

ABSTRACT
T cell-engaging therapies involving bispecific T cell engager (BiTE) and chimeric antigen receptor T (CAR-T) cells have achieved great success in the treatment of hematological tumors. However, the paucity of ideal cell surface molecules that can be targeted on glioblastoma (GBM) partially reduces the immunotherapeutic efficacy. Recently, high expression of Fn14 has been reported in several solid tumors, so the strategy of exploiting this specific antigen for GBM immunotherapy is worth studying. Consequently, we constructed Fn14× CD3 BiTE and Fn14-specific CAR-T cells and investigated their cytotoxic activity against GBM in vitro and in vivo. First, expression of Fn14 was confirmed in glioma tissues and GBM cells. Then, we designed Fn14-specific BiTE and CAR-T cells and tested their cytotoxicity in GBM cell cultures and mouse models of GBM. Fn14 was highly expressed in GBM tissues and cell lines, while it was undetectable in normal brain samples. Fn14× CD3 BiTE, Fn14 CAR-T cells and Fn14 CAR-T/IL-15 cells were antigen-specific and highly cytotoxic, showing good antitumor activity in vitro and causing significant regression of established solid tumors in xenograft models. However, the xenografts treated with Fn14 CAR-T cells regrew, whereas xenografts treated with Fn14 CAR-T/IL-15 cells did not. IL-15 engineering augmented the antitumor activity of Fn14 CAR-T cells and resulted in significant antitumor effects similar to those of Fn14× CD3 BiTE. Our results suggest that Fn14 is an appropriate target for GBM. Anti-Fn14 BiTE and Fn14-specific CAR-T/IL-15 cells may be exciting immunotherapeutic options for malignant brain cancer.
KEYWORDS: GBM, Fn14, BiTE, CAR-T, immunotherapy
Introduction
Glioblastoma (GBM), the highest grade of primary gliomas (WHO grade IV), is the most common and lethal brain tumor in adults. Despite the development of multidisciplinary treatment involving advanced surgical resection, radiotherapy and adjuvant chemotherapy, the median survival for patients with GBM is only 15 months, and the 5-year survival rate is less than 10%.1,2
Tumor immunotherapy has achieved tremendous progress, especially in the field of T cell-engaging therapies. Bispecific T cell engager (BiTE) as well as chimeric antigen receptor T (CAR-T) cells redirect T cells to recognize and kill cancer cells in a targeted manner, making them promising treatments against refractory cancer.3–5 The US Food and Drug Administration has approved products related to BiTE (blinatumomab) and CAR-T (Kymriah and Yescarta) on the basis of their impressive antitumor efficacy against acute lymphoblastic leukemia and relapse as well as refractory B cell non-Hodgkin’s lymphoma.6,7 These hematologic malignancies present uniform antigenic targets, but GBM shows extensive inter- and intratumoral heterogeneity, such that potential antigenic targets are not highly or ubiquitously expressed within tumor tissues. As a result, even promising GBM treatments targeting interleukin-13 receptor alpha 2 (IL-13Ra2), human epidermal growth factor receptor 2 (HER2), and epidermal growth factor receptor variant III (EGFRvIII) show disappointing efficacy, in part because of poor persistence of CAR-T cells and/or antigen loss or modulation, leading to tumor cell resistance.8–12 Therefore, developing effective targeted immunotherapy of GBM requires identifying an ideal, highly tumor-specific antigen.
Fibroblast growth factor-inducible 14 (Fn14), a type I transmembrane protein, is the sole cell surface receptor of tumor necrosis factor–related weak inducer of apoptosis (TWEAK). Activation of the TWEAK/Fn14 molecular pathway contributes to cell proliferation and death, angiogenesis, and inflammation,13 giving the pathway an important role in carcinogenesis in several solid tumors.14–16 Studies have reported that Fn14 is up-regulated in gliomas, and this overexpression can stimulate glioma cell migration and invasion.17,18 Additionally, high Fn14 expression is more common in advanced brain cancers and is associated with higher grade and poorer prognosis.18–20 Therefore, targeting Fn14 to inhibit the TWEAK/Fn14 pathway may be a useful method for treating invasive brain cancer.
The cytokine IL-15 favors adoptive transfer of central memory T cells (TCM), and incorporation of the IL-15 gene into CAR constructs supports CAR-T cell proliferation, survival and effector function.21–23 In this study, we developed Fn14× CD3 BiTE antibody, Fn14-specific CAR-T (Fn14 CAR-T) cells and Fn14-specific CAR-T cells engineered to secrete IL-15 (Fn14 CAR-T/IL-15). We assessed the antitumor activity of the three therapeutic approaches against GBM in vitro and in vivo. Our studies indicated that both the anti-Fn14 BiTE antibody and Fn14-redirected CAR-T cells exerted significant cytotoxic effects against GBM in vitro and in vivo. IL-15 production augmented the antitumor effects of CAR-T cells and resulted in longer remission and survival in mouse models of GBM.
Materials and methods
Cells and mice
HEK-293 T and four GBM cell lines (U87, U251, A172, and T98G) were purchased from the American Type Culture Collection (ATCC) and cultured in DMEM (Gibco). Normal human fetal lung fibroblasts (HFL1) were a kind gift from Jiang Yu in our laboratory and were cultured in HFL1 cell medium (Procell). All the media used above contained 10% fetal calf serum and 1.0 mmol/L penicillin–streptomycin combination (Hyclone), and all cell lines were cultured in a humidified incubator with 5% CO2 at 37 ◦C.
In our study, female NSG mice 6–8 weeks old were purchased from the Model Animal Resource Information Platform of Nanjing University (Nanjing, P. R. China) and kept under pathogen-free conditions in the animal vivarium at the State Key Laboratory of Biotherapy, Sichuan University.
Immunohistochemistry (IHC)
In order to assess the expression of CD3 or Fn14 in tumor-infiltrating T/CAR-T cells and to detect tumor cells, tumor tissues were harvested, embedded in paraffin, sectioned, then stained with commercial anti-CD3 rabbit monoclonal antibody (mAb, Cell Signaling Technology; 1:200) or anti-Fn14 rabbit mAb (Cell Signaling Technology; 1:200). Microarrays of human gliomas (low-grade glioma, n = 34; GBM, n = 38) and normal brain tissues (N = 12) were stained with anti-Fn14 rabbit mAb (Cell Signaling Technology; 1:200).
In brief, tissue sections were incubated at 65°C for 1 h, blocked for 30 min at room temperature with phosphate-buffered saline (PBS) containing 10% normal goat serum (Boster, Wuhan, P. R. China), then incubated at 4°C overnight with primary antibody. Bound primary antibodies were detected using secondary goat anti-rabbit antibodies, followed by DAB (ZSGB-BIO, Beijing, P. R. China). Saturation and intensity of immunostained cells were evaluated in four visual fields under a light microscope. Staining intensity was scored as 0 (-, negative), 1 (+, weakly positive), 2 (++, moderately positive) or 3 (+++, strongly positive). Histological scores were quantified using the following formula: histopathology score = [(1 × % of cells showing weak positive staining) + (2 × % of cells showing moderate positive staining) + (3 × % of cells showing strong positive staining)] × 100.
A summary of the clinical characteristics and Fn14 histoscore is provided in Supplementary Table 1.
Bioinformatic analysis
Fn14 microarray data of various gliomas were downloaded from the Sun Brain Dataset in the Oncomine database (http://www.oncomine.org). We also analyzed Fn14 expression and survival of patients with low-grade glioma (LGG) or GBM using the web-based Gene Expression Profiling Interactive Analysis (GEPIA) (http://gepia.cancer-pku.cn/), which has access to The Cancer Genome Atlas (TCGA). Fn14 expression was also analyzed in GBM tissues encoding wild-type or mutant IDH-1 and in the four pathological types of GBM, based on TCGA data.
Knockout of Fn14 in U87 and U251 cell lines
Fn14 was knocked out from U87 and U251 cells using the CRISPR-Cas9 system and gRNAs that were designed using the online server CHOPCHOP (http://chopchop.cbu.uib.no). The gRNAs were subcloned into lentiCRISPR_v2 (catalog no. 52961, Addgene), and the harvested lentiviral particles were transduced into U87 and U251 cells. After 24 hours, transfected cells were selected using puromycin, and the desired knockdown was confirmed 5 days later by sequencing and fluorescence-activated cell sorting (FACS). The forward primer flanking the Fn14 target was 5ʹ-AAATCATTCGGGAGGAGGTGGGA-3ʹ, and the reverse primer was 5ʹ-CAGCTGTTTTGTGTGAGCCAGC-3ʹ.
Immunofluorescence staining
HFL1 or GBM cells were incubated in 24-well plates with coverslips for 24 h at 37◦C. Then cells were fixed with 4% paraformaldehyde in PBS for 15 min, blocked with 1% BSA, stained for 1 h with primary antibody or purified Fn14 single-chain variable fragment-mouse fragment crystallizable Fc (Fn14 scFv-mFc), then stained for 1 h with Cy3- or FITC-conjugated secondary antibody (Proteintech). Finally, slides were stained with 4ʹ,6-diamidino-2-phenylindole (DAPI) (Beyotime). Immunostained sections were imaged using confocal microscopy.
Western blotting
GBM cells were lysed in RIPA buffer (Beyotime) supplemented with protease inhibitor cocktail (Beyotime), then centrifuged at 10,000 × g for 15 min at 4◦C. Total protein extracts were quantified using a BCA kit (Beyotime), subjected to 10% SDS-PAGE gel, transferred to polyvinylidene difluoride membranes, then incubated with anti-Fn14 antibody (Cell Signaling Technology; 1:2000) and anti-α tubulin antibody (Cell Signaling Technology; 1:2000). Finally, membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (Beyotime, 1:3,000). Target bands were analyzed digitally based on enhanced chemiluminescence (Clinx Science Instruments, Chemiscope 5300).
