

Abstract
Transcriptome-wide association studies (TWAS) systematically investigate the association of genetically predicted gene expression with disease risk, providing an effective approach to identify novel susceptibility genes. Osteoporosis is the most common metabolic bone disease, associated with reduced bone mineral density (BMD) and increased risk of osteoporotic fractures, whereas genetic factors explain approximately 70% of the variance in phenotypes associated with bone. BMD is commonly assessed using dual-energy X-ray absorptiometry (DXA) to obtain measurements (g/cm2) of areal BMD. However, quantitative computed tomography (QCT) measured 3D volumetric BMD (vBMD) (g/cm3) has important advantages compared with DXA since it can evaluate cortical and trabecular microstructural features of bone quality, which can be used to directly predict fracture risk. Here, we performed the first TWAS for volumetric BMD (vBMD) by integrating genome-wide association studies (GWAS) data from two independent cohorts, namely the Framingham Heart Study (FHS, n = 3,298) and the Osteoporotic Fractures in Men (MrOS, n = 4,641), with tissue-specific gene expression data from the Genotype-Tissue Expression (GTEx) project. We first used stratified linkage disequilibrium (LD) score regression approach to identify 12 vBMD-relevant tissues, for which vBMD heritability is enriched in tissue-specific genes of the given tissue. Focusing on these tissues, we subsequently leveraged GTEx expression reference panels to predict tissue-specific gene expression levels based on the genotype data from FHS and MrOS. The associations between predicted gene expression levels and vBMD variation were then tested by MultiXcan, an innovative TWAS method that integrates information available across multiple tissues. We identified 70 significant genes associated with vBMD, including some previously identified osteoporosis-related genes such as LYRM2 and NME8, as well as some novel loci such as DNAAF2 and SPAG16. Our findings provide novel insights into the pathophysiological mechanisms of osteoporosis and highlight several novel vBMD-associated genes that warrant further investigation.
Keywords: Osteoporosis, volumetric bone mineral density, transcriptome-wide association study (TWAS)
INTRODUCTION
Osteoporosis is the most common metabolic bone disease, associated with clinical manifestations of reduced bone mineral density (BMD), deterioration of bone tissue, and increased susceptibility to low trauma osteoporotic fractures (1). The United States has more than 2 million reports of osteoporotic fractures annually with estimated annual medical expenses over $19 billion (2).
BMD is commonly assessed using dual-energy X-ray absorptiometry (DXA) to obtain measurements (g/cm2) of areal BMD. However, quantitative computed tomography (QCT) measured 3D volumetric BMD (vBMD) (g/cm3) of the trabecular compartment is also an important measurement technique, which has a direct link with osteoporosis and vertebral fracture (3). Some previous studies have also shown a strong significant relationship between vBMD and osteoporosis fracture. For instance, Langsetmo et al. reported that lower total volumetric BMDs at all peripheral bone sites were each associated with a similarly increased risk of incident fracture (4). This can likely be attributed to the fact that QCT imaging takes into consideration the construction of microfinite element models of bone strength, which can be used to predict fracture risk as well as explain confounding factors such as osteophytic or extra-skeletal calcification (5,6).
So far, genome-wide association studies (GWAS) have successfully identified over 500 susceptibility loci for osteoporosis-related traits (7), including five significant loci associated with vBMD identified in a meta-analysis by Nielson et al. (8). However, it remains a challenge to understand the biological functions of these genetic loci and the biological mechanisms underlying the observed genetic associations. One approach to tackle this problem is to directly study the association between gene expression levels in specific tissues and the trait of interest. However, due to the specimen availability and the costs, such studies are usually not feasible or sufficiently powerful for many tissues and traits. The fact that many genetic variants play important roles in gene expression regulations (9-12) has motivated the development of methods for transcriptome-wide association studies (TWASs), which can integrate gene expression data from large transcriptome reference datasets such as Genotype-Tissue Expression project (GTEx) (13) with GWAS association statistics to investigate the association of genetically predicted gene expression with disease risk, providing an effective approach to identify novel susceptibility genes (14).
