

Introduction
Leber hereditary optic neuropathy (LHON) is a rare, blinding, maternally inherited mitochondrial genetic disease in need of effective treatment. LHON is a nonsyndromic optic neuropathy affecting the retinal ganglion cells (RGCs), whose axons form the optic nerve and extend into the brain via the optic chiasm and optic tracts. The physiopathology of LHON is characterized by selective loss of RGCs and their axons, which leads to rapidly progressive bilateral central vision loss.
Molecular Genetics and Disease Mechanisms
LHON was the first identified inherited human disease associated with point mutations in mitochondrial DNA (mtDNA)1 and it is considered the most common mitochondrial genetic disorder.2 Three primary point mutations are responsible for LHON in ~90% of subjects: m.3460G>A, m.11778G>A and m.14484T>C, respectively in the MT-ND1, MT-ND4 and MT-ND6 mitochondrial genes. These genes code for 3 subunits of the nicotinamide adenine dinucleotide: ubiquinone oxidoreductase, complex I of the mitochondrial respiratory chain. Mutations in these subunits of complex I ultimately impair ATP synthesis through oxidative phosphorylation and increase the production of reactive oxygen species. This combination may lead vulnerable cells like RGCs, under triggering conditions, to cross the threshold for apoptosis.3 RGCs appear to be selectively vulnerable to mitochondrial dysfunction possibly due to their peculiar neuronal architecture, as their intra-retinal axons remain unmyelinated, and other metabolic factors.4,5 This vulnerability may trigger a catastrophic wave of degeneration that manifests clinically with acute or rapidly progressive painless bilateral central vision loss, either simultaneously or sequentially in the 2 eyes.6
Clinical Features and Natural History
The prevalence of LHON has been estimated at between 1 in 30,000 to 1 in 50,000 in Northern Europe.4,7,8 The mtDNA mutations are necessary, but not sufficient to cause vision loss, and there is a well-documented incomplete penetrance. It is estimated that ~25% to 50% of males and up to 10% of females who carry one of the primary point mutations will manifest the clinical disease.9
The mitochondrial genotype is the most significant prognostic factor of visual outcome, followed by age at onset of vision loss. Variable visual recovery rates have been reported depending on the criteria used, ranging from 4% to 25% for the m.11778G>A mutation to 22% to 25% for the m.3460G>A mutation, and 37% to 1% for the m.14484T>C mutation.9,10 The m.11778G>A MT-ND4 mutation accounts for about 75% of LHON in North America and Europe,5,11 and an even higher proportion among Asian countries,12 and it is known to cause a severe clinical form of LHON.13 Furthermore, it is well established that the natural history of the disease is different for young-onset patients, especially if they are below the age of 12 years.10,14,15 In a recent meta-analysis focusing on ND4-LHON patients aged at least 15 years at onset of vision loss, only 11.3% showed some spontaneous visual recovery, although the definitions used for recovery varied among studies.13
Asymptomatic carriers of the m.11778G>A MT-ND4 mutation have normal visual function before the expression of the disease.16,17 When triggered, LHON classically manifests as subacute, bilateral, painless central vision loss. The bilateral vision loss is most often sequential, with 1 eye affected first, followed by the second eye within about 6 to 8 weeks after onset in the first eye.9–11 However, 25% to 50% of patients report bilateral involvement at initial presentation.9,11 This likely reflects both true instances of simultaneous bilateral onset, but also cases in which the patient is unaware of vision loss in the first eye until the fellow eye becomes involved.18
Clinical manifestation in the first eye is essentially predictive of bilateral involvement,10,11 and occurrences of long-term unilateral involvement have been very rarely reported in retrospective studies.10 In 1 report of 53 affected LHON patients, 32 of whom carried the m.11778G>A MT-ND4 mutation, 90.6% of patients showed bilateral involvement 6 months after onset.14 In one of the first analyses of a LHON cohort published in the literature, van Senus19 reported that 95.7% of patients were affected bilaterally within 6 months of disease onset. A later study of 107 LHON patients molecularly confirmed to carry one of the 3 primary mtDNA mutations, documented bilateral involvement in 97% of patients within 1 year.10 A more recent prospective natural history study of patients with the m.11778G>A MT-ND4 mutation reported bilateral vision loss within 2 months of onset in 53% of patients, and within 6 months of onset in 80% of patients.20
Vision loss typically progresses to nadir over a median of 6 to 8 weeks.10 In 1 study, 94% of patients reached the nadir within 8 weeks of the onset of vision loss.14 In an earlier study, the average time to visual stabilization was 3.7 months in a sample of 87 eyes.11
Current Management of LHON
It is important to establish a molecular diagnosis to help guide genetic counseling. The management of LHON remains largely supportive with the provision of low vision services and in some countries, patients can be registered as legally blind, providing them with access to relevant social services.
