

ABSTRACT
Pediococcus acidilactici is commonly used for pediocin production and lactic acid fermentation. However, a high-efficiency genome editing tool is unavailable for this species. In this study, we constructed endogenous subtype II-A CRISPR-Cas system-based genome interference plasmids which carried a “repeat-spacer-repeat” cassette in the pMG36e shuttle vector. These plasmids exhibited self-interference activities in P. acidilactici LA412. Then, the genome-editing plasmids were constructed by cloning the upstream/downstream donor DNA into the corresponding interference plasmids to exert high-efficiency markerless gene deletion, gene integration, and point mutation in P. acidilactici LA412. We found that endogenous CRISPR-mediated depletion of the native plasmids enhanced the cell growth and that integration of an l-lactate dehydrogenase gene into the chromosome enhanced both cell growth and lactic acid production.
IMPORTANCE A rapid and precise genome editing tool will promote the practical application of Pediococcus acidilactici, one type of lactic acid bacterium with excellent stress tolerance and probiotic characteristics. This study established a high-efficiency endogenous CRISPR-Cas system-based genome editing tool for P. acidilactici and achieved different genetic manipulations, including gene deletion, gene insertion, mononucleotide mutation, and endogenous plasmid depletion. The engineered strain edited by this tool showed significant advantages in cell growth and lactic acid fermentation. Therefore, our tool can satisfy the requirements for genetic manipulations of P. acidilactici, thus making it a sophisticated chassis species for synthetic biology and bioindustry.
KEYWORDS: Pediococcus acidilactici, endogenous CRISPR-Cas, genome editing, lactate dehydrogenase gene, lactic acid
INTRODUCTION
Lactic acid bacteria (LAB) have been widely used in different fields due to their economic value and functional characteristics over the past decades (1). LAB can be used for producing organic acids and fermented food (2–6), and some of them can also promote animal intestinal barrier repair and growth (7, 8). However, LAB are unable to meet increasing production demands due to potential limitations such as incomplete metabolism pathway and poor stability (9–11). With the development of genome-editing approaches, different strategies have been used for improving the beneficial properties of LAB (12). The CRISPR-Cas system has been well studied, and based on this system, some flexible and high-efficiency genome engineering tools have been developed for bacteria, archaea, and eukaryotes (13–17). Various genome editing goals have been successfully achieved in several LAB strains based on CRISPR-Cas systems (18). For example, the edited cells can be identified from Lactobacillus reuteri with the assistance of the CRISPR-Cas9 system (19). The CRISPR-Cas9 system has also been applied successfully in other Lactobacillus species, including Lactobacillus casei (20) and Lactobacillus plantarum (21).
Notably, there are many disadvantages in applying heterogenous CRISPR-based editing tools for genetic engineering, such as low transformation efficiency of the editing plasmids, weak activity, and potential protein toxicity of Cas nucleases (22–24). Many LAB strains harbor different subtypes of the CRISPR-Cas system in their genomes to provide native CRISPR endonucleases for genetic engineering (18, 25). For example, the endogenous subtype I-E CRISPR-Cas system is applied for efficient chromosomal targeting and genetic manipulation in Lactobacillus crispatus (26). The endogenous subtype II-A CRISPR-Cas system (CRISPR-Cas9) is employed to enhance the production of exopolysaccharides in Streptococcus thermophilus after a single nucleotide mutation in the polymerase epsC gene (27).
Pediococcus acidilactici has been studied as an industrial starter strain and potential probiotic that generally exhibits excellent properties such as thermotolerance and stress resistance (28). This species can secret pediocin, which is an effective bacteriocin with a broad antibacterial spectrum (29–31). Due to its probiotic properties, P. acidilactici has recently been used as a feed additive worldwide (32–34). Although plasmid-based homologous recombination (classic double-crossover) strategy has been successfully used for engineering lactate metabolism pathways in P. acidilactici (35), it is difficult for this method to meet the requirement for high-efficiency genome editing. In this study, we developed an endogenous subtype II-A CRISPR-Cas system-based genome editing tool for high-efficiency markerless gene deletion, gene insertion, point mutation, and native plasmid depletion in P. acidilactici. Using this method, we have obtained a native plasmid-deleted strain and an l-lactate dehydrogenase gene-integrated strain, and these two strains showed higher growth rates or/and lactic acid (LA) production efficiency.
RESULTS
Function verification of endogenous CRISPR-Cas system.
We recently isolated P. acidilactici LA412, which contains a complete II-A CRISPR-Cas system consisting of a CRISPR array and some Cas protein genes, including cas1, cas2, csn2, and cas9 (Fig. 1A). However, we found a translation stop codon introduced into cas9, thus resulting in mutation of the Cas9 protein (Fig. 1A). The interference activity of the disrupted Cas9 endonuclease in P. acidilactici LA412 was tested using the challenging plasmid (pMG36e-PS1-NGG), which carried a common protospacer with different protospacer-adjacent motif (PAM) sequences (AGG, TGG, GGG, and CGG) adjacent to its 3′ end (Fig. 1A). The results revealed that all challenging plasmids showed lower transformation efficiencies than the control empty plasmid pMG36e and the plasmid pMG36e-PS1 with the protospacer, but without a PAM sequence (Fig. 1B), indicating that the disrupted Cas9-based CRISPR-Cas system had the interference activities in LA412.
FIG 1.

Function validation of the endogenous CRISPR-Cas system in P. acidilactici LA412. (A) Schematic of the endogenous CRISPR-based genome editing in P. acidilactici LA412. On the upper part of schematic are the CRISPR array, tracrRNA gene, and cas genes (cas1, csn2, cas2, and disrupted cas9, including cas9-N and cas9-C). N(87) represents 87 bp between the ATG and TAA codons. The challenging plasmids with the cassette of “protospacer-PAM” were transformed into the cells and cleaved by the endogenous CRISPR-Cas system. Red, blue, and brown bars on the challenging plasmid indicated the protospacer which matched with spacer 1 (PS1) of the CRISPR array, the NGG PAM (in the sequence, N corresponds to A, T, G, or C), and erythromycin resistance gene emr, respectively. (B) Relative transformation efficiencies of the challenging plasmids with or without NGG PAM. The data are expressed as mean ± SD with three technical replicates. ***, P < 0.001.
High-efficiency gene deletion based on the endogenous CRISPR-Cas system.
In this study, pyrE (a native locus in LA412) encoding the orotate phosphoribosyl transferase was used as the target for testing the interference and gene deletion efficiencies on the genomic DNA of the endogenous CRISPR-Cas system-based tool. The protospacers with different PAM sequences in the pyrE gene were targeted by the guide RNAs (gRNAs) derived from the interference plasmids or the editing plasmids (Fig. 2A). All four interference plasmids were identified as having cleavage activities at the pyrE gene locus since they exhibited lower transformation efficiencies than the empty plasmid pMG36e (Fig. 2B). Moreover, the transformation efficiencies of the editing plasmids were significantly lower than those of the corresponding interference plasmids (Fig. 2B). A total of 16 single colonies were randomly selected from transformants. The target gene was amplified using the primers located upstream/downstream of donor DNA on the chromosome (Fig. 2A). The agarose gel analysis of the PCR products showed the presence of short DNA bands, suggesting deletion of the pyrE gene (Fig. 2C). The editing plasmids targeting the protospacer with adjacent AGG, CGG, and GGG PAMs showed 50%, 50%, and 75% deletion efficiencies on pyrE, respectively (Fig. 2B and C), whereas the editing plasmid targeting the protospacer with adjacent TGG PAM displayed 100% deletion efficiency on pyrE (Fig. 2B and C). The presence of the wild-type band and pyrE-deleted band in PCR products indicated that all the pyrE-deleted colonies were the mixed genotype (Fig. 2C). After one round of screening on the antibiotic-free plate, a plasmid-cured pure genotype of the pyrE-deleted single colony was obtained (Fig. 2D) and verified by DNA sequencing (Fig. 2E).
