

SUMMARY
Cancer immunotherapies often modulate macrophage effector function by introducing either targeting antibodies that activate Fcγ receptors (FcγRs) or blocking antibodies that disrupt inhibitory SIRPα-CD47 engagement. However, how these competing signals are integrated is poorly understood, raising questions about how to effectively titrate immune responses. Here, we find that macrophage phagocytic decisions are regulated by the ratio of activating ligand to inhibitory ligand over a broad range of absolute molecular densities. Using both endogenous and chimeric receptors, we show that activating:inhibitory ligand ratios of at least 10:1 are required to promote phagocytosis of model antibody-opsonized CD47-inhibited targets and that lowering that ratio reduces FcγR phosphorylation because of inhibitory phosphatases recruited to CD47-bound SIRPα. We demonstrate that ratiometric signaling is critical for phagocytosis of tumor cells and can be modified by blocking SIRPα, indicating that balancing targeting and blocking antibodies may be important for controlling macrophage phagocytosis in cancer immunotherapy.
In brief
Suter et al. use reconstituted cell-like particles to quantitatively probe how macrophages integrate simultaneous signals from activating Fc receptors and inhibitory SIRPα. The authors show that the ratio of antibody:CD47 on the target changes relative enrichment of competing Syk kinase and SHP1 phosphatase at the interface, ultimately dictating phagocytosis.
Graphical Abstract
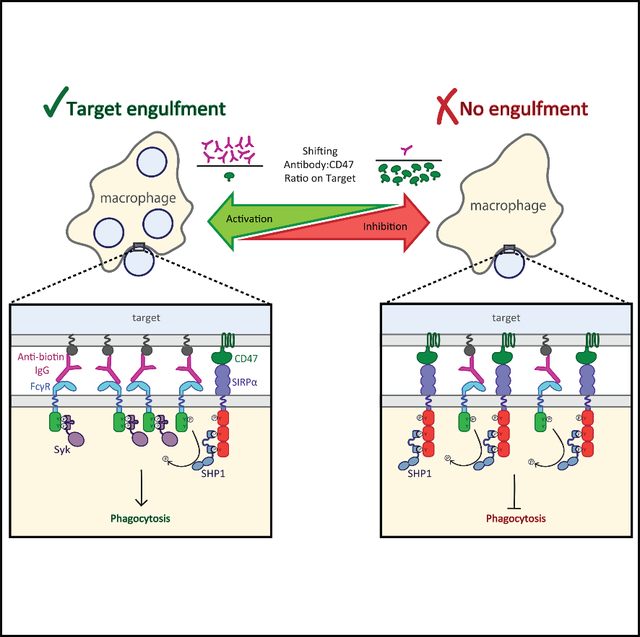
INTRODUCTION
Macrophages play a critical role in cancer immunotherapy by recognizing and destroying antibody-opsonized cells. The clinical success of therapeutic antibodies, including anti-CD20 clearance of chronic lymphocytic leukemia cells (Chu et al., 2018; VanDerMeid et al., 2018) and anti-CD38 clearance of multiple myeloma (van de Donk and Usmani, 2018), has been attributed, in part, to macrophage destruction of tumor cells via antibody-dependent cellular phagocytosis (ADCP). However, potent inhibitory molecules found on tumor cells, such as the ubiquitous “marker of self” CD47, can dampen the pro-phagocytic effect of therapeutic antibodies (Okazawa et al., 2005; Oldenborg et al., 2000). Although the SIRPα-CD47 inhibitory checkpoint is critically important for avoiding the destruction of healthy cells, it is commonly hijacked by tumor cells, which can upregulate CD47 expression from two to six times normal surface expression to avoid recognition and destruction (Willingham et al., 2012).
Disrupting the SIRPα-CD47 inhibitory checkpoint has become an important immunotherapeutic target in recent years (Barclay and Van den Berg, 2014; Feng et al., 2019; Russ et al., 2018), with antibodies that block SIRPα and CD47 intended to increase macrophage effector function. Promising clinical and mouse studies have shown that CD47 blockade therapies can bolster the efficacy of therapeutic antibodies (Chao et al., 2010; Majeti et al., 2009; Theocharides et al., 2012; Weiskopf et al., 2016; Yanagita et al., 2017), although concerns remain about the potential for off-target effects. Because high concentrations of a blocking antibody are best for disrupting the SIRPα-CD47 inhibitory checkpoint, but low antibody concentrations are best for minimizing off-target effects, doses must be found that are neither ineffective nor dangerous. Although this can be done empirically (Feng et al., 2019), understanding the molecular mechanisms underlying macrophage decision-making is important for designing effective and safe combination immunotherapies.
Recent work on macrophage Fcγ receptor (FcγR) activation has helped to clarify the molecular mechanisms responsible for antibody-dependent phagocytosis (Bakalar et al., 2018; Freeman et al., 2016), but how CD47 binding activates SIRPα and how SIRPα counteracts FcγR signaling remain active areas of research. Recently, Morrissey et al. (2020) demonstrated that SIRPα-CD47 binding inhibits macrophage integrin activation. Previously, Tsai and Discher (2008) showed that dephosphorylation of non-muscle myosin IIA was a key component of CD47-mediated shutdown of macrophages. However, it remains possible that a more direct interaction between activating and inhibitory receptors could dictate phagocytic signaling.
Here, we quantitatively evaluate macrophage phagocytic decision-making by systematically varying pro-phagocytic antibody and anti-phagocytic CD47 in a reconstituted target system. We find that the ratio of activating and inhibitory ligands, rather than their absolute number, determines macrophage phagocytosis, with an antibody:CD47 ratio of 10:1 necessary to overcome inhibition in the model system. We show that this ratio-dependent behavior is exhibited by both endogenous and chimeric receptors and that shifting this ratio changes SIRPα-mediated FcγR phosphorylation. Finally, we demonstrate that the activation:inhibition ratio is critical for tumor cell phagocytosis, indicating the effectiveness of inhibitory checkpoint immunotherapies will depend on the relative number of antigens and CD47 on target tumor cells.
RESULTS
Ratio of antibody:CD47 drives phagocytosis in a reconstituted target assay
To study competition between inhibitory SIRPα and activating FcγR signals, we used reconstituted, cell-like target particles consisting of silica beads coated in fluorescent supported lipid bilayers (SLBs). Briefly, Ni-NTA-conjugated lipids in the SLB enabled controlled attachment of His-tagged CD47 (inhibitory ligand), and the target particles were opsonized by binding anti-biotin immunoglobulin G (IgG) to biotinylated lipids (activating ligand) (Figure 1A). Importantly, the fluidity of the SLB facilitates the binding and subsequent enrichment of membrane-bound proteins on the target (Joffe et al., 2020). After protein attachment, target particles were added to RAW 264.7 macrophage-like cell line, and phagocytosis was measured by quantifying internalized target particle fluorescence using confocal microscopy. We found that target particles coated only in antibodies were engulfed, but the addition of CD47 reduced phagocytosis to background (no protein) levels, confirming that this inhibitory axis can be reconstituted in our model system (Figure 1B).
Figure 1. Phagocytosis is dependent on ratio of activating antibody to inhibitory CD47.

(A) Experimental setup of the phagocytosis assay of cell-like target particles by RAW 264.7 macrophages.
(B) Quantification of average internalized, SLB-coated target particle fluorescence shows phagocytosis of protein-coated targets as compared with empty (lipid-only) targets. Each condition is the average of three independent experiments representing a total of >300 cells. Bars represent means ± SEM. Conditions were compared with two-tailed Student’s t test. ***p < 0.001.
(C) Experimental setup of target particles with different anti-biotin IgG and CD47 densities. SLB-coated particles were incubated with 15 different combinations of anti-biotin IgG and CD47 concentrations, in addition to a no-protein (lipid-only) control.
(D) Internalized target particle fluorescence quantification for conditions outlined in (C) (left panel). Data replotted as a function of CD47:antibody ratio (right panel). Each condition is the average of three independent experiments representing a total of >300 cells. Bars represent means ± SEM.
(E) Different anti-biotin IgG to CD47 ratios were created at high concentration, then serially diluted 5-fold, three times, creating a total of four concentrations for each ratio. Internalized target-particle fluorescence was quantified for phagocytosis of each particle ratio. Each condition is the average of three independent experiments representing a total of >300 cells. Bars represent means ± SEM.
We next investigated how macrophage phagocytosis depends on ligand density and whether phagocytosis stops when CD47 reaches a threshold density on the target surface. Antibody and CD47 are attached to the target particle orthogonally, enabling us to precisely control the numbers of activating and inhibitory ligands presented to the macrophage. To verify those numbers, we measured surface densities of fluorescentlylabeledCD47andantibodies on target particles using flow cytometry before every assay (Figure S1A). Importantly, estimated macrophage SIRPα and FcγR receptors are in excess of their corresponding ligands on the target particle surface.
We first measured sensitivity to ligand density by creating a panel of target particles combining three different antibody surface densities (150, 300, and 600 molecules/μm2), each with five different CD47 surface densities (0, 4, 10, 20, 50 molecules/μm2; Figure 1C). For comparison, CD47 surface density on red blood cells is approximately 250 molecules/μm2 (Mouro-Chanteloup et al., 2003). We found that phagocytosis scaled inversely with CD47 surface density and did not exhibit a step function at a specific CD47 density, as would be expected for threshold-governed behavior (Figure 1D). Target particles with the highest antibody density overcame CD47 inhibition better than those with lower antibody densities, indicating that phagocytosis was responding to both CD47 and antibody densities. Indeed, by replotting the data as a function of CD47:antibody ratio, we see that phagocytosis is highly sensitive to small changes in relative CD47:antibody densities, rather than to absolute molecule numbers (Figure 1D).
To more directly test that ratio hypothesis, we mixed antibody and CD47 in ratios varying from 1:1 to 50:1 and then serially diluted them before adding proteins to SLB-coated beads (Figure 1E). This enabled us to create target particles with a fixed ratio of molecules while varying the absolute numbers of those molecules by more than 100-fold (Figure S1B). We find that target particles with the same ratio of molecules yield similar levels of phagocytosis across a wide range of surface densities. We note, however, that phagocytosis can be sensitive to absolute molecule numbers when the measured densities of the antibody or CD47 become extremely low (<20 molecules/μm2 anti-biotin antibody and <2 molecules/μm2 CD47). For dilution series in which the first dilution shows lower phagocytosis than subsequent dilutions, we hypothesized that an excess of antibody saturates FcγRs, effectively lowering the bound antibody:CD47 ligand ratio and reducing phagocytosis.
SIRPα-CD47 binding drives exclusion of CD45 and co-localizes with bound FcγRs
We next investigated how the ratio of antibody:CD47 governs phagocytosis by studying the composition of the interface between a macrophage and target particle. To improve quantification of interfacial organization and density, we used total internal reflection fluorescence (TIRF) microscopy to image macrophages undergoing frustrated phagocytosis on SLB-coated glass coverslips containing the same tunable concentrations of antibody and CD47 as the target particles used above (Figure 2A).
Figure 2. SIRPα-CD47 co-localizes with FcγR-antibody complexes and excludes CD45.

(A) Experimental setup of TIRF imaging of macrophages on SLB.
(B) Workflow to quantify enrichment of CD47 and anti-biotin IgG on an SLB within a RAW 264.7 macrophage cell footprint. SLBs with variable concentrations of anti-biotin IgG (magenta) and CD47 (green) were created, and cell footprints were identified from reflection interference contrast microscopy (RICM) images. Average ratio of pixel intensities (anti-biotin IgG:CD47) was quantified inside and outside of footprints.
(C) Average ratio of anti-biotin IgG and CD47 pixel intensities for SLBs of varying composition. Each condition is the average of three independent experiments representing a total of >200 footprints. Bars represent means ± SEM.
(D) Representative TIRF fluorescent images of RAW 264.7 macrophages or GPMVs generated from RAW 264.7 cells on a planar SLB. SLBs were coated with1 nM of anti-biotin IgG (magenta) and 5 nM of CD47 (green). Line scans of representative images show co-localization of anti-biotin IgG and CD47 in cells and GPMV footprints. Scale bar is 5 μm for GPMVs and 10 μm for cell footprints.