The first randomized controlled trial for a mitochondrial genetic disorder (RHODOS) was conducted to evaluate the safety and efficacy of idebenone—a synthetic analog of coenzyme Q10—in patients with visual loss from LHON for up to 5 years.21 A total of 85 patients were recruited and randomized in a 2:1 ratio to either idebenone at a dose of 300 mg 3 times per day for 6 months, or placebo. Although the primary endpoint was not met, there was evidence of visual benefit in a subgroup of patients. Idebenone was found to be safe and the visual benefit was maintained following cessation of treatment.22 A concurrent retrospective study of LHON patients treated with variable doses of idebenone also found evidence of benefit, especially in those treated within 1 year of disease onset.23 On the basis of the cumulative data, including an expanded access program, idebenone (Raxone; Chiesi Farmaceutici, Parma, Italy) was approved in June 2015 by the European Medicines Agency under exceptional circumstances for the treatment of LHON.24 Raxone is not approved for the treatment of LHON in North America. In a recently published open-label study of 87 patients affected with one of the 3 major LHON mtDNA mutations and treated with idebenone, a clinically relevant recovery [either a gain of −0.2 logMAR equivalent to 10 letters, or an improvement from off-chart to on-chart best-corrected visual acuity (BCVA) by at least 1 full line on the ETDRS chart (5 letters)] was reported in 46% of patients with an average treatment duration of 25.6 months.25 The current consensus guidelines recommend that all patients with LHON who are within 1 year of disease onset should be offered treatment with idebenone at a dose of 300 mg 3 times per day for a minimum period of 1 year.26 As the treatment response to idebenone is partial and limited to a subgroup of patients, other therapeutic strategies are being explored to improve the visual prognosis in LHON.
Gene Therapies for LHON—Preclinical Validation and Clinical Studies
The eye is one of the few immune-privileged compartments of the body, and, as such, constitutes an ideal candidate for local gene therapy. Furthermore, RGCs are located on the inner surface of the retina and should be in theory the most easily accessed using drug delivery via standard intravitreal (IVT) injection. These considerations have led researchers to investigate gene therapy approaches for the treatment of vision loss due to LHON.
Adenoassociated viruses (AAV) are nonpathogenic viruses that can infect nondividing human cells. Without its Rep gene, the vector DNA persists in the host nucleus as a nonintegrative episome, thereby preventing oncogenesis. AAV of serotype 2 are the most common viral vectors used in clinical trials of gene therapy. Three different research groups have developed AAV2-based viral vectors encoding the human wild-type ND4 gene, with the goal of restoring complex I activity and preventing RGC degeneration in LHON patients. This gene therapy approach is based on the allotopic expression of the therapeutic ND4 transgene, initially developed in yeast27,28 and mammalian cells,29–31 and set as proof of principle for LHON by Guy et al32 in cybrid cells carrying the m.11778G>A mutation (Fig. 1). This approach has been refined in terms of efficiency in mitochondrial import and further developed at the preclinical level by Drs Corral-Debrinski, Sahel and colleagues at the Institut de la Vision, Paris, France,34,37,38 and by Dr Guy’s group.39
Figure 1.
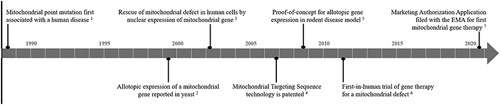
Key breakthroughs in the development of allotopic gene expression to treat mitochondrial defects. This timeline highlights the key breakthroughs in the development of allotopic gene therapies for the treatment of mitochondrial diseases. Leber hereditary optic neuropathy is the first mitochondrial disease for which a gene therapy was assessed in clinical trials and a market authorization was requested from the European Medicines Agency (EMA). Wallace et al.1; Roucou et al.27; Manfredi et al.29; Corral-Debrinski et al.33; Ellouze et al.34; ClinicalTrials.gov.35; GenSight Biologics website.36
The IVT injection of viral vectors containing the therapeutic wild-type ND4 gene allows for the preferential targeting of macular RGCs. In targeted cells, the mutated ND4 gene encoded by the multicopy mitochondrial genome is expressed in the mitochondrial matrix; and its translation coupled with protein folding and assemblage within complex I occurs in the mitochondrial inner membrane. Upon cell transfection, the AAV2 gene therapy vector carries the therapeutic ND4 transgene into the cell nucleus where it is episomically transcribed into a messenger RNA (mRNA), which is later translated by ribosomes associated with the external mitochondrial membrane. The 2 mitochondrial targeting sequences flanking the therapeutic ND4 transcript are necessary not only to address the mRNA directly to the external mitochondrial membrane, but also to optimize the translocation of the newly translated ND4 protein through the inner mitochondrial membrane into the mitochondrial matrix,40 where it can finally integrate within complex I and restore its function, competing with the endogenous mutant ND4 protein. Preclinical studies have demonstrated that recombinant adeno-associated virus 2 (rAAV2)/2-ND4 could effectively rescue ATP production in cultured fibroblasts isolated from ND4-LHON patients,37 and that the therapeutic ND4 protein could successfully integrate into complex I in induced LHON murine models, preventing RGC apoptosis and optic nerve atrophy.37,41,42
A summary of the clinical trials of ocular gene therapy for LHON is presented in Table 1, based on the records listed in the database ClinicalTrials.gov.
Table 1.
