

Abstract
Background
Hereditary sensory and autonomic neuropathies (HSANs) are a group of genetic disorders affecting the peripheral nervous system. Two different associated variants have been identified in dogs: 1 in Border Collies and 1 in Spaniels and Pointers.
Objectives
Clinically and genetically characterize HSAN in a family of mixed breed dogs.
Animals
Five 7‐month‐old mixed breed dogs from 2 related litters were presented for evaluation of a 2‐month history of acral mutilation and progressive pelvic limb gait abnormalities.
Methods
Complete physical, neurological, electrodiagnostic, and histopathological evaluations were performed. Whole genome sequencing of 2 affected dogs (1 from each litter) was used to identify variants that were homozygous or heterozygous in both cases, but wild type in 217 control genomes of 100 breeds. Immunohistochemistry was used to assess protein expression.
Results
Complete physical, neurological, electrodiagnostic, and histopathological evaluations confirmed a disorder affecting sensory and autonomic nerves. Whole genome sequencing identified a missense variant in the RETREG1 (reticulophagy regulator 1) gene (c.656C > T, p.P219L). All affected dogs were homozygous for the variant, which was not detected in 1193 dogs from different breeds. Immunohistochemistry showed no expression of RETREG1 in the cerebellum of affected dogs. One of the affected dogs lived for 5 years and showed gradual progression of the clinical signs.
Conclusions and Clinical Importance
We confirmed the diagnosis of HSAN in a family of mixed breed dogs and identified a novel and possibly pathogenic RETREG1 variant. Affected dogs experienced gradual deterioration over several years.
Keywords: canine, electrodiagnostics, genetics, neurology, peripheral nervous system
Abbreviations
- CSF
cerebrospinal fluid
- DNA
deoxyribonucleic acid
- EDTA
ethylenediaminetetraacetic acid
- EMG
electromyography
- GDNF
glial cell‐derived neurotrophic factor
- HSAN
hereditary sensory and autonomic neuropathy
- IBD
identical by descent
- OMIA
Online Mendelian Inheritance in Animals
- PROVEAN
protein variation effect analyzer
- RETREG1
reticulophagy regulatory 1
- RNA
ribonucleic acid
- SIFT
sorting intolerant from tolerant
- SNAP
sensory nerve action potential
- SNV: single nucleotide variants; SSPCA
Scottish Society for Prevention of Cruelty to Animals; SV, structural variant
- WGS
whole genome sequencing
1. INTRODUCTION
Hereditary sensory and autonomic neuropathies (HSANs) are a heterogeneous group of inherited neurodegenerative diseases affecting peripheral sensory and autonomic neurons.1, 2, 3, 4 Severe distal sensory loss, acral mutilation, and variable autonomic signs characterize HSANs.1, 4 In humans, 8 forms have been described based on phenotype, and genetic variants in more than 20 genes have been reported.1, 4 The pathophysiology of these disorders remains poorly understood.1, 2, 3, 4
Although HSANs have been reported in different dog breeds including Border Collie,5, 6, 7, 8 Border Collie cross,9 Long‐haired Dachshund,10, 11 Miniature Pincher,12 Jack Russell Terrier,13 Pointer,14, 15, 16, 17 English Springer Spaniel,17 French Spaniel,17, 18 and Fox Terrier,4 only 2 causal variants have been identified to date. The first is an inversion disrupting RETREG1 (reticulophagy regulator 1), encoding a Golgi protein, in Border Collies and Border Collie crosses (Online Mendelian Inheritance in Animals [OMIA] 002032‐9615).5, 9 The second is a point variant in a lincRNA upstream of the GDNF gene encoding glial cell‐derived neurotrophic factor in the Pointer, English Springer Spaniel, and French Spaniel (OMIA 001514‐9615).17
Over a 2‐year period, 5 mixed breed dog puppies from 2 different litters were presented to the hospital. All had a history of severe acral mutilation and progressive pelvic limb gait abnormalities. Here we describe the clinical presentation, electrodiagnostic findings, histopathological features, and long‐term outcome of a novel RETREG1 variant in a family of mixed breed dogs.
2. MATERIALS AND METHODS
2.1. Animals
Five related mixed breed dogs were evaluated separately, each at the age of approximately 7 months (Figure 1A). They were from 2 different litters from the same reportedly healthy dam; 1 litter consisting of 2 females was born in December 2009 (2009 litter) and the other consisting of 3 males was born in December 2010 (2010 litter), but no other parentage information was available. These puppies had been relinquished to the Scottish Society for Prevention of Cruelty to Animals (SSPCA) because they were visibly not healthy. We do not know if there were healthy littermates in addition to the 5 affected dogs. Residual blood samples in ethylenediaminetetraacetic acid (EDTA) were retained from 4 dogs for genetic investigations. Samples could not be obtained from the dam or sire(s). Additional diagnostic tests were performed on 2 dogs (2009 litter), including blood testing and cerebrospinal fluid (CSF) analysis, and urinalysis in 1 dog.
FIGURE 1.
