

Abstract
Neurofibrillary tangles, one of the neuropathologic hallmarks of Alzheimer's disease, have a dynamic lifespan of maturity that associates with progressive neuronal dysfunction and cognitive deficits. As neurofibrillary tangles mature, the biology of the neuron undergoes extensive changes that may impact biomarker recognition and therapeutic targeting. Neurofibrillary tangle maturity encompasses three levels: pretangles, mature tangles, and ghost tangles. In this review, we detail distinct and overlapping characteristics observed in the human brain regarding morphologic changes the neuron undergoes, conversion from intracellular to extracellular space, tau immunostaining patterns, and tau isoform expression changes across the lifespan of the neurofibrillary tangle. Post‐translational modifications of tau such as phosphorylation, ubiquitination, conformational events, and truncations are discussed to contextualize tau immunostaining patterns. We summarize accumulated and emerging knowledge of neurofibrillary tangle maturity, discuss the current tools used to interpret the dynamic nature in the postmortem brain, and consider implications for cognitive dysfunction and tau biomarkers.
Keywords: Alzheimer's disease, neurofibrillary tangles, neuropathology, tau positron emission tomography, tau
1. NARRATIVE
There are many ways to approach science and even more ways to answering the unanswered questions of the brain. A particular fascination is how specific cells of the brain are selectively vulnerable to the devastation of disease, but do not become affected until later in life. Alzheimer's disease (AD) is a prime example, as targeted populations of neurons undergo degenerative changes that lead to cognitive dysfunction and brain volume loss. To study these changes in the postmortem brain, an array of specialized stains and antibody‐based methods are used to uncover lesion types and patterns of involvement. AD is defined by the abnormal accumulation of two proteins, amyloid beta (Aβ) and hyperphosphorylated tau. Microscopic inspection of Aβ plaque pathology is observed to accumulate outside of neurons, where plaques interfere with their ability to communicate. Aβ plaque deposition is highly predictable and is quite useful in monitoring the disease course using targeted positron emission tomography (PET) or cerebrospinal fluid (CSF) measures. In contrast, hyperphosphorylated tau accumulates inside of neurons through a series of maturity levels. During this maturation process, neurofibrillary tangles (NFTs) form inside the affected neuron resulting in axonal instability that impairs transport of nutrients and cell signal communication. Advances in PET imaging allow for the detection of abnormal tau accumulation in the living brain. This is complemented by blood and CSF biomarker studies on tau that hold great promise for identifying early changes in asymptomatic individuals and mild cognitive impairment patients.
Several PET radioligands and fluid biomarkers have recently emerged that target various aspects of abnormal tau accumulation. To understand differences in tau PET patterns using different radioligands or the temporal sequence of fluid biomarkers, it may be helpful to gain a broad perspective of NFT maturity and tau biology. Neurofibrillary references the appearance of tau fibers that form in the neuron and tangle refers to the twisted nature of these fibers into a mass that builds up inside the neuron. Tau functions to stabilize microtubules, which enables smooth transport of nutrients and messages passed from neuron to neuron. As abnormal tau undergoes many modifications, it accumulates and becomes unable to stabilize microtubules, leading to the death of the neuron. NFTs have a lifespan that is defined through three levels of maturity, which mark the changes that the neuron undergoes as abnormal tau accumulates. At first, the abnormal tau deposits in tiny granular accumulations outside the nucleus of the cell. There is debate whether this initial maturity level, called pretangles, may even be a protective state during which the neuron is defending itself from becoming imbalanced. The neuron's attempt to offset the imbalance may eventually lead to an ill‐fated rescue attempt as abnormal tau begins to overwhelm the neuron. The granular accumulations form into fibers that fill the neuron, which develops into the mature tangle. The nucleus of the mature tangle‐bearing neuron becomes displaced and shrunken as transport machinery fails. As time passes the neuron dies, but the tau fibers remain as a tombstone forming a ghost tangle.
Although the process of NFT maturity is dynamic, investigating the postmortem brain under the microscope is like seeing a still frame of a movie. The goal of histologic evaluation, much like Mary Shelley's Dr. Frankenstein, is to bring life back to the postmortem brain. Although not in the literal sense, the “life” brought to the histologic observer in the context of NFT maturity is the understanding of where in the lifespan of maturity was a neuron at the time of the patient's death. This may be especially important in neuroimaging studies investigating the relationship between tau pathology and structural changes. Although one may observe tau accumulation in the medial temporal lobe, atrophy may not be expected if all that is observed is pretangle pathology. In contrast, if a tau PET radioligand only recognizes ghost tangles, the tau PET signal would not be expected early in a patient's disease course.
The goal of this review is to highlight the dynamic nature of the NFT by discussing changes that occur along its lifespan. Morphologic changes at each maturity level are first discussed to provide distinct aspects of pretangles, mature tangles, and ghost tangles. Next, commonly used and historically relevant visualization techniques are reviewed to provide confidence when examining changes under the microscope. Importantly, these visualization techniques recognize a small window in the NFT lifespan and there are currently no markers that recognize a single NFT maturity level. One key element of NFT maturity is that it occurs over time, suggesting a therapeutic window of opportunity early in the development. To outline the changes in tau biology in the context of NFT maturity, post‐translational events and isoform dominance are reviewed. Last, the implications of NFT maturity in disease progression are brought into the context of neuroimaging and fluid biomarkers. A holistic knowledge of the dynamic changes to the neuron, rather than considering a tangle as a binary object that exists or does not exist, may have far‐reaching implications for the development of treatments to aid in preventing AD and associated syndromes.
2. INTRODUCTION
Alzheimer's disease (AD) is a multiproteinopathy that is characterized by the abnormal accumulation of two hallmark neuropathologies: amyloid beta (Aβ) plaques and NFTs composed of tau. 1 , 2 Aβ plaques form in the extracellular space and contribute to synaptic dysfunction, 3 whereas NFTs accumulate progressively in the intracellular space and associate with the death of the neuron. 4 , 5 , 6 Studies in transgenic mice showed Aβ plaques forming within 24‐hour time periods using in vivo multiphoton imaging 7 and generally remained morphologically stable for up to 5 months. 8 In contrast, modeling in human brains showed neurons can survive 20 years with NFTs 6 with functional capabilities that suggest a protracted viability of tangle‐bearing neurons. 9 The inherent cross‐sectional design of human autopsy studies somewhat limits our ability to interpret the dynamic nature of NFTs. Thus, a neuropathologist relies upon morphologic distinctions paired with histology and immunohistochemical markers to interpret the still frame captured at the time of death.
Three distinct, but overlapping forms of NFT maturities exist along a continuum: intracellular pretangles, intracellular mature tangles, and extracellular ghost tangles. 10 This suggests the existence of a window in which the AD brain may have (1) different molecular markers to identify various NFT maturities, (2) different peripheral and central biomarkers present throughout the NFT lifespan, and (3) the potential for targeted treatment early in tangle formation to minimize neurodegenerative effects and minimize neuronal loss. The major goal of this review is to provide a historic and modern perspective on NFT maturity in the context of AD. We will emphasize the utility of histologic methods and tau markers used to reveal the level of NFT maturity. As such, we will provide an overview of concepts that demonstrate the dynamic nature of NFT pathology for the tau biomarker community and those new to the AD field. For further information regarding Aβ neuropathology in AD, we direct the reader to the review by Sadigh‐Eteghad et al. 11
2.1. Alzheimer's disease
AD is a progressive neurodegenerative disease first reported by Dr. Alois Alzheimer in a patient who experienced dementia, memory loss, and delusions. 1 , 2 Postmortem analysis of the brain revealed mature tangles and Aβ plaques, 12 which became the recommended hallmark neuropathologies for the eponymous disorder, as first discussed by Khachaturian criteria in 1985. 13 Currently, AD is confirmed postmortem using guidelines provided by the National Institute on Aging and Alzheimer's Association (NIA‐AA). 14 , 15 The NIA‐AA recommends the use of three different measures including Thal amyloid phase (A; Aβ plaques 16 ), Braak tangle stage (B; NFTs 17 ), and modified Consortium to Establish a Registry for Alzheimer's Disease (CERAD, C; neuritic plaques 18 ) to obtain an “ABC” score. 14 , 15 Ante mortem diagnoses of AD dementia can be made using medical history, family history, cognitive testing, biofluid testing, and neuroimaging. 19
2.2. Microtubule‐associated protein tau
The tau protein was first discovered in 1975 (Figure 1) and plays a major role in microtubule stabilization. 20 The gene encoding the protein tau, microtubule associated protein tau (MAPT), was later localized to chromosome 17q21 in 1986 21 (Figure 1). Tau is an intrinsically disordered protein that generally does not adopt a secondary structure (for a review, see Avila et al. 22 ). Through regulation by phosphorylation, the tau protein's major functions are to promote tubulin polymerization and to stabilize microtubules. 23 , 24 Once tau is phosphorylated, it is released from the microtubules and promotes microtubule disassembly. 25 Diseases that are characterized by accumulations of filamentous and hyperphosphorylated tau are termed tauopathies, first coined by Dr. Bernardino Ghetti in collaboration with Dr. Maria Grazia Spillantini. 26 Tauopathies include AD and frontotemporal lobar degeneration with tau (e.g., Pick's disease, corticobasal degeneration, progressive supranuclear palsy; for a review, see Murray and DeTure, 27 Kovacs,28,29 Götz et al.,30 and Murray et al. 31 ).
FIGURE 1.

Historical overview of special stains, antibody development, biomarkers, and Alzheimer's disease (AD)/neurofibrillary tangle maturity research milestones from 1875 to 2020. Blue, staining technique developed; purple, milestone research for AD; orange, antibody development; green, biomarker development. Tick marks denote 1‐year interval.
MAPT mRNA (hereafter referred to as tau mRNA) is alternatively spliced at the amino terminus (N‐terminus) and carboxy‐terminus (C‐terminus). Zero, one, or two inserts (0N, 1N, 2N) may be alternatively spliced at the N‐terminus of tau mRNA 32 (Figure 2). Additionally, exon ten at the C‐terminus of tau mRNA can be alternatively spliced, resulting in the inclusion or exclusion of the second microtubule‐binding repeat region (3R, 4R). 33 4R tau protein binds to microtubules with higher affinity compared to 3R tau 34 and turns over faster than 3R tau. 35 In the human adult brain, 3R and 4R tau proteins are found at approximately equal levels. 36 However, 3R tau mRNA is between two and three times more abundant than 4R tau mRNA. 33 Alternative splicing of tau is a developmentally regulated process as fetal brain contains only 0N3R tau, while the adult brain contains all six isoforms. 32 Isoforms are differentially expressed within different neuronal types in the nervous system. For example, in the hippocampus granule cells in the dentate gyrus express only 3R tau mRNA, whereas some pyramidal cells of Ammon's horn expressed 3R and 4R tau mRNA. 33
FIGURE 2.

Tau structure 196 mapped with tau antibody epitopes. Diagonal stripe denotes alternatively spliced region. Within each subheading the named tau antibody is placed corresponding to its recognition motif with associated references.
2.3. Nomenclature
We will use the term “NFT” to encompass all maturity levels including pretangles, mature tangles, and ghost tangles. Historically, the term “stage” is used to describe NFT maturities. 10 , 12 However, to disentangle “stage” from Braak tangle staging, we instead recommend the use of “levels” to describe the NFT maturities across their lifespan. We acknowledge that although NFT maturity levels can be morphologically distinguished, they encompass a dynamic range in which intermediaries exist between each defined maturity level.
HIGHLIGHTS
Neurofibrillary tangles (NFTs) mature through three levels: pretangles, mature tangles, and ghost tangles.
NFT maturity levels differ in their morphology, tau isoform predilection, and post‐translational modifications.
Visualization techniques recognize a range of the NFT maturity levels and rarely if ever capture the entire NFT lifespan.
RESEARCH IN CONTEXT
Systematic review: The authors searched PubMed and Google Scholar for literature on Alzheimer's disease (AD), tau, neurofibrillary tangles (NFTs), and tau biomarkers.
Interpretation: The NFT is a dynamic neuronal lesion that transforms through three defined maturity levels in AD: pretangle, mature tangle, and ghost tangle. There is a wide array of staining techniques used to interpret tau pathology that are reflective of earlier or advanced aspects of NFT maturity. Based on accumulated knowledge, we hypothesize that tau biomarker recognition of early or advanced maturation levels will impact their interpretation.
Future directions: We recommend quantitative characterization of tau markers in the context of NFT maturity to guide interpretation of functional deficits both in and outside of the medial temporal lobe. Future studies are needed to test the hypothesis that the pretangle begins as a protective response to identify whether a therapeutic window exists to prevent the death of tangle‐bearing neurons.
3. MORPHOLOGIES
3.1. Pretangles
The concept of a NFT maturity level that precedes mature tangles was discussed during the characterization of the monoclonal tau antibody Alz‐50. 37 , 38 Further characterization of “stage 0 tangles” preceding mature tangles was provided by Bancher et al. in 1989, 10 but it was not until 1991 that Bancher et al. coined the term “pretangles” 39 (Figure 1). Pretangles reside in morphologically normal neurons with a healthy nucleus and diffuse or fine granular cytoplasmic and/or perinuclear immunostaining of tau in the neuron 10 , 39 , 40 (Figure 3). Ultrastructural studies demonstrated that pretangles contain diffusely scattered abnormal fibers including paired helical filaments (PHF), straight filaments, and small bundles of aggregated filaments 10 (for more discussion, refer to section 4.2). Correlative light and electron microscopy (EM) also revealed perinuclear tau was composed of straight filaments that were associated with the nuclear membrane. 41 Additionally, pretangles are devoid of ubiquitin immunostaining. 10 , 39 Pretangles are generally not argyrophilic, 10 , 42 although Gallyas silver staining was reported to visualize pretangles. 43 , 44 Gallyas silver staining has a predilection for tau including exon 10 (4R tau, for more discussion, refer to section 4.3) 45 of which pretangles are primarily composed. 43 , 44 Using AT8 immunohistochemistry, early accumulation of pretangle neuropathology in brainstem nuclei was observed in individuals younger than 30 years of age. 46 , 47
FIGURE 3.
