

Abstract
Immunosuppressive drugs can alleviate debilitating symptoms of autoimmune diseases, but, by the same token, excessive immune suppression can result in an increased risk of infection. Despite the dangers of a compromised immune system, clear definitions of what constitutes excessive suppression remain elusive. Here we review the most common infections associated with primary antibody deficiencies (PADs), such as agammaglobulinemia, common variable immunodeficiency (CVID), and IgA deficiency, as well as infections that are associated with drug-induced or secondary antibody immunodeficiencies (SADs). We identify a number of bacterial, viral, and fungal infections (e.g., Listeria monocytogenes, Staphylococcus sp., Salmonella spp., Escherichia coli, influenza, varicella zoster virus, and herpes simplex virus) associated with both PADs and SADs, and suggest that diagnostic criteria for PADs could be used as a first-line measure to identify potentially unsafe levels of immune suppression in SADs. Specifically, we suggest that, based on PAD diagnostic criteria, IgG levels should remain above 2–3 g/L, IgA levels should not fall below 0.07 g/L, and IgM levels should remain above 0.4 g/L to prevent immunosuppressive drugs from inducing mimicking PAD-like effects. We suggest that these criteria could be used in the early stages of drug development, and that pharmacokinetic and pharmacodynamic modeling could help guide patient selection to potentially improve drug safety. We illustrate the proposed approach using atacicept as an example and conclude with a discussion of the applicability of this approach for other drugs that may induce excessive immune suppression.
Supplementary Information
The online version contains supplementary material available at 10.1007/s13318-021-00706-z.
Key Points
| The occurrence of similar infections in patients with primary (PADs) and secondary (SADs) antibody deficiencies suggests there may be value to using diagnostic criteria for PADs to increase the safety of SADs. |
| Atacicept can be used as a retrospective example to demonstrate the potential of the mathematical modeling of early clinical data to increase drug safety by refining the inclusion criteria for patient selection. |
Introduction
Disease management for severe autoimmune disorders, such as rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), and myasthenia gravis, frequently relies on the use of immunosuppressive drugs that both reduce symptoms and attempt to short circuit the disease cycle [1]. For instance, in SLE, immunosuppressive drugs such as atacicept and belimumab are administered to prevent B cell maturation, while drugs such as anifrolumab are designed to interfere with the signaling of inflammatory cytokines such as IFN-γ [2]. While these approaches may alleviate some of the disease symptoms, they can also result in significant immune suppression, especially when administered in addition to nonspecific immune suppressants such as corticosteroids, which is often the standard of care [3–5]. This oversuppression can cause immune deficiency, increasing patient susceptibility to opportunistic infections [2].
There are two recognized types of immunodeficiencies: primary and secondary. Primary antibody deficiencies (PADs) are the most common form of primary antibody deficiency. PADs are typically caused by genetic defects that result in either loss of immunoglobulins (Ig) or their malfunction due to the disruption of B cell differentiation. PADs include common variable immunodeficiency (CVID) and pan-agammaglobulinemia (the absence of IgM, IgG, and IgA), among others. A detailed review of PADs can be found in the literature [6–9]. Unfortunately, there are currently no cures for these conditions, so therapeutic interventions are primarily palliative, aimed at managing infections and boosting the immune system [6, 8, 10].
Secondary antibody deficiencies (SADs) are acquired as a result of disease (e.g., HIV) or drug treatment, such as chemotherapy or immunosuppressive drugs. Mechanisms of action of the latter can involve the depletion of B cells, which occurs for example when rituximab [11] is used for the treatment of lupus nephritis [12, 13] or milatuzumab is used for the treatment of non-Hodgkin lymphomas, multiple myeloma, and chronic lymphocytic leukemia [14–16]. Other mechanisms of action include inhibition of B cell survival (this occurs with, for example, belimumab [17] and atacicept [18]), inhibition of B cell activation (this occurs with imatinib for chronic myelogenous leukemia [19] or with bortezomib for multiple myeloma [20], for example), as well as inhibition of T cell/B cell interactions (this occurs with, for example, abatacept for rheumatoid arthritis [21]). A detailed review of SADs can be found in [22–24].
Both PADs and SADs are associated with increased infection risk, particularly of the upper respiratory tract. Despite this increased susceptibility to infections of patients undergoing immunosuppressive therapy, the exact identification of safety thresholds with regards to acceptable levels of immune suppression remain elusive, due in particular to high interpatient variability. While some adverse events can only be discovered in late-stage clinical trials, many can be identified in preclinical or in early-stage clinical studies when new drugs are first tested on healthy subjects or in patients to assess safety. If it was known and benchmarked earlier in the drug discovery/development process, this information could be quite beneficial, as it could be factored into the quantitative analysis of the drug before it is introduced to clinics.
Here we propose using diagnostic criteria for PADs to inform safety thresholds for SADs. We first summarize known common diagnostic criteria for PAD patients, placing a particular focus on CVID and agammaglobulinemia as diseases that are symptomatically closest to drug-induced antibody deficiencies. We suggest that preclinical pharmacokinetic and pharmacodynamic modeling could be used to guide patient selection to improve drug safety. We illustrate the proposed approach using atacicept as an example. We conclude with a discussion of how this approach could be used to potentially increase the safety of future immunosuppressive drugs.