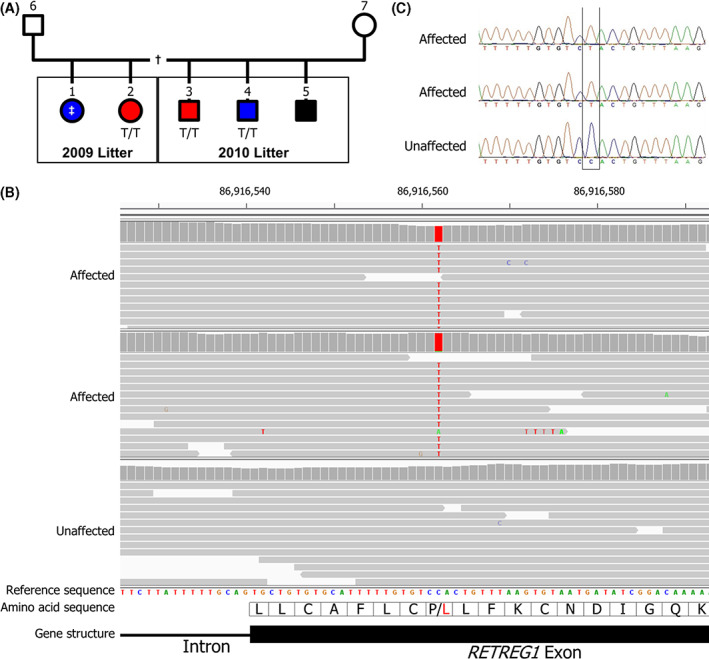
A. Pedigree of the affected family. Five affected dogs (filled symbols) were born to the same dam; 3 males in 1 litter and 2 females in a separate litter. The identity of the sires of both litters is unknown, but relationship testing shows they are likely to be the same dog. One affected dog from each litter was whole genome sequenced (red symbols) and DNA samples were obtained from a further 2 dogs (blue symbols). Three of the cases were genotyped homozygous for the RETREG1 variant (T/T) and DNA from 1 case never yielded any successful genotyping results (‡). Males are denoted by square symbols and females by circles. This pedigree may be incomplete (†)—the existence of healthy littermates is unknown. B. Whole genome sequence viewed with Integrative Genomics Viewer (IGV). Both affected dogs were homozygous for the variant (T) allele and an unaffected dog was homozygous for the wild type (C) allele. The variant is in exon 6 of RETREG1 and alters the amino acid from a Proline (P) to a Leucine (L). C. The RETREG1 C > T variant was confirmed by Sanger sequencing in all 3 affected dogs
2.2. Electrodiagnostic testing
Two affected puppies (2009 litter) were anesthetized to perform a complete electrodiagnostic examination including electromyography (EMG) and measurement of mean motor and sensory nerve conduction velocities (Medelec Synergy, Viasys Healthcare, Old Woking, UK). Insulated stainless steel needle electrodes were used for nerve stimulation, muscle recording, and ground. Motor nerve conduction velocities of the peroneal and ulnar nerves were calculated, and sensory nerve conduction studies were performed on the peroneal and ulnar nerves as previously described.19, 20, 21
2.3. Histopathology and immunohistochemistry
Owner consent was given at presentation for euthanasia and complete necropsy in 3 of the affected dogs (2010 litter). The other 2 dogs (2009 litter) had muscle (gastrocnemius and biceps femoris) and nerve (common peroneal) biopsy samples taken under general anesthesia. In addition to routine samples taken during necropsy (including brain, cervical, thoracic, and lumbar spinal cord), representative samples from multiple nerves (sciatic, common peroneal, ulnar, radial, cutaneous sural, and vagosympathetic nerve trunk), dorsal root ganglia (cervical, thoracic, and lumbar), and autonomic ganglia were collected and fixed in 10% buffered formalin. Slices of formalin‐fixed samples were embedded in paraffin before staining with hematoxylin and eosin and luxol fast blue.
Immunohistochemistry for RETREG1 protein was performed using a commercially available RETREG1 polyclonal antibody (RETREG1 polyclonal antibody [PA5‐42647], ThermoFisher Scientific, Waltham, Massachusetts). Antibody dilution was 1:200 and antigen retrieval using pH 8 EDTA buffer was employed.22 Paraffin blocks containing cerebellum from 3 affected dogs and 2 dogs of similar age from archived samples (normal controls) were stained. Cerebellum was used because it was the central nervous system tissue available for affected and control cases.
2.4. Sequencing and genotyping
Deoxyribonucleic acid from the affected animals (n = 3) was successfully extracted from residual EDTA blood using a DNeasy Blood and Tissue kit (Qiagen, Crawley, UK). Whole genome sequencing (WGS) of 2 affected dogs, 1 from each litter, was outsourced to Edinburgh Genomics, UK; a TruSeq Nano DNA Library (Illumina) was sequenced using the Illumina HiSeq X platform, generating 150 bp paired‐end reads and at least 30X coverage. Data analysis was performed using standard freely available software. Sequence reads were aligned to the dog genome reference (CanFam3.1) using Burrows‐Wheeler Aligner (BWA) and variants were called using Genome Analysis Toolkit (GATK) using GATK best practices.23, 24 Genomes are available from the European Nucleotide Archive (Accession no. PRJEB39868).