Clinical Trials of Ocular Gene Therapy for the Treatment of ND4-LHON
| Sponsor | Gene Therapy Product | Clinical Phase—Study Design—Study Population |
|---|---|---|
| Huazhong University of Science and Technology, China | rAAV2-ND4 with mitochondrial targeting sequence | Phase 1 |
| Open-label study | ||
| 9 ND4-LHON patients who did not experience spontaneous recovery during 12 mo of observation before treatment administration in their worse-seeing eye | ||
| Phase 2/3 | ||
| Open-label study | ||
| 149 ND4-LHON patients treated in their worse-seeing eye with 5E11 vg/μL | ||
| Bascom Palmer Eye Institute, University of Miami, USA | scAAV2-P1ND4v2 | Phase 1 |
| Open-label, dose escalation study | ||
| 12 ND4-LHON patients treated in their worse-seeing eye: 6 with <1 y of vision loss, 6 with >1 y of vision loss | ||
| 2 ND4-LHON patients with unilateral vision loss | ||
| GenSight Biologics, France | rAAV2/2-ND4 with mitochondrial targeting sequence | Phase 1/2 |
| Open-label, dose escalation study | ||
| 15 ND4-LHON patients treated in their worse-seeing eye | ||
| Phase 3 (2 separate trials) | ||
| Double-masked, randomized, sham-controlled studies | ||
| RESCUE: 39 ND4-LHON patients at least 15 y old at onset, with 0-6 mo of vision loss | ||
| REVERSE: 37 ND4-LHON patients at least 15 y old at onset, with 6-12 mo of vision loss | ||
| Right eye randomized to either 9E10 vg or a sham IVT injection, left eye received treatment not allocated to right eye | ||
| Phase 3 | ||
| Double-masked, randomized, placebo-controlled study | ||
| REFLECT: 98 ND4-LHON patients at least 15 y old at onset, with 0-12 mo of vision loss | ||
| 1:1 patient randomization in 2 treatment arms: | ||
| Bilateral IVT injection of 9E10 vg | ||
| or | ||
| First-affected eye received 9E10 vg, second-affected eye received a placebo IVT injection |
IVT indicates intravitreal; LHON, Leber hereditary optic neuropathy; rAAV2, recombinant adeno-associated virus 2 vg, viral genomes.
In 2011, a phase 1 clinical trial of rAAV2-ND4 for the treatment of ND4-LHON was initiated by the Huazhong University of Science and Technology (ClinicalTrials.gov identifier: NCT01267422). This open-label study included 9 patients with ND4-LHON who were first followed for 12 months, then received a single IVT injection of gene therapy in their worse-seeing eye at a dose of 5E9 viral genomes (vg) if they were below the age of 12, and 1E10 vg if they were older. Unexpectedly, 4 of 9 patients showed a clinically meaningful improvement of BCVA (ie, a change better than −0.3 logMAR, equivalent to at least +15 ETDRS letters) in both eyes. In patients who had <2 years of vision loss at treatment administration, the mean change in BCVA from baseline to 36 months was −0.3 logMAR in the injected eyes and −0.35 logMAR in the uninjected eyes, respectively, equivalent to gains of +15 and +18 ETDRS letters.43 In patients who had >2 years of vision loss at treatment administration, the mean change in BCVA from baseline to 36 months was −0.4 logMAR in the injected eyes and −0.25 logMAR in the uninjected eyes, respectively, equivalent to gains of +20 and +13 ETDRS letters. These results were sustained up to 7 years after treatment administration.44 Of note, 7 of 9 (78%) patients treated in this study were children below the age of 15 at onset of vision loss (from 7 to 14 y of age), and the 2 patients who were above the age of 15 years at onset did not show a sustained improvement in BCVA.
In 2014, a phase 1/2a dose-finding clinical trial was initiated by GenSight Biologics to assess the safety and efficacy of rAAV2/2-ND4 gene therapy for the treatment of ND4-LHON (ClinicalTrials.gov identifier: NCT02064569). Fifteen adult patients with a molecularly confirmed diagnosis of ND4-LHON were enrolled in 4 cohorts treated at increasing doses: 3 patients received a dose of 9E9 vg, 3 patients received a dose of 3E10 vg, 6 patients received a dose of 9E10 vg, and 3 patients received a dose of 18E11 vg.45 Two years after treatment administration, the unilateral injection of rAAV2/2-ND4 gene therapy was well tolerated. The dose 9E10 vg/eye was defined as the dose level with the best benefit/risk ratio for the subjects and was therefore chosen for the ensuing pivotal studies.
Also in 2014, a phase 1 clinical trial of scAAV2-P1ND4v2 for the treatment of ND4-LHON was initiated at the Bascom Palmer Eye Institute of the University of Miami (ClinicalTrials.gov identifier: NCT02161380). The open-label dose escalation study included 3 groups of patients: 6 patients with chronic LHON (>1 y of vision loss) treated at 2 different doses [low (1.18E9 vg) and medium (5.81E9 vg)]; 6 patients with acute LHON (<1 y of vision loss) treated at 2 different doses [low (1.18E9 vg) and medium (5.81E9 vg)]; and 2 patients with unilateral vision loss treated at the low dose (1.18E9 vg). Each patient was administered a single IVT injection of gene therapy in their worse-seeing eye. Preliminary results reported signs of efficacy in 2 patients of the chronic group, and 4 patients of the acute group, with follow-up periods varying from 3 to 24 months post treatment administration.46 Changes in BCVA compared with baseline ranged from −0.08 to −0.45 logMAR, equivalent to gains between +4 and +23 ETDRS letters. Interestingly, bilateral improvement of BCVA was reported in 4 of the 6 patients who showed signs of efficacy. No dose response was observed in the study cohorts.