Primary Antibody Deficiencies
PADs are a heterogeneous group of disorders characterized by various degrees of dysfunctional antibody production resulting from a disruption of B cell differentiation at different stages of B cell development [25, 26]. Clinical manifestations range from an almost complete absence of B cells and serum immunoglobulins to selectively impaired antibody responses to specific antigens in the presence of normal total serum immunoglobulin concentrations. All of these disorders lead to an increased susceptibility to infections that predominantly affect the respiratory tract [25].
Agammaglobulinemia
Agammaglobulinemias are rare antibody deficiencies caused by defects during early B cell development [24]. They are characterized by low to undetectable numbers of B cells and significantly decreased concentrations of immunoglobulins. At the time of diagnosis, most patients have low serum immunoglobulin concentrations; specifically, IgG concentrations below 2 g/L, IgA concentrations below 0.15 g/L, and IgM concentrations below 0.4 g/L [27]. Furthermore, almost all patients have markedly decreased B cell numbers, with under 2% CD19- or CD20-positive lymphocytes in the blood [25, 27]. Agammaglobulinemia patients are often prone to serial bacterial and viral infections [27].
Common Variable Immunodeficiency (CVID)
CVID is a disorder of B cell differentiation and maturation with dysfunctional antibody production [25, 28]. According to the immunologic analysis of a European multicenter cohort of CVID patients [28], usually several immunoglobulin classes—particularly IgG and IgA—are reduced. Hypogammaglobulinemia [29] is typically defined as a serum IgG level below 6 g/L; it is further stratified into mild (4–5.99 g/L), moderate (2–3.99 g/L), and severe (0–1.99 g/L). For CVID patients, serum IgG concentrations are in the range of 2.1 ± 1.65 g/L (normal range 7–17 g/L), IgA concentrations are 0.2–0.7 g/L (normal range 0.7–4 g/L), and IgM concentrations are below the norm of 0.4–2.3 g/L. T cell numbers and function are also reduced in some patients. Furthermore, reduced numbers of switched memory B cells (less than 2% of total B cells) have been shown to correlate with disease-associated complications such as splenomegaly and granulomatous disease [25, 28]. Patients often come to medical attention due to acute or chronic bacterial and, less frequently, viral infections [25].
IgA Deficiency
Selective IgA deficiency occurs when concentrations of serum IgA fall below 0.07 g/L in the presence of normal serum concentrations of IgG and IgM; it is diagnosed after other causes of hypogammaglobulinemia have been ruled out [25, 30]. Notably, the IgA deficiency is often compensated for by increased concentrations of IgM. Some IgA-deficient patients progress over time to CVID. A subset of IgA-deficient patients develop recurrent pulmonary and gastrointestinal infections [25].
Commonly Occurring Infections in PAD Patients
Pneumonia is a common infection in PAD patients. For instance, at least two-thirds of CVID patients had one or more episodes of pneumonia prior to diagnosis. In fact, almost all patients with PADs suffer from upper and lower respiratory tract bacterial infections. Haemophilus influenzae, Streptococcus pneumoniae, and Moraxella catarrhalis are present in the nasal cultures of the majority of patients, and frequently more than one pathogen is present [25]. Chronic lung disease and gastrointestinal complications are frequently observed in these patients. PAD patients sometimes also get chronic infectious arthritis, chronic enteroviral meningoencephalitis, acute and/or recurrent bacterial conjunctivitis, and occasional cutaneous infectious complications [25].
Infections Commonly Associated with Immunosuppressive Therapy (SADs)
Orlicka et al. [31] summarized the infections commonly associated with immunosuppressive therapy. These include Listeria monocytogenes, Staphylococcus sp., Salmonella spp., Escherichia coli, influenza, varicella zoster virus, and herpes simplex virus, among others. The most common of these infections are summarized in Fig. 1.
Fig. 1.

Infections that are commonly associated with secondary antibody deficiencies [SADs] (data adapted from [31]), and infections that are also common in primary antibody deficiencies [PADs] (data adapted from [25]). Bacterial infections are colored red, viral inflection are in blue, fungal infections are in brown, and parasitic infections are in green. The occurrence of similar infections in patients with PADs and SADs suggests there may be value to using diagnostic criteria from PADs to inform safety thresholds for SADs
A summary of criteria that correlate with increased infections in PADs patients is given in Fig. 2.
Fig. 2.
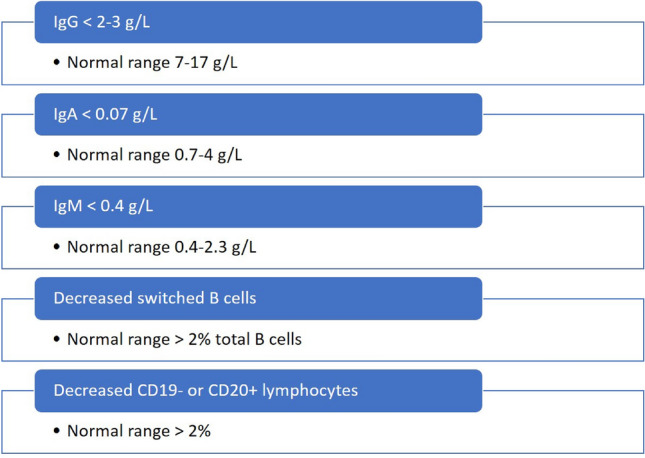
Summary of criteria that correlate with increased infections in patients with primary antibody deficiencies, as described in Sect. 2; it is proposed that these criteria could be used as safety thresholds for secondary antibody deficiencies
Due to the fact that both PAD and SAD patients succumb to similar infections associated with a suppressed immune system, in the absence of a more rigorous understanding of SADs, the levels of immune cells and immunoglobulins commonly observed in PAD patients could be used to inform potential safety thresholds for immunosuppressive therapies.