Samples of DNA from the affected dogs used for WGS (1 case from each litter) also were submitted for a commercially available dog breed ancestry test (Wisdom Panel, Mars Veterinary, Vancouver, Washington) to determine breed composition and relatedness of the affected dogs. Specific breeds identified were designated “ancestral breeds.” To assess relatedness, windows comprising approximately 20 markers and encompassing 1877 markers in total were compared between the 2 cases, to determine the proportion of windows that shared both haplotypes or 1 haplotype, identical by descent (IBD), IBD2, and IBD1, respectively (personal communication, Stephen Davison).
Initially, existing WGS data obtained for other studies from 217 dogs of 100 breeds and crossbreeds were used as controls for the genetic analysis, including 8 Border Collies, 6 Labrador Retrievers, and 1 German Shepherd (the “ancestral breeds”). Two sets of analyses were conducted, based on an autosomal recessive or autosomal dominant mode of inheritance. For autosomal recessive inheritance, variants that were homozygous in both cases, heterozygous or homozygous for the reference allele in the 15 “ancestral” genomes, and homozygous for the reference allele in remaining 202 genomes were retained for further investigation. For autosomal dominant inheritance, variants that were homozygous or heterozygous in both cases and homozygous for the reference allele in all 217 other genomes were retained for further investigation. These retained candidate variants also were screened in dog genomes from 616 genomes: 598 dogs (excluding 32 dogs also in the initial analysis) of 121 breeds, 10 mixed breed dogs, and 8 wolves from the Dog Biomedical Variant Database Consortium (DBVDC)25 to assess their frequency. In addition, all annotated genes (ie, genes annotated with a gene symbol, but not genes annotated with only an ENSCAF ID number) in which these retained variants were found were prioritized for further investigation using the VarElect NGS Phenotyper (https://ve.genecards.org/#input) and based on the keywords “sensory autonomic neuropathy,” “auto‐mutilation,” “sensory ganglion,” “autonomic ganglion,” “insensitivity to pain,” and “joint deformity.”26 In parallel, variants segregating with an autosomal recessive mode of inheritance and predicted to change a protein also were investigated further.
Remaining candidate variants then were validated by Sanger Sequencing in 3 cases (2 of which had already undergone WGS) and 2 unrelated, unaffected dogs (1 Large Munsterlander and 1 Alaskan Malamute). The following sequencing primers were used: RETREG1: GTTGATTTGGTGGCAGGTCTT, ATAAAGAGCCAGCTGACGTCT; KLRG2: TCAGGAAGATTTGGAAGCCTA, TTCCTGAGCAGATACCCAGTC; MUC19: GACCCACCATTCTTGAGTCAC, AGCAGAGGATCCATTTCAAAG. In addition, RETREG1 was manually interrogated for structural variants (SVs) using Integrative Genomics Viewer (IGV).27 Finally, the RETREG1 candidate variant was genotyped in 360 dogs from 122 breeds by TaqMan assay (primers: CCCACACTCCTCCCTTCTTAT, GCAGAACTGACTTGATTTTGCT; wild type probe: 5′6‐FAM‐ACACTTAAACAGTGGACACAAAAATGCAC, variant probe: 5′HEX‐ACACTTAAACAGTAGACACAAAAATGCAC). Primers and probes were designed using Primer3 (http://bioinfo.ut.ee/primer3-0.4.0/) and obtained from Integrated DNA Technologies (IDT, Leuven, Belgium).28
The protein amino acid change caused by the candidate variant was assessed using multiple in silico prediction tools: SIFT, Polyphen‐2, PROVEAN, MutPred2, and SNPs & Go.29, 30, 31, 32, 33 The Genome Aggregation Database (gnomAD) also was interrogated to assess the frequency of variants at human loci that are the equivalent of candidate variants.34 Canine gene and protein annotations are based on the reference genome CanFam3.1, reference sequence NC_006586 (chromosome 4 of CanFam3.1), gene NM_001314111.1 (RETREG1). Human gene and protein annotations are based on reference sequence NC_000005.9 (Chromosome 5, genome build GRCh37.p13) and NP_001030022.1 (RETREG1 isoform 1).
3. RESULTS
3.1. Clinical description
Five related mixed breed puppies from 2 litters were presented over a 2‐year period to the Small Animal Hospital of the University of Glasgow. All had a previous history of severe mutilation of the distal pelvic limbs and chronic progressive pelvic limb gait abnormalities. In all dogs, physical examination findings included multiple areas of trauma on the distal aspect of both pelvic limbs (mainly digits) with some of the dogs having had surgical digit amputations before referral (Figure 2). Neurological examination identified normal mentation and cranial nerve function. Gait assessment identified severe proprioceptive ataxia in all limbs with the pelvic limbs more severely affected, including occasional knuckling of the pelvic limb paws and hyperextension of both tarsi (Figure 2). Paw positioning and hopping were decreased to absent in the pelvic limbs and normal in the thoracic limbs. Withdrawal reflex was decreased and patellar reflex increased in both pelvic limbs, but perineal and cutaneous trunci reflexes were intact. Superficial and deep pain perception was absent distal to the carpi and tarsi but present proximally, and in the tail, trunk, and head (as illustrated in Video S1). Findings were consistent with peripheral sensory neuropathy.
FIGURE 2.