(E) FcγR ITAM and SIRPα ITIM phosphorylation by Lyn kinase upon CD45 phosphatase exclusion.
(F) Representative TIRF image of GPMVs from RAW 264.7 macrophages labeled with anti-CD45 (red) on an SLB with CD47 (green). Line scan of representative image shows CD45 exclusion from interface. Scale bar is 5 μm.
(G) Pearson’s correlation coefficient comparing CD47 localization with anti-biotin IgG (D) and CD47 with localization with CD45 (F) in RAW 264.7 macrophage GPMV footprints. Each condition represents >30 GPMV footprints. Conditions were compared with two-tailed Student’s t test. ***p < 0.001.
(H) Fluorescent confocal images of fixed RAW 264.7 macrophages interacting with target particles. Top row shows macrophages interacting with particles coated only in 1 nM of anti-biotin IgG (magenta). Middle row shows particles coated in 1 nM of anti-biotin antibody plus 50 nM CD47, and the bottom row shows particles coated only in 50 nM of CD47. Phosphorylation visualized via anti-phosphotyrosine (cyan). Cell outlines are shown by white dotted lines. Scale bar is 10 μm.
(I) Quantification of anti-phosphotyrosine signal at particle-macrophage interfaces. Each condition is the average of three independent experiments representing a total of >100 cell-bead interfaces. Bars represent means ± SEM. Conditions were compared with two-tailed Student’s t test. **p < 0.01, ***p < 0.001.
We first tested whether receptor engagement at the interface was proportional to the ligand ratio by dropping cells on to planar SLBs with varying antibody:CD47 ratios and measuring enrichment within the macrophage footprint (Figure 2B). We found that average enrichment within the footprint scales with the ratio of ligands present on the SLB, indicating that binding of the two ligand-receptor pairs is independent of each other (i.e., local enrichment of one does not inhibit enrichment of the other) (Figure 2C).
Next, we examined how the size of bound SIRPα-CD47 compared with the size of bound antibody-FcγR. We previously found that macrophage phagocytosis is dependent on size-based segregation of the inhibitory phosphatase CD45 from the antibody-bound FcγR (Bakalar et al., 2018). Specifically, FcγR binding to an antibody targeting the membrane, which creates a membrane-membrane gap of approximately 12 nm (estimated from crystal structures) (Bakalar et al., 2018; Lu et al., 2011), results in efficient phagocytosis because of maximum exclusion of CD45 (Bakalar et al., 2018). This activating interface gap matches with the approximate size of SIRPα-CD47 interaction (~13 nm estimated by crystal structure (Hatherley et al., 2009)). This would allow bound SIRPα-CD47 to be remain in the gap formed by a short phagocytic interface after CD45 is excluded, potentially inhibiting phagocytosis. To test that prediction, we assessed co-localization of the two binding pairs, FcγR-antibody and SIRPα-CD47, using TIRF microscopy of RAW 264.7 cells and giant plasma membrane vesicles (GPMVs) from RAW cells (Sezgin et al., 2012), which do not have an active cytoskeleton. In both the RAW cells and the GPMVs, SIRPα-CD47 and FcγR-antibody were enriched and co-localized in the footprint, indicating that these two opposing pathways can co-exist at an interface because of their similarity in size (Figures 2D and 2G).
Size-dependent segregation of CD45 from the small gap created by FcγR engagement with antibodies is required for sustained phosphorylation of tyrosine residues in immunoreceptor tyrosine-based activation motifs (ITAMs) on activating FcγRs (Bakalar et al., 2018). ITAMs are phosphorylated by cytoplasmic membrane-anchored kinase Lyn—a member of the Src family kinases—which remains present within an FcγR-antibody interface because of its lack of an extracellular domain (Figure 2E). Lyn is also believed to phosphorylate tyrosines on the immunoreceptor tyrosine-based inhibition motifs (ITIMs) of SIRPα, given previous evidence that Lyn can phosphorylate other ITIM-containing receptors and that depletion of Lyn leads to decreased SIRPα phosphorylation (Abram and Lowell, 2008; Scapini et al., 2009). However, the mechanism by which SIRPα phosphorylation is regulated is not well understood.
We hypothesized that phosphorylation of SIRPα ITIMs when engaged with CD47 is similarly regulated by size-dependent exclusion of CD45, which would prevent dephosphorylation of Lyn-phosphorylated ITIMs. To investigate that, we imaged GPMVs from RAW 264.7 cells on SLBs containing CD47, but not antibody, and saw dramatic exclusion of CD45 from interfaces established by only SIRPα-CD47, as quantified by Pearson’s correlation analysis (Figures 2F and 2G). This observation indicates that SIRPα-CD47 binding creates a small enough gap to exclude CD45 and that exclusion of CD45 does not require FcγR engagement.
We next tested whether CD45 exclusion results in increased tyrosine phosphorylation at the interface between macrophages and target particles coated in antibody, CD47, or both ligands (Figure 2H). Using phosphotyrosine immunostaining (not specific to ITAMs or ITIMs), we expected to detect an increase in phosphorylation at target interfaces with both CD47 and antibody, compared with antibody alone, because of the presence of both phosphorylated ITAMs and ITIMs. Surprisingly, phosphorylation at the target particle interface dropped significantly with the addition of CD47, despite similar levels of antibody on the two target-particle populations (Figure 2I). This unexpected result raised the question of why phosphorylation is reduced, rather than increased, when CD45 is excluded in the presence of CD47.
SIRPα enrichment decreases phosphorylation at the macrophage-target interface
Our phosphorylation results indicated that not only were phosphorylated SIRPα ITIMs not contributing to the total phosphorylation at the particle interface but also that phosphorylation of the antibody-bound FcγR ITAMs decreased. To better quantify that, we immunostained macrophages for phosphotyrosine after allowing them to interact with target particles coated with increasing antibody:CD47 ratios. Normalizing the average pixel intensity of the phosphotyrosine antibody by the anti-biotin antibody intensity, we find that the level of phosphorylation per anti-biotin antibody at the interface goes down with increasing CD47 (Figure 3A), indicating that there is proportionally less phosphorylation for each ITAM at the macrophage-target interface when CD47 is present. We confirmed that this decrease in phosphorylation is not caused by diminished particle engagement due to CD47, and the average level of phosphorylation is not dependent on the number of particles engaged to a cell (Figures S2A–S2C). Although our results suggest that the phosphotyrosine antibody reports ITAM phosphorylation, we note that immunostaining may also pick up phosphorylation of additional downstream components of the activation pathway. Despite that, our phosphorylation data shows a titratable decrease in the activation-pathway phosphorylation with increasing CD47.
Figure 3. SIRPα-bound SHP1 dephosphorylates activation pathway to inhibit phagocytosis.

(A) Quantification of the ratio of anti-phosphotyrosine to anti-biotin IgG fluorescence at macrophage-bead interfaces. RAW 264.7 macrophages interacting target particles coated with a fixed density of anti-biotin IgG (300 molecules/μm2) and variable CD47 (0–1250 molecules/μm2). Each condition is the average of three independent experiments representing a total of >150 particle-cell interfaces. Bars represent means ± SEM. Conditions were compared with two-tailed Student’s t test. *p < 0.05, **p < 0.01, ***p < 0.001.
(B) Schematic of SHP1-GFP sensor for detection of phosphorylated SIRPα (left panel). Average SHP1-GFP fluorescence signal within cell footprints was quantified for cells on SLBs coated with different CD47 densities (0, 250, and 2,500 molecules/μm2) with and without anti-biotin IgG (300 molecules/μm2) (right panel). Each condition is the average of three independent experiments representing a total of >100 macrophage footprints. Bars represent means ± SEM. Conditions were compared with two-tailed Student’s t test. *p < 0.05, **p < 0.01.
(C) Schematic of Syk-mCh sensor for detecting phosphorylated FcγRs. RAW 264.7 cells expressing Syk-mCh sensor were imaged on SLBs coated with anti-biotin IgG (300 molecules/μm2) and a range of CD47 densities (0, 250, and 2,500 molecules/μm2). Average fluorescence signal within the cell footprint was measured for anti-biotin IgG and Syk-mCh, and the ratio of Syk-mCh to anti-biotin IgG was quantified for each cell. Cell footprint fluorescence was measured for cells with no pre-treatment (light gray bars) and with treatment with SHP1/2 phosphatase inhibitor NSC-87877 (dark gray bars). Each condition is the average of three experiments representing a total of >200 cell footprints. Bars represent means ± SEM. Conditions were compared with two-tailed Student’s t test. *p < 0.05, **p < 0.01.
(D) Normalized average Syk-EOS (left) and SHP1-EOS (right) binding events per cell for RAW macrophages on SLBs containing anti-biotin IgG (300 molecules/μm2), CD47 (2500 molecules/μm2), or both. Each condition is the average of three independent experiments representing a total of >50 macrophage footprints. Bars represent means ± SEM. Conditions were compared with two-tailed Student’s t test. *p < 0.05, **p < 0.01.
(E) Schematic of computational model combining activating (top) and inhibitory (bottom) pathways. SIRPα-SHP1 dephosphorylation of FcγR captured in the black dotted inset represents the “interacting” version of the model.
(F) “Interacting” and “non-interacting” models were initiated with a fixed-antibody concentration and variable CD47 concentrations, yielding a range of CD47:antibody ratios. Each model for each ratio condition was run until the steady state was reached, and FcγR-bound Syk was plotted.
(G) Ratio of anti-phosphotyrosine to anti-biotin IgG at macrophage-target interfaces was measured for a range of CD47:antibody ratios on target particles, as described in (A). Quantification was done with and without pre-treatment of macrophages with SHP1/2 phosphatase inhibitor NSC-87877. Each condition is the average of three independent experiments representing a total of >150 particle-cell interfaces. Dots represent means ± SEM. Conditions were compared with two-tailed Student’s t test. *p < 0.05, ***p < 0.001.
(H) Schematic of SHP1-facilitated shutoff of activation pathway. In the absence of CD47, FcγR ITAMs are phosphorylated and bind Syk kinase, promoting phagocytosis. Upon CD47 engagement and SIRPα ITIM phosphorylation, SHP1 phosphatase is brought in close proximity to phosphorylated ITAMs, enabling their dephosphorylation.
To test whether we see a similar result over the entire macrophage-target interface, we fixed and stained macrophages on SLBs with and without CD47. Again, we saw proportionally less phosphorylation per antibody enriched in the interface in the presence of CD47 (Figures S2D and S2E).
Close proximity of ITIM-associated phosphatases decreases FcγR phosphorylation
We speculated that decreasing phosphorylation in the presence of CD47 did not occur because CD45 was less excluded at the interface where bound SIRPα-CD47 and antibody-FcγR co-localize but, rather, because soluble phosphatases bind to phosphorylated ITIM domains at the interface (Kharitonenkov et al., 1997; Veillette et al., 1998). Previous work has shown that recruitment of phosphatases SHP1, SHP2, and SHIP to phosphorylated ITIMs is a key regulator of their phosphatase activity (Binstadt et al., 1996; Burshtyn et al., 1997; Neel et al., 2003). SHP1—the most widely studied and directly implicated in downregulation of macrophage activation—is recruited to phosphorylated SIRPα via its tandem N-terminal SH2 domains and has been shown to dephosphorylate a range of targets, including ITAMs and other substrates of Syk kinase (Frank et al., 2004; Kant et al., 2002). In synthetic peptide studies, SHP1 preferentially dephosphorylates ITAM-like targets (Frank et al., 2004), and in cells, overexpression of SHP1 reduces phosphorylation of activating FcγRIIA, dramatically decreasing Syk activation (Huang et al., 2003).