FIG 2.
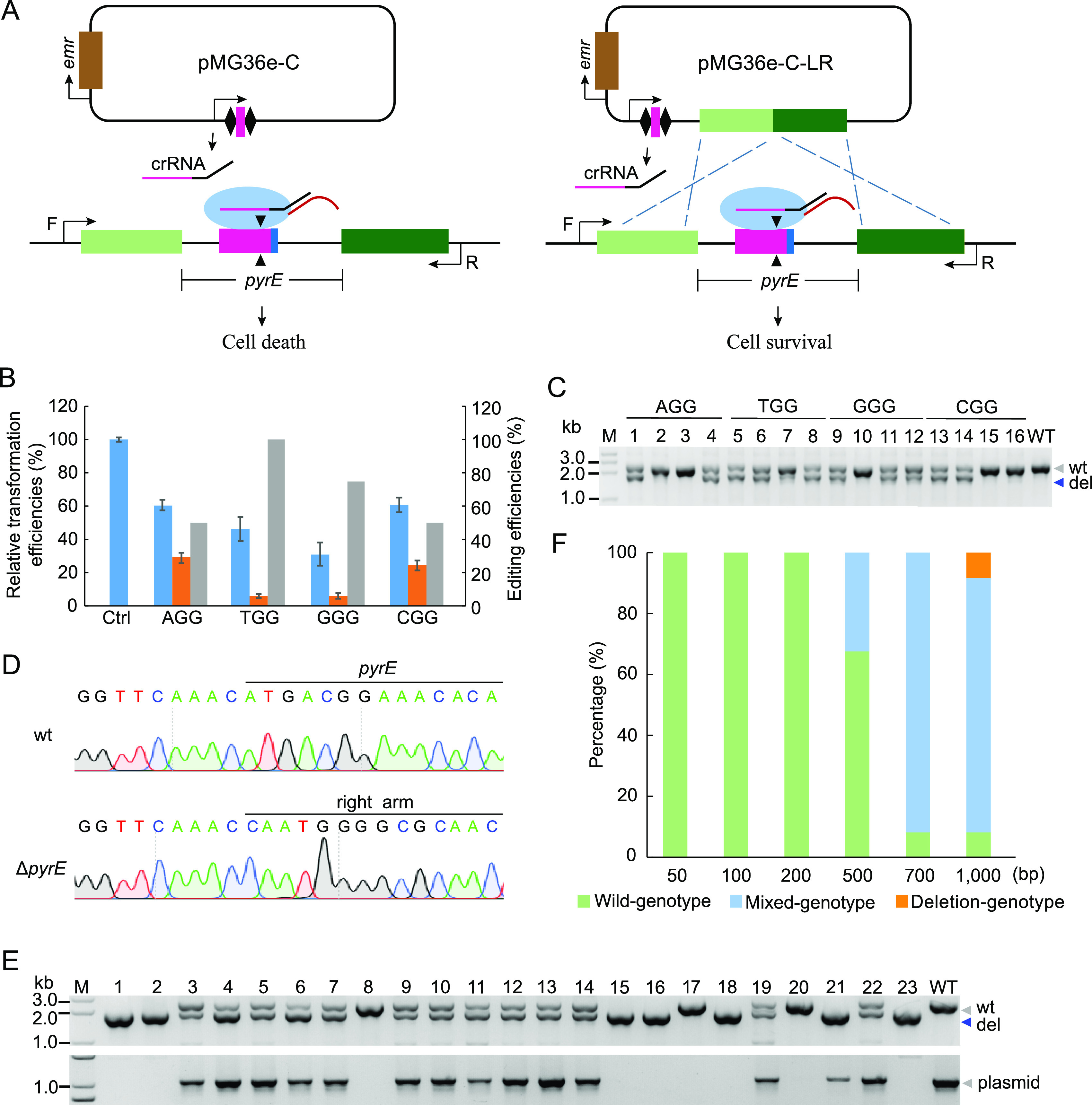
Gene deletion via the endogenous CRISPR-Cas system in P. acidilactici LA412. (A) Schematic of the endogenous CRISPR-based genome interference (left) and genome deletion (right). Transformation of pMG36e plasmid carrying the repeat-spacer-repeat expression cassette led to cleavage of the target region matching with the spacer, further resulting in the deletion of the target gene in the case of transformation of editing plasmid. F and R, respectively, represent the forward and reverse primers used to verify the target gene deletion. (B) Blue columns indicate relative transformation efficiencies of the interference plasmids targeting pyrE gene, orange columns indicate those of corresponding editing plasmids, and gray columns denote pyrE deletion efficiencies. The data are expressed as mean ± SD with three technical replicates. Ctrl, empty plasmid pMG36e as the control. (C) The pyrE gene locus of the randomly selected colonies carrying the editing plasmids was amplified, and the PCR products were verified through agarose gel analysis. M, DNA ladder; WT, control group with P. acidilactici total DNA as the template; “wt” and “del,” wild-type band and the pyrE-deleted band, respectively. (D) DNA sequencing peaks of PCR products of the wild-type (wt) and the pyrE-deleted strain (ΔpyrE). Partial sequences of the pyrE gene and the recombination right arm are presented. (E) PCR for screening pure-deletion genotype (top gel) and validating editing plasmid loss (bottom gel) of 23 colonies on the plate. wt indicates control group with P. acidilactici total DNA (top gel) and pMG36e (bottom gel) as the templates, respectively. (F) Effects of donor length on gene deletion efficiency. Wild genotype, mixed genotype, and deletion genotype indicate wild-type, both wild-type and pyrE gene deletion, and only pyrE gene deletion, respectively. A total of 12 colonies were selected from each editing plasmid group for PCR test.
Considering that the single colonies showed the wild-type gene locus and the deleted gene locus (Fig. 2C), we further studied whether longer donor DNA could improve the editing efficiency. Here, we constructed editing plasmids with different lengths (50, 100, 200, 500, 700, and 1,000 bp) of donor DNA. PCR analysis revealed that when the length of donor DNA of editing plasmids was 50 bp, 100 bp, or 200 bp, the randomly selected colonies exhibited only wild-type bands (Fig. S1 in the supplemental material and Fig. 2F), indicating that it was hard for the donor DNA fragments shorter than 200 bp to induce homologous recombination after CRISPR interference. When the length of donor DNA was 500 bp or 700 bp, single colonies displayed both short DNA bands and the wild-type bands, indicating pyrE gene deletion (Fig. S1 and Fig. 2F). Importantly, ∼10% single colonies showed only pyrE gene-deleted band (pure genotype) when the transformed editing plasmid carried 1,000-bp donor DNA (Fig. S1 and Fig. 2F).
Then, the counterselection function of the pyrE gene was verified. The results demonstrated that the addition of 5-fluoroorotic acid (5-FOA) into MRS medium strongly inhibited cell growth of wild-type strain LA412 (Fig. S2A and B). However, there were no obvious growth differences between wild-type and pyrE-deleted colonies on the MRS plus 5-FOA and uracil (U) plate (Fig. S2B and D).
Depletion of native plasmids increased cell growth based on the endogenous CRISPR-Cas system.