Photos of affected dogs. A and B demonstrate the hyperextension of the tarsi and knuckling when standing. C, D, and E demonstrate different severity of lesions in the pelvic limb digits secondary to acral mutilation
Complete blood cell count and serum biochemistry results were normal in all cases. Lumbar puncture for CSF collection was performed in 2 cases (2009 litter) and nucleated cell count and protein concentration were normal. Serology testing for Toxoplasma gondii and Neospora caninum was performed in the same 2 dogs and was negative. A urine sample was collected by cystocentesis from 1 dog and had normal organic acid concentrations.
3.2. Electrodiagnostic testing
Electrodiagnostic testing was performed in the same 2 dogs, and abnormalities were limited to the absence of sensory nerve action potentials (SNAP) during sensory nerve stimulation.
3.3. Histopathology
No macroscopic changes were observed. Histopathological abnormalities were limited to the peripheral nerves, dorsal root ganglia, and autonomic ganglia. Nerve histology indicated moderate myelinated fiber loss and axonal features compatible with Wallerian degeneration with some dystrophic changes. These changes were more severe in purely sensory nerves (cutaneous sural nerve) and autonomic nerves (vagus) than in mixed nerves (motor and sensory). Dorsal root ganglia and autonomic ganglia (cervical, celiac, and mesenteric) had moderate to severe neuronal necrosis and degeneration with moderate infiltration of lymphocytes and plasma cells (Figure 3A,B). No changes were observed in the muscles sampled.
FIGURE 3.

Microscopic (hematoxylin and eosin, ×100 magnification) section of a dorsal nerve root ganglion of a normal dog (A) and an affected dog (B). There are reduced numbers of neurons and the presence of neuronal degeneration (asterisk). Microscopic sections of the granular cell layer of the cerebellum (immunohistochemistry RETREG1 antibody, ×110 magnification) of a normal dog (C) and an affected dog (D). There is a lack of expression of RETREG1 (brown pigment) in the affected dog
3.4. Follow‐up
Three dogs were euthanized at presentation (2010 litter). Two dogs (2009 litter) were managed with protective collars and bandages to prevent further self‐mutilation. They both received a 2‐month course of gabapentin (10 mg/kg PO q8h; Gabapentin 100 mg capsules, Actavis Ltd, Dublin, Ireland, UK) with no clinically relevant improvement. Slow deterioration in the gait was observed in both cases with gradual involvement of the thoracic limbs. One dog was euthanized at 2 years of age because of urinary incontinence. The other dog was euthanized at 5 years of age when it developed megaesophagus (confirmed radiographically) and secondary aspiration pneumonia. At this time, marked proprioceptive ataxia was present in all limbs and persistent knuckling of both pelvic limbs with secondary malformation was observed in both tarsi. No superficial or deep pain perception in the distal or proximal portion of the limbs could be detected (as illustrated in Video S2).
3.5. Sequencing, genotyping, and protein expression
Because of incomplete availability of pedigree information, pedigree analysis could not be used to determine the mode of inheritance. However, comparison of the Wisdom Panel data between the 2 cases showed that 17% of windows were IBD2 (the 2 cases had identical haplotypes on both chromosomes) and 66% were IBD1 (the 2 cases had identical haplotypes on 1 chromosome). This finding is consistent with the IBD expected between full siblings, suggesting the 2 affected litters share the same sire and dam. Alternatively, it is possible there are 2 different sires that are closely related to each other, or that the sire(s) are closely related to the dam. In addition, both males and females were affected, and 2 separate litters were affected, but the female dam was not affected. Taken together, these findings suggest an autosomal recessive mode of inheritance is more likely, although we cannot exclude the possibility of some other mode of inheritance. Variants identified in the 2 affected genomes were filtered first based on autosomal recessive segregation and next on autosomal dominant segregation. There were 570 variants that met the recessive segregation criteria and 12 026 that met the dominant segregation criteria described in the methods, of which 435 met both recessive and dominant criteria.
All of the annotated genes (excluding genes only annotated with ENSCAF IDs) implicated among the recessive variants (137 variants in 73 genes) and dominant variants (3887 variants in 1706 genes) were analyzed using the VarElect online tool and the phenotype keywords provided in the methods. Only 1 of the recessive genes was a direct match (RETREG1) and 38 of the dominant genes were a direct match, although 2 (RETREG1 and WNK1) had much higher scores (11.09 and 11.19 respectively) than the remainder (≤2.88). The keywords “sensory autonomic neuropathy” and “insensitivity to pain” matched phenotypes associated with both RETREG1 and WNK1, making any variants in these genes promising candidate variants. Only 1 variant (CFA4:86916562, C > T), in the RETREG1 gene, was shared between the recessive and dominant lists of variants and is predicted to be a missense variant, located in exon 6 of RETREG1. Similarly, only 1 variant (CFA27:42850851) was in the WNK1 gene, although it is intronic and approximately 6670 nucleotides away from the nearest exon. On closer examination, this variant is 1 of 5 alleles of a trinucleotide (GTT) repeat element (microsatellite) represented among the 219 WGS. The predominant allele “a” (335/436 alleles) contains 10 GTT repeats, allele “b” (25/436 alleles) contains 11 repeats, allele “c” (71/436 alleles) contains 12 repeats, allele “d” (3/436 alleles) contains 13 repeats, and allele “e” (2/436 alleles) contains 14 repeats. The 2 cases both have the genotype a/e. This finding is consistent with a polymorphic, but benign, microsatellite marker, and this WNK1 variant therefore was excluded from further examination. RETREG1 was reported previously to cause sensory neuropathy in dogs5, 9 and humans.31 The variant in RETREG1 therefore remains the strongest candidate variant and was retained for further investigation.
