

Abstract
Evolutionary processes, including mutation, migration and natural selection, have influenced the prevalence and distribution of various disorders in humans. However, despite a few well-known examples, such as the APOL1 variants — which have undergone positive genetic selection for their ability to confer resistance to Trypanosoma brucei infection but confer a higher risk of chronic kidney disease — little is known about the effects of evolutionary processes that have shaped genetic variation on kidney disease. An understanding of basic concepts in evolutionary genetics provides an opportunity to consider how findings from ancient and archaic genomes could inform our knowledge of evolution and provide insights into how population migration and genetic admixture have shaped the current distribution and landscape of human kidney-associated diseases. Differences in exposures to infectious agents, environmental toxins, dietary components and climate also have the potential to influence the evolutionary genetics of kidneys. Of note, selective pressure on loci associated with kidney disease is often from non-kidney diseases, and thus it is important to understand how the link between genome-wide selected loci and kidney disease occurs in relation to secondary nephropathies.
Subject terms: Genomics, Kidney diseases, Evolution
Here, the authors provide an overview of the evolutionary processes that have implications for our understanding of kidney disease development and progression. They describe data derived from studies of ancient and archaic genomes and how population migration and genetic admixture have shaped the current landscape of human kidney-associated diseases, as well as the potential impact of environmental influences on evolutionary genetics and the adaptation of kidneys.
Key points
Kidney diseases that have an age of onset after the reproductive age (that is, most forms of chronic kidney disease) are unlikely to act as agents of natural selection for the elimination of disease-causing alleles because those alleles are successfully transmitted to successive generations.
Several loci under natural selection owing to the protection they provide against infectious agents (for example, Trypanosoma brucei and Plasmodium falciparum) have an effect on the risk of kidney disease (APOL1-associated chronic kidney disease and sickle cell nephropathy, respectively).
Loci that are under natural selection and that increase the risk of disorders that damage the kidney (such as diabetes mellitus and hypertension) may contribute to the risk of secondary nephropathies.
Only a handful of genetic loci identified by genome-wide association studies as being associated with complex kidney diseases show evidence of natural selection in primates; however, many more of these loci show evidence of natural selection in human populations, suggesting that most selective events at kidney-associated loci occurred relatively recently in the evolution of the human species.
Introduction
An in-depth appreciation of evolution and the constellation of adaptive forces that have shaped biology is critical to understanding how humans have survived and thrived in different environments1. The human kidneys maintain fluid and electrolyte homeostasis by filtering waste and surplus water from the blood. The structure and function of the human kidneys that we observe today reflect our evolution as a species and are largely the result of legacies of adaptations that have accumulated as our ancestors moved from a marine littoral environment to freshwater to a terrestrial environment. These migrations required the evolution of glomeruli that were capable of supporting a high filtration rate followed by the evolution of tubules capable of reabsorbing the majority of filtrate2–4. Selective pressure to maintain a high glomerular filtration rate (GFR) was possibly provided by the need to excrete urea once our ancestors began consuming a meat-rich diet during the Pleistocene epoch5 Since then, other evolutionary pressures have acted to refine human kidney function to its current level.
The nephron is the basic functional unit of the kidney. In humans, nephrogenesis is completed by 36 weeks’ gestation, at which point the nephron numbers for each kidney are established6,7. On average, each fully developed kidney has around one million nephrons, but this number can vary between 0.2 million to 3 million8,9. No nephrogenesis takes place after birth and therefore, the decline in kidney function that occurs as a result of the ageing process or in response to injury leaves those with low nephron endowment at birth — for example, those born prematurely or exposed to nutritional deficiency in utero — vulnerable to kidney disease10,11. When confronted with limited resources in the intrauterine environment, the fetal kidney is vulnerable to compromised development12. By contrast, complex mechanisms have evolved to preferentially protect the fetal brain when challenged similarly13. This hierarchy represents an evolutionary trade-off in resource-limited situations that favours fetal survival, as the effects of compromised kidney development and the consequential reduction in nephron number are generally only observed after reproductive age, when additional pressures such as chronic disorders and environmental insults are present.
Here, we provide insights into the evolutionary genetics of human kidneys that have implications for understanding disease development and progression in children and adults. Our understanding of these processes has been facilitated by the availability of affordable high-throughput genotyping and sequencing technologies, the increasing diversity of populations enrolled in genomic studies, the development of techniques to reliably extract and analyse DNA from ancient genomes and the expansion of statistical toolsets to infer evolutionary pressures acting on the genome. We describe relevant data derived from studies of ancient and archaic genomes and how population migration and genetic admixture have shaped the current landscape of human kidney-associated diseases. We also discuss the impact that infections, environmental toxins, diet and climate may have had on the evolutionary genetics and adaptation of kidneys, and finally, highlight gaps in our knowledge of the evolutionary biology of kidney disease, as well as challenges and opportunities for further research.
Evolutionary forces and processes
Evolution is commonly defined as a change in allele frequencies. It underlies the process by which an organism adapts to its environment to increase the probability of survival and reproductive success. New alleles enter a population through either mutation (that is, a heritable change in the nucleotide sequence of a genome), or migration (that is, from individuals carrying new alleles moving into the population). In addition to mutation and migration, four other forces can drive genetic variation within and between populations: population growth, recombination, random genetic drift and natural selection (Fig. 1). Population growth refers to a change in the number of individuals in the population and can be positive or negative. Recombination is the process by which segments of DNA are broken and recombined; in particular, reciprocal crossing over of genetic material during meiosis can create new combinations of alleles in the offspring. Recombination disrupts genetic linkage and creates new haplotypes, but neither population growth nor recombination can change the expected frequencies of selectively neutral alleles across generations and are therefore not considered further.
Fig. 1. Evolutionary forces and processes.

Common evolutionary forces and processes are represented as acting on alleles within a population. Different alleles at a locus are represented by coloured circles. Exterior rings depict changes in the proportions of the alleles, given the evolutionary processes acting on the original population (middle ring). a | Genetic drift involves a shift in the frequency of alleles as a result of stochastic sampling. b | In natural selection, some alleles affect the success of passing along genetic material to the next generation, causing changes in the distribution of alleles. c | In recombination, different parental alleles combine to form a new allele in the offspring, which contains sections of DNA from each parent. d | Mutations that involve a novel alteration within the DNA have potential to produce a new allele. e | Migration involves the introduction of an allele from another population into a new population. f | Population growth involves the expansion of a population, resulting in a larger population size, but without changes in allele frequencies.
Random genetic drift refers to changes in allele frequencies that occur in finite populations owing to the random transmission of alleles from one generation to the next. Random genetic drift is directionless, meaning that allele frequencies are equally likely to increase or decrease. Natural selection causes allele frequencies to change in a directed manner, such that alleles conferring a fitness advantage increase in frequency. In this context, fitness refers to the likelihood that an allele will be passed on to the next generation, meaning that individuals carrying the allele must survive until reproductive age. The absolute fitness of a genotype is the ratio of the number of individuals with the genotype after selection to the number of individuals with the genotype before selection, whereas the relative fitness of a genotype is the ratio of the average number of surviving offspring with that genotype to the average number of surviving offspring of all genotypes after a single generation.
Studies of natural selection often focus on a single locus. With the accumulation of genome sequence data, it is possible to study polygenic adaptation, which focuses on the net effect of small changes in allele frequencies at potentially hundreds or thousands of loci. When studying small changes in allele frequencies distributed across the genome, it is critical to not misinterpret population structure (that is, systematic differences in the frequencies of neutral alleles between subgroups in a population resulting from a restriction of gene flow) as polygenic adaptation14,15. It is also now possible to study polygenic adaptation of archaic alleles in modern humans, although such studies are limited by a lack of data on the genetic diversity of early versus late archaic humans and on the structure of archaic populations that once lived in Asia, Europe and the Middle East.
In contrast to population growth and recombination, natural selection and migration can change the expected frequencies of selected alleles across generations, which means that they can have an effect on survival and reproductive success in subsequent generations. In this Review, we describe how the processes of natural selection and migration have influenced factors that affect kidney health. Of note, the evolutionary context of kidney disease is complex, and much remains to be understood. For example, consider the relationship between natural selection and congenital anomalies of the kidney and urinary tract (CAKUTs), chronic kidney disease (CKD) and kidney failure. Severe CAKUTs usually lead to early mortality, implying that any underlying genetic variants are filtered out of the population by negative selection. On the other hand, most forms of CKD and kidney failure develop after the reproductive years, meaning that their underlying genetic risk variants are not selected against. Evolutionary forces that select for resistance against a phenotype can occur only if the phenotype develops before reproductive age. Therefore, it is likely that loci associated with CKD or kidney failure that are under natural selection have been selected for or against by non-kidney factors (or possibly by effects on the kidney that are beneficial in early life). A good example of this scenario is APOL1, for which selective pressure is exerted by the ability of variants to protect against Trypanosoma brucei infection. Thus, it is important to note at the outset that any kidney disorder (whether primary or secondary) that has an age of onset after the reproductive age is unlikely to be an agent of natural selection. However, as exemplified by APOL1, several loci under natural selection from non-kidney influences often have an effect on the risk of kidney disorders. It is important to note that although genetic differences between populations can be driven by evolutionary forces with implications for health outcomes, most between-population differences arise from environmental and social factors such as diet, inequitable access to health care, lack of healthcare infrastructure and social issues such as racism.
Ancient and archaic genomes
The study of ancient and archaic genomes has seen tremendous advances in the past decade. Technological advances coupled with an increase in the number and diversity of archaeological finds of human remains means that we now know more about the genomes of our ancestors than ever before. Most studies of ancient human specimens have provided only limited insights into the evolutionary genetics of kidney phenotypes. However, the availability of increasing amounts of archaic genome data from other Homo sub-species, in particular Neanderthals (Homo sapiens neanderthalensis), which existed between 400 thousand years ago (Kya) and 40 Kya, and Denisovans (Homo sapiens denisova) who lived between 195 Kya and 52 Kya, have potential to provide new insights (Fig. 2). Some Neanderthals lived in Europe 300 Kya to 200 Kya — a period characterized by long glacial periods with extreme cold conditions and long winters, during which the main source of food was the hunting of large game. This environment required the inhabitants to adapt to a high-protein, high-fat diet. It has been hypothesized that the large size of the inferior thoracic cavity and the wide pelvis of the Neanderthal were adaptations to provide the space needed for large kidneys that developed following the adoption of a high-protein diet based on meat consumption16,17. Analysis of tooth proteins showed that the rs4236 variant in the MGP gene (which introduces a Thr127Ala mutation into the Matrix Gla protein), which is associated with a reduced risk of nephrolithiasis in individuals of Han Chinese or Japanese ancestry18,19, is also found in Denisovans and Neanderthals. Thus, some genetic variants associated with kidney phenotypes existed in extinct human lineages. It should be noted that greater Neanderthal and Denisovan introgression seems to have occurred in European and Asian ancestry populations than in African populations20 (Fig. 3). Further studies of the introgressed Neanderthal segments in modern humans indicate that these have a functional impact as indicated by evidence of cis-regulatory effects, and effects of the segments on gene expression changes, the regulation of steady-state gene expression and transcriptional responsiveness to immune (in particular virus) challenges, as well as phenotypic effects on traits, including skin tone, hair colour, height, sleep and mood21–25. Ongoing investigations into the functional consequences of introgressed variants may identify effects on aspects of kidney health.