Using Safety Thresholds in Predictive Pharmacokinetic and Pharmacodynamic Modeling
An understanding of safety thresholds for immunosuppressive drugs can be particularly useful in preclinical and early clinical pharmacokinetic and pharmacodynamic (PKPD) modeling. To illustrate this approach, we first describe a general PK model that describes the key mechanisms that drive the absorption, distribution, metabolism, and elimination of a drug upon its administration into the body. The PK model is then coupled with an indirect response PD model, which captures the impact of the change in drug concentration over time on a biomarker, in this case an immunoglobulin. We then illustrate how such a model can be used to increase drug safety, using the immunosuppressive drug atacicept as an example.
First, consider the following two-compartment model that describes the dynamics and interactions over time of the drug, its target, and the drug–target complex:
Drug is absorbed at a rate into the plasma.
Once in the plasma, drug can be distributed into the tissue; it can also be cleared from the plasma, or the drug can redistribute between tissue and plasma.
Once in the tissue, the drug can interact with its target .
The dynamics of the drug–target complex are governed by association/dissociation reactions between and ; the drug–target complex can also be cleared.
Finally, the target has natural turnover rates of synthesis (ksyn) and degradation (kdeg) to allow for the maintenance of the steady-state concentration of the target.
These dynamics can be captured using the following system (Eq. 1) of ordinary differential equations that allow the concentrations of the drug in plasma and in tissue as well as the kinetics of the target and the drug–target complex to be tracked over time:
| 1 |
A summary of the parameters used in system (1) is given in Table 1. The initial conditions are , . The initial concentration of the target is determined by its baseline level such that . A detailed investigation of such models—called site of action (SoA) models—and their properties is provided in, for instance, [32]. A schematic of the processes described by system (1) is given in Fig. 3.
Table 1.
Summary of the parameters used in system (1)
| Parameter | Description | Units |
|---|---|---|
| Physiological parameters | ||
| Volume of the plasma compartment | L | |
| Volume of the tissue (site of action) compartment | L | |
| Drug pharmacokinetics | ||
| Rate of drug clearance from the plasma and tissue compartments | L/day | |
| Rate of drug absorption | 1/day | |
| Drug distribution rate from tissue to plasma | 1/day | |
| Drug distribution rate from plasma to tissue | 1/day | |
| Rate of drug elimination from plasma, | 1/day | |
| Target properties | ||
| Equilibrium dissociation constant for drug–target binding | nM | |
| Second-order rate constant of drug–target binding | nM/day | |
| First-order rate constant for drug dissociation; | 1/day | |
| Homeostatic baseline target concentrations (determined experimentally or obtained from the literature) | nM | |
| or | Rate of target internalization, degradation, or clearance (this can also be calculated from the half-life of the target as ) | 1/day |
| Target synthesis rate where is the target-specific baseline target concentration | nM/day | |
Fig. 3.

A schematic diagram of the sample model described in system (1). Parameter descriptions and units are given in Table 1
This baseline model can be used to quantify the effect of the drug on a particular biomarker, such as a generic immunoglobulin, here denoted as IgX. This is typically done using indirect response models [33], which capture increases or decreases in the level of a particular biomarker over time under the influence of the concentration of the drug. These drug interactions occur either in the plasma or at the SoA [32, 34]. A schematic representation of the output of such a PKPD model is shown in Fig. 4.
Fig. 4.

A schematic representation of the effect of a drug on a biomarker, such as a generic immunoglobulin IgX, as captured by indirect response models (note that the graph does not depict a specific compound; it is used for illustrative purposes)
As an example, consider the turnover dynamics of IgX, which, in their simplest form, can be described as a difference between IgX production and clearance resulting in a baseline (steady-state) IgX concentration, as shown in the following equation (Eq. 2):
| 2 |
where I(t) is concentration of IgX, and kin and kout are the IgX synthesis and clearance rates, respectively. In disease, the IgX concentration could be elevated if either is too high or is too low. Therefore, some mechanisms of therapeutic intervention could involve decreasing the production of IgX, which can be captured as follows (Eq. 3):
| 3 |
It is clear that larger values of as calculated from system (1) will cause the overall term to decrease, resulting in lower IgX production and thus lower IgX levels.
Correspondingly, the effects of increasing the clearance of IgX can be captured by the following equation (Eq. 4):
| 4 |
In this case, if is large, the overall clearance term is increased, thereby increasing the clearance of IgX. Both of these mechanisms would produce the curve depicted in Fig. 4, even though the mechanisms of action are different. Notably, terminology and correspond to maximum inhibition and stimulation, respectively; in the context of this work, either the inhibition of drug production or the stimulation of drug clearance. A thorough description of indirect response models can be found in [33].
These types of indirect response models can be useful for predicting the effect of a drug on biomarker concentrations and thus preventing them from dropping below potentially unsafe levels (Fig. 4). Having a priori guidelines for safety thresholds can thus help us to predict the dose and frequency of administration that will keep immunoglobulin concentrations above unsafe levels, potentially improving the drug safety profile. We propose the following steps to achieve this goal:
Identify the impact of the drug on the biomarker levels; in the analysis presented below, percent reductions were obtained from clinical data (from the phase IIb atacicept study ADDRESS II [35]), but they could also be obtained from preclinical models.
Simulate the predicted human PK and the expected impact on biomarker levels from step 1 to identify the minimum acceptable baseline biomarker levels needed to ensure that the thresholds summarized in Fig. 2 are not crossed during treatment.
