

Introduction
Choroideremia is a rare X-linked recessive retinal degeneration of photoreceptors, retinal pigment epithelium (RPE), and choroid that progresses in a centripetal manner.1 Choroideremia results from pathologic genetic variants of the CHM gene on the X chromosome with nearly all being loss-of-function null mutations.1 The CHM gene product, Rab escort protein 1 (REP-1), is expressed in all human cells and plays an important role in intracellular protein trafficking.2 REP has 2 isoforms, REP-1 and REP-2, where the autosomal inherited REP-2 can compensate for REP-1 deficiency in most tissues except in the RPE, photoreceptors, and choroid.3
All males harboring disease-causing CHM mutations are affected with progressive retinal degeneration from early childhood. A family history of X-linked retinal degeneration may or may not be present. In contrast, the vast majority of female carriers of CHM mutations are asymptomatic although female carriers may show patchy fundus pigmentation due to mosaicism, where 1 X chromosome is randomly inactivated in each cell in fetal development.4 Depending on the proportion of retinal cells that contain the active mutant gene, female carriers may rarely exhibit severe retinal degeneration. The estimated prevalence of males with choroideremia ranges from 1 in 100,000 to 200,000 persons, and choroideremia may be underdiagnosed because of overlapping symptoms with retinitis pigmentosa (RP) and other retinal dystrophies.1,5
Clinical Assessment
On initial presentation in choroideremia, peripheral visual field loss with corresponding areas of degeneration of the choroid and retina along with a normal-appearing fovea are typically found. The areas of chorioretinal degeneration are typically demarcated. A few islands of hyperpigmentation representing RPE hyperplasia may be present.
Choroideremia is often misdiagnosed as RP given similar symptoms of night visual impairment and early peripheral visual loss. However, choroideremia has relatively demarcated areas of chorioretinal atrophy with mild retinal vascular attenuation while RP typically has diffuse peripheral retinal degeneration with severe vascular attenuation. Choroideremia may have rare islands of hyperpigmentation, and in contrast a hallmark of RP is the formation of pigmentary clumping (“bone spicules” appearance) likely in part related to RPE migration along retinal vessels toward the inner retina reflecting capillary network at the time of formation. Mild optic disc pallor may develop in advance stage of choroideremia whereas severe diffuse optic disc pallor is common in advanced RP.6
Genetic testing to include detection of CHM (REP-1) gene mutations is essential to confirm the diagnosis of choroideremia. Chorioretinal degeneration mimicking choroideremia may occur in a variety of other IRD genotypes including gyrate atrophy and C2orf71-associated inherited retinal disease as well as other IRDs.7
A combination of optical coherence tomography (OCT) and fundus autofluorescence (FAF) identifies areas of intact RPE and photoreceptor layers and are helpful to determine choroideremia severity and disease progression. Microperimetry, preferably performed after partial dark adaptation, measures retinal sensitivities at specific loci and determines visual function of the viable retina as well as the retinal fixation point. In areas of end-stage degeneration, OCT reveals complete loss of RPE and choroid with residual retinal tissue. In areas of active degeneration, OCT shows areas of thinning of the retinal layer with loss of ellipsoid zone (EZ) layer, outer retinal tubulations, decreased RPE reflectance, and choroidal thinning. Some patients may develop cystoid macular edema and rarely schisis of the retina or choroidal neovacular membrane. The FAF visualizes the remaining viable retina where areas of intact RPE layer are hyperfluorescent. The remaining islands with preserved RPE appear as areas with normal or increased autofluorescence intensity due to lipofuscin accumulation while areas with RPE atrophy are characterized by hypoautofluorescence. Thus FAF intensity is a good indicator for lipofuscin content and RPE integrity.
Natural History
Males with choroideremia typically experiences subtle nyctalopia starting in childhood with progressive peripheral areas of visual field loss from degeneration of the choroid and retina that gradually coalesce and progress toward the macula. Given these early symptoms are insidious, children and young adolescent males with choroideremia are mostly asymptomatic. Most patients are diagnosed in their late teens or 20s when the central island of vision becomes constricted to <30 degrees with progressively fewer islands of peripheral vision, resulting in difficulties with activities of daily living. Central visual acuity is typically unaffected or mildly affected until advanced stages of the disease when chorioretinal atrophy encroaches into the fovea. Variability of disease severity occurs among affected persons; however, in general, severe visual acuity loss is present by middle age and complete loss of vision can occur in some patients in later life.