P. acidilactici LA412 carries two native plasmids (plasmid 1, 57.6 kb, and plasmid 2, 41.9 kb). We found that the replication protein-coding gene GE00014 on plasmid 1 and gene GE00039 on plasmid 2 shared 53.80% DNA sequence identity. Therefore, we selected an identical protospacer on both gene GE00014 and gene GE00039, cloned it into the plasmid pMG36e-C, and obtained an interference plasmid pMG36e-Srep (Fig. 3A). Transformation of this interference plasmid led to simultaneous cleavage of the two native plasmids. PCR amplification of GE00014 and GE00039 genes confirmed the existence of both or one of two native plasmids (Fig. 3B), whereas the PCR amplification results of GE00037 gene revealed the deletion of plasmid 1 (Fig. 3B). After one round of passage in modified MRS medium containing 5 μg/ml erythromycin, the GE00037 gene on plasmid 1 became undetectable in several transformants (Fig. 3C), but the GE00033 gene on plasmid 2 remained detectable in all the transformants (Fig. 3C). After another round of passage, a single colony (P. acidilactici LA412-ΔP1P2) was obtained in which both two endogenous plasmids were depleted (Fig. 3D).
FIG 3.

Endogenous CRISPR-mediated depletion of native plasmids increased cell growth. (A) Schematic of the interference plasmid pMG36e-Srep, which simultaneously targets the replication protein genes on both native plasmid 1 (P1) and plasmid 2 (P2). These replication protein genes (GE00014 and GE00039) share one identical protospacer to be targeted. (B) Colony PCR amplification of the target gene, GE00037 gene on P1, and GE00033 gene on P2 from 14 randomly selected transformants (lanes 1 to 14). M, DNA ladder; WT, control group with P. acidilactici total DNA as the template. (C) PCR amplification of target gene, GE00037, and GE00033 from 14 transformants (lanes 1 to 14 in panel B) after one passage in modified MRS medium containing 5 μg/ml erythromycin. (D) PCR amplification of the above 3 genes from the native plasmid-depleted single colony. Δ, WT, and “−” indicate the native plasmid-depleted strain, wild-type strain, and double-distilled water (ddH2O) as the template. (E) Growth curves of wild-type (wt) and the native plasmid-depleted strain (ΔP1P2). Data are expressed as the mean ± SD with three technical replicates. *, P < 0.05; **, P < 0.01.
We further investigated the effect of native plasmid depletion on the growth of strain LA412. We found that the native plasmid-depleted strain grew significantly faster than the wild-type strain throughout the exponential phase (Fig. 3E). After the exponential phase, the wild-type and the native plasmid depletion strains showed no significant difference in the maximum optical density at 600 nm (OD600) value (Fig. 3E).
Introduction of point mutation via the endogenous CRISPR-Cas system.
Point mutation was performed at the genomic target site by introducing a mutated nucleotide to the homology arm on the genome-editing plasmid, with the mpi gene (encoding mannose phosphate isomerase) as the target (Fig. 4A). The PCR product of the mpi wild-type gene harbored no XbaI restriction site (Fig. 4B). When a single nucleotide mutation (T→C) was introduced into the mpi gene, one XbaI restriction site (TCTAGA) would be created (Fig. 4B). In order to introduce point mutation, P. acidilactici cells were transformed with the editing plasmid, and PCR amplification products of the mpi gene from the transformants were digested with XbaI. The digested PCR product analysis results indicated that the desirable point mutation occurred in 9 out of 10 transformants (Fig. 4C). Of them, one transformant belonged to the pure mutation genotype, and the other eight transformants belonged to the mixed genotype (Fig. 4C). PCR sequencing further verified that the mutation was introduced in the desired gene locus (Fig. 4D).
FIG 4.

Introduction of point mutation into the P. acidilactici genome. (A) Schematic of point mutation introduction based on endogenous CRISPR-Cas system. The editing plasmid cleaved the target region matching with the spacer, further inducing crossover between the target gene and the donor DNA with a desired nucleotide. (B) Schematic of point mutation detection. PCR amplification product of the wild-type mpi gene in LA412 was not digested by XbaI restriction endonuclease. However, the PCR amplification product of the mpi gene with an introduced point mutation was digested by XbaI to obtain two DNA fragments. (C) PCR products of the mpi gene amplified from 10 randomly selected transformants (lanes 1 to 10) carrying the editing plasmid or from the wild-type strain (wt) were digested by XbaI and analyzed by agarose gel. M, DNA ladder; WT, control group using mpi gene of wild-type strain as the amplification template; undigested, bands of undigested PCR products; fragments 1 and 2, digested bands. (D) Sequencing peaks of the PCR products amplified from the wild-type and point-mutated mpi gene loci. The red rectangle marks the mutation site. (E) Sequencing peaks of the PCR products amplified from the wild type (disrupted) and the corrected cas9 gene locus. The red rectangle indicates TAA stop codon and GAA codon. (F) Blue columns indicate relative transformation efficiencies of the interference plasmids targeting pyrE gene, orange columns indicate those of corresponding editing plasmids, and gray columns denote pyrE deletion efficiencies of the editing plasmids in P. acidilactici LA412cas9 strain. Data are expressed as the mean ± SD with three technical replicates.
As mentioned above, this strain encodes a disrupted Cas9 endonuclease (Fig. 1A). Here, we corrected the mutation of the disrupted cas9 gene by converting the TAA stop codon into GAA codon via the editing tool (Fig. 4E). We further examined the interference efficiencies of the interference plasmids in the cas9-corrected strain (LA412cas9) and found that interference efficiencies in LA412cas9 were similar to those in the wild-type strain LA412 (Fig. 2B and Fig. 4F). However, the transformation efficiencies of four editing plasmids in strain LA412cas9 were significantly increased compared with those in strain LA412 (Fig. 2B and Fig. 4F). Moreover, the proportion of pyrE-deleted bands in mixed-genotype colonies was much higher than that in wild-type strain LA412 (Fig. 2C and Fig. S3).
Integration of l-lactate dehydrogenase gene into chromosome enhanced growth performance and lactic acid fermentation.
The genome of P. acidilactici LA412 encodes two l-lactate dehydrogenases (l-ldh) and one d-lactate dehydrogenase (d-ldh), of which NADH-dependent l-ldh is encoded by the GE00594 gene. Here, we constructed the l-ldh-integrating plasmid pMG36e-Sun-LR::P0594ldh so as to insert one l-ldh-expressing cassette into the noncoding region (between GE01508 and GE01509 gene loci) of the strain LA412 genome. After electroporation, almost all of the colonies were identified as the mixed genotype since they had both wild type- and ldh-integrated bands (Fig. 5A). One pure-genotype strain (wt::ldh) was obtained after one round of screening (Fig. 5B), which was further verified by DNA sequencing (Fig. 5C). Then, Western blotting showed that the ldh gene was efficiently expressed in the transformant (Fig. 5D). Next, the growth performance of the ldh-overexpressing strain wt::ldh was examined. Result revealed that the wt::ldh strain showed a significant growth advantage over the wild-type strain and a higher final cell density, with an OD600 of 3.40 (Fig. 5E). Furthermore, the wt::ldh strain produced significantly more LA with higher titer (24.50 g/liter) and productivity (0.34 g/liter/h) than the wild-type strain (Fig. 5F).
FIG 5.