Based on that, we hypothesized that SHP1 dephosphorylates adjacent ITAMs after binding to SIRPα, suppressing FcγR activation. To test whether SHP1 is recruited to the macrophage-target particle interface when CD47 is present, we created an ITIM phosphorylation sensor by replacing the phosphatase domain of SHP1 with GFP (Figure S3A). This sensor serves to both visualize SHP1 recruitment and also to demonstrate SIRPα phosphorylation, which was hard to capture with phosphotyrosine immunostaining. We first dropped RAW cells stably expressing this sensor onto SLBs coated in different concentrations of CD47 or antibody and quantified the SHP1-GFP sensor intensity within the footprint (Figures 3B and S3B). We found that SHP1 sensor recruitment to SLB interfaces containing only CD47 was greater than those with only antibody, with SHP1 increasing as CD47 surface density increased from 250 to 2,500 molecules/μm2. As expected, SHP-1 sensor recruitment decreased upon addition of Src family kinase inhibitor PP2, which should prevent Lyn kinase phosphorylation of ITIMs (Figure S3C).
We next investigated whether co-localization of ITIMs and ITAMs at the interface between the macrophage and target particle reduced ITAM signaling. We speculated that the presence of SHP1 on nearby ITIMs could dephosphorylate ITAMs and reduce FcγR activation by shutting down recruitment of Syk kinase, which drives phosphorylation of downstream targets, leading to phagocytosis (Crowley et al., 1997; Fütterer et al., 1998; Tsang et al., 2008). To test that, we added macrophages to SLBs with different antibody:CD47 ratios and measured the signal from a fluorescent Syk sensor, which we previously developed to monitor phosphorylated ITAMs in real time (Bakalar et al., 2018) (Figure 3C, left). Adding 250 molecules/μm2 of CD47 to the bilayer decreased the amount of Syk sensor recruited, and adding 2,500 molecules/μm2 decreased recruitment even further (Figure 3C, right, light bars).
To investigate whether the presence of CD47 on the target surface altered Syk and SHP1 binding kinetics, we created a second version of each sensor that replaced the GFP or mCherry with photoactivatable mEOS, which is converted from GFP to RFP with UV stimulation. The ability to convert only a small number of mEOS sensor molecules to RFP reduces background of the sensor and enables single molecule imaging. Macrophages expressing either the Syk-EOS or SHP1-EOS sensor were imaged while bound to SLBs coated in antibody, CD47, or both, and images were then analyzed to quantify molecule dwell times (Figure S3E; Videos S1 and S2). We found that dwell times did not change significantly for either Syk-EOS or SHP1-EOS on different SLB compositions (Figures S3F and S3G). However, the number of binding events dropped significantly for Syk-EOS on CD47-coated SLBs and for SHP1-EOS on antibody-coated SLBs, despite similar sensor expression levels (Figures 3D and S3H). Importantly, Syk-EOS binding events also decreased significantly on SLBs containing both antibody and CD47, providing further evidence that CD47 engagement reduces local FcγR phosphorylation.
Model of ITIM-ITAM negative feedback captures ratio-dependent FcγR phosphorylation
We next developed a model of SHP1 recruitment, FcγR phosphorylation, and Syk recruitment to examine whether the observed ratio-dependent phagocytosis (as observed in Figure 1D) could be explained by negative feedback. The simple kinetic model includes parallel pathways of activation and inhibition (Figure 3E; Table S1) that are based on receptor-ligand binding, which drives receptor phosphorylation and subsequent binding of downstream effectors, SHP1 or Syk. Our model assumes that binding drives receptor enrichment and CD45 exclusion, enabling receptor phosphorylation.
We tested two versions of the model, one in which activation and inhibition interact and one in which they do not. In the “non-interacting” version, the pathways proceed independently, with no SHP1 negative feedback on FcγR ITAM phosphorylation. In the “interacting” version, activation and inhibition pathways are connected by a single integration point, dephosphorylation of FcγR ITAMs by SIRPα-bound SHP1 (Figure 3E). Constants used in the model were obtained from the literature, when available (Barua et al., 2012; Brooke et al., 2004; Li et al., 2007; Ren et al., 2011; Selner et al., 2014), and others were estimated (Table S2). The model is initiated with defined concentrations of unbound CD47, antibody, SIRPα, FcγR, SHP1, and Syk. FcγR-bound Syk is used as a proxy for phagocytosis because its localization to the target interface is known to be required for activation pathway signaling (Crowley et al., 1997; Kiefer et al., 1998). As expected, the non-interacting model shows that FcγR-bound Syk is insensitive to varying of the ratio of CD47:antibody concentrations (Figure 3F), whereas the interacting model shows that SHP1 is able to dephosphorylate FcγR ITAMs, resulting in a sharp decrease of FcγR-bound Syk with an increasing CD47:antibody ratio.
To experimentally test the non-interacting model in cells, we incubated macrophages with target particles coated in a range of CD47:antibody ratios (as in Figure 3A) either with or without SHP1/2 inhibitor NSC-87877, which has previously been shown to be specific (Chen et al., 2006). Addition of the inhibitor should prevent SHP1 from dephosphorylating FcγRs and other activation pathway components, allowing phosphorylation to remain high at the target interface. Consistent with our non-interacting model, we found that the amount of phosphorylation per antibody is higher in the presence of inhibitor for each CD47:antibody ratio (Figure 3G), highlighting the key role SIRPα-bound phosphatases has in shutting down phagocytosis. We next tested the effect of SHP1/2 inhibition on Syk recruitment by dropping cells onto SLBs with varying CD47:antibody ratios in the absence or presence of NSC-87877. With the inhibitor, Syk sensor recruitment stayed constant with or without CD47 on the SLB, as expected from the non-interacting model (Figure 3C, right panel, dark bars). To test the functional effect of SHP1 inhibition, we compared phagocytosis by wild-type macrophages to those expressing low or high levels of our SHP1-GFP sensor and found that cells expressing high SHP1-GFP phagocytose more efficiently in the presence of CD47 than do either wild-type or SHP1-GFP low-expressing macrophages (Figure S3D). Expression of the phosphatase-free SHP1-GFP sensor reduced the amount of active phosphatase at the target interface, thereby reducing local dephosphorylation of FcγRs.
Both our ITIM phosphorylation and Syk recruitment data point to a model of macrophage decision-making in which small changes in the CD47:antibody ratio drive shifts in relative enrichment of SIRPα and FcγRs (Figure 3H). This changing SIRPα enrichment alters the relative density of ITIM-bound phosphatases, which can then rapidly and effectively dephosphorylate nearby ITAMs to stop phagocytosis.
The ITAM:ITIM ratio dictates phagocytosis levels, independent of the extracellular binding domain
The critical parameter in our model is the number of antibody-bound FcγRs relative to the number of CD47-bound SIRPα, which we abstract as the ratio of ITAM:ITIM enriched at a target interface. We wondered whether this ratio-dependent phagocytosis was specific to the competition between FcγRs and SIRPα or whether any receptor with an ITAM or ITIM would behave similarly.
To test that, we created a chimeric receptor containing the intracellular ITAM-containing portion of FcγRIIA and the Syn18 leucine zipper sequence as the extracellular binding domain (Thompson et al., 2012) (Figure S4A). As a ligand for the chimeric receptor, we used the complementary SynZip motif, Syn17, attached to three repeats of the FNIII domain (Fibcon), which we used previously as a tool for modulating extracellular protein height (Bakalar etal., 2018),along with a His-tag for attachment to the SLB (Syn17-F3L). Upon binding, the chimeric receptor should create a membrane gap similar to that of SIRPα-CD47, approximately 13 nm. Using target particles coated in Syn17-F3L, we found that the chimeric receptors drove efficient phagocytosis, which was specific to the FcγRIIA ITAM motif (Figure S4B). When CD47 and Syn17-F3L were added in varying ratios on target particles, we found that the chimeric receptor responded in a titratable way to the activation:inhibition ratio, very similar to the endogenous receptors (Figure 4A).
Figure 4. Ratio of activation to inhibition governs phagocytosis in a synthetic context.

(A) Target particles coated in varying ratios of CD47 to Syn17-F3L were incubated with RAW 264.7 macrophages stably expressing the synthetic activating receptor Syn18-F1L-TM-FcγRIIA-GFP. Each condition is the average of three independent experiments representing a total of >300 cells. Bars represent means ± SEM.
(B) RAW 264.7 macrophages stably expressing synthetic receptors containing three different ITAM-containing signaling motifs: FcγRIIA, CD3ζ, and FcεRI γ-chain. Phagocytosis was quantified for target particles with a range of CD47:Syn17-F3L ratios. Each condition is the average of three independent experiments representing a total of >300 cells. Dots represent means ± SEM.
(C) Target particles containing varying ratios ofSyn17-F3L to anti-biotin IgG were incubated with RAW 264.7 macrophages stably expressing synthetic inhibitory receptor Syn18-F1L-TM-SIRPα-GFP. Each condition is the average of three independent experiments representing a total of >300 cells. Bars represent means ± SEM.
(D) RAW 264.7 macrophages stably expressing synthetic receptors containing four different ITIM-containing signaling motifs: FcγRIIB, SIRPα, PD-1, and CD22. Phagocytosis was quantified for target particles with a range of Syn17-F3L:anti-biotin IgG ratios. Each condition is the average of three independent experiments representing a total of >300 cells. Dots represent means ± SEM.
To investigate whether ITAMs from other receptors also exhibit ratio-dependent signaling, we created additional chimeric receptors featuring the intracellular signaling regions of the CD3 ζ-chain of the T cell receptor complex and FcεRI γ chain (Figure S4A). Each receptor construct was identical, with the exception of the signaling domain. We found that macrophages expressing each chimeric receptor responded similarly to Syn17–3L alone on the target surface (Figure S4C) and that each receptor responded similarly to a titration of CD47, suggesting that the ratio dominates phagocytosis, independent of receptor identity (Figure 4B). Interestingly, the ratio of Syn17 to CD47 required to achieve phagocytosis over the background rate was nearly identical to that of the antibody to CD47, approximately 10:1, indicating that ratiometric signaling through synthetic receptors mimics that through endogenous FcγRs.
To determine whether similar rules exist for ITIM-containing domains, we next created a chimeric SIRPα receptor to compete against the activation signal of endogenous FcγRs. The inhibitory receptor design was the same as the activation chimeric receptors, except for replacing ITAM-containing domains with SIRPα’s ITIM-containing intracellular region (Figure S4A). We found that the chimeric SIRPα response is also titratable with a ratio similar to that of the endogenous SIRPα (Figure 4C). Additional inhibitory chimeric receptors featuring the ITIMs of CD22, FcγRIIB, and PD-1 also showed ratio-dependent phagocytosis behavior (Figure 4D). Interestingly, we observed that macrophage phagocytosis with the chimeric SIRPα never fully decreased to zero in response to the now inhibitory Syn17-F3L ligand. We attribute that elevated background engulfment to the high affinity of Syn17-F3L to the Syn18 receptor, independent of the inhibitory signaling (Figure S4E). Taken together, these experiments with chimeric receptors demonstrate that ratio-dependent phagocytic signaling is generalizable to other ITAM and ITIM signaling domains.
We used the same chimeric SIRPα receptor to test how spatial segregation of activation from inhibition influenced phagocytic decision making. To do so, we measured receptor co-localization and compared phagocytosis of particles coated in antibody plus different concentrations of either a short inhibitory ligand (Syn17-F3L, as used previously) or a tall ligand (Syn17-F5L, which includes two additional Fibcon repeats). Using SLB experiments, we found that Syn17-F5L was significantly segregated from the antibody, unlike the Syn17-F3L ligand, which colocalized with the antibody (Figures S4F and S4G). When normalized to cells expressing a non-signaling Syn18-GFP control, particles coated in a low density of Syn17-F5L were phagocytosed more efficiently than were those with Syn17-F3L, indicating that segregation of the inhibition rendered it less potent (Figure S4H) and that co-localization of activation and inhibition is important for shutting down phagocytosis in certain density regimes.
Ratio-dependent phagocytosis of tumor cells can be shifted using blocking antibodies
Finally, we tested whether our model of ratio-dependent signaling was applicable to live tumor cells. Tumor cell surfaces are notably more complex than the two-component, reconstituted target particles used in our assays so far and, therefore, may be subject to additional inhibitory interactions that complicate efforts to overcome inhibitory signaling (Feng et al., 2019).
