGS and RNA-seq analysis pipeline (Fig.1a and Fig. S2a-e) was established to exclude naturally occurring single nucleotide polymorphisms and enable detection of previously unreported editing sites in the setting of deaminase activation. To detect inflammatory pathway activation during MPN progression, RNA-seq analyses was performed on 113 FACS-purified stem cell (CD34+CD38-Lin) and progenitor (CD34+CD38+Lin−) populations from 54 MPN and AML patients and 24 young and aged healthy controls (Fig. 1a, Fig. S2a and Fig. S2b). A gene expression signature emerged that distinguished MPN and AML progenitors from stem cells in normal young and aged samples (Fig. S2b). A comparison between AML and MF samples elucidated 987 and 678 differentially expressed genes in stem cells and progenitors, respectively (Fig. S2d). Notably, transcripts involved in regulation of inflammation, including CTSA (cathepsin A) and inflammatory cytokine receptor genes (CD97 and EFHD2), were increased in AML stem cells and progenitors relative to MF, suggesting that deregulated inflammatory pathways may contribute to pre-LSC transformation into LSCs (Fig. S2d and Fig. S3a-h).
MPN stem cells harbored only 24 common differentially expressed genes relative to healthy aged bone marrow (ABM) (Fig. S3a). Notably, interferon stimulated gene (ISG) activators of ADAR1, such as IRF9 and IFITM1, were overexpressed in PV stem cells (Fig. S3b). In MF progenitors, expression of CSNK1γ2, a WNT-β-catenin self-renewal pathway regulator, was elevated relative to ABM (Fig. S3d, h). In AML stem and progenitor cells, both IER2 and CSF1R were upregulated, which have been associated with increased cytokine responsiveness as well as release of pro-inflammatory chemokines promoting invasion and metastasis (Fig. S3b, d, g, h). Lastly, comparative RNA-seq analyses revealed that the top enriched signaling pathways in sAML compared with MF were related to inflammation, autoimmunity and WNT signaling further underscoring the importance of inflammatory cytokine signaling and WNT/β-catenin self-renewal pathway activation in pre-LSC evolution to LSC (Fig. S3b-f).
Inflammation-Dependent ADARp150 and APOBEC3C Promote Pre-LSC Evolution
The predominance of inflammatory signatures detected in pre-LSC led us to examine the combined roles of inflammatory cytokine-inducible ADAR1 and APOBEC3 deaminases in MPN progression to AML. Initial hierarchical clustering of RNA-seq analyses, including the top 1% of genes ranked by variance across the dataset, revealed distinct gene (Fig. S2b) and splice isoform expression patterns between normal, MPN and AML hematopoietic stem cell (HSC) and progenitor samples (Fig. 2a). A predominance of interferon- or inflammatory cytokine-related transcripts, including a pro-inflammatory IL6ST isoform, was observed in MPN and sAML progenitors relative to ABM controls (Fig. 2b). Moreover, MPN and AML progenitors displayed a splice isoform switch favoring expression of the inflammatory cytokine-responsive ADAR1 p150 isoform over the constitutively active ADAR1 p110 isoform (Fig. 2c).
Fig. 2. Isoform Switching Favoring ADAR1p150 Expression Drives Pre-LSC Evolution.

A. Heatmap of RNA-Seq expression of splicing isoforms for the top 1% of genes ranked by variance. Annotation for each sample is presented as a stack of colored bars representing phenotype, cell type, source tissue, mutation status, and the treatment type (for MF samples only). Samples without a known JAK2 V617F mutation status are colored in gray. B. A boxplot representing the internally normalized expression of IL6ST isoforms (ENST00000381298 and ENST00000503773 in stem cells) and (ENST00000381298 and ENST00000336909 in the progenitors) in each MPN phenotype. Black dots represent expression values in lowest 2.5% or highest 97.5% of the distribution. C. Ratio of ADAR1 isoforms (p150/p110) in each MPN disease type using RNA-Seq expression data from stem cells and progenitors. D. Signaling Pathway Impact Analysis (SPIA) was performed for ET, PV, MF and AML compared to ABM progenitors. Listed are the top 5 activated pathways based on the NDE (number of differentially expressed genes per pathway)/pSize (number of genes in the pathway) in percent. E. SPIA in cord blood lentivirally transduced with ADAR1 WT (top) or RNA deaminase deficient mutant ADAR1E912A (bottom) compared to pCDH backbone controls (n=3). Listed are the top 6 activated pathways based on the NDE/pSize in percent. F. Correlation of normalized and Log2-transformed counts per million (CPM) data for APOBEC3C with ADAR1 p150 isoform in stem cells (top) and progenitors (bottom). Points are colored by phenotype. The MF risk-group is indicated by point shape with open shapes representing deceased patients. G. Western blot probed for ADAR1 p150 after co-immunoprecipitation with ADAR1 and APOBEC3C-FLAG. H. Colocalization of APOBEC3C and ADAR1 in TF1a cells. Immunofluorescence of anti-APOBEC3C (green) and anti-ADAR1 p150-specific (red) antibodies in TF1a shADAR1 and TF1a shControl knockdown cells demonstrate a colocalization (yellow) of APOBEC3C and ADAR1 p150 proteins in the shControl cells. TF1a shADAR1 cells show ablation of ADAR1 protein.
Because of the observed upregulation of inflammatory cytokine-related genes capable of activating innate immune deaminases in MPNs, we interrogated the differentially expressed genes by performing Signaling Pathway Impact Analysis (SPIA) comparing normal ABM with MPN and AML progenitors (Fig. 2d). Comparison of MPN with ABM revealed activation of pathways involved in regulation of chemokine signaling, RNA transport and transcriptional deregulation in cancer (Fig. 2d). Notably, approximately 70% of the genes in the influenza A pathway were differentially expressed in ET, PV and AML when compared to normal samples (Fig. 2d). Moreover, both Epstein Barr virus (EBV) and Influenza A viral infection-related pathways were activated in AML compared with ABM indicating that anti-viral pathway activation is associated with LSC generation (Fig. 2d). To investigate whether ADAR1 contributed to pre-LSC transformation (Jiang et al., 2017), we performed RNA-seq following lentiviral transduction of cord blood stem or progenitor cells with ADAR1 wild-type (ADAR1 WT) or a deaminase-inactive mutant (ADAR1E912A) (Jiang et al., 2019). SPIA analysis revealed that two KEGG pathways activated by ADAR1 WT overexpression in both cell types were involved in cancer and viral carcinogenesis (Fig. 2e) thereby mirroring the viral pathway activation signature typical of ADAR1-overexpressing AML progenitors (Fig. 2d). Together, these data suggest that inflammatory cytokine-inducible ADAR1p150 expression contributes to pre-LSC maintenance and LSC generation.
While activation of ADAR1 has been linked to cancer progression (Jiang et al., 2013a; Jiang et al., 2017; Peng et al., 2018), the combinatorial role of APOBEC3C and ADAR1p150 in pre-LSC evolution to LSC had not been examined. Both ADAR1p150 and APOBEC3C transcripts were elevated by RNA-seq analyses in high-risk MPN stem cells (Fig. 2f and Table S1). Co-immunoprecipitation analysis revealed that APOBEC3C and ADAR1p150 bind to each other in HEK293T cells (Fig. 2g). Moreover, confocal fluorescence microscopy revealed that co-localization of ADAR1p150 and APOBEC3C in AML cells was reduced by robust lentiviral ADAR1 shRNA knockdown (Fig. 2h and Fig. S2C). These data suggest that activation of both APOBEC3C and ADAR1p150 fuel pre-LSC evolution.
A-to-I RNA Editing Signatures Distinguish Pre-LSC from LSC
To further evaluate the impact of ADAR1 activation on pre-LSC evolution to LSC, we analyzed epitranscriptomic alterations observed in our samples that are present in the REDIportal atlas of A-to-I RNA editing that contains over 16 million events curated from RNAseq analyses derived from 549 individuals (https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkaa916/5940507). We identified known and previously unknown RNA editing events in MPN and AML (secondary and de novo) as well as normal young bone marrow (YBM) and ABM samples. Each MPN subtype and untreated sAML possessed significantly elevated levels of RNA editing compared to ABM controls as measured by the median variant allele frequency (VAF) (Fig. 3a). Inflammatory cytokine-inducible ADAR1p150 expression positively correlated with editing activity in MPN and sAML progenitors suggesting that inflammatory cytokine signaling promotes malignant editing in pre-LSC and LSC (Fig. 3b, Fig. S3a-h, Fig. S4a). To determine the frequency of RNA editing in different transcriptomic regions, we analyzed the VAF associated with A-to-I editing changes in ABM, YBM, MPN and AML progenitors (Fig. 3c). Compared with ABM and YBM, MPN progenitors harbored higher levels of A-to-I editing that resulted in non-synonymous changes (Fig.3d). Notably, 3’ UTR editing increased in MF and persisted in untreated sAML (Fig. 3d). Thus, ADAR1p150 induces A-to-I editing events that may prevent transcript targeting by microRNAs in 3’ UTR regions and thereby enhance transcript stability in MF and sAML(Jiang et al., 2019).
Fig. 3. A-to-I Hyper-editing Distinguishes pre-LSC and LSC from Normal Progenitors.

A. Violin plot of overall RNA editing variant allele frequency (VAF) by MPN subtype and YBM and ABM controls. The overall VAF is statistically significantly elevated in PV, ET, MF, CML, de novo AML and sAML primary patient samples compared to the normal ABM counterpart. B. Correlation of mean A-to-I RNA editing level with normalized and Log2-transformed ADAR1 p150 isoform CPM level in both stem cells (square) and progenitors (triangle). Each color represents a distinct MPN disease stage. C. Box plots comparing VAF of each MPN progenitor subtype and YBM and ABM controls stratified by genomic region. D. Statistical comparison of data from (C). The p-value values are derived from comparing the VAFs of each MPN stage and ABM at each variant classification by the Kolmogorov Smirnov test. E. Top 25 ranked genes by occurrence of nonsynonymous RNA edit mutations broken down by known non-Alu and Alu region, and previously unknown non-Alu and Alu regions stratified by MPN phenotype, treatment and cell type. F. Normalized Log2 transformed RNA-Seq expression data for CDK13 in the stem and progenitor population plotted by MPN phenotype. The results of t-tests (ns = not significant; p < 0.05 = *; p < 0.01 = **, p < 0.005 = ***) between each phenotype and the ABM) group are shown. G. Expression of normalized ADAR1 RNA-Seq expression data compared with expression normalized CDK13 in stem (left) and progenitor (right) populations. The significance of the Pearson correlation (relative to R = 0) is shown along with a trendline of the data. H. Colocalization of CDK13 and ADAR1 in sAML cells by immunostaining of anti-CDK13 (green) and anti-ADAR1 (red) antibodies.