Barbeira et al. recently presented a TWAS method termed MultiXcan, which uses multivariate regression to test the joint effects of gene expression variation from multiple tissues and thus may have increased power compared with traditional single-tissue TWAS methods (15). While a few previous TWAS studies have been conducted for osteoporosis related traits (15-18), including a multi-tissue TWAS study for areal BMD and fracture by us (19), no TWAS study has been reported for vBMD.
In this study, we performed the first TWAS for vBMD by integrating FHS and MrOS, with tissue-specific gene expression data from the GTEx project. First, we performed a stratified linkage equilibrium (LD) score regression to identify relevant tissues where the phenotypic heritability is enriched using 53 tissues from GTEx. Focusing on these vBMD relevant tissues, we subsequently leveraged GTEx expression reference panels to predict tissue-specific gene expression levels based on the genotype data for individuals in FHS and MrOS. MultiXcan, an innovative TWAS method that integrates information available across multiple tissues, was used to test the associations between genetically predicted gene expression and vBMD variation. Lastly, we made functional annotations about significant genes by Protein-protein interaction (PPI) network analysis, Gene ontology (GO) and KEGG pathway analysis. An overview of the study is provided in Figure 1. In our study, we identified 70 significant genes associated with vBMD, including some previously identified osteoporosis-related genes such as LYRM2 and NME8, as well as some novel loci such as DNAAF2 and SPAG16. Our findings provide some novel insights into the functional mechanisms of osteoporosis and highlight some promising candidate genes for future investigation.
Figure 1.
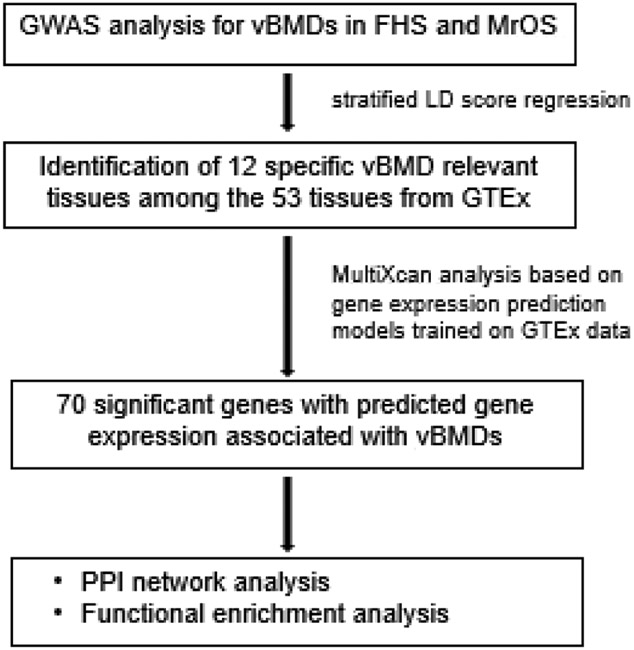
Overview of the analyses.
MATERIALS AND METHODS
GWAS dataset
GWAS datasets for vBMD were obtained from two independent studies, FHS (phs000007.v30.p11) and MrOS (phs000373.v1.p1), through the database of genotype and phenotype (dbGaP) portal. We included 3,298 unrelated individuals of European from FHS with lumbar vertebral (L2 - L4) integral vBMD and trabecular vBMD, and 4,641 unrelated male individuals of European (aged 65 years or older) from MrOS samples with femoral neck trabecular vBMD, integral vBMD, and cortical vBMD. All measures of vBMD were performed using QCT (20,21). Details of the study design and data collection procedures for FHS and MrOS were described previously (22-24).
Genotype quality control (QC) was implemented with PLINK (25). Specifically, SNPs violating the Hardy-Weinberg equilibrium (HWE) rule (p value < 1 × 10−5) and/or with minor allele frequency (MAF) < 0.01 in the sample were removed. Individuals with imputed sex inconsistent with reported sex or of ambiguous imputed sex were removed. When Mendelian errors were detected in the FHS samples, the corresponding genotype values were set to missing. Genotype-derived principal components were examined to remove population outliers by using the program smartpca from the Eigensoft package with the default settings (26).
