

SUMMARY
A small fraction of cancer patients with advanced disease survive significantly longer than patients with clinically comparable tumors. Molecular mechanisms for exceptional responses to therapy have been identified by genomic analysis of tumor biopsies from individual patients. Here, we analyzed tumor biopsies from an unbiased cohort of 111 exceptional responder patients using multiple platforms to profile genetic and epigenetic aberrations as well as the tumor microenvironment. Integrative analysis uncovered plausible mechanisms for the therapeutic response in nearly a quarter of the patients. The mechanisms were assigned to four broad categories—DNA damage response, intracellular signaling, immune engagement, and genetic alterations characteristic of favorable prognosis—with many tumors falling into multiple categories. These analyses revealed synthetic lethal relationships that may be exploited therapeutically and rare genetic lesions that favor therapeutic success, while also providing a wealth of testable hypotheses regarding oncogenic mechanisms that may influence the response to cancer therapy.
Graphical Abstract
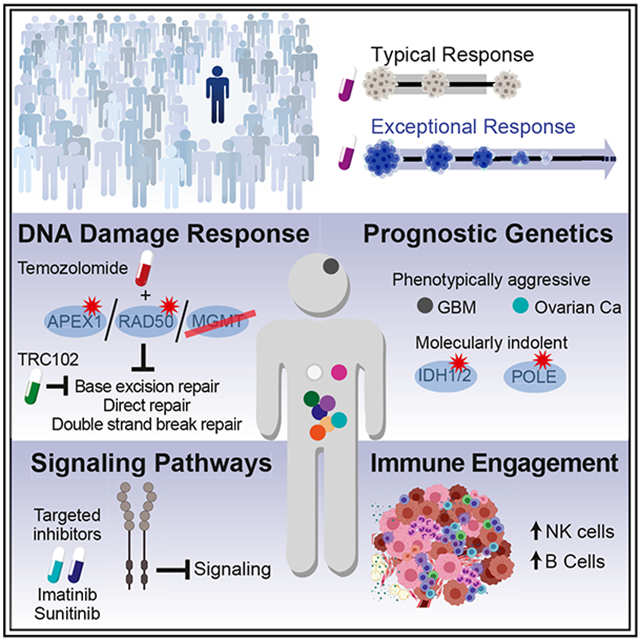
In Brief
Profiling multi-platform genomics of 110 cancer patients with an exceptional therapeutic response, Wheeler et al. identify putative molecular mechanisms explaining this survival phenotype in ~23% of cases. Therapeutic success is related to rare molecular features of responding tumors, exploiting synthetic lethality and oncogene addiction.
INTRODUCTION
Treatment response is assumed to depend on a combination of molecular features of the tumors, including their somatic mutational and epigenetic landscapes, germline polymorphisms, and the tumor microenvironment. Often, cancer therapies only produce meaningful responses in a small subset patients, referred to as “exceptional responders” (ERs), but the molecular basis for these striking therapeutic successes has not been systematically investigated. Nonetheless, early proof-of-concept studies show that genetic analysis of tumors from such patients can yield insights into oncogenic processes that influence the response to therapy. For example, among 14 patients in a phase 2 clinical trial of everolimus in bladder cancer, one had a complete response and that patient’s tumor had mutations inactivating TSC1 and NF2, two negative regulators of the drug target, mTOR (Iyer et al., 2012).
Exceptional responses in cancer are, by definition, rare, and thus the genetic lesions that may be causative would also be expected to be rare. A priori, genetic events conferring therapeutic responsiveness fall into two broad categories: oncogene addiction (Weinstein, 2002) and synthetic lethality (Hartwell et al., 1997). In oncogene addiction, a genetic change in a tumor cell renders it exceptionally dependent on the encoded protein and sensitive to therapies targeting the protein or downstream pathways. An oncogenic aberration that renders a tumor responsive to a therapy can be much more common in some cancer types than others. Nonetheless, this oncogene may confer therapeutic sensitivity when it occurs sporadically. A prime example is the response of some non-melanoma tumors harboring BRAF V600E to vemurafinib (Hyman et al., 2015).
The concept of synthetic lethality, as it applies to cancer, is based on the fact that mammalian cells have redundant regulatory circuits that control a particular phenotype important for malignancy (e.g., proliferation, survival). If a tumor has a genetic alteration that inactivates one of two parallel signaling pathways that control cell survival, for example, a drug targeting the second pathway could be exceptionally effective against that tumor. However, there can be more than two redundant pathways controlling an essential cellular function and, in such cases, a therapeutic response may only occur in tumors that have inactivated more than one of those parallel pathways. If tumors with this constellation of genetic aberrations are uncommon, such a response could be deemed exceptional.
The malignant cells in a tumor coexist with immune and stromal cells in the microenvironment, implying a failure in immune surveillance. Nonetheless, spontaneous regression of melanoma, follicular lymphoma, metastatic renal carcinoma, and certain types of neuroblastoma have been documented, which may indicate that the immune system can eradicate established tumors. This rare and presumably stochastic “awakening” of immune cells could conceivably play a role in exceptional responses to cancer therapy.
Whether these or other hypotheses account for the phenomenon of exceptional responses in cancer is unknown due to the limited number of ER cases that have been profiled molecularly. Previous studies of ER tumors have been largely based on analysis of somatic mutations, whereas it is well known that responses to cancer therapy can be predicted by RNA expression profiling (Alizadeh et al., 2000), analysis of DNA copy number alterations (Slamon et al., 2001), methylation (Figueroa et al., 2010; Mack et al., 2014), and rearrangements (Druker et al., 2001; Kwak et al., 2010), as well as by the abundance of particular immune subpopulations in the microenvironment (Dave et al., 2004; Muntasell et al., 2019; Cabrita et al., 2020; Helmink et al., 2020; Hollern et al., 2019; Petitprez et al., 2020). We therefore sought to investigate the molecular basis for exceptional responses to cancer therapy using multi-platform genomic profiling of tumor biopsies from a large, unbiased cohort of ER patients.
RESULTS
The NCI ER Cohort
To systematically investigate the molecular basis for exceptional responses in cancer, we conducted a multi-platform genomic study of tumors from a large cohort of ER patients, defined operationally as patients for whom a complete or partial response was expected in less than 10% of similarly treated patients or whose duration of response lasted three times the published median or longer (Conley et al., 2020). This cohort allowed us to determine how often integrative genomic analysis can offer an explanation for exceptional responses and whether mechanistic themes would emerge.
Tumor specimens from 111 ER patients (Table S1) were examined using multiple genomic methodologies to detect mutations, copy number changes, aberrant methylation, outlier gene expression, and the cellular makeup of the tumor microenvironment. Integrative analyses of these data allowed us to propose a plausible “level 1” mechanistic explanation (see the STAR Methods) for the exceptional response in 26 (23.4%) cases (Tables 1 and S2). These included cancers of the brain (8); GI tract (6); breast (4); bile duct system and lung (2 each); pancreas, endometrium, ovary, and bladder (1 each). DNA-damaging agents were used to treat 17 (65%) patients, either alone or in 7 (27%) cases together with targeted agents (Figure S1). Targeted therapies were used to treat 14 (54%) patients.
Table 1.
Cases with Strong Hypothesis for Exceptional Response
| Case Number |
Cancer Type | Treatment Associated with Exceptional Response |
Response | Duration (months) |
Exceptional Responder Category |
Key Molecular Findingsb | |||
|---|---|---|---|---|---|---|---|---|---|
| DDR | Signaling Pathway |
Immunea | Prognostic Genetics |
||||||
| 0072 | oligoastrocytoma (G3) | irinotecan | CR | 57 | t | + | • IDH1 p.R132H; 1p, 19q loss (prognostic genetics) | ||
| 0151 | astrocytoma (G3) | bevacizumab, irinotecan | PR | 96 | n | + | • IDH1 p.R132H, ATRX p.D497fs (prognostic genetics) | ||
| 0187 | glioblastoma multiforme | cediranib, cilengitide (NCT00979862) | CR | 111 | b | + | • IDH1 p.R132H (prognostic genetics) | ||
| 0305 | astrocytoma (G3/4) | cabozantinib (NCT01068782) | CR | 60 | b | + | • IDH1 p.R132L, ARTX p.I2050N (prognostic genetics) | ||
| 0394 | glioblastoma multiforme | RT, TMZ | CR | 117 | + | + | • IDH1 p.R132H, ATRX p.M1839K (prognostic genetics) • MGMT, DDB2 promoter me (DDR) |
||
| 0486 | astrocytoma, grade 3 | RT, TMZ, irinotecan (NCT00099125) | CR | 145 | + | + | • IDH1 p.R132C, ATRX p.E991fs (prognostic genetics) • MGMT, DDB2, POLE4 promoter me (DDR) |
||
| 0366 | glioblastoma multiforme | gliadel wafer, RT, TMZ | CR | 135 | + | • inactivating translocation APEX1 (DDR) • MGMT, EXO5 promoter methylation (DDR) • low MGMT and APEX1 expression (DDR) |
|||
| 0256 | astrocytoma anaplastic | bevacizumab, irinotecan | CR | 103 | + | n/a | + | • MSI: MLH1 p.R100*; POLE p.V411L (DDR, prognostic genetics, immune) | |
| 0075 | breast ductal adenocarcinoma, ER− PR− Her2+ | trastuzumab, carboplatin, docetaxel | CR | 76 | + | t | • BRCA2 p.W563*, BRIP1 p.S601*, TOP1 p.F329fs; BRCA1c del (DDR) | ||
| 0197 | breast ductal adenocarcinoma, ER− PR− Her2+ | trastuzumab, capecitabine | CR | 72 | + | + | • germline POLQ p.S1632* (DDR) • high ERBB2 expression (signaling pathway) |
||
| 0512 | breast ductal adenocarcinoma, ER+ PR− Her2+ | anastrazole, trastuzumab | PR | 30 | + | tnb | • high CYP19A; Low ERBB2 expresssion (signaling pathway) • ERBB2 amplification (signaling pathway) |
||
| 0513 | breast ductal adenocarcinoma, ER+ PR+ Her2+ | trastuzumab, pertuzumab (NCT01615068) | CR | 37 | + | t | • high ERRB2, ERBB3 expression (signaling pathway) • ERBB2 amplification (signaling pathway) |
||
| 0399 | cholangiocarcinoma | gemcitabine, cisplatin | CR | 18 | + | b | • TP53 p.R248Q, BRCA2 p.A1648fsc (DDR) | ||
| 0493 | cholangiocarcinoma | gemcitabine, cisplatin | CR | 12 | + | • TP53 p.C135F, extensive chromosome instability, mTOR p.T2380A (DDR) | |||
| 0349 | colon adenocarcinoma | irinotecan | CR | 56 | + | nb | • ATM p.R337C (DDR) • high TOP1 expression (DDR) |
||
| 0474 | colon adenocarcinoma | TMZ, TRC102 (NCT01851369) | PR | 45 | + | • MGMT promoter me, low MGMT expression germline RAD50 p.N1238N (DDR) | |||
| 0483 | rectal adenocarcinoma | bevacizumab, capecitabine, oxaliplatin | CR | 53 | + | b | • BRCA1 exons 7–8 del,c POLN splice site mutation (DDR) | ||
| 0454 | endometrial carcinoma | temsirolimus, carboplatin, taxol (NCT00977574) | CR | 70 | + | + | t b | + | • MSI: MLH1 promoter me (DDR, immune) • PIK3CA p.Y1021C, PTEN fs (signaling pathway) • BRCA2 p.T3033fs, SLX4 p.G142fs, WRN p.G327fs (DDR) |
| 0096 | gastrointestinal stromal | sunitinib | CR | 9 | + | • KIT exon 11 del: YEVQ (signaling pathway) • KDR, FLT1, FLT3 expression (signaling pathway) |
|||
| 0214 | gastrointestinal stromal | imatinib | CR | 70 | + | tnb | • KIT exon 11 del: EVQWKVVE (signaling pathway) • high KIT; deficient SDHB expression (signaling pathway) |
||
| 0392 | GE junction adenocarcinoma | EOX | CRs | 32 | + | + | • TP53 p.G245S, germline EXO1 p.D249N (DDR) | ||
| 0190 | lung, non-small cell | afatinib | CR | 6 | + | t | • EGFR del exon 19 (signaling pathway) | ||
| 0428 | Lung, squamous cell | carboplatin, taxol | CR | 25 | + | tn | • PALB2 p.W898*, DDB1 p.Q466fs, (2) TP53 p.R158H, fs | ||
| 0009 | ovarian carcinoma, clear cell and serous features | bevacizumab | PR | 41 | + | tbn | + | • POLE p.V411L (prognostic genetics) • high VEGFA expression (signaling pathway) |
|
| 0396 | pancreatic adenocarcinoma | FOLFOX | CR | 10 | + | • TP53 splice site, germline BRCA1 p.K339fs (DDR) | |||
| 0401 | urothelial carcinoma | nivolumamb (NCT023387996) | CR | 16 | t | • high PD1, PDL1; highest IFNG expression (immune)d | |||
Abbreviations and treatments: afatinib, tyrosine kinase inhibitor targeting EGFR; cilengitide, binds α-integrins inhibiting angiogenesis; cediranib, inhibitor of VEGF receptor; CR, complete response; PR, partial response; DDR, DNA damage repair, which includes the following pathways: homology-dependent recombination; nucleotide excision repair; base excision repair; nonhomologous end joining; EOX, epirubicine, oxaloplatin, capecitabin; FOLFOX, folinic acid, 5FU, oxaliplatin; gliadel wafer, infused with carmustine (bis-chloroethylnitrosourea [BCNU]), an alkylating agent; imatinib, tyrosine kinase inhibitor targeting PDGFR, ABL, KIT, DDR1, ABL2, NQO2 (N-ribosyldihydronicotinamide:quinone reductase; and oxidoreductase); me, DNA methylation; TMZ, temozolomide; TRC102, methoxyamine, inhibitor of BER through binding to apurinic/apyrimidinic sites. Causes TOP2-dependent irreversible strand breaks and apoptosis; RT, radio therapy.