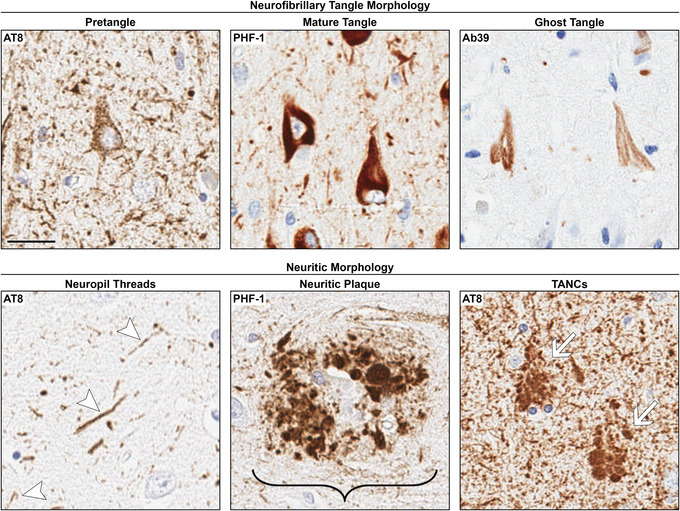
Neurofibrillary tangle (NFT) maturity level and neuritic morphologies in the CA1 subsector of the hippocampus. Pretangles have perinuclear accumulation of tau, in addition to a diffuse or granular staining pattern. Mature tangles assume the shape of the neuron in which they reside (e.g., “flame‐shaped”) and are composed of tightly packed bundles of fibers. The neuron has a dislocated and/or shrunken nucleus. Ghost tangles are loosely arranged bundles of fibers with no associated nucleus as they are the remnants of mature tangles once the neuron has died. Neuropil threads are the thread‐like staining pattern outside of the soma. Neuritic plaques are the accumulation of dystrophic neurites and are larger than a neuron. Tangle associated neuritic clusters (TANCs) are the accumulation of neurites around a ghost tangle and are around the same size as a neuron. AT8 was used for pretangle, neuropil threads, and TANCs. PHF‐1 was used for mature tangles and neuritic plaques. Ab39 was used for ghost tangles. It is important to note that these antibodies recognize a range of the NFT maturity levels. Immunohistochemistry was counterstained with hematoxylin. Arrows point to the labeled pathology. Bracket includes the entire neuritic plaque structure. Scale bar measures 25 μm.
3.2. Mature tangles
Although yet to be termed, mature tangles (Figure 3) were first reported by Alzheimer in 1907 as thick argyrophilic fibers in neurons. 1 , 2 Mature tangles reside in neurons with a shrunken and/or displaced nucleus, with argyrophilic fibrils that appear in a “basket‐like or sling‐like” shape replacing the cytoplasm. 12 NFTs assume the shape of the neuron they occupy, thus mature tangles in pyramidal neurons generally have a flame‐like shape. In addition to their argyrophilic nature, 10 , 17 , 42 mature tangles are ubiquitin positive 48 , 49 and are readily stained by a variety of tau antibodies, which is further discussed in section 3.
3.3. Ghost tangles
Ghost tangles, the more advanced level of NFT maturity (Figure 3), were first described by Alzheimer as the remaining fibrils after disintegration of the neuron and nucleus. 1 , 2 These extracellular tangles are less argyrophilic and appear more loosely bundled than mature tangles. 2 , 10 , 12 Additionally, ghost tangles appear eosinophilic by hematoxylin and eosin (H&E) compared to the basophilic properties of intracellular mature tangles. 50 Ghost tangles are ubiquitin positive 10 and can also be infiltrated with glial processes. 12 , 50 As there is more neuronal death than ghost tangles in AD brains, 51 contribution of co‐existing pathologies should be considered when evaluating the relationship between NFT accumulation and neuronal death. 41 , 52
3.4. Morphology in context
NFTs mature through three defined levels: pretangles, mature tangles, and ghost tangles (Figure 3). Pretangles and mature tangles form intracellularly, whereas ghost tangles become remnants in the extracellular space. While pretangle‐bearing neurons appear morphologically normal with an intact nucleus, there is evidence of transcriptional changes compared to healthy neurons. Protein quality control genes are upregulated in pretangle‐bearing neurons, suggesting a disruption of protein homeostasis. 53 This may parallel the observation of increased phospho‐ubiquitin and granulovacuolar degeneration accumulation in pretangle‐bearing neurons, which becomes less frequently observed in mature tangle‐bearing neurons and may implicate a disruption in the protective role of the mitochondrial quality control pathway. 54 These early changes may initiate a protective response to the accumulation of misfolded proteins, as the unfolded protein response (UPR) becomes activated in pretangle‐bearing neurons, yet these UPR activation markers (e.g., pPERK) are diminished as the mature tangle forms. 55 However, UPR activation induces kinase activity (i.e., GSK‐3β) that may overwhelm the neuron as tau becomes hyperphosphorylated leading to inhibition of anterograde (kinesin‐dependent) fast axonal transport. 56 , 57 GSK‐3β immunoreactivity is strongly present in pretangles, present to a lesser extent in mature tangles, but is not visible in ghost tangles suggesting a precipitous decline in hyperphosphorylation as the neuron dies. 58 Further support for a functional distinction between pretangle‐ and mature tangle–bearing neurons is the emergence of ubiquitin‐labeled proteins in neurons with mature tangles and ghost tangles, but not pretangles suggesting proteolytic degradation attempts are made later in tangle formation. It is important to note that these NFT levels are not static, and intermediaries exist between pretangles, mature tangles, and ghost tangles.
4. VISUALIZATION OF NFTs
For more than a century, many stains and antibodies were developed and used to visualize neuropathology in AD brains. Here we review staining techniques that were historically relevant in identifying AD neuropathology prior to the discovery of the tau protein. 20 We will further expand upon tau immunohistochemical methods that are particularly relevant to NFT maturity, many of which are now commercially available. While there are numerous tau antibodies available, here we review those of historical relevance and those that are of particular importance to our growing understanding of NFT maturity levels.
4.1. Staining techniques pre‐dating the discovery of the tau protein
Hematoxylin, created in the 1840s, 59 is a positively charged basic dye. 60 Eosin, created in 1871, 59 is a negatively charged acidic dye. 60 H&E were first used together between 1875 and 1878 59 (Table 1, Figure 1) as a morphologic stain in which hematoxylin stains nuclei blue (basophilic) and eosin stains the cytoplasm and extracellular matrix pink (eosinophilic; 60 Figure 4). H&E is a routine stain used by many labs to visualize neuronal loss, as well as mature tangles and ghost tangles in AD. 50 , 61 , 62 , 63 Mature tangles appear basophilic compared to unaffected neurons, whereas ghost tangles are eosinophilic due to infiltration by astroglial processes. 50
TABLE 1.
Tinctorial dye, silver stains, and fluorescent dyes used to identify neurofibrillary tangles with references
| Neuropil threads | Pretangles | Mature tangles | Ghost tangles | Reference(s) | |
|---|---|---|---|---|---|
| Tinctorial dye | |||||
| H&E | No | No | Yes | Yes | 50 |
| Congo red | Yes | No | Yes | Yes | 68 |
| Silver staining | 70 | ||||
| Bielschowsky | Yes | No | Yes | Yes | 70 , 195 |
| Bodian | Yes | No | Yes | Yes | 70 |
| Gallyas | Yes | No/Yes | Yes | Yes | 70 |
| Campbell‐Switzer | Yes | No | Yes | Yes | 70 |
| Fluorescent dye | |||||
| Thioflavin‐S | Yes | No | Yes | Yes | 73 |
| Thiazin red | Yes | No | Yes | Yes | 91 , 92 |
FIGURE 4.
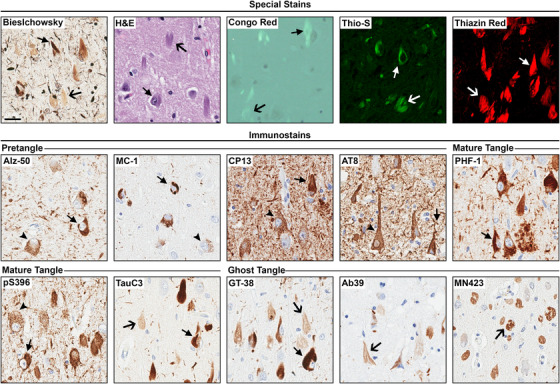
Special stains and tau immunohistochemistry for neurofibrillary tangle (NFT) maturity levels. Representative images for each staining technique were taken at the CA1 sector of the hippocampus in Alzheimer's disease (AD) brains. Routine and special stains include Bielschowsky silver stain, H&E, Congo red, thioflavin‐S (Thio‐S), and thiazin red. All routine and special stains recognized mature tangles and ghost tangles. For immunohistochemistry, Alz‐50, MC‐1, CP13, AT8, PHF‐1, pS396, TauC3, GT‐38, Ab39, and MN423 were used. Tau antibodies recognized a range of NFT maturity levels. Immunohistochemistry was counterstained with hematoxylin. We thank Dr. García‐Sierra for the thiazin red image. Arrow head, pretangles; closed arrow, mature tangles; open arrow, ghost tangles. Scale bar measures 25 μm.
Congo red (Table 1, Figure 4) is a tinctorial stain originally created for the textile industry as a cotton dye in 1884 59 (Figure 1). This dye will exhibit birefringence, in which there are two refractive indices depending upon the orientation of polarized light. 64 Thus, using polarized light microscopy, Congo red shows anomalous colors that are traditionally “apple green,” under cross‐polarization. 64 In 1886, Congo red was first used to stain axons. 59 By 1922, Congo red was used to visualize amyloid in tissue sections 59 where it binds to β‐pleated sheets. 65 , 66 This is due to Congo red's ability to self‐assemble into ribbons that are compatible with the β‐conformation of these protein structures. 67 For over 50 years, this dye was used to visualize AD neuropathology, including mature tangles and ghost tangles. 68 Congo red does not stain pretangles likely owing to the lack of fibrillar structure.
Silver staining (Table 1) was the first technique used to visualize AD neuropathology. Specifically, the Bielschowsky silver staining method was first used by Alzheimer to visualize the neuropathology of his first patient 1 , 2 , 12 (Figure 4). This method was created by Bielschowsky in 1902 69 (Figure 1), and has endured through the modern era, with modified Bielschowsky silver stain protocols routinely used all over the world. Additional silver staining techniques were developed, including Bodian, Gallyas, and Campbell‐Switzer (for a review, see Uchihara 70 ), which are all also used to this day. Positive silver staining is termed argyrophilic and will appear darker compared to non‐argyrophilic structures. This is due to the reduction of silver ions to metallic particles, which can then be visualized through light microscopy (for a review, see Uchihara 70 ). Although both mature tangles and ghost tangles are argyrophilic, ghost tangles are less intensely stained than mature tangles 12 (Figure 4). There are a variety of modifications to silver staining methods, which influences the predilection for 3R/4R neuropathology. 45 Gallyas silver staining has a predilection for 4R tau neuropathology, whereas Campbell‐Switzer has a preference for 3R tau neuropathology. 45 This may explain why Gallyas silver staining reportedly stained pretangles, 43 , 44 because these are predominantly composed of 4R tau. 44 , 63 , 71 , 72
Thioflavins are a group of fluorescent dyes developed in the 1950s (Figure 1) that, like Congo red, will bind to β‐pleated sheets. 73 Thioflavin‐S (Table 1, Figure 4) is excited at 440 nm and is visualized over a wavelength range of 455 to 600 nm allowing detection of Aβ plaques and advanced tangles in the AD brain. 74 As Aβ plaques and NFTs are both visualized by thioflavin‐S, morphologic characteristics are used to differentiate between the pathologies. Specifically, Aβ plaques are round, extracellular accumulations that may contain dystrophic neurites that appear either bulbous or thread‐like; whereas NFTs take the shape of the neuron in the intracellular domain and, once the neuron has died, may remain as a remnant in the extracellular space. Comparing modified Bielschowsky and Bodian silver staining methods with thioflavin‐S, all techniques identified both plaques and NFTs. 75 However, the modified Bielschowsky silver stain and thioflavin‐S performed better than Bodian silver staining at recognizing neuritic plaques. 75 Pretangles are not readily visualized with thioflavin‐S as they do not contain β‐pleated sheets.
4.2. Staining techniques developed after the discovery of the tau protein
In April 1985, Yen et al. reported the mouse monoclonal antibody Ab39, which was developed in an effort to identify NFT pathology (Table 2, Figure 1). The study reported that the Ab39 antibody recognized the majority of mature tangles in AD brains. 76 The epitope is currently unknown, but it is believed to recognize a conformational epitope specific to NFTs. 77 Ab39 has since been used to identify ghost tangles in addition to mature tangles 78 (Figure 4). This antibody was available through Dr. Shu‐Hui Yen, but is no longer produced or distributed. In October of the same year, Binder et al. reported the mouse monoclonal Tau‐1 antibody (Table 2, Figure 1), which was developed to determine the localization of tau in the mammalian central nervous system. 79 The epitope for this antibody is amino acids 162–210 80 (Table 2, Figure 2) with a core sequence encompassing amino acids 192–204 81 , 82 and is available through Millipore Sigma (catalog # MAB3420). Tau‐1 recognizes mature tangles and to a lesser extent ghost tangles and pretangles after dephosphorylation of AD brain tissue sections. 10 , 83
TABLE 2.