Comprehensive genome-wide genotype data for FHS and MrOS individuals were imputed using the Michigan Imputation Server (27) based on the Haplotype Reference Consortium (HRC Version r1.1 2016) reference panel (28). All variants were mapped to GRCh37.
At last, we used PLINK (25) to test the genetic association of 415,792 SNPs with lumbar vertebral trabecular and integral vBMDs in FHS, and 691,126 SNPs with femoral neck integral , trabecular , and cortical vBMDs in MrOS under an additive model of inheritance, accounting for age, gender, height and weight as covariates.
Identification of significant tissues associated with vBMDs
To assess the gene expression enrichment in a tissue for a given trait, we followed the procedure described earlier (29). Briefly, we downloaded the gene expression profiling data in 53 tissues (Supplementary Table 1 (30)) from GTEx (28,29), with an average of 161 samples per tissue. For each gene, we computed a t-statistic to compare the expression level between a given tissue and all other tissues. We first constructed a design matrix X where each row represents a sample either in the target tissue or not in the target tissue. The first column of X has a 1 for every sample in the specific tissue and a −1 for every sample not in the target tissue. The remaining columns are an intercept and covariates. The outcome Y in our model is expression. We fit this model using ordinary least squares, and computed a t-statistic for the first explanatory variable (the first column of X):
where MSE is the mean squared error of the fitted model, i.e.,
where N is the number of rows in X. This process was repeated to compute a t-statistic summarizing the importance of each gene in each tissue.
We then ranked all of the genes by their t-statistic within each tissue and defined the top 10% of genes with the highest t-statistic to be the specifically expressed genes that are representative of a given tissue. A 100-kb window was added on either side of the transcribed region of each gene in the set of specifically expressed genes to construct a genome annotation that corresponded to the specific tissue. Next, we applied stratified LD score regression, in which we jointly modeled the annotation of the given tissue, a genome annotation that corresponded to all genes, as well as the 52 annotations in the ‘baseline model’ (31). Here, the baseline model includes genic regions, enhancer regions and conserved regions in mammals, which were conserved and not specific to any cell types (31). Stratified LD score regression is a method for stratifying the trait heritability according to the contribution of the genome annotation in each specific tissue (32). We first linearly regressed the phenotype Y on genotype X with Y = X · β + ε, where X was a vector of SNPs for an individual, and each SNP has been standardized to mean 0 and variance 1 in the population. All SNPs can be categorized into one of genomic annotations (C1, …, Ck). Given genomic annotations C1, …, Ck, we modeled the effect of SNP i on phenotype Y as drawn from a normal distribution with mean 0 and variance
| (1) |
We can call Var(βi) the per-SNP heritability of SNP i.
Under this model, the expected marginal association statistic at SNP i reflects the contribution of both SNP i and SNPs in LD with SNP i. Specifically,
where N is the GWAS sample size, a is a constant that measures confounding biases,(33) and ℓ(i, k) is the LD score of SNP i to category Ck, defined as ℓ(i, k) = Σjr2(i, j)1{j ∈ Ck}, where r2(i, j) is the squared correlation between SNPs i and j in the population. To estimate the τk, we first downloaded ℓ(i, k) calculated from the 1000 Genomes project reference panel (34), and regressed on ℓ(i, k) (35).
The regression coefficient τk quantifies the contribution of annotation Ck to vBMD trait heritability, conditioning on the set of all genes and the baseline model; τk will equal to 0, if Ck is not enriched; τk will be positive if belonging to Ck increases per-SNP heritability, accounting for the set of all genes and the baseline model, and vice versa. Standard errors of τk, the contribution of annotation Ck to vBMD trait heritability, can be computed with a block jackknife(36) and then used to calculate P-values that tested whether τk was positive. Tissues for which its specific genome annotations Ck showed significant associations (p value < 0.05) with vBMD were deemed as putative vBMD relevant tissues. We applied a lenient threshold in order to be inclusive and ensure that potentially relevant tissues were not excluded at this very initial stage. (When reporting quantitative results, we normalized the coefficient τk by our estimate of the mean per-SNP heritability ΣiVar(βi)/M to make it comparable across phenotypes, where M was the total number of SNPs (32). The normalized coefficient can be interpreted as the proportion by which the per-SNP heritability of an average SNP would increase if τk were added to it. In addition, we defined the enrichment of a category to be the proportion of SNP-heritability in the category divided by the proportion of SNPs. Given that Var(βi) is the per-SNP heritability of SNP i, the total heritability is defined as ΣiVar(βi) and the heritability in genomic annotation Ck is defined as Σi∈CkVar(βi). The total heritability and the heritability in category Ck can be estimated by plugging estimates of τk into Equation (1). We calculated the proportion of heritability, Σi∈CkVar(βi)/ΣiVar(βi), and compared it with the proportion of SNPs, ∣Ck∣/M.)