To conduct tumor-cell phagocytosis assays, we used mouse bone-marrow-derived macrophages (BMDMs) and the mouse colon adenocarcinoma cell line MC38, both of which have been used in previously published tumor cell eating experiments (Morrissey and Vale, 2019; Xu et al., 2017). MC38 cells stably expressed human HER2 protein (MC38-hHER2), a well-characterized tumor antigen overexpressed in 15%–30% of breast cancers (Iqbal and Iqbal, 2014). We opsonized the MC38-hHER2 cells with a mouse anti-hHER2 antibody, which was modified from the clinical hHER2-targeting antibody trastuzumab (McKeage and Perry, 2002). The opsonized tumor cells were incubated with BMDMs for 30 min before imaging (Figure 5A). Importantly, we estimate that macrophage FcγR and SIRPα receptors are in excess of their corresponding ligands on MC38 cells.
Figure 5. Tumor cell phagocytosis is dictated by activation-inhibition ratio.

(A) Representative confocal images of bone-marrow-derived macrophages (BMDMs, magenta) incubated with MC38-hHER2 cells (green), either with or without tumor targeting anti-hHER2 antibody opsonization. White arrows within insets indicate phagocytosed MC38-hHER2 cells. Scale bar is 50 μm.
(B) Quantification of MC38-hHER2 tumor cell phagocytosis by BMDMs. MC38-hHER2 tumor cells were opsonized with a range of anti-hHER2 antibody concentrations (0–5 μg/mL). Each condition is the average of three independent experiments representing >150 total cells. Bars represent means ± SEM. Conditions were compared with two-tailed Student’s t test. **p < 0.01, ***p < 0.001.
(C) Fluorescent confocal images of fixed BMDMs engaging with MC38-hHER2 cells (green) opsonized with different concentrations of anti-hHER2 antibody (0,0.5, and 5 μg/mL, magenta). Phosphorylation visualized via anti-phosphotyrosine (red). BMDM outlines are shown in white dotted lines. Areas of high anti-hHER2 enrichment are highlighted with white arrows. Scale bar is 5 μm.
(D) Anti-SIRPα blocking antibody 18a interrupting SIRPα binding to CD47 (left panel). MC38-hHER2 cells incubated with a range of anti-hHER2 antibody concentrations (0–5 μg/mL) were added to BMDMs in the presence or absence of 5 μg/mL of 18a antibody. Average tumor cell phagocytosis was quantified (right panel). Each condition is the average of three independent experiments representing >150 total cells. Bars represent means ± SEM. Conditions were compared with two-tailed Student’s t test. *p < 0.05, **p < 0.01, ***p < 0.001.
To shift the activation:inhibition ratio we chose to vary the activation signal by titrating the anti-hHER2 antibody, whereas the CD47-SIRP interaction stayed constant. This strategy was informed by clinical evidence that the efficacy of tumor-growth inhibition is correlated to HER2 opsonization levels (McLarty et al., 2009). Over an anti-hHER2 concentration range of 0.01–5 μg/mL, we see a clear increase in tumor cell phagocytosis, consistent with an increased activation:inhibition ratio (Figure 5B). The highest concentration (5 μg/mL) showed a slight decrease in phagocytosis, potentially because of excess anti-hHER2 antibody in the solution saturating unbound FcγRs and preventing their engagement with anti-hHER2 on the tumor cell. To confirm that increased opsonization is accompanied by changes in receptor phosphorylation, we fixed BMDMs interacting with MC38-hHER2 cells and immunostained them for phosphotyrosine. Consistent with our reconstituted target particle experiments, we find that phosphorylation is concentrated at the interface and that regions of antibody enrichment correlate with high levels of phosphorylation (Figure 5C).
Both BMDMs and RAW 264.7 cells exhibit a similar sensitivity to antibody:CD47 ratios on reconstituted target particles (Figure S5A), consistent with BMDMs and RAW cells having similar ratios of FcγR:SIRPα, roughly 2:3 and 1:2, respectively, despite having different absolute numbers of receptors (Figure S5B). From surface measurements of MC38-hHER2 cells, we estimated 26,100 hHER2 molecules/cell and 30,100 CD47 molecules per cell. Surprisingly, at antibody saturation of hHER2, that yields an activation:inhibition ratio of approximately 1:1, much lower than the 10:1 required for our reconstituted target particles. This difference in ratio may be due to many reasons, including differences in antibody affinity and partial segregation of SIRPα-CD47 from antibody-bound FcγR resulting from the size of HER2, suggesting that the optimal ratio must be determined for each specific antibody and antigen.
Finally, we asked whether we could shift the activation-inhibition ratio to improve phagocytic efficiency at lower anti-hHER2 antibody concentrations by blocking SIRPα-CD47 interactions and tipping the balance toward activation. To test that, we used SIRPα antibody 18a, which was developed by Aduro Biotech to bind mouse SIRPα and block CD47 engagement. We found that adding 5 μg/mL of 18a antibody over multiple dilutions of anti-hHER2 antibody systematically raised phagocytosis levels for each concentration of anti-hHER2 (Figure 5D), indicating that phagocytosis of tumor cells is, like the reconstituted target particles, dependent on antibody:CD47 ratios. These data also suggest that SIRPα-CD47 is a primary means of inhibition for these tumor cells because ratiometrically blocking this interaction increases phagocytosis significantly.
DISCUSSION
Immune cells, including macrophages, make important effector function decisions by directly interacting with target cells. Receptor engagement, enrichment, and signaling at cell-cell contacts are critical for determining the response to a target, and understanding those steps at a mechanistic level has the potential to inform efforts to harness macrophage effector functions for therapeutic purposes. This is especially the case when there is competition between inhibitory and activating cues. Here, we ask how macrophages process activating signals from antibodies and inhibitory signals from CD47 on target particles. We show quantitatively that the activating and inhibitory signals are integrated in a ratio-dependent manner, with approximately 10 antigen-bound antibodies required to overcome one CD47. Because each antibody-bound FcγR brings one or two ITAMs to the synapse, whereas each CD47-bound SIRPα brings three ITIMs, our findings estimate that phagocytosis proceeds when there are at least 2–3 ITAMs for every ITIM.
To obtain a mechanistic picture of how macrophages respond to complex stimuli, we examined ligand densities across a regime in which macrophages transition between inhibition and activation of phagocytosis. Ultimately, macrophage decision making comes down to a race between phosphorylation and dephosphorylation of receptors, with Syk and SHP1 in direct competition. Subtle changes in ligand ratio drive changes in relative recruitment of SHP1 (and other phosphatases, such as SHP2 and SHIP) to ITIMs, which rapidly shuts down activation by dephosphorylating nearby ITAMs. Importantly, SHP1 likely dephosphorylates ITIMs as well, as evidenced by low phosphorylation measurements at the interface between macrophages and targets, even in the presence of high SIRPα-CD47 engagement. This suggests that the phosphatase feedback acts not only to shut down activation but also to quickly turn off inhibition, returning the cell to a neutral state, poised to respond to the next potential target. A similar negative-feedback model was proposed for PD-1, in which SHP2 phosphatase recruited to phosphorylated PD-1 clusters suppresses T cell receptor activation (Yokosuka et al., 2012). As such, we speculate that trans-dephosphorylation of activating pathways by proximal phosphatases may be a shutoff mechanism universal to all ITIM motifs.
Interestingly, it appears that the SIRPα-CD47 inhibitory checkpoint recruits SHP1 when CD45 is excluded from the interface, presumably because the gap formed drives size-dependent exclusion of CD45, as originally proposed for T cell receptor activation (Davis and van der Merwe, 2006). Our previous work showed that antigen size is critical for effective ADCP, with shorter antigens more efficiently excluding CD45, which, in turn, allows increased ITAM phosphorylation (Bakalar et al., 2018). We speculate that the similarity in height of SIRPα-CD47 and FcγR-antibody-antigen is not coincidental but, rather, has evolved specifically to shut down phagocytosis at its most efficient point if markers of self are detected. Our findings point to the possibility that CD45’s main role is to maintain non-interacting macrophages in an “off” state by preventing both activating and inhibitory signaling. Exclusion of CD45 upon close contact with a target then enables engaged and enriched receptors of either pathway to signal, with FcγR and SIRPα signaling interpreted ratiometrically.
In our model of macrophage FcγR activation, co-localization of SIRPα-CD47 and FcγR-antibody binders not only reinforces CD45 exclusion but also provides SHP1 access to phosphorylated ITAMs. Our data using different heights of Syn17 ligand suggest that size-dependent segregation of inhibitory signaling can decrease its effectiveness. Consistent with that idea, a study in natural killer (NK) cells found that the inhibitory KIR2DL1-HLAC interaction was most potent when its height was matched to that of the activating NKG2D-MICA receptor-ligand pair (Köhler et al., 2010).
Our data also imply that the most important node for integrating activating, versus inhibitory, signaling is the receptors themselves. In NK cells, it has been suggested that Vav1 acts as an integrative node for inhibition triggered by inhibitory killer cell immunoglobulin-like receptors (KIRs) (Stebbins et al., 2003). However, macrophages lacking Vav1 are still able to phagocytose (Hall et al., 2006). In fact, many commonly cited players in downstream phagocytosis signaling can be removed without preventing productive, FcγR-mediated phagocytosis, including WAVE and Rho proteins (Caron and Hall, 1998; Kheir et al., 2005). This suggests that phagocytosis, which makes use of a complex activation network, is not dependent on a single integrator downstream of the receptors. Instead, signal integration at the membrane enables more direct and localized response to engaged receptors, which may be particularly important when a macrophage is simultaneously interacting with multiple target cells (Dushek et al., 2012). Dephosphorylation of FcγR ITAMs by phosphatases recruited to SIRPα ITIMs may be further modulated by downstream processes that have been previously associated with CD47 on targets, including myosin phosphorylation and integrin activation (Morrissey and Vale, 2019; Tsai and Discher, 2008).
To show the relevance of our ratiometric model beyond reconstituted target particles, we demonstrated that live tumor-cell phagocytosis follows the same requirement for ratiometric activating:inhibitory signaling. Although tumor cells have a far more complex cell surface, our study indicates that careful characterization of receptor and ligand densities on both immune and tumor cells may be important considerations for immunotherapies. More specifically, designing individualized therapeutic dosing informed by surface densities of tumor antigen, CD47, SIRPα, and FcγR could help to optimize results for specific cancers that involve macrophage effector function.
Interestingly, we saw that the ratio of activating:inhibitory ligand necessary for phagocytosis was much less in tumor cell experiments than it was in our reconstituted system, approximately a 1:1 ratio versus a 10:1 ratio, respectively. This difference could be due to variation in antibody affinity toward the antigen or the FcγR, leading to different enrichment profiles and potentially higher activation. CD47 may also be less accessible to SIRPα in the crowded environment of tumor cell membranes than it is in our reconstituted target particle experiments, leading to less receptor engagement and reduced inhibition. In addition, hHER2 is estimated to stand 11 nm above the cell membrane (Bakalar et al., 2018), meaning hHER2-antibody-FcγR would be expected to have a greater height than bound CD47-SIRPα, potentially leading to size-dependent spatial separation of activating and inhibitory signals and reducing the ability of SHP1 to dephosphorylate adjacent ITAMs. Our tumor cell eating experiments may also involve more stable macrophage-target interface facilitated by receptor-ligand interactions not present in the reconstituted system. Although T cell immunological synapse adhesion and stability has been studied extensively (Dustin, 2007; Graf et al., 2007; Philipsen et al., 2013), the effect of macrophage interface stability on signaling efficiency has not yet been completely characterized.