Tau antibodies and their specifications discussed in this review. The antibodies are either historically relevant or important in understanding the makeup of the neurofibrillary tangle maturity levels
| Antibody | Epitope | Immunogen | Species | Clonality | Isotype | Source Catalog # | Ref. |
|---|---|---|---|---|---|---|---|
| 4R‐tau | aa 275‐291 VQIIDKKLDLSNVQSKC | synthetic N279D 4R‐tau peptide VQIIDKKLDLSNVQSKC | Rabbit | Polyclonal | IgG | Cosmo Bio CAC‐TIP‐4RT‐P01 | 112 |
| Ab39 | Conformational | Tangle pellets from AD brain homogenate | Mouse | Monoclonal | IgG1 | Shu‐Hui Yen | 76 , 77 |
| Alz‐50 | aa 2‐10, 312‐342 | AD ventral forebrain homogenate | Mouse | Monoclonal | IgM | (Davies) | 37 , 80 |
| AT8 | pS202, pT205 | PHF‐containing pellet from AD frontal and temporal homogenate | Mouse | Monoclonal | IgG1 | Thermo Fisher Scientific MN1020 | 96 |
| CP13 | pS202 | Mouse | Monoclonal | IgG1 | (Davies) | 105 | |
| GT‐38 | Conformational | Tau PHFs from AD brains | Mouse | Monoclonal | IgG1 | Abcam ab246808 | 114 |
| MC‐1 | aa 7‐9, 312‐322 | Alz‐50 immunoaffinity purified PHFs from AD brain homogenate | Mouse | Monoclonal | IgG1 | (Davies) | 84 |
| MN423 | aa 387‐391 DHGAE, truncation at E391 | Pronase treated neurofibrillary tangle preparations | Mouse | Monoclonal | IgG2b | Nicholas Kannan (Novak) | 85 , 86 , 87 , 120 |
| PHF‐1 | pS396, pS404 | Non‐aggregated PHFs from AD brain | Mouse | Monoclonal | IgG1 | (Davies) | 94 |
| pS396 | pS396 | Synthetic corresponding to human tau aa 350 to the C‐terminus. | Rabbit | Monoclonal | IgG | Abcam ab109390 | |
| RD3 | aa 267‐274, 306‐316 continuous KHQPGGGKVQIVYKPV | KHQPGGGKVQIVYKPV | Mouse | Monoclonal | IgG | Millipore Sigma 05‐803 | 104 |
| RD4 | aa 275‐291 VQIINKKLDLSNVQSKC | VQIINKKLDLSNVQSKC | Mouse | Monoclonal | IgG | Millipore Sigma 05‐804 | 104 |
| TauC3 | aa 412‐421 CSSTGSIDMVD | CSSTGSIDMVD peptide | Mouse | Monoclonal | IgG1 | Nicholas Kannan (Binder) | 113 |
| Tau‐1 | aa 162‐210 | Bovine heat‐stable microtubule associated proteins | Mouse | Monoclonal | IgG2a | Millipore Sigma MAB3420 | 79 , 80 , 81 , 82 |
| Tau‐66 | aa 155‐244, 305‐314 | Recombinant 2N3R polymers tau | Mouse | Monoclonal | IgM | Nicholas Kannan (Binder) | 109 |
| TG‐3 | pT231 | Purified PHFs | Mouse | Monoclonal | IgM | (Davies) | 106 |
Abbreviations: AD, Alzheimer's disease; IgG, immunoglobulin; PHF, paired helical filaments.
In 1986, Wolozin et al. reported the mouse monoclonal antibody Alz‐50 (Table 2, Figure 1), which was designed to investigate NFT pathology in the cholinergic neurons of the ventral forebrain. 37 The discontinuous epitope maps to amino acids 2–10, and 312–342 84 (Figure 2). Alz‐50 immunopositive neurons were interpreted to “recognize a precursor to tangle formation” (i.e., pretangles) as they lacked thioflavin‐S co‐staining 37 (Figure 4). Alz‐50 also recognizes mature tangles, but does not label ghost tangles (Figure 4). Thus, Alz‐50 is considered useful as an early marker of NFT maturity. In 1997, Jicha et al. generated the mouse monoclonal antibody MC‐1 (Figure 1) to create an antibody with a similar epitope to Alz‐50 that would not react with the Alz‐50 positive protein, FAC1. 84 The epitope for MC‐1 is amino acids 7–9 and 312–322 84 (Figure 2). Similar to Alz‐50, MC‐1 recognizes primarily pretangles and mature tangles (Figure 4). Alz‐50 and MC‐1 were previously produced by Dr. Peter Davies.
In 1988, Wischik et al. reported the mouse monoclonal antibody 423 (mAb 423, MN423) (Table 2, Figure 1), which was developed to identify the PHF core. 85 This antibody recognizes amino acids 387–391 of tau truncated at glutamic acid (E) 391 in the core of PHFs. 86 , 87 MN423 primarily identifies ghost tangles and mature tangles. 88 , 89 , 90 MN423 recognizes even advanced forms of ghost tangles in the hippocampus that display a spherical, end‐stage morphology (Figure 4). MN423 is significant in the scientific understanding of AD in that it identified truncated tau in the PHF core. This antibody was originally produced by Dr. Michal Novak.
Thiazin red (Table 1) is a fluorescent stain that binds to β‐pleated sheets 91 and was first used to identify plaques and tangles in AD in 1988 92 (Figure 1). Thiazin red visualizes mature tangles and ghost tangles, 93 but not pretangles because they do not contain fibrils (Figure 4). Additionally, ghost tangles stain less intensely than mature tangles. 93 When excited at 418 nm, thiazin red is visualized at wavelengths > 520 nm. 93 Unlike thioflavin‐S, thiazin red can be used to stain frozen‐thawed AD brain tissue. 92
PHF‐1 is a mouse monoclonal antibody developed in 1989 94 (Table 2, Figure 1) in an effort to recognize PHF proteins. This is a commonly used antibody that recognizes tau phosphorylated at serine (S) 396 and S404 95 (Figure 2) and predominantly immunostains mature tangles, with reduced immunostaining of pretangles and ghost tangles (Figure 4). PHF‐1 was previously produced by Dr. Peter Davies; however, Abcam's pS396 antibody (catalogue # ab109390) may be an acceptable commercial alternative as it shares the phosphorylated S396 epitope with PHF‐1 (Table 2, Figure 2). Like PHF‐1, this antibody primarily stains mature tangles and ghost tangles (Figure 4).
In 1992, Mercken et al. generated the mouse monoclonal antibody AT8 96 (Table 2, Figure 1), which recognizes tau phosphorylated at S202 97 , 98 and threonine (T) 205 99 (Figure 2). AT8 is an important antibody in the AD field due to its versatility for multiple methods including immunoblotting, 97 , 98 immunohistochemistry, 98 and immuno‐EM. 98 AT8 generally immunostains pretangles and mature tangles 42 , 43 , 44 , 46 , 72 , 100 , 101 , 102 , 103 , 104 (Figure 4). Ghost tangles are not readily recognized by AT8; however, neuritic pathology can accumulate around them in the form of tangle‐associated neuritic clusters (TANCs; 42 Figure 3). In contrast, there are previous reports of AT8 immunostaining primarily in mature tangles and ghost tangles. 40 , 63 AT8 is a commercially available antibody (Thermo Fisher Scientific, catalogue # MN1020) with a comparable non‐commercial mouse monoclonal antibody previously produced by Dr. Peter Davies, CP13. CP13 was first reported in 1999 by Jicha et al. 105 (Table 2, Figure 1). This widely used antibody recognizes tau phosphorylated at S202 (Figure 2) and primarily stains pretangles and mature tangles (Figure 4).
TG‐3 is an antibody first reported in 1996 by Vincent et al. 106 (Figure 1). This antibody recognizes a regional conformational change with a phosphorylation site at T231 107 (Table 2). A study by Luna‐Muñoz et al. characterized TG‐3 immunostaining and found this antibody to recognize both fibrillar and non‐fibrillar tau, as reported with thiazin red co‐staining. 108 This antibody recognizes pretangles and mature tangles, but with mixed reports on predilection for ghost tangles. 40 , 108 TG‐3 was previously produced by Dr. Peter Davies.
Tau‐66 is a mouse monoclonal antibody reported in 2001 by Ghoshal et al. in an effort to investigate other conformational changes observed in tau 109 (Table 2, Figure 1). This antibody recognizes a conformational change of tau in which the proline‐rich region interacts with the microtubule binding domain repeat 3 (discontinuous epitope of amino acids 155–244, 305–314) 109 (Figure 2). Upon initial characterization, mature tangles were immunostained by Tau‐66 in the superior temporal gyrus and hippocampus. 109 The Tau‐66 antibody has played a crucial role in the development of the hypothesis of sequential conformational states of tau pathology 90 (for more discussion, refer to section 4.1). This antibody was originally produced by Dr. Lester Binder at Northwestern University and provided by Dr. Nicholas Kanaan at Michigan State University.
De Silva et al. developed mouse monoclonal antibodies RD3 (Millipore Sigma, catalogue # 05‐803) and RD4 (Millipore Sigma, catalogue # 05‐804; Table 2) in 2002 (Figure 1), which recognize 3R and 4R tau, respectively. 104 The putative epitope for RD3 contains continuous amino acids 267–274 and 306–316 and the RD4 epitope contains amino acids 275–291 110 (Figure 2). Pretangles predominately immunostain with RD4, mature tangles immunostain with RD3 and RD4, and ghost tangles predominately immunostain with RD3. 44 , 63 , 71 , 72 , 103 , 111 To simplify double immunostaining procedures, a rabbit polyclonal 4R‐tau antibody was generated in 2013 112 (Cosmo Bio, catalogue # CAC‐TIP‐4RT‐P01). The rabbit polyclonal 4R‐tau antibody shares the same epitope as RD4 112 (Table 2, Figure 1 and 4), and enabled comparison to RD3 by eliminating host species redundancy. 103 Overall, RD3 and RD4 antibodies are the first highly specific antibodies to 3R and 4R isoforms, allowing investigators to study isoform changes in AD.
Mouse monoclonal antibody TauC3 was developed in 2003 113 (Figure 1) and originally produced by Dr. Lester Binder at Northwestern University. This antibody labels tau truncated at aspartic acid (D) 421 recognizing amino acids 412–421 113 (Table 2, Figure 2). This antibody immunostained both mature tangles and ghost tangles 92 (Figure 4), providing evidence that NFTs contain tau that is truncated at a cleavage site.
Gibbons et al. reported the mouse monoclonal antibody GT‐38 (Table 2) in 2018 (Figure 1) in an effort to identify AD‐specific tau neuropathology. 114 The exact epitope is unknown, but is considered to recognize a conformational species of tau. This antibody immunostains both mature tangles and early ghost tangles (Figure 4). GT‐38 did not recognize tau pathology in the context of corticobasal degeneration and progressive supranuclear palsy. 114 Instead, the antibody is shown to be specific for NFT pathology in mixed 3R/4R tauopathies, such as AD, instead of only 3R or 4R primary tauopathies, such as Pick's disease and progressive supranuclear palsy. 114 GT‐38 is now available through Abcam (catalogue # ab246808).
4.3. NFT visualization in context
NFTs are visualized through a variety of special stains, as well as antibody‐based immunohistochemistry. H&E can be used to identify mature tangles and ghost tangles by their tinctorial properties: mature tangles are basophilic and appear blue, whereas ghost tangles are eosinophilic and appear pink. 50 The majority of the special stains discussed (thioflavin‐S, Congo red, thiazin red) will bind to the β‐pleated structure formed by fibrils of the mature tangles and ghost tangles. 65 , 66 , 73 , 91 Additionally, mature tangles and ghost tangles are argyrophilic. 12 Immunohistochemistry is a highly effective method of visualizing NFTs. 37 , 76 , 79 , 85 , 94 , 96 , 104 , 105 , 109 , 113 , 114 , 115 Pretangles are primarily visualized with AT8, CP13, Alz‐50, and MC‐1. Mature tangles are primarily visualized with PHF‐1, pS396, Tau‐66, and TauC3, and can also be visualized with AT8, CP13, Alz‐50, and MC‐1. Ghost tangles are primarily visualized with GT‐38, Ab39, TauC3, and MN423. Importantly, antibodies do not solely recognize one NFT maturity level, but will recognize a small range on the spectrum along the NFT lifespan (Figure 5). A point of consideration with any antibody‐based approach relates to the loss of the epitope, whether an effect of conformational events or other post‐translational modifications that affect antibody recognition.
FIGURE 5.
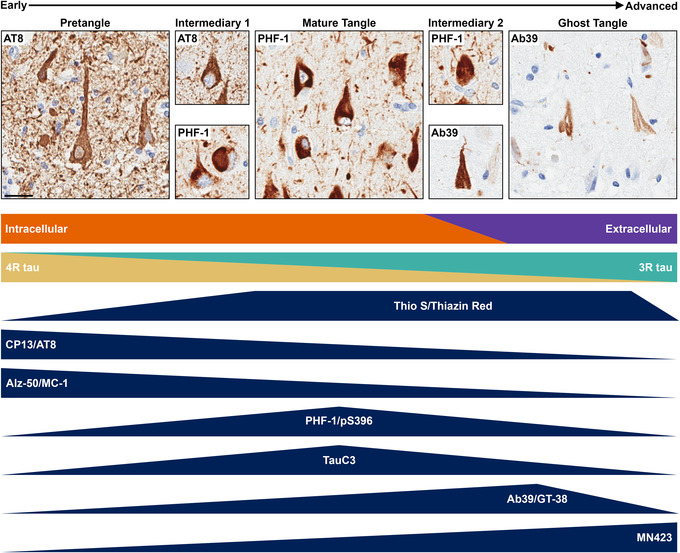
Summary of neurofibrillary tangle (NFT) maturity levels from early to advanced forms. Intermediaries exist between the defined NFT maturity levels. NFTs in intermediary 1 will have focal accumulation of fibrillar tau that does not completely fill the neuron, which was previously speculated to be nucleation sites. 108 Intermediary 2 will have intense staining of the NFT without a nucleus. There is a shift in NFT localization, as pretangles and mature tangles are intracellular and ghost tangles are extracellular. 40 Additionally, there is a shift in isoform expression, as pretangles predominantly express 4R tau and ghost tangles predominantly express 3R tau. 44 Staining methods will label a range of NFT maturity levels, as identified by the dark blue triangles. Thioflavin‐S (Thio‐S) and thiazin red recognize primarily mature tangle sand ghost tangles. CP13, AT8, Alz‐50, and MC‐1 primarily recognize primarily earlier NFT maturity levels. PHF‐1, pS396, and TauC3 primarily recognize a middling NFT maturity level. Finally, Ab39, GT‐38, and MN423 primarily recognize advanced NFT maturity levels. Scale bar measures 25 μm.
Immunohistochemistry and light microscopy are powerful tools to investigate AD neuropathology; however, these methods are not without their limitations. One of the defining criteria to determine NFT maturity level is the presence and shape of the nucleus. Upon sectioning, nuclei can be out of plane to the neuropathology observed, complicating the classification of the NFT maturity level. As previously mentioned, another limitation to microscopic methods is that there are currently no markers available to recognize a single NFT maturity level. This is because the NFT maturity levels are not static entities, but rather dynamic structures that exist along a spectrum of the NFT lifespan. For example, the intermediary between pretangles and mature tangles (intermediary 1, Figure 5) shows focal puncta or fibrils that are intensely stained for tau, but does not fill the entire neuron. This level was described previously as neurons with bead‐shaped structures that were speculated to be nucleation sites. 108 The intermediary between mature tangles and ghost tangles (intermediary 2, Figure 5) shows intense immunostaining of tau throughout the neuron without a nucleus. With the advent of multiplexing technology to reveal multiple protein targets, future studies are encouraged to evaluate shifting profiles along the dynamic spectrum of NFT maturity.