TWAS analyses by MultiXcan
We applied a multi-tissue analysis method, called MultiXcan (15), to test the joint effects of gene expression on vBMD in multiple tissues. The transcriptomics panels from the GTEx Consortium (V7) for gene expression were downloaded from PredictDB (http://predictdb.org/) and the prediction models were trained in a total of 48 tissues. For a given gene in one tissue, gene expression was characterized as an additive inheritance model using PrediXcan (17):
where
Yg is the expression of gene g,
wk,g is the effect size of marker k for gene g,
Xk is the number of reference alleles of marker k,
ϵ is the contribution of other factors that determine the expression.
The effect sizes (wk,g) were estimated with elastic net regularization (37). The expression levels of each gene were estimated by the SNPs in the neighborhood of the corresponding gene (within 1 Mb of the gene start or end, minor allele frequency > 0.05, Hardy-Weinberg equilibrium P value > 0.05). The detailed description on how to construct prediction models could be found in the published studies (38,39) and on the website of PredictDB (http://predictdb.org/).
To integrate the information across tissues, mainly gene expression levels, MultiXcan regresses the phenotype of interest on the predicted expression in multiple tissues as follows:
where
a is the phenotype vector,
gj is the effect size of the predicted gene expression in tissue j,
tj is the predicted gene expression in tissue j,
ϵ is the contribution of other factors that determine the phenotype,
p is the number of available tissue models.
Expression predictions across tissues can be highly correlated. To avoid numerical issues caused by collinearity, we use principal components of the predicted expression data matrix as explanatory variables and discard the axes of smallest variation (namely, PCA regularization). In this study, we chose the first 30 principal components and considered linear regression for modeling so the condition number of the covariance matrix of the predicted expression across tissues was below 30. Other settings were as default. In summary, we used a machine learning-based method PrediXcan to train genotype and gene expression data on the same individuals in the large reference panels in order to predict the gene expressions based on the genotype from our new samples. The predicted gene expressions were used in gene-trait association studies to identify significant genes. False discovery rate (FDR) p values were also calculated for the selection of vBMD-significant genes.
Functional annotation analysis
The conversion from Ensemble ID to gene symbol was conducted in the R packages, clusterProfiler (40) and org.Hs.eg.db (16). Protein-protein interaction (PPI) network analysis was performed with the web-based tool STRING (https://string-db.org/) (41). Gene ontology (GO) and KEGG pathway analyses were executed by the tool Database for Annotation, Visualization and Integrated Discovery (DAVID) (https://david.ncifcrf.gov/) (42,43).
RESULTS
Characteristics of FHS and MrOS
The sample sizes for different vBMD traits varied in the FHS and MrOS dataset (Table 1). In FHS, trabecular and integral vBMDs at lumbar L2 had relatively small sample sizes of approximate 800; whereas the lumbar L3 – L4 trabecular and integral vBMDs had sample sizes of ~2,000-2,600. In MrOS, about 2,500 samples had data for femoral neck cortical vBMD, integral vBMD and trabecular vBMD.
Table 1.