Immune categories. Three cell types were of interest (see text) with single letter designations: t, T cell; n, NK cell CD56dim; b, B cell. Each letter indicates when an ER tumor was in the upper quartile for infiltration by the given immune cell (see the STAR Methods), n/a, not available.
All reported genetic alterations are clonal.
See also Figure S3A and S3B.
See Figure S3C and S3D.
The hypothesized level 1 mechanisms for exceptional responses divided broadly into four categories—DNA damage response (15), intracellular signaling pathway (9), prognostic genetics (9), and immunologic engagement (16)—with many cases involving two or more mechanisms (Table 1). We detail some of the most illustrative cases in the following.
DNA Damage Response
The predominance of plausible DNA damage response mechanisms parallels the frequent use of cytotoxic chemotherapy in routine cancer treatment and in this cohort. One glioblastoma multiforme (GBM) patient (ER0366) was treated sequentially with surgery, localized carmustine, and radiation (Figure 1A). On recurrence, temozolomide was administered, achieving a complete response that has lasted for over 10 years (Figure 1B). Two parallel pathways exist for repair of temozolomide-induced DNA methylation: direct repair (DR) of O6-methylguanine by MGMT (O-6-methylguanine-DNA methyltransferase) and base excision repair (BER) for the more common N3-methyladenine (N3mA) and N7-methylguanine (N7mG) adducts (Erasimus et al., 2016) (Figure 1C). In BER, the methylated purine base is removed by MPG (N-methylpurine DNA glycosylase) to create an abasic site, which is a substrate for APEX1 (apurinic/apyrimidinic endonuclease 1), resulting in a single-stranded gap that is repaired by DNA polymerase β (Erasimus et al., 2016). In the ER0366 tumor, MGMT promoter methylation (Figures S2A and S2B; see the STAR Methods, Clustering of ER and TCGA Brain Tumors) resulted in low MGMT mRNA expression relative to other ER tumors (Figure 1D, left panel), thereby inactivating the DR pathway. This ER tumor also inactivated the BER pathway as a consequence of a translocation that disrupted the third exon of APEX1 on chromosome 14, joining it to the first intron of ACTN4 on chromosome 19 and silencing APEX1 mRNA expression in the process (Figures 1C, iv; 1D, right panel; and 1E). Inactivating APEX1 alterations were observed in only 0.02% of patients in The Cancer Genome Atlas (TCGA) cohort (Knijnenburg et al., 2018) and APEX1 translocations were reported in only three TCGA tumors (Kim and Zhou, 2019), none of which were GBMs. Thus, we attribute the exceptional response to temozolomide in this patient to a rare genetic vulnerability that simultaneously inactivated the two primary pathways that repair temozolomide-induced DNA modifications, DR and BER.
Figure 1. Exceptional Responders with Genetic Disruption of DNA Damage Response Pathways.

(A) Treatment timeline for ER0366 with GBM. Gray shaded bars depict treatments and duration. XRT, radiation treatment. The broad blue arrow indicates the duration of the exceptional response.
(B) MRI before and 11 years after treatment.
(C) The predominant DNA methylation damage by temozolomide (i) with the direct repair and base excision pathways that repair these damaged bases. Both ER0366 and ER0474 exhibited promoter methylation and silencing of MGMT preventing the removal of O6-methylguanine (O6mG) (ii). After MPG (N-methylpurine DNA glycosylase) removes modified adenine (N3mA) and guanine (N7mG) bases (iii), ER0366 (iv), and ER0474 (v–viii) diverge in their details. In ER0366, APEX1 is inactivated by a translocation event (iv). In ER0474, TRC102 binding to the abasic sites created by MPG, inhibits the action of APEX1 (vi). During DNA replication, TOP2 creates double-strand breaks in the DNA, but at TRC102-bound sites, TOP2 cannot reseal the breaks (vii). An inactivating mutation in RAD50 prevents double-strand break repair from resolving these breaks (viii) blocking double-strand break repair.
(D) Expression of MGMT (left) and APEX1 (right) mRNA in GBM (n = 9), colorectal carcinoma (COAD, n = 17), and other ER cases (n = 88). The box marks the 25th and 75th quartiles and the whiskers are 1.5 times the inter-quartile range. Purple asterisks indicate ER0366 and ER0474. See also Figure S2.
(E) Chromosomal translocation fusing ACTN4 to APEX1 in ER0366; the genomic coordinates on hg38 of the breakpoints joined by translocation are given in base pairs (bp).
(F) Treatment timeline for ER0474 with colorectal carcinoma on ClinicalTrials.gov: NCT01851369 (https://clinicaltrials.gov/ct2/show/NCT01851369). On day 2,515 tumor could no longer be visualized by CT; however, a PET scan exhibited minimal fluorodeoxyglucose uptake near the site of the primary tumor, which was not biopsied, and therefore remains classified as PR.
(G) Location of the germline missense mutation, D1238N, in RAD50 in ER0474. When the homologous residue in yeast was mutated, it abolished the action of RAD50 (see text for details).
(H) Waterfall plot showing response of patients in a colorectal cancer expansion cohort in trial NCT01851369 and corresponding MGMT protein expression by IHC. For each subject, 100 tumor cells were scored for MGMT immunofluorescence on a scale of 0–3:0 meaning no tumor cells staining and +3 meaning >60% of the tumor cells positive. All patients that failed to respond, expressed MGMT protein robustly (3+, as indicated); the only patient responding to treatment failed to express MGMT protein (0, as indicated). Bx, biopsy; CT, computed tomography scan; CR, complete response; FOLFOX, 5-fluorouracil, folinic acid, oxaliplatin; FOLFIRI, 5-fluorouracil, folinic acid, irinotecan; GBM, glioblastoma multiforme; mets, metastases; MRI, magnetic resonance imaging; NGS, next-generation sequencing; PD, progressive disease; PET, positron emission tomography scan; Sx, surgery; XRT, X-ray treatment.
A patient with metastatic colon adenocarcinoma (ER0474) achieved an ongoing, nearly complete response lasting 45 months at last follow-up after receiving temozolomide in combination with the investigational drug TRC102 in a phase 1 clinical trial (Figure 1F). In the ER0474 tumor, as in ER0366, MGMT expression was silenced by promoter methylation but, unlike ER0366, there was no genetic lesion in the BER pathway. However, TRC102 binds covalently to abasic sites created by MPG (Figure 1C, v), thereby preventing endonucleolytic cleavage of the damage site by APEX1 (Wilson and Simeonov, 2010) (Figure 1C, vi), and blocking the BER pathway. Although TRC102 DNA adducts allow topoisomerase II DNA double-strand cleavage, they act like a topoisomerase II poison, triggering a DNA double-strand repair response and apoptosis (Yan et al., 2007) (Figure 1C, vii). The MRN (MRE11-RAD50-NBS1) protein complex is required for DNA double-strand break repair (Stracker and Petrini, 2011). Of note, the ER0474 tumor harbored a rare germline heterozygous missense mutation targeting the MRN subunit RAD50 (D1238N), which was rendered homozygous in the tumor through copy number neutral loss of heterozygosity on chromosome 5q. This particular RAD50 mutation (Figure 1G) was previously shown to abrogate the DNA repair activity of the MRN complex (Al-Ahmadie et al., 2014), suggesting that the exceptional response of this tumor may have stemmed from apoptosis that was triggered by unrepaired, topoisomerase II-induced double-strand breaks (Figure 1C, viii). Given the above, we hypothesize that the combination of temozolomide and TRC102 was effective in this patient because all necessary DNA repair pathways were compromised genetically, epigenetically, or pharmacologically.
The clinical trial testing temozolomide plus TRC102 was expanded to include an additional cohort of 16 patients with colon adenocarcinoma, among whom there was one more partial response. MGMT expression was measured by immunohistochemistry in biopsies from 11 patients in this expansion cohort. The tumor associated with the partial response did not express MGMT, whereas 10 tumors that did not respond to this therapy expressed this enzyme robustly (Figure 1H). These findings are in keeping with the mechanism outlined above for ER0474 and suggest that this therapeutic combination will be most effective in patients whose tumors lack MGMT expression. However, the partial response observed in this patient contrasts with the complete response in ER0474, suggesting that the RAD50 lesion in ER0474 may have augmented the therapeutic effect by crippling DNA double-strand break repair.
Platinum compounds were used in 8 of the 17 level 1 cases treated with DNA-damaging agents. Tumors from six of these eight cases had somatic or germline mutations in BRCA1, BRCA2, or PALB2. BRCA pathway mutations sensitize tumors to platinum-based antineoplastic drugs (Isakoff et al., 2015), but these drugs rarely produce exceptional responses in metastatic cases. ER0075 was a patient with Her2+ metastatic breast cancer who had a complete response to the combination of trastuzumab, carboplatin, and docetaxel, which has lasted 7 years (Figure 2A). This patient’s tumor had a homozygous deletion of BRCA1, inactivating somatic mutations in BRCA2 and BRIP1 (Figure 2B), and a high-level amplification and overexpression of ERBB2 (Figures 2C and 2D). BRIP1 physically interacts with BRCA1, and these two proteins function with BRCA2 to promote homologous recombination and the resolution of stalled replication forks that occur during replicative stress (Feng and Jasin, 2017). The exceptional response of this tumor may thus be ascribed to a triple deficiency in the BRCA pathway, addressed by carboplatin, and to ERBB2 amplification, addressed by trastuzumab.
Figure 2. Genetic Disruption of Multiple Members of Double-Stranded Break Repair Pathway Combined with HER2 Amplification.

(A) Treatment timeline for ER0075 with metastatic breast adenocarcinoma.
(B) Truncating mutations in BRCA2 and BRIP1 in ER0075.
(C) Relative copy number levels in ER0075. Purple dots indicate homozygous deletion of BRCA1.
(D) Expression of ERBB2 mRNA in ER breast adenocarcinomas (BRCA, n = 10), all other ER (n = 104) and TCGA (n = 4) cohorts. Purple asterisk indicates ER0075; the box marks the 25th and 75th quartiles and the whiskers are 1.5 times the inter-quartile range.
(E) Treatment timeline for ER0399, cholangiocarcinoma.
(F) Location of BRCA2 frameshift mutation (see also Table 1) in case ER0399.
(G) Treatment timeline for ER0483, rectal adenocarcinoma.
(H) Intragenic deletion of exons 7–8 in BRCA1 in case ER0483. BRCA, breast cancer; Ca, carcinoma; Dx, diagnosis; Mb, megabases; PR, partial response; NLS, nuclear localization signal; see legend to Figure 1 for all other abbreviations.
We identified inactivating somatic BRCA2 or BRCA1 genetic lesions in ER0399, a metastatic cholangiocarcinoma, and ER0483, a metastatic rectal adenocarcinoma, respectively, both of which had an exceptional response when treated with platinum-based therapies (Table 1; Figures 2E-2H). BRCA lesions are rarely observed in these types of tumors (Figures S3A and S3B) but could plausibly render these malignancies susceptible to platinum-based antineoplastic drugs as in other cancer types.
Intracellular Signaling Pathways
Agents targeting oncogenic signaling pathways, most commonly trastuzumab or bevacizumab, were used to treat 14 of the level 1 cases (Table 1). A patient with metastatic, estrogen receptor-positive breast cancer (ER0512) was treated with trastuzumab because of a high-level ERBB2 amplification, together with anastrozole, resulting in an ongoing 2.4-year partial response (Figure 3A). While the patient was clinically HER2+ (as defined by DNA amplification), the tumor had exceedingly low expression of ERBB2 mRNA in comparison with the full TCGA breast cancer cohort (Figure 3B). Indeed, molecular profiling classified this tumor into the basal-like subtype (Figure S4) rather than the HER2-enriched subtype, making it unlikely that trastuzumab contributed to the exceptional response. The expression of estrogen receptor in this tumor prompted treatment with anastrozole, an inhibitor of aromatase (CYP19A), that converts testosterone into estradiol. In the TCGA and ER breast cancer cohorts, the expression of CYP19A1 in this patient’s tumor was in the top 1.5% of all samples (Figure 3B). Thus, the exceptional response in this patient may have been elicited by anastrozole, potentially reflecting an extreme addiction of this tumor to aromatase-dependent estrogen synthesis. While plausible, the relationship between exceptionally high CYP19A1 expression and response to anastrozole requires validation in other cohorts.