5. NEUROFIBRILLARY CHANGES IN AD
5.1. Post‐translational modifications of tau
Post‐translational modifications are alterations that occur after mRNA has been translated to protein and can affect a protein's function and/or downstream signaling. Examples include phosphorylation, ubiquitination, truncation, and conformational changes. 116 For a detailed review of post‐translational modifications of tau in AD, please refer to Martin et al. 117 Below, we highlight the utility of immunohistochemical markers of post‐translational modifications that provide keen insight into our understanding of the temporal sequence of events related to NFT maturity.
Phosphorylation occurs naturally to release tau from microtubules and to promote depolymerization. 25 All six isoforms of tau in PHFs exhibit abnormal phosphorylation 118 on S, T, and tyrosine sites. Phosphorylated tau is observed at each level of NFT maturity. 10 In comparison, ubiquitinated tau is found in mature tangles and ghost tangles, but not in pretangles. 10 , 39 Additionally, dystrophic neurites radiating from the neuritic plaques are largely found to be ubiquitin‐positive, although a subset of ubiquitin‐negative neurites was also reported. 48 , 119
The truncated form of tau was first identified in the PHF core in 1988. 85 , 115 This truncation event occurs at E391 87 and is recognized by the antibody MN423 85 , 115 , 120 (Table 2). Truncation at E391 is identified primarily in mature tangles and ghost tangles. 89 , 121 In 2003, another C‐truncation event at D421, recognized by the antibody TauC3 (Table 2), was identified 113 and occurs at a caspase cleavage site. 113 , 122 , 123 , 124 , 125 Truncation at D421 was identified in pretangles 123 and mature tangles 121 , 123 providing a framework for the interpretation in the context of NFT maturity.
C‐terminal truncations correlate with N‐terminal truncations. 126 Immunoblotting of the insoluble fraction containing mature tangles and ghost tangles identified the lack of an amino terminal that is recognized by the Alz‐50 antibody. 127 Additionally, immunohistochemistry and immunofluorescence experiments also provide evidence for a truncated N‐terminus of tau in mature tangles and ghost tangles. 88 , 89 In vitro experiments identified a caspase‐6 cleavage site at D13 126 and antibodies that span across this site recognized tau in pretangles and mature tangles, but not ghost tangles. 126 This provides further evidence that the N‐terminus is truncated in advanced levels of NFT maturity. The “post‐translational sequence of events” hypothesis is summarized in Figure 6 (adapted from Binder et al. 90 ).
FIGURE 6.
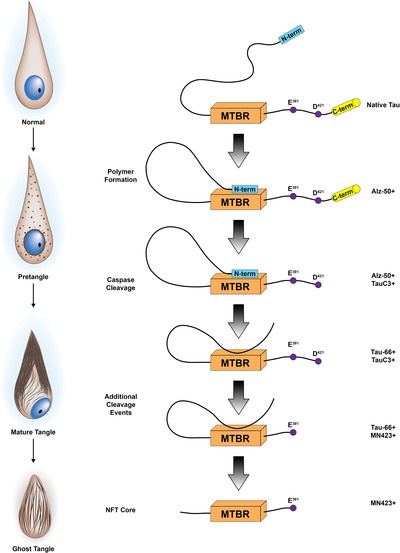
Proposed tau conformational and truncation events hypothesized along the neurofibrillary tangle (NFT) lifespan. First, the N‐terminus binds to the microtubule binding region (MTBR), as recognized by Alz‐50. Next, the C‐terminus is truncated at D421, as recognized by Tau‐C3. Next, the N‐terminus is truncated, and the proline‐rich region will bind to the microtubule binding region, as recognized by TauC3. Then, tau is truncated at E391, as recognized by MN423. Finally, the proline‐rich region is further cleaved. These conformational and truncation events occur through the NFT lifespan. For example, pretangles are predominantly Alz‐50 positive, but not MN423 positive. Mature tangles are positive for Alz‐50, TauC3, Tau‐66, and MN423. Ghost tangles are predominantly positive for TauC3, Tau‐66, and MN423. Sequence of events figure reprinted from Biochim Biophys Acta, 1739, Binder et al., Tau, tangles, and Alzheimer's disease, 216‐223, Copyright (2005), with permission from Elsevier.
The earliest known structural conformational change in tau is identified by both the Alz‐50 and MC‐1 antibodies. The discontinuous epitope of these antibodies recognizes both the N‐terminus and the microtubule binding domain 80 , 84 (Table 2, Figure 2). Alz‐50 and MC‐1 were used to identify the earliest conformational change as it recognizes pretangles and mature tangles. 37 , 38 , 84 The Tau‐66 antibody (Table 2) recognizes a subsequent conformational change, specific to the interaction of the proline‐rich region and the beginning of the third microtubule binding repeat 109 (Figure 2). This antibody primarily recognizes mature tangles and ghost tangles 88 , 109 and may also recognize some pretangles determined by punctate immunostaining. 109 However, Alz‐50 and Tau‐66 do not co‐localize, indicating these antibodies recognize different populations of mature tangles. 88
Many additional post‐translational modifications, including glycosylation, 128 , 129 glycation, 130 , 131 nitration, 132 and acetylation 133 , 134 are found on tau (for a review, see Martin et al. 117 ). We have focused on available evidence for a sequence of conformational and truncation events especially as it pertains to phosphorylation, but there is a need to better understand the relevance of post‐translational modifications to NFT maturity. Does the slow accumulation of hyperphosphorylated tau prior to N‐terminal truncation account for the resilience of dendritic spines in the brains of individuals who died without overt cognitive symptoms? 135 Do C‐terminal truncations of tau lead to defects in axonal transport machinery that are ultimately responsible for the death of the neuron? These questions and others are especially important in the context of neuronal health, synaptic function, and axonal transport as future studies are encouraged to examine these sequences of events in the context of NFT maturity.
5.2. Ultrastructural changes
In 1963, ultrastructural studies of mature tangles demonstrated accumulation of PHFs (Figure 1) composed of double helical structures. 136 , 137 X‐ray diffraction studies showed that PHFs conform to a β‐pleated sheet structure, 138 which is a secondary structure of proteins composed of amino acid chains that are bound by hydrogen bonds to form sheets (for a review, see Taylor et al. 139 ). The major component of PHFs is tau 140 , 141 , 142 , 143 , 144 , 145 , 146 with a minor component composed of ubiquitin. 48 , 49 Immunostaining for both tau and ubiquitin mimicked observations of argyrophilic NFTs on silver stained sections. Straight filaments were also identified in mature NFTs by EM. However, straight filaments are not as abundant as PHFs in AD brains. 147 Using cryogenic‐EM, Fitzpatrick et al. found that PHF cores and straight filament cores were composed of identical protofilaments; however, PHF and straight filament cores differ in how the protofilaments are packed. 148 As cryo‐EM studies continue to advance our understanding of tau biology, it will be of great interest to characterize how protofilament packaging develops from pretangles to mature tangles and onto ghost tangles. Through careful structural visualization of filamentous changes in NFT maturity we may be able to better understand what antigen–antibody interface becomes unmasked sufficient for therapeutic targeting.
5.3. Tau isoforms
AD is considered a 3R/4R tauopathy, meaning the NFTs are composed of both isoforms with no predominance of one over the other. This is in contrast to primary tauopathies, which are classified as 3R‐predominant (e.g., Pick's disease) or 4R‐predominant (e.g., progressive supranuclear palsy, corticobasal degeneration). 27 , 28 Although AD is classified as a 3R/4R tauopathy, evidence suggests a shift in the tau isoform expression through the NFT maturity levels. Both 3R and 4R tau pathology have been mapped in the hippocampus according to NFT maturity using immunohistochemistry 63 , 72 , 149 and immunofluorescence. 44 , 71 Pretangles contain predominantly 4R tau, mature tangles contain both 3R and 4R tau, and ghost tangles contain predominantly 3R tau. 44 , 63 , 71 , 72 , 150 Even though there is a predominance of the tau isoforms in pretangles and ghost tangles, 3R and 4R tau can also be expressed in the opposite end of the NFT lifespan as well. There is also heterogeneity of tau isoform expression in neurons themselves, with 3R tau expressed in the dendrites and 3R/4R tau expressed in the soma of mature tangle‐bearing neurons. 111 An increase in 3R tau through disease progression was reported in the brainstem of AD brains. 103 The change from 4R to 3R tau is not a post‐translational modification, which indicates that possible mechanisms for this change in isoform expression in NFTs may be due to altered alternative splicing regulation or metabolism of the isoforms. In contrast, no change in tau protein isoform expression was observed in the frontal cortex, a region that accumulates NFTs later on in the disease course, of AD brains compared to controls. 33 , 103 Interestingly, some reports show the ratio between 4R:3R tau mRNA was increased in AD brains compared to controls, 151 , 152 , 153 which may suggest an overcompensation of generating 4R tau mRNA due to the increased proportion of 3R tau protein in the neuron.
There are currently no MAPT mutations identified that cause AD; however, several mutations were identified as causative for frontotemporal dementia (for a review, see Ghetti et al. 154 ). Interestingly, different mutations confer predominance of 3R or 4R tau inclusions. For example, tau pathology in frontotemporal dementia patients with V337M and R406W mutations contain both 3R and 4R tau. 36 Patients with these two mutations have PHFs and straight filaments, as well as pretangles, mature tangles, neuropil threads, and neuritic plaques that are similar to neuropathology observed in AD. 155 , 156 In contrast, there are numerous mutations that result in predominantly 3R or 4R tau pathology. The P301L and N279K are two examples of mutations causing predominantly 4R tau pathology in frontotemporal dementia. 36 Considerations of mutation effects on tau isoform expression are important, especially for modeling tauopathies or biomarker evaluation of tau markers.
5.4. Neuritic pathology
Although the focus of this review is on NFTs, we would be remiss not to discuss neuritic pathologies as they are a consistent observation in AD brains. Neuritic pathologies encompass several forms including neuropil threads, neuritic plaques, and TANCs. Neuropil threads (Figure 3) were first described in 1986 (Figure 1) as argyrophilic, thread‐like structures outside of cell bodies (i.e., neuropil). 157 Neuropil threads were identified in dendrites of tangle‐bearing neurons in AD brains using 100 μm thick silver stained sections, 158 as well as serial tau immunostained sections. 102 Additionally, neuropil threads were identified in axons by immuno‐EM on AD brains. 159 Thus, neuritic pathology owing to axonal versus dendritic accumulation is not typically specified. Neuropil threads are observed in proximity to pretangles, mature tangles, and ghost tangles. They are primarily composed of PHFs and straight filaments, but may also include neurofilament. 159 In fact, evidence suggests that PHFs and straight filaments replace neurofilament in the neurites. 159 Multiple antibodies recognize neuropil threads, but this pathology is revealed to a much greater extent by antibodies that recognize hyperphosphorylated tau (e.g., CP13, AT8, and PHF‐1; Figure 3). Neuropil threads are not readily observed on H&E, but can be observed on silver‐stained sections and with Congo red or other fluorescent dyes (Table 1). Various subsets of neuropil threads can be revealed depending upon the antibody, stain, or dye used, which we speculate may reflect the maturity level of the originating NFT.
Neuritic plaques (Figure 3) are generally larger than a neuron and are principally composed of extracellular Aβ deposits that contain bulbous and thread‐like dystrophic neurites. Neuritic plaques are composed of both dendrites and axons 160 and preferentially form near microvasculature. 161 , 162 , 163 Neuritic plaques are argyrophilic and can be stained with a variety of tau antibodies. The often‐overlooked form of neuritic pathology, TANCs (Figure 3) were first defined in 1992 (Figure 1) and are composed of neurites surrounding a ghost tangle. 164 However, not all ghost tangles are associated with TANCs. TANCs are identified by silver staining and multiple tau antibodies, like AT8 and Alz‐50 42 , 165 as well as other markers as exampled by the synaptic protein chromogranin A. 164 Awareness of neuritic pathologies is especially relevant to digital pathology studies that use quantitative software to measure burden of pathology. Depending upon the tau marker of interest, neuritic pathologies may occupy a sizable portion of the annotated area. The specific contribution of increasing neuritic pathology may be difficult to disentangle from accumulation of NFTs themselves. Caution is warranted not to overinterpret their contribution as above and beyond that of NFTs depending on the marker used; as exampled by AT8, which may demonstrate rich staining of neuritic pathology disproportionate to the number of tangles as they may have matured past the point of being AT8‐immunopositive (e.g., ghost tangles).
6. CLINICOPATHOLOGIC CORRELATIONS
In 1991, Braak and Braak described neuropathologic staging of neurofibrillary changes in AD, which has become a widely accepted neuropathologic measure termed Braak tangle stage 17 (Figure 1). They used a variety of silver staining methods to evaluate mature tangles, ghost tangles, and neuropil threads. Braak tangle stages I and II display initial neurofibrillary changes in the transentorhinal region in the pre‐α neurons and entorhinal cortex with minimal changes in the CA1 subsector of the hippocampus. 17 Braak stages III and IV have further involvement of the entorhinal cortex and hippocampal subsectors with involvement of temporal cortex. 17 During Braak stages V and VI ghost tangles are present in the transentorhinal region and mature tangles are present throughout the hippocampus and striatum. 17 Association cortices become involved, with eventual involvement of primary cortices. 17
A strong correlation exists between clinical symptoms of AD dementia and NFT accumulation. 5 , 166 , 167 , 168 , 169 Cognitive deficits are not readily observed with the minimal neuropathology of Braak tangle stages I and II. However, by Braak stages III and IV with the involvement of the limbic system and temporal cortices, cognitive impairment and personality changes begin to show. 166 , 170 This agrees well with clinicopathologic observations from Delacourte biochemical staging of AD that identified mild cognitive changes in cases with hippocampal and temporal cortices involvement (stages 3‐6). 168 By Braak stages V and VI, mature tangles and ghost tangles are identified throughout much of the brain, which is reflective of global cognitive impairment. 166 Delacourte biochemical staging similarly reported striking cognitive impairment by stage 7, whereupon two or more polymodal association areas are affected. 168 Taken together, the spatiotemporal patterning of these staging systems underscores the detrimental nature of cortical NFT involvement in the AD brain. Hierarchical staging provides a robust measure useful in interpretation of global tangle involvement, but more work is needed to understand the impact of NFT maturity admixture. Careful evaluation of the composition of NFT maturities (e.g., 90% pretangles with only 10% mature tangles) in the context of cognitive impact may have wide‐reaching implications for cognitive reserve. Perhaps the ability of an individual to withstand NFT pathology exists within the context of how fast or how slow pretangles become mature tangles. Primary age‐related tauopathy (PART) cases are a good example of how NFT admixture may affect cognition, 171 as cases lie along a spectrum of severity. The majority of PART cases demonstrate mild cognitive impairment, but a subset with significant ghost tangles in medial temporal lobe structures make up the entity tangle‐predominant dementia. 150 , 172 As the name belies, the extensive involvement of ghost tangles even in the absence of significant cortical involvement confers a dementing syndrome.