In 2016, GenSight Biologics initiated 2 phase 3, randomized, double-masked, sham-controlled pivotal studies, RESCUE and REVERSE, that evaluated the efficacy and safety of a unilateral IVT injection of rAAV2/2-ND4 (9E10 vg) in recently affected ND4-LHON patients (up to 12 mo of vision loss) with a follow-up of 2 years. Patients had to be at least 15 years old at the time of onset of vision loss. The right eye of each subject was randomly allocated to receive either rAAV2/2-ND4 or a sham treatment in a 1:1 allocation ratio. The fellow (left) eye received the treatment not allocated to the right eye. rAAV2/2-ND4 was administered once via a single IVT injection. Sham IVT injection was performed once by applying pressure to the eye at the location of a typical procedure using the blunt end of a syringe without a needle. The 2 studies had an identical study design, the only difference being the vision loss duration at screening, with the RESCUE study (ClinicalTrials.gov identifier: NCT02652767) including patients who had a duration of vision loss ≤6 months in the first-affected eye, and the REVERSE study (ClinicalTrials.gov identifier: NCT02652780) including patients who had a duration of vision loss between 6 and 12 months at screening in both eyes.
RESCUE and REVERSE included 39 and 37 patients, respectively. The primary endpoint, defined as a 15-letter difference in the change in BCVA from baseline to week 48 between the group of eyes that received rAAV2/2-ND4 and the group of eyes that received a sham injection, was not met in either trial due to an unexpected and sustained improvement in the contralateral sham-treated eyes.47,48 Two years after treatment administration, the mean improvement from nadir BCVA (worst measure of visual acuity) in REVERSE and RESCUE studies was respectively +28 and +26 ETDRS letters equivalent in drug-treated eyes, and +24 and +23 ETDRS letters equivalent in sham-treated eyes. Consequently, a bilateral improvement in BCVA was evidenced in study patients who had been unilaterally injected with rAAV2/2-ND4. This contralateral therapeutic effect on sham-treated eyes was sustained and clinically relevant, mirroring the improvement in eyes treated with rAAV2/2-ND4. REVERSE and RESCUE patients are currently followed in an extension study up to 5 years after injection (NCT03406104). In this study, the bilateral treatment effect of rAAV2/2-ND4 was sustained up to 3 years after gene therapy administration (last available observation).49
In 2017, an open-label clinical trial of rAAV2-ND4 for the treatment of ND4-LHON was initiated by the Huazhong University of Science and Technology (ClinicalTrials.gov identifier: NCT03153293). A total of 149 patients with ND4-LHON received a single dose of gene therapy (5E11 vg) in their worse-seeing eye. Mean age for this cohort was 19±7.1 years, and average disease duration at time of treatment administration was 40.56±49.99 months, varying from 1 to 312 months (26 y). A rapid and significant improvement of BCVA (by at least −0.3 logMAR) was reported within 3 days in at least 1 eye of 54/149 (36.2%) of patients and in both eyes in 17/147 (11.4%) of patients.50 Although patient age at onset is not indicated, the reported mean age at study enrollment of 19±7.1 years would imply that a fair proportion of patients treated in this open-label clinical trial were children.
In 2018, GenSight Biologics initiated a third phase 3 pivotal clinical study, REFLECT (ClinicalTrials.gov identifier: NCT03293524), a randomized, double-masked, placebo-controlled trial. The ongoing REFLECT study evaluates the efficacy and safety of bilateral IVT injection of rAAV2/2-ND4 (9E10 vg/eye) in ND4-LHON patients with bilateral vision loss within 1 year. Ninety-eight patients received an IVT injection of rAAV2/2-ND4 in the first-affected eye and were randomly allocated to an IVT injection of either rAAV2/2-ND4 or placebo in the second-affected eye. The study primary endpoint will be the difference in change from baseline of BCVA between the group of second-affected eyes that received the drug product, and the group of second-affected eyes that received a placebo, at 1.5 years after treatment administration.51,52 The results of the primary endpoint will be available mid-2021.
All these pivotal studies have confirmed that ocular gene therapy for LHON via IVT injection has an overall good safety profile with excellent systemic tolerability and mostly mild ocular side effects, always responsive to conventional ophthalmologic treatments.47,48 The most common adverse events were intraocular inflammation and elevated intraocular pressure, mostly mild and resolving with conventional treatment (topical and/or oral corticosteroids for inflammation, and intraocular pressure lowering agents for increased intraocular pressure).45,47,48 Moreover, the IVT injection of rAAV2/2-ND4 showed limited biodissemination, with the vector DNA mostly detected in the visual system. Patients’ systemic humoral and cellular immunologic responses were mild and did not correlate with intraocular inflammation, acknowledging the local ocular nature of the immune response.53
Contralateral Effect of Gene Therapy in LHON
All 3 research groups assessing the efficacy and safety of unilateral IVT gene therapy in ND4-LHON patients reported an unexpected, sustained, and clinically meaningful improvement of BCVA in the untreated contralateral eyes not consistent with the reported natural history of the disease.13 One possible mechanism for the contralateral effect observed with rAAV2/2-ND4 gene therapy was investigated in a non-human primate study.47 Data from that study demonstrated the presence of viral vector DNA in not only the injected eye, but also in tissues of the uninjected eye in cynomolgus monkeys (rAAV2/2-ND4 DNA was detected and/or quantified in contralateral uninjected eyes/visual tissues), potentially explaining the bilateral treatment effect reported in LHON patients in the phase 3 REVERSE and RESCUE trials and those of the other 2 groups. Since rAAV2/2-ND4 DNA was detected and quantified in the optic chiasm of injected animals, the anatomic route taken by the viral vector DNA to transfer from the treated eye to the nontreated eye is hypothesized to be via the optic nerve and chiasm (through anterograde and subsequent retrograde transport along the optic projections). A systemic transfer of rAAV2/2-ND4 DNA cannot be excluded but is less likely given that biodissemination studies have shown limited and transient presence of rAAV2/2-ND4 DNA in blood.