Prospective longitudinal natural history studies of choroideremia are scarce. The 2-year, prospective, multicenter, observational study (NIGHT study: NSR-CHM-OS1, NCT03359551) is the largest and examines over 300 adult males with genetically confirmed choroideremia and active disease visible within the macula with measurable best-corrected visual acuity (BCVA) in at least one eye using the Early Treatment of Diabetic Retinopathy Study (ETDRS) letter chart.8,9 Preserved area of EZ, preserved area of autofluorescence (PAF), microperimetry, contrast sensitivity, color vision, and reading-speed tests were assessed every 4 months over a period of 20 months. Participants slowly lost BCVA at ~0.5 letters annually. However, older patients with more advanced disease may experience greater decline (Lam et al,10 IOVS 2021;62;ARVO E-Abstract 3545798). PAF area, preserved EZ area, and mean retinal sensitivity on microperimetry consistently decreased at each visit and were generally symmetric. In contrast, bilateral asymmetry for BCVA is evident with a mean inter-eye difference of nearly 20 ETDRS letters with only 26% of individuals showed <5-letter differences between right eye and left eye. This study reinforces the importance of BCVA, preserved EZ area, and PAF as measures of natural CHM progression and the necessity of long-term follow-up of individuals with choroideremia for understanding disease progression and its response to treatment.
A meta-analysis of visual acuity in eyes with choroideremia from 23 studies not including the NIGHT study showed BCVA as a function of age followed a 2-phase decline of slow followed by rapid decline, with an estimated transition age of 39 years.11 Similarly to the NIGHT study, BCVA differences between the right eye and left eye increased as BCVA worsened owing to differences of foveal involvement between the 2 eyes with advance disease.
Gene Therapy Rationale and Goal
The goals of the macular-targeted subretinal gene therapy for choroideremia are to slow down the progressive chorioretinal degeneration in the macular region and to improve the function of the remaining viable retina in the macular region. Treatment of the residual peripheral islands of functional retina at present is not feasible. Given the gene therapy targets are the photoreceptors and the RPE, subretinal gene therapy injection is the most direct method to deliver the adeno-associated viral (AAV) vector encoding REP1 (AAV-REP1).
Given the small size of the CHM protein coding sequence (1.9 kb) and the preservation of the macular region until advance stages of choroideremia, gene therapy using an AAV-REP1 injected subretinally and targeting the macular region is quite feasible. The AAV2 expression cassette used in subretinal clinical trials to date consists of a chicken β actin promoter, shown to have long-term transduction of the RPE in the approved gene therapy for RPE65-associated Leber congenital amaurosis12 and include a Woodchuck hepatitis virus posttranslational regulatory element (WPRE) downstream of the CHM cDNA encoding REP1. WPRE is known to enhance AAV-mediated transgene expression.13
Gene Therapy Planning, Surgery, and Perioperative Care
Preoperative planning consists of determining the treatment target zone, preferred injection sites, and preexisting areas of severe retinal thinning which may lead to a retinal hole formation. FAF allows determination of treatment zone by mapping viable hyperfluorescent RPE areas onto the color photo of the macula using vascular landmarks and selection of preferred injection sites near conspicuous vascular landmarks (Figs. 1A, B). Selecting potential injection sites nearby the preserved RPE may facilitate entering subretinal space while reducing a possibility of suprachoroidal injection. Microperimetry helps to determine the central visual field and preferred retinal locus of fixation (Fig. 1C), which may be eccentric to the fovea in advanced degeneration and should be included in the treatment zone. OCT allows foveal assessment and localization (Fig. 1D) as well as review of the retinal morphology and choroidal thickness near the potential injection sites to avoid areas of excessive atrophy, epiretinal membranes, or thick nerve fiber layer.14,15 Injection site at least 2 mm from the fovea is preferred since injecting closer increases foveal stretch and the risk of macular hole formation.
Figure 1.