Introduce variability using population PKPD modeling with the minimum acceptable baseline concentrations from step 2 to refine patient selection criteria to minimize adverse events associated with immune suppression.
This approach is summarized in Fig. 5.
Fig. 5.

Using modeling and safety thresholds to guide initial patient selection to minimize the risk of adverse events associated with immunosuppressive drugs. PK pharmacokinetic(s), PD pharmacodynamic(s)
Example: Atacicept
Consider the following example, which focuses on the immunosuppressive drug atacicept, a fully human, recombinant, soluble fusion protein that targets the B cell stimulating factor (BLyS, also known as BAFF [36]) and a proliferation-inducing ligand (APRIL). Atacicept showed evidence of efficacy in SLE in the phase II/III study APRIL-SLE and the phase IIb study ADDRESS II [18, 35]. A summary of the PK parameters for atacicept as estimated from data are provided in the Supplementary Information; analysis of three clinical trials with the population PK model for atacicept can additionally be found in [37, 38].
ADDRESS II included 306 participants (n = 100 received placebo, n = 102 received atacicept 75 mg, n = 104 received atacicept 150 mg), and the treatment duration was 24 weeks. Median reductions in baseline serum IgG were reported to be ~ 25 and 30% with atacicept 75 and 150 mg, respectively, with a maximum IgG reduction of approximately 40% [31]. Additionally, the median percentage reductions in the serum levels of IgA (~ 45% with 75 mg and ~ 50% with 150 mg) and IgM (~ 60% with 75 mg and ~ 70% with 150 mg) compared with the baseline were reported. With regards to treatment-emergent adverse events, the most common adverse events were increases in upper respiratory infections, including nasopharyngitis (4.9% for the 75 mg group and 6.7% for the 150 mg group), bronchitis (1.0% for the 75 mg group and 3.8% for the 150 mg group), and influenza (2.9% for the 75 mg group and 2.9% for the 150 mg group). Pneumonia occurred in 1 patient (1.0%) in each atacicept group.
The proposed approach can be applied to this example as follows.
For step 1 outlined in Fig. 5, consider the following system, which describes an indirect response model with inhibition of IgG production:
| 5 |
where IgG(t) is the concentration of immunoglobulin G, Ctot is the concentration of total atacicept, IgG0 is the baseline IgG concentration, kin is the zero-order IgG production rate, kout is the first-order elimination rate of IgG, Imax is the maximum effect of the drug on IgG synthesis as estimated from the data, and IC50 is the concentration of total atacicept that is required to produce 50% of the maximum effect atacicept can induce. In the model, the decrease in IgG synthesis was related to the (total) plasma atacicept concentration as shown in Fig. 6.
Fig. 6.
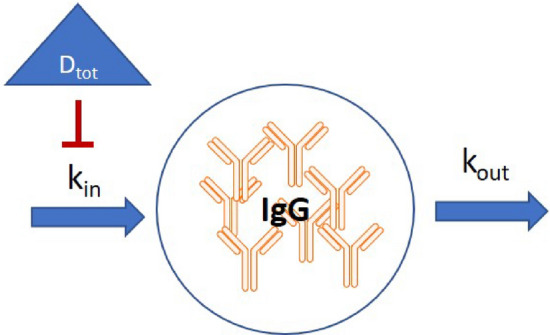
Schematic representation of the IgG indirect response model used to model the effect of atacicept on IgG production
For step 2 outlined in Fig. 5, we couple system (1) with Eq. (5), simulating the PK for a 150 mg dose of atacicept administered weekly for 24 weeks (to match the duration of the trial) as well as the projected reductions in IgG for various initial levels of IgG (see Fig. 7). Different baseline levels of IgG reflect the fact that SLE therapy is often administered to a pretreated patient population, resulting in lower initial IgG levels. As one can see in the scenario analysis reported in Fig. 7B, the IgG concentrations of individuals with baseline levels of IgG below 5.5 g/L are predicted to fall below the threshold of 3 g/L after 12 weeks, potentially increasing the risk of adverse events (the threshold of 3 g/L was taken as a tentative safety threshold because it is a mid-range value for moderate hypogammaglobulinemia as defined in [29]). Similar analyses can be performed for IgM and IgA.
Fig. 7.

Scenario analysis of the variation in the drug concentration and IgG concentration with the administration of 150 mg of atacicept weekly for 24 weeks. Pharmacokinetic parameters are defined in Table 1. The treatment schedule and the maximum IgG reduction of up to 40% were obtained from [35]. A Simulated pharmacokinetics (PK) of 150 mg of atacicept administered weekly for 24 weeks (to mimic the design of the ADDRESS II clinical trial of atacicept). B Simulated dynamics of the safety biomarker IgG for various baseline levels
This type of initial analysis can be performed at the preclinical stage to identify patient eligibility criteria, which in this case corresponds to the lowest baseline level of IgG that still allows the risk of infection due to immune suppression to be mitigated. Notably, no preclinical values of Imax (the maximum fractional IgG reduction due to the drug) were available for these simulations, so we estimated that Imax corresponds to an approximately 40% reduction in IgG, as observed in phase 1 trials. In the absence of preclinical estimates, phase 1 data can be used to evaluate the maximum IgG reduction in healthy subjects, which can then be used to refine the patient selection criteria for subsequent clinical trial phases.