Other mechanisms could also contribute to the contralateral effect of ocular gene therapy in LHON. Transfer of mitochondrial material (eg, mRNA or proteins) cannot be excluded at this stage, especially given the extensive literature on this topic.54 Furthermore, a recent study of an induced model of neurodegeneration in glaucoma reported transorbital exchange of astrocyte-derived metabolites from a healthy eye to a stressed contralateral eye via the optic chiasm.54 Finally, brain plasticity could partially account for the improvement of visual function in the contralateral eye, as reported in some blind subjects implanted with a retinal prosthesis.55
Conclusions
LHON is a rare mitochondrial blinding disease with an unmet medical need for efficacious therapies. Gene therapies have been designed to compensate for the mitochondrial defect by transfecting cells of the inner retina with the functional wild-type gene, thereby restoring the activity of the respiratory chain and rescuing RGCs. Three research groups have developed gene therapy for the treatment of ND4-LHON, the most prevalent and severe genotype of LHON. Across trials and clinical developments, published studies report a sustained and clinically meaningful bilateral benefit beyond the expected natural history of the disease in a substantial proportion of ND4 patients who were unilaterally treated. A non-human primate study demonstrating the transfer of viral vector DNA from the injected to the noninjected eye provides insights into the possible mechanisms of this apparent contralateral therapeutic effect. IVT administration targeting the RGCs constitutes a safe and easy way to administrate the product, avoiding the complications of subretinal surgery. Furthermore, biodissemination and systemic humoral and cellular immunologic responses have been shown to be limited. Gene therapy has an overall good safety profile with excellent systemic tolerability and mostly mild ocular side effects, responsive to conventional ophthalmologic treatments. The results of late-phase clinical trials are now available and could have major implications for gene therapy clinical trial design and outcome measures, paving the way to potentially expand this treatment to the other LHON mutations.
Footnotes
J.-A.S. is supported by the Agence Nationale de la Recherche within the Programme Investissements d’Avenir, Institut Hospitalo Universitaire FOReSIGHT (ANR-18-IAHU-0001) and LabEx LIFESENSES (ANR-10-LABX-65). J.-A.S. is a co-founder and shareholder of GenSight Biologics, and a patent co-author on allotopic transport. N.J.N. is supported in part by an ophthalmology department core grant from the NIH/NEI (P30 EY006360). N.J.N. is a consultant for GenSight Biologics, Santhera Pharmaceuticals and Stealth BioTherapeutics, has received research support from GenSight and Santhera Pharmaceuticals, served on the Data Safety Monitoring Board for the Quark NAION study and is a medical legal consultant. P.Y.-W.-M. is supported by a Clinician Scientist Fellowship Award (G1002570) from the Medical Research Council (UK), and also receives funding from Fight for Sight (UK), the Isaac Newton Trust (UK), Moorfields Eye Charity, the Addenbrooke’s Charitable Trust, the National Eye Research Centre (UK), the UK National Institute of Health Research (NIHR) as part of the Rare Diseases Translational Research Collaboration, and the NIHR Biomedical Research Centre based at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. P.Y.-W.-M. is a consultant for GenSight Biologics and Stealth BioTherapeutics and has received research support from GenSight and Santhera Pharmaceuticals. C.V.C. is a consultant for Santhera Pharmaceuticals and GenSight Biologics. V.C. is supported by grants from the Italian Ministry of Health (RF-2018-12366703), the Italian Ministry of Research (20172T2MHH), and Telethon-Italy (GUP15016). V.C. is also supported by patients’ organizations MITOCON and IFOND, and patients’ donations. V.C. is a consultant for Santhera Pharmaceuticals, GenSight Biologics and Stealth BioTherapeutics, and has received research support from Santhera Pharmaceuticals and Stealth BioTherapeutics. V.B. is supported in part by an ophthalmology department core grant from the NIH/NEI (P30 EY006360). V.B. is a consultant for GenSight Biologics, Santhera Pharmaceuticals and Stealth BioTherapeutics and has received research support from GenSight and Santhera Pharmaceuticals. M.L.M. is a consultant for GenSight Biologics and has received research support from GenSight. R.S. is a consultant for GenSight Biologics. T.K. is supported by the German Federal Ministry of Education and Research (BMBF, Bonn, Germany) through grants to the German Network for Mitochondrial Disorders (mitoNET, 01GM1906A) and to the E-Rare project GENOMIT (01GM1920B). T.K. is a consultant for Santhera Pharmaceuticals and GenSight Biologics, and has received research support from Santhera Pharmaceuticals and GenSight Biologics. A.A.S. is a consultant for Stealth BioTherapeutics. L.B. and M.T. are GenSight Biologics employees. B.K. is a consultant for GenSight Biologics.