Current RNA editing databases are primarily based on cell line or bulk tumor cell RNA-seq data that may mask the cell type and context-specific RNA editing events that trigger pre-LSC evolution into LSC. To identify RNA editing sites specific to pre-LSC, we compared RNA-seq variants with matching WGS data (Fig. 1a-c) and quantified non-synonymous editing events using REDIportal and two other established RNA editing databases (Kiran and Baranov, 2010; Ramaswami and Li, 2014) (Fig. 3e). We observed a strikingly different editing pattern between MPN stem cells and progenitors and ABM and YBM control samples. While missense editing of CDK13, a splicing and cell cycle regulatory gene, occurred in 84% of MPN samples, missense editing was not detected in ABM and YBM (Fig. 3e). Moreover, CDK13 transcript levels were elevated in MF (p<0.0001) and sAML stem cells (p<0.0001) and progenitors (Fig. 3f). Additionally, increased CDK13 transcript levels correlated with ADAR1 overexpression (Fig. 3g). Furthermore, confocal fluorescence microscopy revealed CDK13 upregulation and increased ADAR1 in myeloid leukemia cells consistent with CDK13 transcript stabilization and subsequent increased translation as a result of ADAR1 upregulation (Fig. 3h). While A-to-I RNA editing and stabilization of CDK13 transcripts have been linked to a worse prognosis in hepatocellular carcinoma (Dong et al., 2018), CDK13 targeting with a covalent inhibitor, THZ531, has potent anti-tumor activity suggesting that this approach may have further utility in LSC targeting (Iniguez et al., 2018). Conversely, a SUMF2 missense (recoding) editing was more prevalent in normal controls than MPN samples (Fig. 3e and Fig. S4b, h-j). While previous studies indicate that ADAR1-mediated A-to-I editing events occur predominantly in Alu repetitive element containing dsRNA structures (Jiang et al., 2017), our MPN stem and progenitor cell-enriched RNA-seq analysis of the nonsynonymous editing events reveal that recoding events also occur in non-Alu regions (Fig. 3e). Together these data suggest that distinctive RNA editing events predict MPN initiation and progression.
Subsequently, we examined the expression of all 1295 differentially edited genes in MPN, AML and ABM stem and progenitor cells. Hierarchical clustering of the gene expression values for differentially edited genes revealed that MPN samples clustered together when compared with normal young and aged controls (Fig. 4a). Notably, AML samples tended to cluster closer to normal samples indicative of reversion to stemness typical of aggressive malignancies. Further analysis of the differentially edited genes propagated over the STRING (Szklarczyk et al., 2017) interactome and clustered with Louvain clustering revealed that in both AML and MF progenitors A-to-I RNA edited transcripts were significantly enriched for genes involved in chromatin organization, transcription and mRNA splicing (Fig. 4b-c). Interestingly, AML progenitors demonstrated differential editing of ribosomal regulatory genes compared to ABM thereby suggesting that disruption of translational control by ADAR1p150 activation may fuel pre-LSC transformation.
Fig. 4. The RNA Editome Distinguishes Pre-LSC from LSC.

A. Heatmap based on gene expression z-scores of 1295 differentially edited genes across all comparisons with Aged Bone Marrow (ABM). B-C. Network analysis of differentially edited genes between (B) normal aged samples (ABM) and MF, and (C) normal aged samples (ABM) and AML. In MF (B), out of the 834 significantly differentially edited genes, 690 were found in the interactome and used as seeds for network propagation on the STRING high confidence interactome. In AML (C), out of the 757 significantly differentially edited genes, 642 were found in the interactome and used as seeds for network propagation on the STRING high confidence interactome. Differential expression log fold change is mapped to node color: blue - significantly down in MF compared to ABM; red - significantly up in MF compared to ABM. Gray nodes were not significantly differentially expressed (fdr < 0.05).
To further investigate the role of ADAR1 in clonal evolution, we performed single cell RNA-seq (scRNA-seq) of CD34+ cord blood cells transduced with a lentiviral backbone control (shCTRL) or lentiviral shRNA targeting ADAR1 (shADAR1) (Fig. S4c and Table S2). In keeping with a ribosomal regulatory role for ADAR1, tSNE analysis revealed differential ribosomal gene editing following ADAR1 knockdown (Fig. S4d)(Solomon et al., 2017). Notably, ribosomal transcripts were also differentially edited in MF and AML compared with normal age-matched progenitors (Fig. 4b-c and Fig. S4e-g). These editing-induced changes in ribosomal gene expression suggest that ADAR1 activation may alter protein turnover rates in LSC. These observations correspond with previous reports showing disruption of proteostasis (i.e. protein turnover) as a driver of LSC propagation in mouse models of leukemia (Signer et al., 2014) and ADAR1-induced proteomic diversity as a contributor to therapeutic resistance in a broad array of malignancies (Chua et al., 2020; Peng et al., 2018).
RNA Editing-Induced STAT3 Splice Isoform Switching Induces Pre-LSC Evolution to LSC
While the overall A-to-I RNA editing events increased in MPN progenitors compared with their normal counterparts, progression of MF and sAML was marked by increased editing of specific regions, including in lincRNA and in 3’UTR (Fig. 3d), as well as of selective transcripts (Fig. 4a-c). Notably, STRING interactome analysis revealed that a transcriptional activator of ADAR1 and embryonic self-renewal agonist, STAT3, was differentially edited and overexpressed in AML compared to normal progenitors (Fig. 4c). Previously, we reported that the JAK/STAT signaling pathway activates malignant A-to-I RNA editing in stem cell regulatory transcripts and increases LSC self-renewal capacity in CML (Zipeto et al., 2016). Since A-to-I RNA editing can remove the 3’ splice acceptor adenosine, we investigated whether ADAR1 activity is linked to pre-mRNA splicing changes and expression of alternative STAT3 isoforms.
Alternative splicing in STAT3 exon 23 generates two isoforms, STAT3α and STAT3β (Fig. 5a and Fig. S5a-h). A previous report showed that intronic RNA editing of STAT3 favored increased expression of the relatively rare STAT3β splice isoform (Goldberg et al., 2017). In AML, 4 out of 7 samples harbored A-to-I RNA editing at previously validated STAT3 intronic editing sites whereas only one ABM sample possessed these STAT3 RNA editing events (Fig. 5b). The expression of alternatively spliced STAT3β increased in MPN and AML stem cells and progenitors with elevated ADAR1p150 expression compared to ABM controls (Fig. 5c and Fig.S5a-b). Following exposure to inflammatory cytokines, Western blot analyses revealed that ADAR1p150 levels were elevated in sAML (pt.255 and pt.705) CD34+ cells compared to MF (pt. 749) or normal CD34+ cord blood cells (Fig. 5d). Notably, expression of the ADAR1 RNA editing-induced phospho-STAT3β isoform increased in sAML compared to normal controls (Fig. 5d and Fig. S5a-b). In keeping with ADAR1 induction of alternative STAT3 splice isoform usage, lentiviral ADAR1 overexpression in primary MPN progenitors was associated with increased STAT3β splice isoform expression (Fig.5f and Fig. S5h). Conversely, treatment with selective JAK2 inhibitors, ruxolitinib or fedratinib, in the presence of inflammatory cytokines, reduced the levels of ADAR1p150 and phospho-STAT3 in sAML stem cells (Fig. 5e, Fig. S5f). Following interferon-alpha treatment of myeloid leukemia cells, lentiviral ADAR1 shRNA knockdown reduced phosho-STAT3β expression (Fig. S5e,g). Thus, STAT3 editing increases overall phospho-STAT3, which can bind to the ADAR1 promoter and activate ADAR1 transcription. This feedback loop contributes to LSC generation and can be disrupted by pharmacologic JAK2/STAT3 inhibition or ADAR1 shRNA knockdown.
Fig. 5. ADAR1-Induced STAT3 Intronic Editing and Splice Isoform Switching in LSC.

A. Diagram of STAT3 isoform generation by intronic RNA editing of STAT3 transcripts. B. Intronic A-to-I RNA editing locations (Goldberg et al., 2017) in ABM and AML as determined by RNA-seq analysis. C. Correlation of normalized Log2-transformed CPM data of the STAT3β isoform and the ADAR1 p150 isoform in stem cells and progenitors of ABM, YBM, MPN and AML samples. The MF risk-group is indicated by the diamond shape. D. Western blot analysis of cord blood CD34+ cells (left, n = 2), high-risk MF (pt. 705 & 749) and sAML (pt. 255) CD34+ cells (right, n = 3). E. Western blot analysis of sAML (pt. 255) CD34+ cells treated with FDA-approved JAK2 inhibitors (ruxolitinib and fedratinib) compared with a JAK3 inhibitor (FM-381) at concentrations of 1nM, 10nM, and 100 nM. F. Correlation of ADAR1 p150 expression with the expression of STAT3β isoform. The CD34+ cells from cord blood (n = 3), sAML and high-risk MF samples (n = 5) were transduced with pCDH, ADAR1 overexpressing vectors. The relative gene expression was measured by RT-qPCR and normalized to HPRT values. G. pSTAT3 levels measured by flow cytometry in CD34+ populations of two sAML patients (2008–5 and 50261). H. Self-renewal capacity, as measured by colony replating assays, in MF CD34+ cells transduced with pCDH backbone or ADAR1 WT. The error bar shows SEM and significance determined by Student’s t test.
As a self-renewal agonist, STAT3 represses GSK3β via ARID1 and prevents phosphorylation as well as subsequent degradation of β–catenin (Bowman et al., 2001; Hirai et al., 2011; Nusse and Clevers, 2017; Wu et al., 2016). In keeping with the LSC-propagating effects of STAT3 upregulation, engrafted sAML progenitors expressed phospho-STAT3 as measured by flow cytometric evaluation of humanized sAML mouse model bone marrow (Fig. 5g). Moreover, lentivirally enforced ADAR1 expression in pre-LSC increased colony replating to a level more typical of LSC in sAML (Fig. 5h, Fig. S5c-d). Together, these data suggest that ADAR1 A-to-I editing-induced STAT3 splice isoform switching promotes transformation of pre-LSC into LSC that drive therapy-resistant sAML transformation.
DISCUSSION
In addition to environmental mutagens, cumulative data suggest that APOBEC3 DNA and ADAR1 RNA deaminases serve as enzymatic drivers of cancer evolution (Alexandrov et al., 2013a; Alexandrov et al., 2013b; Burns et al., 2013a; Chua et al., 2020; Jiang et al., 2017). Specifically, patterns of C-to-T deamination induced by aberrant activation of APOBEC3 family members, have been identified by whole-exome sequencing in many human malignancies (Burns et al., 2013b). Moreover, inflammatory cytokine-induced hyperactivation of ADAR1p150 results in A-to-I deamination of self-renewal and cell cycle regulatory transcripts thereby fueling therapeutic resistance in leukemia (Jiang et al., 2013b; Jiang et al., 2019; Lazzari et al., 2017; Zipeto et al., 2016). However, the combinatorial roles of APOBEC3 and ADAR1 in primary human pre-cancer stem cell evolution to therapy-resistant cancer stem cells had not been examined.
In this study, we focused on characterizing the combined capacity of enzymatic DNA and RNA deamination to induce pre-LSC evolution to LSCs that fuel sAML transformation. To this end, we established 1) primary human stem and progenitor cell-based WGS and RNA-seq editome analysis pipelines that enabled identification of pre-LSC and LSC-specific DNA and RNA editing sites, 2) lentiviral APOBEC3C wild-type and mutant functional impact analysis in hematopoietic stem and progenitor cells, and 3) lentiviral ADAR1 wild-type, mutant, and shRNA knockdown analysis of RNA editing-related recoding events and splicing alterations. Using these combined human hematopoietic stem and progenitor cell-focused molecular and functional analysis strategies, we discovered that APOBEC3C activation fuels C-to-T mutagenesis and expansion of MPN progenitors with increased ADAR1p150 A-to-I RNA editing capacity thereby initiating LSC generation. Confocal fluorescence microscopic analyses showing APOBEC3C and ADAR1p150 co-localization together with co-immunoprecipitation of APOBEC3C and ADAR1p150 data suggest that these enzymes may function as a complex to increase deamination of cytosolic single-stranded DNA (ssDNA) and dsRNA in response to increased systemic inflammatory cytokine signaling or in response to viral replication.