Construction of Fn14-mFc recombinant protein and Fn14× CD3 BiTE
The sequence of the anti-Fn14 scFv (comprising VL and VH) that was used in Fn14 scFv-mFc recombinant protein, BiTE and CAR vectors was derived from a Fn14 monoclonal antibody P4A8 (patent EP2294089A2). The antibody is affinity-matured and has been shown to bind invasive human glioma cells.18 The anti-CD3 domain used in BiTE was derived from Blinatumomab (Blincyto, Amgen).24 The cDNAs encoding Fn14 and CD3-specific scFv were synthesized by Genewiz, and a recombinant single-chain Fn14× CD3 BiTE was constructed with a G4S linker between the two scFv’s. Then, the recombinant cDNAs of Fn14 scFv-mFc and BiTE were subcloned into eukaryotic expression vectors with a His tag at the C-terminus to facilitate protein purification.
HEK293T cells were transfected with the expression vectors described above and cultured in FreeStyle serum-free medium (Thermo Fisher Scientific, Waltham, MA, USA) at 37°C with 5% CO2 in a humidified incubator. One week later, the supernatants were harvested, and the recombinant Fn14 scFv-mFc and Fn14× CD3 BiTE were purified on Ni-NTA affinity columns and further purified using a Superdex 200 Increase 10/300 GL Column (GE). The purified Fn14 scFv-mFc and Fn14× CD3 BiTE were analyzed on SDS–PAGE gel after staining with Coomassie brilliant blue for size estimation and quality control.
Construction of the CAR vector
The original Fn14-targeted CAR consisted of CD8α signal peptide, Fn14-specific scFv, hinge, CD8 transmembrane, the cytoplasmic domain of human CD28, 4–1BB and CD3ξ. Control CAR was constructed by swapping the Fn14 scFv sequence with the CD19 scFv. IL-15 autocrine CAR was constructed by adding the IL-15 sequence directly after 4–1BB/CD3ξ. A truncated, nonfunctional human EGFR (tEGFR) was used for CAR detection and control of unwanted expansion.25–27
Manufacture of lentivirus and T cell transduction
Lentivirus particles of all the CARs mentioned above were produced in HEK293T cells. In brief, HEK-293 T cells were cultured in DMEM and co-transfected with the target plasmid as well as two packaging plasmids (psPAX2 and pMD.2 G) in a ratio of 4:3:2 using polyethyleneimine (PEI, Sigma). After transfection, supernatants were collected at 24 and 48 h and concentrated by ultracentrifugation at 25,000 × g for 2 h at 4°C. Concentrated viruses were resuspended in RPMI-1640 and stored at – 80◦C.
For T cell generation, peripheral blood mononuclear cells (PBMCs) from healthy donors were isolated by gradient centrifugation at 800 × g for 15 min. Harvested PBMCs were cultured in X–vivo media (Lonza) containing 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin in a 37°C incubator with an atmosphere of 5% CO2. T cells were stimulated for 48 h with 200 ng/mL of anti-CD3 monoclonal Ab (OKT3, BioLegend), 100 ng/mL anti-CD28 monoclonal Ab (CD28.2, BioLegend) and 100 U/mL recombinant human interleukin (IL)-2 (Life Science). Then, activated T lymphocytes were transduced using lentivirus (CD19 CAR, Fn14 CAR or Fn14 CAR/IL15) at a multiplicity of infection of 10 using hexadimethrine bromide (polybrene) (Sigma) in the presence of IL-2. After 12 h, the T cells were collected and maintained in X–vivo medium (Lonza) containing 100 U/ml IL-2. Lentivirus was titrated using the TCID50 assay. Control T cells were not transduced (hereafter “NT cells”).
T-cell phenotype, activation and proliferation
At 10 days after transduction, T cells were collected and stained with antibodies against CD4, CD8, CD45RO, and CD62L to characterize phenotype.
In activation studies, U87 cells (1 × 105) were seeded in 24-well plates overnight and co-cultured for 48 h at 37◦C with 4 × 105 effector cells (NT, CD19 CAR-T, Fn14 CAR-T, or Fn14 CAR-T/IL15 cells). After that, T cells were collected and stained with antibody against CD69 for activation analysis.
In proliferation studies, 1 × 105 U87 cells and 4 × 105 CytoTel Red-labeled effector cells were co-cultured at 37◦C for three days, then all cells were transferred into new wells and 1 × 105 U87 cells were added. After three co-culture cycles, T cells were collected and detected by flow cytometry based on CytoTel Red signal. In these tests, all analyses were conducted using ACEA NovoCyte (Agilent Biosciences), and 0.1 μg/ml Fn14× CD3 BiTE was added to the NT+BiTE group every day.
Cytotoxicity of Fn14× CD3 BiTE and Fn14-redirected CAR-T cells
A 12-h luciferase-based assay was performed to detect the antigen-specific cytotoxicity of Fn14× CD3 BiTE and Fn14-redirected CAR-T cells in a two-dimensional (2D) model. For testing Fn14× CD3 BiTE, U87, U251, U87KO, U251KO, and HFL1 tumor cells (2 × 104) expressing firefly luciferase (FFluc) were plated per well in a 96-well black plate (Corning, USA). At 24 h later, effector cells (NT cells) were added at an effector: tumor cell (E:T) ratio of 4:1 to assess whether Fn14× CD3 BiTE showed Fn14-redirected cytotoxicity based on differences between with or without 0.1 μg/ml Fn14× CD3 BiTE. Next, tumor-FFluc cells were incubated with Fn14× CD3 BiTE at concentrations ranging from 10−4 to 1 μg/ml and an E:T of 4:1.
For testing Fn14-redirected CAR-T cells, CD19 CAR-T, Fn14 CAR-T, or Fn14 CAR-T/IL15 cells were co-cultured together with HFL1-FFluc or tumor-FFluc cells at E:T ratios of 0:1, 1:1, 4:1, 8:1 and 16:1. Luciferin was added to each well, and luminescence was quantified using an Infinite® M200plate reader (Tecan). The number of viable HFL1-FFluc or tumor-FFluc cells in each well was calculated based on standard curves generated from serial dilutions of the target cells. Cytotoxicity was calculated as a percentage as follows: [(Cell number in untreated well − Cell number in assay well)/(Cell number in untreated well)] × 100%.28
We also assessed cytotoxicity using a three-dimensional (3D) spheroid model. Briefly, U87-GFP cells (5 × 104) were seeded in ultra-low attachment 24-well dishes (Corning, USA) and grown in serum-free DMEM/F12 medium (Gibco, USA) supplemented with 2% B27 hormone mixture (Gibco), 20 ng/mL human recombinant epidermal growth factor (Sino Biological), 10 ng/mL human recombinant fibroblast growth factor 2 (Sino Biological) and 1 μg/mL heparin for 21 days. Fresh medium was added every 48 h. Effector cells were stained with CytoTel Red (AAT Bioquest) according to the manufacturer’s instructions, then stained cells (2 × 105) were added to the wells at an E:T of 4:1. The maximal diameters of tumor spheres were measured daily under a fluorescence microscope. Where indicated, 0.1 μg/ml Fn14× CD3 BiTE was added daily to the NT+BiTE group.
Cytokine production assays
Tumor cells (U87, U251, U87KO, U251KO) or HFL1 cells (1 × 105 in all cases) were co-cultured with 4 × 105 effector cells (CD19 CAR-T, Fn14 CAR-T, Fn14 CAR-T/IL15 and NT cells) in 24-well plates at 37◦C without exogenous cytokines. Culture medium was assayed for the cytokines IFN-γ and TNF-α at 24 h later, and for IL-15 at 24, 48 and 72 h. Where noted, 0.1 μg/ml Fn14× CD3 BiTE was added to the NT+BiTE group. All cytokines were measured using ELISA kits (BioLegend).
Flow cytometry
Briefly, cells (2 × 105) were washed twice with PBS, then incubated in 1% BSA solution with Fc blocker (Biolegend) for 30 min. Fluorochrome-conjugated antibodies were purchased from Biolegend (CD3, CD4, CD8, CD45RO, CD62L, CD69, TWEAK/Fn14 and PD-1) or BD (Annexin-V, 7-AAD). Purified Fn14 scFv-mFc was used as primary antibody and anti-mouse IgG-FITC (BioLegend) as second antibody to verify the Fn14-specific scFv. CAR-T cells were stained with primary antibody against EGFR (C225), followed by PE-labeled anti-human-IgG antibody (BioLegend) for CAR detection.
In this study, T cell phenotype and activation were analyzed using FlowJo 9.3.2 software. The transduction efficiency of CAR-T cells, TWEAK/Fn14 expression on GBM cells, and proliferation of cells were measured using ACEA NovoCyte (Agilent Biosciences).
Patient-derived xenografts (PDXs)
We collected a primary patient-derived GBM sample (Supplementary Fig. S1 C) and modified the tumor cells with luciferase gene (GBM-FFluc) for later use. Then PDXs were established by intracranially injecting GBM-FFluc cells (2 × 105) into each mouse using the stereotactic apparatus. On day 7, mice were randomly divided into five groups (n = 4/group), and two groups were used to assess the cytotoxicity of Fn14× CD3 BiTE. Concretely, T cells (5 × 105) and Fn14× CD3 BiTE (10 ng) in 5 μl X–vivo medium were injected directly into the lesion.29 Control mice were treated with identical amounts of NT cells suspended in 5 μl X–vivo medium. Treatments were carried out using a stereotactic device once every two days for a total of four times. For cytotoxicity evaluation of Fn14-redirected CAR-T cells in the other three groups, CD19 CAR-T, Fn14 CAR-T or Fn14 CAR-T/IL-15 cells (1 × 107) were administered once via the tail vein. In this study, tumors were imaged based on bioluminescence using the IVIS system (Caliper Life Sciences). Body position strongly influences bioluminescence intensity, so every mouse was measured three times in different body positions in order to reduce measurement error. Mouse death served as the endpoint of the experiment.
Statistical analysis
All statistical analyses were performed using GraphPad Prism 8.0. Data were presented as mean ± SD. Comparisons of continuous variables between two groups were performed using the two-sided unpaired t-test. Comparisons of continuous variables among three groups were performed using one-way analysis of variance (ANOVA), followed by Dunnett’s test of multiple comparisons.