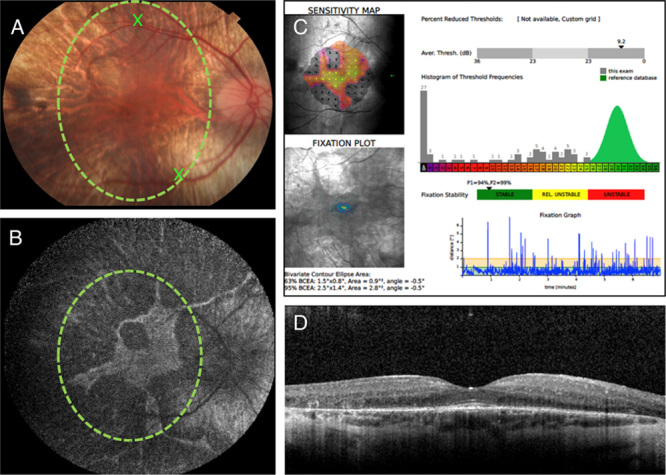
A, A 30-degree color photograph of the right eye of a 25-year-old male with genetically proven choroideremia. The green circle shows treatment target zone based on the preserved RPE areas seen on the fundus autofluorescence shown in (B). The 2 Xs indicate 2 potential injection sites located near the retinal vascular landmarks, at least 3 mm away from the fovea. B, A 55-degree fundus autofluorescence image showing hyperfluorescent area of preserved RPE and retina. The green circle shows the treatment target zone, which was mapped to the fundus color photo shown in (A), using vascular landmarks. C, Microperimetry image showing fundus sensitivity map, fixation plot, the average threshold value (dB), and fixation stability plot. D, Optical coherence tomography through the fovea shows thin outer retinal layer with retinal tubulation temporally, preserved subfoveal outer retinal layers and retinal pigment epithelium, and thin choroid.
Surgery consists of pars plana vitrectomy; elevating adherent posterior hyaloid; creating a subretinal prebleb with balanced salt solution (BSS) to assure fluid propagates toward the fovea; injecting the viral vector into the BSS prebleb through the same injection hole in order to minimize vector reflux; irrigating the vitreous cavity with BSS to remove refluxed virus; and suturing all sclerotomies to avoid postoperative hypotony.14,15 Surgeon’s goal is to deliver the virus into subretinal space over the treatment zone with minimal reflux while avoiding suprachoroidal injection, excessive foveal stretch or dehiscence. A larger BSS prebleb is typically needed to detach the tightly adherent fovea. Intraoperative microscope-integrated OCT is an essential tool that allows to closely watch the fovea, monitor tissue planes during BSS and the virus injections, and assess the extent of the delivered subretinal treatment.15 If a macular hole is seen intraoperatively after some or all virus has been delivered, macular hole surgery will likely cause the treatment to fail. Inner limiting membrane peeling and gas injection will likely squeeze the virus out of surbretinal space, thus further manipulation should be avoided. The hole may close spontaneously and repair can be performed as needed at a later date.16 On the other hand, peripheral retinal breaks or schisis areas should be treated intraoperatively to minimize the risk of retinal detachment.16 Immediately after surgery, the patient is maintained on his back for 2 hours to facilitate virus absorption without bleb migration. Subretinal fluid typically disappears by the next day or by 1-week follow-up.
Postoperative care consists of monitoring patients for intraocular inflammation, which may occur within the anterior chamber, vitreous cavity or subretinal space and may lead to vision loss if not treated promptly. To prevent inflammation oral prednisone is administered for a total of 21 days starting at 1 mg/kg/d, up to 80 mg, 2 days before surgery with a taper after a total of 10 days on the high dose. Routine topical corticosteroids are prescribed with a taper adjusted to patient’s response. Patients should be observed closely for prolonged or recurrent inflammation which may manifest itself as decreased microperimetry response, recurrent anterior chamber or vitreous cells, cystoid macular edema, subretinal hyperreflective densities, and thickened choroid. Patients should be managed with prompt administration of topical, subtenon, intravitreal or oral steroids.