Fig. 2. Relationships between anatomically modern humans, Neanderthals and Denisovans.

Note that there are probably multiple admixture events from distinct Denisovan populations to various Southeast Asian island populations167.
Fig. 3. Archaic humans and anatomically modern humans.

Fossil archaic human specimens that have yielded sequence data have come from archaeological sites ranging across Asia and Europe168,169. Interbreeding between Neanderthals (red dots on the figure) and anatomically modern humans (AMHs) occurred on multiple occasions, based on observed patterns of divergence and heterozygosity and modelling of gene flow170. The percentage of Neanderthal DNA in AMHs is low, at around 1–4%, in Asians, Europeans, Native Americans, North Africans and Oceanians171. Neanderthal DNA is present in sub-Saharan Africans in even lower amounts, as a result of the gene flow of Neanderthal lineages into ancestral Europeans after the split of Europeans and East Asians, their migration to Africa and subsequent gene flow into Africans20. Denisovans (blue dots) have been identified in archaeological sites in Siberia172 and the Tibetan Plateau169. The Siberian site also has Neanderthal specimens, and a first-generation hybrid has been identified, with a Neanderthal mother and a Denisovan father173. The percentage of Denisovan DNA in AMHs is up to 4–6% in Melanesians and negligible in African and Western Eurasians172. Neanderthal DNA sequences in AMH are dispersed throughout approximately 20% of the human genome, with evidence that some Neanderthal sequences have been positively selected and that some regions of the genome are depleted of Neanderthal sequences, consistent with the notion of purifying (negative) selection174. In particular, two derived mutations in APOL1 confer protection against Trypanosoma brucei, the cause of sleeping sickness, at the cost of an increased risk of kidney disease39. The APOL1 haplotype present in the human reference genome generated by the Human Genome Project175 is shared with both Neanderthals and Denisovans. It may have been reintroduced into the AMH gene pool via gene flow from archaic humans and might have conferred a selective advantage176. An archaic SLC16A11 haplotype that affects hepatic lipid metabolism may have introgressed from Neanderthals into ancestors of Amerindians and conferred an advantage in the context of diet29. Admixture with Neanderthals also introduced into European genomes multiple variants that regulate immune-responsive gene expression, specifically to viral challenges22. It is conceivable, although unproven, that the kidneys might be affected by these variants.
One example of an archaic genome variant that is indirectly linked to kidney disease is a five-SNP haplotype in SLC16A11, a locus associated with an increased risk of type 2 diabetes mellitus (T2DM) that was first described in Mexican and other Latin American populations26. This diabetes risk haplotype has a frequency of up to 50% in Native Americans and about 10% in East Asians, but it is rare to absent in other populations, including in Africans. Interestingly, evidence indicates that the risk haplotype introgressed into modern human populations outside Africa within the last 250 Kya through admixture with Neanderthals or close Neanderthal relatives26. Hence, one could speculate that introgression potentially occurred in North or East Asia, after out-of-Africa migration. Notably, studies of SLC16A11 gene expression in the HeLa cell line (which does not express SLC16A11) indicate a role for this variant in lipid metabolism. Expression of SLC6A11 led to several changes in lipid metabolites, including an increase in intracellular levels of triacylglycerol — a lipid species that is associated with an increased risk of T2DM and insulin resistance26. T2DM is an important risk factor for CKD and thus we can posit that the SLC16A11 haplotype acquired via archaic admixture has an indirect effect on kidney phenotypes via the development of T2DM (Fig. 4).
Fig. 4. Examples of loci under selection that can influence phenotypes in which kidney disease is a feature or complication.
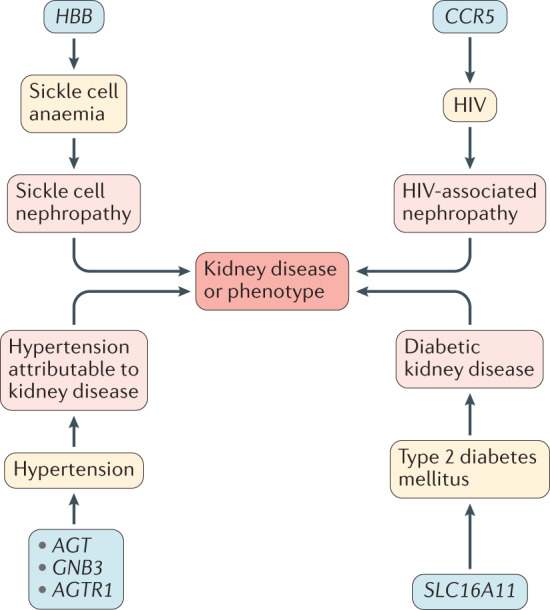
For example, HBB is a locus under selection owing to falciparum malaria, but is also the locus for sickle cell anaemia, which can be characterized by kidney disease (sickle cell nephropathy).
Another example of archaic genetic variants that exert indirect effects on diabetic kidney disease (DKD) are those that encode fatty acid desaturases (FADs). These proteins are important for converting short-chain polyunsaturated fatty acids (SC-PUFAs) to long-chain fatty acids (LC-PUFAs). The genes that encode FADs are associated with multiple metabolic traits, including T2DM27–29. Notably, studies of FADS1 in archaic human genomes showed that the Neanderthal and Denisovan sequences clustered with the ancestral haplotype found in Eurasians and not with a derived haplotype found in high frequencies in Africans. The derived haplotype originally evolved 500–100 Kya on the lineage leading to modern humans after the split from Neanderthals, and present-day geographic distributions of both haplotypes suggest that both haplotypes were present in Africa at the time of the Out-of-Africa migrations30. Unusually, the FAD genes show signatures of positive selection (that is, high frequencies of derived alleles and extended haplotype homozygosity) in multiple populations, including populations of African, South Asian, European and Inuit ancestry29–34. Of note, different alleles seem to be under selection pressure in different modern populations, with differences in the direction of the effect on the selected alleles. For example, the selected alleles in the Greenland Inuits are associated with increased concentrations of SC-PUFAs (including linoleic acid) and decreased concentrations of LC-PUFAs, in contrast to non-Inuit European population groups, in which the selected alleles are associated with a decrease in serum levels of the SC-PUFA linoleic acid and an increase in arachidonic and eicosapentaenoic acids29,31. These effects reflect the adaptation of each population to their historical diets. The Inuit diet is characterized by very high levels of LC-PUFAs from fish and marine mammals but a low intake of some SC-PUFAs, whereas the variants present in Europeans likely arose because of a change in the dietary composition of fatty acids following the transition to agriculture. Thus, dietary adaptations in different populations have placed the FAD genes under strong selection pressure. How these variants contribute to metabolic disorders, such as T2DM, which is a major risk factor for CKD, remains to be explored.
APOL1 evolution
The geographic migration of peoples and genetic admixture with other populations have played major roles in the evolutionary history of humanity. The impact of these forces on kidney disease is most clearly observed in terms of the effects of APOL1 variants on the risk of various kidney diseases35,36 in people of African ancestry37–39. At the global level, the APOL1 recessive risk haplotype (two APOL1 risk alleles) is found in appreciable to high frequencies in West Africans and populations of admixed West African ancestry, including Hispanic Americans, Afro-Caribbeans (for example, Jamaican, Barbadian, Grenadian, Trinidadian, Panamanian, Honduran, Haitian) and Brazilians from Salvador, but is absent from populations of European and Asian ancestry40–42. For individuals with two APOL1 risk alleles, the lifetime attributable risk of idiopathic focal segmental glomerulosclerosis (FSGS) is ~4% and of HIV-associated nephropathy is ~50%, in contrast to 0.2% and 2.5%, respectively, for individuals without the risk alleles40. Although natural selection at the APOL1 locus has been attributed to protection against sleeping sickness caused by infection with T. brucei subspecies43, the frequency distributions of and levels of protection afforded by APOL1 variants do not match the present-day geographic distributions of known trypanosomal subspecies; moreover, some evidence suggests that the protection provided by the APOL1 variants may extend to other infectious agents such as Leishmania parasites44. The present-day global distribution of APOL1 variants are a result of the forced migration of Africans during the trans-Atlantic slave trade and their admixture with other populations42. As a consequence, alleles that arose through natural selection in Africa currently represent the most important genetic risk factor for kidney failure in North America.
The APOL1 gene is one of six genes in the APOL family45. Of the six APOL proteins, APOL1 is the only one that is secreted into the bloodstream, where it circulates as a component of lipoproteins. Expression of APOL1 mRNA is high in placenta, liver and lung, and low in heart and kidney46. Circulating APOL1 exists as a complex with HDL — specifically the densest subfractions HDL3c and HDL3b — and APOA-I, the major apoprotein of HDL47,48. In humans, APOL1 is localized to the cytosol, where it acts to facilitate cell death through autophagy49. In the kidneys of mice, podocytes show a high level of autophagy under basal conditions; glomerular injury triggers autophagy to prevent glomerular disease whereas inhibition of autophagy leads to late-onset glomerulosclerosis50. Furthermore, podocyte-specific expression of the APOL1 risk variant blocks autophagic flux and reduces endosomal trafficking to induce kidney disease51.
Of note, APOL1 risk variants are not associated with an increased risk of DKD. In liver, high levels of glucose induce expression of the insulin-sensitive transcription factor USF1 and promotes USF1-mediated transactivation of genes involved in glucose and lipid metabolism, such as LIPC, which encodes hepatic lipase52–54. LIPC is involved in the delipidation of HDL2 to HDL3 (refs55,56). Although HDL levels are reduced in individuals with T2DM compared with levels in non-diabetic individuals, this reduction reflects reductions in levels of HDL2; by contrast, HDL3b and HDL3c levels are elevated57,58. USF1 also transactivates HNF4A53, which in turn transactivates APOA1, APOL1 and HPR59. Given the ability of APOL1 risk variants to reduce autophagic flux51, the absence of an association between APOL1 variants and DKD may suggest a protective effect of hyperglycaemia on autophagic flux.
Genetic factors and CKD
Mendelian kidney diseases
The explosion in high-throughput next-generation sequencing technologies and improvements in analytical pipelines over the past decade have led to the discovery of multiple Mendelian causes of CKD60–64. Broadly speaking, these discoveries can be divided into five categories. First, variants that cause CAKUTs, including renal dysplasia or aplasia, multicystic dysplastic kidneys and obstructive uropathies; most of the genes in this category are important in kidney development62. Second are variants that cause cystic kidney diseases such as autosomal recessive and autosomal dominant polycystic kidney diseases and microcystic kidney disease such as nephronopthisis60,61; this category is enriched for genes associated with cilia — evolutionarily conserved organelles that are found on the surface of most mammalian cells. Third are variants that cause glomerular diseases that are associated with proteinuria; most of the genes in this category localize to the podocyte63,64. Fourth are variants that cause kidney tubule defects61; and fifth are variants that cause metabolic disorders that result in kidney disease such as hyperoxaluria. A catalogue of these genes and the clinical implications of identifying the genetic defects can be found in recent reviews61,65,66. In evolutionary terms, it is important to note that many of the causal mutations are filtered out of the population by natural selection because the associated disorders are not compatible with successful reproduction and transmission of the alleles to the next generation. For those disorders that are compatible with a near-normal lifespan, the underlying mutations may persist subject to genetic drift, admixture and other population genetic processes.