Figure 3. Exceptional Responders with Genetic Disruption of Signaling Pathways.

(A) Treatment timeline for ER0512 with metastatic breast adenocarcinoma.
(B) CYP19A1 and ERBB2 mRNA expression in ER and TCGA tumors. Pink asterisks, HER2+ ER tumors; black asterisks, HER2− ER tumors. ER0512 has high CYP19A1 but low ERBB2 expression. See also Figure S4.
(C) Treatment timeline for ER0096 with gastrointestinal stromal tumor (GIST).
(D) Relative mRNA expression of indicated genes in GIST (n = 3) and other ER cases (n = 111). Pink asterisk, ER0096; the box marks the 25th and 75th quartiles and the whiskers are 1.5 times the inter-quartile range.
The gastrointestinal stromal tumor from patient ER0096 had a deletion of KIT exon 11, resulting in a constitutively active KIT isoform. This patient relapsed after an initial response to the imatinib, which targets KIT, but then achieved a complete response to sunitinib (Figure 3C). Gene expression profiling revealed high expression of KIT, as expected, but also of genes encoding several tyrosine kinases that are targeted by sunitinib (KDR, FLT1, and FLT3) (Lemmon and Schlessinger, 2010), potentially accounting for the exceptional complete response (Figure 3D). Of note, previous studies have associated high expression of these targets with response to sunitinib. For example, a patient with refractory B-ALL was treated with sunitinib based on high expression of FLT3 in the leukemic cells and achieved a near complete molecular remission (Griffith et al., 2016). In a randomized phase 3 trial, sunitinib prolonged progression-free and overall survival in patients with pancreatic neuroendocrine tumors (Raymond et al., 2011), which are characterized by high expression of FLT1 (VEGFR1) and/or KDR (VEGFR2) in the absence of recurrent mutations in either gene (La Rosa et al., 2003).
Immune Microenvironment
Given the role of immune surveillance in cancer, we examined immune cell infiltration in ER tumors and control TCGA tumors using a NanoString immune-oncology gene expression profiling panel and immunohistochemistry. Among 18 pre-defined signatures of immune subpopulations (see the STAR Methods, Evaluation of Immune Cell-Specific Gene Expression), signatures of B cells and activated (CD56dim) natural killer (NK) cells were higher in ER tumors than in TCGA tumors (false discovery rates of 0.0016 and 0.0001, respectively; Figure 4A; Table S3; see also the STAR Methods). Similar results were obtained when comparing ER and TCGA tumors from the same cancer subtype (Figure S5; Table S3). Of note, emerging studies have associated therapeutic response with increased abundance of tumor-infiltrating B cells and/or activated NK cells (Cabrita et al., 2020; Helmink et al., 2020; Hollern et al., 2019; Muntasell et al., 2019; Petitprez et al., 2020). The immune microenvironment of ER tumors is summarized in Table 1.
Figure 4. Immune Microenvironment of Exceptional Responders.

(A) Comparison of B cell- and CD56dim cell-type-specific expression scores between ER (n = 93) and TCGA (n = 35) cases. The box marks the 25th and 75th quartiles and the whiskers are 1.5 times the inter-quartile range. See also Figure S5 and Table S3.
(B) Treatment timeline for ER0401 with metastatic bladder carcinoma. p values represent FDR-adjusted t test.
(C) Relative mRNA expression of PDCD1, encoding PD-1, and CD274, encoding PD-L1, in bladder cancer from ER (n = 3), other ER cancers (n = 111), and BLCA from TCGA (n = 3). Red asterisk is ER0401; the box is defined as in (A). CDDP, cisplatin; Mit-C, mitomycin C.
(D) DNA copy number at the MDM2 locus in ER0401 showing high-level amplification of IFNG, encoding interferon-γ.
(E) Relative mRNA expression levels of IFNG and MDM2. This case expresses a higher level of IFNG mRNA than any other ER case. n, number of cases; the box is as in (A).
(F) In situ analysis of IFNG mRNA expression in top panel, CD3 immunofluorescence (green) labeling T cells. DAPI labeling (blue) of cell nuclei, most of which are tumor cells. Middle panel, IFNG mRNA detected by in situ hybridization using a fluorescently labeled IFNG probe (red), with anti-CD3 co-staining (green). Most IFNG mRNA foci were detected in the malignant bladder cancer (Ca) cells but were also present in T cells, as indicated. The tissue section was DNase treated to abrogate hybridization due to amplified IFNG DNA. Bottom panel, high-power image showing IFNG mRNA foci as indicated.
We uncovered an unusual immune mechanism while investigating the exceptional response of a patient with metastatic urothelial cancer (ER0401) who was treated with the immune checkpoint inhibitor nivolumab following failure of chemotherapy, radiation, and surgery. Nivolumab produced a complete response lasting 7 months, an outcome observed in only ~3% of bladder cancer patients (Kim and Seo, 2018) (Figure 4B). This tumor expressed high mRNA levels of PDCD1, encoding the nivolumab target PD-1, and CD274, encoding the PD-1 ligand PD-L1 (Figure 4C). It also harbored approximately 32 copies of an amplicon encompassing MDM2 and IFNG (encoding interferon-γ; Figure 4D), both of which were expressed at exceptionally high levels (Figure 4E). Amplification of MDM2 was detected in ~5% of TCGA urothelial cancers but was never associated with IFNG mRNA upregulation, as in this ER case (Figures S3C and S3D). Immunohistochemical staining of this tumor revealed a relatively low abundance of CD3+ T cells (Figure 4F, top panel), although in moderate proportion relative to tumor-infiltrating lymphocytes in other ER and TCGA samples (Table S4). CD3+ T cells typically produce interferon-γ. We therefore used in situ mRNA hybridization to investigate whether the high-level IFNG amplification might result in high IFNG mRNA expression by the malignant bladder cells. Indeed, IFNG mRNA was robustly expressed by both the malignant bladder cancer cells and the sparse CD3+ T cells (Figure 4F, middle panel, and magnified T cells in Figure 4F, bottom panel). While the amplification of IFNG in this case could represent either a homogeneously staining region or an extrachromosomal DNA circle (ecDNA), the high degree of amplification (32×) is more consistent with ecDNA (Turner et al., 2017), as is the extreme expression of IFNG mRNA, which is consistent with an altered chromatin structure in ecDNA (Wu et al., 2019). Interferon-γ plays an important, although complex, role in promoting anti-cancer immunity, and its expression has been associated with favorable response to checkpoint blockade (Benci et al., 2019; Garris et al., 2018), providing a potential explanation for the exceptional response in this case.
Prognostic Genetics
We defined a “prognostic genetics” category that included ER tumors with genetic lesions that are now known to be associated with a favorable prognosis but were not part of routine cancer diagnosis at the time these patients presented clinically (Table 1). Although ER patients in the prognostic genetics category relapsed following front-line therapy, their exceptional survival following salvage therapy could have been abetted by their prognostically favorable genomic characteristics.
Several of the ten high-grade GBMs and astrocytomas had genetic lesions that are much more common in low-grade glioma (LGG) than in high-grade glioma and have been associated with an indolent clinical course following standard therapy (Cancer Genome Atlas Research et al., 2015). Specifically, six had activating IDH1 R132 mutations, including one that also co-deleted chromosome arms 1p and 19q, and four had ATRX mutations (Tables 1 and S5). Three of the IDH1 mutant tumors exhibited a DNA methylation pattern characteristic of LGG, whereas the three tumors with wild-type IDH1 had a DNA methylation pattern resembling GBM (Figure 5A). Two ATRX mutations were truncating while two introduced missense substitutions in functionally important ATRX domains and were predicted to be deleterious. In the TCGA cohort of LGG tumors, these domains preferentially accumulated missense mutations while, in contrast, truncating mutations were distributed throughout the gene (p = 0.0065, Fisher’s exact text, two-sided). Recent reports demonstrated that several missense mutations targeting one of these domains (SNF2_N) are loss-of-function (Mitson et al., 2011), leading us to hypothesize that the missense variants observed in these two ER patients may well be pathogenic.
Figure 5. Mutations in ER GBM Cases Prognostic for Favorable Outcome.

(A) IDH1 mutation correlates with DNA methylation pattern in LGG and GBM. A heatmap showing unsupervised clustering of cancer-associated DNA hypermethylation profiles of brain tumors from ER (n = 6), TCGA LGG (n = 511), and TCGA GBM (n = 131) cases as indicated in tracks above the heatmap. The DNA methylation β values are represented by using a color scale from dark blue (low DNA methylation) to red (high DNA methylation). Four ER cases (ER0072, ER0151, ER0187, and ER0256) were excluded from this analysis, as described in the STAR Methods (see “Joint unsupervised clustering of ER and TCGA brain tumors”). See also Figure S6 and Table S5.
(B) Treatment timeline for case ER0394.
(C) Treatment timeline for ER case ER0305.
(D) Missense mutations in ATRX in ER0305 and ER0394 are likely to be functional. The distribution of missense (green lollipops) and truncating mutations (gray lollipops) in gliomas are shown. The two missense mutations found in the ER gliomas (orange squares), M1839K and I2050N, are in key functional domains of the proteins, SNF2_N and Helicase (He), respectively. SNF2_N domain, amino acid positions 1,536–1,889; helicase domain, amino acid positions 2,018–2,155. Darker green shading depicts the density of missense mutations; light gray shading is the distribution of truncating mutations. The increase in missense mutation density over the SNF2 and helicase domains suggests that these mutations may be enriched for functional mutations in glioma. (Compare to distributions of missense and truncating mutations in lung adenocarcinoma from the TCGA Pan Lung Cancer, where ATRX plays a much smaller role and inactivating mutations are rare, see Figure S6B.)
(E) Treatment timeline for case ER0256.
(F) Treatment timeline for case ER0009. Doxil, doxorubicin, liposomal.
(G) Schematic depiction of POLE (left) and MLH1 (right) genes accounting for the ultramutated phenotype in cases ER0256 and ER0009.
Another prognostically favorable genetic lesion is a hotspot mutation targeting DNA replication polymerase-ε (POLE V411L), which was detected in one refractory anaplastic astrocytoma (ER0256) and one poorly differentiated ovarian carcinoma with clear cell and serious features (ER0009; Figures 5E-5G). Additionally, this ER ovarian tumor had microsatellite instability due to a nonsense mutation in MLH1 (Figure 5G, right panel). POLE V411L alters the exonuclease proofreading domain of this polymerase, causing an ultramutated genome with a distinctive mutational signature (Temko et al., 2018). Consistent with this, ER0256 and ER0009 had 380 and 240 mutations/Mb, respectively, ~100-fold higher than tumors without this mutation. These rare POLE mutations are present in less than 1% of gliomas (Erson-Omay et al., 2015) and were not detected among 398 TCGA ovarian serous carcinomas (Cancer Genome Atlas Research Network, 2011). The prolonged survival of these two ER patients may reflect the favorable prognosis of this POLE mutation in other cancer types, possibly involving immune recognition of mutant neoantigens (see Temko et al., 2018 and references therein).
Compound ER Mechanisms
Sixteen level 1 cases (62%) harbored molecular changes that suggested classification into two or more ER categories. One exemplar was a patient with stage IVb endometrial cancer (ER0454) who achieved a complete response with paclitaxel, carboplatin, and temsirolimus and was alive without evidence of disease 5.8 years after diagnosis (Figure 6A). Exome sequencing revealed inactivating mutations targeting three proteins involved in DNA repair—BRCA2, SLX4, and WRN—that could have sensitized the tumor to carboplatin therapy. In addition, an activating PIK3CA mutation and an inactivating PTEN mutation could foster addiction to PI3 kinase/mTOR signaling and sensitivity to the mTORC1 inhibitor temsirolimus (Meric-Bernstam et al., 2012) (Figure 6B). Tumor sequencing also revealed microsatellite instability (MSI) due to methylation and silencing of MLH1 (Figure 6C). MSI endometrial cancers are associated with a relatively favorable prognosis and abundant microenvironmental immune cells, attributed to their elevated mutational burden (Cosgrove et al., 2017), as was observed in this ER tumor (Figure 6D). Thus, this tumor exhibited features of all four ER categories, which individually or together could explain the observed favorable outcome.
Figure 6. Exceptional Responders Often Exhibit Vulnerabilities in Multiple Categories.

(A) Treatment timeline for ER0454 with metastatic endometrial carcinoma.
(B) Activating PIK3CA mutation and truncating PTEN mutation in ER0454.