Integration and overexpression of ldh gene enhanced cell growth and LA production. (A) Agarose gel analysis of PCR amplification products of ldh expression cassette in 12 randomly selected colonies. M, DNA ladder; WT, control group with LA412 total DNA as the template. (B) Agarose gel analysis of PCR amplification products of ldh expression cassette in the wild-type (wt) and in pure-genotype P. acidilactici strains obtained after one round of screening. Blue and gray arrows indicate the integrated and the wild-type bands, respectively. (C) DNA sequencing peaks of PCR products of right arm amplified from wt and ldh expression cassette amplified from wt::ldh, respectively. (D) Western blotting of the expression ldh gene. M, Protein ladder. (E) Growth curve of wt and wt::ldh strains, respectively. (F) LA fermentation of wt and wt::ldh strains.
DISCUSSION
P. acidilactici is the ideal species for LA and probiotic production. However, no high-efficiency genome editing method is available except the traditional homologous recombination method (35). Recently, CRISPR-based genome editing tools have been developed for engineering industrial microorganisms to achieve high production efficiency and integrate metabolism pathways (36). The endogenous CRISPR-based genome editing methods especially showed much higher editing efficiency in prokaryotes (23). Therefore, the endogenous CRISPR-Cas system in P. acidilactici genomes was analyzed. We found that 21 out of all 71 publicly available P. acidilactici genomes encode the complete II-A CRISPR-Cas system. However, P. acidilactici LA412 encodes a disrupted Cas9 endonuclease gene, which is broken into two parts, namely, cas9-N and cas9-C (Fig. 1A). It could be speculated that this disruption might be caused by the mutation of GAA into TAA, based on the cas9 gene sequences in other publicly available P. acidilactici strains. Interference experiments targeting challenge plasmids in wild-type strain LA412 showed that the interrupted Cas9 in endogenous subtype II-A CRISPR-Cas system was functional (Fig. 1B) and that high editing efficiency and proportion of edited bands in LA412cas9 suggested an increase in the activities of the corrected system (Fig. 2B and C, Fig. 4F, and Fig. S3 in the supplemental material), although they were still much lower than those in Streptococcus thermophilus (12) and Streptococcus pyogenes (37). In addition, high knockout efficiency of the pyrE gene further demonstrated that the endogenous subtype II-A CRISPR-Cas system even with the interrupted cas9 gene in P. acidilactici LA412 could be employed for high-efficiency gene deletion (Fig. 2C). Since long donor DNA contributed to high editing efficiency, long donor DNA should be used for genome editing in P. acidilactici species.
A counterselection marker is important for genetic manipulation. The pyrE gene encoding the orotate phosphoribosyl transferase was widely used for counterscreening in selectable genetic systems (38). Inhibition of wild-type cell growth in 5-FOA-containing plates (Fig. S1A and B) suggested that the pyrE gene could probably be used as a counterscreening marker in P. acidilactici species. Therefore, we used the pyrE as the target gene to test the editing efficiency first, and we further tested the resistance of the pyrE-deleted cells to 5-FOA. Unexpectedly, the pyrE-deleted strain showed sensitivity to 5-FOA (Fig. S1B and D). Based on this result, we speculated that in addition to the pyrE gene, there might be other gene products converting 5-FOA into toxic 5-fluorouracil (5-FU), thus resulting in incomplete growth recovery of ΔpyrE strain on the MRS with 5-FOA and U plate. Meanwhile, we found that loss of the editing plasmid could be achieved by one round of screening on the antibiotic-free plate (Fig. 2E), thus reducing the dependence on counterselection marker.
The endogenous CRISPR-based editing tool could be used to delete toxic genes or to enhance pathways of beneficial products in P. acidilactici species. In wild-type strain LA412, the native plasmid 2 encodes addiction module killer protein (GE00032), which is annotated as phage-derived Gp49-like protein (DUF891). This protein exhibits 67.52% amino acid sequence identity with type II toxin-antitoxin system RelE/ParE family toxin RelE, and RelE with mRNA endonuclease activity can inhibit protein synthesis (39, 40). Considering this, we hypothesized that removing native plasmid 2 might be beneficial for cell growth. As shown in Fig. 3E, the depletion of native plasmids indeed enhanced cell growth, confirming the value of the genetic tool established in this study. This plasmid-curing strain has high industrial application value due to its growth advantage, shorter production cycle, and lower production costs. Moreover, the lactic acid production pathway was strengthened via the endogenous CRISPR-based tool in LA412. The NADH-dependent LDHs contribute to NAD+ regeneration and the intracellular NADH/NAD+ balance during sugar catabolism (41). Therefore, an NADH-dependent ldh gene expression cassette was integrated onto the chromosome based on the genetic tool we established. The result indicated that the integration and overexpression of the NADH-dependent ldh gene in strain LA412 significantly enhanced cell growth and LA production (Fig. 5E and F). Overall, the endogenous CRISPR-based genome editing tool could make P. acidilactici a sophisticated chassis species for synthetic biology and bioindustry.
MATERIALS AND METHODS
Strains, culture conditions, and transformation.
P. acidilactici LA412 was recently isolated from the fermented soybean product in our laboratory. Strain LA412 and the derived strains (Table 1) were cultured in the modified MRS medium (5 g/liter glucose, 8 g/liter beef powder, 10 g/liter peptone, 4 g/liter yeast extract, 3 g/liter sodium acetate, 2 g/liter K2HPO4, 2 g/liter ammonium citrate dibasic, 1 g/liter Tween 80, 0.20 g/liter MgSO4·7H2O, and 0.05 g/liter MnSO4·H2O) at 37°C. Uracil (U) and 5-fluoroorotic acid (5-FOA) were added into the MRS medium when required. To prepare the competent cells, a single colony of strain LA412 on the modified MRS medium plate was transferred into 5 ml modified MRS liquid medium and cultured overnight at 37°C to obtain the seed culture. Then, 20 ml modified MRS liquid medium containing d/l-threonine (3%) inoculated with the seed culture (2%) was cultured to OD600 of 1.50 under the conditions of 37°C and 180 rpm. Then, cells were centrifuged, and the pellets were resuspended with 20 ml prechilled buffer I (205.37 g/liter sucrose, 0.20 g/liter MgCl2·6H2O, and 1.86 g/liter K3PO4·3H2O, pH 7.5). This process was conducted twice. Then, the pellets were resuspended in 1 ml prechilled buffer I containing prechilled lysozyme at a final concentration of 0.1 mg/ml. After incubation at 37°C for 30 min, cells were centrifuged and resuspended with 20 ml prechilled buffer solution I twice. Finally, the cells were resuspended with 1 ml prechilled buffer solution II (171.15 g/liter sucrose, 10% glycerin), and aliquots (80 μl) were stored at −80°C or immediately used for electroporation. Centrifugation was conducted at 6,000 × g at 4°C for 5 min.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Characteristic | Source |
|---|---|---|
| Strains | ||
| E. coli Trans10 | Host for plasmid construction | TransGen Biotech |
| P. acidilactici LA412 | P. acidilactici containing a complete II-A CRISPR-Cas system | Lab storage |
| P. acidilactici LA412-ΔpyrE | P. acidilactici LA412 with pyrE gene deletion | This work |
| P. acidilactici LA412-ΔP1P2 | P. acidilactici LA412 with native plasmid depletion | This work |
| P. acidilactici LA412-mpi(XbaI) | P. acidilactici LA412 with point mutation at the mpi gene locus | This work |
| P. acidilactici LA412cas9 | P. acidilactici LA412 containing the corrected cas9 gene | This work |
| P. acidilactici LA412::ldh | P. acidilactici LA412 integrated with ldh expression cassette | This work |
| Plasmids | ||
| pMG36e | Expression vector for P. acidilactici | Lab storage |
| pMG36e-C | Interference plasmid backbone with one mini-CRISPR cassette | This work |
| pMG36e-PS1 | Challenging plasmid without PAM sequence | This work |
| pMG36e-PS1-NGG | Challenging plasmid containing a protospacer followed by NGG PAM (N = A, T, G, or C) | This work |
| pMG36e-SpyrE (NGG) | Interference plasmid targeting a protospacer followed by an adjacent NGG PAM (N = A, T, G, or C) at the pyrE gene locus | This work |
| pMG36e-SpyrE-LR (NGG) | Gene deletion plasmid targeting a protospacer followed by an adjacent NGG PAM (N = A, T, G, or C) at the pyrE gene locus | This work |
| pMG36e-Srep | Interference plasmid targeting two native plasmids | This work |
| pMG36e-Smpi-LR | Editing plasmid for point mutation at mpi gene locus | This work |
| pMG36e-Scas9-LR | Editing plasmid for cas9 gene correction | This work |
| pMG36e-Sun-LR::P0594ldh | Editing plasmid for integration of ldh expression cassette | This work |
For electroporation, 500 ng plasmids were added into the cuvette with 80 μl competent cells. Then, the electroporation was conducted under the conditions of 2,500 V, 25 μF, and 200 Ω. Immediately after electroporation, 900 μl prechilled modified MRS liquid medium supplemented with 171.15 g/liter sucrose was added into the cuvette and placed in an ice bath for 5 min. Then, the cells were transferred into a 1.50-ml centrifuge tube and incubated for 6 h at 37°C with shaking at 180 rpm. Finally, 100 μl culture was spread on agar plate with modified MRS medium containing 5 μg/ml erythromycin, and the plates were incubated at 37°C for 2 days.