For survival analysis of orthotopic GBM models, the survival curve was obtained by Kaplan-Meier plot, and survival differences were assessed for significance using the log rank test.
In vitro experiments to measure cytokine release and cytotoxicity were conducted using T cells from two donors. The cells from each donor were tested in triplicate in two independent experiments. Differences associated with P < .05 were considered significant (*P < .05; **P < .01; ***P < .001).
Ethical approval
All procedures involving animals were in accordance with the ethical standards of West China Hospital of Sichuan University, and were approved by our institutional Biomedical Ethics Committee. The Committee also approved the sampling and analysis of tumor tissue and blood from GBM patients and healthy donors (approval 2018–061). Written informed consent was obtained from all donors.
Results
Evaluation of Fn14 expression in gliomas
First, bioinformatic analysis was performed to examine Fn14 expression in gliomas. In the Oncomine database, Fn14 microarray data were downloaded from the Sun Brain Dataset for 23 normal brain, 57 LGG and 81 GBM samples. Fn14 was highly expressed in LGG and even more so in GBM (Figure 1A). In GEPIA, we also analyzed Fn14 expression in LGG and GBM samples as well as survival of the corresponding patients. We found that Fn14 was highly expressed in LGG and GBM (Figure 1B), and higher expression correlated with worse disease-free survival (Figure 1C). Then, IHC, immunofluorescence, western blotting and flow cytometry showed that Fn14 was expressed in human gliomas (Figure 1D) and the GBM cell lines U87, U251, T98G and A172 (Figure 2A–C). The intensity of anti-Fn14 staining was 179.1 ± 35.9 in GBM and 140.3 ± 45.3 in LGG. Fn14 was extremely low or undetectable in normal brain tissues (Figure 1E). The histopathology score of Fn14 was higher for recurrent GBM than for primary GBM (p = .0121; Supplementary Fig. 1A). Fn14 expression was higher in GBM involving wild-type IDH-1 (Supplementary Fig. 1B).
Figure 1.
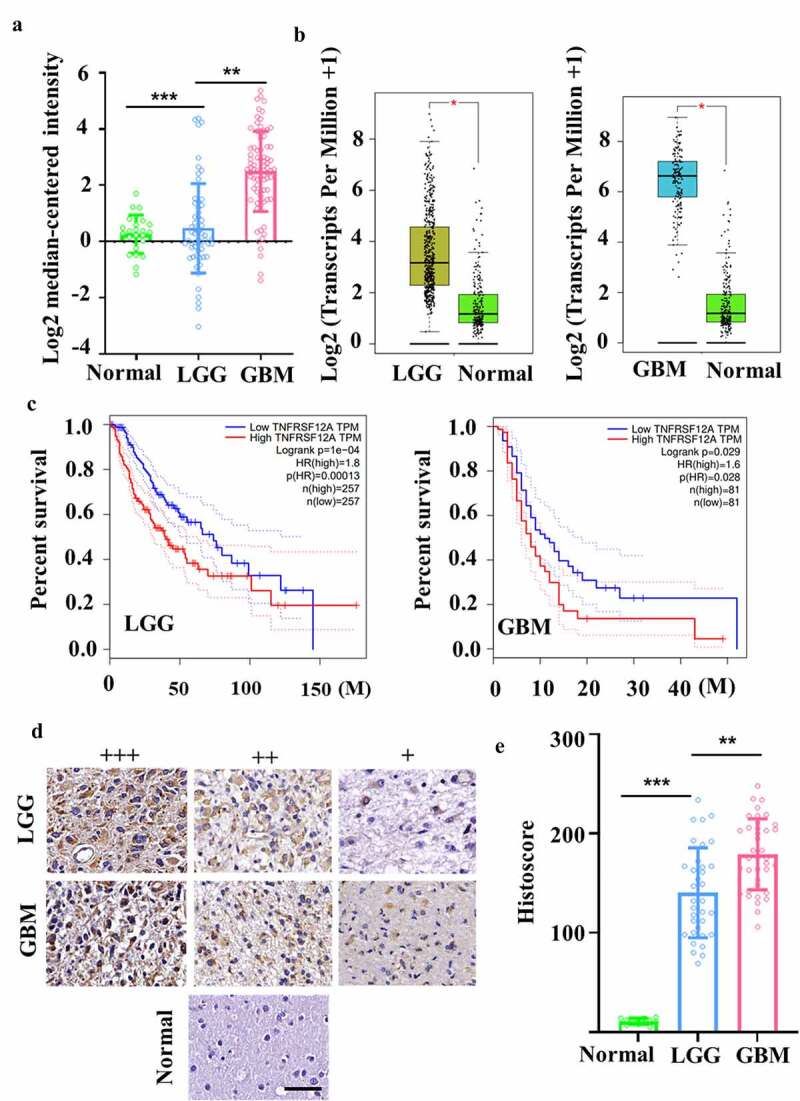
Evaluation of Fn14 expression in gliomas. (A, B) Differential expression of Fn14 in normal, LGG and GBM tissues was analyzed in microarray and RNA-seq datasets from Oncomine and TCGA databases. (C) Correlational analysis between disease-free survival and Fn14 expression in LGG and GBM based on GEPIA. (D) Representative staining of Fn14 in normal, LGG and GBM tissue. Scale bar, 20 μm. (E) Histopathology scores of Fn14 in normal, LGG and GBM tissue. *P < 0.05; **P < 0.01; ***P < 0.001
Figure 2.
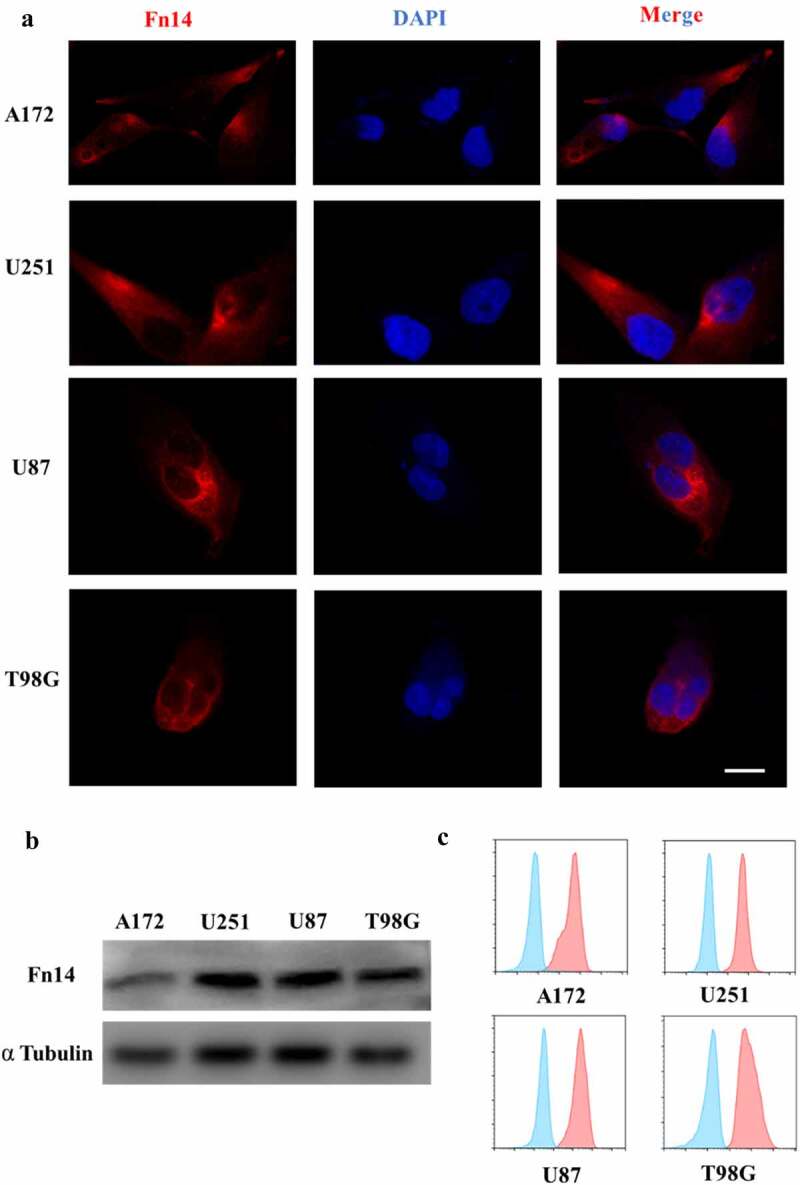
Fn14 expression in four human GBM cell lines. (A) Immunofluorescence (IF), (B) western blotting and (C) flow cytometry showing surface expression of Fn14 in the cell lines A172, U251, U87 and T98G. Scale bar, 10 μm
These experiments suggest that Fn14 may be a useful target in therapies mediated by effector cells, especially in therapies against recurrent GBM and GBM involving wild-type IDH-1.
Generation and characterization of Fn14-mFc recombinant protein, Fn14× CD3 BiTE and CAR-modified T cells
The Fn14-specific scFv derived from P4A8 mAb was combined with mFc or CD3 scFv to construct Fn14-mFc or Fn14× CD3 BiTE in order to confirm binding and assess cytotoxicity (Figure 3A). The resulting Fn14-mFc and tandem single-chain Fn14× CD3 BiTE were expressed in HEK-293 T cells and verified by SDS–PAGE (Figure 3C). Before experiments, we also developed Fn14-knockout GBM cells (U87KO and U251KO) using CRISPR/Cas9 (Supplementary Fig. 1D, E). The Fn14-mFc recombinant protein showed obvious affinity for U87 and U251 cell lines, but not for FACS-sorted U87KO or U251KO cell lines (Figure 3D and E).
Figure 3.