(C) mRNA expression of MLH1 (left panel) and promoter methylation of MLH1 (right panel) in endometrial carcinomas (UCEC) in the ER and TCGA cohorts. Right panel, top are color-coded DNA methylation levels from 18 probes in the MLH1 promoter region (see heatmap color key to the right) for ER0454 and, negative control, ER0521. Right panel, bottom, are methylation data from probe cg00893636 versus MLH1 gene expression levels.
(D) Left panel, relative CD8 and CD3 mRNA expression levels in ER UCEC, all other ER, and TCGA cases. Purple asterisk is ER0454. Right panel, immunohistochemical analysis of CD3 and CD8 expression in tissue sections of ER0454. In both (C and D), box plots mark the 25th and 75th quartiles and the whiskers are 1.5 times the inter-quartile range; ER UCEC, n = 3; all other ER, n = 111; UCEC TCGA, n = 3.
UCEC, endometrial cancer.
DISCUSSION
By multi-platform genomic analysis of tumors from a large cohort of ER patients we were able to generate plausible mechanisms accounting for the favorable clinical outcomes of nearly one-quarter of these patients. We identified ER cases in our cohort with presumptive mechanistic explanations invoking oncogene addiction, synthetic lethality, the tumor immune microenvironment, and the compound action of multiple genetic aberrations in the same tumor. Our study provides plausible and testable hypotheses that will need to be confirmed in larger cohorts of ERs. Nonetheless, our study has implications for the use and utility of comprehensive molecular profiling in cancer diagnosis and therapy.
Common mechanistic themes emerged among pathologically diverse ER tumors treated with similar agents, highlighting opportunities to exploit synthetic lethal vulnerabilities created by genomic abnormalities in cancer. This was best exemplified by the response of a GBM and a colorectal carcinoma to temozolomide, which exerts its antineoplastic effect by methylating adenine and guanine bases throughout the genome. Tumors resist temozolomide using both the DR and BER pathways, which were dually inactivated in these tumors genetically, epigenetically, or by treatment with the investigation drug TRC102. In addition to inactivating the BER pathway, TRC102 poisons topoisomerase 2, leaving double-strand DNA breaks that are repaired by the MRN complex. The ER tumor that was treated with TRC102 harbored an inactivating mutation targeting the MRN subunit RAD50, potentially fostering DNA damage-induced apoptosis. Of note, temozolomide plus TRC102 also induced a partial remission in one additional patient with colorectal carcinoma, and this patient’s tumor was the only one of 11 tested in which the DR pathway was inactivating by MGMT silencing. Together, our results suggest a synthetic lethal relationship between the DR and BER pathways in the context of temozolomide treatment, a concept supported by pre-clinical studies (MacLeod et al., 2019; Montaldi et al., 2015). These findings suggest a precision medicine strategy to develop the temozolomide/TRC102 combination in MGMT-inactivated tumors and support the development of inhibitors of MGMT (Wang et al., 2015) and MRN (Shibata et al., 2014) for use in this context.
Our analysis of ER tumors with lesions in the BRCA pathway demonstrates both the need for multi-platform genomic profiling as part of cancer diagnosis and the sophistication that is needed to interpret the findings correctly. One ER tumor mutationally inactivated BRCA2 and its interacting partner BRIP1, and also deleted BRCA1, suggesting that compound damage to the homologous recombination pathway should be considered when weighing treatment with platinum compounds and/or PARP inhibitors. We detected inactivating BRCA1 and BRCA2 mutations in two ER tumors representing cancer subtypes in which such events are extremely rare. Both tumors responded exceptionally to cisplatin, suggesting that the detection of such rare events could provide clinical benefit to such patients.
The prognostic genetics category emphasizes the limitations of histopathological cancer diagnosis and the need for rapid integration of relevant genomic findings into routine clinical practice. The POLE-mutant cases in this category illustrate that driver mutations with known prognostic value in some cancer subtypes can also be clinically relevant when they occur sporadically in other cancers. It is arguable whether patients in the prognostic genetics category should be considered ERs, since it is possible that they would have had a favorable outcome no matter what treatment they received. Nonetheless, our analysis suggests that it is important to recognize such prognostic genetic alterations in tumors from patients on clinical trials to correctly understand the efficacy of an investigational drug. A broad implication for all cancer patients is that molecular diagnoses should include screening for such prognostic genetic features since this information could inform therapeutic decisions.
Notwithstanding the mechanistic insights reported here regarding exceptional responses to therapy in cancer, we were unable to solve these fascinating clinical puzzles in many cases, leaving ample room to investigate these ER tumors by alternative analytical methods in the future. To foster reanalysis of these ER cases, all of their molecular profiling data and clinical information have been deposited in the NCI Genomic Data Commons, laying a foundation for even larger studies of exceptional responses in cancer in the future. The need for further study is highlighted by several of our ER cases, in which the proposed mechanistic explanation involved the co-occurrence of multiple genomic abnormalities targeting the same pathway, a tumor genotype that would be rare in unselected cancer series. Other exceptional responses were apparently explained by a constellation of genomic aberrations falling into distinct mechanistic categories, again likely to be uncommon overall in cancer and even among ER cases. These considerations imply that much larger cohorts of ER patients will need to be genomically profiled to establish which rare co-occurring genetic aberrations foster therapeutic sensitivity. In this regard, other large retrospective population-based genomic profiling efforts, such as AACR Project GENIE (Consortium, 2017), should prove helpful in identifying additional exceptional responder patients whose genomic and clinical data can be aggregated to understand the actionability of rare genetic lesions. A final important observation is that the majority of ER cases could not have been “solved” by analysis of DNA mutations alone, emphasizing the need for multi-platform genomic analyses of additional ER cases in the future. Such analyses may solidify, modify, or reject the hypotheses we have proffered, arguing for an international effort to study large cohorts of these fascinating patients.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Louis M. Staudt, MD, Ph.D. (lstaudt@mail.nih.gov)
Materials Availability
There are no tangible materials produced by this study that are available for distribution.
Data and Code Availability
All DNA and RNA sequence BAM files, DNA methylation files, mutation data, NanoString expression tables, and clinical information gathered in this study are available in the NCI, Genomic Data Commons under project ID: EXCEPTIONAL_RESPONDERS-ER available at https://gdc.cancer.gov/about-data/publications/exceptional_responders_2020. The Exceptional Responders project is registed in dbGaP under accession number Phs001145.v1.p1 at https://www.ncbi.nlm.nih.gov/gap/.
All computer code used in this study is available in the public domain or commercially available as noted in the Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat monoclonal Anti-CD3 | Bio Rad | MCA1477 |
| Rabbit monoclonal Anti-CD8 | Abcam | Ab101500 |
| Rabbit monoclonal Anti-CD45 | Cell Signaling Technology | 13917 |
| Rabbit monoclonal. Anti-PDL1 | Abcam | B228462 |
| Mouse monoclonal Anti PD-1 | Abcam | Ab52587 |
| Rabbit polyclonal Anti-cytokeratin, wide spectrum | DAKO | Z0622 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Critical Commercial Assays | ||
| Library Amplification Readymix: with KAPA HiFi DNA Polymerase | Kapa Biosystems, Inc., | Cat# KK2612 |
| Agencourt AMPure XP beads | Beckman Coulter | A63882 |
| SeqCap EZ HGSC VCRome, 96 Reactions | Roche NimbleGen | Cat# (06465676001) |
| xGen Blocking Oligos | Integrated DNA technologies | Cat# 1016184, Cat#1016186 |
| TruSeq RNA Exome (previously marketed as TruSeq RNA Library Prep for Enrichment (Cat. No. 20020189), TruSeq RNA Enrichment (Cat. No. 20020490), Exome Panel (Cat. No. 20020183) | Illumina | 20020490 |
| TruSeq Rapid SBS Kit-HS (200 cycles) | Illumina | FC-402-4001 |
| TruSeq SBS kit v3-HS (200 cycles) | Illumina | FC-401-3001 |
| HiSeq Rapid SBS Kit v2 (200 cycles) | Illumina | FC-402-4021 |
| XT_HS_IO360_CSO + Standard | NanoString | 115000222 |
| Low RNA Input Kit | NanoString | 1220000219 |
| XT_IO360 Primers | NanoString | 115000219 |
| Deposited Data | ||
| TCGA data sets | TCGA legacy archive | https://portal.gdc.cancer.gov/legacy-archive/ |
| RNAseq BAM files | This paper | Phs001145.v1.p1 at https://www.ncbi.nlm.nih.gov/gap/ and EXCEPTIONAL_RESPONDERS-ER at https://portal.gdc.cancer.gov/ |
| DNA BAM files | This paper | Phs001145.v1.p1 at https://www.ncbi.nlm.nih.gov/gap/ and EXCEPTIONAL_RESPONDERS-ER at https://portal.gdc.cancer.gov/ |
| Mutation tables for SNV and indels, and for CNA | This paper | Phs001145.v1.p1 at https://www.ncbi.nlm.nih.gov/gap/ and EXCEPTIONAL_RESPONDERS-ER at https://portal.gdc.cancer.gov/ |
| DNA Methylation IDAT files | This paper | Phs001145.v1.p1 at https://www.ncbi.nlm.nih.gov/gap/ and EXCEPTIONAL_RESPONDERS-ER at https://portal.gdc.cancer.gov/ |
| NanoString expression tables | This Paper | Phs001145.v1.p1 at https://www.ncbi.nlm.nih.gov/gap/ and EXCEPTIONAL_RESPONDERS-ER at https://portal.gdc.cancer.gov/ |
| Immunohistochemistry immune cell type quantification (see also Supplementary Table 4) | This paper | Phs001145.v1.p1 at https://www.ncbi.nlm.nih.gov/gap/ and EXCEPTIONAL_RESPONDERS-ER at https://portal.gdc.cancer.gov/ |
| Clinical information | This paper | Phs001145.v1.p1 at https://www.ncbi.nlm.nih.gov/gap/ and EXCEPTIONAL_RESPONDERS-ER at https://portal.gdc.cancer.gov/ |
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| RNAscope 2.5 LS Probe - Hs-IFNG-C3 | Advanced Cell Diagnostics | Cat# 310508-C3 |
| Recombinant DNA | ||
| Software and Algorithms | ||
| R v3.6. 2006 | R Development Core Team | https://www.r-project.org |
| function HotellingsT2 in R package ICSNP, v1.1-1 | Nordhausen K, Sirkia S, Oja H, Tyler DE, 2018 | https://CRAN.R-project.org/package=ICSNP |
| functions anova, lm, p.adjust, and t.test in the R package stats (incorporated into base R, v3.6) | R Development Core Team | https://www.r-project.org |
| function gls in R package nlme, v3.1-137 | Pinheiro J, Bates D, DebRoy S, Sarkar D, R Development Core Team, 2018 | https://CRAN.R-project.org/package=nlme |
| Function RUVr in RUVseq package v1.22.0 | PMID: 25150836 | https://www.bioconductor.org/packages/release/bioc/html/RUVSeq.html |
| Function varianceStabilizingTransformation in DESeq2 Bioconductor package v1.28.1 | PMID: 25516281 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Function betweenLaneNormalization in EDAseq Bioconductor package v2.22.0 | PMID: 22177264 | https://bioconductor.org/packages/release/bioc/html/EDASeq.html |
| SeSAMe v1.3.2 | PMID: 30085201 | https://www.bioconductor.org/packages/release/bioc/html/sesame.html |
| Other | ||
METHOD DETAILS
Overview of Genomic Data Analysis
Genomic data from all platforms, as described below, were aggregated and evaluated by genomic data analysts and presented to a molecular tumor board consisting of clinicians and cancer biologists. Cases were assigned priority levels based on evidence for ER mechanisms as follows: Level 1: Novel or rare molecular alterations that could be associated through literature support and/or mechanistic plausibility with pathways relevant to disease prognosis and/or response to therapy; Level 2: Molecular alterations that were plausibly associated with therapeutic response, but not supported by evidence in the literature; Level 3: No cancer driver genetic alterations were observed.
Tumor Sample Processing
Tumor samples (primary, metastatic, and/or recurrent) with or without matched germline controls (blood or uninvolved solid tissue) were obtained from 132 patients. FFPE specimens and fresh blood samples were shipped ambient overnight. Frozen specimens were shipped overnight to the Biospecimen Core Resource using a cryoport that maintained an average temperature of less than −180°C.
Pathology quality control was performed on each tumor specimen to confirm the specimen was histologically consistent the reported histology, as well as to assess the percent tumor nuclei, percent necrosis, and other pathology features. Tumor samples with ≥5% tumor nuclei were submitted for nucleic acid extraction.
DNA and RNA were co-extracted from solid tissue using a modification of the DNA/RNA AllPrep kit (Qiagen). For frozen tissues, the flow-through from the Qiagen DNA column was processed using a mirVana miRNA Isolation Kit (Ambion). FFPE tissues were extracted with a Qiagen FFPE DNA column and a HighPure miRNA RNA column (Roche). DNA was extracted from blood (when available) using the QiaAmp DNA Blood Midi kit (Qiagen).