Gene Therapy Results
Approved treatment for choroideremia is currently not yet available although clinical trials to treat male choroideremia patients have been well under way. The first human phase 1 gene therapy clinical trial in choroideremia was reported in 2014 (NCT01461213).17 MacLaren and colleagues administered AAV2-REP1 1010 genome particles (gp) in 0.1 mL subfoveally in 1 eye of 6 patients with advanced disease. Two patients with poor BCVA at baseline gained 11 and 21 letters in the treated eyes at 6 months posttreatment and the improvement was sustained at 3.5 years of follow-up.18 Baseline BCVA at baseline was good in 3 patients and was maintained in the treated eyes at 3.5 years posttreatment.18 One patient had a surgical complication leading to a lower dose of vector and had decline in BCVA from 6 months to 3.5 years likely due to degeneration in the fovea.18
Subsequently, phase 2 open-label gene therapy clinical trials using the same AAV-REP1 construct were conducted in Miami, USA (NCT02553135),19 Tubingen, Germany (NCT02671539),20 and Edmonton, Canada (NCT02077361)21 with 1011 gp in 0.1 mL administered subfoveally in one eye of each patient with an inclusion criteria of baseline BCVA of 78 to 34 letters (Snellen equivalent 20/32 to 20/300 logMAR 0.14 to 1.02). Taken together, the results from the phase 1 and 2 clinical trials showed a favorable safety profile with few serious adverse events including 2 cases of retinal hole over nonseeing retina19 and 1 case of localized intraretinal immune response associated with a small amount of subretinal, intraretinal, and vitreous hemorrhage associated with several air bubbles up to 1000 mm in diameter injected into the subretinal space.21 Given the primary outcome measure was improvement of BCVA in the treated eye, some of the treated eyes achieved BCVA improvement up to >15 ETDRS letters with maintenance of the BCVA gain up to 12 months (Nightstar Topline Results). There was no difference detected in the rate of decline of preserved FAF area in treated and control eyes over the study period of 24 months although subfoveal choroidal thickness which is associated with better BCVA declined at a lower rate in the treated versus control eyes from baseline (−5% vs. −24%, respectively).20
Clinical trials using the same AAV2-REP1 vector [Nightstar/Biogen timrepigene emparvovec (BIIB111/AAV2-REP1)] include the GEMINI open-label phase 2 trial to assess safety of bilateral, sequential administration (NCT03507686) and the STAR phase 3 trial to assess efficacy and safety with unilateral randomization to low dose, high dose, and control treatment groups (NCT03496012). The phase 3 STAR trial did not meet its primary endpoint of proportion of participants with a ≥15 ETDRS letter improvement from baseline in BCVA at month 12 in the interventional group in comparison to the noninterventional control group and did not demonstrate efficacy on key secondary endpoints (Biogen topline results June 14, 2021, http://media.biogen.com/news-releases/news-release-details/biogen-announces-topline-results-phase-3-gene-therapy-study), Safety results from the phase 3 STAR study were consistent with previous studies. Of interest, Spark Therapeutics (Philadelphia, PA) initiated a phase 1/2 study of unilateral subretinal administration of AAV2-hCHM vector in subjects with choroideremia and BCVA of better than 20/200 in the study eye (NCT02341807). In addition, 4D molecular therapeutics has initiated a dose escalating phase 1 trial of unilateral intravitreal administration of AAV capsid variant (4D-100) carrying a transgene encoding a codon-optimized human CHM gene in subjects with choroideremia and BCVA of better than 20/200 in the study eye (NCT04483440). Initial clinical safety data at both of the 2 dose levels in the 4D molecular therapeutics trial indicate that 4D-110 was well-tolerated and did not result in any dose-limiting toxicity (n=6; all patients followed between 1 and 9 mo) (4D Molecular Therapeutics topline results June 24, 2021, https://ir.4dmoleculartherapeutics.com/news-releases/news-release-details/4d-molecular-therapeutics-announces-rare-disease-ophthalmology).
Gene Therapy Future
Given the gene therapy results as well as the progressive nature of the chorioretinal atrophy in choroideremia and the significant unmet need, slowing down the disease progression may be more of an attainable treatment goal than improvement in visual function. Macular-targeted subretinal gene therapy for choroideremia improves BCVA in some treated eyes and the improvement may continue at least up to a few years. However, the phase 3 STAR trial did not meet its primary endpoint of proportion of participants with a ≥15 ETDRS letter improvement from baseline in BCVA at month 12 in the interventional group in comparison to the noninterventional control group and did not demonstrate efficacy on key secondary endpoints. Whether and to what extent macular subretinal gene therapy for choroideremia may slow down disease progression in the macular region requires further study. Intravitreal gene therapy for choroideremia is in early clinical trial testing and its safety and efficacy remain to be determined.
Footnotes
Supported by James V. Bastek, M.D. Hereditary Retinal Disease Research Program, Bascom Palmer Eye Institute, Research to Prevent Blindness Unrestricted Award.
B.L.L., J.L.D., N.Z.G. have received clinical trial funding from Biogen. B.L.L. is a consultant for Biogen.
Contributor Information
Byron L. Lam, Email: blam@med.miami.edu.
Janet L. Davis, Email: jdavis@med.miami.edu.
Ninel Z. Gregori, Email: NGregori@med.miami.edu.