Genomic DNA extraction, sequencing, and annotation.
Five milliliters of strain LA412 cell culture were centrifuged, and the pellets were resuspended with 180 μl prechilled lysozyme (20 mg/ml) and then placed in the 37°C incubator for 2 h. Total DNA extraction was next conducted with the DNA extraction kit (Tiangen), and the DNA extracts were sequenced by Nanopore high-throughput sequencing technology. A total of 1,195,170 sequence reads were quality controlled and assembled using Canu v1.5 software (42). Then, the coding gene and CRISPR array on the assembled genome were predicted using Prodigal (43) and CRT software (44), respectively. Furthermore, the gene function annotation results were obtained by comparing the predicted gene sequences against the nr database (45) using BLAST (46).
Plasmid construction.
To construct the challenging plasmids to be targeted and cleaved by the host, the cassettes of PS1-NGG in which the PS1 (protospacer 1) matched the first spacer (spacer 1) at the CRISPR locus in LA412 were cloned between XbaI and HindIII restriction sites on the pMG36e shuttle vector (Table 1). The construction of interference plasmids (targeting and cleaving genomic DNA) and the editing plasmids (targeting and editing genomic DNA such as knockout, knock-in, and point mutation) were based on pMG36e. A DNA fragment containing a mini-CRISPR (repeat-BbsI-repeat-terminator) was artificially synthesized and cloned between XbaI and PstI restriction sites on pMG36e to obtain pMG36e-C. To construct the interference plasmids, 30-bp spacers (which matched with the target DNA sequences followed by an NGG PAM) were cloned between two BbsI sites on pMG36e-C. To construct the gene deletion plasmids, the upstream/downstream donor DNA sequences of the target sites were cloned between PstI and HindIII sites on the interference plasmids, respectively. To construct the integration plasmids, the upstream donor DNA, the DNA sequences to be inserted into the genome, and the downstream donor DNA were amplified using splicing by overhang extension PCR (SOE PCR), and then amplification products were cloned between PstI and HindIII sites on the interference plasmids. Specifically, the ldh gene expression cassette in LA412, including its promoter and the open reading frame, was integrated into the genome for overexpression of lactate dehydrogenase. To construct the editing plasmids for point mutation, the donor DNAs with the desired nucleotide covering the target regions were cloned into the interference plasmids. All the primers used were listed in Table 2.
TABLE 2.
Primers used in this study
| Primer | Sequences (5′–3′) |
|---|---|
| Primers for cloning the challenging plasmids with different PAMs | |
| PS1-F | CTAG CCCTTCATCATTATCAACTTGAAAGGGGTA |
| PS1-R | AGCT TACCCCTTTCAAGTTGATAATGATGAAGGG |
| PS1-AGG-F | CTAG CCCTTCATCATTATCAACTTGAAAGGGGTA AGG |
| PS1-AGG-R | AGCT CCT TACCCCTTTCAAGTTGATAATGATGAAGGG |
| PS1-TGG-F | CTAG CCCTTCATCATTATCAACTTGAAAGGGGTA TGG |
| PS1-TGG-R | AGCT CCA TACCCCTTTCAAGTTGATAATGATGAAGGG |
| PS1-GGG-F | CTAG CCCTTCATCATTATCAACTTGAAAGGGGTA GGG |
| PS1-GGG-R | AGCT CCC TACCCCTTTCAAGTTGATAATGATGAAGGG |
| PS1-CGG-F | CTAG CCCTTCATCATTATCAACTTGAAAGGGGTA CGG |
| PS1-CGG-R | AGCT CCG TACCCCTTTCAAGTTGATAATGATGAAGGG |
| Primers for cloning the interference plasmids targeting the pyrE gene | |
| SpyrE (AGG)-F | GATC CAAACCCGCGTTACGTAAACAAATTGCCAC |
| SpyrE (AGG)-R | AAAC GTGGCAATTTGTTTACGTAACGCGGGTTTG |
| SpyrE (TGG)-F | GATC GGCAACGGTGATCGGCGGGGTGGCAACGGC |
| SpyrE (TGG)-R | AAAC GCCGTTGCCACCCCGCCGATCACCGTTGCC |
| SpyrE (GGG)-F | GATC CGTCCGTTCTAAACCCAAGGATCACGGAGC |
| SpyrE (GGG)-R | AAAC GCTCCGTGATCCTTGGGTTTAGAACGGACG |
| SpyrE (CGG)-F | GATC AACAACAACTAACTCCTGCCCAGCTAGATT |
| SpyrE (CGG)-R | AAAC AATCTAGCTGGGCAGGAGTTAGTTGTTGTT |
| Primers for deletion of the pyrE gene | |
| L(pyrE, 50)-F | AA CTGCAG TTTTTGTACCCAAAATAGCGA |
| R(pyrE, 50)-R | CCC AAGCTT GATTTAATTGAAACTAAATCTTATT |
| L(pyrE, 100)-F | AA CTGCAG AGTAGGTGGAATGGACGCG |
| R(pyrE, 100)-R | CCC AAGCTT TTGGAAGGTAGCCTAAAAAAGC |
| L(pyrE, 200)-F | AA CTGCAG GTGGCTTGACATTTAATTTCAGG |
| R(pyrE, 200)-R | CCC AAGCTT CGTCCGCTTCGTTAAAGTTT |
| L(pyrE, 500)-F | AA CTGCAG ATCACAAAATTGGACAACAACC |
| R(pyrE, 500)-R | CCC AAGCTT CTAAATTCTCGTCCAAAATTGG |
| L(pyrE, 700)-F | AA CTGCAG GGCTGGGGGGCATGGAC |
| R(pyrE, 700)-R | CCC AAGCTT ACCCGCAAAGTAGACTTGTGC |
| L(pyrE, 1,000)-F | AA CTGCAG CCCTTAAACCGGCCTTACTG |
| R(pyrE, 1,000)-R | CCC AAGCTT GGTCATTTGTTTAAAGATCCGTG |
| L(pyrE)-R | AGTTGCGCCCCATTG GTTTGAACCCCTTTATTTGGATTTC |
| R(pyrE)-F | TAAAGGGGTTCAAAC CAATGGGGCGCAACTACAAC |
| pyrE-F | AACATGGTGCCTAGTGACGCC |
| pyrE-R | GCATTCCACCAGTTTGCTTTC |
| Primers for depletion of native plasmids | |
| Srep-F | GATC CTACTTATACGGCACCATGCAAAACCTATT |
| Srep-R | AAAC AATAGGTTTTGCATGGTGCCGTATAAGTAG |
| rep-F | CCAAATAGGTTTTGCATGGT |
| rep-R | CATGCTAATGACTTCTTAACTGAT |
| 0037-F | ATGAGCACCACTATTTTATCATT |
| 0037-R | TTAATGACGTAAATTAAGCAAGA |
| 0033-F | TTGGCAGTTAAGAGTATTGGATC |
| 0033-R | TCACAAACATTGAACATTTTTAGC |
| Primers for point mutation at the mpi gene | |
| Smpi-F | GATC GTACCATAATGCCCTTGCCCAACGCATGAA |