Contributor Information
José-Alain Sahel, Email: j.sahel@gmail.com.
Nancy J. Newman, Email: ophtnjn@emory.edu.
Patrick Yu-Wai-Man, Email: py237@cam.ac.uk.
Catherine Vignal-Clermont, Email: cvignal@for.paris.
Valerio Carelli, Email: valerio.carelli@unibo.it;valerio.carelli@unibo.it.
Valérie Biousse, Email: vbiouss@emory.edu.
Mark L. Moster, Email: markmoster@gmail.com.
Robert Sergott, Email: rcs220@comcast.net.
Thomas Klopstock, Email: thomas.klopstock@med.uni-muenchen.de.
Alfredo A. Sadun, Email: alfredo.sadun@gmail.com.
Laure Blouin, Email: lblouin@gensight-biologics.com.
Barrett Katz, Email: bkatzmd@yahoo.com.
Magali Taiel, Email: mtaiel@GENSIGHT-BIOLOGICS.COM.
References
- 1.Wallace DC, Singh G, Lott MT, et al. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science. 1988;242:1427–1430. [DOI] [PubMed] [Google Scholar]
- 2.Yu-Wai-Man P, Griffiths PG, Brown DT, et al. The epidemiology of Leber hereditary optic neuropathy in the north east of England. Am J Hum Genet. 2003;72:333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carelli V, Ross-Cisneros FN, Sadun AA. Mitochondrial dysfunction as a cause of optic neuropathies. Prog Retin Eye Res. 2004;23:53–89. [DOI] [PubMed] [Google Scholar]
- 4.Yu-Wai-Man P, Griffiths PG, Chinnery PF. Mitochondrial optic neuropathies - disease mechanisms and therapeutic strategies. Prog Retin Eye Res. 2011;30:81–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu-Wai-Man P, Votruba M, Burté F, et al. A neurodegenerative perspective on mitochondrial optic neuropathies. Acta Neuropathol. 2016;132:789–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sadun A, La Morgia C, Carelli V. Leber’s hereditary optic neuropathy. Curr Treat Options Neurol. 2011;13:109–117. [DOI] [PubMed] [Google Scholar]
- 7.Puomila A, Hämäläinen P, Kivioja S, et al. Epidemiology and penetrance of Leber hereditary optic neuropathy in Finland. Eur J Hum Genet. 2007;15:1079–1089. [DOI] [PubMed] [Google Scholar]
- 8.Spruijt L, Smeets HJ, Hendrickx A, et al. A MELAS-associated ND1 mutation causing Leber hereditary optic neuropathy and spastic dystonia. Arch Neurol. 2007;64:890. [DOI] [PubMed] [Google Scholar]
- 9.Yu-Wai-Man P, Turnbull DM, Chinnery PF. Leber hereditary optic neuropathy. J Med Genet. 2002;39:162–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Riordan-Eva P, Sanders MD, Govan GG, et al. The clinical features of Leber’s hereditary optic neuropathy defined by the presence of a pathogenic mitochondrial DNA mutation. Brain. 1995;118:319–337. [DOI] [PubMed] [Google Scholar]
- 11.Newman NJ, Lott MT, Wallace DC. The clinical characteristics of pedigrees of Leber’s hereditary optic neuropathy with the 11778 mutation. Am J Ophthalmol. 1991;111:750–762. [DOI] [PubMed] [Google Scholar]
- 12.Hotta Y, Fujiki K, Hayakawa M, et al. Clinical features of Japanese Leber’s hereditary optic neuropathy with 11778 mutation of mitochondrial DNA. Jpn J Ophthalmol. 1995;39:96–108. [PubMed] [Google Scholar]
- 13.Newman NJ, Carelli V, Taiel M, et al. Visual outcomes in leber hereditary optic neuropathy patients with the m.11778G>A (MTND4) mitochondrial DNA mutation. J Neuroophthalmol. 2020;40:547–557. [DOI] [PubMed] [Google Scholar]
- 14.Nikoskelainen EK, Huoponen K, Juvonen V, et al. Ophthalmologic findings in Leber hereditary optic neuropathy, with special reference to mtDNA mutations. Ophthalmology. 1996;103:504–514. [DOI] [PubMed] [Google Scholar]
- 15.Majander A, Bowman R, Poulton J, et al. Childhood-onset Leber hereditary optic neuropathy. Br J Ophthalmol. 2017;101:1505–1509. [DOI] [PubMed] [Google Scholar]
- 16.Guy J, Feuer WJ, Porciatti V, et al. Retinal ganglion cell dysfunction in asymptomatic G11778A: Leber hereditary optic neuropathy. Invest Ophthalmol Vis Sci. 2014;55:841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hwang TJ, Karanjia R, Moraes-Filho MN, et al. Natural history of conversion of Leber’s hereditary optic neuropathy. Ophthalmology. 2017;124:843–850. [DOI] [PubMed] [Google Scholar]
- 18.Newman NJ. Hereditary optic neuropathies: from the mitochondria to the optic nerve. Am J Ophthalmol. 2005;140:517.e1–517.e9. [DOI] [PubMed] [Google Scholar]
- 19.van Senus AHC. An Investigation of the occurrence of Leber’s disease in the Netherlands. Ophthalmologica. 1962;144:415–419. [DOI] [PubMed] [Google Scholar]
- 20.Lam BL, Feuer WJ, Schiffman JC, et al. Trial end points and natural history in patients with G11778A Leber hereditary optic neuropathy: preparation for gene therapy clinical trial. JAMA Ophthalmol. 2014;132:428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klopstock T, Yu-Wai-Man P, Dimitriadis K, et al. A randomized placebo-controlled trial of idebenone in Leber’s hereditary optic neuropathy. Brain. 2011;134:2677–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klopstock T, Metz G, Yu-Wai-Man P, et al. Persistence of the treatment effect of idebenone in Leber’s hereditary optic neuropathy. Brain. 2013;136:e230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carelli V, La Morgia C, Valentino ML, et al. Idebenone treatment in Leber’s hereditary optic neuropathy. Brain. 2011;134:e188. [DOI] [PubMed] [Google Scholar]
- 24.EMA European Public Assessment Report [EMA website]. 2015. Available at: www.ema.europa.eu/en/documents/assessment-report/raxone-epar-public-assessment-report_en.pdf. Accessed December 18, 2020.