References
- 1.Coussa RG, Traboulsi EI. Choroideremia: a review of general findings and pathogenesis. Ophthalmic Genet. 2012;33:57–65. [DOI] [PubMed] [Google Scholar]
- 2.Gordiyenko NV, Fariss RN, Zhi C, et al. Silencing of the CHM gene alters phagocytic and secretory pathways in the retinal pigment epithelium. Invest Ophthalmol Vis Sci. 2010;51:1143–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang AY, Mysore N, Vali H, et al. Choroideremia is a systemic disease with lymphocyte crystals and plasma lipid and rbc membrane abnormalities. Invest Ophthalmol Vis Sci. 2015;56:8158–8165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Syed R, Sundquist SM, Ratnam K, et al. High-resolution images of retinal structure in patients with choroideremia. Invest Ophthalmol Vis Sci. 2013;54:950–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bertelsen M, Jensen H, Bregnhøj JF, et al. Prevalence of generalized retinal dystrophy in denmark. Ophthalmic Epidemiol. 2014;21:217–223. [DOI] [PubMed] [Google Scholar]
- 6.Jaissle GB, May CA, van de Pavert SA, et al. Bone spicule pigment formation in retinitis pigmentosa: insights from a mouse model. Graefes Arch Clin Exp Ophthalmol. 2010;248:1063–1070. [DOI] [PubMed] [Google Scholar]
- 7.Gerth-Kahlert C, Tiwari A, Hanson JV, et al. C2orf71 mutations as a frequent cause of autosomal-recessive retinitis pigmentosa: clinical analysis and presentation of 8 novel mutations. Invest Ophthalmol Vis Sci. 2017;58:3840–3850. [DOI] [PubMed] [Google Scholar]
- 8.Hariri AH, Velaga SB, Girach A, et al. Measurement and reproducibility of preserved ellipsoid zone area and preserved retinal pigment epithelium area in eyes with choroideremia. Am J Ophthalmol. 2017;179:110–117. [DOI] [PubMed] [Google Scholar]
- 9.Hariri AH, Ip MS, Girach A, et al. Macular spatial distribution of preserved autofluorescence in patients with choroideremia. Br J Ophthalmol. 2019;103:933–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lam BL, MacLaren RE, Fischer MD, et al. NIGHT study: natural progression of choroideremia. Association for Research in Vision and Ophthalmology Annual Meeting, Virtual, 2021. Invest Ophthalmol Vis Sci. 2021;62:3545798. [Google Scholar]
- 11.Shen LL, Ahluwalia A, Sun M, et al. Long-term natural history of visual acuity in eyes with choroideremia: a systematic review and meta-analysis of data from 1004 individual eyes. Br J Ophthalmol. 2021;105:271–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maguire AM, Simonelli F, Pierce EA, et al. Safety and efficacy of gene transfer for leber’s congenital amaurosis. N Engl J Med. 2008;358:2240–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loeb JE, Cordier WS, Harris ME, et al. Enhanced expression of transgenes from adeno-associated virus vectors with the woodchuck hepatitis virus posttranscriptional regulatory element: implications for gene therapy. Hum Gene Ther. 1999;10:2295–2305. [DOI] [PubMed] [Google Scholar]
- 14.Davis JL, Gregori NZ, MacLaren RE, et al. Surgical technique for subretinal gene therapy in humans with inherited retinal degeneration. Retina. 2019;39:S2–S8. [DOI] [PubMed] [Google Scholar]
- 15.Gregori NZ, Lam BL, Davis JL. Intraoperative use of microscope-integrated optical coherence tomography for subretinal gene therapy delivery. Retina. 2019;39:S9–S12. [DOI] [PubMed] [Google Scholar]
- 16.Gregori NZ, Davis JL. Surgical observations from the first 120 cases of subretinal gene therapy for inherited retinal degenerations. Retina. 2020. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 17.MacLaren RE, Groppe M, Barnard AR, et al. Retinal gene therapy in patients with choroideremia: initial findings from a phase 1/2 clinical trial. Lancet. 2014;383:1129–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Edwards TL, Jolly JK, Groppe M, et al. Visual acuity after retinal gene therapy for choroideremia. N Engl J Med. 2016;374:1996–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lam BL, Davis JL, Gregori NZ, et al. Choroideremia gene therapy phase 2 clinical trial: 24-month results. Am J Ophthalmol. 2019;197:65–73. [DOI] [PubMed] [Google Scholar]
- 20.Fischer MD, Ochakovski GA, Beier B, et al. Efficacy and safety of retinal gene therapy using adeno-associated virus vector for patients with choroideremia: a randomized clinical trial. JAMA Ophthalmol. 2019;137:1247–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dimopoulos IS, Hoang SC, Radziwon A, et al. Two-year results after aav2-mediated gene therapy for choroideremia: The Alberta experience. Am J Ophthalmol. 2018;193:130–142. [DOI] [PubMed] [Google Scholar]