| Smpi-R | AAAC TTCATGCGTTGGGCAAGGGCATTATGGTAC |
| L(mpi)-F | AA CTGCAG GCCAGGCTCGCTAACGA |
| L(mpi)-R | GACTAAAATTCTAGA TGCTAACCAAGACTTGTCCG |
| R(mpi)-F | AAGTCTTGGTTAGCA TCTAGAATTTTAGTCAATAAAGGA |
| R(mpi)-R | CCC AAGCTT CGCCTCCCTGGGAGCAC |
| mpi-F | AAAGTGGTCACCGGAAAC |
| mpi-R | AGCTCACCCAGAGCACG |
| Primers for correction of the cas9 gene | |
| Scas9-F | GATC CGCACCTTGCCGAGGTGGACCCCACCTTTT |
| Scas9-R | AAAC AAAAGGTGGGGTCCACCTCGGCAAGGTGCG |
| L(cas9)-F | AA CTGCAG GGATTGATGAATTTAGTGCTAATAA |
| L(cas9)-R | TGGTTTAGCCGGTCA AATAACGTATCAAGCTCAATATTT |
| R(cas9)-F | GCTTGATACGTTATT TGACCGGCTAAACCAGCTTTATGA |
| R(cas9)-R | GG GGTACC TGCCCCATTTTGCCCAGT |
| Primers for integration of the ldh expression cassette | |
| Sun-F | GATC TCAAATTCGCAAAATATCTTGTTATCTAAA |
| Sun-R | AAAC TTTAGATAACAAGATATTTTGCGAATTTGA |
| L(un)-F | GG GGTACC CAATTTCAGCATTGCTTAACAAA |
| L(un)-R | CTCAAGCTGAAGGGG AATAAACAAAAACGGGCATACTG |
| 0594ldh-F | CCGTTTTTGTTTATT CCCCTTCAGCTTGAGCTC |
| 0594ldh-R | TATGTATCACAAGAT TTAGTGGTGGTGGTGGTGGTG TTTGTCTTGTTTTTCAGCAAGA |
| R(un)-F | CACCACCACCACTAA ATCTTGTGATACATAGGGGGGA |
| R(un)-R | GG GGTACC TCGCTTAGGAGGCGATTG |
| ldh-F | TCCGCAAAACGATTACGG |
| ldh-R | CCTTTGTTATTAGCGTTTCAAGC |
| Primers for validation of plasmid curing | |
| emr-F | TCGACCCATATTTAAAAAGC |
| emr-R | AGTTTATGCATCCCTTAACTTA |
Measurement of the interference and editing efficiency.
The empty plasmid pMG36e, challenging plasmids, interfering plasmids, and editing plasmids were electroporated into P. acidilactici LA412 competent cells. The colonies on the plates containing 5 μg/ml erythromycin were counted after 48 h incubation. The colony number in the control group (pMG36e) was defined as X1, and that in the challenging plasmid group and interference plasmid group was defined as X2. The number of colonies carrying the editing plasmid was defined as X3, and the number of colonies with the edited gene was defined as X4. Each electroporation was performed in triplicate. Interference efficiency and editing efficiency were calculated in the following formulas.
Pure-genotype strain screening and plasmid curing.
The mixed-genotype colonies carrying both the wild-type and edited gene were isolated from selection plates. These cells were transferred into modified MRS liquid medium without antibiotics every 6 h. After several passages, cell culture was streaked on agar plate containing modified MRS medium without antibiotics. Then, the targeted genes of several single colonies were identified through colony PCR. DNA sequencing was conducted to further confirm the gene-edited single colonies (pure genotype). In addition, the erythromycin resistance gene emr in the pMG36e plasmid was amplified to determine the presence of the editing plasmid in the cells. Generally, the pure-genotype cells without editing plasmid were obtained after 1 to 3 rounds of screening. The verification primers are listed in Table 2.
Native plasmid depletion.
One spacer which matched with two identical 30-bp protospacers followed by a TGG PAM on two replication protein genes (GE00014 on plasmid 1 and GE00039 on plasmid 2) was cloned into pMG36e-C to obtain interference plasmid pMG36e-Srep. The resultant pMG36e-Srep was transformed into P. acidilactici LA412 to cleave both native plasmids. To verify the presence of the native plasmids, GE00014 and GE00039 genes on two endogenous plasmids were amplified with primer pair rep-F/rep-R. The GE00037 gene on plasmid P1 and GE00033 gene on plasmid P2 were also amplified with primer pairs (0037-F/0037-R and 0033-F/0033-R) for further verification. The verification primers are presented in Table 2.
Lactic acid fermentation.
Lactic acid fermentation medium contained 25 g/liter glucose, 5 g/liter yeast extract, 5 g/liter CH3COONa, 2 g/liter ammonium citrate dibasic, 2 g/liter K2HPO4, 0.58 g/liter MgSO4·7H2O, 0.25 g/liter MnSO4·H2O, and 0.6 g CaCO3/g glucose. Fermentation was performed in 100 ml MRS medium inoculated with 2% seed solution (OD600 of 2.50) at 37°C and 180 rpm. The fermentation cultures were sampled every 6 to 12 h.
High-performance liquid chromatography analysis.
Detection of glucose and LA concentrations was conducted as described previously (47).
Growth curve measurement.
The fresh cell culture was inoculated into 20 ml modified MRS and incubated at 37°C overnight to obtain the seed culture. Until optical density at 600 nm (OD600) reached 1.50, 1% seed culture was then inoculated into 100 ml modified MRS medium and incubated at 37°C. The OD600 value of the culture was determined every hour. The experiment was conducted in triplicate.
Western blotting.
Cells (20 ml) were cultured to the end of the logarithmic phase (OD600, of 2.50) and then centrifuged. The cell pellets were resuspended with 350 μl lysozyme solution (20 mg/ml) and incubated at 37°C for 2 h. Afterward, 1.70 ml 2×HEPES buffer was added. Next, the mixture was centrifuged (10,000 × g, 4°C, 30 min) after ultrasonic wave treatment, and the supernatant was separated through 12% SDS-PAGE. Protein was transferred to the nitrocellulose membrane, and antibody reaction and visualization were conducted as reported previously (48).