RNA samples were quantified by measuring Abs260 with a UV spectrophotometer and DNA quantified by PicoGreen assay. DNA specimens were resolved by 1% agarose gel electrophoresis to confirm size of molecular weight fragments. A custom Sequenom SNP panel or AmpFISTR™ Identifiler™ (ThermoFisher) was utilized to verify that all DNA samples from a case were derived from the same patient and that the molecular sex matched the reported clinical sex. RNA was analyzed via the RNA6000 Nano assay (Agilent) for determination of an RNA Integrity Number (RIN) for frozen samples or DV200 for FFPE samples. The BCR received samples from a total of 132 patients, of which 119 cases were sent for genomic analysis. Of the 13 that disqualified, 4 samples were too small to perform pathology review, 6 had low nucleic acid yields, 1 had fewer than 5% tumor nuclei, and 2 were ineligible due to other pathology findings.
DNA and RNA samples were sent to the Human Genome Sequencing Center (HGSC) at Baylor College of Medicine (Houston, TX) for sequencing and DNA from tumor samples with greater than 15% tumor nuclei were sent to Foundation Medicine (Cambridge, MA) for targeted deep sequencing. When nucleic acid quantities were a limiting factor, nucleic acids were prioritized for sequencing at the HGSC. When residual DNA (96 cases) was available, 250-500 ng of DNA was sent to the Bellvitge Biomedical Research Institute (Barcelona, Spain) for DNA methylation.
For cases with residual tumor (71 cases), 5 unstained slides were prepared at 5-micron thickness and shipped to MD Anderson (Houston, TX) or the Oregon Health & Science University (Portland, OR) for immunohistochemistry analysis.
Pathology and Image Analysis
Digital slides containing sections of tumor stained for CD3, CD45, CD8, PD-1, and PD-L1 using chromogenic immunohistochemistry (IHC) were evaluated. Whole slide images were evaluated for quantification and characterization of immune cell infiltrates and PD-L1 expression on neoplastic cells. Serial sections from formalin-fixed paraffin embedded tumors were stained using IHC for PD-L1, CD3, CD45, CD8, and PD1. All slides were digitally scanned and scored using semi-quantitative grading and digital image analysis.
The number of positive cells per mm2 (PD1, CD3, CD45, CD8, and PDL1) and the intensity of positive staining (PDL1) were calculated using QuPath (McCarty et al., 1985). CD3, CD45, CD8, and PD1 expression was found on scattered immune cells that were individualized within the tumor and in the perivascular compartment. The average area evaluated for PD1, PDL1, CD3, CD45, and CD8 expression was 16.2 mm2.
In addition to digital cell counts, each sample was scored using a semi-quantitative score as follows: 0 for no positive cells, 1 for minimal positive cells, 2 for low numbers of infiltrating positive cells which are predominantly along periphery of tumor or perivascular, 3 for moderate infiltrates within tumor with or without significant cells along periphery of tumor, and 4 for marked infiltrate of positive cells within tumor sample. PDL1 expression was evaluated on neoplastic cells and tumor-infiltrating immune cells throughout viable regions of tumor including the invasive front. The staining intensity was scored using a scale of 0-3 as follows: 0 for no staining, 1 for mild staining, 2 for moderate, and 3 for strong staining and tumor H-score (Bankhead et al., 2017) was calculated using QuPath. Tumors were considered PDL1 positive when > 5% of the tumor cells and/or tumor-infiltrating immune cells were moderately or strongly PD-L1 positive(Patel and Kurzrock, 2015).
Multiple melanoma samples were evaluated which appeared amelanotic (lacking melanin pigment) and therefore melanin bleach was not performed. If present, melanin pigment is chromogenically similar to the DAB (3,3’-diaminobenzidine) chromogen used for IHC staining in this study and can falsely increase staining estimates. Intracellular black pigment (pneumoconiosis) was present in multiple pulmonary samples and was distinguished from brown pigment resulting from IHC DAB pigment using deconvolution during quantifications.
IFNG In Situ Hybridization
Interferon gamma (IFNG) expression was detected by staining 5 um FFPE tissue sections with RNAscope 2.5 LS Probe - Hs-IFNG-C3 (ACD, Cat# 310508-C3) and the RNAscope® LS Multiplex Fluorescent Assay (ACD, Cat# 322800) using the Bond RX auto-stainer (Leica Biosystems) with a tissue pretreatment of 15 minutes at 95°C with Bond Epitope Retrieval Solution 2 (Leica Biosystems), 15 minutes of Protease III (ACD) at 40°C, and 1:750 dilution of TSA-Cyanine 5 Plus (PerkinElmer).
To confirm if IFNG expression was within activated T-cells, sections where subsequently stained by IHC with rat anti-human CD3 antibody (Bio Rad Ref# MCA1477) at a 1:100 dilution for 60 minutes followed with a 1:100 dilution of rabbit anti-rat IgG secondary antibody (Vector Laboratories, Inc., Ref# BA-4001) using the Bond Polymer Refine Kit (Leica Biosystems) minus the post primary reagent, DAB and Hematoxylin. Antibody binding was detected with anti-HRP antibody conjugated with Alexa 488 (Jackson ImmunoResearch Laboratories, Inc., Cat# 123-545-021) diluted to 13.6 ug/ml for 30 min. Confirmation of tumor cells was determined by subsequent Cytokeratin IHC staining. Following a normal donkey serum block, sections were incubated with Pan Cytokeratin (AE1/AE3 Invitrogen# 41-9003-82) at a 1:25 dilution for 30 minutes. Antibody binding was detected with Donkey anti-mouse IgG Alexa 594 (Invitrogen# A-21203).
The RNAscope® 3-plex LS Multiplex Negative Control Probe (Bacillus subtilis dihydrodipicolinate reductase (dapB) gene in channels C1, C2, and C3, Cat# 320878) followed by IHC with rat anti-IgG1κ Isotype antibody (BD Pharmingen, Cat# 559072) and mouse IgG1 (BD Bioscience# 550878) was used as a negative control for in situ hybridization and IHC negative control. The RNAscope® LS 2.5 3-plex Positive Control Probe, Hs, was used as a technical control to ensure the RNA quality of tissue sections was suitable for staining. Slides were digitally imaged using an Aperio ScanScope FL Scanner (Leica Biosystems).
Nuclease Treatment
Tissue sections were deparaffinized twice in xylene for 5 minutes each and then twice in 100% ethanol for 3 minutes each. Tissue pretreatment consisted of peroxidase quenching with RNAscope® Hydrogen Peroxide for 10 minutes at room temperature, antigen retrieval by boiling in 1X RNAscope® Target Retrieval Reagent for 15 minutes. DNase treatment of sections was performed before protease digestion by incubating slides with an 8-fold dilution of DNase I (Qiagen) in Buffer RDD (Qiagen) for 30 minutes at 37°C. Tissue was permeabilized with a 15-minute incubation of Protease Plus (ACD) at 40°C. RNase treatment of tissue was performed after the protease digestion by incubating slides in 5 mg/ml RNase A (Sigma, Cat#R6513) in PBS for 30 minutes at 40°C. Slides were thoroughly rinsed in nuclease-free water. Slides were stained (without tissue pretreatment) with a duplex of RNAscope human PPIB and IFNG probes as described above with 1:750 dilutions of TSA-Cyanine 3 Plus and TSA-Cyanine 5 Plus (PerkinElmer), respectively.
IFNg and CD3 Immunofluorescence
RNAscope IFNG staining was performed as described above. Slides were then treated with a peroxidase block and subsequently IHC stained. Normal donkey serum was applied to the sections, followed by incubation with IFN-gamma (1:25 30’, abcam #ab218426). Reaction was detected with donkey anti-mouse IgG Alexa Fluor 594 (Invitrogen). Multiplex staining continued on the Bond RX auto-stainer using the Bond Polymer Refine Detection Kit for CD3 (with omission of peroxidase block, post primary reagent, DAB, and hematoxylin). Sections were incubated with CD3 (1:100 60’, BioRad #MCA1477), followed by Rabbit anti-Rat IgG (Vector Labs), Polymer anti-rabbit HRP (Bond Refine Kit) and Alexa Fluor 488 AffiniPure Goat anti-Horseradish Peroxidase (Jackson ImmunoResearch Laboratories). For negative controls, appropriate isotype reagents (mouse IgG2b or rat IgG1) were applied to sections in place of primary antibodies. Cover slips with Prolong Gold antiFade Mountant (Invitrogen) were placed on slides.
NanoString Gene Expression
Data Production
RNA from FFPE tumor and adjacent normal samples were evaluated for gene expression using the nCounter PanCancer IO 360 Gene Expression Panel, which includes 750 genes related to the microenvironment and immune response in cancer and 20 housekeeping control genes. Starting with 10 ng of total RNA, we used the Nanostring Low RNA Input Amplification kit (MAN-10046-02) for multiplexed target enrichment. Input RNA was initially converted to cDNA prior to amplification with target-specific primers.
In addition, total RNA from 48 TCGA samples, purified from FFPE tissue matched to patients whose fresh frozen tumors had also been studied, were obtained from the BioSpecimen Core Resource. Of these 35 were successfully sequenced on the NanoString Platform. These include: BLCA, 3; BRCA, 4; COAD, 8; LUAD, 12; RCC, 4; UCEC, 4.
Amplified cDNA was hybridized with probes from the NanoString Technologies nCounter PanCancer IO360 Profiling Panel including additional custom probes using hybridization protocol (MAN-10023-13). Overnight hybridization occurred for 20 hours at 65°C after which the reactions were transferred to the nCounter Prep Station. Removal of excess probes with magnetic bead purification was performed on the nCounter Prep Station (software v4.0.11.2). Once unbound reporter and capture probes were washed away, the ternary target-probe complexes were immobilized to the streptavidin-coated cartridge and aligned by an electric current.
The cartridge was transferred to the nCounter Digital Analyzer (software v3.0.1.4) and scanned at 555 field of view (FOV). An epifluorescent microscope and CCD camera identified sets of fluorescent spots, which were tabulated for data output. Quality control metrics were recorded using the nSolver Analysis Software v3.0.22.
Data Analysis
NanoString gene expression values were normalized with the method of Bhattacharya et al. (2020) with upper-quartile normalization (Bullard et al., 2010) and Remove Unwanted Variation using Residuals (RUVr) using the RUVr function from the RUVSeq Bioconductor package(Risso et al., 2014). We estimated unwanted technical variation using the set of housekeeping genes that had 1) expression above background in >97% of samples and 2) the highest correlation with expression of other housekeeping genes (Spearman coefficient ≥0.8), considering the deviance residuals from tumor and adjacent normal samples. We removed 2 dimensions of unwanted variation with RUVr and counts were variance-stabilizing transformation (VST)-scaled using the DESeq2 Bioconductor package (Love et al., 2014).
Evaluation of Immune Cell Markers
A 77-gene immune signature representative of 16 individual immune cell types and two cytokines was curated based on work from Danaher et al., 2017 and Bindea et al., 2013. The cell-types and genes composing the signatures were as follows: B-cells (9 genes) BLK, CD19, FCRL2, HLA-DOB, MS4A1, PNOC, SPIB, TCL1A, TNFRSF17; CD45 cells (1 gene) PTRPC; T-cells (12 genes) CD2, CD28, CD3D, CD3E, CD3G, CD6, CD96, IL2RB, LCK, SH2D1A, TRAT1, ZAP70; Th1 cells (1 gene) TBX21; Treg cells (3 genes) FOXP3, HAVCR2, CD274; CD8 T cells (3 genes) CD8A, CD8B, GZMM; T helper cells (2 genes) ICOS, CD4; Exhausted CD8 cells (4 genes) LAG3, CD244, EOMES, PTGER4; Tfh cells (2 genes) CXCL13, PDCD1; Cytotoxic cells (10 genes) CTSW, GNLY, GZMA, GZMB, GZMH, KLRB1, KLRD1, KLRK1, NKG7, PRF1; Dendritic Cells (3 genes) CCL13, CD209, HSD11B1; Macrophages (7 genes) CD68, CD84, CXCL5, CD163*, MS4A4A, CCL7, CYBB; Mast cells (3 genes) CPA3, MS4A2, HDC; Neutrophils (8 genes) FPR1, SI-GLEC5, CSF3R, FCAR, CEACAM3, S100A12, CXCR2, LILRB2; NK CD56dim cells (4 genes)KIR2DL3, KIR3DL1, KIR3DL2, IL21R; NK cells (3 genes) XCL1, XCL2, NCR1; Cytokines (2 genes) IFNG, TNF. Immune signatures were evaluated in 128 samples with expression results available on the NanoString platform and include 35 TCGA tumors and 93 exceptional responders. Individual cell and cytokine scores (18 total, see Table S3) were calculated as the median standardized gene expression of marker genes for each cell-type or cytokine. Statistical analyses to compare immune profiles, characterized by the 18 different immune scores, between ER and TCGA tumor samples were performed in the R statistical computing environment (R Core Team, 2018, see Key Resources Table). A Hotelling T2 test (Johnson and Wichern, 2007) was performed using the function HotellingsT2 in the R package ICSNP (Nordhausen et al., see Key Resources Table) to assess for an overall difference in the mean 18-score immune profile for ER versus TCGA cases (see Table S3, and main text Figure 4A).