Complex kidney diseases
The study of genetics of complex kidney diseases has benefited immensely from genome-wide association studies (GWAS), designed to test the association between genotype and phenotype. GWAS are powered to find associations for common alleles that tend to exist at similar frequencies across populations. By contrast, admixture mapping is powered to detect alleles that differ in frequency between populations. Differentiated alleles are generally divided into two types: alleles that are evolutionarily old and are common, meaning that they are present in nearly all populations across the globe, but with large differences in allele frequency; and evolutionarily young alleles that have low to rare frequencies and tend to be population specific. If an allele is deleterious and has a large effect size, then the causal allele will tend to be eliminated. However, a rare allele has little impact from a population or public health perspective because the fraction of disease attributable to an allele is a function of the square of the allele’s frequency.
Many GWAS have attempted to elucidate the genetic architecture of kidney traits, and over 260 loci have been associated with estimated GFR67. Although actual causal genes have been identified for only a few such loci68, it should be noted that these studies represent a major advance towards the goal of identifying the complete catalogue of genes that influence kidney traits. Of the 276 genes and/or loci that are associated with CKD69 in the National Human Genome Research Institute–European Bioinformatics Institute GWAS Catalogue (version 12-18-2019), only 5 (BSND, C9, CCDC57, PLG and SP3) show evidence of positive selection in simian primates — an infraorder of mammals that includes 5 hominoids (human, chimpanzee, gorilla, orangutan and gibbon), 1 New World monkey (marmoset) and 3 Old World monkeys (macaque, baboon and vervet)70 (Table 1). On the other hand, 84 CKD-associated loci show signatures of selection in one or more of the 26 modern human populations in the 1000 Genomes Project71, and 48 of these 84 loci (57%) show signs of selection in only one population. This observation suggests that most of the selective pressure exerted on CKD-associated loci occurred after the human–chimpanzee divergence72. These findings illustrate that natural selection events can occur over a variety of timescales and demonstrate how our increasingly rich knowledge of genetic associations can facilitate the identification of selected loci that confer disease risk.
Table 1.
CKD-associated loci under positive selection in primates
| HGNC name | Full name of description | GWAS catalogue genes | Mapped trait | Refs |
|---|---|---|---|---|
| BSND | Barttin CLCNK-type chloride channel accessory β-subunit | BSND | CKD, diabetic kidney disease, T2DM | 162 |
| C9 | Complement component 9 | C9, DAB2 | CKD, serum creatinine level | 163,67 |
| CCDC57 | Coiled-coil domain containing 57 | CCDC57, SLC16A3 | CKD, proteinuria | 164 |
| PLG | Plasminogen | PLG, LPA | CKD, APOL1 risk genotype carrier status | 165 |
| SP3 | SP3 transcription factor | SP3, CDCA7 | CKD, T1DM | 166 |
Of the 276 loci that are associated with CKD (EFO-0003884) in the National Human Genome Research Institute–European Bioinformatics Institute (NHGRI-EBI) GWAS Catalogue, only these five loci show evidence of positive selection in simian primates. In cases where the GWAS Catalogue loci spans more than one gene, all the genes in the locus are listed. CKD, chronic kidney disease; T1DM, type 1 diabetes mellitus; T2DM, type 2 diabetes mellitus.
Of the 84 CKD-associated loci that show signatures of selection in modern human populations, 66 (79%) are associated with either positive selection (that is, an increase in new advantageous alleles in the population) or purifying selection (that is, the selective removal of alleles that are deleterious; also known as negative selection). However, about one-fifth (21%) are associated with more than one form of selection or have more than one region in the gene showing signatures of selection (Supplementary Table 1). These observations suggest that CKD-associated loci in modern human populations have been the target of a wide variety of evolutionary forces. For example, we have found that among CKD-associated loci that are under selection, there is statistical over-representation of members of the class C/3 (metabotropic glutamate/pheromone receptors) reactome pathway (C.N. R., unpublished observations). This pathway is mainly involved in signal transduction by G protein-coupled receptors (GPCRs). GPCRs are involved in nearly all physiological processes, including the regulation of water and electrolyte transport in kidney tubules, the maintenance of acid–base balance, and the regulation of kidney blood flow and filtration73,74. Several kidney disorders are associated with dysregulated GPCR signalling, including kidney fibrosis and CKD. The class C/3 GPCRs include the metabotropic glutamate receptors and taste receptors75. Although these genes show evidence of natural selection and past demographic events (such as bottlenecks and population expansions), they also have functions in several systems beyond their involvement in taste, thereby making it difficult to speculate about the factors responsible for the selective pressures76.
Gene expression studies have shown differences in the transcriptional profiles of specific nephron compartments. For instance, tubule-specific genes are enriched for genes associated with the regulation of serum metabolites, whereas glomerulus-specific genes are enriched for CKD-GWAS significant hit variants and variants associated with blood pressure68. Gene network analyses of tubules have also provided insights into the mechanisms by which genetically regulated functional pathways can drive disease, for example, implicating a role for the endo-lysosomal pathway in the development of CKD68 and T cell and collagen pathways in tubulointerstitial fibrosis77. As of yet, no evidence exists, either from studies of individual loci or from studies of many loci in aggregate, for a role of natural selection in adaptive changes in the expression of genes in the kidney.
Immune factors
The human immune system is responsible for distinguishing self from non-self and is a critical defence against disease-causing microorganisms. The adaptive immune system is continuously challenged and has been shaped by exposure to infectious agents78 and introgression from archaic humans21. Therefore, it is not surprising that immune genes have been a target of evolutionary selection. Nowhere is this more apparent than in genes in the MHC region, particularly the HLA genes. Kidney diseases that have associations with immune genes generally fall into three broad categories. First, those associated with autoimmunity, in which an immune response is mounted against kidney-specific tissue or tissue components (for example, Goodpasture disease and membranous nephropathy) or affects multiple organ systems (for example, systemic lupus erythematosus). Second, those kidney diseases for which no evidence of autoimmunity currently exists but that have recognizable immune components in their aetiopathogenesis, including infection-related glomerulonephritis, sepsis-induced acute kidney injury, minimal change disease, primary FSGS and DKD. Third, drug-induced kidney diseases associated with commonly prescribed drugs, including abacavir, allopurinol, sulfonamides, penicillins, penicillamine and NSAIDs79, which can induce inflammation in various kidney tissue components, including the glomerulus, proximal tubules and the interstitium. For example, drugs such as rifampicin and NSAIDs can induce acute interstitial nephritis, whereas lithium and calcineurin inhibitors can cause chronic interstitial nephritis. Carriers of the HLA-B*57:01 allele are at a high risk of hypersensitivity reactions to treatment with the antiretroviral agent abacavir, which often damages the kidney80,81.
Strong purifying selection has been observed for variants of some immunity genes including those in the Toll-like receptor (TLR) pathway (for example, TLR3, MYD88 and IRAK4) owing to its essential role in protecting the host against life-threatening infections82. On the other hand, positive selection has been observed in nearly 200 other genes that mediate differences in disease susceptibility or outcome. Well-known examples include variants in HBB, G6PD and DARC that confer protection against P. falciparum and/or P. vivax-associated malaria and, as mentioned earlier, against variants in APOL1 that confer resistance to T. brucei. Pathogen-driven selection at these immune loci have confirmed probable implications for kidney disease. Studies over the past decade indicate that humans have acquired genetic diversity from archaic humans at several immune genes, including HLA genes (such as the HLA B*73 allele), TLR1 and the O-acetyl-l-serine (OAS) cluster of genes83,84. A 2016 study showed that regulatory variants that affect steady-state gene expression and transcriptional responsiveness to immune challenges were preferentially introduced into European genomes via admixture with Neanderthals and that some of these variants may have conferred a selective advantage to modern European populations22. Therefore, pathogen-driven selection at immune genes and introgression of functional variants from Neanderthal into modern humans likely have an evolutionary role in kidney disorders through either direct or indirect mechanisms.
Exposure to environmental factors
Over the course of human history, our species has been challenged by marked shifts in environmental exposures, and adaptation to those challenges has been key to our success in populating the diverse ecosystems of the world. In the context of kidney health, environmental factors that have been particularly challenging include infectious agents, toxins, dietary components and climate. Each of these factors is considered below.
Infectious agents
The kidneys are affected in many diseases caused by exposure to bacterial, viral, fungal and parasitic agents85. In some infections, kidney involvement is minor; however, kidney failure can occur in some instances. Bacterial infections can lead to septicaemia or toxic shock, with symptoms including inflammation due to systemic infection or the release of toxins, ischaemia or hypoperfusion. Infections caused by Staphylococcus aureus and Leptospira interrogans, and to a lesser extent Mycobacteria tuberculosis, Legionella and Rickettsia, can localize to the kidneys. Viruses such as arenaviruses, flaviviruses and hantaviruses are associated with haemorrhagic fever and have variable but generally low kidney involvement, typically presenting as vasomotor nephropathy. However, hepatitis B virus, hepatitis C virus, and HIV are clearly associated with kidney disease, which can be severe. For example, HIV-associated nephropathy is characterized by both glomerular and tubular damage and classically presents as collapsing FSGS. In immunocompromised individuals, cytomegalovirus and polyomaviruses are also associated with nephropathy.
Infections with coronaviruses have also been reported to involve the kidneys. Various case series of patients infected with the novel SARS-CoV-2 virus — the cause of the COVID-19 pandemic — have demonstrated kidney involvement manifesting as proteinuria, haematuria and acute kidney injury (AKI)86,87. SARS-CoV-2 preferentially infects the respiratory tract but some evidence suggests that it might infect the kidney, along with a broad array of other organs and tissues88. Viral RNA and protein have been detected in all compartments of the kidney, with preferential localization to glomerular cells. The associated histological features include diffuse proximal tubular injury and collapsing FSGS89,90. Although the development of AKI in patients with COVID-19 is associated with poor prognosis and high mortality87,91, SARS-CoV-2 is unlikely to have a major role as an agent of natural selection as the resulting disease — COVID-19 — disproportionately affects the elderly (that is, individuals beyond reproductive age).
Among parasitic diseases, malaria has known associations with glomerular disease, whereas other diseases such as schistosomiasis, filariasis and leishmaniasis (caused by Schistosoma, Filarioidea and Leishmania parasites, respectively) can lead to nephritis or nephrosis. Fungal infections typically affect the kidneys only in immunocompromised individuals. Amyloid deposition in the kidney can occur in the context of primary amyloidosis or in chronic conditions such as rheumatoid arthritis, ankylosing spondylitis, inflammatory bowel disease and chronic infection. The immune response to such insults, including the release of inflammatory mediators and the trapping or deposition of immune complexes, involves a number of genes in which mutations may modify the severity of kidney damage.
One example of an infectious agent with evolutionary implications for the kidney is the protozoan parasite Plasmodium falciparum, which is responsible for most malaria deaths worldwide. The distribution of the sickle cell mutation (rs334, c.20A>T) in the HBB subunit of haemoglobin, which arose ~7,300 years ago, is the result of balancing selection between protection against P. falciparum infection and mortality from sickle cell disease92. Consistent with the malaria hypothesis, the geographical distribution of the sickle cell mutation includes the Arabian Peninsula, parts of the Mediterranean (including Greece and southern Turkey) and parts of India93. Sickle cell nephropathy is a well-known complication of sickle cell anaemia (SCA) and accounts for the 5–18% prevalence of kidney failure observed in patients with SCA94. As would be expected, SCA and sickle cell nephropathy are common in West and Central Africa where the selection pressure from P. falciparum malaria is highest. However, both conditions are also observed in North America, Brazil, the Caribbean and other regions where Africans have migrated through voluntary or involuntary means. The sickle cell mutation has been associated with increased albumin-to-creatinine ratio in self-identified Hispanics and Latinx in the USA95, and with eGFR in non-Hispanic Black individuals in the Million Veteran Program96. Thus, the currently observed distribution of sickle cell nephropathy is a result of geographic migration and admixture following natural selection of a malaria-associated allele.