In addition to analyzing variants in candidate genes as indicated by VarElect, we also focused on homozygous autosomal recessive variants predicted to alter a protein, of which there were 4. A missense variant in the gene CAST also was observed in 2/616 DBVDC genomes (1 homozygous and 1 heterozygous Labrador Retriever, 1 of these was of German ancestry, was submitted for research at the age of 14 months with skeletal dysplasia type 2 (SD2) and was 1 of the cases previously reported.35 There was no report of sensory neuropathy at the time of sample submission (personal communication, Tosso Leeb); further clinical information about the second dog was unavailable (personal communication, Oliver Forman) and was therefore excluded as likely benign. The remaining 3 variants were absent from all 616 DBVDC genomes. One variant (CFA3:1083274, G > A) initially was thought to be in a gene homologous to human KLRG2, but the canine Ensembl gene (ENSCAFG00000031165) has since been retired and the variant is now considered intergenic. The second variant (CFA27:13231303, C > G) was in a gene orthologous to MUC19 and the third was the same variant in the RETREG1 gene identified through the VarElect workflow. The intergenic/KLRG2 variant was found to be heterozygous in 1 of the cases by Sanger sequencing and (because KLRG2 has not been associated with any disorders to our knowledge) it, therefore, was considered benign and excluded from further investigation. The MUC19 and RETREG1 variants were homozygous in all 3 cases by Sanger sequencing, and homozygous for the wild type allele in 2 control dogs. However, MUC19 (Mucin 19, oligomeric) encodes a member of the gel‐forming mucin protein family, responsible for the gel‐like properties of mucus. MUC19 is expressed primarily in salivary glands and testes, and this variant therefore was considered benign. Finally, the RETREG1 variant was retained and prioritized for further investigation because of its previous association with sensory neuropathy. Multiple attempts at amplifying DNA to screen these 3 variants from the fourth case (Figure 1A) were unsuccessful.
Comparison of the CanFam3.1 RETREG1 annotation (NM_001314111.1) with the human RETREG1 annotation (NP_001030022.1) indicated similar structure of 9 exons, very similar amino acid sequence with a difference in the number of amino acid residues: 497 in humans and 485 in dogs. The observed missense variant in RETREG1 is a C > T variant at position 656 of the coding sequence in exon 6 (c.656C > T, p.P219L; Figure 1) and is predicted to be deleterious by all prediction algorithms used (Table S1). This variant also was not found in 360 dogs of 120 breeds, making it absent from a total of 1193 canids (1175 dogs of 192 breeds, 10 mixed breed dogs, and 8 wolves). This variant is different from the RETREG1 variant reported in Border Collies. The ancestry test (Figure S1) indicated that 1 of the parents of both litters was 25% German Shepherd and 25% Parson Russell Terrier. Border Collie also was detected in the same parent for 1 puppy and Labrador Retriever was detected in the other parent for the other puppy, but both with low confidence. All 4 of these breeds were represented among the 1193 dogs homozygous for the wild type allele (3 Parson Russell Terriers, 23 German Shepherds, 48 Border Collies, and 20 Labrador Retrievers).
The amino acid affected (P219 in the dog and P231 in humans) is conserved in 94 species. The equivalent human nucleotide (5:16479075 in GRCh37) is not represented at all in the gnomAD dataset, although P231 is represented. Three variants are observed in only 1 or 2 individuals each (of almost 250 000) and in the heterozygous state only (Table S2).
Immunohistochemistry for RETREG1 showed normal expression in the cerebellum of 2 control dogs and no expression in the cerebellum of 3 of the affected dogs (Figure 3C,D). We therefore concluded that the c.656C > T, p.P219L RETREG1 variant is likely pathogenic.
4. DISCUSSION
We confirmed a HSAN in a family of mixed breed dogs and identified a novel variant in the canine RETREG1 gene. Our study had some limitations that warrant further discussion. First, because of the lack of pedigree and phenotypic data for relatives, we cannot exclude the possibility that the mode of inheritance may be autosomal dominant or some other more complex form of inheritance, such as dominant with incomplete penetrance or polygenic/complex. However, the finding that the 2009 litter and 2010 litter are likely full siblings adds support to the hypothesis of autosomal recessive inheritance. Second, because we did not look for structural variants (SVs) on a genome‐wide level we cannot exclude the possibility that the disease is caused by a SV. However, because SVs are usually in linkage disequilibrium with single nucleotide variants (SNV) and all SNVs in or near functional candidate genes were examined (using VarElect), this possibility is unlikely. Third, we cannot exclude the possibility that 1 of the variants designated as intergenic or in or near an ENSCAF annotated gene is causal. Finally, because we have not genotyped large numbers of dogs of the ancestral breeds, we cannot exclude the possibility that the variant is in fact a rare but benign variant that segregates in 1 of these breeds. We confirmed that the amino acid affected (P219 in the dog and P231 in humans) is conserved in 94 species, the equivalent human nucleotide is not represented at all in the gnomAD dataset, and amino acid changes at P231 are rare and heterozygous only. These findings suggest that variants at this locus are more likely to be pathogenic. Taken with the observation that RETREG1 protein expression is absent in the affected dogs, this finding suggests that RETREG1 is likely the gene causing disease. However, we still cannot exclude the possibility that the RETREG1 variant is not causal but is rather in linkage disequilibrium with the causal variant, or simply happens to segregate with the disease by chance.