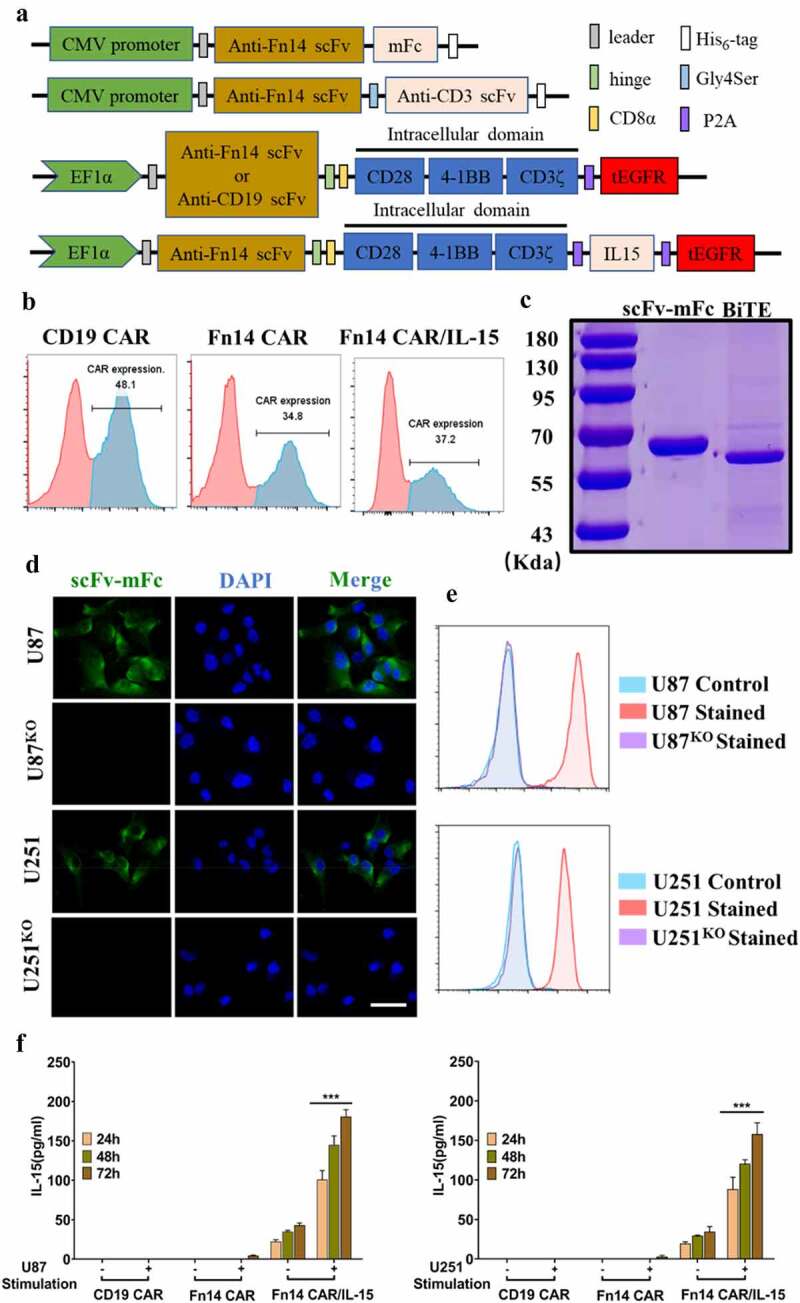
Construction of Fn14×CD3 BiTE and Fn14-specific CAR-T cells with or without IL-15. (A) Schematic representations of Fn14 scFv-mFc, Fn14×CD3 BiTE and Fn14-specific CAR-T cells with or without IL-15. (B) After transduction, expression of CD19 CAR, Fn14 CAR and Fn14 CAR/IL-15 on T cells was analyzed by flow cytometry. (C) SDS–PAGE of purified Fn14 scFv-mFc and Fn14×CD3 BiTE. (D, E) Fn14 scFv-mFc was used as primary antibody to validate the ability of Fn14-specific scFv to bind Fn14 on U87 and U251 cells. FACS-sorted U87KO and U251KO cells served as controls. Scale bar, 20 μm. (F) T lymphocytes transduced with the Fn14 CAR/IL-15 construct released IL-15 after antigen stimulation. ***P < 0.001
We also constructed CD19 CAR (control CAR), Fn14 CAR, and Fn14 CAR/IL-15 (Figure 3A). Primary human T cells from healthy donors transduced with these CAR transgenes showed respective transduction efficiencies of 48.1%, 34.8% and 37.2% (Figure 3B). At 10 days after transduction, the ratios of CD4+/CD8+ T cells were 6:5 in NT cells, 3:5 in Fn14 CAR-T cells and 1:2 in Fn14 CAR-T/IL-15 cells. The percentage of memory T cells, defined as CD45RO+ and CD62L+ cells, was 85.2% among Fn14 CAR-T/IL-15 cells, higher than the corresponding percentages among NT cells (61.5%) and Fn14 CAR-T (60.9%) (Supplementary Fig. 3A).
Fn14× CD3 BiTE activates T cells and mediates the killing of T cells against GBM cell lines in vitro
We tested the ability of Fn14× CD3 BiTE to activate T cells in vitro. When U87 cells were incubated with Fn14× CD3 BiTE for 48 h, 99.6% of T cells were activated, based on expression of the marker CD69 (Supplementary Fig. 2A).
We also tested the ability of Fn14× CD3 BiTE to promote proliferation of T cells. Effector cells were stained with CytoTell Red, then incubated for three cycles with Fn14× CD3 BiTE and target cells. Based on flow cytometry, CytoTell Red was more diluted in the BiTE group (86.15%) than in the comparison group (39.82%), suggesting that T cells incubated with Fn14× CD3 BiTE underwent more cell divisions (Supplementary Fig. 2B).
Next, we evaluated the ability of Fn14× CD3 BiTE to elicit antigen-specific cytotoxic responses in vitro. Tumor-FFluc cells were co-cultured with effector cells at an E:T of 4:1 for 12 h (Fn14× CD3 BiTE, 0.1 μg/ml). T cells that were redirected by Fn14× CD3 BiTE were highly cytotoxic against wild-type parental U87 and U251 cells, but they had no obvious effect against FACS-sorted U87KO or U251KO tumor cells or normal HFL1 cells (Figure 4A, Supplementary Fig. 4B-D). Fn14× CD3 BiTE also improved T-cell–mediated cytotoxicity at much lower concentrations, giving EC50 values of 17.6 ng/ml against U87 cells and 20.9 ng/ml against U251 cells (Figure 4C).
Figure 4.
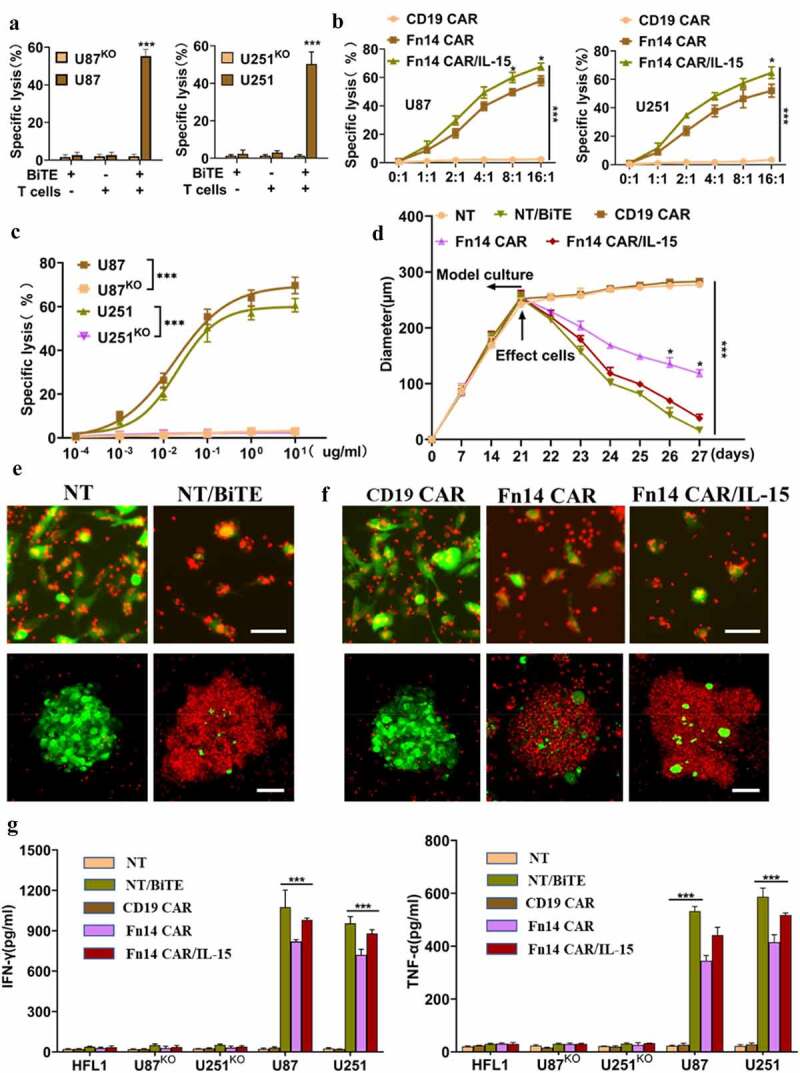
Fn14×CD3 BiTE, Fn14 CAR and Fn14 CAR/IL-15 redirect T cells to kill Fn14-expressing gliomas in vitro. (A) Luciferase-based cytotoxicity assay demonstrating specific lysis of U87 cells and U251 cells, but not the corresponding Fn14-knockout (KO) cell lines, by T cells and Fn14×CD3 BiTE. E:T ratio, 4:1; incubation time, 12 h; Fn14×CD3 BiTE, 0.1 μg/mL. (B) T cells transduced with Fn14 CAR or Fn14 CAR/IL-15 constructs showed cytotoxicity against U87 and U251 cells at different E:T ratios. Incubation time, 12 h. (C) Specific lysis of U87 and U251 cell lines and the corresponding KO lines varied with Fn14×CD3 BiTE concentration. E:T ratio, 4:1; incubation time, 12 h. (D) Fn14×CD3 BiTE, Fn14 CAR as well as Fn14 CAR/IL-15 cells redirected T cells to lyse 3D spheroids of U87 cells. (E, F) Representative images showing the ability of Fn14×CD3 BiTE and Fn14-specific CAR-T cells to kill 2D tumor models (upper panels) and 3D tumor models (lower panels). Scale bars, 20 μm (upper) or 100 μm (lower). (G) Concentrations of IFN-γ and TNF-α in culture medium, as measured by ELISA. *P < 0.05; ***P < 0.001
The redirecting cytotoxicity of Fn14× CD3 BiTE against GFP-expressing U87 cells was assessed in 2D and 3D co-culture models. The 2D model (upper panels) showed obvious tumor-lytic activity (Figure 4E), while the 3D cancer spheroid model (lower panels) showed lysis of tumor spheres and smaller average maximal diameters (Figure 4D and E). Consistent with its potent cytotoxicity in vitro, Fn14× CD3 BiTE favored secretion of the cytokines IFN-γ and TNF-α > 10-fold in culture medium (Figure 4G). These results indicate that Fn14× CD3 BiTE can significantly improve T-cell function.