A 32-base pair deletion in the CC-chemokine receptor 5 gene (CCR5Δ32) is another example of evolutionary pressure on a locus that has consequences for the kidney. CCR5 is expressed in immune cells (including T cells, macrophages, dendritic cells and eosinophils) and is required, along with CXCR4 and CD4, for HIV-1 to gain entry into cells. Individuals who are homozygous for the CCR5Δ32 mutation are resistant to HIV-1 infection97. Interestingly, this mutation is present in up to 10% of individuals of European ancestry but is virtually absent in populations of African, East Asian and indigenous American ancestry98,99 — a difference that probably accounts in part for the disparities in HIV infection and, by extension, HIV-related comorbidities such as HIV-associated nephropathy in different populations (although it should be noted that most of these differences are due to public health measures and access to health care). The mutation has been dated to at least 1,500–2,000 years ago and, given its geographic distribution, must have arisen after the out-of-Africa migration99–102. As the mutation arose long before the emergence of HIV, the selection pressure that allowed it to spread to such a high frequency within an evolutionary short time frame must have been an agent other than HIV. The selective agent may be Yersinia pestis (the bacterium responsible for the bubonic plague), as the CCR5Δ32 mutation also protects against Y. pestis infection and would have conferred survival benefits during the bubonic plague epidemics of the Middle Ages. It has been suggested that other infectious agents, including the variola virus, which causes smallpox, and flaviviruses, may be better candidates than Y. pestis to explain global frequencies of CCR5Δ32 as the geographical distribution and clinical effect of the allele are a better fit with the current geographical distribution of the CCR5Δ32 mutation103.
Environmental toxins
Owing to their role in blood filtration, the kidneys are exposed to myriad toxins, most of which are efficiently removed through the urine by glomerular filtration, passive diffusion or active transport processes. However, some toxic substances are difficult for the kidneys to efficiently filter and may accumulate, causing damage. Particularly challenging substances are those to which humans have been exposed only in modern times, such as glyphosphate-based herbicides. These herbicides have been associated with AKI following exposure to high doses104, with CKD following occupational exposure among individuals in Sri Lanka105, and with kidney damage in animal studies106,107. Other substances that are toxic to the kidneys are present in a variety of sources in the modern environment. Exposure to lead, cadmium, mercury and uranium can occur in the context of poor infrastructure whereby these substances are incorrectly disposed of; exposure to cadmium, mercury and melamine can occur during industrial practices; and exposure to arsenic, cadmium and aristolochic acid can be traced to food contamination108.
In the context of evolution, the relationship between the kidneys and arsenic may be of most interest. Exposure to high levels of arsenic is associated with a variety of serious effects, including kidney damage and reduced kidney function109–113. High mortality from kidney diseases has been observed in areas with high arsenic levels in drinking water, including Utah and Michigan, USA114,115. Despite the observed toxic effects of arsenic exposure, some indigenous Andean populations live in areas with arsenic-contaminated drinking water but do not experience the expected harmful outcomes. Among these populations, efficient arsenic metabolism has been reported, such that inorganic arsenic is rapidly metabolized to the less toxic and more readily excretable dimethylarsinic acid116,117. For example, protective variants of AS3MT (which encodes the metabolic enzyme arsenite methyltransferase and is associated with efficient arsenic metabolism) are frequent (68%) in the Camarones people of Northern Chile — the region with the highest arsenic levels in the Americas118. High frequencies of protective variants are also found in indigenous Argentinean populations, with allele frequencies inversely associated with distance from arsenic exposure119. A GWAS of mono- and de-methylated arsenic levels in urine samples from an Argentinean Andes population identified AS3MT as the major gene influencing arsenic metabolism in humans116. The high frequency of protective AS3MT variants among individuals with a long ancestral history in this region with arsenic-contaminated drinking water and the evidence of a selective sweep around AS3MT suggest natural selection among these indigenous populations for efficient arsenic metabolism116, perhaps dating from an ancient Andean common ancestor118. This example represents the first known human adaptation to a toxic chemical116. As arsenic is a cause of kidney damage, this finding also represents an example of a positively selected locus that confers protection from nephrotoxicity via reduced susceptibility to the toxic effects of arsenic.
Diet
The need to excrete urea increased once our human ancestors began consuming a meat-rich diet following a transition from a diet of plants and fruits5, which probably placed selective pressure on the kidneys to maintain a high GFR. Interest in the ancestral diets of humans has grown in recent years, based on the premise that chronic disease might in some instances stem from a mismatch between modern dietary trends and that which our ancestors evolved to eat120, which primarily comprised wild animals and uncultivated plants121. However, the concept of a single ancestral diet is flawed, in that much of the success of our species stems from an ability to adapt to a wide range of environments and concomitant dietary diversity122. However, the over-riding consensus is that industrialization has facilitated the current high intakes of animal protein, refined grains and processed foods (with associated high sodium intake), whereas fruit and vegetable intake has decreased. These shifts are consequential in terms of the risk of chronic diseases, including kidney disease, particularly as related to maintaining the acid–base balance in the body.
The relationship between dietary acid load and kidney disease risk is supported by epidemiological data123–128. Dietary sodium intake predicts acid–base status independently of dietary acid load, with a predicted effect of similar magnitude129. US adults have an average dietary sodium intake of 3,584 mg per day130; the majority of salt intake comes from processed foods, whereas table salt contributes only a minor proportion of daily sodium intake131. The systematic use of salt has been dated to the Neolithic period132, the onset and duration of which varies by geographic region but encompassed the period ~10,000 BCE to 2,000 BCE. As a comparison, the average sodium intake of pre-Neolithic individuals is estimated to have been 400–1,200 mg133. A meta-analysis of dietary sodium and CKD risk found an association between the highest versus lowest sodium intake and increased risk of CKD (relative risk 1.09 [95% CI 1.01–1.19]), but with inconsistent results across geographic groups or study type134. The increased contribution of dietary salt resulting from a reliance on processed foods, coupled with the replacement of base-producing fruits and vegetables with acid-producing or neutral animal foods and grains, has resulted in a modern diet that chronically challenges the kidneys, leading to increased risk of functional decline. Although the kidney undergoes physiological changes in response to these challenges, maladaptation does not necessarily lead to mortality before the reproductive age and thus any underlying genetic variants that may influence this process are subject to minimal or no negative selection. The important role of the kidney in regulating body sodium levels is highlighted by the genetics of SLC12A1. This gene encodes Na–K–Cl cotransporter 2 (NKCC2), which is central to the reabsorption of sodium, potassium and chloride from the urine. Rare homozygous variants in SLC12A1 lead to neonatal Bartter syndrome, a disease characterized by an excess loss of salt in the urine, with accompanying polyuria, polydipsia, hypercalciuria and hypokalaemia. Heterozygotes for disease-causing SLC12A1 variants experience reduced blood pressure, presumably through reduced blood volume secondary to increased salt excretion135. Loss-of-function variants in SLC12A1 are under purifying selection136, which is consistent with the importance of renal sodium and potassium reabsorption in maintaining homeostasis, particularly in the African savanna — the home of early humans.
Climate, latitude and temperature
The African environment in which modern humans arose is characterized by a hot and wet climate. With salt scarcity being the norm, the thermoregulatory mechanisms needed for survival included a high capacity to sweat, efficient renal conservation of sodium and rapid vascular reactivity to blood volume changes. Indeed, the evolutionary prediction is that natural selection on the African continent would have favoured salt avidity and effective heat loss137. As humankind spread to other regions with different climates, these mechanisms are thought to have become maladaptive in certain environments. Indeed, evidence suggests that heat adaptation is associated with latitude, temperature and rainfall138. Of note, geographic latitude, ambient temperature, solar radiation and other similar measures are often confounded. In relation to health and disease, these phenomena have often been studied in relation to blood pressure regulation and hypertension. In this context, it is important to note that genes associated with climate adaptation, including ATP1A1, AQP2, CSK, AGTR1, AGT, CYP3A5 and GNB3, are both under natural selection and associated with blood pressure138–142. ATP1A1 and AQP2 (encoding the sodium/potassium-transporting ATPase subunit α1 and aquaporin 2, respectively) are involved in osmoregulation, whereas functional alleles in the other genes affect volume avidity (that is, water retention to maintain cardiovascular volume) and cardiovascular reactivity (that is, changes in heart rate, blood pressure and other cardiovascular measures in response to external stressors), and correlate with geographic latitude, rainfall and temperature. A 2016 study from Chile suggested that hypertension is more strongly associated with geographic latitude than with solar radiation and ambient temperatures143. This finding suggests that genetic variants influencing hypertension risk may be a characteristic of populations in those regions, rather than being due to climate adaptation-related genes or to physiological adaptation.
Insights into the effect of the environment on human kidney function in our evolutionary past might be gleaned from studies of men in Central America working in heavy manual labour occupations in hot environments, in whom the incidence of CKD is high144–146. This form of kidney disease has been termed MesoAmerican nephropathy or chronic interstitial nephritis in agricultural communities and is characterized by an absence of traditional risk factors such as hypertension, diabetes and proteinuria147–149. Environmental exposures to nephrotoxic pesticides and herbicides (glyphosate, organochlorine, alachlor, atrazine, metolachlor, pendimethalin, paraquat) have been implicated in its aetiology150. In addition, these individuals are also commonly exposed to extreme heat stress, with repeated episodes of dehydration151. One longitudinal study showed the rapidity and impact of these environmental conditions, with young men (aged 17–38 years) developing a marked reduction in kidney function, characterized by a 20% increase in serum creatinine level and a 9% reduction in estimated GFR, after only 9 weeks152. In evolutionary terms, it is possible that the human kidney is maladapted to conditions of repeated dehydration and heat stress and that over time, genetic variants that confer protection from such stress will be selected for in the exposed populations. To our knowledge, the role of genetic risk variants is currently unknown.
Insight into renal adaptations to the environment can also be derived from populations that currently live subsistence lifestyles in deserts and similar environments, characterized by heat stress and water scarcity. These populations include the KhoeSan peoples of Southern Africa and Australian Aborigines. Notably, studies of kidney function in Australian Aboriginal populations have demonstrated much higher rates of CKD and kidney failure than in non-Aboriginal Australians153,154. Although risk factors such as cardiovascular disease, metabolic disorders and reduced access to health care contribute to this observed disparity, autopsy studies have demonstrated that kidneys of adult Australian Aborigines have on average 30% fewer glomeruli than those of non-Aboriginal Australians, associated with an observed compensatory increase in glomerular volume153. Analyses of kidney biopsy samples have been unable to identify a single histological lesion that explains these findings, implying that the excessive rates of CKD and kidney failure among Australian Aborigines may be due to low nephron endowment at birth. Small body size and low nephron endowment are a consequence of poor nutritional environmental conditions. However, whether these changes are examples of acclimatization or adaptation in Australian Aborigines is unclear. Current evidence suggests that these changes may reflect acclimatization, given the particularly high rates of kidney disease among Australian Aborigines living in remote or very remote areas155, which likely reflects inadequate access to care, and given that nephron number was positively associated with height156, highlighting the link to nutritional status. However, the genetic influence on nephron number remains unclear and until genetic studies are available, we cannot rule out a possible role for loci under natural selection that may underlie these changes.