Notably, while the results of simulations of systems (1) and (5) suggest that the average IgG concentration at baseline should be no lower than 5.5–6 g/L in order to minimize the possibility of crossing the IgG safety threshold, these results are deterministic and do not capture interpatient variability. To address this, a population PKPD model was used to simulate predicted IgG concentrations in a population treated with atacicept (step 3 in Fig. 5). While the baseline IgG levels in healthy individuals are in the 7–17 g/L range [38, 39], with typical values in the 10–12 g/L range, patients undergoing standard of care treatment can have significantly lower baseline IgG levels. Modeling is a simple way to implicitly take this variability into account by changing the initial level of IgG at the beginning of the simulation. A detailed description of the PKPD model of atacicept and the corresponding model parameter values can be found in the Supplementary Information.
A simulation of the PK and IgG profiles of 4000 SLE subjects was performed for weekly administration of a 150 mg flat dose of atacicept for 24 weeks, replicating the setup of the clinical trials. The covariate information used for the simulation (body weight, baseline BlyS, and ethnicity) corresponded to the combined APRIL-SLE and ADDRESS II modified intention-to-treat populations, from which over 1500 subjects with a baseline between 5 and 7 g/L (median concentration 6 g/L) were sampled with replacement. Simulated IgG profiles are shown in Fig. 8; these include median profiles together with the corresponding 90% prediction intervals for the entire simulated population. The proportion of subjects for whom IgG is predicted to drop below the safety threshold of 3 g/L at any time point is 3.8%; additional simulations can be found in Supplementary Materials.
Fig. 8.
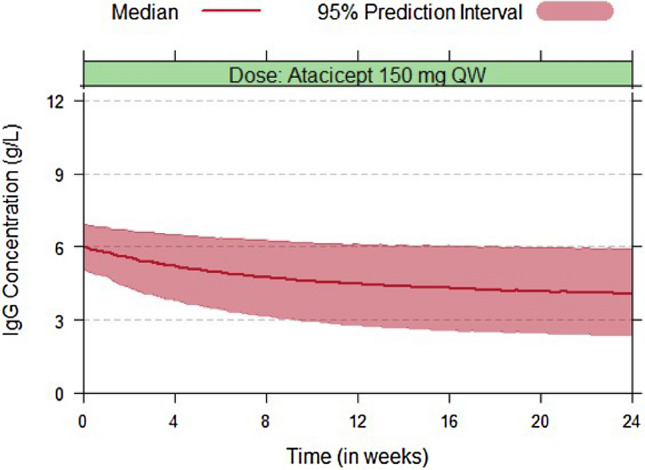
Simulated IgG profiles following 150 mg weekly (QW) administration of atacicept in systemic lupus erythematosus (SLE) subjects for 24 weeks. The baseline IgG was taken to be between 5 and 7 g/L, with a median concentration of 6 g/L. The proportion of subjects for whom IgG is predicted to drop below the safety threshold of 3 g/L at any time point is 3.8%
Notably, in the ADDRESS II clinical trial of atacicept, an exclusion criterion of IgG < 6 g/L was instituted to minimize the possibility of adverse events. The analysis presented here, which was conducted using only preclinical and early clinical data applied to simple PK and PD models, was able to rediscover this criterion.
Notably, with the 6 g/L threshold exclusion criterion in place in the long-term extension (LTE) part of the ADDRESS II study [35] (which was designed to gather long-term safety and tolerability data), 6 participants (approximately 2% of all patients) developed severe hypogammaglobulinemia (IgG < 3 g/L) during or after week 12.
Overall, the proposed analysis corresponds well with the results observed in clinical trials of atacicept, highlighting the value of implementing this analysis from the earliest stages. Specifically, these results suggest that incorporating safety thresholds into indirect response models and combining them with population simulations can help guide initial patient selection to potentially minimize the risk of adverse events associated with immunosuppressive drugs from the earliest stages of clinical development.
Discussion
It is proposed in this work that using diagnostic criteria for PADs can help guide the establishment of safety thresholds for immunosuppressive therapies to ensure that these therapies do not oversuppress the immune system, resulting in SADs. These criteria can be used with PKPD modeling to improve patient safety by decreasing the likelihood of SAD development.
The main premise of this approach is to maximize the utility of preclinical and early clinical data (however sparse) to improve compound safety in the clinical setting by refining the criteria for patient selection in relation to baseline biomarker levels. For the initial assessment, only two pieces of data are necessary: the longitudinal PK and the data that connect the PK to biomarker reduction. For biomarkers with short half-lives, in vitro assays, which are typically run in the early stages of drug development, could provide an initial estimate of this link. In the case of immunoglobulins, with a half-life measured in days, this link can realistically only be estimated from longitudinal in vivo studies in nonhuman primates or phase I clinical studies that report maximum levels of biomarker inhibition, as in the aforementioned case of atacicept.
The key assumption that must hold for this approach to work is that the relationship between drug concentration (PK) and its effect on biomarker levels translates between species, or that there is a known factor by which this relationship translates. One can then use simulations to assess the impact of the projected human PK on biomarker levels and to identify baseline IgG (or other biomarker) levels that can serve to identify exclusion criteria aimed at preventing excessive IgG reduction. Notably, since the key to this analysis is the relationship between PK and biomarker reduction, any PK model that appropriately describes the pharmacokinetics of the drug can be used. Since the underlying indirect response model is correlative and nonmechanistic, it can be applied to a variety of biomarkers and indications. The projections should of course be refined as clinical PK data become available to improve the utility of the output.