Although extracellular binding domains of immune receptors have been honed for target specificity and affinity, we think that the ratiometric signaling model we propose here may be broadly relevant to ITAM-dependent signaling in different cell types. In previous work in NK cells, overexpression of inhibitory Ly49 ligands on model tumor cells was able to shut down NKG2D-mediated activation in an expression-dependent manner, suggesting that relative amounts of the activating and inhibitory ligands were important in that system (Malarkannan, 2006). Additionally, previous work in microglia has shown that complement receptor CD11b requires ITAM-containing adaptor protein DAP12 (Wakselman et al., 2008), indicating that the ITAM:ITIM ratio is important not only in complement-mediated phagocytosis but also in non-immunoreceptor (e.g., integrin) signaling as well. Probing whether ratio-dependent competition of activating and inhibitory signaling is widely applicable to diverse immune cell types is a question for future investigation that will require measurement of effector function over a range of absolute receptor-surface densities.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Daniel Fletcher (fletch@berkeley.edu).
Materials availability
Plasmids generated in this study can be obtained from the lead contact.
There are restrictions to the availability of reagents obtained through Aduro Biotech. These reagents MC38 cells expressing hHER2, as well as anti-SIRPα antibody clone 18a and mouse IgG1 anti-hHER2 antibody. Anti-SIRPα clone p84 and anti-CD47 antibody MIAP 301 were obtained from Aduro Biotech, but are commercially available.
Data and code availability
Data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All cell lines tested negative for mycoplasma as verified by Mycoalert detection kit (Lonza).
RAW 264.7 macrophage cell culture
RAW 264.7 murine male macrophage-like cell line was obtained from the UC Berkeley Cell Culture Facility. Cells were cultured in RPMI 1640 media (Corning) supplemented with 10% heat-inactivated fetal bovine serum (HI-FBS, Thermo Fisher Scientific) and 1% Pen-Strep (Thermo Fisher Scientific) in non-tissue culture-treated 10 cm dishes (VWR) at 37°C, 5% CO2.
Bone marrow derived macrophage cell culture
Bone marrow derived macrophages (BMDMs) from male C57BL/6 (B6) mice were a kind gift from the Portnoy Lab (UC Berkeley). BMDMs were grown in RPMI 1640 media supplemented with 10% HI-FBS and 1% Pen-Strep at 37°C. BMDMs were used in experiments within 24 hours of thawing.
MC38 cell culture
MC38 female murine colon adenocarcinoma cells stably expressing human HER2 (referred to as MC38-hHER2) were obtained from Aduro Biotech. MC38-hHER2 cells were grown in RPMI 1640 media supplemented with 10% HI-FBS and 1% Pen-Strep at 37°C.
HEK293T cell culture
Human embryonic kidney (HEK) 293T cells were obtained from the Cell Culture facility at UC San Francisco. Cells were cultured in DMEM media (Thermo Fisher Scientific) supplemented with 10% HI-FBS and 1% Pen-Strep at 37°C.
METHOD DETAILS
Preparation of cell-like reconstituted target particles
Cell-like reconstituted target-particles were generated according to previously published protocol (Joffe et al., 2020), summarized in the following sections.
Formation of small unilamellar vesicles (SUVs)
SUVs were prepared by rehydrating a lipid film composed primarily of POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine, Avanti Polar Lipids), doped with 0.5% of Biotinyl Cap PE (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(cap biotinyl), Avanti Polar Lipids), 1% DGS-Ni-NTA (1,2-dioleoyl-sn-glycero-3-[(N-(5-amino1-carboxypentyl)iminodiacetic acid)succinyl] with nickel salt, Avanti Polar Lipids), and 0.4% LISS-Rhodamine (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl), Avanti Polar Lipids) in pure deionized H2O. The rehydrated solution was vortexed briefly and then sonicated using a tip-sonicator at 20% power pulsing off and on for 3 minutes. SUVs were then filtered through a 200 nm PTFE filter (Millipore). SUV solutions were stored at 4°C and used within 48 hours of preparation to avoid phospholipid oxidization.
Formation of cell-like target particles
40 μL of 4.07 μm silica beads (Bangs Laboratories) were cleaned using a 3:2 mixture of H2SO4:H2O2 (Piranha) for 30 minutes while sonicating. Cleaned beads were spun down at 1000 × g and washed 3 times with 1 mL pure water before being resuspended in 400 μL of water. Cleaned beads were stored at room temperature. To form SLBs, 40 μL of SUVs was added to 160 μL of MOPS buffer (25 mM MOPS (3-(N-morpholino)propanesulfonic acid), Thermo Fisher Scientific), 125 mM NaCl, pH 7.4), along with 20 μL of clean bead slurry. The mixture was incubated at room temperature for 15 minutes while rotating continuously. Coated beads (also referred to as target particles) were spun down at 50 × g for 1 minute, and 200 μL of the solution was removed and replaced with PBS (Corning).
Addition and quantification of protein on particles
AlexaFluor647-labeled anti-biotin mouse IgG (clone BK-1/39, Thermo Fisher Scientific) and AlexaFluor488-labeled His-tagged recombinant mouse CD47 (SinoBiological) were diluted in PBS to appropriate concentrations for target particle experiments. 50 μL of SLB-coated beads was added to 50 μL of protein dilution, and beads were incubated at room temperature for 20 minutes with continuous rotation.
SLB and protein-coated target particles were diluted in PBS and analyzed using an Attune NxT flow cytometer (Thermo Fisher Scientific). Target particles were compared to calibrated beads with known numbers of AlexaFluor488 and AlexaFluor647 fluorophores (Quantum MESF Kits, Bangs Laboratories), which enabled calculate of protein surface density.
Fluorescent labeling of proteins
His-tagged recombinant mouse CD47 (as well as proteins and antibodies noted later) was labeled using AlexaFluor488 NHS Ester (Succinimidyl Ester, Thermo Fisher Scientific) reconstituted in anhydrous DMSO (dimethylsulfoxide, Sigma Aldrich). Dye was mixed with protein at a 5x molar ratio (dye:protein ratio was 5:1) and incubated at room temperature for 1 hour. Excess dye was removed by purifying protein over NAP-5 columns (GE Healthcare). Labeling was confirmed using NanoDrop 2000c (Thermo Fisher).
Imaging techniques
All live cells were maintained at 37°C, 5% CO2 with a stage top incubator (Okolab) during imaging. For confocal microscopy, cells were imaged with a spinning disk confocal microscope (Eclipse Ti, Nikon) with a spinning disk (Yokogawa CSU-X, Andor), CMOS camera (Zyla, Andor), and either a 20x objective (Plano Fluor, 0.45NA, Nikonor a 60x objective (Apo TIRF, 1.49NA, oil, Nikon). For total internal reflection fluorescence (TIRF) microscopy, cells were imaged with TIRF microscope (Eclipse Ti, Nikon), 60x objective (Apo TIRF, 1.49NA, oil, Nikon) and EMCCD camera (iXON Ultra, Andor). Both microscopes were controlled with Micro-Manager. Images were analyzed and prepared using FIJI (Schindelin et al., 2012).
Phagocytosis assays
Target particle phagocytosis assay
35,000 macrophages were seeded in wells of a tissue-culture flat-bottom 96-well plate (Falcon) in 100 μL of RPMI 1640 medium. Post-seeding, cells were incubated at 37°C for 3–4 hours prior to target particle addition. 100 μL of target particles were prepared for each well with appropriate protein concentrations, and 95 μL of the 100 μL were added. Macrophages were incubated with target particles at 37°C for exactly 20 minutes. During those 20 minutes, the remaining 5 μL were diluted in 250 μL of PBS and immediately measured using flow cytometry as previously described.
After incubation, wells were washed twice with PBS to remove excess particles. PBS containing 1 μM of CellTracker Green (CMFDA, Thermo Fisher Scientific) and 10 μM Hoechst 33342 (Thermo Fisher Scientific) was added to wells to stain the cytoplasm and nuclei, respectively. Wells were imaged after 10 minutes of staining using a spinning-disk confocal microscope (Nikon) at 20x. Images were acquired in an automated grid pattern at the same location within each well to reduce bias in image acquisition. For each well, at least 100 cells were imaged. Images were than analyzed using a custom CellProfiler (v2.1.1, Broad Institute) (Carpenter et al., 2006) project program to identify single cells and quantify the internalized target particle fluorescence intensity (of LISS-Rhodamine lipid) within each cell. Average internalized fluorescence per cell was calculated per condition. Independent replicates were conducted on different days, and replicates were normalized to average eating for all conditions to control for day-to-day variation.
Target particle binding assay
To assess particle engagement, macrophages were seeded in 96-well plates and target particles were prepared as with the phagocytosis assay. Upon addition of target particles to macrophage wells, the 96-well plate was incubated at 4°C for 30 minutes to enable binding but prevent engulfment. Cells were then rinsed of excess beads and stained with 1 μM of CellTracker Green (CMFDA, Thermo Fisher Scientific) and 10 μM Hoechst 33342 (Thermo Fisher Scientific) while kept on ice. Images were acquired and analyzed using a custom CellProfiler (v2.1.1) project program to identify bound beads. Average number of bound beads per cell was calculated per condition. Independent replicates were conducted on different days, and replicates were normalized to average eating for all conditions to control for day-to-day variation.
Tumor cell phagocytosis assay
After thawing and resuspending in RPMI 1640 medium, BMDMs were incubated with 1 μM of CellTracker Deep Red (Thermo Fisher Scientific) at room temperature for 10 minutes. Cells were washed 2 times in PBS to remove excess dye, resuspended in RPMI 1640 medium and seeded at 40,000 cells per well in a tissue-culture flat-bottom 96-well plate. Cells were incubated at 37°C overnight (approximately 18 hours prior to tumor cell addition).
MC38-hHER2 cells were incubated in 1 μM Cell Tracker Green in PBS for 10 minutes at room temperature. Cells were washed 2 times in PBS to remove excess dye, and then resuspended in RPMI 1640 media. AlexaFluor647-labeled anti-hHER2 antibody (mouse IgG2a isotype targeting clinical Trastuzumab hHER2 epitope, Aduro Biotech) was serially diluted with MC38-hHER2 cells to achieve appropriate final concentrations for opsonization. MC38-hHER2 cells were coated in anti-hHER2 antibody dilutions 20 minutes prior to addition to BMDMs wells. When applicable, 18a antibody (SIRPα-blocking antibody, Aduro Biotech) was added directly to seeded BMDMs 20 minutes prior to MC38-hHER2 cell addition. 40,000 MC38-hHER2 cells (in 100 μL) were added to each well of 40,000 BMDMs to achieve a 1:1 ratio.
MC38-hHER2 cells were incubated with BMDMs for exactly 30 minutes. Wells were washed 2 times with PBS to remove excess MC38-hHER2 cells. Wells were imaged using the same microscope and automated technique as the target particle phagocytosis assay. Images were analyzed using a CellProfiler program that identified single BMDM cells and quantified the internalized MC38-hHER2 fluorescence intensity (CellTracker Green) within each BMDM cell. Average internalized fluorescence per cell was calculated per condition. Independent replicates were conducted on different days, and replicates were normalized to average eating for all conditions to control for day-to-day variation.
Generation of cell lines
Design of synthetic receptors
The alpha helical SYNZIP Syn18-Syn17 binding pair was chosen due to its antiparallel orientation (Thompson et al., 2012). For each synthetic receptor design, the 41 amino acid sequence of Syn18 was placed N-terminally to one FNIII domain (Jacobs et al., 2012) to control the height above the membrane of the synthetic receptor protein (Figure S2A). The extracellular part was followed by the transmembrane domain of mouse SIRPα (Accession #P97797, amino acid 374–394), followed by intracellular regions of FcγRIIA (Accession #P12318, amino acid 240–317), CD3ζ (Accession #24161, amino acid 52–164), FcεRI γ-chain (Accession #P20491, amino acid 45–86), SIRPα (Accession #P97797, amino acid 395–513), CD22 (Accession #P35329, amino acid 722–862), PD-1 (Accession #Q02242, amino acid 191–288), and FcγRIIB (Accession #P08101, amino acid 232–330). A C-terminal GFP facilitated cell sorting and validated receptor localization. The entirety of each of the annotated intracellular domains was ordered as a separate gene fragment (Integrated DNA Technologies) and each complete receptor was cloned into pHR lentiviral expression vector (Clontech). Each region of the protein was amplified using PCR and fragments were combined using Gibson assembly.