The protein encoded by RETREG1 is responsible for regulating turnover of the endoplasmic reticulum by selective phagocytosis.36, 37 Its inhibition impairs proteostasis in the endoplasmic reticulum because of accumulation of misfolded proteins, which in turn compromises neuronal survival and causes neurodegeneration.36, 37 The mechanism by which only sensory and autonomic neurons are affected remains unknown, because the RETREG1 protein is expressed in many other neurons and the changes do not seem to cause histopathological abnormalities (as in the cerebellum used for immunohistochemistry in our study).36 Human P231 is in the fourth transmembrane domain and in a reticulon homology domain (RHD) of the RETREG1.38 Proline residues within transmembrane domains cause kinks in the alpha helices, which in turn affects the structure of the protein.39 The structure and topology of the RHD of RETREG1 affect the ability of the protein to sense and induce endoplasmic reticulum curvature during autophagy.38 It therefore follows that a possible effect of the variant described here is a change in the structure of the protein, which in turn could be affecting normal and healthy autophagy. Deregulation of autophagy is a feature of various neurodegenerative diseases in humans.40
This variant is the second reported in RETREG1 that is associated with HSAN in dogs. Previously, an inversion disrupting this gene was described in Border Collies and in mixed breed dogs with Border Collie ancestry.5, 9 Interestingly, there are strong similarities in some of the clinical signs between the previously reported Border Collies and the cases presented here, including similar age of onset, the pelvic limbs being more severely affected than the thoracic limbs, and the presence of self‐mutilation and absent nociception in the distal portion of the limbs.5, 6, 7, 8, 9 There are also some differences, with the primary difference being that none of the dogs described here experienced autonomic signs until later in life, whereas some of the previously reported Border Collies had urinary incontinence from a young age. Also, some of the previously reported Border Collies had decreased motor nerve conduction velocities, a feature not observed in the 2 dogs in our report that had electrodiagnostic studies performed.5, 6, 7, 8, 9
Long‐term outcome has only been reported in the literature in 1 of the Border Collies that lived for almost 2 years, and the affected dog developed severe regurgitation and marked muscle atrophy.8 The 2 dogs with long‐term outcome in our study developed gradual deterioration of the proprioceptive ataxia, malformation of both tarsi, and developed autonomic signs (urinary incontinence at 2 years of age in 1 dog and megaesophagus in another dog at 5 years of age), but no signs of motor involvement or muscle atrophy. Interestingly, 1 of the dogs lived for 5 years, which could indicate that this family of mixed breed dogs had a less severe form of the disease than the Border Collies, but it is difficult to be certain because of the low number of long‐term outcomes reported. The autonomic deficits appeared to take longer to develop than did the sensory deficits in our cases.
Although HSANs have been reported in different dog breeds, they remain rare. The forms of HSAN reported in the long‐haired Dachshund,10, 11 Jack Russell Terrier13 and in Border Collies have more similarities to that reported here because they all presented with proprioceptive deficits and signs of autonomic dysfunction. This finding is in contrast to HSANs reported in the Miniature Pincher,12 Pointer,14, 15, 16, 17 English Springer Spaniel,17 French Spaniel,17, 18 and Fox Terrier,4 which present with loss of pain sensation, but no proprioceptive deficits or autonomic signs. Recently, a variant in the GDNF gene was reported as the cause of insensitivity to pain in the Pointer, English Springer Spaniel, and French Spaniel.17
Histopathologically, the severity of changes was variable depending on the nerve and ganglion analyzed with purely sensory nerves being more affected than mixed (motor and sensory) and autonomic nerves, and sensory ganglia showing more severe changes than autonomic ganglia. The presence of lymphoplasmocytic infiltrates in some of the ganglia was surprising, but inflammation has been described in some hereditary neuropathies in humans.36 The lack of expression of RETREG1 on immunohistochemistry in the affected dogs suggests that the amino acid change affects protein conformation and antibody binding.
In humans, a growing number of HSANs have been assigned to associated gene defects in recent years.1, 2, 4 A variant in RETREG1 was first reported in 2009 as the cause of severe sensory and autonomic neuropathy in a Saudi‐Arabian family.36 Several neuropathy genes have been identified so far, with some involved in cytoskeleton formation, transcriptional regulation, intracellular transport, endoplasmic reticulum function, sphingolipid biosynthesis, and interaction with the extracellular environment.1, 2 This variety of genes illustrates how many different components are required for building and maintaining the peripheral nervous system and the fact that the central and peripheral nervous systems are particularly vulnerable to dysfunctions in multiple pathways.2 Despite approximately 20 genes having been implicated in HSAN in humans, some cases remain unexplained and the availability of spontaneous models such as the dog can help explain the pathophysiology of these diseases.4
To our knowledge, our study is the second description of HSAN associated with a RETREG1 variant in dogs. The identity of the sire(s) of the 2 litters is unknown, but analysis of Wisdom Panel data supports the hypothesis that they are likely to be the same dog, could be closely related to the dam, or could be related to each other (if not a single sire). It is not currently known whether the causal variant arose de novo in the recent ancestry of the affected dogs described in our study or whether it arose more distantly, in which case the possibility exists it is segregating in the wider canine population. The affected dogs were crossbred, but do have German Shepherd, Parson Russell Terrier, and possibly also Labrador Retriever and Border Collie in their recent ancestry. The variant was not detected in these breeds during our study, but the number of dogs tested was small. Future studies of these breeds searching for the RETREG1 variant described here should be considered. With good management (use of a protective collar and bandages to prevent self‐mutilation), some affected dogs can have a longer lifespan than previously reported in the Border Collie despite gradual deterioration and later appearance of autonomic signs.