In MPN stem and progenitor cells, APOBEC3C induces proliferation and C-to-T mutagenesis that sets the stage for inflammatory cytokine-induced ADAR1p150 activation resulting in CDK13 missense editing and transcript instability, which has been linked to decreased survival of patients with therapeutically recalcitrant malignancies (Dong et al., 2018). Ultimately, inflammation-responsive ADAR1p150 activation in APOBEC3C-overexpressing MPN pre-LSC induces STAT3 intronic editing, which increases expression of STAT3β. In a previous study, STAT3β was found to repress GSK3β and ARID1B thereby preventing phosphorylation and subsequent degradation of β-catenin (Wu et al., 2016), which is frequently deregulated in LSC (Jamieson et al., 2004; Zipeto et al., 2016). In keeping with this report, we found that ADAR1p150 upregulation was associated with increased STAT3β splice isoform expression in LSC. Moreover, ADAR1p150 induction could be reduced by pharmacologic JAK2/STAT3 inhibition with fedratinib or ruxolitinib and could be phenocopied by lentiviral ADAR1 shRNA knockdown. In the setting of increased APOBEC3C expression by high risk MF pre-LSC, lentiviral ADAR1 overexpression enhanced colony replating efficiency, as an in vitro surrogate measure of self-renewal, suggesting that APOBEC3C and ADAR1 work in concert to induce pre-LSC evolution to LSC. If a pro-inflammatory microenvironment in the marrow is conducive to the accumulation of additional somatic mutations driving pre-LSC transformation into LSC, this could explain observed clinical benefits of MPN therapy. Indeed, the decreases seen in disease progression in MPNs, both from PV to MF by therapy with long-acting interferons (Jager et al., 2020), as well as decreases in progression from MF to AML with JAK inhibition {Verstovsek, 2017 #14028; Vannucchi et al., 2017; Yacoub et al., 2019) may well originate from decreases in an inflammatory microenvironment in the marrow.
In addition to providing a robust framework for predicting and preventing pre-cancer stem cell evolution, the discovery of combined base deaminase deregulation is particularly relevant for identifying the potentially pre-malignant consequences of clinical gene therapy strategies (Komor et al., 2016) involving APOBEC3 and ADAR base editors. Because APOBEC3 and ADAR1 can be activated by cytosolic ssDNA, dsRNA structures, and lentiviral transduction, they may contribute to DNA mutations and RNA alterations induced by CRISPR-Cas guided base editing technologies as well as lentivirally delivered therapeutic gene correction strategies. Also, the differential effects of APOBEC3 and ADAR1 on stem cell fate specification will need to be considered prior to implementation of stem cell gene therapy approaches involving base editing technologies. The potential for induction of both genomic and epitranscriptomic instability provides a strong rationale for deciphering the oncogenic potential of combinatorial APOBEC3 and ADAR1 activation (Grunewald et al., 2019).
While we focused on the C-to-T DNA mutational impact of APOBEC3C overexpression, other APOBEC3 enzymes, such as APOBEC3A, can also induce DNA editing in response to interferon thereby promoting genomic instability (Sharma et al., 2015). Moreover, the RNA editing capacity of APOBEC3C and other APOBEC3 enzymes has not been clearly elucidated in stem cells and forms the basis for launching whole-transcriptome and single stem cell RNA sequencing analyses as well as functional stem cell impact studies. Moreover, C-to-T deamination by APOBEC3C could remove cytosine thereby preventing cytosine methylation. Because cytosine demethylation represents a major cancer mutational signature, the role of APOBEC3C in the induction of malignant genetic modifications that determine expression patterns for a large set of genes will need to be further studied.
In contrast to normal HSPC, inflammatory cytokines induce both APOBEC3C and ADAR1 expression in pre-LSC thereby promoting evolution to LSC in sAML. Further investigation into whether sustained ADAR1 activation occurs as a result of downregulation of an ADAR repressor, like AIMP2, which enhances degradation of ADAR proteins (Tan et al., 2017) or if ADAR1 hyperactivation promotes malignant reprogramming of pre-LSC into LSC by altering ER stress responses (Guallar et al., 2020), may provide additional insights into the cell type and context-specific causes and functional consequences of deaminase deregulation.
Both ADAR1 and APOBEC3 play important roles in the intrinsic responses to viral infection and protect the human genome from retrotransposition. They also play important roles in innate and adaptive immunity by controlling the response to inflammatory cytokine signals. In keeping with the induction of deaminases by inflammatory cytokines, we found that the top activated genes in pre-LSC compared with normal HSPC controls corresponded with anti-viral signaling pathways and chemokine signaling. The most common anti-viral signature was related to EBV infection (ET, PV, and AML), which is associated with viral oncogenesis. These data suggest that inflammation-dependent deaminases induced by viral infection; human endogenous retroviral activation; LINE element retrotransposition; or chronic cytokine signaling, promote MPN pre-LSC transformation into LSC in sAML and will need to be further studied with viral transcriptome analysis pipelines. Also, recent studies suggest that deletion of ADAR1 sensitizes malignant cells to PD-1 immune checkpoint blockade (Ishizuka et al., 2019). Thus, early detection and targeted inhibition of combined APOBEC3C and ADAR1 activation may have important implications for preventing human pre-cancer stem cell evolution to cancer stem cells that promote therapeutic resistance and disease progression.
STAR METHODS
Key Resource Table
| REAGENT OR RESOURCE | SOURCE | IDENTIFIER | |
|---|---|---|---|
| Antibodies | |||
| b-actin | Abcam | Cat #ab8227; RRID: AB_2305186 | |
| ADAR1 | Abcam | Cat #ab126745; RRID: AB_11145661 |
|
| APOBEC3C | Abcam | Cat # ab72652; RRID: AB_ 1523141 |
|
| pSTAT3 (Y705)-FITC | eBioscience | Cat # 11–9033-42; RRID: AB_2572522 |
|
| CD34 BV421 | BD | Cat # 740081; RRID: AB_2739844 |
|
| ADAR1 | Abcam | Cat # 126745; RRID: AB_ 11145661 |
|
| ADAR1 | Cell Signaling | Cat # 14175; RRID: AB_ 2722520 |
|
| STAT3 (124H6) | Cell Signaling | Cat # 9139; RRID: AB_ 331757 |
|
| p-STAT3 Y705 | Cell Signaling | Cat # 9131; RRID: AB_ 2331586 |
|
| GAPDH | Millipore | Cat # MAB374; RRID: AB_ 2107445 |
|
| CDK13 | Abcam | Cat # ab251955 | |
| ADAR1-APC | Abcam | Cat # ab168809 | |
| APOBEC3C | Abcam | Cat # ab221874; RRID: AB_ 2722520 |
|
| GAPDH | Abcam | Cat # ab181602; RRID: AB_2630358 |
|
| Goat pAb Rb IgG AF568 | Abcam | Cat # ab175471; RRID: AB_2576207 |
|
| Goat pAb Rb IgG AF568 | Abcam | Cat # ab150077; RRID: AB_2630356 |
|
| HRP-linked anti-rabbit IgG | Cell Signaling | Cat # 7074; RRID: AB_2099233 |
|
| HRP-linked anti-mouse IgG | Cell Signaling | Cat # 7076; RRID: AB_330924 |
|
| CD45-Alexa405 | Invitrogen | Cat #MHCD4526; RRID: AB_10372211 | |
| CD38-Alexa647 | Ab Serotec | Cat #MCA1019A647; RRID: AB_324854 | |
| CD34-Biotin | Invitrogen | Cat #CD3458115; RRID: AB_2536503 | |
| Strepavidin-Alexa488 | Invitrogen | Cat #S32354; RRID: AB_2315383 | |
| 7AAD | Invitrogen | Cat #A1310 | |
| Alexa Fluor® 594 Goat Anti- Rabbit IgG (H+L) Antibody |
Invitrogen | Cat #A11012; RRID: AB_10562717 |
|
| CD45-APC | Invitrogen | Cat #MHCD4505; RRID: AB_10372216 | |
| CD45-BB515 | BD | Cat #564585; RRID: AB_2732068 | |
| CD34-BV 421 | BD | Cat # 562577; RRID: AB_2687922 | |
| CD38-PE-Cy7 | BD | Cat #335790; RRID: AB_399969 | |
| CD123-PE | BD | Cat #554529; RRID: AB_395457 | |
| CD45RA-FITC | Invitrogen | Cat #MHCD45RA01; RRID: AB_10373858 | |
| CD8-PE-Cy 5.5 | BD | Cat #555368; RRID: AB_395771 | |
| CD56- PE-Cy 5.5 | BD | Cat #555517; RRID: AB_395907 | |
| CD4- PE-Cy 5.5 | BD | Cat #555348; RRID: AB_395753 | |
| CD3- PE-Cy 5.5 | BD | Cat #555334; RRID: AB_395741 | |
| CD19- PE-Cy 5.5 | BD | Cat #555414; RRID: AB_395814 | |
| CD2- PE-Cy 5.5 | BD | Cat #555328; RRID: AB_395735 | |
| CD14- PerCP-Cy5.5 | BD | Cat # 550787; RRID: AB_393884 | |
| CD8-PE-Cy 5.5 | BD | Cat #555368; RRID: AB_395771 | |
| CD56- PE-Cy 5.5 | BD | Cat #555517; RRID: AB_395907 | |
| CD3-FITC | BioLegend | Cat #300306; RRID: AB_314042 | |
| Bacterial and Virus Strains | |||
| DH5alpha Competent E. Coli | Invitrogen | Cat #12297016 | |
| Stbl2 Competent E. Coli | Invitrogen | Cat #10268019 | |
| Biological Samples | |||
| Aged normal and MPN Patient samples | Obtained through patients consenting at UCSD Moores Cancer Center according to Institutional Review Boardapproved protocols. | N/A | |
| Young Cord blood CD34+ cells | Purchased from AllCells or StemCell Technologies |
Cat #CB008F-S Cat #70008.5 Cat #70008.2 |
|
| Critical Commercial Assays | |||
| SYBR GreenER qPCR SuperMix |
Invitrogen | Cat #11761–500 | |
| SuperScript III First-Strand Synthesis SuperMix for qRT- PCR |
Invitrogen | Cat #11752–250 | |
| Prolong Gold antifade reagent with DAPI | Invitrogen | Cat # P36935 | |
| Live/Dead Fixable Near IR Dead Cell Stain kit |
Invitrogen | Cat #L10119 | |
| Secrete-Pair Dual Luminescence Assay Kit |
GeneCopoeia | Cat #LF033 | |
| Experimental Models: Cell Lines | |||
| K562 | ATCC | Cat #CCL-243; RRID: CVCL_0004 | |
| TF1a | ATCC | Cat #CRL-2451; RRID: CVCL_3608 | |
| 293T | ATCC | Cat #CRL-3216; RRID: CVCL_0063 | |