- 25.Catarino CB, von Livonius B, Priglinger C, et al. Real-world clinical experience with idebenone in the treatment of Leber hereditary optic neuropathy. J Neuroophthalmol. 2020;40:558–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carelli V, Carbonelli M, de Coo IF, et al. International consensus statement on the clinical and therapeutic management of Leber hereditary optic neuropathy. J Neuroophthalmol. 2017;37:371–381. [DOI] [PubMed] [Google Scholar]
- 27.Roucou X, Artika IM, Devenish RJ, et al. Bioenergetic and structural consequences of allotopic expression of subunit 8 of yeast mitochondrial ATP synthase. The hydrophobic character of residues 23 and 24 is essential for maximal activity and structural stability of the enzyme complex. Eur J Biochem. 1999;261:444–451. [DOI] [PubMed] [Google Scholar]
- 28.Sylvestre J, Vialette S, Corral Debrinski M, et al. Long mRNAs coding for yeast mitochondrial proteins of prokaryotic origin preferentially localize to the vicinity of mitochondria. Genome Biol. 2003;4:R44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manfredi G, Fu J, Ojaimi J, et al. Rescue of a deficiency in ATP synthesis by transfer of MTATP6, a mitochondrial DNA-encoded gene, to the nucleus. Nat Genet. 2002;30:394–399. [DOI] [PubMed] [Google Scholar]
- 30.Kaltimbacher V, Bonnet C, Lecoeuvre G, et al. mRNA localization to the mitochondrial surface allows the efficient translocation inside the organelle of a nuclear recoded ATP6 protein. RNA. 2006;12:1408–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sylvestre J, Margeot A, Jacq C, et al. The role of the 3’ untranslated region in mRNA sorting to the vicinity of mitochondria is conserved from yeast to human cells. Mol Biol Cell. 2003;14:3848–3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guy J, Qi X, Pallotti F, et al. Rescue of a mitochondrial deficiency causing Leber Hereditary Optic Neuropathy. Ann Neurol. 2002;52:534–542. [DOI] [PubMed] [Google Scholar]
- 33.Corral-Debrinski M, Sahel JA, Kaltimbacher V, et al. Importation d’une protéine mitochondriale au moyen d’une méthode allotopique améliorée [Mitochondrial protein import via an optimized allotopic method]. Institut National de la Propriété intellectuelle (INPI) [National Institute of Intellectual Property], patent application number WO2006117250. 2006.
- 34.Ellouze S, Augustin S, Bouaita A, et al. Optimized allotopic expression of the human mitochondrial ND4 prevents blindness in a rat model of mitochondrial dysfunction. Am J Hum Genet. 2008;83:373–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.ClinicalTrials.gov Identifier: NCT01267422. Sponsor: Bin Li, Huazhong University of Science and Technology. Safety and Efficacy Study of rAAV2-ND4 Treatment of Leber Hereditary Optic Neuropathy (LHON). Trial initiation. 2011.
- 36.GenSight Biologics Submits EU Marketing Authorisation Application for LUMEVOQ® Gene Therapy to Treat Vision Loss due to Leber Hereditary Optic Neuropathy (LHON) [GenSight Biologics website]. 2020. Available at: https://www.gensight-biologics.com/2020/09/15/gensight-biologics-submits-eu-marketing-authorisation-application-for-lumevoq-gene-therapy-to-treat-vision-loss-due-to-leber-hereditary-optic-neuropathy-lhon/. Accessed December 18, 2020.