Conclusions and opportunities
The evolutionary context of kidney disease is complex and much remains to be understood. Major forms of CAKUT often lead to early mortality, and thus responsible genetic variants would be filtered out of the population by negative selection. On the other hand, most forms of CKD and kidney failure have an age of onset after the reproductive years, meaning that their underlying genetic risk variants are probably not selected against (that is, they are not under purifying selection). Therefore, it is likely that kidney-associated loci that are under natural selection have been selected for or against by non-renal factors, as in the case of APOL1. An unusual feature of CKD and kidney failure is that they are complications, comorbidities, or end points of several other disorders (such as diabetes mellitus and hypertension), which are often more prevalent in the population. Therefore, apart from loci directly associated with CKD and kidney failure, other loci may be indirectly linked through their intermediate phenotypes (Fig. 4). Currently, more data exist on the evolutionary context of the latter set of loci than on directly associated renal loci. Although the available evidence on the role of specific genes in evolutionary nephrology is presented here, it should be noted that the effect on disease risk may involve more than a single mutation or gene. Nonetheless, some selected variants in specific genes have potential clinical application. For example, APOL1 high-risk variants are potential markers not only for FSGS but also for other forms of kidney disease such as sickle cell nephropathy and lupus nephritis157, whereas the CCR5Δ32 mutation has been associated with reduced inflammation and lower cardiovascular mortality in a European study158 and a clinical trial of CCR5 inhibitors is currently underway in kidney transplant recipients. It is important for such studies to be as diverse as possible, not just to avoid race-based assumptions about allele frequencies but also to facilitate better generalizability of findings and ensure equity in realizing the benefits of the research159.
A number of gaps exist in our current knowledge of evolutionary genetics and nephrology. The pathogenesis of many apparently non-genetic kidney disorders is not yet fully understood and thus the contribution of any specific risk factor to disease is difficult to estimate at the present time. Our understanding of genetic factors that influence variation of nephron number remains limited. Similarly, the genetics of blood volume is under-studied. Another major challenge in the study of kidney health and disease is the lack of clinical data from large numbers of individuals for variables other than GFR estimated from serum creatinine level. This paucity of data limits the genomic and genetic studies that can be done. Measures or markers of proteinuria or albuminuria and tubulointerstitial fibrosis that can be easily and inexpensively assessed at the population level are needed before large-scale genetic studies can be performed. The availability of tools and methods for single-cell sequencing and single-cell transcriptional profiling presents an opportunity for the monitoring of gene expression at the single-cell level. These technologies will provide greater insights into kidney cell types and cell-type specific gene expression patterns, potentially enabling the mapping of genetic diseases of the kidney to single cell types160,161 and further enriching our knowledge about the evolutionary genetics of kidney disease.
Supplementary information
Glossary
- Pleistocene epoch
A period from about 2.58 million years ago to 11,700 years ago. It is sometimes referred to as the Ice Age because it spanned the world’s most recent period of repeated glaciations.
- Populations
In the context of this manuscript, groups of individuals with historical limitations to gene flow, for example, imposed by geography, such that they can now be distinguished genetically from other groups.
- Genetic admixture
Interbreeding between individuals from two or more previously isolated populations, typically referred to as hybridization in non-human species.
- Genetic linkage
Tendency of DNA sequences that are physically close on a chromosome to be inherited together owing to a low probability of recombination during meiosis.
- Haplotypes
Sets of alleles in a region of DNA that are tightly linked and likely to be inherited together.
- Archaic humans
A grouping of genus Homo excluding anatomically modern Homo sapiens sapiens.
- Ancient human
Homo sapiens sapiens predating modern populations.
- Adaptation
Genetically determined trait that has evolved by natural selection to maintain or increase fitness in response to environment.
- Ancestry
Genetic similarity of individuals to previously isolated populations.
- Introgression
Transfer of alleles from one species to another when matings produce fertile hybrids.
- Ancestral haplotype
Haplotype prior to a specific mutational event.
- Derived haplotype
Haplotype after a specific mutational event.
- Admixture mapping
A technique for finding regions of the genome associated with diseases or traits that show differential risk by ancestry.
- Bottlenecks
An event in which the census population size is greatly reduced, resulting in a loss of low-frequency alleles.
- Population expansions
An event in which the census population size is increased, resulting in an excess of low-frequency alleles.
- O-acetyl-l-serine (OAS) cluster
A cluster of genes involved in the de novo biosynthesis of l-cysteine that are responsive to sulfur starvation.
- Vasomotor nephropathy
Decrease in glomerular capillary pressure that leads to acute kidney failure.
- Acclimatization
Physiological, anatomical or morphological changes within an individual made in response to environmental change.
Author contributions
The authors contributed equally to all aspects of the article.
Competing interests
The authors declare no competing interests.
Footnotes
Peer review information
Nature Reviews Nephrology thanks P. O’Connell, who co-reviewed with J. Li, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Adebowale A. Adeyemo, Email: adeyemoa@mail.nih.gov
Charles N. Rotimi, Email: rotimic@mail.nih.gov
Supplementary information
The online version contains supplementary material available at 10.1038/s41581-021-00483-7.
References
- 1.Boyd, R. & Silk, J. B. How Humans Evolved (WW Norton & Company, 2014).
- 2.Cohen JJ. Relationship between energy requirements for Na+ reabsorption and other renal functions. Kidney Int. 1986;29:32–40. doi: 10.1038/ki.1986.5. [DOI] [PubMed] [Google Scholar]
- 3.Chevalier RL. Evolutionary nephrology. Kidney Int. Rep. 2017;2:302–317. doi: 10.1016/j.ekir.2017.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith, H. W. From Fish to Philosopher: the Story of our Internal Environment (Little, Brown, and Co., 1953).
- 5.Brenner BM, Meyer TW, Hostetter TH. Dietary protein intake and the progressive nature of kidney disease: the role of hemodynamically mediated glomerular injury in the pathogenesis of progressive glomerular sclerosis in aging, renal ablation, and intrinsic renal disease. N. Engl. J. Med. 1982;307:652–659. doi: 10.1056/NEJM198209093071104. [DOI] [PubMed] [Google Scholar]
- 6.Wood-Bradley RJ, Barrand S, Giot A, Armitage JA. Understanding the role of maternal diet on kidney development; an opportunity to improve cardiovascular and renal health for future generations. Nutrients. 2015;7:1881–1905. doi: 10.3390/nu7031881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gurusinghe S, Tambay A, Sethna CB. Developmental origins and nephron endowment in hypertension. Front. Pediatr. 2017;5:151. doi: 10.3389/fped.2017.00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bertram JF, Douglas-Denton RN, Diouf B, Hughson MD, Hoy WE. Human nephron number: implications for health and disease. Pediatr. Nephrol. 2011;26:1529–1533. doi: 10.1007/s00467-011-1843-8. [DOI] [PubMed] [Google Scholar]
- 9.Hoy WE, et al. Distribution of volumes of individual glomeruli in kidneys at autopsy: association with age, nephron number, birth weight and body mass index. Clin. Nephrol. 2010;74:S105–S112. doi: 10.5414/cnp74s105. [DOI] [PubMed] [Google Scholar]
- 10.Luyckx VA, et al. Effect of fetal and child health on kidney development and long-term risk of hypertension and kidney disease. Lancet. 2013;382:273–283. doi: 10.1016/S0140-6736(13)60311-6. [DOI] [PubMed] [Google Scholar]
- 11.Luyckx VA, Brenner BM. Birth weight, malnutrition and kidney-associated outcomes — a global concern. Nat. Rev. Nephrol. 2015;11:135. doi: 10.1038/nrneph.2014.251. [DOI] [PubMed] [Google Scholar]
- 12.Woodman AG, Bourque SL. Developmental programming of renal function: nephron endowment and beyond. J. Physiol. 2018;596:5495–5496. doi: 10.1113/JP276318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Giussani DA. The fetal brain sparing response to hypoxia: physiological mechanisms. J. Physiol. 2016;594:1215–1230. doi: 10.1113/JP271099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sohail M, et al. Polygenic adaptation on height is overestimated due to uncorrected stratification in genome-wide association studies. eLife. 2019;8:e39702. doi: 10.7554/eLife.39702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berg JJ, et al. Reduced signal for polygenic adaptation of height in UK Biobank. eLife. 2019;8:e39725. doi: 10.7554/eLife.39725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ben-Dor M, Gopher A, Barkai R. Neandertals’ large lower thorax may represent adaptation to high protein diet. Am. J. Phys. Anthropol. 2016;160:367–378. doi: 10.1002/ajpa.22981. [DOI] [PubMed] [Google Scholar]
- 17.Zanolli C, Hourset M, Esclassan R, Mollereau C. Neanderthal and Denisova tooth protein variants in present-day humans. PLoS ONE. 2017;12:e0183802. doi: 10.1371/journal.pone.0183802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao B, et al. A polymorphism of matrix Gla protein gene is associated with kidney stones. J. Urol. 2007;177:2361–2365. doi: 10.1016/j.juro.2007.01.118. [DOI] [PubMed] [Google Scholar]
- 19.Lu X, et al. A polymorphism of matrix Gla protein gene is associated with kidney stone in the Chinese Han population. Gene. 2012;511:127–130. doi: 10.1016/j.gene.2012.09.112. [DOI] [PubMed] [Google Scholar]
- 20.Chen L, Wolf AB, Fu W, Li L, Akey JM. Identifying and Interpreting Apparent Neanderthal Ancestry in African Individuals. Cell. 2020;180:677–687.e16. doi: 10.1016/j.cell.2020.01.012. [DOI] [PubMed] [Google Scholar]
- 21.Luo Y. Neanderthal DNA highlights complexity of COVID risk factors. Nature. 2020;587:552–553. doi: 10.1038/d41586-020-02957-3. [DOI] [PubMed] [Google Scholar]
- 22.Quach H, et al. Genetic adaptation and neandertal admixture shaped the immune system of human populations. Cell. 2016;167:643–656.e17. doi: 10.1016/j.cell.2016.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McCoy RC, Wakefield J, Akey JM. Impacts of neanderthal-introgressed sequences on the landscape of human gene expression. Cell. 2017;168:916–927.e12. doi: 10.1016/j.cell.2017.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dannemann M, Kelso J. The contribution of Neanderthals to phenotypic variation in modern humans. Am. J. Hum. Genet. 2017;101:578–589. doi: 10.1016/j.ajhg.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dannemann M, Prufer K, Kelso J. Functional implications of Neandertal introgression in modern humans. Genome Biol. 2017;18:61. doi: 10.1186/s13059-017-1181-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Consortium STD, et al. Sequence variants in SLC16A11 are a common risk factor for type 2 diabetes in Mexico. Nature. 2014;506:97–101. doi: 10.1038/nature12828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bokor S, et al. Single nucleotide polymorphisms in the FADS gene cluster are associated with delta-5 and delta-6 desaturase activities estimated by serum fatty acid ratios. J. Lipid Res. 2010;51:2325–2333. doi: 10.1194/jlr.M006205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Glaser C, Lattka E, Rzehak P, Steer C, Koletzko B. Genetic variation in polyunsaturated fatty acid metabolism and its potential relevance for human development and health. Matern. Child Nutr. 2011;7:27–40. doi: 10.1111/j.1740-8709.2011.00319.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fumagalli M, et al. Greenlandic Inuit show genetic signatures of diet and climate adaptation. Science. 2015;349:1343–1347. doi: 10.1126/science.aab2319. [DOI] [PubMed] [Google Scholar]