| Experimental Models: | |||
| Recombinant DNA | |||
| pCDH-EF1-MCS-T2A-copGFP | SBI System Biosciences | Cat #CD521A-1 | |
| pCDH-ADAR1 WT | Zipeto et al, 2016 | N/A | |
| pCDH-ADAR1E912A | Zipeto et al, 2016 | N/A | |
| pCDH-CMV-EF1-copGFP | SBI System Biosciences | Cat #CD511B-1 | |
| shADAR1-pLKO.1 (CCGGACCTCCTCACGAGCCCA AGTTCGTTTACCAAGCAAAA) |
This paper | N/A | |
| shScramble -pLKO.1 (CCTAAGGTTAAGTCGCCCTCG ) |
This paper | N/A | |
| Equipment | |||
| Olympus FluoView FV10i | Jamieson laboratory | N/A | |
| MACSQuoant 10 Analyzer | Jamieson laboratory | N/A | |
| Deposited Data | |||
| RNA-sequencing dataset | This paper | dbGAP: PHS002228.v1.p1. |
|
| DNA-sequencing dataset | This paper. | dbGAP: PHS002228.v1.p1. |
|
| Analysis codes | This paper | Github: https://github.com/ucsdccbb/MPN_atlas_methods | |
| Software and Algorithms | |||
| Burrows-Wheeler Aligner | Li H., 2013 | https://github.com/lh3/bwa | |
| VarScan2 | Koboldt et al., 2012 | https://dkoboldt.github.io/varscan/ | |
| Strelka2 | Kim et al., 2018 | https://github.com/Illumina/strelka | |
| MuSE | Fan et al. 2016 | https://bioinformatics.mdanderson.org/publicsoftware/muse/ | |
| Mutect2 | Benjamin et al., 2019 | https://gatk.broadinstitute.org/hc/en-us/articles/360037593851-Mutect2 | |
| SigProfilerMatrixGenerator | Bergstrom et al., 2019 | https://github.com/AlexandrovLab/SigProfilerMatrixGenerator | |
| Cutadapt v1.15 | Martin, 2011 | https://github.com/marcelm/cutadapt | |
| samblaster | Faust, et al., 2014 | https://github.com/GregoryFaust/samblaster | |
| Sambamba v0.4.7 | Tarasov, et al., 2015 | https://lomereiter.github.io/sambamba/ | |
| Samtools, v1.1 | Li et al., 2009 | http://www.htslib.org/ | |
| PicardTools v1.96 | https://broadinstitute.github.io/picard/ | ||
| Genome Analysis Tool Kit v2.4–9; v3.8 |
McKenna et al., 2010 | http://www.broadinstitute.org/gsa/wiki/index.php/The_Genome_Analysis_Toolkit | |
| bwa-mem v0.7.12 | Li and Durbin, 2009 | http://biobwa.sourceforge.net/bwa.shtml)) | |
| STAR v2.5.1a; v2.5.2b | Dobin et al., 2013 | https://github.com/alexdobin/STAR | |
| RSEM v1.3.0 | Li & Dewey, 2011 | https://deweylab.github.io/RSEM/ | |
| ENCODE long RNA-seq Pipeline |
https://github.com/ENCODE-DCC/long-rna-seqpipeline | ||
| FastQC | Andrews, et al., 2012 | https://www.bioinformatics.babraham.ac.uk/projects/fastqc// | |
| REDItools | Picardi & Pesole, 2013 | http://srv00.recas.ba.infn.it/reditools/ | |
| CrossMap | Zhao et a., 2014 | http://crossmap.sourceforge.net/ | |
| Mutect v1.1.5 | Cibulskis, et al., 2013 | https://software.broadinstitute.org/cancer/cga/mutect | |
| Bedtools v2.22.1; v2.26.0 | Quinlan & Hall, 2010 | http://bedtools.readthedocs.io/en/latest/ | |
| EdgeR | Robinson et al., 2010 | http://bioconductor.org/packages/release/bioc/html/edgeR.html | |
| Vcftools | Danecek, et al., 2011 | https://vcftools.github.io/index.html | |
| GENE-E | https://software.broadinstitute.org/GENE-E/ | ||
| Cirrus-ngs | https://github.com/ucsdccbb/cirrus-ngs | ||
| R v3.4.3 | https://cran.r-project.org/ | ||
| limma | Ritchie et al., 2015 | https://www.bioconductor.org/packages/release/bioc/html/limma.html | |
| limma-voom | Law et al., 2014 | https://www.bioconductor.org/packages/release/bioc/html/limma.html | |
| ANNOVAR v2017Jun01 | Wang, et al., 2010 | http://annovar.openbioinformatics.org/ | |
| SPIA | Tarca, et al., 2020 | https://bioconductor.org/packages/release/bioc/html/SPIA.html | |
| VariantAnnotation v1.24.5 | Obenchain, et al., 2014 | https://bioconductor.org/packages/release/bioc/html/VariantAnnotation.html | |
| SNPiR | https://github.com/rpiskol/SNPiR | ||
| Oncotator v1.9.8 | Ramos, et al., 2015 | https://software.broadinstitute.org/cancer/cga/oncotator | |
| bcl2fastq | https://support.illumina.com/sequencing/sequencing_software/bcl2fastqconversion-software.html | ||
| GProfiler | |||
| Cytoscape v3.7.1 | https://cytoscape.org/ | ||
| VisJS2Jupyter | Rosenthal, et al., 2018 | https://github.com/ucsdccbb/visJS2jupyter | |
| Rtsne, v0.15 | https://github.com/jkrijthe/Rtsne | ||
| Rcircos | Zhang, et al. 2013 | https://cran.r-project.org/web/packages/RCircos/index.html | |
| CNVkit | Talevich, et al. 2014 | https://cnvkit.readthedocs.io/en/stable/ | |
| Lumpy | Layer, et al. 2014 | https://github.com/arq5x/lumpy-sv | |
| Manta | Chen, et al., 2016 | https://github.com/Illumina/manta | |
| AnnotSV | Geoffrey, et al. 2018 | https://lbgi.fr/AnnotSV/ | |
| SURVIVOR | Jeffares, et al. 2017 | https://github.com/fritzsedlazeck/SURVIVOR | |
| Databases/Reference Datasets | |||
| 1000 Genomes Project | 1000G_phase1.snps.high _confidence.hg19.sites.vcf | ||
| dbSNP hg19 v138 | dbsnp_138.hg19.vcf | ||
| Hg19 fasta | ucsc.hg19.fasta | ||
| GRCh37v87 gtf | Homo_sapiens.GRCh37.8 7.gtf.gz |
||
| STRING high confidence interactome |
Szklarczyk et al., 2015 | 9606.protein.links.v11.0.txt | |
| ExAC | Lek, et al. 2016 | http://exac.broadinstitute.org/ | |
| gnomAD | https://gnomad.broadinstitute.org/ | ||
| DARNED | Kiran & Baranov, 2010 | rnaEditDB.txt | |
| RADAR v2 hg19 | Ramaswami & Li, 2014 | Human_AG_all_hg19_v2.t | |
| xt | |||
| GSVA | Hanzelmann et al, 2013 | https://bioconductor.org/packages/release/bioc/html/GSVA.html | |
| ICGC PCAWG MPN dataset | Campbell et al, 2020 | http://dcc.icgc.org/pcawg/ | |
| Other | |||
| Normal HSPC, CML CP progenitor, and BC CML progenitor gene expression |
Jiang et al, 2013 | BioProject: PRJNA214016 | |
| Cord blood CD34+ transduced with pCDH, ADAR1 WT, or ADAR1E912A |
Zipeto et al, 2016 | BioProject: PRJNA319866 | |
Resource Availability.
Lead Contact.
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Dr. Catriona Jamieson (cjamieson@health.ucsd.edu).
Materials Availability.
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
The sequencing datasets generated during this study are available at dbGAP, accession number PHS002228.v1.p1. The analysis code and documentation of computational analyses are available through Github: https://github.com/ucsd-ccbb/MPN_atlas_methods. Data from previous studies are available. Normal HSPC, CML CP progenitor, and BC CML progenitor gene expression are available at PRJNA214016. Cord blood CD34+ transduced with pCDH, ADAR1 WT, or ADAR1E912A are available at PRJNA319866.
Experimental Model and Subject Details
Animal
All mouse studies were completed in accordance with University Laboratory Animal Resources and Institutional Animal Care and Use Committee of the University of California regulations. Immunocompromised RAG2−/−γc−/− mice were bred and maintained in the Sanford Consortium vivarium according to IACUC approved protocols. Newborn mice (2–3 days old) of both sexes were used in the study.
Human Subjects
Primary adult non-leukemic blood and bone marrow as well as patient samples were obtained from consenting patients at the University of California in accordance with a UC San Diego human research protections program Institutional Review Board approved protocol (#131550). The IRB reviewed this protocol and found that it meets the requirements as stated in 45 CFR 46.404 and 21 CFR 50.51. Human cord blood and normal aged-match samples were purchased as purified CD34+ cells (AllCell or StemCell Techologies).
Primary cell cultures
All human cell lines (293T and K562) were cultured in 37°C in DMEM supplemented with 10% FBS and 2 mM L-glutamine and maintained according to ATCC protocols. All cell lines were confirmed to be mycoplasma-free with repeated testing.
Experimental Model and Subject Details
Primary human subjects are as the following: 4 healthy individuals and total of 78 MPN patients. The subjects were allocated to experimental groups based on the healthy history. The health status, whether the subjects were involved in previous procedures, and previous treatment history can be found in the Supplemental Table 1. All mice used in this study were housed and bred under specific pathogen-free conditions at University of California, San Diego in accordance with all the guidelines of the Institutional Animal Care and Use Committee (IACUC).
Method Details
Patient Sample Processing and Preparation for Whole-Genome Sequencing
CD34+ cells: Peripheral blood mononuclear cells were isolated by Ficoll-paque density centrifugation and cryopreserved in liquid nitrogen. CD34+ cells were selected from peripheral blood mononuclear cells from both MPN patients and normal controls by magnetic bead separation (MACS; Miltenyi, Bergisch Gladbach, Germany) as previously described(Jiang et al., 2013b) with minor modification using a different kit for magnetic bead separation: Catalog 130–100-453. DNA from the peripheral blood CD34+ population was extracted according to manufacturer recommendations using QIAamp DNA Blood Mini Kit (Qiagen, Catalog number 51104).
Saliva cells: Subjects abstained from eating at least 1 hour prior to saliva donation and rinsed their mouths with water to remove food residue immediately prior to saliva donation. Subjects then deposited 1 mL of saliva into the collection device, which was stabilized immediately afterwards (Biomatrica, Catalog number 97021–011A). Stabilized saliva was passed through 70–100 micron strainers to further remove food residues. DNA was extracted using the QIAamp DNA Blood Mini Kit (Qiagen, Catalog number 51104) described above with minor modifications. Both peripheral blood (90X) and saliva (30X) cell samples were sequenced on the Illumina HiSeq X sequencer using a 150-base paired-end single-index read format.