Design of Syk and SHP1 sensors
mCh-Syk live phosphosensor was designed and utilized as previously published (Bakalar et al., 2018).
SHP1 interacts with phosphorylated SIRPα through interaction with its tandem SH2 domains. We created a sensor that specifically localizes to phosphorylated ITIMs by fusing the two SH2 domains of SHP1 to GFP to enable visualization of intracellular localization of the expressed protein. SH2 domain sequences were obtained from Addgene (pGEX-SHP-1(NC)-SH2, A #46496) (Machida et al., 2007) and a linker region (GGGSGGGS) was placed between the SH2 domains and GFP. The sensor was cloned into pHR lentiviral expression vector under control of low-expression UBC promoter and high-expression SFFV promoter.
Additional mEOS constructs were created using Syk and SHP1 SH2 domains as described above. Each was cloned into pHR lentiviral expression vector under control of low-expression UBC promoter.
Generation of stable cell lines
HEK293T cells were grown to 70% confluency in a 6-well tissue-culture plate, and 160 ng pDM2.G (Clontech), 1.3 μg CMV 8.91 (Clontech), and 1.5 μg of target vector were transfected using TransIT-293T transfection reagent (Mirus Bio). Viral supernatant was collected 60 hours after transfection and spun at 1000 × g to remove HEK293T cells and used immediately. 1 mL of lentiviral supernatant was added to 5×105 RAW 264.7 macrophages with 4 μg/mL of hexadimethrine bromide (Millipore) and cells were spinoculated at 300 × g for 30 minutes at room temperature. Cells were resuspended and plated into a 10 cm non-tissue-culture dish. Cells were sorted via fluorescence-activated cell sorting using an Aria Fusion cell sorter (Becton Dickinson) and the sorted population was expanded for use. For cell lines expressing synthetic receptors, function of Syn18 receptors was confirmed using AlexaFluor647-labeled soluble Syn17-F3L (see Purification of Syn17-F3L).
Purification of Syn17-F3L
Design of Syn17-F3L and Syn17-F5L
To create the Syn18 receptor ligands, the 42-amino acid sequence of Syn17 (Thompson et al., 2012) was followed by three or five repeats of the synthetic FNIII domain (Jacobs et al., 2012). Three Fibcon repeats were used to achieve a receptor-ligand interaction height that corresponds to FcγR-antibody, and five repeats were used to intentionally increase receptor-ligand height. Finally, a C-terminal His-10 allowed for attachment to Ni-NTA lipids. Each region of the protein was amplified via PCR and fragments were combined using Gibson assembly into pET28 vector (Millipore) for expression in E. coli.
Syn17-F3L and Syn17-F5L protein expression and purification
Syn17-F3L or Syn17-F5L was expressed in Rosetta DE3 competent cells (Millipore). Cells were grown at 37°C to OD = 0.8 and expression was induced with 0.3 mM IPTG (Isopropyl b-D-1-thiogalactopyranoside, Calbiochem) overnight at 18°C. Cells were pelleted and resuspended in 25 mM HEPES pH 7.4 (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, Thermo Fisher Scientific), 150 mM NaCl, 0.5 mM TCEP (tris(2-carboxyethyl)phosphine, Thermo Fisher Scientific) and 10 mM imidazole (Thermo Fisher Scientific), and lysed. After centrifugation, lysate was affinity purified using Cobalt-charged His-Trap Chelating column (GE Healthcare) through imidazole gradient elution. Peak fractions were gel-filtered using Superdex 200 column (GE Healthcare) into 25 mM HEPES pH 7.4, 150 mM NaCl, 0.5 mM TCEP. Protein was then labeled with AlexFluor647 NHS Ester (Thermo Fisher Scientific) as described in “Fluorescent labeling of proteins.”
SLB TIRF imaging and quantification
Formation of planar SLBs
To image footprints of macrophages and GPMVs, planar SLBs were formed onto coverslips via fusion of SUVs to RCA-cleaned glass coverslips. PDMS (Polydimethylsiloxane, Sylgard) placed atop RCA-cleaned glass formed the imaging chamber, and 50 μL of MOPS buffer and 50 μL of non-fluorescent SUV solution were added. The SUVs were incubated for 20 minutes at room temperature. Excess SUVs were then removed by gently washing 5x with 60 μL of PBS. Appropriate antibody or CD47 dilutions were prepared, 60 μL were added to the washed SLB and incubated for 20 minutes. Excess protein was removed by gently washing the SLB 2x with PBS. The fluidity of proteins on the SLB was confirmed by using a spinning-disk confocal (Nikon) to examine diffusion of labeled molecules after photobleaching a small region of interest.
GPMV formation
GPMVs were made according to the protocol outlined by Sezgin et al. (2012). RAW 264.7 macrophages were seeded in a 6-well plate (Falcon). After adhering, they were rinsed with 1 mL GPMV buffer (10 nM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), 150 mM NaCl, 2 mM CaCl2, pH 7.4) before addition of 1 mL of vesiculation buffer (GPMV buffer plus 25 mM PFA (paraformaldehyde, Electron Microscopy Services) and 2 mM DTT (1,4-dithiothreitol, Anaspec). Cells were incubated for 1 hour at 37°C and GPMVs were collected by removing supernatant from cells.
TIRF imaging of GPMV and cell interfaces
Macrophages or GPMVs were added directly to planar SLBs containing AlexaFluor647-labeled anti-biotin IgG, AlexaFluor488-labeled CD47, or both. Cells or GPMVs were allowed to settle for 20 minutes prior to imaging. Engaged GPMVs or cells were located and identified using reflection interference contrast microscopy (RICM). Images were acquired using RICM and TIRF microscopy for all relevant fluorescent channels. When applicable, CD45 was labeled with 10 nM anti-CD45 antibody (clone 30-F11 Brilliant Violet 421, BioLegend).
Quantification of TIRF images
Images were quantified by identifying ROIs in RICM so as to not bias analysis by segmenting on a fluorescent channel. RICM images were flattened using a background (no cells or GPMV) image, then outlines of SLB-bound cells or GPMVs were identified. Fluorescence inside and outside these ROI outlines was measured and averaged for each fluorescent channel. The outside background measurement was subtracted from the average signal inside of the footprint. This was implemented using an automated custom macro in FIJI (Schindelin et al., 2012). Data was processed and analyzed using Python 3.5 (http://python.org).
Single molecule imaging and quantification
Macrophages expressing SHP1-EOS and Syk-EOS were incubated on planar SLBs containing unlabeled anti-biotin IgG, CD47, or both. Cells were allowed to settle for 20 minutes prior to imaging. Cells were imaged for EOS expression using 488 nm excitation in TIRF. Single molecules were then converted to RFP using 50 ms pulse of 405 nm light. Timelapse images were acquired every 30 ms using a 561 nm excitation. Timelapse images were then analyzed using MATLAB TrackNTrace package (Stein and Thiart, 2016) and a custom MATLAB script to identify binding events and quantify molecule dwell times.
Staining and quantification of phosphotyrosine
Fixation of macrophages with target particles
To stain for phosphotyrosine at macrophage-target interfaces, macrophages were seeded into 8-well imaging chambers with a coverslip glass bottom (Cellvis) and allowed to adhere for at 3–4 hours. If applicable, cells were treated with 3 μM SHP1/2 phosphatase inhibitor (NSC-87877, Sigma-Aldrich) for 20 minutes prior to target particle addition. Target particles coated in non-fluorescent SLBs plus AlexaFluor647-labeled anti-biotin IgG and/or AlexaFluor488-labeled CD47 were added to the seeded cells. After exactly 10 minutes at 37°C, cells were fixed for 10 minutes with 4% PFA in PBS. Cells were then permeabilized using 0.2% Tween-20 (Thermo Fisher Scientific) for 10 minutes and rinsed twice with PBS. Non-specific binding was blocked with 3% (w/v) bovine serum albumin (BSA, ChemCruz) in PBS with 0.5 g/mL Fc Block (BD Biosciences) for 30 minutes. Phosphotyrosine antibody (P-Y-1000 MultiMab, Cell Signaling Technology 8954S) was added to cells at 1:500 dilution in 1% BSA and incubated at room temperature for 1 hour. Cells were rinsed twice with 1% BSA, then incubated with a secondary antibody (AlexaFluor546-labeled goat anti-rabbit IgG, Invitrogen A11010) at a dilution of 1:1000 in 1% BSA for 1 hour at room temperature. Cells were washed twice with PBS before fluorescence and brightfield imaging with spinning-disk confocal microscope.
Interfaces between cells and bound (but not internalized) target particles were manually identified using brightfield images. Average pixel intensity within the interface was quantified using an automated custom macro in ImageJ.
Fixation and quantification of macrophages on SLB
To stain phosphotyrosine within footprints, macrophages were dropped onto a planar SLB and incubated at 37°C for 10 minutes. After exactly 10 minutes, cells were fixed and stained according to “Fixation of macrophages with beads.” Cells were imaged using RICM and TIRF imaging, and fluorescence within the footprint was measured as described in “Quantification of TIRF images.”
Fixation of BMDMs with tumor cells
To stain phosphotyrosine at BMDM-MC38 interfaces, 60,000 BMDMs were seeded into 8-well imaging chambers with a coverslip glass bottom. 40,000 MC38-hHER2 cells were incubated with various concentrations of AlexaFluor647-labeled anti-hHER2 antibody for 20 minutes at room temperature with continuous rotation. Opsonized MC38-hHER2 cells were added to BMDMs and incubated at 37°C for exactly 10 minutes. Cells were fixed and stained according to “Fixation of macrophages with beads.”
Modeling
Activation and inhibition pathways were modeled using a set of 13 differential equations. Each equation represented a molecular species in the model, and the mass balance equations calculated species concentration change per unit time. Equations were implemented using Python 3.5 using SciPy software package functions for numerically solving ODEs (scipy.org). Simulations were run for 200 time points, a sufficient time to consistently reach steady state values for all species. Antibody concentrations were initiated from 1000 to 5000, and CD47 concentrations between 0 and 10000.