T lymphocytes transduced with the Fn14 CAR/IL-15 construct release IL-15 after antigen stimulation, which enhances the expansion, anti-tumor activity and survival of CAR-modified T cells in vitro
We separately co-cultured CD19 CAR, Fn14 CAR and Fn14 CAR/IL-15 T cells with U87 or U251 cells to model antigen stimulation or the corresponding cell lines without antigen stimulation (U87KO, U251KO). IL-15 was nearly undetectable from stimulated or unstimulated CD19 CAR and Fn14 CAR cells (Figure 3F). It was produced in small amounts in unstimulated Fn14 CAR/IL-15 cells, with levels ranging from 21 to 41 pg/ml in U87 cells or from 18 to 39 pg/ml in U251 cells. Notably, its production significantly increased after antigen stimulation, with levels ranging from 93 to 187 pg/ml in U87 cells, or from 78 to 168 pg/ml in U251 cells (p < .001).
In the U87 stimulation group, high levels of TNF-α and IFN-γ were detected only in stimulated Fn14 CAR and Fn14 CAR/IL-15 cells, and levels were similar in the two cases (p = .45; Supplementary Fig. 2D). In the U87KO, U251KO and HFL1 stimulation groups (Supplementary Fig. 2C), similarly small amounts of IL-15 were detected in stimulated and unstimulated Fn14 CAR/IL-15 cells.
The proportions of CAR-T cells expressing the activation marker CD69 were 4.4% for control CAR-T cells, 25.4% for Fn14 CAR-T cells, and 45.3% for Fn14 CAR-T/IL-15 cells (Supplementary Fig. 3B). Fn14 CAR-T/IL-15 cells showed more diluted CytoTell Red signal intensity (46.34%) than Fn14 CAR-T cells (24.16%), and they showed a lower rate of 7-AAD+/Annexin V+ staining (19.7% vs 30.5%) and higher rate of 7-AAD−/Annexin V− (56.1% vs 48.3%) (Supplementary Fig. 3C, D).
Finally, we also assessed the antitumor activities of Fn14 CAR-T and Fn14 CAR-T/IL-15 cells in vitro. Fn14 CAR-T and Fn14 CAR-T/IL-15 cells efficiently eliminated tumor cells at an E:T ratio of 2:1, and the specific antitumor effects positively correlated with E:T (Figure 4B). Fn14 CAR-T/IL-15 cells showed stronger antitumor activity against U87 cells at E:T ratios of 8:1 (p = .031) and 16:1 (p = .016), while they showed stronger antitumor activity against U251 cells at E:T of 16:1 (p = .024). Vehicle-transfected control T cells were not cytotoxic against U87 cells, nor were transduced T cells cytotoxic against normal HFL1 cells (Supplementary Fig. 4E, F). Cytotoxicity against U87 cells was confirmed in 2D and 3D co-culture models, and the results were similar to Fn14× CD3 BiTE-redirected cytotoxicity. In the last two days before the end of co-culture, a larger number of Fn14 CAR-T/IL-15 cells than Fn14 CAR-T cells were clustered and involved in tumor killing, leading to more lysis. Fn14 CAR-T/IL-15 cells also led to spheres of smaller average maximal diameter (Figure 4D and F) as well as higher levels of IFN-γ and TNF-α in culture medium (Figure 4G).
These experiments suggest that secretion of IL-15 strengthens the anti-tumor activity of CAR-T cells by enhancing cell proliferation, activation and survival. This translates to greater killing ability with co-culture time.
Fn14× CD3 BiTE represses established tumors, and transgenic IL-15 increases antitumor activity, expansion and persistence of Fn14 CAR-T/IL-15 cells in vivo
In order to assess antitumor activity in vivo, we established glioblastoma PDXs in NSG mice (Figure 5A and E). For Fn14× CD3 BiTE evaluation, T cells (5 × 105 per mouse) and Fn14× CD3 BiTE (10 ng per mouse) were administered intra-tumorally every two days starting on day 7 after PDX inoculation, for a total of four times. These treatments significantly suppressed tumor growth and increased tumor cell-killing ability (Figure 5B and C). Survival was 100% in the NT/BiTE group on day 60, while mice treated with NT cells were dead within 40 days (Figure 5D).
Figure 5.
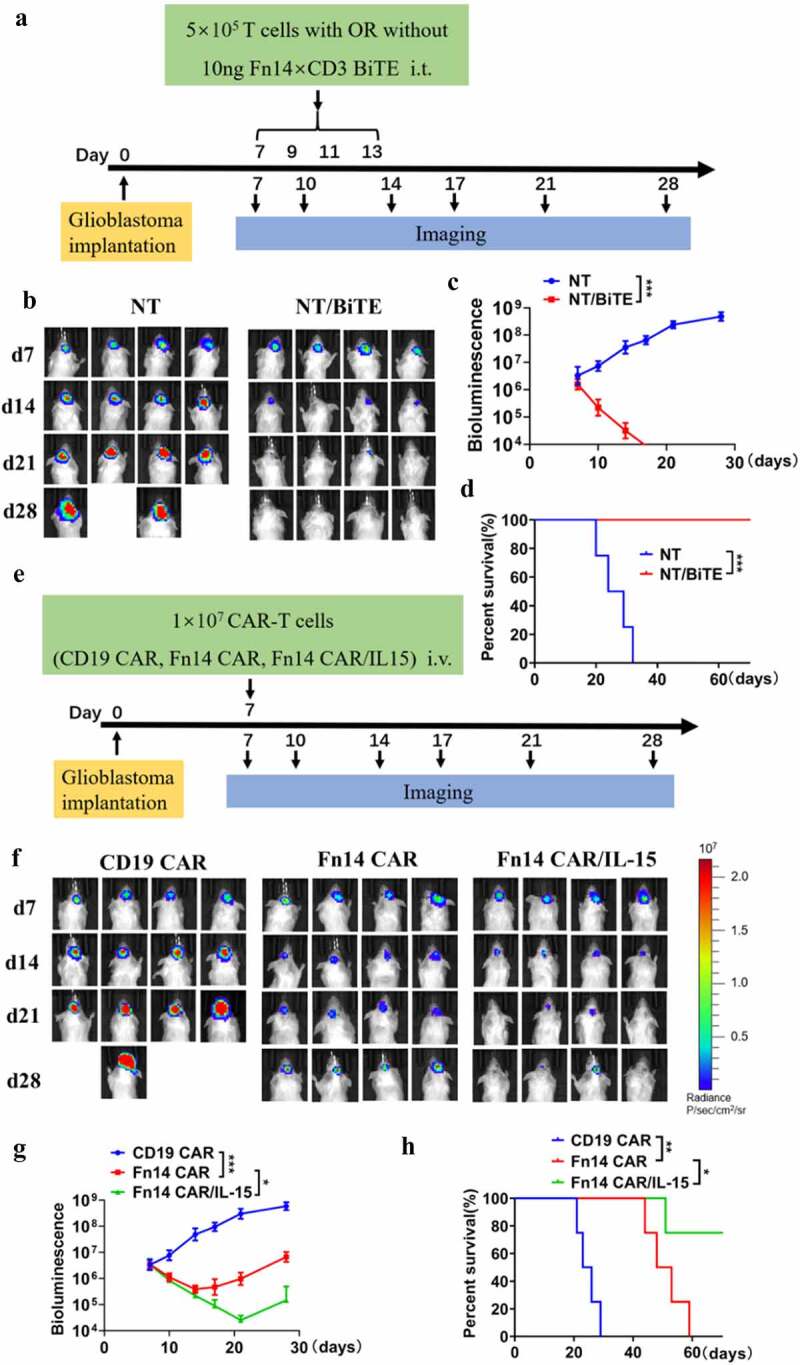
Antitumor effects of Fn14-redirected BiTE, CAR-T and CAR-T/IL-15 cells in PDXs. (A, E) Schematic showing how NSG mouse models were treated with Fn14-specific BiTE and modified T cells. (B, F) Representative images of xenograft bioluminescence after different treatments over time (n=4). (C, G) Tumor total flux (p/s) was calculated using Living Image software. (D and H) Overall survival of tumor-bearing mice, compared using the log rank test. *P < 0.05; **P < 0.01; ***P < 0.001
To assess cytotoxic effects of Fn14-redirected CAR-T cells, we administered a single dose of 1 × 107 CAR-T cells per mouse (Figure 5F and G). As predicted, tumors grew rapidly in mice following control T-cell therapy, while Fn14 CAR-T and Fn14 CAR-T/IL-15 cells were similarly efficient at controlling tumor growth during the first 7 days after administration. However, tumor regression was only transient in the case of Fn14 CAR-T cells, with relapse in mice by day 28 and death of all tumor-bearing mice within 60 days.