The proposed approach could also be adopted for other compounds that can reduce IgG levels and therefore mimic primary antibody deficiencies. For instance, the administration of rituximab, an anti-CD20 B-cell-depleting monoclonal antibody used for lymphoma and rheumatoid arthritis, has resulted in clinically significant hypogammaglobulinemia; according to a meta-analysis conducted by Roberts et al. [40], out of 288 patients who received rituximab, 56% had IgG hypogammaglobulinemia during follow-up, with IgG levels falling below 5 g/L in 22% and below 3 g/L in 4% of patients and IgM ≤ 0.3 g/L observed in 58% of patients. Furthermore, IgG concentrations prior to and at the time of treatment correlated with the IgG nadir post rituximab, which predictably confirms the need to refine patient inclusion criteria before the trials begin so that patient safety is improved with regards to excessive immunoglobulin depletion during the trials. Notably, the results also apply to pediatric populations [41], where applying a similar approach to similar drugs might improve safety profiles as those drugs move into the clinical setting.
Another example of a drug that has resulted in significant immunoglobulin reductions is tabalumab, a B-cell activating factor (BAFF) inhibitor. Administration of this drug led to a twofold drop in IgG levels from baseline in end-stage renal disease patients [42]; if the approach proposed in the present work had been implemented for tabalumab, the safety of this treatment may have been improved. Yet another example is blisibimod, a selective BAFF inhibitor [43] that induced a relatively modest IgG reduction (10%) and thus would not necessitate patient selection criteria refinement based on baseline IgG levels. While the application of the proposed methodology to these particular drugs is no longer necessary given that they are at advanced stages of development, they do highlight the relevance of this approach to compounds beyond atacicept that can significantly affect immunoglobulin levels.
Conclusions
We have described a complementary tool that can help leverage preclinical data for a drug to maximally improve patient safety using a totality of evidence approach before the drug enters clinics. The proposed guidelines are preliminary, and a more rigorous understanding of SAD-related infections is needed. Notably, drug-induced immune suppression is of course not solely dependent on concentrations of various immunoglobulins or any individual subsets of immune cells. Patients with SADs often have complex underlying diseases and conditions, in contrast to PAD patients, so focusing solely on these biomarkers would be insufficient to avoid all the potential issues that come with the administration of immunosuppressive drugs. Nevertheless, the aforementioned application of early-stage PKPD modeling can be helpful in the quantitative analysis of immunosuppressive drugs, which has the potential to increase patient safety.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
The authors would like to thank Matthias Boedding for valuable and insightful comments, as well as Amy Kao for information provided on atacicept clinical trials.
Declarations
Author contributions
Writing and critical review of the manuscript: all authors. Study design: IK. Population PK modeling of atacicept: OP. Clinical information on atacicept: CVM.
Funding
This research was supported by EMD Serono Research and Development Institute, Inc., a business of Merck KGaA, Darmstadt, Germany.
Conflict of interest
The authors are employees of EMD Serono Research and Development Institute, Inc., a business of Merck KGaA, Darmstadt, Germany. In 2020 Merck out-licensed atacicept to Vera Therapeutics.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Code availability
Available upon request.
Footnotes
The original online version of this article was revised due to a retrospective Open Access order.
Change history
9/28/2021
A Correction to this paper has been published: 10.1007/s13318-021-00719-8
References
- 1.Yu T, Enioutina EY, Brunner HI, Vinks AA, Sherwin CM. Clinical pharmacokinetics and pharmacodynamics of biologic therapeutics for treatment of systemic lupus erythematosus. Clin Pharm. 2017;56(2):107–125. doi: 10.1007/s40262-016-0426-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morand EF, Furie R, Tanaka Y, Bruce IN, Askanase AD, Richez C, et al. Trial of anifrolumab in active systemic lupus erythematosus. NEJM Mass Med Soc. 2020;382(3):211–221. doi: 10.1056/NEJMoa1912196. [DOI] [PubMed] [Google Scholar]
- 3.Hougardy D, Peterson G, Bleasel M, Randall C. Is enough attention being given to the adverse effects of corticosteroid therapy? J Clin Pharma Ther. 2000;25(3):227–234. doi: 10.1046/j.1365-2710.2000.00284.x. [DOI] [PubMed] [Google Scholar]
- 4.Fardet L, Flahault A, Kettaneh A, Tiev K-P, Généreau T, Tolédano C, et al. Corticosteroid-induced clinical adverse events: frequency, risk factors and patient’s opinion. Brit J Dermatol. 2007;157(1):142–148. doi: 10.1111/j.1365-2133.2007.07950.x. [DOI] [PubMed] [Google Scholar]