7. BIOMARKER IMPLICATIONS
Understanding the lifespan of NFT maturity is critical to the study of how the disease progresses. In addition to investigating underlying molecular processes, NFT maturity also has implications for neuroimaging biomarkers. Neuroimaging techniques have become more common in assisting ante mortem diagnosis of AD dementia, especially within the context of a research framework. 173 Since the early 2000s, multiple positron emission tomography (PET) tracers were developed to visualize tau in vivo. 31 These tracers include FDDNP, 174 PBB3, 175 flortaucipir (T807, AV‐1451), 176 and THK‐5351 177 (Figure 1). Flortaucipir is a Food and Drug Administration‐approved first‐generation tau PET tracer that can differentiate patients with AD from other tauopathies and healthy controls 178 , 179 , 180 (Figure 7). MK‐6240 is a more recent tau PET tracer first characterized in 2016 181 that was identified through in vitro binding to PHFs from AD brain homogenates and closely reflects flortaucipir binding. 182 Autoradiographic studies of flortaucipir demonstrate binding to more mature levels of NFT pathology, using disease states to make broad comparisons. 183 As exampled by PART cases that can lie along a spectrum of mild pretangle involvement in the hippocampus to end‐stage ghost tangle involvement, 171 flortaucipir binding was readily observed in mild PART cases, but binding was minimally observed in areas where extensive ghost tangles were identified. 183 This and ongoing studies suggest that the composition of NFT maturity levels may influence tau PET signal. The field of tau PET tracers is expanding rapidly with new compounds being evaluated each year 184 , 185 (for a review of related tauopathies, please refer to Leuzy et al. 186 ). Given the complex nature of tau, researchers have high hopes that PET tracers will also demonstrate some specificity for different isoforms of tau other than 3R/4R tau. Flortaucipir and PI‐2620, a more recently developed tau PET tracer, 187 among others, have shown some PET signal in 4R predominant tauopathies. 188 It is too early to know the full extent of tau PET tracer profiles for both tau severity and NFT maturity, but the opportunities are intriguing.
FIGURE 7.
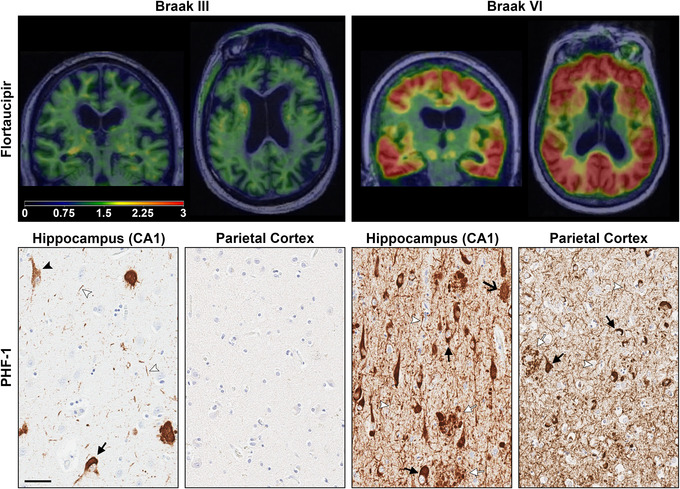
Tau positron emission tomography (PET) imaging of Alzheimer's type tau pathology and corresponding hippocampal tau pathology. Coronal and axial flortaucipir PET scans shown in the top panel were overlaid onto structural magnetic resonance imaging of two study participants who later came to autopsy. 179 The left panel from a Braak stage III study participant diagnosed with diffuse Lewy body disease shows minimal flortaucipir uptake (below threshold) throughout the brain with slightly more prominence in white matter that is likely nonspecific. The corresponding histology image from PHF‐1 immunostained hippocampus revealed scattered pretangles, mature tangles, and rare ghost tangles. In contrast, the Braak VI study participant diagnosed with AD in the right panel demonstrated striking flortaucipir uptake with more intense tau PET signal (warmer red and orange color overlay) throughout the brain. The corresponding PHF‐1 immunostained hippocampus and parietal cortex reveals frequent mature tangles and ghost tangles compared to the Braak III patient. Neuritic pathology including neuropil threads and neuritic plaques is also readily observed in areas of tangle pathology. As the neuropathologic underpinnings of tau PET signal remains an intense area of investigation, the comparative contribution of tangles and neuritic pathology is still unknown. Immunohistochemistry was counterstained with hematoxylin. Black closed arrow, mature tangles; black open arrow, ghost tangles; white arrow head, neuropil thread; white closed arrow, neuritic plaque. Scale bar measures 50 μm.
Further understanding of NFT maturity may also be useful in identifying other blood or cerebrospinal fluid (CSF) biomarkers to aid diagnosis. As the technology continues to advance, molecular detection of above‐mentioned tau truncations and conformational events may be a viable target in CSF and plasma. Recent work demonstrates the hypothesized associations of serum and CSF biomarkers and AD neuropathology. Plasma tau pT181 is strongly associated with both Aβ and tau PET. 189 Plasma tau pT181 better predicted elevated brain Aβ than total tau and was equal or enhanced compared to age and apolipoprotein E (APOE) ε4 carrier status combined. 190 Others have shown an association between baseline CSF Aβ1‐42 and changes in CSF tau pT181 191 that was strongest in female APOE ε4 carriers, emphasizing the potential role of sex differences in AD pathology. 192 Extension of NFT maturities to the temporal sequence of CSF tau or plasma‐based tau measures will be an invaluable contribution to our understanding of how to predict neuropathologic changes and their impact on cognition in preclinical patients. Does early detection of pT217 and pT181 correspond with the slow accumulation of pretangle pathology, whereas later recognition of pT205 corresponds to accumulation of significant mature tangle pathology? 193 As the technology continues to advance, molecular detection of multiplexed tau truncations and conformational events in CSF and plasma may become the targeted biomarker capable of predicting change before neuronal death has occurred.
8. SPECULATION AND FUTURE DIRECTIONS
It may not be enough to have potential treatments remove NFT lesions. It may be more prudent to prevent them from maturing. To move forward, we must close this gap of knowledge concerning NFT maturity. This largely starts with deeper characterizations of the antibodies used to better answer scientific questions. There are many tau antibodies available to researchers that recognize different populations of tangles. For example, AT8 is widely used and suggested as a means to obtain Braak staging. 14 , 15 AT8 predominately visualizes pretangles and mature tangles, and rarely recognizes ghost tangles, excluding an entire maturity level from its studies. While AT8 may be an ideal antibody to study NFT formation, it lacks utility in neurons that are dying/dead from NFTs.
Overwhelming evidence supports the concept of NFT maturation. However, given the cross‐sectional nature of postmortem studies, it is difficult, if not impossible, to observe NFTs maturing in the human brain in vivo. A critical knowledge gap concerning the shift in 4R to 3R tau isoform expression may represent differential dysregulation of tau throughout the NFT lifespan. 53 , 71 , 150 , 153 Because 4R positive pretangles lack fibrillar structure, it seems unlikely that they will serve as a seed through which PHF will form and aggregate. As such, we cannot exclude the possibility that pretangles, mature tangles, and ghost tangles are distinct lesions that occur in isolation. As discussed by Luna‐Muñoz et al., bead‐like structures were observed in neurons between pretangles and mature tangles. 108 They speculated these structures to be nucleation sites, which may provide evidence for the maturation of pretangles into mature tangles through an intermediary 1 level (Figure 5). 108 Regardless, there is a need to further understand pretangles and how mature tangles form as a distinct or minimally related structure compared to ghost tangles.
AD researchers must also work to establish a more complete sequence of post‐translational events through the maturity levels. Some conformational and truncation events have been heavily investigated; however, phosphorylation at different sites, ubiquitination, and other post‐translational events have been less well characterized. Even though it is likely that many of these events occur outside of a temporal sequence, understanding the order of post‐translational events may allow for more targeted treatment options.
There is a need for more studies concerning functional consequences of the NFT maturity levels, both within the neuron and in the neuronal network. More clarity is needed to determine why pretangles form, and if they are protective or toxic. Studies in mouse models have shown that neurons with NFTs remain functional in networks for a time, but do they become non‐functional? How long do NFT‐burdened neurons remain functional compared to not?
We also suggest scientists in the biomarker field work more closely with neuropathologists. For neuroimagers, collaborating with a neuropathologist may lead to a better understanding under the microscope of what the PET ligand may be binding to. We cannot forget the lessons learned from tau antibody‐based studies that demonstrate post‐translational events will remove or add epitopes that allows for less or more binding of an antibody through the NFT lifespan. Why would the same not be true for PET ligands? It is possible that post‐translational events of tau in NFTs will open up or remove ligand binding sites that can influence PET imaging results. More must be done to better understand what these PET ligands are binding to and is signal lost as the NFT matures. For fluid biomarkers, working with a neuropathologist will allow a better understanding of where in the NFT lifespan the targets exist. These targets have been known for a significant number of years, yet we lack understanding of their temporal distribution in the NFT lifespan.
For many years, we have known that certain populations of neurons are predominately vulnerable to NFT pathology, with many compelling follow‐up questions. Do NFTs in certain regions or neuronal populations in the brain mature at different rates? Additionally, does the tau isoform expression of neuronal populations predispose neurons to NFTs? Do NFT maturity levels affect the neuropil surrounding tangle‐burdened neurons?
With the accumulated knowledge, we speculate that early events in the lifespan of NFT maturity begin as protective. The maladaptive response leads to an ill‐fated rescue attempt that overwhelms the neuron driving it to form a mature tangle. Neuroimaging biomarkers designed to target NFTs from tissue homogenates likely recognize the middling form whereupon the neuron begins to die. This may result in a lack of binding until sufficient tau has accumulated, which should correlate well with functional and structural changes. Circulating tau markers will likely benefit early diagnostics. As circulating markers do not provide regional specificity, pairing with neuroimaging measures should provide deeper insight as not all AD cases accumulate tau in the expected topographic pattern.
9. CONCLUSIONS
NFTs exist on a spectrum from early to advanced forms of three distinct, but overlapping, maturity levels (Figure 5). This spectrum spans the intracellular domain to the extracellular space. Pretangles are found in morphologically normal neurons and have a diffuse or granular tau immunostaining pattern that may also be hyperphosphorylated. Mature tangles occur in neurons that may have a dislocated or shrunken nucleus, and are composed of tightly packed fibrils of phosphorylated tau. Ghost tangles occur after the neuron has died, and are composed of loosely packed bundles of fibrils. As NFTs are not static entities, intermediaries of these levels can also be identified in patient tissue. The predominance of tau isoform changes from 4R in pretangles, to 3R/4R in mature tangles, and 3R in ghost tangles. Post‐translational modifications, including phosphorylation, acetylation, glycosylation, and ubiquitination, are present across NFT maturity levels. Truncation and conformational changes of tau occur throughout the lifespan of NFTs, which can be visualized using immunohistochemical techniques (Figure 4). A variety of other staining techniques are used to identify NFT maturity levels, including tinctorial dyes such as H&E and Congo red, and special stains such as silver staining and thioflavin‐S (Figure 4).
There is more than 100 years of research on AD, 1 , 12 yet a large gap in knowledge regarding tau and NFTs still exists. It is well known that tau is hyperphosphorylated in AD, but the reason for this event is still unclear. It is also currently unknown why PHFs are not degraded even though they are ubiquitinated. Questions still remain as to whether therapeutic intervention could halt neuropathology at the pretangle level, thereby preventing neuronal death and cognitive decline. Much of our accumulated knowledge regarding NFT maturity derives from the hippocampus and was thus the focus of this review. NFT maturity studies in the entorhinal cortex 40 , 42 and nucleus basalis of Meynert 53 , 101 have greatly informed the biology. Future studies should consider evaluation of these critical areas and evaluation throughout the cortex to examine similarities and differences in the molecular profile of NFT maturities given heterogeneity of cell types. Moreover, the effect of underlying transcript differences that may confer selective vulnerability deserves further investigation in the context of NFT maturity to uncover timing of mRNA changes prior to formation of lesions or as a result.
The accumulated knowledge and use of immunohistochemical techniques provides us with the ability to infer the dynamic aspects of NFT pathology. Molecular insights gleaned from animal modeling and postmortem human studies provide a broad perspective of the lifespan of NFT maturity. Coupled with modern biomarker readouts, the future holds great promise for identifying mechanisms of early accumulation of tau pathology that can be targeted prior to structural change and the eventual death of the neuron. Thus, the historic perspective of morphology has provided the basis for our understanding of NFT maturity. Through multi‐disciplinary approaches integrating next generation sequencing and innovative neuropathology techniques, 194 we are well positioned to uncover the meaning of the lifespan of NFTs in AD.
CONFLICTS OF INTEREST
Dr. Moloney reports no disclosures. Dr. Lowe serves as a consultant for Bayer Schering Pharma, Philips Molecular Imaging, Piramal Imaging, AVID Radiopharmaceuticals, Eisai Inc., and GE Healthcare and receives research support from GE Healthcare, Siemens Molecular Imaging, AVID Radiopharmaceuticals, the NIH (NIA, NCI), and the MN Partnership for Biotechnology and Medical Genomics. Dr. Murray served as a paid consultant for AVID Radiopharmaceuticals.
ACKNOWLEDGMENTS
This review is dedicated to the memory of the late, great Dr. Peter Davies. Without his altruism, so many of the studies cited in this manuscript may not have been possible. The investigators are supported by grants from the Alzheimer's Association (AARG‐17‐533458); National Institute on Aging (R01 AG054449, U01 AG57195, P30 AG062677); the Florida Department of Health, and the Ed and Ethel Moore Alzheimer's Disease Research Program (8AZ06, 20a22); and a kind gift from David and Frances Strawn. We are grateful to Virginia Phillips, Ariston Librero, Jo Landino, and Monica Castanedes‐Casey for histologic support during figure development. We would like to thank Dr. Francisco García‐Sierra (Cinvestav) for sharing the thiazin red image; Dr. Benjamin Wolozin (Boston University) for sharing Alz‐50 antibody; Dr. Peter Davies (Northwell Health's Feinstein Institutes for Medical Research) for sharing the CP13 and PHF‐1 antibody; and Dr. Nicholas Kannan (Michigan State University) for sharing tau antibodies (TauC3 originally from Dr. Lester Binder at Northwestern University and MN423 originally from Dr. Michal Novak).