Not all tumor types in the ER set (24 tumor types, 93 cases) were represented in the TCGA set (6 tumor types, 35 cases), therefore a second Hotelling T2 test was performed restricted to only the subset of six tumor types represented in both case sets (Table S3, and Figure S5). Additionally, a two-factor multivariate analysis of variance (MANOVA) (Johnson and Wichern, 2007) was performed using the R functions lm and anova in the packages stats (incorporated into base R) applied to the data for the six overlapping tumor types to evaluate overall difference in the mean 18-score immune profile between ER and TCGA cases after adjustment for tumor type. Analyses were also conducted for the individual immune scores, and p-values were adjusted for multiple testing (18 scores), controlling the false discovery rate (FDR) at a maximum of 10% using the method of Benjamini and Hochberg (Benjamini and Hochberg, 1995) as implemented in the R function p.adjust. For an individual score type, comparisons of mean score unadjusted for tumor type were based on t-tests; each adjusted comparison was based on a likelihood ratio test in the context of a two-factor Gaussian linear model evaluating the significance of ER vs TCGA after adjustment for tumor type using the functions gls as implemented in R package nlme and anova in base R.
Whole Exome Sequencing
Library Preparation
Tumor DNA samples were predominantly derived from FFPE except 3 ovarian samples, which were fresh frozen (FF); and Normal DNA samples, which were derived from the peripheral blood. DNA samples were constructed using Illumina paired-end pre-capture libraries following the protocol previously described for FF samples(Rokita et al., 2019). DNA input for libraries was 250 ng of DNA and Pre-capture Ligation Mediated-PCR (LM-PCR) was performed for 8-10 cycles using the Library Amplification Readymix containing KAPA HiFi DNA Polymerase (Kapa Biosystems, Inc., Cat # KK2612)
Exome Capture
For exome capture, FFPE, FF and normal samples were pooled separately as 4 libraries per pool using 250 ng of library for each sample. These pools of libraries were then hybridized separately in solution to the HGSC VCRome 2.1 design (Bainbridge et al., 2011) (42Mb, NimbleGen) according to the manufacturer’s protocol NimbleGen SeqCap EZ Exome Library SR User’s Guide (Version 2.2) form samples enriched between 2015-2017. Starting in December 2017, probes for exome coverage across >3,500 clinically relevant genes that are previously <20X (~2.72Mb) were supplemented with PKv1 and PKv2 into the VCRome 2.1 probe to enhance capture performance of low coverage regions. Blocking oligonucleotides from Sigma (individually sequence specifically synthesized) or xGen Universal Blocking oligonucleotides (Integrated DNA Technologies) were added into the hybridization to block the adaptor sequences. Hybridization for FF and normal samples was performed at 56°C for ~16h and for FFPE samples hybridization was at 42°C for 72h. Post-capture LM-PCR amplification was performed using the Library Amplification Readymix containing KAPA HiFi DNA Polymerase (Kapa Biosystems, Inc., Cat # KK2612) with 12 cycles of amplification. After the final AMPure XP bead purification, quantity and size of the capture library was analyzed using the Agilent Bioanalyzer 2100 DNA Chip 7500.
Sequencing and Analysis
The library pools were then loaded onto a HiSeq flow cell lane, and following amplification with Illumina’s cBot cluster generation system, sequencing runs were performed in paired-end mode (2 X 100-bp reads) using the Illumina HiSeq 2000 or 2500 platforms. On average, the data yielded 10.5 Gb data and 92.6% target bases were covered to 20x or greater depth per sample.
The sequencing pipeline for mapping reads and calling somatic variation was described previously(Wang et al., 2016). Nucleotide substitutions were called using CARNAC (Consensus and Repeatable Nucleotide Alterations in Cancer), and small indels were detected using PinDel(Ye et al., 2009). Variant annotation was performed with Oncotator(Ramos et al., 2015), with mutation frequencies taken from COSMIC(Forbes et al., 2017), dbSNP(Sayers et al., 2019), and ExAC(Lek et al., 2016) databases. Tumor purity was estimated using the variant allele fraction of the somatic clonal variants of driver genes. Somatic mutations are tabulated in Table S6. Putative rare and novel germline variation was defined as SNVs or indels with variant allele fraction ranging from 0.35 to 0.65, and with population frequency < 0.001 in Thousand Genomes or ExAC databases (Table S7). Copy number alterations, including focal amplification, deletion, arm-level amplification, and arm-level deletion analyzed using VarScan2(Koboldt et al., 2012). Copy number segmentation files from VarScan2 were processed using the Nexus package (BioDiscovery, Inc.) to yield relative copy number plots showing genomic regions of gain, loss or focal amplification and deletion for each tumor. Table S8 tabulates the genes involved in each regional copy number event.
RNA Sequencing
Total RNA was quantified using either Caliper Gx or Bioanalyzer 7500 (Agilent) and RNA integrity was determined based on- RIN and DV200 values. To access the quality of the RNA isolated from FFPE, DV200 was considered as a priority over RIN as previously described(Greytak et al., 2018). For CTS sequencing, total RNA requirements were 50ng and DV200 ≥30%. Illumina’s TruSeq RNA Library Prep for Enrichment (Cat. No. 20020189), formally known as TruSeq RNA Access Library Prep Kit (Cat # RS-301-2001) was used to prepare cDNA, libraries following manufacturer’s instructions. Libraries were quantified using Agilent Bioanalyzer 2100 DNA Chip 7500 and 4- libraries were pool for capture enrichment using the TruSeq RNA Enrichment (Cat# 20020490) and Exome Panel (Cat# 20020183). Enriched library pools were sequenced on Illumina HiSeq 2000 or 2500 platforms to generate 2X100-bp reads.
In addition to the ER samples, total RNA from 48 TCGA samples, purified from FFPE tissue matched to patients whose fresh frozen tumors had also been studied, were obtained from the BioSpecimen Core Resource. Library preparation and sequencing were exactly has described for the ER samples. Expression levels for comparing TCGA and ER data shown in all figures pertain to this data set, except for Figures 4A and S5 which are derived from NanoString data.
On average, 108M reads were generated per sample. RNA-Seq reads were aligned to the human reference genome (hg38) with the STAR algorithm(Dobin et al., 2013). Gene expression levels determined by HTSeq(Anders et al., 2015) were normalized to FPKM-UQ, using gene models from GENCODE v22 (Harrow et al., 2012). Fusion transcripts were identified using the DeFuse algorithm(McPherson et al., 2011).
DNA Methylation
Data Production
The DNA methylation status of the Exceptional Responders patients was determined using bisulfite-converted DNA processed by the Infinium FFPE restoration process and then hybridized on an Infinium MethylationEPIC BeadChip array (~850,000 CpG sites) following the manufacturer’s instructions (Illumina Infinium HD Methylation Assay Experienced User Card, Automated Protocol 15019521 v01), as previously described(Moran et al., 2016).
The signal intensities corresponding to methylated (M) and unmethylated (U) alleles were extracted from the IDAT files by the read-IDATpair function in the R package SeSAMe (https://github.com/zwdzwd/sesame). A detection P-value for each probe was calculated using pOOBAH (P-value with Out-Of-Band probes for Array Hybridization), which is based on the empirical cumulative distribution function of the out-of-band signal from all Type-I probes(Zhou et al., 2017). The signal intensities were further processed with background correction and dye-bias correction. The background correction is based on the noob method(Triche et al., 2013). The dye-bias is corrected using a non-linear quantile interpolation-based method using the dyeBiasCorrTypeINorm function(Zhou et al., 2017). β values, defined as SM/(SM+SU) for each locus where SM and SU represent signal intensities for methylated and unmethylated alleles, were computed using the getBetas function. β values range from zero to one, with scores of zero indicating no DNA methylation and scores of one indicating complete DNA methylation. Probes with a detection P-value greater than 0.05 in a given sample were masked as not available (NA). Additional experiment-independent masking of probes subject to cross-hybridization and genetic polymorphism (N=105,454) was implemented according to the probe manifest (release 20180909) downloaded from http://zwdzwd.github.io/InfiniumAnnotation(Zhou et al., 2018). Further information on the EPIC array, including detailed annotation of transcription association for each probe, was obtained from the same source. Probe success rates for each sample calculated after removing the aforementioned probes masked independently of an experiment are provided in Table S9. GBM (Astrocytoma G4) 0187 was excluded from the subsequent DNA methylation analysis due to a very low tumor purity.
Epigenetic Silencing in DNA Damage Repair Pathways (DDR)
We examined epigenetic silencing in 276 genes involved in major DDR pathways(Knijnenburg et al., 2018). For each gene, we first selected CpG sites in the promoter region, defined as the 3 kb region spanning from 1,500 bp upstream to 1,500 bp downstream of the annotated transcription start site (TSS), that are typically unmethylated (median β value across tumors < 0.2). We then identified tumors having at least one methylated (β value >0.3) CpG site within that region. Evaluating using a heatmap, we generally observed DNA methylation at multiple CpG sites across the region in a hypermethylated tumor (MLH1, Figure 3I, right panel; and MGMT, Figure S2A). The remainder of the cases in which we did not observe a methylated CpG site were left as not having evidence of DNA hypermethylation, given various numbers of missing data in each sample and a limited number of CpG sites covered in the array. To identify cases of epigenetic silencing, we first selected the optimal probe for calling epigenetic silencing among TCGA tumors of the same type by evaluating scatter plots of DNA methylation versus gene expression. We used HM450 data to include additional probes not examined in the TCGA Pan-Cancer study which was restricted to the probe sets represented on the older generation HM27 and HM450 array(Knijnenburg et al., 2018). We called epigenetic silencing among Exceptional Responders based on whether the DNA methylation level fell within the range of TCGA tumors exhibiting epigenetic silencing. We reported six genes with evidence for epigenetic silencing (Tables 1 and S5). Five genes (EXO5, DDB2, MGMT, MLH3 and MLH1) have been described previously as frequently hypermethylated in a variety of cancers(Knijnenburg et al., 2018). We also identified POLE4 epigenetic silencing, which was not examined in the TCGA study due to lack of shared CpG island probes between HM27 and HM450 arrays. MGMT promoter DNA methylation β-value (cg12981137) in each tumor sorted according to the heatmap analysis shown in Figure S2A are included in Table S10.
Clustering of ER and TCGA Brain Tumors
To examine DNA hypermethylation profiles in brain tumors from Exceptional Responders, we performed joint unsupervised clustering analysis of Exceptional Responder (N=6), TCGA LGG (N=511), and TCGA GBM (N=131) cases in which IDH1 and ATRX mutation status have been determined (Figure S6C). Four Exceptional Responder cases were excluded from the analysis for the following reasons: No DNA methylation data available (0256); Poor DNA methylation data quality (0072 and 0151 with a probe success rate 0.057 and 0.055, respectively); A sample aliquot with little or no tumor cells (0187). Unsupervised clustering was performed based on CpG sites that did not exhibit tissue-specific DNA methylation but acquired cancer-associated DNA hypermethylation. We used DNA methylation data from TCGA histologically normal tissues (720 samples from 22 different tissue types) and leukocytes (3 samples obtained using the sesameDataGet function in the R package sesameData). We selected 100,405 CpG sites that lacked tissue-specific DNA methylation (Methylated at < 10% frequency in any tissue type using a β-value of > 0.2 to define positive DNA methylation). To minimize the influence of variable tumor purity levels on a clustering result, we dichotomized the tumor DNA methylation data using a β-value of ≥ 0.3 to define positive DNA methylation and < 0.3 a lack of methylation. The dichotomization not only ameliorated the effect of tumor sample purity on the clustering but also removed a great portion of residual batch effects that are mostly reflected in small variations near the two ends of the range of β-values. For clustering analysis, we used 5,604 CpG sites that had data in more than 80% of the samples and were methylated in more than 5% of the tumors. We applied hierarchical clustering with Ward’s method to cluster the distance matrix computed with the Jaccard index. A heatmap was generated based on the original β-values.
QUANTIFICATION AND STATISTICAL ANALYSIS
Immune Cell Markers. Eighteen individual immune cell-type and cytokine scores were calculated as the median standardized gene expression of marker genes for each cell-type or cytokine.
Statistical analyses to compare immune profiles between ER (24 tumor types, 93 cases) and TCGA (6 tumor types, 35 cases) tumor samples were performed in the R statistical computing environment (R Core Team, 2018, see Key Resources Table).
The function HotellingsT2 (R package ICSNP,see Key Resources Table) assessed the overall difference in the mean 18-score immune profile for ER versus TCGA cases (see Table S3, and main text Figure 4A).