Patient Sample processing and Preparation for Whole-transcriptome Sequencing
Whole-transcriptome sequencing (RNA-seq) was performed on 78 samples distributed as follows: PV (n=6), ET (n=2), MF (n=29), CML (n=5), AML (n=12), and non-MPN control individuals (n=24). These samples can further be broken down based on tissue of collection (peripheral blood or bone marrow) and cell types (stem cells and progenitor). In summary, from 54 subjects and 24 non-MPN controls, 113 samples were included in the RNA sequencing cohort. Mononuclear cells from peripheral blood and bone marrow were purified, cryopreserved, and enriched for CD34+ cells as described above. Enriched CD34+ fractions were stained with fluorescent antibodies against human CD45, CD34, CD38, Lineage markers (BD Pharmingen; CD2 PE-Cy5, 1:20, cat 555328, CD3 PE-Cy5, 1:20, cat 555334, CD4 PE-Cy5, 1:10, cat 555348, CD8 PE-Cy5, 1:50, cat 555368, CD14 PerCP-Cy5.5, 3:100, cat 550787, CD19 PE-Cy5, 1:50, cat 555414, CD20 PE-Cy5, 1:20, cat 555624, CD56 PE-Cy5, 1:10, cat 555517, CD45 APC, 1:50, cat 335790, CD34 BV421, 1:100, cat 562577, CD38 PE-Cy7, 1:50, cat 335790), and propidium iodide. Cells were FACS-purified using a FACS Aria II (Sanford Consortium Stem Cell Core Facility) into hematopoietic stem cell (Lin−CD45+CD34+CD38−) and progenitor (Lin−CD45+CD34+CD38+) populations directly into RLT lysis buffer (Qiagen) for RNA extraction followed by RNA-Seq (The Scripps Research Institute Next Generation Sequencing Core) on Illumina HiSeq platforms.
| Antibody | Supplier | Catalog Number | Clone | Lot |
|---|---|---|---|---|
| CD2 PE-Cy5 | BD Pharmingen | 555328 | RPA-2.10 | 6070653 |
| CD3 PE-Cy5 | BD Pharmingen | 555334 | UCHT1 | 5349958 |
| CD4 PE-Cy5 | BD Pharmingen | 555348 | RPA-T4 | 6036632 |
| CD8 PE-Cy5 | BD Pharmingen | 555368 | RPA-T8 | 5219728 |
| CD14 PerCP-Cy5.5 | BD Pharmingen | 550787 | M5E2 | 6070674 |
| CD19 PE-Cy5 | BD Pharmingen | 555414 | HIB19 | 6126777 |
| CD20 PE-Cy5 | BD Pharmingen | 555624 | 2H7 | 6126778 |
| CD56 PE-Cy5 | BD Pharmingen | 555517 | B159 | 7177552 |
| CD45 APC | Life Technologies | MHCD4505 | HI30 | 1966219A |
| CD34 BV421 | BD Pharmingen | 562577 | 581 | 7153978 |
| CD38 PE-Cy7 | BD Biosciences | 335790 | HB7 | 8002648 |
Bioinformatics analysis
The analysis code and documentation for the computational analyses are available through Github: https://github.com/ucsd-ccbb/MPN_atlas_methods.
Whole genome sequencing (WGS) and mutation calling without matched normal
Whole genome sequencing of 44 saliva samples was performed at 30X coverage. The samples were distributed among PV (n=5), ET (n=4), MF (n=28), CML (n=3) and non-MPN control individuals (n=4, including 3 healthy volunteers and 1 CLL). In parallel, whole genome sequencing of 43 peripheral blood CD34+ stem and progenitor cell enriched samples was performed at 90X coverage for the following sample distribution: PV (n=6), ET (n=4), MF (n=26), CML (n=3) and non-MPN control individuals (n=4, including 1 CLL). WGS analysis was performed on 82 samples, with matching 41 peripheral blood CD34+ samples. We performed sequence alignment and variant calling using the Genome Analysis Toolkit (GATK) best practice pipeline. The reference genomes were realigned to the human 1000 genomes v37(Genomes Project et al., 2015), which contains the autosomes, X, Y and MT but without haplotype sequence or EBV. BWA-mem v.0.7.12.(Li and Durbin, 2009) was used for mapping short reads against the human 1000 genomes v37. Subsequent processing was carried out with SAMtools v.1.1(Cibulskis et al., 2013; Lai et al., 2016; Li et al., 2009; McKenna et al., 2010), Picard Tools v1.96, Genome Analysis Toolkit (GATK) v2.4–9(McKenna et al., 2010), which consisted of the following steps: sorting and splitting of the BAM files, marking of duplicate reads, local realignment, indel realignment and recalibration of base quality scores, outputting reads coverage files in bed format for each individual, and calling germline and somatic variants.
Ensemble Variant Calling from Whole Genome Sequencing Data
Short-read sequences from paired blood and saliva sample were each mapped against human genome build GRCh38d1.vd1 using BWA-mem v0.7.17 and sorted by SAMtools(Li et al., 2009). No minimum mapping quality score was required for mapping. Duplicate reads were annotated using Picard MarkDuplicate(Heldenbrand et al., 2019) with a validation stringency set to “STRICT”. Variant calling was performed on the paired mapped reads using four independent variant callers: GATK4 Mutect2 v4.1.4.1(Heldenbrand et al., 2019), Strelka2 v2.9.10(Kim et al., 2018), Varscan2 v2.4.3(Koboldt et al., 2012), and MuSE v.1.0rc(Fan et al., 2016). Any mutation identified by at least 2 of the variant callers was considered genuine. The ensemble variant calling pipeline was validated against 10 previously characterized ICGC PCAWG whole-genome sequenced samples exhibiting over 95% concordance in each sample (Consortium, 2020). Each of the variant callers used the gnomAD hg38 dbSNP file for filtering(Lek et al., 2016). For Mutect2, paired reads were allowed to independently support different haplotypes during initial variant calling and the expected frequency of alleles not found in the germline resource was 0.00003125 as per best-practices approach(Heldenbrand et al., 2019). Contamination table and read orientation models were built from the paired samples and was subsequently used for filtering. For VarScan2, the initial variant calling expected a tumor purity of 0.8 and the subsequent filtering required a minimum coverage of 10 reads and at least 3 alternative reads in tumor with a minimum alternative allele frequency of 0.2. For Strelka2 and MuSE, the default setting for whole genome sequence was used to produce a list of raw and filtered variants.
Analysis of Mutational Signatures and Mutational Patterns
Mutational patterns were generated using SigProfilerMatrixGenerator (Bergstrom et al., 2019) and mutational signatures analysis was performed using our well-established SigProfiler computational framework (Alexandrov et al., 2020). Briefly, the framework identifies the set of mutational signatures that optimally explain the observed mutational patterns without overfitting these mutational patterns. The analysis revealed that clock-like signatures SBS1 and SBS5 were sufficient to recapitulate the patterns observed in MPN samples from both CD34+ stem cells and bulk blood.
Variant Annotation & Filtering
Peripheral blood variants were annotated with Oncotator (Ramos et al., 2015) from a multisample VCF file. We filtered variants using the strategy of (Sukhai et al., 2019) to obtain somatic variants from tumor only samples, retained insertions, deletions, and nonsynonymous variants with ExAC, 1000 Genomes, and gnomad population allele frequency < 0.002. Variants with ClinVar clinical significance of “benign” ere removed. We also removed variants present in three normal controls.
Structural Variant and Copy Number Analysis
Lumpy (Layer et al., 2014) and Manta (Chen et al., 2016) were used to call SV structural variants. SVs not annotated as imprecise but present in both callers (Jeffares et al., 2017) were annotated and prioritized with AnnotSV (Geoffroy et al., 2018) SVs were subsequently filtered to exclude those present in 1000 Genomes Project and gnomad, and ranked 1–4 by AnnotSV. SVs present in the three normal controls were also removed from all samples.
CNVkit was used to discover somatic copy number variants with the batch command and -m wgs parameter. The three normal controls were pooled together for use as a normal panel. Circos plots of variations were created using circlize (Gu et al., 2014; Zhang et al., 2013).
RNA-sequencing read preprocessing
RNA-Seq was performed on Illumina’s NextSeq 500 sequencer with 150bp paired-end reads. Sequencing data were de-multiplexed and output as fastq files using Illumina’s bcl2fastq (v2.17).
RNA editing analysis
RNA reads were aligned using 2-pass alignment with STAR 2.5.2b 2-pass alignment. Alignment deduplication was performed with Picard MarkDuplicates followed for SortSam. Alignments were then processed sequentially according to GATK best practices for calling RNA-Seq variants with tools SplitNCigarReads, RealignerTargetCreator, IndelRealigner, BaseRecalibrator, PrintReads. Variants were called with HaplotypeCaller and filtered with VariantFiltration for FS < 30, QD > 2, QUAL > 20(79). Mismatches in first 6 base pairs of each read were discarded. Alu sites were identified and kept from RepeatMasker. Non-Alu variants were further processed: We removed those in repetitive regions based on the RepeatMasker annotation. Intronic sites within 4bp of splicing junctions were removed. Next, we filtered variants in homopolymer runs. All sites were then kept if there were a minimum of three alternative allele carrying reads and ten total reads and a minimum allele frequency of 0.10. We then identified known RNA editing sites according to RADAR (Ramaswami and Li, 2014) and DARNED (Kiran and Baranov, 2010). For patients without matched whole genome sequencing data, there is a non-zero probability that copy number changes could result in false positive editing sites, but the extensive filtering steps should minimize these instances. To filter mismatches to ADAR specific RNA edits, we kept A to G variants in genes on the positive strand and T to C variants on the negative strand (Kiran and Baranov, 2010; Piskol et al., 2013; Ramaswami and Li, 2014; Ramaswami et al., 2012; Ramaswami et al., 2013). Previously, unreported editing sites were predicted with patient data where there were matching RNA and DNA samples. Notably, we only predicted RNA editing sites with sufficient DNA coverage (> 10 reads/site) to compare mismatches and excluded sites that were identified as DNA variants. RNA edits were annotated with Oncotator and further filtered to remove sites that exist in ExAC, 1000 Genomes Project, and dbSNP. Sites were annotated with variant classification (3’UTR, 5’UTR, 5’ Flank, nonsynonymous, synonymous, Silent, Intron, ncRNA, IGR. Differential editing analysis was performed using a Chi-Square test compare the differences in editing in each gene for each variant classification (i.e. MDM2–3’UTR MF vs AN). Significance was set at p < 0.05. The contingency table for each test was set up as follows:
| Condition 1 | Condition 2 | |
|---|---|---|
| Edited | N sites | N sites |
| Not Edited | N possible sites N sites | N possible sites N sites |
N sites is the number of aggregated sites where N possible sites is the number of uniquely edited coordinates within a variant classification * number of samples. Genes with only intergenic differentially editing events were removed. To account for multiple testing, adjusted p-values were calculated using the Benjamini-Hochberg procedure and genes with events below an adjusted p-value of 0.05 were called significant and retained in the final lists.
Transcript and gene quantification and differential expression
Quality control of the raw fastq files was performed using the software tool FastQC (Andrews, S. & Others. FastQC: a quality control tool for high throughput sequence data. (2010). Sequencing reads were aligned to the human genome (hg19) using the STAR v2.5.1a aligner (Dobin et al., 2013). Read and transcript quantification was performed with RSEM(Li and Dewey, 2011) v1.3.0 and GENCODE annotation (genocode.v19.annotation.gtf). The R BioConductor packages edgeR (Robinson et al., 2010) and limma (Ritchie et al., 2015) were used to implement the limma-voom(Law et al., 2014) method for differential expression analysis at both the gene and transcript levels. The experimental design was modeled upon disease and tissue type (~0 + disease; ~0 +tissue; ~0 + disease + tissue). Significance was defined by using an adjusted p-value cut-off of 0.05 after multiple testing correction using a moderated t-statistic in Limma. Genes or transcripts with an adjusted p-value of < 0.05 (based on the moderated t-statistic using the Benjamini-Hochberg (BH) method for multiple testing correction [27]) were considered significantly differentially expressed (DE) (Benjamini et al., 2001). Functional enrichment of the differentially expressed genes/transcripts was performed using Signaling Pathway Impact Analysis with the Bioconductor package SPIA (Tarca et al., 2009). Gene Set Enrichment Analysis was performed with the Bioconductor package GSVA (Hanzelmann et al., 2013).
Network analysis of differentially edited genes
Significantly differentially edited genes were used as seeds for network propagation (Cowen et al., 2017) on the STRING high confidence interactome (Szklarczyk et al., 2015) for three comparisons (AML vs MF, AML vs Aged Normal, MF vs Aged Normal). The most proximal genes to the seed set were identified using a network propagation method, using degree-matched sampling to generate proximity z-scores for each gene in the network. Genes with a z-score >2 were retained in the network and used for visualization and downstream analysis. A graph-based modularity maximization clustering algorithm was used to identify groups of genes within the most proximal genes which were highly interconnected. Genes in the entire network and within each of these clusters were annotated with associated pathways identified by functional enrichment analysis, with the gprofiler tool (Reimand et al., 2007) using the proximal gene set as the background gene list for enrichment of the clusters and the STRING interactome genes as the background for the entire network enrichment.