Fn14 CAR-T/IL-15 cells, in contrast, led to superior anti-tumor activity: three of four mice showed persistent tumor regression and survived through the end of observation (Figure 5F–H). More Fn14 CAR-T/IL-15 cells were detected in peripheral blood on days 14 and 21, and they continuously produced IL-15 and proliferated, consistent with in vitro experiments (Figure 6E and G). In addition, fewer PD-1+ T cells and higher levels of inflammatory cytokines (IFN-γ, TNF-α) were detected in the Fn14 CAR/IL-15 group on days 14 and 21 (Figure 6F and H). Mice treated with Fn14 CAR-T/IL-15 cells weighed more than control mice starting from day 18 (Supplementary Fig. 2E). These results indicate that IL-15 secretion can reduce exhaustion of T cells and strengthen anti-tumor activity in vivo.
Figure 6.
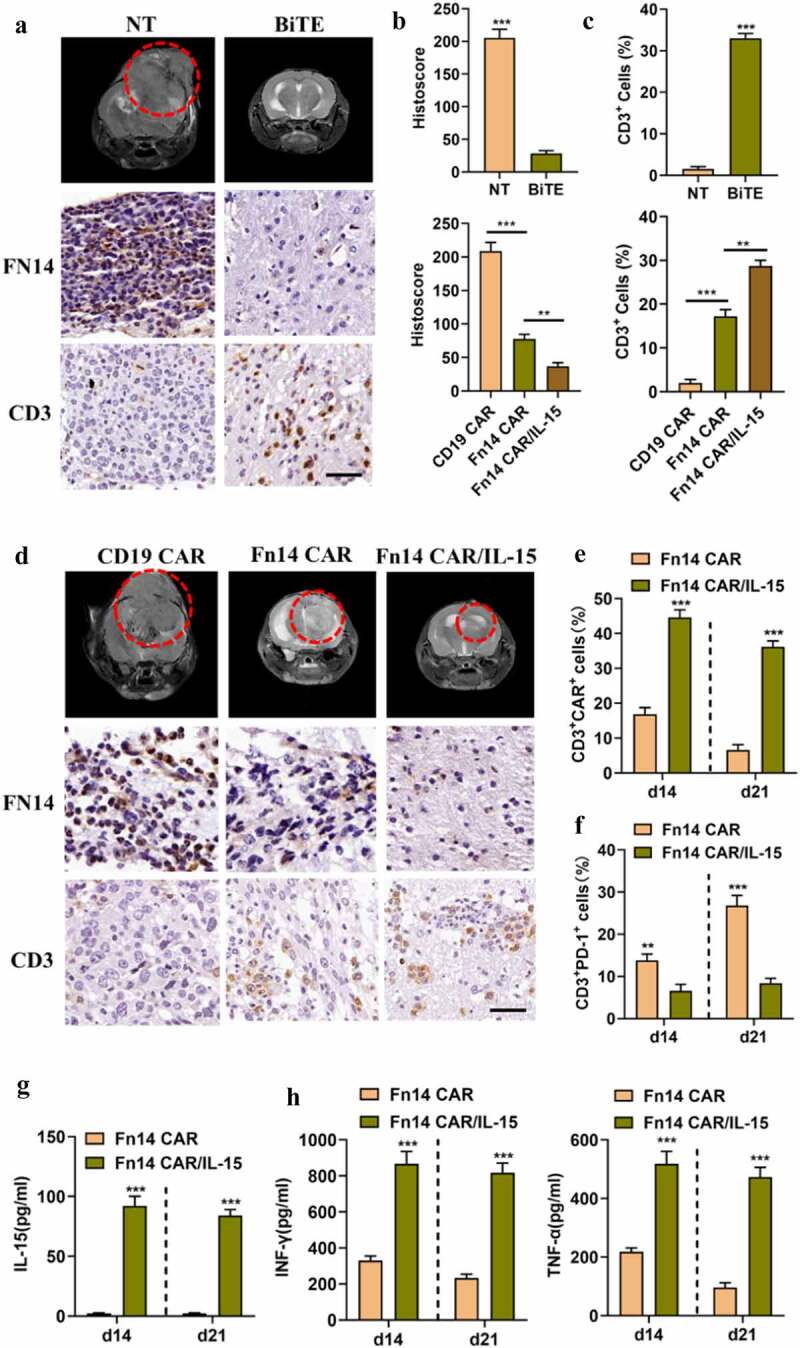
Fn14×CD3 BiTE mediates the distant migration of T cells, and IL-15 augments proliferation and persistence of Fn14 CAR-T/IL-15 cells in vivo. (A, D) Mouse tumors were subjected to MRI on day 28 (red circle), then specimens were harvested and stained for Fn14 and CD3 in order to evaluate the tumor-lytic activity and distant T cell infiltration. Scale bar, 20 μm. (B, C) Fn14 staining intensity and average percentage of CD3+ cells per field in different groups. (E-G) The peripheral blood of Fn14 CAR-T/IL-15-treated mice showed a higher percentage of CAR+ cells, fewer PD-1+ T cells, and higher level of IL-15 than Fn14 CAR-T cells on days 14 and 21. (H) Fn14 CAR-T/IL-15 cells secreted larger amounts of the inflammatory cytokines IFN-γ and TNF-α than Fn14 CAR-T cells did. **P < 0.01; ***P < 0.001
Expression of Fn14 and infiltration of T cells in xenografts
Based on magnetic resonance imaging (MRI), tumor-bearing mice treated with NT/BiTE or Fn14 CAR-T/IL-15 cells showed significant tumor repression by day 28 in comparison to their counterparts treated with NT or CD19 CAR-T cells. In contrast, mice treated with Fn14 CAR-T cells showed only moderate repression (Figure 6A and D).
Consistent with the MRI results, IHC of tumor sections showed that NT/BiTE and Fn14 CAR-T/IL-15 treatment obviously reduced Fn14 expression (Figure 6A, B and D). Furthermore, NT/BiTE treatment induced substantially more T cell infiltration of tumor tissue away from the puncture site than NT treatment, based on immunostaining for CD3. These results suggest that Fn14× CD3 BiTE shows good tissue-penetrating ability, which allows it to promote distant migration by T cells, leading to tumor killing. In addition, massive numbers of Fn14 CAR-T/IL-15 cells and relatively few Fn14 CAR-T cells infiltrated tumor tissues and caused corresponding levels of tumor regression (Figure 6A, C and D). These experiments suggest that intralesional application of NT/BiTE or systemic administration of Fn14 CAR-T/IL-15 cells can induce excellent responses against GBM.
Discussion
Advanced T cell-based therapies including BiTE and CAR-T therapy have become research hotspots because of their promise in treating hematologic malignancies. However, treating solid tumors with therapies based on T cells still faces major challenges, especially in the case of GBM, which shows extensive inter- and intratumoral heterogeneity. As a result, many GBM cells lack ideal, targetable antigens.30 These considerations highlight the need for identifying a specific, ubiquitous antigen within tumors that is persistently expressed and whose targeting does not trigger cytotoxicity.
Fn14, the sole transmembrane surface receptor of TWEAK, is expressed at low levels in normal brain tissue and upregulated in advanced brain tumors. Fn14-targeted therapies exert highly specific antitumor activity against GBM or some solid tumors; these therapies include Fn14-specific mAbs (P4A8, PDL192), anti-Fn14 antibody conjugated nanoparticles, and Fn14-targeted fusion constructs.31–34 Therefore, we hypothesized that Fn14-redirected BiTE and CAR-T strategies would be feasible and effective for developing targeted GBM treatments. We first verified Fn14 expression in GBM tissue, based on the Oncomine and TCGA databases. Fn14 staining intensity and histoscore varied among GBM patients. This indicates that Fn14 expression is not completely uniform or ubiquitous, like the expression of the tumor-associated antigens Her2, IL13Rα2 and EGFRvIII.8–10 Nevertheless, Fn14 is highly expressed in GBM and may be a suitable tumor-associated antigen for targeted therapy. Since we found that Fn14 is expressed more often in recurrent GBM and GBM involving wild-type IDH-1, we designed Fn14-targeted BiTE and CAR-T cells and tested their antitumor efficacy in vitro and in vivo.
In line with our hypothesis, Fn14× CD3 BiTE showed remarkable tumor-lytic activity in vitro and in a preclinical model. At the same time, it had no cytotoxic effects on Fn14-knockout U87 or U251 cell lines or on normal HFL1 cells. This suggests that T cell-mediated effects occur in a Fn14 antigen-restricted fashion and are relatively safe. Our observation of an upregulated activation marker, good proinflammatory cytokine secretion and enhanced T-cell proliferation in vitro are consistent with reports that bispecific antibody recruits, activates and expands T cells to strengthen antitumor ability.35,36
One problem with our approach is that Fn14 is expressed to some extent in normal tissues.13 Thus, even though Fn14× CD3 BiTE can pass through the blood-brain barrier and enter the central nervous system, the presence of TWEAK/Fn14 in normal tissues can lead to cross-reactivity and compete with uptake by the tumor. In addition, the Fn14× CD3 BiTE in our study contains a tandem scFv and non-IgG; as a result, it has a shorter plasma half-life than IgG-like bispecific antibodies with an Fc region, and it cannot perform additional effector functions, such as Ab-dependent cellular cytotoxicity and complement-dependent cytotoxicity.37 Given these circumstances and to ensure a high level of Fn14× CD3 BiTE in xenografts, we injected it within the lesions of an intracranial orthotopic model as reported,29 rather than administering it systemically. Others have reported significant therapeutic responses of intracerebral GBM tumors to a tandem bispecific antibody that targeted EGFRvIII and was delivered systemically,38,39 in contrast to our approach. That efficacy likely reflects the strictly tumor-specific expression of EGFRvIII and strong uptake by the tumors. Nevertheless, EGFRvIII×CD3 BiTE in those studies also accumulated in off-target tissues, limiting therapeutic efficacy.