- 30.Ameur A, et al. Genetic adaptation of fatty-acid metabolism: a human-specific haplotype increasing the biosynthesis of long-chain omega-3 and omega-6 fatty acids. Am. J. Hum. Genet. 2012;90:809–820. doi: 10.1016/j.ajhg.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buckley MT, et al. Selection in Europeans on fatty acid desaturases associated with dietary changes. Mol. Biol. Evol. 2017;34:1307–1318. doi: 10.1093/molbev/msx103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kothapalli KS, et al. Positive selection on a regulatory insertion-deletion polymorphism in FADS2 influences apparent endogenous synthesis of arachidonic acid. Mol. Biol. Evol. 2016;33:1726–1739. doi: 10.1093/molbev/msw049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mathias RA, et al. Adaptive evolution of the FADS gene cluster within Africa. PLoS ONE. 2012;7:e44926. doi: 10.1371/journal.pone.0044926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mathieson I, et al. Genome-wide patterns of selection in 230 ancient Eurasians. Nature. 2015;528:499–503. doi: 10.1038/nature16152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kopp JB, et al. MYH9 is a major-effect risk gene for focal segmental glomerulosclerosis. Nat. Genet. 2008;40:1175–1184. doi: 10.1038/ng.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kao WHL, et al. MYH9 is associated with nondiabetic end-stage renal disease in African Americans. Nat. Genet. 2008;40:1185–1192. doi: 10.1038/ng.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pollak MR, Genovese G, Friedman DJ. APOL1 and kidney disease. Curr. Opin. Nephrol. Hypertens. 2012;21:179–182. doi: 10.1097/MNH.0b013e32835012ab. [DOI] [PubMed] [Google Scholar]
- 38.Freedman BI, et al. The apolipoprotein L1 (APOL1) gene and nondiabetic nephropathy in African Americans. J. Am. Soc. Nephrol. 2010;21:1422–1426. doi: 10.1681/ASN.2010070730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Genovese G, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science. 2010;329:841–845. doi: 10.1126/science.1193032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kopp JB, et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J. Am. Soc. Nephrol. 2011;22:2129–2137. doi: 10.1681/ASN.2011040388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Limou S, Nelson GW, Kopp JB, Winkler CA. APOL1 kidney risk alleles: population genetics and disease associations. Adv. Chronic Kidney Dis. 2014;21:426–433. doi: 10.1053/j.ackd.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nadkarni GN, et al. Worldwide frequencies of APOL1 renal risk variants. N. Engl. J. Med. 2018;379:2571–2572. doi: 10.1056/NEJMc1800748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cooper A, et al. APOL1 renal risk variants have contrasting resistance and susceptibility associations with African trypanosomiasis. eLife. 2017;6:e25461. doi: 10.7554/eLife.25461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Samanovic M, Molina-Portela MP, Chessler AD, Burleigh BA, Raper J. Trypanosome lytic factor, an antimicrobial high-density lipoprotein, ameliorates Leishmania infection. PLoS Pathog. 2009;5:e1000276. doi: 10.1371/journal.ppat.1000276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Page NM, Butlin DJ, Lomthaisong K, Lowry PJ. The human apolipoprotein L gene cluster: identification, classification, and sites of distribution. Genomics. 2001;74:71–78. doi: 10.1006/geno.2001.6534. [DOI] [PubMed] [Google Scholar]
- 46.Monajemi H, Fontijn RD, Pannekoek H, Horrevoets AJG. The apolipoprotein L gene cluster has emerged recently in evolution and is expressed in human vascular tissue. Genomics. 2002;79:539–546. doi: 10.1006/geno.2002.6729. [DOI] [PubMed] [Google Scholar]
- 47.Duchateau PN, et al. Apolipoprotein L, a new human high density lipoprotein apolipoprotein expressed by the pancreas: identification, cloning, characterization, and plasma distribution of apolipoprotein L. J. Biol. Chem. 1997;272:25576–25582. doi: 10.1074/jbc.272.41.25576. [DOI] [PubMed] [Google Scholar]
- 48.Davidson WS, et al. Proteomic analysis of defined HDL subpopulations reveals particle-specific protein clusters: relevance to antioxidative function. Arterioscler. Thromb. Vasc. Biol. 2009;29:870–876. doi: 10.1161/ATVBAHA.109.186031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wan G, et al. Apolipoprotein L1, a novel Bcl-2 homology domain 3-only lipid-binding protein, induces autophagic cell death. J. Biol. Chem. 2008;283:21540–21549. doi: 10.1074/jbc.M800214200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hartleben B, et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J. Clin. Invest. 2010;120:1084–1096. doi: 10.1172/JCI39492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Beckerman P, et al. Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice. Nat. Med. 2017;23:429–438. doi: 10.1038/nm.4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Corre S, Galibert M-D. Upstream stimulating factors: highly versatile stress-responsive transcription factors. Pigment Cell Res. 2005;18:337–348. doi: 10.1111/j.1600-0749.2005.00262.x. [DOI] [PubMed] [Google Scholar]
- 53.Rada-Iglesias A, et al. Whole-genome maps of USF1 and USF2 binding and histone H3 acetylation reveal new aspects of promoter structure and candidate genes for common human disorders. Genome Res. 2008;18:380–392. doi: 10.1101/gr.6880908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Deursen D, Jansen H, Verhoeven AJ. Glucose increases hepatic lipase expression in HepG2 liver cells through upregulation of upstream stimulatory factors 1 and 2. Diabetologia. 2008;51:2078–2087. doi: 10.1007/s00125-008-1125-6. [DOI] [PubMed] [Google Scholar]
- 55.Jansen H, Verhoeven AJM, Sijbrands EJG. Hepatic lipase: a pro- or anti-atherogenic protein? J. Lipid Res. 2002;43:1352–1362. doi: 10.1194/jlr.R200008-JLR200. [DOI] [PubMed] [Google Scholar]
- 56.Deeb SS, Zambon A, Carr MC, Ayyobi AF, Brunzell JD. Hepatic lipase and dyslipidemia: interactions among genetic variants, obesity, gender, and diet. J. Lipid Res. 2003;44:1279–1286. doi: 10.1194/jlr.R200017-JLR200. [DOI] [PubMed] [Google Scholar]
- 57.Syvänne M, et al. High density lipoprotein subfractions in non-insulin-dependent diabetes mellitus and coronary artery disease. J. Lipid Res. 1995;36:573–582. doi: 10.1016/S0022-2275(20)39891-6. [DOI] [PubMed] [Google Scholar]
- 58.Krauss RM. Lipids and lipoproteins in patients with type 2 diabetes. Diabetes Care. 2004;27:1496–1504. doi: 10.2337/diacare.27.6.1496. [DOI] [PubMed] [Google Scholar]
- 59.Consortium TEP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Braun DA, Hildebrandt F. Ciliopathies. Cold Spring Harb. Perspect. Biol. 2017;9:a028191. doi: 10.1101/cshperspect.a028191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hildebrandt F. Genetic kidney diseases. Lancet. 2010;375:1287–1295. doi: 10.1016/S0140-6736(10)60236-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.van der Ven AT, et al. Whole-exome sequencing identifies causative mutations in families with congenital anomalies of the kidney and urinary tract. J. Am. Soc. Nephrol. 2018;29:2348–2361. doi: 10.1681/ASN.2017121265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Varner JD, et al. Genetic testing for steroid-resistant-nephrotic syndrome in an outbred population. Front. Pediatr. 2018;6:307. doi: 10.3389/fped.2018.00307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Warejko JK, et al. Whole exome sequencing of patients with steroid-resistant nephrotic syndrome. Clin. J. Am. Soc. Nephrol. 2018;13:53–62. doi: 10.2215/CJN.04120417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Groopman EE, Rasouly HM, Gharavi AG. Genomic medicine for kidney disease. Nat. Rev. Nephrol. 2018;14:83–104. doi: 10.1038/nrneph.2017.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Groopman EE, Povysil G, Goldstein DB, Gharavi AG. Rare genetic causes of complex kidney and urological diseases. Nat. Rev. Nephrol. 2020;16:641–656. doi: 10.1038/s41581-020-0325-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wuttke M, et al. A catalog of genetic loci associated with kidney function from analyses of a million individuals. Nat. Genet. 2019;51:957–972. doi: 10.1038/s41588-019-0407-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Qiu C, et al. Renal compartment-specific genetic variation analyses identify new pathways in chronic kidney disease. Nat. Med. 2018;24:1721–1731. doi: 10.1038/s41591-018-0194-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.European Molecular Biology Laboratory. Chronic kidney disease. EMBL-EBIhttp://www.ebi.ac.uk/efo/EFO_0003884 (2021).
- 70.van der Lee R, Wiel L, van Dam TJP, Huynen MA. Genome-scale detection of positive selection in nine primates predicts human-virus evolutionary conflicts. Nucleic Acids Res. 2017;45:10634–10648. doi: 10.1093/nar/gkx704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Murga-Moreno J, Coronado-Zamora M, Bodelon A, Barbadilla A, Casillas S. PopHumanScan: the online catalog of human genome adaptation. Nucleic Acids Res. 2019;47:D1080–D1089. doi: 10.1093/nar/gky959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Moorjani P, Amorim CE, Arndt PF, Przeworski M. Variation in the molecular clock of primates. Proc. Natl Acad. Sci. USA. 2016;113:10607–10612. doi: 10.1073/pnas.1600374113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Weiss RH. G protein-coupled receptor signalling in the kidney. Cell Signal. 1998;10:313–320. doi: 10.1016/S0898-6568(97)00137-X. [DOI] [PubMed] [Google Scholar]
- 74.Kamal FA, Travers JG, Blaxall BC. G protein-coupled receptor kinases in cardiovascular disease: why “where” matters. Trends Cardiovasc. Med. 2012;22:213–219. doi: 10.1016/j.tcm.2012.07.023. [DOI] [PubMed] [Google Scholar]
- 75.Brauner-Osborne H, Wellendorph P, Jensen AA. Structure, pharmacology and therapeutic prospects of family C G-protein coupled receptors. Curr. Drug Targets. 2007;8:169–184. doi: 10.2174/138945007779315614. [DOI] [PubMed] [Google Scholar]
- 76.Dobon B, Rossell C, Walsh S, Bertranpetit J. Is there adaptation in the human genome for taste perception and phase I biotransformation? BMC Evol. Biol. 2019;19:39. doi: 10.1186/s12862-019-1366-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Beckerman P, et al. Human kidney tubule-specific gene expression based dissection of chronic kidney disease traits. EBioMedicine. 2017;24:267–276. doi: 10.1016/j.ebiom.2017.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nédélec Y, et al. Genetic ancestry and natural selection drive population differences in immune responses to pathogens. Cell. 2016;167:657–669.e21. doi: 10.1016/j.cell.2016.09.025. [DOI] [PubMed] [Google Scholar]
- 79.Robson KJ, Ooi JD, Holdsworth SR, Rossjohn J, Kitching AR. HLA and kidney disease: from associations to mechanisms. Nat. Rev. Nephrol. 2018;14:636–655. doi: 10.1038/s41581-018-0057-8. [DOI] [PubMed] [Google Scholar]
- 80.Hetherington S, et al. Genetic variations in HLA-B region and hypersensitivity reactions to abacavir. Lancet. 2002;359:1121–1122. doi: 10.1016/S0140-6736(02)08158-8. [DOI] [PubMed] [Google Scholar]
- 81.Mallal S, et al. Association between presence of HLA-B*5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1 reverse-transcriptase inhibitor abacavir. Lancet. 2002;359:727–732. doi: 10.1016/S0140-6736(02)07873-X. [DOI] [PubMed] [Google Scholar]
- 82.Barreiro LB, Quintana-Murci L. From evolutionary genetics to human immunology: how selection shapes host defence genes. Nat. Rev. Genet. 2010;11:17–30. doi: 10.1038/nrg2698. [DOI] [PubMed] [Google Scholar]
- 83.Dannemann M, Andres AM, Kelso J. Introgression of neandertal- and denisovan-like haplotypes contributes to adaptive variation in human toll-like receptors. Am. J. Hum. Genet. 2016;98:22–33. doi: 10.1016/j.ajhg.2015.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Deschamps M, et al. Genomic signatures of selective pressures and introgression from archaic hominins at human innate immunity genes. Am. J. Hum. Genet. 2016;98:5–21. doi: 10.1016/j.ajhg.2015.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Herberg, J., Pahari, A., Walters, S. & Levin, M. in Pediatric Nephrology: Sixth Completely Revised, Updated and Enlarged Edition Ch. 52 (eds Avner, E. D., Harmon, W. E., Niaudet, P. & Yoshikawa, N.) 1235–1273 (Springer, 2009).