CONFLICT OF INTEREST DECLARATION
Authors declare no conflict of interest.
OFF‐LABEL ANTIMICROBIAL DECLARATION
Authors declare no off‐label use of antimicrobials.
INSTITUTIONAL ANIMAL CARE AND USE COMMITTEE (IACUC) OR OTHER APPROVAL DECLARATION
Approved by the local ethical committee of the University of Glasgow, School of Veterinary Medicine.
HUMAN ETHICS APPROVAL DECLARATION
Authors declare human ethics approval was not needed for this study.
Supporting information
Video S1 Neurological examination in one of the affected cases.
Video S2. Gait at presentation (7 months of age) and at long‐term follow‐up (5 years of age) in one of the affected cases.
Figure S1 Wisdom Panel results.
Table S1. Variant pathogenicity prediction results.
Table S2. Proline231 variants from gnomAD.
ACKNOWLEDGMENT
Funding was provided by the University of Glasgow Small Animal Hospital Fund. Preliminary results were presented as oral communication and abstract at the 32nd Annual Symposium of the European Society of Veterinary Neurology, Wroclaw, Poland, September 2019. The authors thank Oliver Forman and Stephen Davison at Wisdom Panel for additional relationship testing.
Gutierrez‐Quintana R, Mellersh C, Wessmann A, et al. Hereditary sensory and autonomic neuropathy in a family of mixed breed dogs associated with a novel RETREG1 variant. J Vet Intern Med. 2021;35(5):2306‐2314. 10.1111/jvim.16242
Funding information University of Glasgow
REFERENCES
- 1.Rotthier A, Baets J, Timmerman V. Mechanisms of disease in hereditary sensory and autonomic neuropathies. Nat Rev Neurol. 2012;8:73‐85. [DOI] [PubMed] [Google Scholar]
- 2.Weis J, Claeys KG, Roos A, et al. Towards a functional pathology of hereditary neuropathies. Acta Neuropathol. 2017;133:493‐515. [DOI] [PubMed] [Google Scholar]
- 3.Sapio MR, Goswami SC, Gross JR, et al. Transcriptomic analyses of genes and tissues in inherited sensory neuropatheis. Exp Neurol. 2016;238:375‐395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Correard S, Plassais J, Lagoutte L, et al. Canine neuropathies: powerful spontaneous models for human hereditary sensory neuropathies. Hum Genet. 2019;138:455‐466. [DOI] [PubMed] [Google Scholar]
- 5.Forman OP, Hitti RJ, Pettitt L, et al. An inversion disrupting FAM134B is associated with sensory neuropathy in the Border Collie dog breed. G3 (Bethesda). 2016;6:2687‐2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wheeler SJ. Sensory neuropathy in a Border Collie puppy. J Small Anim Pract. 1987;28:281‐289. [Google Scholar]
- 7.Vermeersch K, Van Ham L, Braund KG, et al. Sensory neuropathy in two Border Collie puppies. J Small Anim Pract. 2005;46:295‐299. [DOI] [PubMed] [Google Scholar]
- 8.Harkin KR, Cash WC, Shelton GD. Sensory and motor neuropathy in a Border Collie. J Am Vet Med Assoc. 2005;227:1263‐1265. [DOI] [PubMed] [Google Scholar]
- 9.Amengual‐Batle P, Rusbridge C, José López R, et al. Two mixed breed dogs with sensory neuropathy are homozygous for an inversion disrupting FAM134B previously identified in Border Collies. J Vet Intern Med. 2018;32:2082‐2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duncan ID, Griffiths IR. A sensory neuropathy affecting Long‐haired Dachshund dogs. J Small Anim Pract. 1982;23:381‐390. [DOI] [PubMed] [Google Scholar]
- 11.Duncan ID, Griffiths IR, Munz M. The pathology of a sensory neuropathy affecting Long Haired Dachshund dogs. Acta Neuropathol. 1982;58:141‐151. [DOI] [PubMed] [Google Scholar]
- 12.Bardagí M, Montoliu P, Ferrer L, Fondevila D, Pumarola M. Acral mutilation síndrome in a Minature Pinscher. J Comp Pathol. 2011;144:235‐238. [DOI] [PubMed] [Google Scholar]
- 13.Franklin RJM, Olby NJ, Targett MP, Houlton JEF. Sensory neuropathy in a Jack Russell terrier. J Small Anim Pract. 1992;33:402‐404. [Google Scholar]
- 14.Cummings JF, de Lahunta A, Winn SS. Acral mutilation and nociceptive loss in English pointer dogs. A canine sensory neuropathy. Acta Neuropathol. 1981;53:119‐127. [DOI] [PubMed] [Google Scholar]
- 15.Cummings JF, de Lahunta A, Braund KG, Mitchell WJ Jr. Hereditary sensory neuropathy. Nociceptive loss and acral mutilation in Pointer dogs: canine hereditary sensory neuropathy. Am J Pathol. 1983;112(1):136‐138. [PMC free article] [PubMed] [Google Scholar]
- 16.Cummings JG, de Lahunta A, Simpson ST, et al. Reduced substance P‐like immunoreactivity in hereditary sensory neuropathy of Pointer dogs. Acta Neuropathol. 1984;63(1):33‐40. [DOI] [PubMed] [Google Scholar]