- 37.Bonnet C, Augustin S, Ellouze S, et al. The optimized allotopic expression of ND1 or ND4 genes restores respiratory chain complex I activity in fibroblasts harboring mutations in these genes. Biochim Biophys Acta. 2008;1783:1707–1717. [DOI] [PubMed] [Google Scholar]
- 38.Bonnet C, Kaltimbacher V, Ellouze S, et al. Allotopic mRNA localization to the mitochondrial surface rescues respiratory chain defects in fibroblasts harboring mitochondrial DNA mutations affecting complex I or v subunits. Rejuvenation Res. 2007;10:127–144. [DOI] [PubMed] [Google Scholar]
- 39.Qi X, Sun L, Lewin AS, Hauswirth WW, et al. The mutant human ND4 subunit of complex I induces optic neuropathy in the mouse. Invest Ophthalmol Vis Sci. 2007;48:1–10. [DOI] [PubMed] [Google Scholar]
- 40.Bykov YS, Rapaport D, Herrmann JM, et al. Cytosolic events in the biogenesis of mitochondrial proteins. Trends Biochem Sci. 2020;45:650–667. [DOI] [PubMed] [Google Scholar]
- 41.Cwerman-Thibault H, Augustin S, Lechauve C, et al. Nuclear expression of mitochondrial ND4 leads to the protein assembling in complex I and prevents optic atrophy and visual loss. Mol Ther Methods Clin Dev. 2015;2:15003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koilkonda R, Yu H, Talla V, et al. LHON gene therapy vector prevents visual loss and optic neuropathy induced by G11778A mutant mitochondrial DNA: biodistribution and toxicology profile. Invest Ophthalmol Vis Sci. 2014;55:7739–7753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang S, Ma SQ, Wan X, et al. Long-term outcomes of gene therapy for the treatment of Leber’s hereditary optic neuropathy. EBioMedicine. 2016;10:258–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yuan J, Zhang Y, Liu H, et al. Seven-year follow-up of gene therapy for Leber’s hereditary optic neuropathy. Ophthalmology. 2020;127:1125–1127. [DOI] [PubMed] [Google Scholar]
- 45.Vignal C, Uretsky S, Fitoussi S, et al. Safety of rAAV2/2-ND4 gene therapy for leber hereditary optic neuropathy. Ophthalmology. 2018;125:945–947. [DOI] [PubMed] [Google Scholar]
- 46.Guy J, Feuer WJ, Davis JL, et al. Gene therapy for Leber hereditary optic neuropathy: low- and medium-dose visual results. Ophthalmology. 2017;124:1621–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Newman NJ, Yu-Wai-Man P, Carelli V, et al. Efficacy and safety of intravitreal gene therapy for Leber hereditary optic neuropathy treated within 6 months of disease onset. Ophthalmology. 2020;40:547–557. [DOI] [PubMed] [Google Scholar]
- 48.Yu-Wai-Man P, Newman NJ, Carelli V, et al. Bilateral visual improvement with unilateral gene therapy injection for Leber hereditary optic neuropathy. Sci Transl Med. 2020;12:eaaz7423. [DOI] [PubMed] [Google Scholar]
- 49.GenSight Biologics reports sustained efficacy and safety among LHON patients three years after LUMEVOQ® treatment [GenSight Biologics website]. 2020. Available at: https://www.gensight-biologics.com/2020/07/06/gensight-biologics-reports-sustained-efficacy-and-safety-among-lhon-patients-three-years-after-lumevoq-treatment/. Accessed December 18, 2020.
- 50.Liu HL, Yuan JJ, Zhang Y, et al. Factors associated with rapid improvement in visual acuity in patients with Leber’s hereditary optic neuropathy after gene therapy. Acta Ophthalmol. 2020;98:e730–e733. [DOI] [PubMed] [Google Scholar]
- 51.Efficacy & Safety Study of Bilateral IVT Injection of GS010 in LHON Subjects Due to the ND4 Mutation for up to 1 Year (REFLECT) [ClinicalTrials.Gov website]. 2017. Available at: https://clinicaltrials.gov/ct2/show/NCT03293524?term=NCT03293524&draw=2&rank=1. Accessed December 18, 2020.
- 52.GenSight Biologics completes enrollment of GS010 REFLECT Phase III trial in the treatment of Leber Hereditary Optic Neuropathy ahead of schedule [GenSight Biologics website]. 2019. Available at: https://www.gensight-biologics.com/2019/07/11/gensight-biologics-completes-enrollment-of-gs010-reflect-phase-iii-trial-in-the-treatment-of-leber-hereditary-optic-neuropathy-ahead-of-schedule/. Accessed December 18, 2020.
- 53.Bouquet C, Vignal Clermont C, Galy A, et al. Immune Response and intraocular inflammation in patients with Leber hereditary optic neuropathy treated with intravitreal injection of recombinant adeno-associated virus 2 carrying the ND4 gene: A secondary analysis of a phase 1/2 clinical trial. JAMA Ophthalmol. 2019;137:399–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cooper ML, Pasini S, Lambert WS, et al. Redistribution of metabolic resources through astrocyte networks mitigates neurodegenerative stress. Proc Natl Acad Sci USA. 2020;117:18810–18821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Castaldi E, Cicchini GM, Cinelli L, et al. Visual BOLD response in late blind subjects with argus II retinal prosthesis. PLoS Biol. 2016;14:e1002569. [DOI] [PMC free article] [PubMed] [Google Scholar]