- 86.Pei G, et al. Renal involvement and early prognosis in patients with COVID-19 pneumonia. J. Am. Soc. Nephrol. 2020;31:1157–1165. doi: 10.1681/ASN.2020030276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cheng Y, et al. Kidney disease is associated with in-hospital death of patients with COVID-19. Kidney Int. 2020;97:829–838. doi: 10.1016/j.kint.2020.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Puelles VG, et al. Multiorgan and renal tropism of SARS-CoV-2. N. Engl. J. Med. 2020;383:590–592. doi: 10.1056/NEJMc2011400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kissling S, et al. Collapsing glomerulopathy in a COVID-19 patient. Kidney Int. 2020;98:228–231. doi: 10.1016/j.kint.2020.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Su H, et al. Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China. Kidney Int. 2020;98:219–227. doi: 10.1016/j.kint.2020.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gabarre P, et al. Acute kidney injury in critically ill patients with COVID-19. Intensive Care Med. 2020;46:1339–1348. doi: 10.1007/s00134-020-06153-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shriner D, Rotimi CN. Whole-genome-sequence-based haplotypes reveal single origin of the sickle allele during the holocene wet phase. Am. J. Hum. Genet. 2018;102:547–556. doi: 10.1016/j.ajhg.2018.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Piel FB, et al. Global migration and the changing distribution of sickle haemoglobin: a quantitative study of temporal trends between 1960 and 2000. Lancet Glob. Health. 2014;2:e80–e89. doi: 10.1016/S2214-109X(13)70150-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Saborio P, Scheinman JI. Sickle cell nephropathy. J. Am. Soc. Nephrol. 1999;10:187–192. doi: 10.1681/ASN.V101187. [DOI] [PubMed] [Google Scholar]
- 95.Kramer HJ, et al. African ancestry-specific alleles and kidney disease risk in hispanics/latinos. J. Am. Soc. Nephrol. 2017;28:915–922. doi: 10.1681/ASN.2016030357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hellwege JN, et al. Mapping eGFR loci to the renal transcriptome and phenome in the VA Million Veteran Program. Nat. Commun. 2019;10:3842. doi: 10.1038/s41467-019-11704-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kay MA, Walker BD. Engineering cellular resistance to HIV. N. Engl. J. Med. 2014;370:968–969. doi: 10.1056/NEJMe1400593. [DOI] [PubMed] [Google Scholar]
- 98.Martinson JJ, Chapman NH, Rees DC, Liu YT, Clegg JB. Global distribution of the CCR5 gene 32-basepair deletion. Nat. Genet. 1997;16:100–103. doi: 10.1038/ng0597-100. [DOI] [PubMed] [Google Scholar]
- 99.Stephens JC, et al. Dating the origin of the CCR5-Delta32 AIDS-resistance allele by the coalescence of haplotypes. Am. J. Hum. Genet. 1998;62:1507–1515. doi: 10.1086/301867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Duncan SR, Scott S, Duncan CJ. Reappraisal of the historical selective pressures for the CCR5-Delta32 mutation. J. Med. Genet. 2005;42:205–208. doi: 10.1136/jmg.2004.025346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Drancourt M, Aboudharam G, Signoli M, Dutour O, Raoult D. Detection of 400-year-old Yersinia pestis DNA in human dental pulp: an approach to the diagnosis of ancient septicemia. Proc. Natl Acad. Sci. USA. 1998;95:12637–12640. doi: 10.1073/pnas.95.21.12637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zajac V. Evolutionary view of the AIDS process. J. Int. Med. Res. 2018;46:4032–4038. doi: 10.1177/0300060518786919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Baker JL, Shriner D, Bentley AR, Rotimi CN. Pharmacogenomic implications of the evolutionary history of infectious diseases in Africa. Pharmacogenomics J. 2017;17:112–120. doi: 10.1038/tpj.2016.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mohamed F, et al. Mechanism-specific injury biomarkers predict nephrotoxicity early following glyphosate surfactant herbicide (GPSH) poisoning. Toxicol. Lett. 2016;258:1–10. doi: 10.1016/j.toxlet.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 105.Jayasumana C, et al. Drinking well water and occupational exposure to herbicides is associated with chronic kidney disease, in Padavi-Sripura, Sri Lanka. Environ. Health. 2015;14:6–6. doi: 10.1186/1476-069X-14-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li M-H, et al. Metabolic profiling of goldfish (Carassius auratis) after long-term glyphosate-based herbicide exposure. Aquat. Toxicol. 2017;188:159–169. doi: 10.1016/j.aquatox.2017.05.004. [DOI] [PubMed] [Google Scholar]
- 107.Wunnapuk K, et al. Use of a glyphosate-based herbicide-induced nephrotoxicity model to investigate a panel of kidney injury biomarkers. Toxicol. Lett. 2014;225:192–200. doi: 10.1016/j.toxlet.2013.12.009. [DOI] [PubMed] [Google Scholar]
- 108.Weidemann DK, Weaver VM, Fadrowski JJ. Toxic environmental exposures and kidney health in children. Pediatr. Nephrol. 2016;31:2043–2054. doi: 10.1007/s00467-015-3222-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Prasad GVR, Rossi NF. Arsenic intoxication associated with tubulointerstitial nephritis. Am. J. Kidney Dis. 1995;26:373–376. doi: 10.1016/0272-6386(95)90660-6. [DOI] [PubMed] [Google Scholar]
- 110.Saxena PN, Anand S, Saxena N, Bajaj P. Effect of arsenic trioxide on renal functions and its modulation by Curcuma aromatica leaf extract in albino rat. J. Environ. Biol. 2009;30:527–531. [PubMed] [Google Scholar]
- 111.Sener U, et al. Protective effects of thymoquinone against apoptosis and oxidative stress by arsenic in rat kidney. Ren. Fail. 2016;38:117–123. doi: 10.3109/0886022X.2015.1103601. [DOI] [PubMed] [Google Scholar]
- 112.Liu J, Liu Y, Habeebu SM, Waalkes MP, Klaassen CD. Chronic combined exposure to cadmium and arsenic exacerbates nephrotoxicity, particularly in metallothionein-I/II null mice. Toxicology. 2000;147:157–166. doi: 10.1016/S0300-483X(00)00194-3. [DOI] [PubMed] [Google Scholar]
- 113.Dutta S, Saha S, Mahalanobish S, Sadhukhan P, Sil PC. Melatonin attenuates arsenic induced nephropathy via the regulation of oxidative stress and inflammatory signaling cascades in mice. Food Chem. Toxicol. 2018;118:303–316. doi: 10.1016/j.fct.2018.05.032. [DOI] [PubMed] [Google Scholar]
- 114.Lewis DR, Southwick JW, Ouellet-Hellstrom R, Rench J, Calderon RL. Drinking water arsenic in Utah: a cohort mortality study. Environ. Health Perspect. 1999;107:359–365. doi: 10.1289/ehp.99107359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Meliker JR, Wahl RL, Cameron LL, Nriagu JO. Arsenic in drinking water and cerebrovascular disease, diabetes mellitus, and kidney disease in Michigan: a standardized mortality ratio analysis. Environ. Health. 2007;6:4. doi: 10.1186/1476-069X-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schlebusch CM, et al. Human adaptation to arsenic-rich environments. Mol. Biol. Evol. 2015;32:1544–1555. doi: 10.1093/molbev/msv046. [DOI] [PubMed] [Google Scholar]
- 117.De Loma J, et al. Elevated arsenic exposure and efficient arsenic metabolism in indigenous women around Lake Poopó, Bolivia. Sci. Total Environ. 2019;657:179–186. doi: 10.1016/j.scitotenv.2018.11.473. [DOI] [PubMed] [Google Scholar]
- 118.Apata M, Arriaza B, Llop E, Moraga M. Human adaptation to arsenic in Andean populations of the Atacama Desert. Am. J. Phys. Anthropol. 2017;163:192–199. doi: 10.1002/ajpa.23193. [DOI] [PubMed] [Google Scholar]
- 119.Eichstaedt CA, et al. Positive selection of AS3MT to arsenic water in Andean populations. Mutat. Res. 2015;780:97–102. doi: 10.1016/j.mrfmmm.2015.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ströhle A, Hahn A, Sebastian A. Estimation of the diet-dependent net acid load in 229 worldwide historically studied hunter-gatherer societies. Am. J. Clin. Nutr. 2009;91:406–412. doi: 10.3945/ajcn.2009.28637. [DOI] [PubMed] [Google Scholar]
- 121.Eaton, S. & Konner, M. Paleolithic nutrition: a consideration of its nature and current implications. In eds. Goodman, AH, Dufour, DL & Pelto, GH Nutritional Anthropology: Biocultural Perspectives on Food and Nutrition. Mayfield Publishing Company, CA, 2000.