- 5.McDonough AK, Curtis JR, Saag KG. The epidemiology of glucocorticoid-associated adverse events. Curr Opin Rheumatol. 2008;20(2):131–137. doi: 10.1097/BOR.0b013e3282f51031. [DOI] [PubMed] [Google Scholar]
- 6.Bonilla FA, Khan DA, Ballas ZK, Chinen J, Frank MM, Hsu JT, et al. Practice parameter for the diagnosis and management of primary immunodeficiency. J Allergy Clin Immunol. 2015;136(5):1186–1205. doi: 10.1016/j.jaci.2015.04.049. [DOI] [PubMed] [Google Scholar]
- 7.De Vries E, European Society for Immunodeficiencies (ESID) Members. Patient-centred screening for primary immunodeficiency, a multi-stage diagnostic protocol designed for non-immunologists: 2011 update. Clin Exp Immunol. 2012;167(1):108–19. [DOI] [PMC free article] [PubMed]
- 8.Costa-Carvalho BT, Grumach AS, Franco JL, Espinosa-Rosales FJ, Leiva LE, King A, et al. Attending to warning signs of primary immunodeficiency diseases across the range of clinical practice. J Clin Immunol. 2014;34(1):10–22. doi: 10.1007/s10875-013-9954-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Locke BA, Dasu T, Verbsky JW. Laboratory diagnosis of primary immunodeficiencies. Clin Rev Allergy Immunol. 2014;46(2):154–168. doi: 10.1007/s12016-014-8412-4. [DOI] [PubMed] [Google Scholar]
- 10.Aguilar C, Malphettes M, Donadieu J, Chandesris O, Coignard-Biehler H, Catherinot E, et al. Prevention of infections during primary immunodeficiency. Clin Infect Dis. 2014;59(10):1462–1470. doi: 10.1093/cid/ciu646. [DOI] [PubMed] [Google Scholar]
- 11.Weiner GJ. Rituximab: mechanism of action. Sem Hematol. 2010;47:115–123. doi: 10.1053/j.seminhematol.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kotagiri P, Martin A, Hughes P, Becker G, Nicholls K. Single-dose rituximab in refractory lupus nephritis. Int Med J. 2016;46(8):899–901. doi: 10.1111/imj.13136. [DOI] [PubMed] [Google Scholar]
- 13.Rovin BH, Furie R, Latinis K, Looney RJ, Fervenza FC, Sanchez-Guerrero J, et al. Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: the Lupus Nephritis Assessment with Rituximab study. Arthritis Rheum. 2012;64(4):1215–1226. doi: 10.1002/art.34359. [DOI] [PubMed] [Google Scholar]
- 14.Kaufman J, Niesvizky R, Stadtmauer E, Chanan-Khan A, Siegel D, Horne H, et al. Dose-escalation trial of milatuzumab (humanized anti-CD74 monoclonal antibody) in multiple myeloma. J Clin Oncol Am Soc Clin Oncol. 2009;27(15S):8593–8593. doi: 10.1200/jco.2009.27.15_suppl.8593. [DOI] [PubMed] [Google Scholar]
- 15.Frölich D, Blaβfeld D, Reiter K, Giesecke C, Daridon C, Mei HE, et al. The anti-CD74 humanized monoclonal antibody, milatuzumab, which targets the invariant chain of MHC II complexes, alters B-cell proliferation, migration, and adhesion molecule expression. Arthritis Res Ther BioMed Cent. 2012;14(2):R54. doi: 10.1186/ar3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berkova Z, Tao R-H, Samaniego F. Milatuzumab—a promising new immunotherapeutic agent. Exp Opin Investig Drugs. 2010;19(1):141–149. doi: 10.1517/13543780903463854. [DOI] [PubMed] [Google Scholar]
- 17.Navarra SV, Guzmán RM, Gallacher AE, Hall S, Levy RA, Jimenez RE, et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet. 2011;377(9767):721–731. doi: 10.1016/S0140-6736(10)61354-2. [DOI] [PubMed] [Google Scholar]
- 18.Cogollo E, Silva MA, Isenberg D. Profile of atacicept and its potential in the treatment of systemic lupus erythematosus. Drug Des Dev Ther. 2015;9:1331. doi: 10.2147/DDDT.S71276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hochhaus A, Larson RA, Guilhot F, Radich JP, Branford S, Hughes TP, et al. Long-term outcomes of imatinib treatment for chronic myeloid leukemia. NEJM. 2017;376(10):917–927. doi: 10.1056/NEJMoa1609324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palumbo A, Chanan-Khan A, Weisel K, Nooka AK, Masszi T, Beksac M, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. NEJM. 2016;375(8):754–766. doi: 10.1056/NEJMoa1606038. [DOI] [PubMed] [Google Scholar]
- 21.Schiff M. Abatacept treatment for rheumatoid arthritis. Rheumatology. 2010;50(3):437–449. doi: 10.1093/rheumatology/keq287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duraisingham SS, Buckland M, Dempster J, Lorenzo L, Grigoriadou S, Longhurst HJ. Primary vs. secondary antibody deficiency: clinical features and infection outcomes of immunoglobulin replacement. PLoS One. 2014;9(6):e100324. doi: 10.1371/journal.pone.0100324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duraisingham SS, Buckland MS, Grigoriadou S, Longhurst HJ. Secondary antibody deficiency. Exp Rev Clin Immunol. 2014;10(5):583–591. doi: 10.1586/1744666X.2014.902314. [DOI] [PubMed] [Google Scholar]
- 24.Srivastava S, Wood P. Secondary antibody deficiency—causes and approach to diagnosis. Clin Med. 2016;16(6):571–576. doi: 10.7861/clinmedicine.16-6-571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fried AJ, Bonilla FA. Pathogenesis, diagnosis, and management of primary antibody deficiencies and infections. Clin Microbiol Rev. 2009;22(3):396–414. doi: 10.1128/CMR.00001-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Durandy A, Kracker S, Fischer A. Primary antibody deficiencies. Nat Rev Immunol. 2013;13(7):519–533. doi: 10.1038/nri3466. [DOI] [PubMed] [Google Scholar]