Measurement of cell surface densities
BMDM, RAW 264.7, or MC38-hHER2 cells were resuspended at a density of 300,000 cells/mL in PBS with 5% FBS. On RAW 264.7 cells, Mouse Fc Block (rat anti-mouse CD16/CD32, BD Biosciences 553142) and p84 (provided by Aduro Biotech) were used to label FcγRs and SIRPα, respectively. On BMDMs, Fc Block and 18a with mutant Fc tail to prevent FcγR binding (provided by Aduro Biotech) were used to label FcγRs and SIRPα, respectively. On MC38-hHER2 cells, anti-hHER2 (mouse Fc Trastuzumab) and MIAP 301 (both provided by Aduro Biotech) were used to label hHER2 and CD47, respectively. Antibodies were labeled using AlexFluor647 NHS Ester (Thermo Fisher Scientific) (see Fluorescent labeling of proteins). Each antibody was added at a top concentration of 50 μg/mL, then serially diluted 4-fold into 8 sequential samples. This yielded a final range of 0.01 to 50 μg/mL antibody. Each antibody concentration was mixed with 30,000 cells and incubated at 4°C for 30 minutes to prevent internalization. Cells were washed twice by spinning for 5 minutes at 300 × g, removing supernatant, and replacing with PBS. Fluorescence of antibodies bound to cell surface was immediately measured on an Attune NxT flow cytometer. Surface antibodies were quantified using calibrated fluorescent beads.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analysis was performed in Python 3.5 using SciPy software package functions. The number of cells, particles, and interfaces quantified per experiment are indicated for each figure in the figure legend. In general, significance was based on a two-sample Student’s t test for the mean values of experimental replicates, where *p < 0.05, **p < 0.01, and ***p < 0.001. Statistical details of data presented are indicated in figure legends.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Anti-biotin antibody (clone BK1/39, AlexaFluor647) | Santa Cruz Biotechnology | RRID: AB_2892070 |
| Anti-human HER2 antibody (Trastuzumab epitope target, mouse IgG1) | Aduro Biotech | N/A |
| Anti-SIRPα antibody (clone 18a, D265A Fc mutant) | Aduro Biotech | N/A |
| Anti-phosphotyrosine antibody (P-Y-1000 MultiMab) | Cell Signaling Technology | RRID: AB_2687925 |
| Anti-rabbit secondary antibody (AlexaFluor546 anti-rabbit goat IgG) | Thermo Scientific | RRID:AB_2534077 |
| Anti-CD45 antibody (clone 30-F11, Brilliant Violet 421) | Biolegend | RRID: AB_10899570 |
| Mouse Fc Block | BD Biosciences | Cat# 553142 |
| Anti-CD47 antibody (clone MIAP 301) | Aduro Biotech | N/A |
| Anti-SIRPα antibody (clone p84) | Aduro Biotech | N/A |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| Rosetta2 (DE3) pLysS | UC Berkeley MacroLab | N/A |
| XL1-Blue | UC Berkeley MacroLab | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| TransIT-293 | Mirus Bio | Cat# MIR 2700 |
| POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) | Avanti Polar Lipids | Cat# 850457 |
| Recombinant mouse CD47 (His tagged) | SinoBiological | Cat# 57231-M08H |
| Alexa Fluor 488 NHS Ester | Thermo Fisher | Cat# A20000 |
| CellTracker Deep Red | Thermo Fisher | Cat# C34565 |
| Alexa Fluor 647 NHS Ester | Thermo Fisher | Cat# A37573 |
| DGS-Ni-NTA (1,2-dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl) iminodiacetic acid)succinyl], with nickel salt) | Avanti Polar Lipids | Cat# 709404 |
| NSC-87877, SHP1/2 Inhibtor | Sigma-Aldrich | Cat# 56932–43–5 |
| Biotinyl Cap PE (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(cap biotinyl)) | Avanti Polar Lipids | Cat# 870273 |
| LISS-Rhodamine (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl)) | Avanti Polar Lipids | Cat# 810150 |
| CMFDA (CellTracker Green) | Thermo Fisher | Cat# C2925 |
| Hoechst 33342 | Thermo Fisher | Cat# H3570 |
| PP2, Src family kinase inhibitor | Sigma Aldrich | Cat# 529573 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| MC38 mouse colon carcinoma cell line | Aduro Biotech | N/A |
| RAW 264.7 mouse macrophage-like cell line | UC Berkeley Cell Culture Facility | https://bds.berkeley.edu/facilities/cell-culture |
| Bone marrow derived macrophages, C57BL/6J background | UC Berkeley Portnoy Lab | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pHR-SHP1-SH2-mEOS | this paper | SHP1 SH2 from Addgene (pGEX-SHP-1(NC)-SH2, plasmid #46496); mEOS |
| pHR-Syn18-F1L-TM-FcgRIIA-ITAM-GFP | this paper | Syn18: SIAATLENDLARLENENARLEKDIANLERDLAKLEREEAYF; F1L as in Bakalar et al. (2018); Transmembrane domain of mouse SIRPα (Uniprot #P97797, amino acid 374–394); ITAM of FcγRIIA (Uniprot #P12318, amino acid 240–317); EGFP |
| pHR-Syn18-F1L-TM-CD3z-ITAM-GFP | this paper | As above except ITAM of CD3ζ (Uniprot #24161, amino acid 52–164) |
| pHR-Syn18-F1L-TM-FcERIgamma-ITAM-GFP | this paper | As above except ITAM of FcεRI γ-chain (Uniprot #P20491, amino acid 45–86) |
| pHR-Syn18-F1L-TM-FcgRIIB-ITIM-GFP | this paper | As above except ITIM of FcγRIIB (Uniprot #P08101, amino acid 232–330) |
| pHR-Syn18-F1L-TM-SIRPa-ITIM-GFP | this paper | As above except ITIM of SIRPα (Uniprot #P97797, amino acid 395–513) |
| pHR-Syn18-F1L-TM-CD22-ITIM-GFP | this paper | As above except ITIM of CD22 (Uniprot #P35329, amino acid 722–862) |
| pHR-Syn18-F1L-TM-PD1-ITIM-GFP | this paper | As above except ITIM of PD-1 (Uniprot #Q02242, amino acid 191–288) |
| pHR-Syn18-F1L-TM-GFP | this paper | As above except no ITAM or ITIM |
| pet28-Syn 17-F3L-H10-KCK-H6 | this paper | Syn17: NEKEELKSKKAELRNRI EQLKQKREQLKQKIANLRKEIEAYK; F3L as in Bakalar et al. (2018) |
| pet28-Syn 17-F5L-H10-KCK-H6 | this paper | Syn17: NEKEELKSKKAELRNR IEQLKQKREQLKQKIANLRKEIEAYK; F5L as in Bakalar et al. (2018) |
| pHR-Syk-SH2-mCh | Bakalar et al., 2018 | N/A |
| Second generation Lenti virus packaging plasmids pMD2.G and p8.91 | CloneTech | N/A |
| pHR-SHP1-SH2-GFP | this paper | SHP1 SH2 from Addgene (pGEX-SHP-1(NC)-SH2, plasmid #46496) |
| pHR-Syk-SH2-mEOS | this paper | Syk SH2 from Uniprot ID P48025; mEOS |
|
| ||
| Software and algorithms | ||
|
| ||
| Cell Profiler | Broad Institute | RRID: SCR_007358; https://cellprofiler.org/ |
| MATLAB | Mathworks | RRID: SCR_001622; https://www.mathworks.com/products/matlab.html |
| Python | Python | RRID: SCR_008394; https://www.python.org/ |
| Fiji Image Analysis | Fiji | RRID:SCR_002285; https://imagej.net/software/fiji/ |
|
| ||
| Other | ||
|
| ||
| 8-well glass bottom dishes | Cellvis | Cat# C8–1 |
| Calibrated fluorescent beads, Quantum MESF | Bangs Laboratories, Inc. | Cat# 488, 647 |
| 4 μm non-functionalized silica bead | Bangs Laboratories, Inc. | Cat# SS05002 |
Highlights.
The ratio of activating IgG to inhibitory CD47 dictates macrophage phagocytosis
ITIM-associated phosphatases halt activation by dephosphorylating adjacent FcγRs
Ratio-dependent phagocytosis can be shifted using checkpoint blockade strategies
Ratio dictates phagocytosis of both endogenous and synthetic (CAR) receptors
ACKNOWLEDGMENTS
The authors would like to thank Fletcher Laboratory members, especially Carmen Chan and Aaron Joffe, for useful feedback and technical consultation; the Cancer Research Laboratory Flow Cytometry Facility for cell sorting; and the Portnoy Laboratory of UC Berkeley for providing BMDMs. We are grateful to Aduro Biotech—specifically Andrea van Elsas, Meredith Leong, Sander van Duijnhoven, and Sanne Spijkers—for support, discussions, and supplying a range of reagents used in this study. This work was supported by the NSF Center for Cellular Construction DBI-1548297 (D.A.F.), NIH R01 GM134137 (D.A.F.), the Immunotherapeutics and Vaccine Research Initiative at UC Berkeley (D.A.F.), the Miller Institute for Basic Research (D.A.F.), and the Chan Zuckerberg Biohub (D.A.F.). E.C.S. was funded by an NSF-GRFP fellowship.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109587.
REFERENCES
- Abram CL, and Lowell CA (2008). The diverse functions of Src family kinases in macrophages. Front. Biosci. 13, 4426–4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakalar MH, Joffe AM, Schmid EM, Son S, Podolski M, and Fletcher DA (2018). Size-dependent segregation controls macrophage phagocytosis of antibody-opsonized targets. Cell 174, 131–142.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barclay AN, and Van den Berg TK (2014). The interaction between signal regulatory protein alpha (SIRPα) and CD47: structure, function, and therapeutic target. Annu. Rev. Immunol. 32, 25–50. [DOI] [PubMed] [Google Scholar]
- Barua D, Hlavacek WS, and Lipniacki T (2012). A computational model for early events in B cell antigen receptor signaling: analysis of the roles of Lyn and Fyn. J. Immunol. 189, 646–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binstadt BA, Brumbaugh KM, Dick CJ, Scharenberg AM, Williams BL, Colonna M, Lanier LL, Kinet J-P, Abraham RT, and Leibson PJ (1996). Sequential involvement of Lck and SHP-1 with MHC-recognizing receptors on NK cells inhibits FcR-initiated tyrosine kinase activation. Immunity 5, 629–638. [DOI] [PubMed] [Google Scholar]
- Brooke G, Holbrook JD, Brown MH, and Barclay AN (2004). Human lymphocytes interact directly with CD47 through a novel member of the signal regulatory protein (SIRP) family. J. Immunol. 173, 2562–2570. [DOI] [PubMed] [Google Scholar]
- Burshtyn DN, Yang W, Yi T, and Long EO (1997). A novel phosphotyrosine motif with a critical amino acid at position −2 for the SH2 domain-mediated activation of the tyrosine phosphatase SHP-1. J. Biol. Chem. 272, 13066–13072. [DOI] [PubMed] [Google Scholar]
- Caron E, and Hall A (1998). Identification of two distinct mechanisms of phagocytosis controlled by different Rho GTPases. Science 282, 1717–1721. [DOI] [PubMed] [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, et al. (2006). CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 7, R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao MP, Alizadeh AA, Tang C, Myklebust JH, Varghese B, Gill S, Jan M, Cha AC, Chan CK, Tan BT, et al. (2010). Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell 142, 699–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Sung S-S, Yip MLR, Lawrence HR, Ren Y, Guida WC, Sebti SM, Lawrence NJ, and Wu J (2006). Discovery of a novel shp2 protein tyrosine phosphatase inhibitor. Mol. Pharmacol. 70, 562–570. [DOI] [PubMed] [Google Scholar]
- Chu CC, VanDerMeid KR, Elliott MR, Baran AM, Barr PM, and Zent CS (2018). Antibody-dependent cellular phagocytosis is responsible for efficacy of anti-CD20 monoclonal antibody therapy in chronic lymphocytic leukemia. J. Immunol. 200 (suppl), 56.4. [Google Scholar]
- Crowley MT, Costello PS, Fitzer-Attas CJ, Turner M, Meng F, Lowell C, Tybulewicz VLJ, and DeFranco AL (1997). A critical role for Syk in signal transduction and phagocytosis mediated by Fcgamma receptors on macrophages. J. Exp. Med. 186, 1027–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis SJ, and van der Merwe PA (2006). The kinetic-segregation model: TCR triggering and beyond. Nat. Immunol. 7, 803–809. [DOI] [PubMed] [Google Scholar]
- Dushek O, Goyette J, and van der Merwe PA (2012). Non-catalytic tyrosine-phosphorylated receptors. Immunol. Rev. 250, 258–276. [DOI] [PubMed] [Google Scholar]
- Dustin ML (2007). Cell adhesion molecules and actin cytoskeleton at immune synapses and kinapses. Curr. Opin. Cell Biol. 19, 529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng M, Jiang W, Kim BYS, Zhang CC, Fu Y-X, and Weissman IL (2019). Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat. Rev. Cancer 19, 568–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank C, Burkhardt C, Imhof D, Ringel J, Zschörnig O, Wieligmann K, Zacharias M, and Böhmer F-D (2004). Effective dephosphorylation of Src substrates by SHP-1. J. Biol. Chem. 279, 11375–11383. [DOI] [PubMed] [Google Scholar]