- 122.Ungar PS. The evolution of human diet: the known, the unknown, and the unknowable. Evolut. Anthropol. 2004;13:45–46. doi: 10.1002/evan.10126. [DOI] [Google Scholar]
- 123.Banerjee T, et al. Dietary acid load and chronic kidney disease among adults in the United States. BMC Nephrol. 2014;15:137–137. doi: 10.1186/1471-2369-15-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rebholz CM, et al. Dietary acid load and incident chronic kidney disease: results from the ARIC Study. Am. J. Nephrol. 2015;42:427–435. doi: 10.1159/000443746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Driver TH, et al. Low serum bicarbonate and kidney function decline: the multi-ethnic study of atherosclerosis (MESA) Am. J. Kidney Dis. 2014;64:534–541. doi: 10.1053/j.ajkd.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Goraya N, Simoni J, Jo C, Wesson DE. Dietary acid reduction with fruits and vegetables or bicarbonate attenuates kidney injury in patients with a moderately reduced glomerular filtration rate due to hypertensive nephropathy. Kidney Int. 2012;81:86–93. doi: 10.1038/ki.2011.313. [DOI] [PubMed] [Google Scholar]
- 127.Mahajan A, et al. Daily oral sodium bicarbonate preserves glomerular filtration rate by slowing its decline in early hypertensive nephropathy. Kidney Int. 2010;78:303–309. doi: 10.1038/ki.2010.129. [DOI] [PubMed] [Google Scholar]
- 128.Lin J, Fung TT, Hu FB, Curhan GC. Association of dietary patterns with albuminuria and kidney function decline in older white women: a subgroup analysis from the nurses’ health study. Am. J. Kidney Dis. 2011;57:245–254. doi: 10.1053/j.ajkd.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Frassetto LA, Morris RC, Sebastian A. Dietary sodium chloride intake independently predicts the degree of hyperchloremic metabolic acidosis in healthy humans consuming a net acid-producing diet. Am. J. Physiol. Renal Physiol. 2007;293:F521–F525. doi: 10.1152/ajprenal.00048.2007. [DOI] [PubMed] [Google Scholar]
- 130.Bailey RL, et al. Estimating sodium and potassium intakes and their ratio in the American diet: data from the 2011–2012 NHANES. J. Nutr. 2016;146:745–750. doi: 10.3945/jn.115.221184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.US Institute of Medicine. Strategies to Reduce Sodium Intake in the United States (eds Henney, J. E., Taylor, C. L. & Boon, C. S.) (National Academies, 2010). [PubMed]
- 132.Cordain L, et al. Origins and evolution of the Western diet: health implications for the 21st century. Am. J. Clin. Nutr. 2005;81:341–354. doi: 10.1093/ajcn.81.2.341. [DOI] [PubMed] [Google Scholar]
- 133.Qian Q. Salt, water and nephron: mechanisms of action and link to hypertension and chronic kidney disease. Nephrology. 2018;23:44–49. doi: 10.1111/nep.13465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Liu N, et al. Association between sodium intakes with the risk of chronic kidney disease: evidence from a meta-analysis. Int. J. Clin. Exp. Med. 2015;8:20939–20945. [PMC free article] [PubMed] [Google Scholar]
- 135.Ji W, et al. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat. Genet. 2008;40:592–599. doi: 10.1038/ng.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Welling PA. Rare mutations in renal sodium and potassium transporter genes exhibit impaired transport function. Curr. Opin. Nephrol. Hypertens. 2014;23:1–8. doi: 10.1097/01.mnh.0000437204.84826.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rossier BC, Bochud M, Devuyst O. The hypertension pandemic: an evolutionary perspective. Physiology. 2017;32:112–125. doi: 10.1152/physiol.00026.2016. [DOI] [PubMed] [Google Scholar]
- 138.Young JH, et al. Differential susceptibility to hypertension is due to selection during the out-of-Africa expansion. PLoS Genet. 2005;1:e82. doi: 10.1371/journal.pgen.0010082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Gurdasani D, et al. The African Genome Variation Project shapes medical genetics in Africa. Nature. 2015;517:327–332. doi: 10.1038/nature13997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ji LD, Tang NL, Xu J. AGTR1 has undergone natural selection in Euro-Asian populations in relation to ambient temperature that predisposes Chinese populations to essential hypertension. Int. J. Cardiol. 2016;209:278–280. doi: 10.1016/j.ijcard.2016.02.031. [DOI] [PubMed] [Google Scholar]
- 141.Thompson EE, et al. CYP3A variation and the evolution of salt-sensitivity variants. Am. J. Hum. Genet. 2004;75:1059–1069. doi: 10.1086/426406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Levy D, et al. Genome-wide association study of blood pressure and hypertension. Nat. Genet. 2009;41:677–687. doi: 10.1038/ng.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Cabrera SE, Mindell JS, Toledo M, Alvo M, Ferro CJ. Associations of blood pressure with geographical latitude, solar radiation, and ambient temperature: results from the Chilean health survey, 2009–2010. Am. J. Epidemiol. 2016;183:1071–1073. doi: 10.1093/aje/kww037. [DOI] [PubMed] [Google Scholar]
- 144.Garcia-Trabanino R, et al. Heat stress, dehydration, and kidney function in sugarcane cutters in El Salvador — a cross-shift study of workers at risk of Mesoamerican nephropathy. Env. Res. 2015;142:746–755. doi: 10.1016/j.envres.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 145.Johnson RJ, Wesseling C, Newman LS. Chronic kidney disease of unknown cause in agricultural communities. N. Engl. J. Med. 2019;380:1843–1852. doi: 10.1056/NEJMra1813869. [DOI] [PubMed] [Google Scholar]
- 146.Wesseling C, et al. The epidemic of chronic kidney disease of unknown etiology in Mesoamerica: a call for interdisciplinary research and action. Am. J. Public Health. 2013;103:1927–1930. doi: 10.2105/AJPH.2013.301594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wesseling C, et al. Mesoamerican nephropathy: geographical distribution and time trends of chronic kidney disease mortality between 1970 and 2012 in Costa Rica. Occup. Env. Med. 2015;72:714–721. doi: 10.1136/oemed-2014-102799. [DOI] [PubMed] [Google Scholar]
- 148.Correa-Rotter R, Wesseling C, Johnson RJ. CKD of unknown origin in Central America: the case for a Mesoamerican nephropathy. Am. J. Kidney Dis. 2014;63:506–520. doi: 10.1053/j.ajkd.2013.10.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wesseling C, et al. Resolving the enigma of the Mesoamerican nephropathy: a research workshop summary. Am. J. Kidney Dis. 2014;63:396–404. doi: 10.1053/j.ajkd.2013.08.014. [DOI] [PubMed] [Google Scholar]
- 150.De Broe ME, Vervaet BA. Is an environmental nephrotoxin the primary cause of CKDu (Mesoamerican nephropathy)? PRO. Kidney360. 2020;1:591–595. doi: 10.34067/KID.0003172020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wesseling C, et al. Heat stress, hydration and uric acid: a cross-sectional study in workers of three occupations in a hotspot of Mesoamerican nephropathy in Nicaragua. BMJ Open. 2016;6:e011034. doi: 10.1136/bmjopen-2016-011034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wesseling C, et al. Kidney function in sugarcane cutters in Nicaragua — a longitudinal study of workers at risk of Mesoamerican nephropathy. Env. Res. 2016;147:125–132. doi: 10.1016/j.envres.2016.02.002. [DOI] [PubMed] [Google Scholar]
- 153.Hoy WE, Hughson MD, Singh GR, Douglas-Denton R, Bertram JF. Reduced nephron number and glomerulomegaly in Australian Aborigines: a group at high risk for renal disease and hypertension. Kidney Int. 2006;70:104–110. doi: 10.1038/sj.ki.5000397. [DOI] [PubMed] [Google Scholar]
- 154.Hoy W. Renal disease in Australian Aborigines. Nephrol. Dial. Transpl. 2000;15:1293–1297. doi: 10.1093/ndt/15.9.1293. [DOI] [PubMed] [Google Scholar]
- 155.Hoy WE, et al. CKD in aboriginal Australians. Am. J. Kidney Dis. 2010;56:983–993. doi: 10.1053/j.ajkd.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 156.Hoy WE, et al. Renal biopsy findings among Indigenous Australians: a nationwide review. Kidney Int. 2012;82:1321–1331. doi: 10.1038/ki.2012.307. [DOI] [PubMed] [Google Scholar]
- 157.Freedman BI, Limou S, Ma L, Kopp JB. APOL1-associated nephropathy: a key contributor to racial disparities in CKD. Am. J. Kidney Dis. 2018;72:S8–S16. doi: 10.1053/j.ajkd.2018.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Muntinghe FL, et al. CCR5 deletion protects against inflammation-associated mortality in dialysis patients. J. Am. Soc. Nephrol. 2009;20:1641–1649. doi: 10.1681/ASN.2008040432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT02741323 (2019).
- 160.Park J, et al. Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science. 2018;360:758–763. doi: 10.1126/science.aar2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sullivan KM, Susztak K. Unravelling the complex genetics of common kidney diseases: from variants to mechanisms. Nat. Rev. Nephrol. 2020;16:628–640. doi: 10.1038/s41581-020-0298-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.van Zuydam NR, et al. A genome-wide association study of diabetic kidney disease in subjects with type 2 diabetes. Diabetes. 2018;67:1414–1427. doi: 10.2337/db17-0914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kottgen A, et al. New loci associated with kidney function and chronic kidney disease. Nat. Genet. 2010;42:376–384. doi: 10.1038/ng.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wuttke M, et al. Genetic loci associated with renal function measures and chronic kidney disease in children: the pediatric investigation for genetic factors linked with renal progression consortium. Nephrol. Dial. Transpl. 2016;31:262–269. doi: 10.1093/ndt/gfv342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Langefeld CD, et al. Genome-wide association studies suggest that APOL1-environment interactions more likely trigger kidney disease in African Americans with nondiabetic nephropathy than strong APOL1-second gene interactions. Kidney Int. 2018;94:599–607. doi: 10.1016/j.kint.2018.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Sandholm N, et al. Genome-wide association study of urinary albumin excretion rate in patients with type 1 diabetes. Diabetologia. 2014;57:1143–1153. doi: 10.1007/s00125-014-3202-3. [DOI] [PubMed] [Google Scholar]
- 167.Teixeira JC, et al. Widespread Denisovan ancestry in Island Southeast Asia but no evidence of substantial super-archaic hominin admixture. Nat. Ecol. Evol. 2021;5:616–624. doi: 10.1038/s41559-021-01408-0. [DOI] [PubMed] [Google Scholar]
- 168.Hajdinjak M, et al. Reconstructing the genetic history of late Neanderthals. Nature. 2018;555:652–656. doi: 10.1038/nature26151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Chen F, et al. A late Middle Pleistocene Denisovan mandible from the Tibetan Plateau. Nature. 2019;569:409–412. doi: 10.1038/s41586-019-1139-x. [DOI] [PubMed] [Google Scholar]
- 170.Kuhlwilm M, et al. Ancient gene flow from early modern humans into Eastern Neanderthals. Nature. 2016;530:429–433. doi: 10.1038/nature16544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Green RE, et al. A draft sequence of the Neandertal genome. Science. 2010;328:710–722. doi: 10.1126/science.1188021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Reich D, et al. Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature. 2010;468:1053–1060. doi: 10.1038/nature09710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Slon V, et al. The genome of the offspring of a Neanderthal mother and a Denisovan father. Nature. 2018;561:113–116. doi: 10.1038/s41586-018-0455-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Vernot B, Akey JM. Resurrecting surviving Neandertal lineages from modern human genomes. Science. 2014;343:1017–1021. doi: 10.1126/science.1245938. [DOI] [PubMed] [Google Scholar]
- 175.Lander ES, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 176.Thomson R, et al. Evolution of the primate trypanolytic factor APOL1. Proc. Natl Acad. Sci. USA. 2014;111:E2130–E2139. doi: 10.1073/pnas.1400699111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.