Statistical analyses to compare immune profiles between a subset of six tumor types represented in both ER (38 cases) and TCGA (35 cases) sets (Table S3, and Figure S5) was performed using the same Hotelling T2.
The two-factor multivariate analysis of variance (MANOVA) used the R functions lm and anova in the packages stats (incorporated into base R) applied to the data for the six overlapping tumor types to evaluate overall difference in the mean 18-score immune profile between ER and TCGA cases after adjustment for tumor type.
p-values for individual immune scores were adjusted for multiple testing (18 scores), controlling the false discovery rate (FDR) at a maximum of 10% using the method of Benjamini and Hochberg as implemented in the R function p.adjust.
Comparisons of mean score, for an individual score-type, unadjusted for tumor type were based on t-tests; each adjusted comparison was based on a likelihood ratio test in the context of a two-factor Gaussian linear model evaluating the significance of ER vs TCGA after adjustment for tumor type using the functions gls as implemented in R package nlme and anova in base R.
These methods are described in detail in the STAR Methods, “Evaluation of Immune Cell Markers.”
T-tests and ANOVA analyses are based on the normal distribution theory. We did not test the assumption of normality because even when the raw data are not exactly normally distributed, these analysis methods are very robust to departures from normality as long as the sample sizes are larger than approximately 30.
ADDITIONAL RESOURCES
Study Oversight
This trial, NCT02243592, was approved by the institutional review board/ethics committee at each site and conducted per Good Clinical Practice guidelines, defined by the International Conference on Harmonisation https://www.ich.org/. All patients provided written informed consent in accordance with Declaration of Helsinki principles, https://pubmed.ncbi.nlm.nih.gov/24141714/. Further details surrounding patient accrual are published, https://pubmed.ncbi.nlm.nih.gov/32339229/.
Supplementary Material
Highlights.
Genomics of 110 patients with exceptional response to therapy profiled
Plausible molecular mechanisms related to therapy identified in ~23% of cases
Proposed mechanisms involve DNA damage, signaling, and the immune response
Synthetic lethality with temozolomide in tumors with a defective DNA damage response
ACKNOWLEDGMENTS
Supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, and by the Center for Cancer Genomics, National Cancer Institute. We thank Ned Sharpless for his critique of the manuscript and advice. We thank Daniel Kraushaar, Rene Stainaker, and Ornella Tuekam of the Genomic and RNA Profiling Core Laboratory at Baylor College of Medicine for generating the NanoString data. We also Dong Zhang, currently at Knight Cancer Institute, Oregon Health Sciences University, for expert assistance with immunohistochemical staining of ER tumor samples for immune cell markers. This project has been funded in whole or in part with federal funds from the NCI, NIH, under grants and contracts. BCM was supported by grant no. 5U24CA143843 and then LEIDOS subcontract no. HHSN261200800001E/17X184TO1, Nationwide Children’s Hospital on LEIDOS subcontract no. HHSN261200800001E/14X242TO1, and then NCI contract no. HHSN261201700005I/TO1, IMS on NCI contract no. HHSN26120150002B/TO10, University of Chicago (Genomic Data Center) on NCI contract HHSN261200800001E/17X147TO2. Van Andel Institute was supported by NIH/NCI grant no. U24 CA210969, and University of North Carolina was supported by grant no. 5 U24 CA210988 and funding from the NIH (Program in Translational Medicine T32 GM122741).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.ccell.2020.10.015.
DECLARATION OF INTERESTS
P.W.L. is a member of the Scientific Advisory Board for AnchorDX. H.S.’s husband is part of the Scientific Advisory Board for AnchorDX. N.D. is employed by Foundation Medicine and holds Roche stock. A.J. was an employee of and had ownership interest in Foundation Medicine. V.M. was an employee of and had ownership interest in Foundation Medicine (through 12/31/2019) and is an equity shareholder of Mirati Therapeutics, Inc; on the Board of Directors, compensated by and an equity shareholder of Revolution Medicines, Inc.; a part-time employee and equity shareholder of EqRx (since March 2020); and holds patents with the USPO: 8501413, 8067175 (held by Sloan-Kettering Institute for Cancer Research and licensed to Molecular MD). All other authors declare no competing interests.
REFERENCES
- Al-Ahmadie H, Iyer G, Hohl M, Asthana S, Inagaki A, Schultz N, Hanrahan AJ, Scott SN, Brannon AR, McDermott GC, et al. (2014). Synthetic lethality in ATM-deficient RAD50-mutant tumors underlies outlier response to cancer therapy. Cancer Discov. 4, 1014–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, et al. (2000). Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 403, 503–511. [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bainbridge MN, Wang M, Wu Y, Newsham I, Muzny DM, Jefferies JL, Albert TJ, Burgess DL, and Gibbs RA (2011). Targeted enrichment beyond the consensus coding DNA sequence exome reveals exons with higher variant densities. Genome Biol. 12, R68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankhead P, Loughrey MB, Fernandez JA, Dombrowski Y, McArt DG, Dunne PD, McQuaid S, Gray RT, Murray LJ, Coleman HG, et al. (2017). QuPath: open source software for digital pathology image analysis. Sci. Rep 7, 16878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benci JL, Johnson LR, Choa R, Xu Y, Qiu J, Zhou Z, Xu B, Ye D, Nathanson KL, June CH, et al. (2019). Opposing functions of interferon coordinate adaptive and innate immune responses to cancer immune checkpoint blockade. Cell 178, 933–948 e914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, and Hochberg Y (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57, 289–300. [Google Scholar]
- Bhattacharya A, Hamilton AM, Furberg H, Pietzak E, Purdue MP, Troester MA, Hoadley KA, and Love MI (2020). An approach for normalization and quality control for NanoString RNA expression data. Brief Bioinform. 10.1093/bib/bbaa163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, Angell H, Fredriksen T, Lafontaine L, Berger A, et al. (2013). Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 39, 782–795. [DOI] [PubMed] [Google Scholar]
- Bullard JH, Purdom E, Hansen KD, and Dudoit S (2010). Evaluation of statistical methods for normalization and differential expression in mRNA-seq experiments. BMC Bioinformatics 11, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, Johansson I, Phung B, Harbst K, Vallon-Christersson J, et al. (2020). Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 577, 561–565. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, Network (2011). Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N., Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, Cooper LA, Rheinbay E, Miller CR, Vitucci M, et al. (2015). Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N. Engl. J. Med 372, 2481–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conley BA, Staudt L, Takebe N, Wheeler DA, Wang L, Cardenas MF, Korchina V, Zenklusen JC, McShane LM, Tricoli JV, et al. (2020). The Exceptional Responders initiative: feasibility of a National Cancer Institute pilot study. J. Natl. Cancer Inst 10.1093/jnci/djaa061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium APG (2017). AACR project GENIE: powering precision medicine through an international consortium. Cancer Discov. 7, 818–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosgrove CM, Cohn DE, Hampel H, Frankel WL, Jones D, McElroy JP, Suarez AA, Zhao W, Chen W, Salani R, et al. (2017). Epigenetic silencing of MLH1 in endometrial cancers is associated with larger tumor volume, increased rate of lymph node positivity and reduced recurrence-free survival. Gynecol. Oncol 146, 588–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danaher P, Warren S, Dennis L, D’Amico L, White A, Disis ML, Geller MA, Odunsi K, Beechem J, and Fling SP (2017). Gene expression markers of tumor infiltrating leukocytes. J. Immunother. Cancer 5, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dave SS, Wright G, Tan B, Rosenwald A, Gascoyne RD, Chan WC, Fisher RI, Braziel RM, Rimsza LM, Grogan TM, et al. (2004). Prediction of survival in follicular lymphoma based on molecular features of tumor-infiltrating immune cells. N. Engl. J. Med 351, 2159–2169. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S, and Sawyers CL (2001). Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med 344, 1031–1037. [DOI] [PubMed] [Google Scholar]
- Erasimus H, Gobin M, Niclou S, and Van Dyck E (2016). DNA repair mechanisms and their clinical impact in glioblastoma. Mutat. Res. Rev. Mutat. Res 769, 19–35. [DOI] [PubMed] [Google Scholar]
- Erson-Omay EZ, Caglayan AO, Schultz N, Weinhold N, Omay SB, Ozduman K, Koksal Y, Li J, Serin Harmanci A, Clark V, et al. (2015). Somatic POLE mutations cause an ultramutated giant cell high-grade glioma subtype with better prognosis. Neuro Oncol. 17, 1356–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng W, and Jasin M (2017). Homologous recombination and replication fork protection: BRCA2 and more! Cold Spring Harb Symp. Quant Biol. 82, 329–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa ME, Lugthart S, Li Y, Erpelinck-Verschueren C, Deng X, Christos PJ, Schifano E, Booth J, van Putten W, Skrabanek L, et al. (2010). DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell 17, 13–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, Cole CG, Ward S, Dawson E, Ponting L, et al. (2017). COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 45, D777–D783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garris CS, Arlauckas SP, Kohler RH, Trefny MP, Garren S, Piot C, Engblom C, Pfirschke C, Siwicki M, Gungabeesoon J, et al. (2018). Successful anti-PD-1 cancer immunotherapy requires T cell-dendritic cell crosstalk involving the cytokines IFN-gamma and IL-12. Immunity 49, 1148–1161.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greytak SR, Engel KB, Zmuda E, Casas-Silva E, Guan P, Hoadley KA, Mungall AJ, Wheeler DA, Doddapaneni HV, and Moore HM (2018). National Cancer Institute biospecimen evidence-based practices: harmonizing procedures for nucleic acid extraction from formalin-fixed, paraffin-embedded tissue. Biopreserv Biobank 16, 247–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith M, Griffith OL, Krysiak K, Skidmore ZL, Christopher MJ, Klco JM, Ramu A, Lamprecht TL, Wagner AH, Campbell KM, et al. (2016). Comprehensive genomic analysis reveals FLT3 activation and a therapeutic strategy for a patient with relapsed adult B-lymphoblastic leukemia. Exp. Hematol 44, 603–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. (2012). GENCODE: the reference human genome annotation for the ENCODE Project. Genome Res. 22, 1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell LH, Szankasi P, Roberts CJ, Murray AW, and Friend SH (1997). Integrating genetic approaches into the discovery of anticancer drugs. Science 278, 1064–1068. [DOI] [PubMed] [Google Scholar]
- Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, Yizhak K, Sade-Feldman M, Blando J, Han G, et al. (2020). B cells and tertiary lymphoid structures promote immunotherapy response. Nature 577, 549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollern DP, Xu N, Thennavan A, Glodowski C, Garcia-Recio S, Mott KR, He X, Garay JP, Carey-Ewend K, Marron D, et al. (2019). B cells and T follicular helper cells mediate response to checkpoint inhibitors in high mutation burden mouse models of breast cancer. Cell 179, 1191–1206.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman DM, Puzanov I, Subbiah V, Faris JE, Chau I, Blay JY, Wolf J, Raje NS, Diamond EL, Hollebecque A, et al. (2015). Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N. Engl. J. Med 373, 726–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isakoff SJ, Mayer EL, He L, Traina TA, Carey LA, Krag KJ, Rugo HS, Liu MC, Stearns V, Come SE, et al. (2015). TBCRC009: a multicenter phase II clinical trial of platinum monotherapy with biomarker assessment in metastatic triple-negative breast cancer. J. Clin. Oncol 33, 1902–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer G, Hanrahan AJ, Milowsky MI, Al-Ahmadie H, Scott SN, Janakiraman M, Pirun M, Sander C, Socci ND, Ostrovnaya I, et al. (2012). Genome sequencing identifies a basis for everolimus sensitivity. Science 338, 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RA, and Wichern DW (2007). Applied Multivariate Statistical Analysis, Sixth Edition (Prentice-Hall; ). [Google Scholar]