Network visualization and propagation was performed using Cytoscape (Shannon et al., 2003) and VisJS2jupyter (Rosenthal et al., 2018). The subgraph composed of the most proximal genes is visualized using a modified spring-embedded layout algorithm, modified by cluster membership, so that genes belonging to the same cluster are separated from other clusters. Differential expression log fold change was mapped to the node color, for the significantly differentially expressed genes (FDR<0.05) within the subgraph.
scRNA-seq analysis
tSNE visualization: For each cell, read counts per gene were transformed into count probabilities 𝒑 by dividing by total number of reads detected from that cell. A suitable distance metric defined between any two cells i and j is the Jensen-Shannon divergence (Lin, 1991):
where is the entropy of the count probability distribution for cell i. Index k spans all genes.
We are interested in knowing whether a particular independently defined set of genes discriminates between samples 1 and 2 (control and ADAR knockdown, respectively). Let us denote this gene set by S = {k1, …, kn}. In order to properly calculate distance between cells defined by gene set S, we need to put all the reads from genes not in S into one bin, so that the transformed probabilities become a vector of length n + 1:
It is these marginalized count probabilities with reduced dimension that enter the Jensen-Shannon formula above. Suppose there are N cells in the experiment (N = N1 + N2), where N1 is the number of cells in Sample 1 and N2 is the number of cells in Sample 2. The distance matrix is an N × N matrix of distances in an n-dimensional space, which we project into two dimensions using t-distributed stochastic neighbor embedding (tSNE) (van der Maaten and Hinton, 2008). The two dimensions are called tSNE 1 and tSNE 2. The main parameter of the tSNE method, perplexity, was set to 50. This value is in the range where the results do not visibly depend on perplexity.
Lentiviral overexpression and shRNA knockdown
Lentiviral human wild-type and mutant ADAR1E912A (pCDH-EF1-T2A-copGFP) and shRNA targeting ADAR1 were produced according to published protocols (Zipeto et al., 2016). All lentiviruses were tested by transduction of 293T cells and transduction efficiency was assessed by qRT-PCR. Lentiviral transduction of primary patient samples was performed at a MOI of 100 to 200. The cells were cultured for 3 to 4 days in 96-well plates (2X105-5X105 cells per well) containing StemPro (Life Technologies) media supplemented with human IL-6, stem cell factor (SCF), Thrombopoietin (Tpo) and FLT-3 (all from R&D Systems) (Abrahamsson et al., 2009; Goff et al., 2013; Jiang et al., 2013b; Zipeto et al., 2016). The transduced cells were collected for RNA extraction and cDNA was synthesized according to published methods (Abrahamsson et al., 2009; Goff et al., 2013; Jiang et al., 2013b; Zipeto et al., 2016).
In vivo humanized mouse model of MF
Human CD34+ cells isolated from MF744 (JAK2 V617F+) patient blood were transduced with pCDH lentivirus control or ADAR1-OE lentivirus with a MOI of 100 for 48 hours, followed by intravenous transplantation into adult NSG-S mice (NSG-SGM3), expressing human IL-3, GM-CSF and SCF, 24 hours after 300 cGy of irradiation. Following engraftment at 13 weeks post-transplantation, BM and spleen were collected and processed. Engraftment in BM and spleen of each mouse was analyzed by flow cytometry.
Generation of stable cell lines
TF1a cells were cultured in RPMI medium supplemented with 10% fetal bovine serum. Cells were transduced with pLKO.1 shScrambled or pLKO.1 shADAR1 lentiviral vectors, respectively. Stable knockdown was confirmed via Western Blot and cells were expanded.
INFα Treatment
Cells were treated with a single dose of IFNα (R&D Systems) at 10ng/ml 16hrs before harvest and analysis.
Protein Extraction and Western Blot
Cells were harvested and washed twice with ice-cold PBS before being resuspended in lysis buffer (20mM Tris pH7.5, 150mM NaCl, 5% Glycerol, 0.5% NP-40, freshly added protease inhibitor cocktail). Cells were lysed on ice for 15min, then centrifuged at 16,000*g for 10min to get rid of insoluble material. Supernatant was boiled in 5x SDS buffer (250mM Tris pH6.8, 40% Glycerol, 10% SDS, 0.01% Bromphenol Blue, 5% beta-mercaptoethanol) for 5min, then resolved by SDS-PAGE and transferred onto PVDF membranes. Membranes were incubated with 5% BSA in TBS-T for blocking and probed with primary and secondary antibodies diluted in 5% BSA in TBS-T.
APOBEC3C lentiviral Vectors
Lentiviral human wild-type APOBEC3C (pCDH-EF1-T2A-copGFP) was cloned by Eton Biosciences. Mutant APOBEC3CE68Q lacking catalytic activity was created by introducing a single G-to-C point mutation in the active site of APOBEC3C using the QuikChange II site-directed mutagenesis kit (Agilent). All lentiviruses were tested by transduction of 293T cells and transduction efficiency was assessed by qRT-PCR. Lentiviral transduction of primary patient samples was performed at a MOI of 100 to 200. The cells were cultured for 48 to 72 hours in 96-well plates (2X105–5X105 cells per well) containing StemPro (Life Technologies) media supplemented with human IL-6, stem cell factor (SCF), Thrombopoietin (Tpo) and FLT-3 (all from R&D Systems) (Abrahamsson et al., 2009; Goff et al., 2013; Jiang et al., 2013b; Zipeto et al., 2016). The transduced cells were collected for RNA extraction and cDNA was synthesized according to published methods (Abrahamsson et al., 2009; Goff et al., 2013; Jiang et al., 2013b; Zipeto et al., 2016), or collected into sterile PBS containing 2% FBS for staining and flow cytometry analysis.
APOBE3C WT and mutant flow cytometry
A minimum of 200,000 CD34 selected normal mixed donor cord blood cells lentivirally transduced for 48 or 72 hours with pCDH backbone, APOBEC3C and APOBEC3CE68Q mutant were blocked using anti-human FcR blocking reagents (Miltenyi Biotec) and then subjected to the following stains: Dapi for live cell discrimination, CD34-APC (BD Biosciences, Clone 8G12), CD38-PEcy7 (BD Biosciences, Clone HB7), CD3-APCcy7 (Biolegend, Clone 17A2), CD14-PerCPcy5.5 (Biolegend, Clone HCD14), CD19-PE (BioLegend, CloneHIB14). Cells were analyzed on a Miltenyi Biotec MACS Quant flow cytometer. Upon debris, doublet, and dead cell exclusion, samples were analyzed for abundance of each differentiation marker, including hematopoietic stem cells (CD3-CD14-CD19-CD34+CD38-), and hematopoietic progenitors (CD3-CD14-CD19-CD34+CD38+).
ADAR and APOBEC3C Co-Immunoprecipitation
HEK293T cells were transfected at 90% confluence with either pCDH, ADAR1 + pCDH, APOBEC3C-FLAG, or ADAR1 + APOBEC3C. Cells were collected after 72 hours into non-denaturing lysis buffer for 30 minutes. Collection of starting material (SM) at this point. Lysate was bound to Anti-FLAG M2 magnetic beads overnight at 4°C. Supernatant was removed and saved to check binding efficiency (flow through, FT). Beads boiled in 1X SDS-2-mercaptoethanol loading buffer and loaded into gel. Gels probed for ADAR1 and 𝛽-actin loading control.
Phosph-STAT3 flow cytometry
The samples were incubated with NearIR Live/Dead at 1:1000 at room temperature in the dark for 15 minutes. Samples were then blocked with anti-mouse and anti-human FcR for 20 minutes in the dark at 4C. Afterwards, samples were stained with CD34 BV421 at 1:100 and incubated for 20 minutes in the dark at 4C. Next, samples were fixed with 0.8% PFA and permeabilized with 1x saponin. Samples were incubated with pSTAT3 FITC at 1:10 overnight. Samples were ran on the MACS Quant 10 Analyzer and analyzed utilizing FlowJo.
Immunofluorescence
The slides for immunofluorescence were prepared by diluting cells (4 × 105 cells/ml) in PBS. 200 microliters of cells were spotted on microscope slides by cytospin at 1,000 rpm for 10 minutes at room temperature. After cytospin, the slides were transferred into a coplin jar containing ice-cold PBS incubate for 5 min; transferred into ice-cold CSK buffer (10 mM PIPES, pH 6.8; 100 mM NaCL; 300 mM sucrose; 3 mM MgCl2) incubate 1 min; transferred into ice-cold CSKT buffer incubate for 5 min; transferred into ice-cold CSK buffer for 1 min; transferred into 4% paraformaldehyde in PBS incubate for 10min at room temperature. Immunofluorescence was performed by immersing slides in PBST (1x PBS with 0.1% Tween-20) for several minutes. Slides were overlaid with 250 microliters of blocking solution (1x PBS, 1% fetal bovine serum, 0.1% Tween-20) for 1 hour at room temperature. Blocking solution was removed and 100 microliters of primary antibody was added to the cells and incubated for 3 hours at room temperature. The slides were washed 2 times in coplin jars with PBST for 5 min each at room temperature. Secondary antibody was overlaid to spotted cells for 1 hour in the dark. Slides were washed in coplin jars with PBST 2x at room temperature. DAPI was added and the slides were sealed with a coverslip. Imaging was performed using an Olympus Fluoview confocal microscope.
Quantification and Statistical Analysis
Statistical analyses
Data are shown as mean ± SEM in all graphs and statistical differences were calculated using a two-tailed unpaired Student’s test, unless otherwise indicated. P values < 0.05 were considered significant. The number of n (number of patients or number of experimental repeats) are indicated in each figure legend, All statistical analyses and plots were produced in GraphPad Prism or R (v3.3.3).
Additional Resources
The authors have no additional resources to provide.
Supplementary Material
Table S2. Gene Expression Derived from scRNA-seq Analysis of cord blood CD34+ cells following lentiviral ADAR1 shRNA knockdown (see attached excel sheet). Related to Figure 3 and Supplemental Figure 4.
Table S1. Patient Characteristics. Related to Figure 1.
HIGHLIGHTS.
Inflammation-dependent APOBEC3C C-to-T deaminase fuels human HSPC expansion
C-to-T mutagenesis increases during human MPN pre-LSC evolution
Inflammation induced ADAR1p150 isoform expression promotes hyperediting in pre-LSC
JAK2/STAT3 inhibition and shRNA ADAR1 knockdown prevent STAT3 isoform switching in LSC
ACKNOWLEDGEMENTS
We would like to thank Human Longevity Inc (HLI) for whole-genome sequencing analysis guidance. We would like to thank our funding agencies for their vital support, including NIH/NCI R01CA205944, NIH/NIDDK R01DK114468-01, NIH/NCI 2P30CA023100-28, CIRM TRAN1-10540, MPN Research Foundation, LLS Blood Cancer Discoveries, NASA NRA NNJ13ZBG001N and NIH/NCATS UL1TR001442. Q.J. was supported by NIH/NCI K22 CA229606. F.H. was funded by the Swedish Childhood Cancer Foundation, Barncancerfonden. Also, we would like to thank the Koman Family Foundation, the Strauss Family Foundation, the Sanford Stem Cell Clinical Center, the UC San Diego Moores Cancer Center, and the Moores Family Foundation for their generous support.
Footnotes
Declaration of Interests: The authors declare no competing interests.