Moloney CM, Lowe VJ, Murray ME. Visualization of neurofibrillary tangle maturity in Alzheimer's disease: A clinicopathologic perspective for biomarker research. Alzheimer's Dement. 2021;17:1554–1574. 10.1002/alz.12321
REFERENCES
- 1. Alzheimer A. Über eine eigenartige Erkankung der Hirnrinde. Allg Z Psychiatr Ps. 1907;18:177‐179. [Google Scholar]
- 2. Stelzmann RA, Schnitzlein HN, Murtagh FR. An english translation of Alzheimer's 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin Anat. 1995;8:429‐431. [DOI] [PubMed] [Google Scholar]
- 3. Spires TL, Meyer‐Luehmann M, Stern EA, et al. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J Neurosci. 2005;25:7278‐7287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gomez‐Isla T, Hollister R, West H, et al. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol. 1997;41:17‐24. [DOI] [PubMed] [Google Scholar]
- 5. Giannakopoulos P, Herrmann FR, Bussiere T, et al. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer's disease. Neurology. 2003;60:1495‐1500. [DOI] [PubMed] [Google Scholar]
- 6. Morsch R, Simon W, Coleman PD. Neurons may live for decades with neurofibrillary tangles. J Neuropathol Exp Neurol. 1999;58:188‐197. [DOI] [PubMed] [Google Scholar]
- 7. Meyer‐Luehmann M, Spires‐Jones TL, Prada C, et al. Rapid appearance and local toxicity of amyloid‐beta plaques in a mouse model of Alzheimer's disease. Nature. 2008;451:720‐724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Christie RH, Bacskai BJ, Zipfel WR, et al. Growth arrest of individual senile plaques in a model of Alzheimer's disease observed byin vivomultiphoton microscopy. J Neurosci. 2001;21:858‐864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kuchibhotla KV, Wegmann S, Kopeikina KJ, et al. Neurofibrillary tangle‐bearing neurons are functionally integrated in cortical circuits in vivo. Proc Natl Acad Sci U S A. 2014;111:510‐514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bancher C, Brunner C, Lassmann H, et al. Accumulation of abnormally phosphorylated tau precedes the formation of neurofibrillary tangles in Alzheimer's disease. Brain Res. 1989;477:90‐99. [DOI] [PubMed] [Google Scholar]
- 11. Sadigh‐Eteghad S, Sabermarouf B, Majdi A, Talebi M, Farhoudi M, Mahmoudi J. Amyloid‐beta: a crucial factor in Alzheimer's disease. Med Princ Pract. 2015;24:1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Alzheimer A. Über eigenartige Krankheitsfälle des späteren Alters (On certain peculiar diseases of old age). Z Gesamte Neurol Psy. 1911;4:356‐385. [Google Scholar]
- 13. Khachaturian ZS. Diagnosis of Alzheimer's disease. Arch Neurol. 1985;42:1097‐105. [DOI] [PubMed] [Google Scholar]
- 14. Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement. 2012;8:1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol. 2012;123:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thal DR, Rub U, Orantes M, Braak H. Phases of A beta‐deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791‐800. [DOI] [PubMed] [Google Scholar]
- 17. Braak H, Braak E. Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol. 1991;82:239‐259. [DOI] [PubMed] [Google Scholar]
- 18. Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD): Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479. [DOI] [PubMed] [Google Scholar]
- 19. Alzheimer's Association . 2020 Alzheimer's disease facts and figures. Alzheimers Dement. 2020;16:391‐460. [Google Scholar]
- 20. Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A. 1975;72:1858‐1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Neve RL, Harris P, Kosik KS, Kurnit DM, Donlon TA. Identification of cDNA clones for the human microtubule‐associated protein tau and chromosomal localization of the genes for tau and microtubule‐associated protein 2. Mol Brain Res. 1986;1:271‐280. [DOI] [PubMed] [Google Scholar]
- 22. Avila J, Jimenez JS, Sayas CL, et al. Tau structures. Front Aging Neurosci. 2016;8:262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Drubin DG, Kirschner MW. Tau protein function in living cells. J Cell Biol. 1986;103:2739‐2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cleveland DW, Hwo S‐Y, Kirschner MW. Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J Mol Biol. 1977;116:227‐247. [DOI] [PubMed] [Google Scholar]
- 25. Lindwall G, Cole RD. Phosphorylation affects the ability of tau protein to promote microtubule assembly. J Biol Chem. 1984;259:5301‐5305. [PubMed] [Google Scholar]
- 26. Spillantini MG, Goedert M, Crowther RA, Murrell JR, Farlow MR, Ghetti B. Familial multiple system tauopathy with presenile dementia: a disease with abundant neuronal and glial tau filaments. Proc Natl Acad Sci U S A. 1997;94:4113‐4118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Murray ME, DeTure M. The Neuropathology of Dementia. In: Smith GE, Farias ST, eds. APA Handbook of Dementia. Washington, DC: American Psychological Association; 2018:41‐66. [Google Scholar]
- 28. Kovacs GG. Invited review: neuropathology of tauopathies: principles and practice. Neuropathol Appl Neurobiol. 2015;41:3‐23. [DOI] [PubMed] [Google Scholar]
- 29. Kovacs GG. Tauopathies. In: Kovacs GG, Alafuzoff I. Handbook Clinical Neurology. ed: Elselvier; 2018:355‐368. [DOI] [PubMed] [Google Scholar]
- 30. Götz J, Halliday G, Nisbet RM. Molecular pathogenesis of the tauopathies. Annu Rev Pathol. 2019;14:239‐261. [DOI] [PubMed] [Google Scholar]
- 31. Murray ME, Kouri N, Lin WL, Jack CR, Jr. , Dickson DW, Vemuri P. Clinicopathologic assessment and imaging of tauopathies in neurodegenerative dementias. Alzheimers Res Ther. 2014;6:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule‐associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron. 1989;3:519‐526. [DOI] [PubMed] [Google Scholar]
- 33. Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA. Cloning and sequencing of the cDNA encoding an isoform of microtubule‐associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J. 1989;8:393‐399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Goode BL, Chau M, Denis PE, Feinstein SC. Structural and functional differences between 3‐repeat and 4‐repeat tau isoforms. Implications for normal tau function and the onset of neurodegenetative disease. J Biol Chem. 2000;275:38182‐38189. [DOI] [PubMed] [Google Scholar]
- 35. Sato C, Barthelemy NR, Mawuenyega KG, et al. Tau kinetics in neurons and the human central nervous system. Neuron. 2018;97:1284‐1298 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hong M, Zhukareva V, Vogelsberg‐Ragaglia V, et al. Mutation‐specific functional impairments in distinct tau isoforms of hereditary FTDP‐17. Science. 1998;282:1914‐1917. [DOI] [PubMed] [Google Scholar]
- 37. Wolozin BL, Pruchnicki A, Dickson DW, Davies P. A neuronal antigen in the brains of Alzheimer patients. Science. 1986;232:648‐650. [DOI] [PubMed] [Google Scholar]
- 38. Hyman BT, Van Hoesen GW, Wolozin BL, Davies P, Kromer LJ, Damasio AR. Alz‐50 antibody recognizes Alzheimer‐related neuronal changes. Ann Neurol. 1988;23:371‐379. [DOI] [PubMed] [Google Scholar]
- 39. Bancher C, Grundke‐Iqbal I, Iqbal K, Fried VA, Smith HT, Wisniewski HM. Abnormal phosphorylation of tau precedes ubiquitination in neurofibrillary pathology of Alzheimer disease. Brain Res. 1991;539:11‐18. [DOI] [PubMed] [Google Scholar]
- 40. Augustinack JC, Schneider A, Mandelkow EM, Hyman BT. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer's disease. Acta Neuropathol. 2002;103:26‐35. [DOI] [PubMed] [Google Scholar]
- 41. Tatsumi S, Uchihara T, Aiba I, et al. Ultrastructural differences in pretangles between Alzheimer disease and corticobasal degeneration revealed by comparative light and electron microscopy. Acta Neuropathol Commun. 2014;2:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Braak E, Braak H, Mandelkow EM. A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol. 1994;87:554‐567. [DOI] [PubMed] [Google Scholar]
- 43. Uchihara T, Nakamura A, Yamazaki M, Mori O. Evolution from pretangle neurons to neurofibrillary tangles monitored by thiazin red combined with Gallyas method and double immunofluorescence. Acta Neuropathol. 2001;101:535‐539. [DOI] [PubMed] [Google Scholar]
- 44. Uchihara T. Pretangles and neurofibrillary changes: similarities and differences between AD and CBD based on molecular and morphological evolution. Neuropathology. 2014;34:571‐577. [DOI] [PubMed] [Google Scholar]
- 45. Uchihara T, Tsuchiya K, Nakamura A, Akiyama H. Silver staining profiles distinguish Pick bodies from neurofibrillary tangles of Alzheimer type: comparison between Gallyas and Campbell‐Switzer methods. Acta Neuropathol. 2005;109:483‐489. [DOI] [PubMed] [Google Scholar]
- 46. Braak H, Del Tredici K. The pathological process underlying Alzheimer's disease in individuals under thirty. Acta Neuropathol. 2011;121:171‐181. [DOI] [PubMed] [Google Scholar]
- 47. Attems J, Thal DR, Jellinger KA. The relationship between subcortical tau pathology and Alzheimer's disease. Biochem Soc Trans. 2012;40:711‐715. [DOI] [PubMed] [Google Scholar]
- 48. Perry G, Friedman R, Shaw G, Chau V. Ubiquitin is detected in neurofibrillary tangles and senile plaque neurites of Alzheimer‐disease brains. Proc Natl Acad Sci U S A. 1987;84:3033‐3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mori H, Kondo J, Ihara Y. Ubiquitin is a component of paired helical filaments in Alzheimer's disease. Science. 1987;235:1641‐1644. [DOI] [PubMed] [Google Scholar]
- 50. Yamaguchi H, Morimatsu M, Hirai S, Takahashi K. Alzheimer's neurofibrillary tangles are penetrated by astroglial processes and appear eosinophilic in their final stages. Acta Neuropathol. 1987;72:214‐217. [DOI] [PubMed] [Google Scholar]
- 51. Kril JJ, Patel S, Harding AJ, Halliday GM. Neuron loss from the hippocampus of Alzheimer's disease exceeds extracellular neurofibrillary tangle formation. Acta Neuropathol. 2002;103:370‐376. [DOI] [PubMed] [Google Scholar]
- 52. James BD, Bennett DA, Boyle PA, Leurgans S, Schneider JA. Dementia from Alzheimer disease and mixed pathologies in the oldest old. JAMA. 2012;307:1798‐1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tiernan CT, Ginsberg SD, Guillozet‐Bongaarts AL, et al. Protein homeostasis gene dysregulation in pretangle‐bearing nucleus basalis neurons during the progression of Alzheimer's disease. Neurobiol Aging. 2016;42:80‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hou X, Fiesel FC, Truban D, et al. Age‐ and disease‐dependent increase of the mitophagy marker phospho‐ubiquitin in normal aging and Lewy body disease. Autophagy. 2018;14:1404‐1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hoozemans JJ, van Haastert ES, Nijholt DA, Rozemuller AJ, Eikelenboom P, Scheper W. The unfolded protein response is activated in pretangle neurons in Alzheimer's disease hippocampus. Am J Pathol. 2009;174:1241‐1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. LaPointe NE, Morfini G, Pigino G, et al. The amino terminus of tau inhibits kinesin‐dependent axonal transport: implications for filament toxicity. J Neurosci Res. 2009;87:440‐451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase‐3. Trends Biochem Sci. 2004;29:95‐102. [DOI] [PubMed] [Google Scholar]
- 58. Leroy K, Yilmaz Z, Brion JP. Increased level of active GSK‐3beta in Alzheimer's disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol Appl Neurobiol. 2007;33:43‐55. [DOI] [PubMed] [Google Scholar]
- 59. Cook HC. Origins of tinctorial methods in histology. J Clin Pathol. 1997;50:716‐720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chan JK. The wonderful colors of the hematoxylin‐eosin stain in diagnostic surgical pathology. Int J Surg Pathol. 2014;22:12‐32. [DOI] [PubMed] [Google Scholar]
- 61. Ishii T. Distribution of Alzheimer's neurofibrillary changes in the brain stem and hypothalamus of senile dementia. Acta Neuropathol. 1966;6:181‐187. [DOI] [PubMed] [Google Scholar]
- 62. Ball MJ. Neuronal loss, neurofibrillary tangles and granulovacuolar degeneration in the hippocampus with ageing and dementia. A quantitative study. Acta Neuropathol. 1977;37:111‐118. [DOI] [PubMed] [Google Scholar]
- 63. Kitamura T, Sugimori K, Sudo S, Kobayashi K. Relationship between microtubule‐binding repeats and morphology of neurofibrillary tangle in Alzheimer's disease. Acta Neurol Scand. 2005;112:327‐334. [DOI] [PubMed] [Google Scholar]
- 64. Howie AJ, Brewer DB, Howell D, Jones AP. Physical basis of colors seen in congo red‐stained amyloid in polarized light. Lab Invest. 2008;88:232‐242. [DOI] [PubMed] [Google Scholar]
- 65. Elhaddaoui A, Delacourte A, Pigorsch E, Sajid J, Turrell S. Mechanism of congo red binding to amyloid proteins with a beta pleated sheet structure. Fifth International Conference on the Spectroscopy of Biological Molecules. Loutraki, Greece: Springer; 1993:153‐154. [Google Scholar]
- 66. Elhaddaoui A, Delacourte A, Turrell S. Spectroscopic study of congo red and thioflavin binding to amyloid‐like proteins. J Mol Struct. 1993;294:115‐118. [Google Scholar]
- 67. Stopa B, Piekarska B, Konieczny L, et al. The structure and protein binding of amyloid‐specific dye reagents. Acta Biochim Pol. 2003;50:1213‐1227. [PubMed] [Google Scholar]
- 68. Sarkar S, Raymick J, Cuevas E, Rosas‐Hernandez H, Hanig J. Modification of methods to use Congo‐red stain to simultaneously visualize amyloid plaques and tangles in human and rodent brain tissue sections. Metab Brain Dis. 2020;35:1371‐1383. [DOI] [PubMed] [Google Scholar]
- 69. Ryan NS, Rossor MN, Fox NC. Alzheimer's disease in the 100 years since Alzheimer's death. Brain. 2015;138:3816‐3821. [DOI] [PubMed] [Google Scholar]