Despite local damage from the microinjector, Fn14× CD3 BiTE eliminated the lesion, led to distant T cell infiltration and decreased expression of Fn14. This suggests that Fn14× CD3 BiTE has noteworthy ability to penetrate tissues and induce T lymphocytes to lyse xenografts. Indeed, all tumor-bearing mice treated with Fn14× CD3 BiTE survived more than two months, while NT-treated animals died within 40 days.
Fn14 CAR-T cells displayed high killing efficiency against U87 and U251 cell lines in vitro, but they induced only transient tumor regression in vivo, such that tumors eventually regrew and led to death. Other studies have also reported limited therapeutic efficacy of CAR-T cell infusion against solid tumors,8–10 which those researchers attributed to loss of anti-tumor ability of effector cells because of tumor heterogeneity progression and an immunosuppressive tumor microenvironment.
IL-15 is known to provide a supportive milieu for T cells, enhancing their effector function and antitumor activity, so we developed “armored” CAR-T cells that secrete it. Fn14 CAR-T/IL-15 cells contained higher numbers of central memory T cells and upregulated CD69 after binding to the specific tumor-antigenic Fn14. These results are consistent with reports that IL-15 production promotes T-cell differentiation and activation.23,40 Indeed, Fn14 CAR-T/IL-15 cells displayed stronger antitumor activity than Fn14 CAR-T cells in a 3D co-culture model and in vivo. We also detected high levels of IL-15 along with a higher percentage of CAR+ T cells and fewer PD-1+ T cells in peripheral blood on days 14 and 21 in the Fn14 CAR-T/IL-15 group. These results are in agreement with previous work,22 and they reflect that sustained production of IL-15 can maintain the proliferation and survival of Fn14 CAR-T/IL-15 cells.41–43 These effects of IL-15 were confirmed by our observation of greater T cell infiltration and lower Fn14 expression in tumors treated with Fn14 CAR-T/IL-15 cells. These results suggest that Fn14 can be an effective target in GBM CAR-T therapy.
On the other hand, IL-15 acts as a growth cytokine for T cells, which can endow “stemness” properties on Fn14 CAR-T/IL-15 cells. Nevertheless, we did not observe unwanted proliferation in our experiments. If necessary, uncontrolled expansion can be controlled by using a clinical antibody against the co-engineered surface tag tEGFR used for CAR detection.44 This approach is analogous to the use of inducible caspase-9 to inhibit CAR-T activity.22,45
Our experiments suggest that either use of Fn14× CD3 BiTE or transfer of Fn14-specific CAR-T cells may be effective against GBM, at least in the short term. Fn14 CAR-T/IL-15 treatment led to lower intratumoral Fn14 expression than Fn14 CAR-T treatment. However, the number of tumor cells expressing Fn14 decreased with treatment time, suggesting that in the longer term, antigen escape and heterogeneity may limit therapeutic efficacy. This implies that treating GBM will require a multimodal approach.
Our in vivo experiments should be interpreted with caution because the xenografts were grown in immunodeficient mice, which cannot replicate the immunosuppressive tumor microenvironment and may not allow IL-15 to exert its full range of immunoactivating effects. Therefore, future studies should develop immunocompetent models in which the effects of transgenic expression of IL-15 on immune and other cell types can be evaluated. Despite these limitations, our experiments in vitro and in vivo suggest that anti-Fn14 BiTE and Fn14-specific CAR-T cells can efficiently recognize Fn14 on GBM cells and cause tumor regression. Targeting the tumor antigen Fn14 may be a powerful approach for exploiting T cells to treat GBM.
Supplementary Material
Acknowledgments
We thank all healthy donators for donating the peripheral blood mononuclear cells to this study. We also thank Jiang Yu for the gift of HFL1 cells.
Funding Statement
This work was supported by the National Natural Science Foundation of China (81772693, 82073404 and 82002648), Double Top Construction and Innovation Spark Item of Sichuan University (2082604401047), Incubation project of clinical study, West China Hospital, Sichuan University (2019HXFH020), 1.3.5 project for disciplines of excellence, West China Hospital, Sichuan University (ZYJC18007).
Disclosure statement
The authors report no conflict of interest.
Author contributions
Conceptualization, GWL, ZLZ, LJC; Data Curation and Formal Analysis, GWL, ZLZ; Project Administration, XT, JHH; Resources, Methodology, LYY, GQW; Investigation and Visualization, KHZ; Validation, YC, CL; Writing–original draft, GWL, YLW; Writing–review and editing, APT; Supervision, LXZ. All authors have read and approved the final version of this manuscript.
Data availability statement
Some data were derived from the following resources available in the public domain
https://portal.gdc.cancer.gov/files/da904cd3-79d7-4ae3-b6c0-e7127998b3e6;
https://tcga-xena-hub.s3.us-east-1.amazonaws.com/download/TCGA.GBM.sampleM ap%2FGBM_clinicalMatrix;
https://tcga-xena-hub.s3.us-east-1.amazonaws.com/download/TCGA.GBM.sampleMap%2FHT_HG-U133A.gz
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Louis DN, Perry A, Reifenberger G, Von DA, Figarella D, Webster B, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW, et al. The 2016 World Health Organization classification of tumors of the central nervous system : a summary. Acta Neuropathol. 2016;131(6):803–14. doi: 10.1007/s00401-016-1545-1. [DOI] [PubMed] [Google Scholar]
- 2.Tan AC, Ashley DM, López GY.. Management of glioblastoma : state of the art and future directions. CA Cancer J Clin. 2020:1–14. doi: 10.3322/caac.21613. [DOI] [PubMed] [Google Scholar]
- 3.Velasquez MP, Bonifant CL, Gottschalk S. Redirecting T cells to hematological malignancies with bispecific antibodies. Blood. 2018;131(1):30–38. doi: 10.1182/blood-2017-06-741058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goebeler ME, Bargou RC. T cell-engaging therapies — BiTEs and beyond. Nat Rev Clin Oncol. 2020;17(7):418–434. doi: 10.1038/s41571-020-0347-5. [DOI] [PubMed] [Google Scholar]
- 5.Singh AK, McGuirk JP. CAR T cells: continuation in a revolution of immunotherapy. Lancet Oncol. 2020;21(3):e168–e178. doi: 10.1016/S1470-2045(19)30823-X. [DOI] [PubMed] [Google Scholar]
- 6.Jen EY, Xu Q, Schetter A, Przepiorka D, Shen YL, Roscoe D, Sridhara R, Deisseroth A, Philip R, Farrell AT, et al. FDA approval: blinatumomab for patients with B-cell precursor acute lymphoblastic leukemia in morphologic remission with minimal residual disease. Clin Cancer Res. 2019;25(2):473–477. doi: 10.1158/1078-0432.CCR-18-2337. [DOI] [PubMed] [Google Scholar]
- 7.Calmes-Miller J. FDA approves second CAR T-cell therapy. Cancer Discov. 2018;8:5–6. doi: 10.1158/2159-8290.CD-NB2017-155. [DOI] [PubMed] [Google Scholar]
- 8.Brown CE, Alizadeh D, Starr R, Weng LWJ, Wagner JR, Naranjo A, Ostberg JR, Blanchard MS, Kilpatrick J, Simpson J. Regression of glioblastoma after chimeric antigen receptor T- cell therapy. N Engl J Med. 2016;375(26):2561–2569. doi: 10.1056/NEJMoa1610497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D. HER2-specific chimeric antigen receptor–modified virus-specific t cells for progressive glioblastoma a phase 1 dose-escalation trial. JAMAOncology. 2017;3:1094–1101. doi: 10.1001/jamaoncol.2017.0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9. doi: 10.1126/scitranslmed.aaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salinas RD, Durgin JS, O’Rourke DM. Potential of glioblastoma-targeted chimeric antigen receptor (CAR) T-cell therapy. CNS Drugs. 2020;34(2):127–145. doi: 10.1007/s40263-019-00687-3. [DOI] [PubMed] [Google Scholar]
- 12.Goff SL, Morgan RA, Yang JC, Sherry RM, Paul F, Restifo NP, Feldman SA, Lu Y-C, Lu L, Zheng Z, et al. Pilot trial of adoptive transfer of chimeric antigen receptor transduced T cells targeting EGFRvIII in patients with glioblastoma. J Immunother. 2019;42(4):126–135. doi: 10.1097/CJI.0000000000000260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Winkles JA. The TWEAK-Fn14 cytokine-receptor axis: discovery, biology and therapeutic targeting. Nat Rev Drug Discov. 2008;7(5):411–425. doi: 10.1038/nrd2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Willis AL, Tran NL, Chatigny JM, Charlton N, Vu H, Brown SAN, Black MA, McDonough WS, Fortin SP, Niska JR, et al. The fibroblast growth factor – inducible 14 receptor is highly expressed in HER2-positive breast tumors and regulates breast cancer cell invasive capacity. Mol Cancer Res. 2008;6(5):725–735. doi: 10.1158/1541-7786.MCR-08-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yin J, Liu Y, Tillman H, Barrett B, Hewitt S, Ylaya K, Fang L, Lake R, Corey E, Morrissey C, et al. AR-regulated TWEAK-FN14 pathway promotes prostate cancer bone metastasis. Cancer Res. 2014;74(16):4306–4318. doi: 10.1158/0008-5472.CAN-13-3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dwyer BJ, Jarman EJ, Gogoi-Tiwari J, Ferreira-Gonzalez S, Boulter L, Guest RV, Kendall TJ, Kurian D, Kilpatrick AM, Robson AJ, et al. TWEAK/Fn14 signalling promotes cholangiocarcinoma niche formation and progression. J Hepatol. 2021;74(4):860–872. doi: 10.1016/j.jhep.2020.11.018. [DOI] [PubMed] [Google Scholar]