REFERENCES AND NOTES:
- Abrahamsson AE, Geron I, Gotlib J, Dao KH, Barroga CF, Newton IG, Giles FJ, Durocher J, Creusot RS, Karimi M, et al. (2009). Glycogen synthase kinase 3beta missplicing contributes to leukemia stem cell generation. Proceedings of the National Academy of Sciences of the United States of America 106, 3925–3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, Boot A, Covington KR, Gordenin DA, Bergstrom EN, et al. (2020). The repertoire of mutational signatures in human cancer. Nature 578, 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, et al. (2013a). Signatures of mutational processes in human cancer. Nature 500, 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Campbell PJ, and Stratton MR (2013b). Deciphering signatures of mutational processes operative in human cancer. Cell Rep 3, 246–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao EL, Nandakumar SK, Liao X, Bick AG, Karjalainen J, Tabaka M, Gan OI, Havulinna AS, Kiiskinen TTJ, Lareau CA, et al. (2020). Inherited myeloproliferative neoplasm risk affects haematopoietic stem cells. Nature 586, 769–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Drai D, Elmer G, Kafkafi N, and Golani I (2001). Controlling the false discovery rate in behavior genetics research. Behav Brain Res 125, 279–284. [DOI] [PubMed] [Google Scholar]
- Bergstrom EN, Huang MN, Mahto U, Barnes M, Stratton MR, Rozen SG, and Alexandrov LB (2019). SigProfilerMatrixGenerator: a tool for visualizing and exploring patterns of small mutational events. BMC Genomics 20, 685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman T, Broome MA, Sinibaldi D, Wharton W, Pledger WJ, Sedivy JM, Irby R, Yeatman T, Courtneidge SA, and Jove R (2001). Stat3-mediated Myc expression is required for Src transformation and PDGF-induced mitogenesis. Proceedings of the National Academy of Sciences of the United States of America 98, 7319–7324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buisson R, Langenbucher A, Bowen D, Kwan EE, Benes CH, Zou L, and Lawrence MS (2019). Passenger hotspot mutations in cancer driven by APOBEC3A and mesoscale genomic features. Science 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns MB, Lackey L, Carpenter MA, Rathore A, Land AM, Leonard B, Refsland EW, Kotandeniya D, Tretyakova N, Nikas JB, et al. (2013a). APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 494, 366–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns MB, Temiz NA, and Harris RS (2013b). Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat Genet 45, 977–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Li Y, Lin CH, Chan TH, Chow RK, Song Y, Liu M, Yuan YF, Fu L, Kong KL, et al. (2013). Recoding RNA editing of AZIN1 predisposes to hepatocellular carcinoma. Nature medicine 19, 209–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Kallberg M, Cox AJ, Kruglyak S, and Saunders CT (2016). Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32, 1220–1222. [DOI] [PubMed] [Google Scholar]
- Chua BA, Van Der Werf I, Jamieson C, and Signer RAJ (2020). Post-Transcriptional Regulation of Homeostatic, Stressed, and Malignant Stem Cells. Cell Stem Cell 26, 138–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, and Getz G (2013). Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 31, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium I.T.P.-C.A.o.W.G. (2020). Pan-cancer analysis of whole genomes. Nature 578, 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowen L, Ideker T, Raphael BJ, and Sharan R (2017). Network propagation: a universal amplifier of genetic associations. Nat Rev Genet 18, 551–562. [DOI] [PubMed] [Google Scholar]
- Di Giorgio S, Martignano F, Torcia MG, Mattiuz G, and Conticello SG (2020). Evidence for host-dependent RNA editing in the transcriptome of SARS-CoV-2. Sci Adv 6, eabb5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Chen G, Cai Z, Li Z, Qiu L, Xu H, Yuan Y, Liu XL, and Liu J (2018). CDK13 RNA Over-Editing Mediated by ADAR1 Associates with Poor Prognosis of Hepatocellular Carcinoma Patients. Cell Physiol Biochem 47, 2602–2612. [DOI] [PubMed] [Google Scholar]
- Eide CA, and Druker BJ (2017). Understanding cancer from the stem cells up. Nature medicine 23, 656–657. [DOI] [PubMed] [Google Scholar]
- Fan Y, Xi L, Hughes DS, Zhang J, Zhang J, Futreal PA, Wheeler DA, and Wang W (2016). MuSE: accounting for tumor heterogeneity using a sample-specific error model improves sensitivity and specificity in mutation calling from sequencing data. Genome Biol 17, 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genomes Project C, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, et al. (2015). A global reference for human genetic variation. Nature 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoffroy V, Herenger Y, Kress A, Stoetzel C, Piton A, Dollfus H, and Muller J (2018). AnnotSV: an integrated tool for structural variations annotation. Bioinformatics 34, 3572–3574. [DOI] [PubMed] [Google Scholar]
- Gishizky ML, Johnson-White J, and Witte ON (1993). Efficient transplantation of BCR-ABL-induced chronic myelogenous leukemia-like syndrome in mice. Proc Natl Acad Sci U S A 90, 3755–3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff DJ, Recart AC, Sadarangani A, Chun HJ, Barrett CL, Krajewska M, Leu H, Low-Marchelli J, Ma W, Shih AY, et al. (2013). A Pan-BCL2 inhibitor renders bone-marrow-resident human leukemia stem cells sensitive to tyrosine kinase inhibition. Cell Stem Cell 12, 316–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg L, Abutbul-Amitai M, Paret G, and Nevo-Caspi Y (2017). Alternative Splicing of STAT3 Is Affected by RNA Editing. DNA Cell Biol 36, 367–376. [DOI] [PubMed] [Google Scholar]
- Grinfeld J, Nangalia J, Baxter EJ, Wedge DC, Angelopoulos N, Cantrill R, Godfrey AL, Papaemmanuil E, Gundem G, MacLean C, et al. (2018). Classification and Personalized Prognosis in Myeloproliferative Neoplasms. N Engl J Med 379, 1416–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunewald J, Zhou R, Garcia SP, Iyer S, Lareau CA, Aryee MJ, and Joung JK (2019). Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature 569, 433–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Gu L, Eils R, Schlesner M, and Brors B (2014). circlize Implements and enhances circular visualization in R. Bioinformatics 30, 2811–2812. [DOI] [PubMed] [Google Scholar]
- Guallar D, Fuentes-Iglesias A, Souto Y, Ameneiro C, Freire-Agulleiro O, Pardavila JA, Escudero A, Garcia-Outeiral V, Moreira T, Saenz C, et al. (2020). ADAR1-Dependent RNA Editing Promotes MET and iPSC Reprogramming by Alleviating ER Stress. Cell Stem Cell 27, 300–314 e311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L, Diao L, Yu S, Xu X, Li J, Zhang R, Yang Y, Werner HM, Eterovic AK, Yuan Y, et al. (2015). The Genomic Landscape and Clinical Relevance of A-to-I RNA Editing in Human Cancers. Cancer Cell 28, 515–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanzelmann S, Castelo R, and Guinney J (2013). GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartner JC, Walkley CR, Lu J, and Orkin SH (2009). ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat Immunol 10, 109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldenbrand JR, Baheti S, Bockol MA, Drucker TM, Hart SN, Hudson ME, Iyer RK, Kalmbach MT, Kendig KI, Klee EW, et al. (2019). Recommendations for performance optimizations when using GATK3.8 and GATK4. BMC Bioinformatics 20, 557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai H, Karian P, and Kikyo N (2011). Regulation of embryonic stem cell self-renewal and pluripotency by leukaemia inhibitory factor. Biochem J 438, 11–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iniguez AB, Stolte B, Wang EJ, Conway AS, Alexe G, Dharia NV, Kwiatkowski N, Zhang T, Abraham BJ, Mora J, et al. (2018). EWS/FLI Confers Tumor Cell Synthetic Lethality to CDK12 Inhibition in Ewing Sarcoma. Cancer Cell 33, 202–216 e206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizuka JJ, Manguso RT, Cheruiyot CK, Bi K, Panda A, Iracheta-Vellve A, Miller BC, Du PP, Yates KB, Dubrot J, et al. (2019). Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature 565, 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager R, Gisslinger H, Fuchs E, Bogner E, Milosevic Feenstra JD, Weinzierl J, Schischlik F, Gisslinger B, Schalling M, Zorer M, et al. (2020). Germline Genetic Factors Influence Outcome of Interferon Alpha Therapy in Polycythemia Vera. Blood. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamieson CH, Ailles LE, Dylla SJ, Muijtjens M, Jones C, Zehnder JL, Gotlib J, Li K, Manz MG, Keating A, et al. (2004). Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med 351, 657–667. [DOI] [PubMed] [Google Scholar]
- Jeffares DC, Jolly C, Hoti M, Speed D, Shaw L, Rallis C, Balloux F, Dessimoz C, Bahler J, and Sedlazeck FJ (2017). Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat Commun 8, 14061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Crews LA, Barrett CL, Chun HJ, Court AC, Isquith JM, Zipeto MA, Goff DJ, Minden M, Sadarangani A, et al. (2013a). ADAR1 promotes malignant progenitor reprogramming in chronic myeloid leukemia. Proc Natl Acad Sci U S A 110, 1041–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Crews LA, Holm F, and Jamieson CHM (2017). RNA editing-dependent epitranscriptome diversity in cancer stem cells. Nat Rev Cancer 17, 381–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Crews LA, and Jamieson CH (2013b). ADAR1 promotes malignant progenitor reprogramming in chronic myeloid leukemia. Proceedings of the National Academy of Sciences of the United States of America 110, 1041–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Q, Isquith J, Zipeto MA, Diep RH, Pham J, Delos Santos N, Reynoso E, Chau J, Leu H, Lazzari E, et al. (2019). Hyper-Editing of Cell-Cycle Regulatory and Tumor Suppressor RNA Promotes Malignant Progenitor Propagation. Cancer Cell 35, 81–94 e87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor U, Licht K, Amman F, Jakobi T, Martin D, Dieterich C, and Jantsch MF (2020). ADAR-deficiency perturbs the global splicing landscape in mouse tissues. Genome Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Scheffler K, Halpern AL, Bekritsky MA, Noh E, Kallberg M, Chen X, Kim Y, Beyter D, Krusche P, et al. (2018). Strelka2: fast and accurate calling of germline and somatic variants. Nat Methods 15, 591–594. [DOI] [PubMed] [Google Scholar]
- Kiran A, and Baranov PV (2010). DARNED: a DAtabase of RNa EDiting in humans. Bioinformatics 26, 1772–1776. [DOI] [PubMed] [Google Scholar]
- Kleppe M, Koche R, Zou L, van Galen P, Hill CE, Dong L, De Groote S, Papalexi E, Hanasoge Somasundara AV, Cordner K, et al. (2018). Dual Targeting of Oncogenic Activation and Inflammatory Signaling Increases Therapeutic Efficacy in Myeloproliferative Neoplasms. Cancer Cell 33, 29–43 e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, and Wilson RK (2012). VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22, 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komor AC, Kim YB, Packer MS, Zuris JA, and Liu DR (2016). Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Z, Markovets A, Ahdesmaki M, Chapman B, Hofmann O, McEwen R, Johnson J, Dougherty B, Barrett JC, and Dry JR (2016). VarDict: a novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res 44, e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, and Smyth GK (2014). voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 15, R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layer RM, Chiang C, Quinlan AR, and Hall IM (2014). LUMPY: a probabilistic framework for structural variant discovery. Genome Biol 15, R84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzari E, Mondala PK, Santos ND, Miller AC, Pineda G, Jiang Q, Leu H, Ali SA, Ganesan AP, Wu CN, et al. (2017). Alu-dependent RNA editing of GLI1 promotes malignant regeneration in multiple myeloma. Nat Commun 8, 1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing S (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JH (1991). Divergence Measures Based on the Shannon Entropy. Ieee T Inform Theory 37, 145–151. [Google Scholar]
- Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, Nellaker C, Vesely C, Ponting CP, McLaughlin PJ, et al. (2014). The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep 9, 1482–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisel M, Hinterleitner R, Pacis A, Chen L, Earley ZM, Mayassi T, Pierre JF, Ernest JD, Galipeau HJ, Thuille N, et al. (2018). Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficient host. Nature 557, 580–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesa RA, Jamieson C, Bhatia R, Deininger MW, Fletcher CD, Gerds AT, Gojo I, Gotlib J, Gundabolu K, Hobbs G, et al. (2017). NCCN Guidelines Insights: Myeloproliferative Neoplasms, Version 2.2018. J Natl Compr Canc Netw 15, 1193–1207. [DOI] [PubMed] [Google Scholar]
- Miles LA, Bowman RL, Merlinsky TR, Csete IS, Ooi AT, Durruthy-Durruthy R, Bowman M, Famulare C, Patel MA, Mendez P, et al. (2020). Single-cell mutation analysis of clonal evolution in myeloid malignancies. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusse R, and Clevers H (2017). Wnt/beta-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 169, 985–999. [DOI] [PubMed] [Google Scholar]
- Pardanani A, Finke C, Abdelrahman RA, Lasho TL, and Tefferi A (2013). Associations and prognostic interactions between circulating levels of hepcidin, ferritin and inflammatory cytokines in primary myelofibrosis. Am J Hematol 88, 312–316. [DOI] [PubMed] [Google Scholar]
- Peng X, Xu X, Wang Y, Hawke DH, Yu S, Han L, Zhou Z, Mojumdar K, Jeong KJ, Labrie M, et al. (2018). A-to-I RNA Editing Contributes to Proteomic Diversity in Cancer. Cancer Cell 33, 817–828 e817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petljak M, Alexandrov LB, Brammeld JS, Price S, Wedge DC, Grossmann S, Dawson KJ, Ju YS, Iorio F, Tubio JMC, et al. (2019). Characterizing Mutational Signatures in Human Cancer Cell Lines Reveals Episodic APOBEC Mutagenesis. Cell 176, 1282–1294 e1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piskol R, Ramaswami G, and Li JB (2013). Reliable identification of genomic variants from RNA-seq data. Am J Hum Genet 93, 641–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami G, and Li JB (2014). RADAR: a rigorously annotated database of A-to-I RNA editing. Nucleic Acids Res 42, D109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami G, Lin W, Piskol R, Tan MH, Davis C, and Li JB (2012). Accurate identification of human Alu and non-Alu RNA editing sites. Nat Methods 9, 579–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami G, Zhang R, Piskol R, Keegan LP, Deng P, O’Connell MA, and Li JB (2013). Identifying RNA editing sites using RNA sequencing data alone. Nat Methods 10, 128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos AH, Lichtenstein L, Gupta M, Lawrence MS, Pugh TJ, Saksena G, Meyerson M, and Getz G (2015). Oncotator: cancer variant annotation tool. Hum Mutat 36, E2423–2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimand J, Kull M, Peterson H, Hansen J, and Vilo J (2007). g:Profiler--a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res 35, W193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal SB, Len J, Webster M, Gary A, Birmingham A, and Fisch KM (2018). Interactive network visualization in Jupyter notebooks: visJS2jupyter. Bioinformatics 34, 126–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi DJ, Jamieson CH, and Weissman IL (2008). Stems cells and the pathways to aging and cancer. Cell 132, 681–696. [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Patnaik SK, Taggart RT, Kannisto ED, Enriquez SM, Gollnick P, and Baysal BE (2015). APOBEC3A cytidine deaminase induces RNA editing in monocytes and macrophages. Nat Commun 6, 6881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, Kennedy JA, Schimmer AD, Schuh AC, Yee KW, et al. (2014). Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 506, 328–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signer RA, Magee JA, Salic A, and Morrison SJ (2014). Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature 509, 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon O, Di Segni A, Cesarkas K, Porath HT, Marcu-Malina V, Mizrahi O, SternGinossar N, Kol N, Farage-Barhom S, Glick-Saar E, et al. (2017). RNA editing by ADAR1 leads to context-dependent transcriptome-wide changes in RNA secondary structure. Nat Commun 8, 1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukhai MA, Misyura M, Thomas M, Garg S, Zhang T, Stickle N, Virtanen C, Bedard PL, Siu LL, Smets T, et al. (2019). Somatic Tumor Variant Filtration Strategies to Optimize Tumor-Only Molecular Profiling Using Targeted Next-Generation Sequencing Panels. The Journal of Molecular Diagnostics 21, 261–273. [DOI] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al. (2015). STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43, D447–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, et al. (2017). The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res 45, D362–D368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan MH, Li Q, Shanmugam R, Piskol R, Kohler J, Young AN, Liu KI, Zhang R, Ramaswami G, Ariyoshi K, et al. (2017). Dynamic landscape and regulation of RNA editing in mammals. Nature 550, 249–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarca AL, Draghici S, Khatri P, Hassan SS, Mittal P, Kim JS, Kim CJ, Kusanovic JP, and Romero R (2009). A novel signaling pathway impact analysis. Bioinformatics 25, 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tefferi A, Guglielmelli P, Lasho TL, Gangat N, Ketterling RP, Pardanani A, and Vannucchi AM (2018). MIPSS70+ Version 2.0: Mutation and Karyotype-Enhanced International Prognostic Scoring System for Primary Myelofibrosis. J Clin Oncol 36, 1769–1770. [DOI] [PubMed] [Google Scholar]
- Tefferi A, Vaidya R, Caramazza D, Finke C, Lasho T, and Pardanani A (2011). Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: a comprehensive cytokine profiling study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 29, 1356–1363. [DOI] [PubMed] [Google Scholar]
- van der Maaten L, and Hinton G (2008). Visualizing Data using t-SNE. J Mach Learn Res 9, 2579–2605. [Google Scholar]
- Vannucchi AM, Verstovsek S, Guglielmelli P, Griesshammer M, Burn TC, Naim A, Paranagama D, Marker M, Gadbaw B, and Kiladjian JJ (2017). Ruxolitinib reduces JAK2 p.V617F allele burden in patients with polycythemia vera enrolled in the RESPONSE study. Ann Hematol 96, 1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstovsek S, Gotlib J, Mesa RA, Vannucchi AM, Kiladjian JJ, Cervantes F, Harrison CN, Paquette R, Sun W, Naim A, et al. (2017). Long-term survival in patients treated with ruxolitinib for myelofibrosis: COMFORT-I and -II pooled analyses. J Hematol Oncol 10, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, Catalano JV, Deininger M, Miller C, Silver RT, et al. (2012). A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med 366, 799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Keng VW, Patmore DM, Kendall JJ, Patel AV, Jousma E, Jessen WJ, Choi K, Tschida BR, Silverstein KA, et al. (2016). Insertional Mutagenesis Identifies a STAT3/Arid1b/beta-catenin Pathway Driving Neurofibroma Initiation. Cell Rep 14, 1979–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yacoub A, Mascarenhas J, Kosiorek H, Prchal JT, Berenzon D, Baer MR, Ritchie E, Silver RT, Kessler C, Winton E, et al. (2019). Pegylated interferon alfa-2a for polycythemia vera or essential thrombocythemia resistant or intolerant to hydroxyurea. Blood 134, 1498–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zafra MP, Schatoff EM, Katti A, Foronda M, Breinig M, Schweitzer AY, Simon A, Han T, Goswami S, Montgomery E, et al. (2018). Optimized base editors enable efficient editing in cells, organoids and mice. Nat Biotechnol 36, 888–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Meltzer P, and Davis S (2013). RCircos: an R package for Circos 2D track plots. BMC Bioinformatics 14, 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang WC, and Slack FJ (2016). ADARs Edit MicroRNAs to Promote Leukemic Stem Cell Activity. Cell Stem Cell 19, 141–142. [DOI] [PubMed] [Google Scholar]
- Zhou C, Sun Y, Yan R, Liu Y, Zuo E, Gu C, Han L, Wei Y, Hu X, Zeng R, et al. (2019). Off-target RNA mutation induced by DNA base editing and its elimination by mutagenesis. Nature. [DOI] [PubMed] [Google Scholar]
- Zipeto MA, Court AC, Sadarangani A, Delos Santos NP, Balaian L, Chun HJ, Pineda G, Morris SR, Mason CN, Geron I, et al. (2016). ADAR1 Activation Drives Leukemia Stem Cell Self-Renewal by Impairing Let-7 Biogenesis. Cell Stem Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S (2012). A quality control analysis tool for high throughput sequencing data. [Google Scholar]
- Benjamin DS, Cibulskis T, Getz K, Stewart G, Lichtenstein C (2019). Calling Somatic SNVs and Indels with Mutect2. [Google Scholar]
- Consortium I.T.P.-C.A.o.W.G. (2020). Pan-cancer analysis of whole genomes. Nature 578, 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust GG, and Hall IM (2014). SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics 30, 2503–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoffroy V, Herenger Y, Kress A, Stoetzel C, Piton A, Dollfus H, and Muller J (2018). AnnotSV: an integrated tool for structural variations annotation. Bioinformatics 34, 3572–3574. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Crews LA, Barrett CL, Chun HJ, Court AC, Isquith JM, Zipeto MA, Goff DJ, Minden M, Sadarangani A, et al. (2013). ADAR1 promotes malignant progenitor reprogramming in chronic myeloid leukemia. Proceedings of the National Academy of Sciences of the United States of America 110, 1041–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. [Google Scholar]
- Martin M. (2011). Cutadapt Removes Adapter Sequences From High-Throughput Sequencing Reads. [Google Scholar]
- Obenchain V, Lawrence M, Carey V, Gogarten S, Shannon P, and Morgan M (2014). VariantAnnotation: a Bioconductor package for exploration and annotation of genetic variants. Bioinformatics 30, 2076–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picardi E, and Pesole G (2013). REDItools: high-throughput RNA editing detection made easy. Bioinformatics 29, 1813–1814. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal SB, Len J, Webster M, Gary A, Birmingham A, and Fisch KM (2018). Interactive network visualization in Jupyter notebooks: visJS2jupyter. Bioinformatics 34, 126–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talevich E, Shain AH, Botton T, and Bastian BC (2016). CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput Biol 12, e1004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, and Prins P (2015). Sambamba: fast processing of NGS alignment formats. Bioinformatics 31, 2032–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarca AK, P. Draghici S (2020). SPIA: Signaling Pathway Impact Analysis (SPIA) using combined evidence of pathway over-representation and unusual signaling perturbations. R package version 2.42.0,. [Google Scholar]
- Wang K, Li M, and Hakonarson H (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research 38, e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Sun Z, Wang J, Huang H, Kocher JP, and Wang L (2014). CrossMap: a versatile tool for coordinate conversion between genome assemblies. Bioinformatics 30, 1006–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2. Gene Expression Derived from scRNA-seq Analysis of cord blood CD34+ cells following lentiviral ADAR1 shRNA knockdown (see attached excel sheet). Related to Figure 3 and Supplemental Figure 4.
Table S1. Patient Characteristics. Related to Figure 1.
Data Availability Statement
The sequencing datasets generated during this study are available at dbGAP, accession number PHS002228.v1.p1. The analysis code and documentation of computational analyses are available through Github: https://github.com/ucsd-ccbb/MPN_atlas_methods. Data from previous studies are available. Normal HSPC, CML CP progenitor, and BC CML progenitor gene expression are available at PRJNA214016. Cord blood CD34+ transduced with pCDH, ADAR1 WT, or ADAR1E912A are available at PRJNA319866.