- 70. Uchihara T. Silver diagnosis in neuropathology: principles, practice and revised interpretation. Acta Neuropathol. 2007;113:483‐499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Uchihara T, Hara M, Nakamura A, Hirokawa K. Tangle evolution linked to differential 3‐ and 4‐repeat tau isoform deposition: a double immunofluorolabeling study using two monoclonal antibodies. Histochem Cell Biol. 2012;137:261‐267. [DOI] [PubMed] [Google Scholar]
- 72. Lace G, Savva G, Forster G, et al. Hippocampal tau pathology is related to neuroanatomical connections: an ageing population‐based study. Brain. 2009;132:1324‐1334. [DOI] [PubMed] [Google Scholar]
- 73. Kelényi G. On the histochemistry of azo group‐free thiazole dyes. J Histochem Cytochem. 1967;15:172‐180. [DOI] [PubMed] [Google Scholar]
- 74. Schweers O, Mandelkow EM, Biernat J, Mandelkow E. Oxidation of cysteine‐322 in the repeat domain of microtubule‐associated protein tau controls the in vitro assembly of paired helical filaments. Proc Natl Acad Sci U S A. 1995;92:8463‐8467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Yamamoto T, Hirano A. A comparative Study of Modified Bielschowsky, Bodian and Thioflavin S Stains on Alzheimer's neurofibrillary tangles. Neuropathol Appl Neurobiol. 1986;12:3‐9. [DOI] [PubMed] [Google Scholar]
- 76. Yen SH, Crowe A, Dickson DW. Monoclonal antibodies to Alzheimer neurofibrillary tangles. 1. Identification of polypeptides. Am J Pathol. 1985;120:282‐291. [PMC free article] [PubMed] [Google Scholar]
- 77. Dickson DW, Ksiezak‐Reding H, Liu WK, Davies P, Crowe A, Yen SHC. Immunocytochemistry of neurofibrillary tangles with antibodies to subregions of tau protein: identification of hidden and cleaved tau epitopes and a new phosphorylation site. Acta Neuropathol. 1992;84:596‐605. [DOI] [PubMed] [Google Scholar]
- 78. Murray ME, Przybelski SA, Lesnick TG, et al. Early Alzheimer's disease neuropathology detected by proton MR spectroscopy. J Neurosci. 2014;34:16247‐16255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Binder LI, Frankfurter A, Rebhun LI. The distribution of tau in the mammalian central nervous system. J Cell Biol. 1985;101:1371‐1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Carmel G, Mager EM, Binder LI, Kuret J. The structural basis of monoclonal antibody Alz50's selectivity for Alzheimer's disease pathology. J Biol Chem. 1996;271:32789‐32795. [DOI] [PubMed] [Google Scholar]
- 81. Liu WK, Moore WT, Williams RT, Hall FL, Yen SH. Application of synthetic phospho‐ and unphospho‐ peptides to identify phosphorylation sites in a subregion of the tau molecule, which is modified in Alzheimer's disease. J Neurosci Res. 1993;34:371‐376. [DOI] [PubMed] [Google Scholar]
- 82. Szendrei GI, Lee VM, Otvos L, Jr. . Recognition of the minimal epitope of monoclonal antibody Tau‐1 depends upon the presence of a phosphate group but not its location. J Neurosci Res. 1993;34:243‐249. [DOI] [PubMed] [Google Scholar]
- 83. Grundke‐Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule‐associated protein τ (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 1986;83:4913‐4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Jicha GA, Bowser R, Kazam IG, Davies P. Alz‐50 and MC‐1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res. 1997;48:128‐132. [DOI] [PubMed] [Google Scholar]
- 85. Wischik C, Novak M, Thøgersen H, et al. Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci U S A. 1988;85:4506‐4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Khuebachova M, Verzillo V, Skrabana R, et al. Mapping the C terminal epitope of the Alzheimer's disease specific antibody MN423. J Immunol Methods. 2002;262:205‐215. [DOI] [PubMed] [Google Scholar]
- 87. Novak M, Kabat J, Wischik C. Molecular characterization of the minimal protease resistant tau unit of the Alzheimer's disease paired helical filament. EMBO J. 1993;12:365‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. García‐Sierra F, Ghoshal N, Quinn B, Berry RW, Binder LI. Conformational changes and truncation of tau protein during tangle evolution in Alzheimer's disease. J Alzheimers Dis. 2003;5:65‐77. [DOI] [PubMed] [Google Scholar]
- 89. Bondareff W, Wischik C, Novak M, Amos W, Klug A, Roth M. Molecular analysis of neurofibrillary degeneration in Alzheimer's disease. An immunohistochemical study. Am J Pathol. 1990;137:711. [PMC free article] [PubMed] [Google Scholar]
- 90. Binder LI, Guillozet‐Bongaarts AL, Garcia‐Sierra F, Berry RW. Tau, tangles, and Alzheimer's disease. Biochim Biophys Acta. 2005;1739:216‐223. [DOI] [PubMed] [Google Scholar]
- 91. Resch JF, Lehr GS, Wischik CM. Design and synthesis of a potential affinity/cleaving reagent for beta‐pleated sheet protein structures. Bioorg Med Chem Lett. 1991;1:519‐522. [Google Scholar]
- 92. Luna‐Munoz J, Peralta‐Ramirez J, Chavez‐Macias L, Harrington CR, Wischik CM, Mena R. Thiazin red as a neuropathological tool for the rapid diagnosis of Alzheimer's disease in tissue imprints. Acta Neuropathol. 2008;116:507‐515. [DOI] [PubMed] [Google Scholar]
- 93. Mena R, Edwards P, Prez‐Olvera O, Wischik CM. Monitoring pathological assembly of tau and ß‐amyloid proteins in Alzheimer's disease. Acta Neuropathol. 1995;89:50‐56. [DOI] [PubMed] [Google Scholar]
- 94. Greenberg S, Schein J. The isolation of a relatively specific paired helical filament antibody. Soc Neurosci. 1989:1038. [Google Scholar]
- 95. Otvos L, Jr. , Feiner L, Lang E, Szendrei GI, Goedert M, Lee VM. Monoclonal antibody PHF‐1 recognizes tau protein phosphorylated at serine residues 396 and 404. J Neurosci Res. 1994;39:669‐673. [DOI] [PubMed] [Google Scholar]
- 96. Mercken M, Vandermeeren M, Lubke U, et al. Monoclonal antibodies with selective specificity for Alzheimer Tau are directed against phosphatase‐sensitive epitopes. Acta Neuropathol. 1992;84:265‐272. [DOI] [PubMed] [Google Scholar]
- 97. Biernat J, Mandelkow EM, Schröter C, et al. The switch of tau protein to an Alzheimer‐like state includes the phosphorylation of two serine‐proline motifs upstream of the microtubule binding region. EMBO J. 1992;11:1593‐1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Goedert M, Jakes R, Crowther RA, et al. The abnormal phosphorylation of tau protein at Ser‐202 in Alzheimer disease recapitulates phosphorylation during development. Proc Natl Acad Sci U S A. 1993;90:5066‐5070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Goedert M, Jakes R, Vanmechelen E. Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neurosci Lett. 1995;189:167‐170. [DOI] [PubMed] [Google Scholar]
- 100. Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011;70:960‐969. [DOI] [PubMed] [Google Scholar]
- 101. Sassin I, Schultz C, Thal DRet al. Evolution of Alzheimer's disease‐related cytoskeletal changes in the basal nucleus of Meynert. Acta Neuropathol. 2000;100:259‐269. [DOI] [PubMed] [Google Scholar]
- 102. Togo T, Akiyama H, Iseki E, et al. Immunohistochemical study of tau accumulation in early stages of Alzheimer‐type neurofibrillary lesions. Acta Neuropathol. 2004;107:504‐508. [DOI] [PubMed] [Google Scholar]
- 103. Uematsu M, Nakamura A, Ebashi M, Hirokawa K, Takahashi R, Uchihara T. Brainstem tau pathology in Alzheimer's disease is characterized by increase of three repeat tau and independent of amyloid beta. Acta Neuropathol Commun. 2018;6:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. de Silva R, Lashley T, Gibb G, et al. Pathological inclusion bodies in tauopathies contain distinct complements of tau with three or four microtubule‐binding repeat domains as demonstrated by new specific monoclonal antibodies. Neuropathol Appl Neurobiol. 2003;29:288‐302. [DOI] [PubMed] [Google Scholar]
- 105. Jicha GA, Weaver C, Lane E, et al. cAMP‐dependent protein kinase phosphorylations on tau in Alzheimer's disease. J Neurosci. 1999;19:7486‐7494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Vincent I, Rosado M, Davies P. Mitotic mechanisms in Alzheimer's disease? J Cell Biol. 1996;132:413‐425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Jicha GA, Lane E, Vincent I, Otvos L, Jr. , Hoffmann R, Davies P. A conformation‐ and phosphorylation‐dependent antibody recognizing the paired helical filaments of Alzheimer's disease. J Neurochem. 1997;69:2087‐2095. [DOI] [PubMed] [Google Scholar]
- 108. Luna‐Munoz J, Garcia‐Sierra F, Falcon V, Menendez I, Chavez‐Macias L, Mena R. Regional conformational change involving phosphorylation of tau protein at the Thr231, precedes the structural change detected by Alz‐50 antibody in Alzheimer's disease. J Alzheimers Dis. 2005;8:29‐41. [DOI] [PubMed] [Google Scholar]
- 109. Ghoshal N, Garcia‐Sierra F, Fu Y, et al. Tau‐66: evidence for a novel tau conformation in Alzheimer's disease. J Neurochem. 2001;77:1372‐1385. [DOI] [PubMed] [Google Scholar]
- 110. Togo T, Sahara N, Yen SH, et al. Argyrophilic grain disease is a sporadic 4‐repeat tauopathy. J Neuropathol Exp Neurol. 2002;61:547‐556. [DOI] [PubMed] [Google Scholar]
- 111. Hara M, Hirokawa K, Kamei S, Uchihara T. Isoform transition from four‐repeat to three‐repeat tau underlies dendrosomatic and regional progression of neurofibrillary pathology. Acta Neuropathol. 2013;125:565‐579. [DOI] [PubMed] [Google Scholar]
- 112. Dan A, Takahashi M, Masuda‐Suzukake M, et al. Extensive deamidation at asparagine residue 279 accounts for weak immunoreactivity of tau with RD4 antibody in Alzheimer's disease brain. Acta Neuropathol Commun. 2013;1:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Gamblin TC, Chen F, Zambrano A, et al. Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer's disease. Proc Natl Acad Sci U S A. 2003;100:10032‐10037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Gibbons GS, Banks RA, Kim B, et al. Detection of Alzheimer Disease (AD)‐specific tau pathology in AD and NonAD tauopathies by immunohistochemistry with novel conformation‐selective tau antibodies. J Neuropathol Exp Neurol. 2018;77:216‐228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Wischik C, Novak M, Edwards P, Klug A, Tichelaar W, Crowther R. Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci U S A. 1988;85:4884‐4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Seo J‐W, Lee K‐J. Post‐translational modifications and their biological functions: proteomic analysis and systematic approaches. BMB Rep. 2004;37:35‐44. [DOI] [PubMed] [Google Scholar]
- 117. Martin L, Latypova X, Terro F. Post‐translational modifications of tau protein: implications for Alzheimer's disease. Neurochem Int. 2011;58:458‐471. [DOI] [PubMed] [Google Scholar]
- 118. Goedert M, Spillantini MG, Cairns NJ, Crowther RA. Tau proteins of alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron. 1992;8:159‐168. [DOI] [PubMed] [Google Scholar]
- 119. Dickson DW, Wertkin A, Mattiace LA, et al. Ubiquitin immunoelectron microscopy of dystrophic neurites in cerebellar senile plaques of Alzheimer's disease. Acta Neuropathol. 1990;79:486‐493. [DOI] [PubMed] [Google Scholar]
- 120. Novak M, Wischik CM, Edwards P, Pannell R, Milstein C. Characterisation of the first monoclonal antibody against the pronase resistant core of the Alzheimer PHF. Prog Clin Biol Res. 1989;317:755‐761. [PubMed] [Google Scholar]
- 121. Basurto‐Islas G, Luna‐Munoz J, Guillozet‐Bongaarts AL, Binder LI, Mena R, Garcia‐Sierra F. Accumulation of aspartic acid421‐ and glutamic acid391‐cleaved tau in neurofibrillary tangles correlates with progression in Alzheimer disease. J Neuropathol Exp Neurol. 2008;67:470‐483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Canu N, Dus L, Barbato C, et al. Tau cleavage and dephosphorylation in cerebellar granule neurons undergoing apoptosis. J Neurosci. 1998;18:7061‐7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Rissman RA, Poon WW, Blurton‐Jones M, et al. Caspase‐cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest. 2004;114:121‐130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Chung CW, Song YH, Kim IK, et al. Proapoptotic effects of tau cleavage product generated by caspase‐3. Neurobiol Dis. 2001;8:162‐172. [DOI] [PubMed] [Google Scholar]
- 125. Fasulo L, Ugolini G, Visintin M, et al. The neuronal microtubule‐associated protein tau is a substrate for caspase‐3 and an effector of apoptosis. J Neurochem. 2000;75:624‐633. [DOI] [PubMed] [Google Scholar]
- 126. Horowitz PM, Patterson KR, Guillozet‐Bongaarts AL, et al. Early N‐terminal changes and caspase‐6 cleavage of tau in Alzheimer's disease. J Neurosci. 2004;24:7895‐7902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Endoh R, Ogawara M, Iwatsubo T, Nakano I, Mori H. Lack of the carboxyl terminal sequence of tau in ghost tangles of Alzheimer's disease. Brain Res. 1993;601:164‐172. [DOI] [PubMed] [Google Scholar]
- 128. Takahashi M, Tsujioka Y, Yamada T, et al. Glycosylation of microtubule‐associated protein tau in Alzheimer's disease brain. Acta Neuropathol. 1999;97:635‐641. [DOI] [PubMed] [Google Scholar]
- 129. Wang J‐Z, Grundke‐Iqbal I, Iqbal K. Glycosylation of microtubule–associated protein tau: an abnormal posttranslational modification in Alzheimer's disease. Nat Med. 1996;2:871‐875. [DOI] [PubMed] [Google Scholar]
- 130. Smith MA, Taneda S, Richey PL, et al. Advanced Maillard reaction end products are associated with Alzheimer disease pathology. Proc Natl Acad Sci U S A. 1994;91:5710‐5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Yan SD, Chen X, Schmidt AM, et al. Glycated tau protein in Alzheimer disease: a mechanism for induction of oxidant stress. Proc Natl Acad Sci U S A. 1994;91:7787‐7791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Reynolds MR, Reyes JF, Fu Y, et al. Tau nitration occurs at tyrosine 29 in the fibrillar lesions of Alzheimer's disease and other tauopathies. J Neurosci. 2006;26:10636‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Irwin DJ, Cohen TJ, Grossman M, et al. Acetylated tau, a novel pathological signature in Alzheimer's disease and other tauopathies. Brain. 2012;135:807‐818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Cook C, Carlomagno Y, Gendron TF, et al. Acetylation of the KXGS motifs in tau is a critical determinant in modulation of tau aggregation and clearance. Hum Mol Genet. 2014;23:104‐116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Boros BD, Greathouse KM, Gentry EG, et al. Dendritic spines provide cognitive resilience against Alzheimer's disease. Ann Neurol. 2017;82:602‐614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Kidd M. Paired helical filaments in electron microscopy of Alzheimer's disease. Nature. 1963;197:192‐193. [DOI] [PubMed] [Google Scholar]