- 17.Tran NL, McDonough WS, Savitch BA, Sawyer TF, Winkles JA, Berens ME. The tumor necrosis factor-like weak inducer of apoptosis (TWEAK)-fibroblast growth factor-inducible 14 (Fn14) signaling system regulates glioma cell survival via NFκB pathway activation and BCL-XL/BCL-W expression. J Biol Chem. 2005;280(5):3483–3492. doi: 10.1074/jbc.M409906200. [DOI] [PubMed] [Google Scholar]
- 18.Tran NL, McDonough WS, Savitch BA, Fortin SP, Winkles JA, Symons M, Nakada M, Cunliffe HE, Hostetter G, Hoelzinger DB, et al. Increased fibroblast growth factor-inducible 14 expression levels promote glioma cell invasion via Rac1 and nuclear factor-κB and correlate with poor patient outcome. Cancer Res. 2006;66(19):9535–9542. doi: 10.1158/0008-5472.CAN-06-0418. [DOI] [PubMed] [Google Scholar]
- 19.Hersh DS, Harder BG, Roos A, Peng S, Heath JE, Legesse T, Kim AJ, Woodworth GF, Tran NL, Winkles JA. The TNF receptor family member Fn14 is highly expressed in recurrent glioblastoma and in GBM patient-derived xenografts with acquired temozolomide resistance. Neuro Oncol. 2018;20(10):1321–1330. doi: 10.1093/neuonc/noy063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perez JG, Tran NL, Rosenblum MG, Schneider CS, Connolly NP, Kim AJ, Woodworth GF, Winkles JA. The TWEAK receptor Fn14 is a potential cell surface portal for targeted delivery of glioblastoma therapeutics. Oncogene. 2016;35(17):2145–2155. doi: 10.1038/onc.2015.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giuffrida L, Sek K, Henderson MA, House IG, Lai J, Chen AXY. IL-15 preconditioning augments CAR T cell responses to checkpoint blockade for improved treatment of solid tumors. Mol Ther. 2020;28. doi: 10.1016/j.ymthe.2020.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Y, Sun C, Landoni E, Metelitsa L, Dotti G, Savoldo B, Zhao Z, Dai H, Wang H, Fang Z. Eradication of neuroblastoma by T cells redirected with an optimized GD2-specific chimeric antigen receptor and interleukin-15. Clin Cancer Res. 2019;38(1):25. doi: 10.1158/1078-0432.CCR-18-1811. [DOI] [PubMed] [Google Scholar]
- 23.Hurton LV, Singh H, Najjar AM, Switzer KC, Mi T, Maiti S, Olivares S, Rabinovich B, Huls H, Forget M-A, et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc Natl Acad Sci. 2016;113(48):E7788–E7797. doi: 10.1073/pnas.1610544113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kantarjian H, Stein A, Gökbuget N, Fielding AK, Schuh AC, Ribera J-M, Wei A, Dombret H, Foà R, Bassan R, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376(9):836–847. doi: 10.1056/NEJMoa1609783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang X, Chang W, Wong CW, Colcher D, Sherman M, Ostberg JR, Forman SJ, Riddell SR, Jensen MC. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. 2011;118(5):1255–1263. doi: 10.1182/blood-2011-02-337360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ying Z, Huang XF, Xiang X, Liu Y, Kang X, Song Y. A safe and potent anti-CD19 CAR T cell therapy. Nat Med. 2019;25. doi: 10.1038/s41591-019-0421-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paszkiewicz PJ, Fräßle SP, Srivastava S, Sommermeyer D, Hudecek M, Drexler I, Sadelain M, Liu L, Jensen MC, Riddell SR, et al. Targeted antibody-mediated depletion of murine CD19 CAR T cells permanently reverses B cell aplasia. J Clin Invest. 2016;126(11):4262–4272. doi: 10.1172/JCI84813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shum T, Omer B, Tashiro H, Kruse RL, Wagner DL, Parikh K, Yi Z, Sauer T, Liu D, Parihar R, et al. Constitutive signaling from an engineered IL7 receptor promotes durable tumor elimination by tumor-redirected T cells. Cancer Discov. 2017;7(11):1238–1247. doi: 10.1158/2159-8290.CD-17-0538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grosse-Hovest L, Wick W, Minoia R, Weller M, Rammensee HG, Brem G, Jung G. Supraagonistic, bispecific single-chain antibody purified from the serum of cloned, transgenic cows induces T-cell-mediated killing of glioblastoma cells in vitro and in vivo. Int J Cancer. 2005;117(6):1060–1064. doi: 10.1002/ijc.21294. [DOI] [PubMed] [Google Scholar]
- 30.Bagley SJ, Desai AS, Linette P, June CH, O’Rourke DM. CAR T-cell therapy for glioblastoma: recent clinical advances and future challenges. Neuro Oncol. 2018;20(11):1429–1438. doi: 10.1093/neuonc/noy032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Michaelson JS, Amatucci A, Kelly R, Su L, Garber E, Day ES, Berquist L, Cho S, Li Y, Parr M. Development of an Fn14 agonistic antibody as an anti-tumor agent. MAbs. 2011;3(4):362–375. doi: 10.4161/mabs.3.4.16090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schneider CS, Perez JG, Cheng E, Zhang C, Hanes J, Winkles JA, Winkles JA, Woodworth GF, Kim AJ. Minimizing the non-specific binding of nanoparticles to the brain enables active targeting of Fn14-positive glioblastoma cells. Biomaterials. 2015;42:42–51. doi: 10.1016/j.biomaterials.2014.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De CAA, Cheung LH, Mohamedali KA, Whitsett TG, Winkles JA, Hittelman WN. Therapeutic efficacy and safety of a human fusion construct targeting the TWEAK receptor Fn14 and containing a modified granzyme B. J Immunother Cancer. 2020;8:1–11. doi: 10.1136/jitc-2020-001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Culp PA, Choi D, Zhang Y, Yin J, Seto P, Ybarra SE, Su M, Sho M, Steinle R, Wong MHL, et al. Antibodies to TWEAK receptor inhibit human tumor growth through dual mechanisms. Clin Cancer Res. 2010;16(2):497–508. doi: 10.1158/1078-0432.CCR-09-1929. [DOI] [PubMed] [Google Scholar]
- 35.Bacac M, Fauti T, Sam J, Colombetti S, Weinzierl T, Ouaret D, Bodmer W, Lehmann S, Hofer T, Hosse RJ, et al. A novel carcinoembryonic antigen T-cell bispecific antibody (CEA TCB) for the treatment of solid tumors. Clin Cancer Res. 2016;22(13):3286–3297. doi: 10.1158/1078-0432.CCR-15-1696. [DOI] [PubMed] [Google Scholar]
- 36.Pillarisetti K, Edavettal S, Mendonça M, Li Y, Tornetta M, Babich A, Majewski N, Husovsky M, Reeves D, Walsh E, et al. A T-cell–redirecting bispecific G-protein–coupled receptor class 5 member D x CD3 antibody to treat multiple myeloma. Blood. 2020;135(15):1232–1243. doi: 10.1182/blood.2019003342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H, Er Saw P, Song E. Challenges and strategies for next-generation bispecific antibody-based antitumor therapeutics. Cell Mol Immunol. 2020;17(5):451–461. doi: 10.1038/s41423-020-0417-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Choi BD, Kuan C, Cai M, Archer GE, Mitchell DA. Systemic administration of a bispecific antibody targeting EGFRvIII successfully treats intracerebral glioma. Proc Natl Acad Sci. 2013:110. doi: 10.1073/pnas.1219817110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gedeon PC, Schaller TH, Chitneni SK, Choi BD, Kuan CT, Suryadevara CM, Snyder DJ, Schmittling RJ, Szafranski SE, Cui X, et al. A rationally designed fully human EGFRvIII:CD3-targeted bispecific antibody redirects human T cells to treat patient-derived intracerebral malignant glioma. Clin Cancer Res. 2018;24(15):3611–3631. doi: 10.1158/1078-0432.CCR-17-0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang X, Sun S, Hwang I, Tough DF, Sprent J. Potent and selective stimulation of memory-phenotype CD8+ T cells in vivo by IL-15. Immunity. 1998;8(5):591–599. doi: 10.1016/S1074-7613(00)80564-6. [DOI] [PubMed] [Google Scholar]
- 41.Teague RM, Sather BD, Sacks JA, Huang MZ, Dossett ML, Morimoto J, Tan X, Sutton SE, Cooke MP, Öhlén C, et al. Interleukin-15 rescues tolerant CD8+ T cells for use in adoptive immunotherapy of established tumors. Nat Med. 2006;12(3):335–341. doi: 10.1038/nm1359. [DOI] [PubMed] [Google Scholar]
- 42.Marks-Konczalik J, Dubois S, Losi JM, Sabzevari H, Yamada N, Feigenbaum L, Waldmann TA, Tagaya Y. IL-2-induced activation-induced cell death is inhibited in IL-15 transgenic mice. Proc Natl Acad Sci U S A. 2000;97(21):11445–11450. doi: 10.1073/pnas.200363097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klebanoff CA, Finkelstein SE, Surman DR, Lichtman MK, Gattinoni L, Theoret MR, Grewal N, Spiess PJ, Antony PA, Palmer DC, et al. IL-15 enhances the in vivo antitumor activity of tumor-reactive CD8 + T Cells. Proc Natl Acad Sci U S A. 2004;101(7):1969–1974. doi: 10.1073/pnas.0307298101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kao RL, Truscott LC, Chiou TT, Tsai W, Wu AM, De Oliveira SN. A cetuximab-mediated suicide system in chimeric antigen receptor-modified hematopoietic stem cells for cancer therapy. Hum Gene Ther. 2019;30(4):413–428. doi: 10.1089/hum.2018.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, Heslop HE, Rooney CM, Brenner MK, Dotti G, et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. 2010;24(6):1160–1170. doi: 10.1038/leu.2010.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Some data were derived from the following resources available in the public domain
https://portal.gdc.cancer.gov/files/da904cd3-79d7-4ae3-b6c0-e7127998b3e6;
https://tcga-xena-hub.s3.us-east-1.amazonaws.com/download/TCGA.GBM.sampleM ap%2FGBM_clinicalMatrix;
https://tcga-xena-hub.s3.us-east-1.amazonaws.com/download/TCGA.GBM.sampleMap%2FHT_HG-U133A.gz