Data availability.
The genomic sequence was uploaded to the SRA database (accession number SRR14308572).
ACKNOWLEDGMENTS
This work was supported by the Special Fund for Agro-scientific Research in the Public Interest (no. 201503137 to N.P.), the National Natural Science Foundation of China (nos. 31671291 to N.P., 32070036 to M.H., and 31900400 to T.L.), and the Fundamental Research Funds for the Central Universities (no. 2662019PY028 to N.P.).
We declare that we have no conflict of interest.
Footnotes
Supplemental material is available online only.
Contributor Information
Nan Peng, Email: nanp@mail.hzau.edu.cn.
Edward G. Dudley, The Pennsylvania State University
REFERENCES
- 1.Makarova K, Slesarev A, Wolf Y, Sorokin A, Mirkin B, Koonin E, Pavlov A, Pavlova N, Karamychev V, Polouchine N, Shakhova V, Grigoriev I, Lou Y, Rohksar D, Lucas S, Huang K, Goodstein DM, Hawkins T, Plengvidhya V, Welker D, Hughes J, Goh Y, Benson A, Baldwin K, Lee JH, Diaz-Muniz I, Dosti B, Smeianov V, Wechter W, Barabote R, Lorca G, Altermann E, Barrangou R, Ganesan B, Xie Y, Rawsthorne H, Tamir D, Parker C, Breidt F, Broadbent J, Hutkins R, O'Sullivan D, Steele J, Unlu G, Saier M, Klaenhammer T, Richardson P, Kozyavkin S, Weimer B, Mills D. 2006. Comparative genomics of the lactic acid bacteria. Proc Natl Acad Sci USA 103:15611–15616. 10.1073/pnas.0607117103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kleerebezem M, Kuipers OP, Smid EJ. 2017. Editorial: lactic acid bacteria-a continuing journey in science and application. FEMS Microbiol Rev 41:S1–S2. 10.1093/femsre/fux036. [DOI] [PubMed] [Google Scholar]
- 3.Mokoena MP. 2017. Lactic acid bacteria and their bacteriocins: classification, biosynthesis and applications against uropathogens: a mini-review. Molecules 22:1255. 10.3390/molecules22081255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Vos WM. 2011. Systems solutions by lactic acid bacteria: from paradigms to practice. Microb Cell Fact 10(Suppl 1):S2. 10.1186/1475-2859-10-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Filannino P, Di Cagno R, Gobbetti M. 2018. Metabolic and functional paths of lactic acid bacteria in plant foods: get out of the labyrinth. Curr Opin Biotechnol 49:64–72. 10.1016/j.copbio.2017.07.016. [DOI] [PubMed] [Google Scholar]
- 6.Gaspar P, Carvalho AL, Vinga S, Santos H, Neves AR. 2013. From physiology to systems metabolic engineering for the production of biochemicals by lactic acid bacteria. Biotechnol Adv 31:764–788. 10.1016/j.biotechadv.2013.03.011. [DOI] [PubMed] [Google Scholar]
- 7.Lee Y-S, Kim T-Y, Kim Y, Lee S-H, Kim S, Kang SW, Yang J-Y, Baek I-J, Sung YH, Park Y-Y, Hwang SW, O E, Kim KS, Liu S, Kamada N, Gao N, Kweon M-N. 2018. Microbiota-derived lactate accelerates intestinal stem-cell-mediated epithelial development. Cell Host Microbe 24:833–846.e6. 10.1016/j.chom.2018.11.002. [DOI] [PubMed] [Google Scholar]
- 8.Wu H, Xie S, Miao J, Li Y, Wang Z, Wang M, Yu Q. 2020. Lactobacillus reuteri maintains intestinal epithelial regeneration and repairs damaged intestinal mucosa. Gut Microbes 11:997–1014. 10.1080/19490976.2020.1734423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Pijkeren JP, Barrangou R. 2017. Genome editing of food-grade lactobacilli to develop therapeutic probiotics. Microbiol Spectr 5. 10.1128/microbiolspec.BAD-0013-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chowdhury MY, Li R, Kim JH, Park ME, Kim TH, Pathinayake P, Weeratunga P, Song MK, Son HY, Hong SP, Sung MH, Lee JS, Kim CJ. 2014. Mucosal vaccination with recombinant Lactobacillus casei-displayed CTA1-conjugated consensus matrix protein-2 (sM2) induces broad protection against divergent influenza subtypes in BALB/c mice. PLoS One 9:e94051. 10.1371/journal.pone.0094051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiao H, Wang Q, Bang-Berthelsen CH, Jensen PR, Solem C. 2020. Harnessing adaptive evolution to achieve superior mannitol production by Lactococcus lactis using its native metabolism. J Agric Food Chem 68:4912–4921. 10.1021/acs.jafc.0c00532. [DOI] [PubMed] [Google Scholar]
- 12.Plavec TV, Berlec A. 2019. Engineering of lactic acid bacteria for delivery of therapeutic proteins and peptides. Appl Microbiol Biotechnol 103:2053–2066. 10.1007/s00253-019-09628-y. [DOI] [PubMed] [Google Scholar]
- 13.Bhaya D, Davison M, Barrangou R. 2011. CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annu Rev Genet 45:273–297. 10.1146/annurev-genet-110410-132430. [DOI] [PubMed] [Google Scholar]
- 14.Adli M. 2018. The CRISPR tool kit for genome editing and beyond. Nat Commun 9:1911. 10.1038/s41467-018-04252-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Y, Pan S, Zhang Y, Ren M, Feng M, Peng N, Chen L, Liang YX, She Q. 2016. Harnessing type I and type III CRISPR-Cas systems for genome editing. Nucleic Acids Res 44:e34. 10.1093/nar/gkv1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choudhary E, Thakur P, Pareek M, Agarwal N. 2015. Gene silencing by CRISPR interference in mycobacteria. Nat Commun 6:6267. 10.1038/ncomms7267. [DOI] [PubMed] [Google Scholar]
- 17.Hsu PD, Lander ES, Zhang F. 2014. Development and applications of CRISPR-Cas9 for genome engineering. Cell 157:1262–1278. 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roberts A, Barrangou R. 2020. Applications of CRISPR-Cas systems in lactic acid bacteria. FEMS Microbiol Rev 44:523–537. 10.1093/femsre/fuaa016. [DOI] [PubMed] [Google Scholar]
- 19.Oh JH, van Pijkeren JP. 2014. CRISPR-Cas9-assisted recombineering in Lactobacillus reuteri. Nucleic Acids Res 42:e131. 10.1093/nar/gku623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Song X, Huang H, Xiong Z, Ai L, Yang S. 2017. CRISPR-Cas9(D10A) nickase-assisted genome editing in Lactobacillus casei. Appl Environ Microbiol 83:e01259-17. 10.1128/AEM.01259-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou D, Jiang Z, Pang Q, Zhu Y, Wang Q, Qi Q. 2019. CRISPR/Cas9-assisted seamless genome editing in Lactobacillus plantarum and its application in N-acetylglucosamine production. Appl Environ Microbiol 85:e01367-19. 10.1128/AEM.01367-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vento JM, Crook N, Beisel CL. 2019. Barriers to genome editing with CRISPR in bacteria. J Ind Microbiol Biotechnol 46:1327–1341. 10.1007/s10295-019-02195-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y, Peng N. 2019. Endogenous CRISPR-Cas system-based genome editing and antimicrobials: review and prospects. Front Microbiol 10:2471. 10.3389/fmicb.2019.02471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang J, Zong W, Hong W, Zhang ZT, Wang Y. 2018. Exploiting endogenous CRISPR-Cas system for multiplex genome editing in Clostridium tyrobutyricum and engineer the strain for high-level butanol production. Metab Eng 47:49–59. 10.1016/j.ymben.2018.03.007. [DOI] [PubMed] [Google Scholar]