- 27.Winkelstein JA, Marino MC, Lederman HM, Jones SM, Sullivan K, Burks AW, et al. X-linked agammaglobulinemia: report on a United States registry of 201 patients. Medicine. 2006;85(4):193–202. doi: 10.1097/01.md.0000229482.27398.ad. [DOI] [PubMed] [Google Scholar]
- 28.Wehr C, Kivioja T, Schmitt C, Ferry B, Witte T, Eren E, et al. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood Am Soc Hematol. 2008;111(1):77–85. doi: 10.1182/blood-2007-06-091744. [DOI] [PubMed] [Google Scholar]
- 29.Barmettler S, Ong M-S, Farmer JR, Choi H, Walter J. Association of immunoglobulin levels, infectious risk, and mortality with rituximab and hypogammaglobulinemia. JAMA Netw Open. 2018;1(7):e184169–e184169. doi: 10.1001/jamanetworkopen.2018.4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bonilla FA, Bernstein IL, Khan DA, Ballas ZK, Chinen J, Frank MM, et al. Practice parameter for the diagnosis and management of primary immunodeficiency. Ann Allergy Asthma Immunol. 2005;94(5):S1–S63. doi: 10.1016/S1081-1206(10)61142-8. [DOI] [PubMed] [Google Scholar]
- 31.Orlicka K, Barnes E, Culver EL. Prevention of infection caused by immunosuppressive drugs in gastroenterology. Ther Adv Chronic Dis. 2013;4(4):167–185. doi: 10.1177/2040622313485275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tiwari A, Abraham AK, Harrold JM, Zutshi A, Singh P. Optimal affinity of a monoclonal antibody: guiding principles using mechanistic modeling. AAPS J. 2017;19(2):510–519. doi: 10.1208/s12248-016-0004-1. [DOI] [PubMed] [Google Scholar]
- 33.Sharma A, Jusko WJ. Characteristics of indirect pharmacodynamic models and applications to clinical drug responses. Brit J Clin Pharmacol. 1998;45(3):229–239. doi: 10.1046/j.1365-2125.1998.00676.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chudasama VL, Zutshi A, Singh P, Abraham AK, Mager DE, Harrold JM. Simulations of site-specific target-mediated pharmacokinetic models for guiding the development of bispecific antibodies. J PKPD. 2015;42(1):1–18. doi: 10.1007/s10928-014-9401-1. [DOI] [PubMed] [Google Scholar]
- 35.Merrill JT, Wallace DJ, Wax S, Kao A, Fraser PA, Chang P, et al. Efficacy and safety of atacicept in patients with systemic lupus erythematosus: results of a twenty-four-week, multicenter, randomized, double-blind, placebo-controlled, parallel-arm, phase II b study. Arthritis Rheumatol. 2018;70(2):266–276. doi: 10.1002/art.40360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cancro M. The BLyS/BAFF family of ligands and receptors: key targets in the therapy and understanding of autoimmunity. Ann Rhem Dis. 2006;65(Suppl 3):iii34–6. [DOI] [PMC free article] [PubMed]
- 37.Papasouliotis O, Yalkinoglu Ö, Golob M, Willen D, Girard P. AB0529 population pharmacokinetics of atacicept in systemic lupus erythematosus (SLE) Ann Rheum Dis. 2015;74:1077. doi: 10.1136/annrheumdis-2015-eular.3504. [DOI] [Google Scholar]
- 38.Pitsui M, Papasouliotis O, Farrell C, Girard P, Yalkinoglu O, Vazquez-Mateo C. Population pharmacokinetics of atacicept in systemic lupus erythematosus (SLE)—an analysis of three clinical trials. Arthritis Rheumatol. 2019:71(Suppl 10). https://acrabstracts.org/abstract/population-pharmacokinetics-of-atacicept-in-systemiclupus-erythematosus-sle-an-analysis-of-three-clinical-trials/ [DOI] [PMC free article] [PubMed]
- 39.Willen D, Uhl W, Wolna P, Papasouliotis O, Yalkinoglu Ö. Safety, tolerability, pharmacokinetics, and pharmacodynamics of atacicept in a randomized trial in healthy Caucasian and Japanese subjects. Eur J Drug Metab Pharm. 2020;45(1):27–40. doi: 10.1007/s13318-019-00575-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roberts DM, Jones RB, Smith RM, Alberici F, Kumaratne DS, Burns S, et al. Rituximab-associated hypogammaglobulinemia: incidence, predictors and outcomes in patients with multi-system autoimmune disease. J Autoimmun. 2015;57:60–65. doi: 10.1016/j.jaut.2014.11.009. [DOI] [PubMed] [Google Scholar]
- 41.Khojah AM, Miller ML, Klein-Gitelman MS, Curran ML, Hans V, Pachman LM, Fuleihan RL. Rituximab-associated Hypogammaglobulinemia in pediatric patients with autoimmune diseases. Pediatr Rheumatol Online J. 2019;17(1):61. doi: 10.1186/s12969-019-0365-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mujtaba M, Komocsar W, Nantz E, Samaniego M, Henson S, Hague J, et al. Effect of treatment with tabalumab, a B cell-activating factor inhibitor, on highly sensitized patients with end-stage renal disease awaiting transplantation. Am J Transplant. 2016;16(4):1266–1275. doi: 10.1111/ajt.13557. [DOI] [PubMed] [Google Scholar]
- 43.Merrill JT, Shanahan WR, Scheinberg M, Kalunian KC, Wofsy D, Martin RS. Phase III trial results with blisibimod, a selective inhibitor of B-cell activating factor, in subjects with systemic lupus erythematosus (SLE): results from a randomised, double-blind, placebo-controlled trial. Ann Rheum Dis. 2018;77(6):883–889. doi: 10.1136/annrheumdis-2018-213032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Not applicable.