- 17.Plassais J, Lagoutte L, Correard S, et al. A point mutation in a lincRNA upstream of GDNF is associated to a canine insensitivity to pain: a spontaneous model for human sensory neuropathies. PLoS Genet. 2016;29:e1006482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paradis M, de Jaham C, Page N, et al. Acral mutilation and analgesia in 13 French spaniels. Vet Dermatol. 2005;16(2):87‐93. [DOI] [PubMed] [Google Scholar]
- 19.Walker TL, Redding RW, Braund KG. Motor nerve conduction velocity and latency in the dog. Am J Vet Res. 1979;40:1433‐1439. [PubMed] [Google Scholar]
- 20.Niederhauser UB, Holliday TA, Hyde DM, McQuarrie A, Fisher LD. Correlation of sensory electroneurographic recordings and myelinated fiber diameters of the superficial peroneal nerve of dogs. Am J Vet Res. 1990;51:1587‐1595. [PubMed] [Google Scholar]
- 21.Holliday TA, Ealand BG, Weldon NE. Sensory nerve conduction velocity: technical requirements and normal values for branches of the radial and ulnar nerve of the dog. Am J Vet Res. 1977;38:1543‐1551. [PubMed] [Google Scholar]
- 22.Krenacs L, Harris CA, Raffeld M, Jaffe ES. Immunohistochemical diagnosis of T‐cell lymphomas in paraffin sections. J Cell Pathol. 1996;1:125‐136. [Google Scholar]
- 23.Li H, Durbin R. Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics. 2009;25:1754‐1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McKenna A, Hanna M, Banks E, et al. The genome toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res. 2010;20:1297‐1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jagannathan V, Drögemüller C, Lee T, et al. A comprehensive biomedical variant catalogue on whole genome sequences of 582 dogs and eight wolves. Anim Genet. 2019;50:695‐704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stelzer G, Plaschkes I, Oz‐Levi D, et al. VarElect: the phenotype‐based variation prioritizer of the GeneCards Suite. BMC Genomics. 2016;23:444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robinson JT, Thorvaldsdottir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365‐386. [DOI] [PubMed] [Google Scholar]
- 29.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non‐synonymous variants on protein function using SIFT algorithm. Nat Protoc. 2009;4:1073‐1081. [DOI] [PubMed] [Google Scholar]
- 30.Adzhubei I, Jordan DM, Sunyaey SR. Predicting functional effect of human missesse mutations using PolyPhen‐2. Curr Protoc Hum Genet. 2013; Chapter 7;Unit7.20. 10.1002/0471142905.hg0720s76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choi Y, Chan AP. PROVEAN web server: a toll to predict the functional effect of aminoacid substitutions and indels. Bioinformatics. 2015;31:2745‐2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pejaver V, Urresti J, Lugo‐Martinez J, et al. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat Commun. 2020;11:5918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Calabrese R, Capriotti E, Fariselli P, et al. Functional annotations improve the predictive score of human disease‐related variants in proteins. Human Variant. 2009;30:1237‐1244. [DOI] [PubMed] [Google Scholar]
- 34.Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434‐443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frischknecht M, Niehof‐Oellers H, Jagannathan V, et al. A COL11A2 mutation in Labrador Retrievers with mild disproportionate dwarfism. PLoS One. 2013;8:e60149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kurth I, Pamminger T, Hennings JC, et al. Mutation variants in FAM134B, encoding a newly identified Golgi protein, cause severe sensory and autonomic neuropathy. Nat Genet. 2009;41:1179‐1181. [DOI] [PubMed] [Google Scholar]
- 37.Islam F, Gopalan V, King‐yin LA. RETREG1 (FAM134B): a new player in human diseases: 15 years after the discovery in cancer. J Cell Physiol. 2018;233:4479‐4489. [DOI] [PubMed] [Google Scholar]
- 38.Khaminets A, Heinrich T, Mari M, et al. Regulation of endoplasmic reticulum turnover by selective authophagy. Nature. 2015;522:354‐358. [DOI] [PubMed] [Google Scholar]
- 39.Yohannan S, Faham S, Yang D, Whitelegge JP, Bowie JU. The evolution of transmembrane helix kinks and the structural diversity of G protein‐coupled receptors. PNAS. 2004;101:959‐963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bredesen DE, Rao RV, Mehlen P. Cell death in the nervous system. Nature. 2006;443:796‐802. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1 Neurological examination in one of the affected cases.
Video S2. Gait at presentation (7 months of age) and at long‐term follow‐up (5 years of age) in one of the affected cases.
Figure S1 Wisdom Panel results.
Table S1. Variant pathogenicity prediction results.
Table S2. Proline231 variants from gnomAD.