- 137. Terry RD. The fine structure of neurofibrillary tangles in Alzheimer's disease. J Neuropathol Exp Neurol. 1963;22:629‐642. [DOI] [PubMed] [Google Scholar]
- 138. Kirschner DA, Abraham C, Selkoe DJ. X‐ray diffraction from intraneuronal paired helical filaments and extraneuronal amyloid fibers in Alzheimer disease indicates cross‐beta conformation. Proc Natl Acad Sci U S A. 1986;83:503‐507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Taylor WR, May ACW, Brown NP, Aszódi A. Protein structure: geometry, topology and classification. Rep Prog Phys. 2001;64:517‐590. [Google Scholar]
- 140. Grundke‐Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM. Microtubule‐associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem. 1986;261:6084‐6089. [PubMed] [Google Scholar]
- 141. Delacourte A, Defossez A. Alzheimer's disease: tau proteins, the promoting factors of microtubule assembly, are major components of paired helical filaments. J Neurological Sci. 1986;76:173‐186. [DOI] [PubMed] [Google Scholar]
- 142. Kosik KS, Joachim CL, Selkoe DJ. Microtubule‐associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci U S A. 1986;83:4044‐4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Nukina N, Ihara Y. One of the antigenic determinants of paired helical filaments is related to tau protein. J Biochem. 1986;99:1541‐1544. [DOI] [PubMed] [Google Scholar]
- 144. Goedert M, Wischik C, Crowther R, Walker J, Klug A. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule‐associated protein tau. Proc Natl Acad Sci U S A. 1988;85:4051‐4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Wood JG, Mirra SS, Pollock NJ, Binder LI. Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the axonal microtubule‐associated protein tau (τ). Proc Natl Acad Sci U S A. 1986;83:4040‐4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Brion J‐P, Passareiro H, Nunez J, Flament‐Durand J. Mise en évidence immunologique de la protéine tau au niveau des lésions de dégénérescence neurofibrillaire de la maladie d'Alzheimer. Arch Biol (Liege). 1985;96:229‐235. [Google Scholar]
- 147. Yagishita S, Itoh Y, Nan W, Amano N. Reappraisal of the fine structure of Alzheimer's neurofibrillary tangles. Acta Neuropathol. 1981;54:239‐246. [DOI] [PubMed] [Google Scholar]
- 148. Fitzpatrick AWP, Falcon B, He S, et al. Cryo‐EM structures of tau filaments from Alzheimer's disease. Nature. 2017;547:185‐190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Espinoza M, de Silva R, Dickson DW, Davies P. Differential incorporation of tau isoforms in Alzheimer's disease. J Alzheimers Dis. 2008;14:1‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Jellinger KA, Attems J. Neurofibrillary tangle‐predominant dementia: comparison with classical Alzheimer disease. Acta Neuropathol. 2007;113:107‐117. [DOI] [PubMed] [Google Scholar]
- 151. Glatz DC, Rujescu D, Tang Y, et al. The alternative splicing of tau exon 10 and its regulatory proteins CLK2 and TRA2‐BETA1 changes in sporadic Alzheimer's disease. J Neurochem. 2006;96:635‐644. [DOI] [PubMed] [Google Scholar]
- 152. Yasojima K, McGeer EG, McGeer PL. Tangled areas of Alzheimer brain have upregulated levels of exon 10 containing tau mRNA. Brain Res. 1999;831:301‐305. [DOI] [PubMed] [Google Scholar]
- 153. Ginsberg SD, Che S, Counts SE, Mufson EJ. Shift in the ratio of three‐repeat tau and four‐repeat tau mRNAs in individual cholinergic basal forebrain neurons in mild cognitive impairment and Alzheimer's disease. J Neurochem. 2006;96:1401‐1408. [DOI] [PubMed] [Google Scholar]
- 154. Ghetti B, Oblak AL, Boeve BF, Johnson KA, Dickerson BC, Goedert M. Invited review: Frontotemporal dementia caused by microtubule‐associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging. Neuropathol Appl Neurobiol. 2015;41:24‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Smith R, Puschmann A, Scholl M, et al. 18F‐AV‐1451 tau PET imaging correlates strongly with tau neuropathology in MAPT mutation carriers. Brain. 2016;139:2372‐2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Spina S, Schonhaut DR, Boeve BF, et al. Frontotemporal dementia with the V337M MAPT mutation: tau‐PET and pathology correlations. Neurology. 2017;88:758‐766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Braak H, Braak E, Grundke‐Iqbal I, Iqbal K. Occurrence of neuropil threads in the senile human brain and in Alzheimer's disease: a third location of paired helical filaments outside of neurofibrillary tangles and neuritic plaques. Neurosci Lett. 1986;65:351‐355. [DOI] [PubMed] [Google Scholar]
- 158. Braak H, Braak E. Neuropil threads occur in dendrites of tangle‐bearing nerve cells. Neuropathol Appl Neurobiol. 1988;14:39‐44. [DOI] [PubMed] [Google Scholar]
- 159. Perry G, Kawai M, Tabaton M, et al. Neuropil threads of Alzheimer's disease show a marked alteration of the normal cytoskeleton. J Neurosci. 1991;11:1748‐1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Probst A, Basler V, Bron B, Ulrich J. Neuritic plaques in senile dementia of Alzheimer type: a Golgi analysis in the hippocampal region. Brain Res. 1983;268:249‐254. [DOI] [PubMed] [Google Scholar]
- 161. Kawai M, Kalaria RN, Harik SI, Perry G. The relationship of amyloid plaques to cerebral capillaries in Alzheimer's disease. Am J Pathol. 1990;137:1435‐1446. [PMC free article] [PubMed] [Google Scholar]
- 162. Miyakawa T, Shimoji A, Kuramoto R, Higuchi Y. The relationship between senile plaques and cerebral blood vessels in Alzheimer's disease and senile dementia. Morphological mechanism of senile plaque production. Virchows Arch B Cell Pathol Incl Mol Pathol. 1982;40:121‐129. [DOI] [PubMed] [Google Scholar]
- 163. Hansra GK, Popov G, Banaczek PO, et al. The neuritic plaque in Alzheimer's disease: perivascular degeneration of neuronal processes. Neurobiol Aging. 2019;82:88‐101. [DOI] [PubMed] [Google Scholar]
- 164. Munoz DG, Wang D. Tangle‐associated neuritic clusters: a new lesion in Alzheimer's disease and aging suggests that aggregates of dystrophic neurites are not necessarily associated with ß/A4. Am J Pathol. 1992;140:1167‐1178. [PMC free article] [PubMed] [Google Scholar]
- 165. García‐Sierra F, Wischik CM, Harrington CR, Luna‐Muñoz J, Mena R. Accumulation of C‐terminally truncated tau protein associated with vulnerability of the perforant pathway in early stages of neurofibrillary pathology in Alzheimer's disease. J Chem Neuroanat. 2001;22:65‐77. [DOI] [PubMed] [Google Scholar]
- 166. Braak H, Braak E. Staging of Alzheimer's disease‐related neurofibrillary changes. Neurobiol Aging. 1995;16:271‐278. [DOI] [PubMed] [Google Scholar]
- 167. Nelson PT, Alafuzoff I, Bigio EH, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012;71:362‐381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Delacourte A, David JP, Sergeant N, et al. The biochemical pathway of neurofibrillary degeneration in aging and Alzheimer's disease. Neurology. 1999;52:1158‐1165. [DOI] [PubMed] [Google Scholar]
- 169. Murray ME, Lowe VJ, Graff‐Radford NR, et al. Clinicopathologic and 11C‐Pittsburgh compound B implications of Thal amyloid phase across the Alzheimer's disease spectrum. Brain. 2015;138:1370‐1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Jicha GA, Parisi JE, Dickson DW, et al. Neuropathologic outcome of mild cognitive impairment following progression to clinical dementia. Arch Neurol. 2006;63:674‐681. [DOI] [PubMed] [Google Scholar]
- 171. Crary JF, Trojanowski JQ, Schneider JA, et al. Primary age‐related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 2014;128:755‐766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Janocko NJ, Brodersen KA, Soto‐Ortolaza AI, et al. Neuropathologically defined subtypes of Alzheimer's disease differ significantly from neurofibrillary tangle‐predominant dementia. Acta Neuropathol. 2012;124:681‐692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Jack CR, Jr. , Bennett DA, Blennow K, et al. NIA‐AA research framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14:535‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Shoghi‐Jadid K, Small GW, Agdeppa ED, et al. Localization of neurofibrillary tangles and beta‐amyloid plaques in the brains of living patients with Alzheimer disease. Am J Geriatr Psychiatry. 2002;10:24‐35. [PubMed] [Google Scholar]
- 175. Maruyama M, Shimada H, Suhara T, et al. Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron. 2013;79:1094‐1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Chien DT, Bahri S, Szardenings AK, et al. Early clinical PET imaging results with the novel PHF‐tau radioligand [F‐18]‐T807. J Alzheimers Dis. 2013;34:457‐468. [DOI] [PubMed] [Google Scholar]
- 177. Harada R, Okamura N, Furumoto S, et al. 18F‐THK5351: a novel PET radiotracer for imaging neurofibrillary pathology in Alzheimer disease. J Nucl Med. 2016;57:208‐214. [DOI] [PubMed] [Google Scholar]
- 178. Sander K, Lashley T, Gami P, et al. Characterization of tau positron emission tomography tracer [(18)F]AV‐1451 binding to postmortem tissue in Alzheimer's disease, primary tauopathies, and other dementias. Alzheimers Dement. 2016;12:1116‐1124. [DOI] [PubMed] [Google Scholar]
- 179. Lowe VJ, Lundt ES, Albertson SM, et al. Tau‐positron emission tomography correlates with neuropathology findings. Alzheimers Dement. 2020;16:561‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Marquie M, Normandin MD, Vanderburg CR, et al. Validating novel tau positron emission tomography tracer [F‐18]‐AV‐1451 (T807) on postmortem brain tissue. Ann Neurol. 2015;78:787‐800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Hostetler ED, Walji AM, Zeng Z, et al. Preclinical Characterization of 18F‐MK‐6240, a promising PET Tracer for in vivo quantification of human neurofibrillary tangles. J Nucl Med. 2016;57:1599‐1606. [DOI] [PubMed] [Google Scholar]
- 182. Aguero C, Dhaynaut M, Normandin MD, et al. Autoradiography validation of novel tau PET tracer [F‐18]‐MK‐6240 on human postmortem brain tissue. Acta Neuropathol Commun. 2019;7:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Lowe VJ, Curran G, Fang P, et al. An autoradiographic evaluation of AV‐1451 Tau PET in dementia. Acta Neuropathol Commun. 2016;4:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Teng E, Ward M, Manser PT, et al. Cross‐sectional associations between [(18)F]GTP1 tau PET and cognition in Alzheimer's disease. Neurobiol Aging. 2019;81:138‐145. [DOI] [PubMed] [Google Scholar]
- 185. Sanabria Bohorquez S, Marik J, Ogasawara A, et al. [(18)F]GTP1 (Genentech Tau Probe 1), a radioligand for detecting neurofibrillary tangle tau pathology in Alzheimer's disease. Eur J Nucl Med Mol Imaging. 2019;46:2077‐2089. [DOI] [PubMed] [Google Scholar]
- 186. Leuzy A, Chiotis K, Lemoine L, et al. Tau PET imaging in neurodegenerative tauopathies‐still a challenge. Mol Psychiatry. 2019;24:1112‐1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Kroth H, Oden F, Molette J, et al. Discovery and preclinical characterization of [(18)F]PI‐2620, a next‐generation tau PET tracer for the assessment of tau pathology in Alzheimer's disease and other tauopathies. Eur J Nucl Med Mol Imaging. 2019;46:2178‐2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Utianski RL, Martin PR, Botha H, et al. Longitudinal flortaucipir ([(18)F]AV‐1451) PET imaging in primary progressive apraxia of speech. Cortex. 2019;124:33‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Thijssen EH, La Joie R, Wolf A, et al. Diagnostic value of plasma phosphorylated tau181 in Alzheimer's disease and frontotemporal lobar degeneration. Nat Med. 2020;26:387‐397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Mielke MM, Hagen CE, Xu J, et al. Plasma phospho‐tau181 increases with Alzheimer's disease clinical severity and is associated with tau‐ and amyloid‐positron emission tomography. Alzheimers Dement. 2018;14:989‐997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Buckley RF, Mormino EC, Chhatwal J, et al. Associations between baseline amyloid, sex, and APOE on subsequent tau accumulation in cerebrospinal fluid. Neurobiol Aging. 2019;78:178‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Liesinger AM, Graff‐Radford NR, Duara R, et al. Sex and age interact to determine clinicopathologic differences in Alzheimer's disease. Acta Neuropathol. 2018;136:873‐885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Barthelemy NR, Li Y, Joseph‐Mathurin N, et al. A soluble phosphorylated tau signature links tau, amyloid and the evolution of stages of dominantly inherited Alzheimer's disease. Nat Med. 2020;26:398‐407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Crist AM, Hinkle KM, Wang X, et al. Leveraging selective vulnerability of the hippocampus in Alzheimer's disease subtypes reveals SERPINA5 as a novel tau binding partner. 2020. arXiv.2020.12.18.423469.
- 195. Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease‐associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112:389‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Mukrasch MD, Bibow S, Korukottu J, et al. Structural polymorphism of 441‐residue tau at single residue resolution. PLoS Biol. 2009;7:e34. [DOI] [PMC free article] [PubMed] [Google Scholar]