- Freeman SA, Goyette J, Furuya W, Woods EC, Bertozzi CR, Bergmeier W, Hinz B, van der Merwe PA, Das R, and Grinstein S (2016). Integrins Form an Expanding Diffusional Barrier that Coordinates Phagocytosis. Cell 164, 128–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fütterer K, Wong J, Grucza RA, Chan AC, and Waksman G (1998). Structural basis for syk tyrosine kinase ubiquity in signal transduction pathways revealed by the crystal structure of its regulatory SH2 domains bound to a dually phosphorylated ITAM peptide. J. Mol. Biol. 281, 523–537. [DOI] [PubMed] [Google Scholar]
- Graf B, Bushnell T, and Miller J (2007). LFA-1-mediated T cell costimulation through increased localization of TCR/class II complexes to the cSMAC and exclusion of CD45 from the immunological synapse. J. Immunol. 179, 1616–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AB, Gakidis MAM, Glogauer M, Wilsbacher JL, Gao S, Swat W, and Brugge JS (2006). Requirements for Vav guanine nucleotide exchange factors and Rho GTPases in FcγR- and complement-mediated phagocytosis. Immunity 24, 305–316. [DOI] [PubMed] [Google Scholar]
- Hatherley D, Graham SC, Harlos K, Stuart DI, and Barclay AN (2009). Structure of signal-regulatory protein α: a link to antigen receptor evolution. J. Biol. Chem. 284, 26613–26619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z-Y, Hunter S, Kim M-K, Indik ZK, and Schreiber AD (2003). The effect of phosphatases SHP-1 and SHIP-1 on signaling by the ITIM- and ITAM-containing Fcγ receptors FcgammaRIIB and FcgammaRIIA. J. Leukoc. Biol. 73, 823–829. [DOI] [PubMed] [Google Scholar]
- Iqbal N, and Iqbal N (2014). Human epidermal growth factor receptor 2 (her2) in cancers: overexpression and therapeutic implications. Mol. Biol. Int. 2014, 852748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs SA, Diem MD, Luo J, Teplyakov A, Obmolova G, Malia T, Gilliland GL, and O’Neil KT (2012). Design of novel FN3 domains with high stability by a consensus sequence approach. Protein Eng. Des. Sel. 25, 107–117. [DOI] [PubMed] [Google Scholar]
- Joffe AM, Bakalar MH, and Fletcher DA (2020). Macrophage phagocytosis assay with reconstituted target particles. Nat. Protoc. 15, 2230–2246. [DOI] [PubMed] [Google Scholar]
- Kant AM, De P, Peng X, Yi T, Rawlings DJ, Kim JS, and Durden DL (2002). SHP-1 regulates Fcgamma receptor-mediated phagocytosis and the activation of RAC. Blood 100, 1852–1859. [PubMed] [Google Scholar]
- Kharitonenkov A, Chen Z, Sures I, Wang H, Schilling J, and Ullrich A (1997). A family of proteins that inhibit signalling through tyrosine kinase receptors. Nature 386, 181–186. [DOI] [PubMed] [Google Scholar]
- Kheir WA, Gevrey J-C, Yamaguchi H, Isaac B, and Cox D (2005). A WAVE2-Abi1 complex mediates CSF-1-induced F-actin-rich membrane protrusions and migration in macrophages. J. Cell Sci. 118, 5369–5379. [DOI] [PubMed] [Google Scholar]
- Kiefer F, Brumell J, Al-Alawi N, Latour S, Cheng A, Veillette A, Grinstein S, and Pawson T (1998). The Syk protein tyrosine kinase is essential for Fcγ receptor signaling in macrophages and neutrophils. Mol. Cell. Biol. 18, 4209–4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler K, Xiong S, Brzostek J, Mehrabi M, Eissmann P, Harrison A, Cordoba S-P, Oddos S, Miloserdov V, Gould K, et al. (2010). Matched sizes of activating and inhibitory receptor/ligand pairs are required for optimal signal integration by human natural killer cells. PLoS ONE 5, e15374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Jiang N, Nagarajan S, Wohlhueter R, Selvaraj P, and Zhu C (2007). Affinity and kinetic analysis of Fcγ receptor IIIa (CD16a) binding to IgG ligands. J. Biol. Chem. 282, 6210–6221. [DOI] [PubMed] [Google Scholar]
- Lu J, Ellsworth JL, Hamacher N, Oak SW, and Sun PD (2011). Crystal structure of Fcγ receptor I and its implication in high affinity γ-immunoglobulin binding. J. Biol. Chem. 286, 40608–40613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machida K, Thompson CM, Dierck K, Jablonowski K, Kärkkäinen S, Liu B, Zhang H, Nash PD, Newman DK, Nollau P, et al. (2007). High-throughput phosphotyrosine profiling using SH2 domains. Mol. Cell 26, 899–915. [DOI] [PubMed] [Google Scholar]
- Majeti R, Chao MP, Alizadeh AA, Pang WW, Jaiswal S, Gibbs KD Jr., van Rooijen N, and Weissman IL (2009). CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 138, 286–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malarkannan S (2006). The balancing act: inhibitory Ly49 regulate NKG2-Dmediated NK cell functions. Semin. Immunol. 18, 186–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeage K, and Perry CM (2002). Trastuzumab: a review of its use in the treatment of metastatic breast cancer overexpressing HER2. Drugs 62, 209–243. [DOI] [PubMed] [Google Scholar]
- McLarty K, Cornelissen B, Scollard DA, Done SJ, Chun K, and Reilly RM (2009). Associations between the uptake of 111In-DTPA-trastuzumab, HER2 density and response to trastuzumab (Herceptin) in athymic mice bearing subcutaneous human tumour xenografts. Eur. J. Nucl. Med. Mol. Imaging 36, 81–93. [DOI] [PubMed] [Google Scholar]
- Morrissey MA, and Vale RD (2019). CD47 suppresses phagocytosis by repositioning SIRPΑ and preventing integrin activation. BioRxiv. 10.1101/752311. [DOI] [Google Scholar]
- Morrissey MA, Kern N, and Vale RD (2020). CD47 ligation repositions the inhibitory receptor SIRPΑ to suppress integrin activation and phagocytosis. Immunity 53, 290–302.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouro-Chanteloup I, Delaunay J, Gane P, Nicolas V, Johansen M, Brown EJ, Peters LL, Van Kim CL, Cartron JP, and Colin Y (2003). Evidence that the red cell skeleton protein 4.2 interacts with the Rh membrane complex member CD47. Blood 101, 338–344. [DOI] [PubMed] [Google Scholar]
- Neel BG, Gu H, and Pao L (2003). The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem. Sci. 28, 284–293. [DOI] [PubMed] [Google Scholar]
- Okazawa H, Motegi S, Ohyama N, Ohnishi H, Tomizawa T, Kaneko Y, Oldenborg P-A, Ishikawa O, and Matozaki T (2005). Negative regulation of phagocytosis in macrophages by the CD47-SHPS-1 system. J. Immunol. 174, 2004–2011. [DOI] [PubMed] [Google Scholar]
- Oldenborg P-A, Zheleznyak A, Fang Y-F, Lagenaur CF, Gresham HD, and Lindberg FP (2000). Role of CD47 as a marker of self on red blood cells. Science 288, 2051–2054. [DOI] [PubMed] [Google Scholar]
- Philipsen L, Engels T, Schilling K, Gurbiel S, Fischer K-D, Tedford K, Schraven B, Gunzer M, and Reichardt P (2013). Multimolecular analysis of stable immunological synapses reveals sustained recruitment and sequential assembly of signaling clusters. Mol. Cell. Proteomics 12, 2551–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren L, Chen X, Luechapanichkul R, Selner NG, Meyer TM, Wavreille A-S, Chan R, Iorio C, Zhou X, Neel BG, and Pei D (2011). Substrate specificity of protein tyrosine phosphatases 1B, RPTPα, SHP-1, and SHP-2. Biochemistry 50, 2339–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russ A, Hua AB, Montfort WR, Rahman B, Riaz IB, Khalid MU, Carew JS, Nawrocki ST, Persky D, and Anwer F (2018). Blocking “don’t eat me” signal of CD47-SIRPα in hematological malignancies, an indepth review. Blood Rev. 32, 480–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scapini P, Pereira S, Zhang H, and Lowell CA (2009). Multiple roles of Lyn kinase in myeloid cell signaling and function. Immunol. Rev. 228, 23–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selner NG, Luechapanichkul R, Chen X, Neel BG, Zhang Z-Y, Knapp S, Bell CE, and Pei D (2014). Diverse levels of sequence selectivity and catalytic efficiency of protein-tyrosine phosphatases. Biochemistry 53, 397–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sezgin E, Kaiser H-J, Baumgart T, Schwille P, Simons K, and Levental I (2012). Elucidating membrane structure and protein behavior using giant plasma membrane vesicles. Nat. Protoc. 7, 1042–1051. [DOI] [PubMed] [Google Scholar]
- Stebbins CC, Watzl C, Billadeau DD, Leibson PJ, Burshtyn DN, and Long EO (2003). Vav1 dephosphorylation by the tyrosine phosphatase SHP-1 as a mechanism for inhibition of cellular cytotoxicity. Mol. Cell. Biol. 23, 6291–6299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein SC, and Thiart J (2016). TrackNTrace: A simple and extendable open-source framework for developing single-molecule localization and tracking algorithms. Sci. Rep. 6, 37947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theocharides APA, Jin L, Cheng P-Y, Prasolava TK, Malko AV, Ho JM, Poeppl AG, van Rooijen N, Minden MD, Danska JS, et al. (2012). Disruption of SIRPα signaling in macrophages eliminates human acute myeloid leukemia stem cells in xenografts. J. Exp. Med. 209, 1883–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson KE, Bashor CJ, Lim WA, and Keating AE (2012). SYNZIP protein interaction toolbox: in vitro and in vivo specifications of heterospecific coiled-coil interaction domains. ACS Synth. Biol. 1, 118–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai RK, and Discher DE (2008). Inhibition of “self” engulfment through deactivation of myosin-II at the phagocytic synapse between human cells. J. Cell Biol. 180, 989–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang E, Giannetti AM, Shaw D, Dinh M, Tse JKY, Gandhi S, Ho H, Wang S, Papp E, and Bradshaw JM (2008). Molecular mechanism of the Syk activation switch. J. Biol. Chem. 283, 32650–32659. [DOI] [PubMed] [Google Scholar]
- van de Donk NWCJ, and Usmani SZ (2018). CD38 antibodies in multiple myeloma: mechanisms of action and modes of resistance. Front. Immunol. 9, 2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanDerMeid KR, Elliott MR, Baran AM, Barr PM, Chu CC, and Zent CS (2018). Cellular Cytotoxicity of Next-Generation CD20 Monoclonal Antibodies. Cancer Immunol. Res. 6, 1150–1160. [DOI] [PubMed] [Google Scholar]
- Veillette A, Thibaudeau E, and Latour S (1998). High expression of inhibitory receptor SHPS-1 and its association with protein-tyrosine phosphatase SHP-1 in macrophages. J. Biol. Chem. 273, 22719–22728. [DOI] [PubMed] [Google Scholar]
- Wakselman S, Béchade C, Roumier A, Bernard D, Triller A, and Bessis A (2008). Developmental neuronal death in hippocampus requires the microglial CD11b integrin and DAP12 immunoreceptor. J. Neurosci. 28, 8138–8143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiskopf K, Jahchan NS, Schnorr PJ, Cristea S, Ring AM, Maute RL, Volkmer AK, Volkmer J-P, Liu J, Lim JS, et al. (2016). CD47-blocking immunotherapies stimulate macrophage-mediated destruction of smallcell lung cancer. J. Clin. Invest. 126, 2610–2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willingham SB, Volkmer J-P, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, Wang J, Contreras-Trujillo H, Martin R, Cohen JD, et al. (2012). The CD47-signal regulatory protein alpha (SIRPα) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 109, 6662–6667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu MM, Pu Y, Han D, Shi Y, Cao X, Liang H, Chen X, Li X-D, Deng L, Chen ZJ, et al. (2017). Dendritic cells but not macrophages sense tumor mitochondrial dna for cross-priming through signal regulatory protein α signaling. Immunity 47, 363–373.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagita T, Murata Y, Tanaka D, Motegi SI, Arai E, Daniwijaya EW, Hazama D, Washio K, Saito Y, Kotani T, et al. (2017). Anti-SIRPα antibodies as a potential new tool for cancer immunotherapy. JCI Insight 2, e89140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, and Saito T (2012). Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med. 209, 1201–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.