- Kim HS, and Seo HK (2018). Immune checkpoint inhibitors for urothelial carcinoma. Investig. Clin. Urol 59, 285–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim P, and Zhou X (2019). FusionGDB: fusion gene annotation DataBase. Nucleic Acids Res. 47, D994–D1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knijnenburg TA, Wang L, Zimmermann MT, Chambwe N, Gao GF, Cherniack AD, Fan H, Shen H, Way GP, Greene CS, et al. (2018). Genomic and molecular landscape of DNA damage repair deficiency across The Cancer Genome Atlas. Cell Rep 23, 239–254 e236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, and Wilson RK (2012). VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, Ou SH, Dezube BJ, Janne PA, Costa DB, et al. (2010). Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N. Engl. J. Med 363, 1693–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Rosa S, Uccella S, Finzi G, Albarello L, Sessa F, and Capella C (2003). Localization of vascular endothelial growth factor and its receptors in digestive endocrine tumors: correlation with microvessel density and clinicopathologic features. Hum. Pathol 34, 18–27. [DOI] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon MA, and Schlessinger J (2010). Cell signaling by receptor tyrosine kinases. Cell 141, 1117–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack SC, Witt H, Piro RM, Gu L, Zuyderduyn S, Stutz AM, Wang X, Gallo M, Garzia L, Zayne K, et al. (2014). Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 506, 445–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod G, Bozek DA, Rajakulendran N, Monteiro V, Ahmadi M, Steinhart Z, Kushida MM, Yu H, Coutinho FJ, Cavalli FMG, et al. (2019). Genome-wide CRISPR-Cas9 screens expose genetic vulnerabilities and mechanisms of temozolomide sensitivity in glioblastoma stem cells. Cell Rep 27, 971–986.e9. [DOI] [PubMed] [Google Scholar]
- McCarty KS Jr., Miller LS, Cox EB, Konrath J, McCarty KS Sr, and .. (1985). Estrogen receptor analyses. Correlation of biochemical and immunohistochemical methods using monoclonal antireceptor antibodies. Arch. Pathol. Lab Med 109, 716–721. [PubMed] [Google Scholar]
- McPherson A, Hormozdiari F, Zayed A, Giuliany R, Ha G, Sun MG, Griffith M, Heravi Moussavi A, Senz J, Melnyk N, et al. (2011). deFuse: an algorithm for gene fusion discovery in tumor RNA-seq data. Plos Comput. Biol 7, e1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meric-Bernstam F, Akcakanat A, Chen H, Do KA, Sangai T, Adkins F, Gonzalez-Angulo AM, Rashid A, Crosby K, Dong M, et al. (2012). PIK3CA/PTEN mutations and Akt activation as markers of sensitivity to allosteric mTOR inhibitors. Clin. Cancer Res 18, 1777–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitson M, Kelley LA, Sternberg MJ, Higgs DR, and Gibbons RJ (2011). Functional significance of mutations in the Snf2 domain of ATRX. Hum. Mol. Genet 20, 2603–2610. [DOI] [PubMed] [Google Scholar]
- Montaldi AP, Godoy PR, and Sakamoto-Hojo ET (2015). APE1/REF-1 down-regulation enhances the cytotoxic effects of temozolomide in a resistant glioblastoma cell line. Mutat. Res. Genet. Toxicol. Environ. Mutagen 793, 19–29. [DOI] [PubMed] [Google Scholar]
- Moran S, Arribas C, and Esteller M (2016). Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences. Epigenomics 8, 389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muntasell A, Rojo F, Servitja S, Rubio-Perez C, Cabo M, Tamborero D, Costa-Garcia M, Martinez-Garcia M, Menendez S, Vazquez I, et al. (2019). NK cell infiltrates and HLA class I expression in primary HER2(+) breast cancer predict and uncouple pathological response and disease-free survival. Clin. Cancer Res 25, 1535–1545. [DOI] [PubMed] [Google Scholar]
- Patel SP, and Kurzrock R (2015). PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol. Cancer Ther 14, 847–856. [DOI] [PubMed] [Google Scholar]
- Petitprez F, de Reynies A, Keung EZ, Chen TW, Sun CM, Calderaro J, Jeng YM, Hsiao LP, Lacroix L, Bougouin A, et al. (2020). B cells are associated with survival and immunotherapy response in sarcoma. Nature 577, 556–560. [DOI] [PubMed] [Google Scholar]
- R Core Team (2018). R: A Language and Environment for Statistical Computing, Version 3.5.1 (R Foundation for Statistical Computing) [Google Scholar]
- Ramos AH, Lichtenstein L, Gupta M, Lawrence MS, Pugh TJ, Saksena G, Meyerson M, and Getz G (2015). Oncotator: cancer variant annotation tool. Hum. Mutat 36, E2423–E2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond E, Dahan L, Raoul JL, Bang YJ, Borbath I, Lombard-Bohas C, Valle J, Metrakos P, Smith D, Vinik A, et al. (2011). Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N. Engl. J. Med 364, 501–513. [DOI] [PubMed] [Google Scholar]
- Risso D, Ngai J, Speed TP, and Dudoit S (2014). Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol 32, 896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rokita JL, Rathi KS, Cardenas MF, Upton KA, Jayaseelan J, Cross KL, Pfeil J, Egolf LE, Way GP, Farrel A, et al. (2019). Genomic profiling of childhood tumor patient-derived xenograft models to enable rational clinical trial design. Cell Rep 29, 1675–1689.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayers EW, Agarwala R, Bolton EE, Brister JR, Canese K, Clark K, Connor R, Fiorini N, Funk K, Hefferon T, et al. (2019). Database resources of the national center for biotechnology information. Nucleic Acids Res. 47, D23–D28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata A, Moiani D, Arvai AS, Perry J, Harding SM, Genois MM, Maity R, van Rossum-Fikkert S, Kertokalio A, Romoli F, et al. (2014). DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol. Cell 53, 7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, et al. (2001). Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med 344, 783–792. [DOI] [PubMed] [Google Scholar]
- Stracker TH, and Petrini JH (2011). The MRE11 complex: starting from the ends. Nat. Rev. Mol. Cell Biol 12, 90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temko D, Van Gool IC, Rayner E, Glaire M, Makino S, Brown M, Chegwidden L, Palles C, Depreeuw J, Beggs A, et al. (2018). Somatic POLE exonuclease domain mutations are early events in sporadic endometrial and colorectal carcinogenesis, determining driver mutational landscape, clonal neoantigen burden and immune response. J. Pathol 245, 283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triche TJ Jr., Weisenberger DJ, Van Den Berg D, Laird PW, and Siegmund KD (2013). Low-level processing of Illumina Infinium DNA methylation BeadArrays. Nucleic Acids Res. 41, e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner KM, Deshpande V, Beyter D, Koga T, Rusert J, Lee C, Li B, Arden K, Ren B, Nathanson DA, et al. (2017). Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 543, 122–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Abegg D, Hoch DG, and Adibekian A (2016). Chemoproteomics-enabled discovery of a potent and selective inhibitor of the DNA repair protein MGMT. Angew. Chem. Int. Ed. Engl 55, 2911–2915. [DOI] [PubMed] [Google Scholar]
- Wang L, Ni X, Covington KR, Yang BY, Shiu J, Zhang X, Xi L, Meng Q, Langridge T, Drummond J, et al. (2015). Genomic profiling of Sezary syndrome identifies alterations of key T cell signaling and differentiation genes. Nat. Genet 47, 1426–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein IB (2002). Cancer. Addiction to oncogenes—the Achilles heal of cancer. Science 297, 63–64. [DOI] [PubMed] [Google Scholar]
- Wilson DM 3rd, and Simeonov A (2010). Small molecule inhibitors of DNA repair nuclease activities of APE1. Cell Mol Life Sci 67, 3621–3631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Turner KM, Nguyen N, Raviram R, Erb M, Santini J, Luebeck J, Rajkumar U, Diao Y, Li B, et al. (2019). Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature 575, 699–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L, Bulgar A, Miao Y, Mahajan V, Donze JR, Gerson SL, and Liu L (2007). Combined treatment with temozolomide and methoxyamine: blocking apurininc/pyrimidinic site repair coupled with targeting topoisomerase IIalpha. Clin. Cancer Res 13, 1532–1539. [DOI] [PubMed] [Google Scholar]
- Ye K, Schulz MH, Long Q, Apweiler R, and Ning Z (2009). Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25, 2865–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Laird PW, and Shen H (2017). Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res. 45, e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Triche TJ Jr., Laird PW, and Shen H (2018). SeSAMe: reducing artifactual detection of DNA methylation by Infinium BeadChips in genomic deletions. Nucleic Acids Res. 46, e123. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All DNA and RNA sequence BAM files, DNA methylation files, mutation data, NanoString expression tables, and clinical information gathered in this study are available in the NCI, Genomic Data Commons under project ID: EXCEPTIONAL_RESPONDERS-ER available at https://gdc.cancer.gov/about-data/publications/exceptional_responders_2020. The Exceptional Responders project is registed in dbGaP under accession number Phs001145.v1.p1 at https://www.ncbi.nlm.nih.gov/gap/.
All computer code used in this study is available in the public domain or commercially available as noted in the Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat monoclonal Anti-CD3 | Bio Rad | MCA1477 |
| Rabbit monoclonal Anti-CD8 | Abcam | Ab101500 |
| Rabbit monoclonal Anti-CD45 | Cell Signaling Technology | 13917 |
| Rabbit monoclonal. Anti-PDL1 | Abcam | B228462 |
| Mouse monoclonal Anti PD-1 | Abcam | Ab52587 |
| Rabbit polyclonal Anti-cytokeratin, wide spectrum | DAKO | Z0622 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Critical Commercial Assays | ||
| Library Amplification Readymix: with KAPA HiFi DNA Polymerase | Kapa Biosystems, Inc., | Cat# KK2612 |
| Agencourt AMPure XP beads | Beckman Coulter | A63882 |
| SeqCap EZ HGSC VCRome, 96 Reactions | Roche NimbleGen | Cat# (06465676001) |
| xGen Blocking Oligos | Integrated DNA technologies | Cat# 1016184, Cat#1016186 |
| TruSeq RNA Exome (previously marketed as TruSeq RNA Library Prep for Enrichment (Cat. No. 20020189), TruSeq RNA Enrichment (Cat. No. 20020490), Exome Panel (Cat. No. 20020183) | Illumina | 20020490 |
| TruSeq Rapid SBS Kit-HS (200 cycles) | Illumina | FC-402-4001 |
| TruSeq SBS kit v3-HS (200 cycles) | Illumina | FC-401-3001 |
| HiSeq Rapid SBS Kit v2 (200 cycles) | Illumina | FC-402-4021 |
| XT_HS_IO360_CSO + Standard | NanoString | 115000222 |
| Low RNA Input Kit | NanoString | 1220000219 |
| XT_IO360 Primers | NanoString | 115000219 |
| Deposited Data | ||
| TCGA data sets | TCGA legacy archive | https://portal.gdc.cancer.gov/legacy-archive/ |
| RNAseq BAM files | This paper | Phs001145.v1.p1 at https://www.ncbi.nlm.nih.gov/gap/ and EXCEPTIONAL_RESPONDERS-ER at https://portal.gdc.cancer.gov/ |
| DNA BAM files | This paper | Phs001145.v1.p1 at https://www.ncbi.nlm.nih.gov/gap/ and EXCEPTIONAL_RESPONDERS-ER at https://portal.gdc.cancer.gov/ |
| Mutation tables for SNV and indels, and for CNA | This paper | Phs001145.v1.p1 at https://www.ncbi.nlm.nih.gov/gap/ and EXCEPTIONAL_RESPONDERS-ER at https://portal.gdc.cancer.gov/ |
| DNA Methylation IDAT files | This paper | Phs001145.v1.p1 at https://www.ncbi.nlm.nih.gov/gap/ and EXCEPTIONAL_RESPONDERS-ER at https://portal.gdc.cancer.gov/ |
| NanoString expression tables | This Paper | Phs001145.v1.p1 at https://www.ncbi.nlm.nih.gov/gap/ and EXCEPTIONAL_RESPONDERS-ER at https://portal.gdc.cancer.gov/ |
| Immunohistochemistry immune cell type quantification (see also Supplementary Table 4) | This paper | Phs001145.v1.p1 at https://www.ncbi.nlm.nih.gov/gap/ and EXCEPTIONAL_RESPONDERS-ER at https://portal.gdc.cancer.gov/ |
| Clinical information | This paper | Phs001145.v1.p1 at https://www.ncbi.nlm.nih.gov/gap/ and EXCEPTIONAL_RESPONDERS-ER at https://portal.gdc.cancer.gov/ |
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| RNAscope 2.5 LS Probe - Hs-IFNG-C3 | Advanced Cell Diagnostics | Cat# 310508-C3 |
| Recombinant DNA | ||
| Software and Algorithms | ||
| R v3.6. 2006 | R Development Core Team | https://www.r-project.org |
| function HotellingsT2 in R package ICSNP, v1.1-1 | Nordhausen K, Sirkia S, Oja H, Tyler DE, 2018 | https://CRAN.R-project.org/package=ICSNP |
| functions anova, lm, p.adjust, and t.test in the R package stats (incorporated into base R, v3.6) | R Development Core Team | https://www.r-project.org |
| function gls in R package nlme, v3.1-137 | Pinheiro J, Bates D, DebRoy S, Sarkar D, R Development Core Team, 2018 | https://CRAN.R-project.org/package=nlme |
| Function RUVr in RUVseq package v1.22.0 | PMID: 25150836 | https://www.bioconductor.org/packages/release/bioc/html/RUVSeq.html |
| Function varianceStabilizingTransformation in DESeq2 Bioconductor package v1.28.1 | PMID: 25516281 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Function betweenLaneNormalization in EDAseq Bioconductor package v2.22.0 | PMID: 22177264 | https://bioconductor.org/packages/release/bioc/html/EDASeq.html |
| SeSAMe v1.3.2 | PMID: 30085201 | https://www.bioconductor.org/packages/release/bioc/html/sesame.html |
| Other | ||