- 25.Crawley AB, Henriksen ED, Stout E, Brandt K, Barrangou R. 2018. Characterizing the activity of abundant, diverse and active CRISPR-Cas systems in lactobacilli. Sci Rep 8:11544. 10.1038/s41598-018-29746-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hidalgo-Cantabrana C, Goh YJ, Pan M, Sanozky-Dawes R, Barrangou R. 2019. Genome editing using the endogenous type I CRISPR-Cas system in Lactobacillus crispatus. Proc Natl Acad Sci USA 116:15774–15783. 10.1073/pnas.1905421116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hidalgo-Cantabrana C, O'Flaherty S, Barrangou R. 2017. CRISPR-based engineering of next-generation lactic acid bacteria. Curr Opin Microbiol 37:79–87. 10.1016/j.mib.2017.05.015. [DOI] [PubMed] [Google Scholar]
- 28.Bosma EF, Forster J, Nielsen AT. 2017. Lactobacilli and pediococci as versatile cell factories - evaluation of strain properties and genetic tools. Biotechnol Adv 35:419–442. 10.1016/j.biotechadv.2017.04.002. [DOI] [PubMed] [Google Scholar]
- 29.Chelliah R, Saravanakumar K, Daliri EB, Kim JH, Lee JK, Jo HY, Kim SH, Ramakrishnan SR, Madar IH, Wei S, Rubab M, Barathikannan K, Ofosu FK, Subin H, Eun-Ji P, Yeong JD, Elahi F, Wang MH, Park JH, Ahn J, Kim DH, Park SJ, Oh DH. 2020. Unveiling the potentials of bacteriocin (Pediocin L50) from Pediococcus acidilactici with antagonist spectrum in a Caenorhabditis elegans model. Int J Biol Macromol 143:555–572. 10.1016/j.ijbiomac.2019.10.196. [DOI] [PubMed] [Google Scholar]
- 30.Porto MC, Kuniyoshi TM, Azevedo PO, Vitolo M, Oliveira RP. 2017. Pediococcus spp.: an important genus of lactic acid bacteria and pediocin producers. Biotechnol Adv 35:361–374. 10.1016/j.biotechadv.2017.03.004. [DOI] [PubMed] [Google Scholar]
- 31.Kim WS, Lee JY, Singh B, Maharjan S, Hong L, Lee SM, Cui LH, Lee KJ, Kim G, Yun CH, Kang SK, Choi YJ, Cho CS. 2018. A new way of producing pediocin in Pediococcus acidilactici through intracellular stimulation by internalized inulin nanoparticles. Sci Rep 8:5878. 10.1038/s41598-018-24227-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mehdinejad N, Imanpour MR, Jafari V. 2018. Combined or individual effects of dietary probiotic Pedicoccus acidilactici and nucleotide on growth performance, intestinal microbiota, hemato-biochemical parameters, and innate immune response in goldfish (Carassius auratus). Probiotics Antimicrob Prot 10:558–565. 10.1007/s12602-017-9297-3. [DOI] [PubMed] [Google Scholar]
- 33.Lei Z, Wu Y, Nie W, Yin D, Yin X, Guo Y, Aggrey SE, Yuan J. 2018. Transcriptomic analysis of xylan oligosaccharide utilization systems in Pediococcus acidilactici strain BCC-1. J Agric Food Chem 66:4725–4733. 10.1021/acs.jafc.8b00210. [DOI] [PubMed] [Google Scholar]
- 34.Soltani M, Badzohreh G, Mirzargar S, Farhangi M, Shekarabi PH, Lymbery A. 2019. Growth behavior and fatty acid production of probiotics, Pediococcus acidilactici and Lactococcus lactis, at different concentrations of fructooligosaccharide: studies validating clinical efficacy of selected synbiotics on growth performance of Caspian roach (Rutilus frisii kutum) fry. Probiotics Antimicrob Proteins 11:765–773. 10.1007/s12602-018-9462-3. [DOI] [PubMed] [Google Scholar]
- 35.Yi X, Zhang P, Sun J, Tu Y, Gao Q, Zhang J, Bao J. 2016. Engineering wild-type robust Pediococcus acidilactici strain for high titer L- and D-lactic acid production from corn stover feedstock. J Biotechnol 217:112–121. 10.1016/j.jbiotec.2015.11.014. [DOI] [PubMed] [Google Scholar]
- 36.Donohoue PD, Barrangou R, May AP. 2018. Advances in industrial biotechnology using CRISPR-Cas systems. Trends Biotechnol 36:134–146. 10.1016/j.tibtech.2017.07.007. [DOI] [PubMed] [Google Scholar]
- 37.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821. 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jonuscheit M, Martusewitsch E, Stedman KM, Schleper C. 2003. A reporter gene system for the hyperthermophilic archaeon Sulfolobus solfataricus based on a selectable and integrative shuttle vector. Mol Microbiol 48:1241–1252. 10.1046/j.1365-2958.2003.03509.x. [DOI] [PubMed] [Google Scholar]
- 39.Harms A, Brodersen DE, Mitarai N, Gerdes K. 2018. Toxins, targets, and triggers: an overview of toxin-antitoxin biology. Mol Cell 70:768–784. 10.1016/j.molcel.2018.01.003. [DOI] [PubMed] [Google Scholar]
- 40.Zhang J, Ito H, Hino M, Kimura M. 2017. A RelE/ParE superfamily toxin in Vibrio parahaemolyticus has DNA nicking endonuclease activity. Biochem Biophys Res Commun 489:29–34. 10.1016/j.bbrc.2017.05.105. [DOI] [PubMed] [Google Scholar]
- 41.Kasai T, Suzuki Y, Kouzuma A, Watanabe K. 2019. Roles of d-lactate dehydrogenases in the anaerobic growth of Shewanella oneidensis MR-1 on sugars. Appl Environ Microbiol 85:e02668-18. 10.1128/AEM.02668-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. 2017. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 27:722–736. 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bland C, Ramsey TL, Sabree F, Lowe M, Brown K, Kyrpides NC, Hugenholtz P. 2007. CRISPR Recognition Tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics 8:209. 10.1186/1471-2105-8-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yangyang D, Li J, Wu S, Zhu Y, Chen Y, He F. 2006. Integrated nr database in protein annotation system and its localization. Computer Engineering 32:71. [Google Scholar]
- 46.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hu J, Lin Y, Zhang Z, Xiang T, Mei Y, Zhao S, Liang Y, Peng N. 2016. High-titer lactic acid production by Lactobacillus pentosus FL0421 from corn stover using fed-batch simultaneous saccharification and fermentation. Bioresour Technol 214:74–80. 10.1016/j.biortech.2016.04.034. [DOI] [PubMed] [Google Scholar]
- 48.Liu T, Li Y, Wang X, Ye Q, Li H, Liang Y, She Q, Peng N. 2015. Transcriptional regulator-mediated activation of adaptation genes triggers CRISPR de novo spacer acquisition. Nucleic Acids Res 43:1044–1055. 10.1093/nar/gku1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 to S3. Download AEM.00948-21-s0001.pdf, PDF file, 0.3 MB (316KB, pdf)
Data Availability Statement
The genomic sequence was uploaded to the SRA database (accession number SRR14308572).