Sample sizes of the FHS and MrOS subjects for different types of vBMDs
| vBMD | Number of subjects | Males | Females | |
|---|---|---|---|---|
| FHS | ||||
| Lumbar vertebrae | L2 trabecular vBMD | 806 | 232 | 574 |
| L3 trabecular vBMD | 2,440 | 1,263 | 1,177 | |
| L4 trabecular vBMD | 1,920 | 1,138 | 782 | |
| L2 integral vBMD | 800 | 228 | 572 | |
| L3 integral vBMD | 2,423 | 1,254 | 1,169 | |
| L4 integral vBMD | 1,902 | 1,129 | 773 | |
| MrOS | ||||
| Femoral Neck (FN) | Cortical vBMD | 2,519 | 2,519 | 0 |
| Integral vBMD | 2,520 | 2,520 | 0 | |
| Trabecular vBMD | 2,517 | 2,517 | 0 | |
Tissues relevant to vBMDs and genes associated with vBMD through expression
By applying stratified LD score regression analyses on GWAS summary statistics, we identified several tissues with significant contributions to vBMD heritability (Table 2). For instance, we found that adrenal gland and left ventricle heart were statistically significant with all lumbar vertebral (L2 - L4) integral vBMDs and trabecular vBMDs (p < 0.05). In addition, skeletal muscle was relevant with lumbar L4 integral vBMD (p = 0.034) and trabecular vBMD (p = 0.027). In MrOS, left ventricle heart, atrial appendage heart, stomach, artery tibial and lung all showed significant relevance to femoral neck cortical vBMD, integral vBMD and trabecular vBMD (p < 0.05).
Table 2.
Significant tissues relevant to vBMDs
| Trait | Name | Coefficient | Coefficient P value |
|---|---|---|---|
| L2 integral vBMD | Esophagus Mucosa | 1.73E-11 | 0.010 |
| Skin Not Sun Exposed (Suprapubic) | 1.38E-11 | 0.015 | |
| Heart Left Ventricle | 1.35E-11 | 0.020 | |
| Adrenal Gland | 1.42E-11 | 0.025 | |
| Heart Atrial Appendage | 9.88E-12 | 0.038 | |
| Brain - Spinal cord (cervical c-1) | 9.94E-12 | 0.040 | |
| L2 trabecular vBMD | Esophagus Mucosa | 1.84E-11 | 0.003 |
| Adrenal Gland | 1.39E-11 | 0.017 | |
| Heart Left Ventricle | 1.21E-11 | 0.021 | |
| Skin Not Sun Exposed (Suprapubic) | 1.21E-11 | 0.022 | |
| Transverse Colon | 1.19E-11 | 0.045 | |
| L3 integral vBMD | Heart Left Ventricle | 1.66E-12 | 0.011 |
| Adrenal Gland | 1.67E-12 | 0.014 | |
| Esophagus Mucosa | 1.48E-12 | 0.020 | |
| Transverse Colon | 2.08E-12 | 0.021 | |
| L2 trabecular vBMD | Heart Left Ventricle | 1.87E-12 | 0.004 |
| Adrenal Gland | 2.01E-12 | 0.009 | |
| Esophagus Mucosa | 1.75E-12 | 0.013 | |
| Transverse Colon | 1.97E-12 | 0.026 | |
| Heart Atrial Appendage | 1.14E-12 | 0.040 | |
| Skin Not Sun Exposed (Suprapubic) | 1.28E-12 | 0.049 | |
| L4 integral vBMD | Transverse Colon | 2.88E-12 | 0.017 |
| Adrenal Gland | 2.05E-12 | 0.019 | |
| Heart Left Ventricle | 1.81E-12 | 0.027 | |
| Esophagus Mucosa | 1.85E-12 | 0.034 | |
| Skeletal Muscle | 1.69E-12 | 0.034 | |
| L4 trabecular vBMD | Transverse Colon | 3.68E-12 | 0.007 |
| Heart Left Ventricle | 2.50E-12 | 0.011 | |
| Esophagus Mucosa | 2.57E-12 | 0.015 | |
| Adrenal Gland | 2.21E-12 | 0.024 | |
| Skeletal Muscle | 2.10E-12 | 0.027 | |
| Skin Not Sun Exposed (Suprapubic) | 2.30E-12 | 0.031 | |
| Esophagus Muscularis | 1.88E-12 | 0.041 | |
| FN cortical vBMD | Heart Left Ventricle | 8.20E-12 | <0.001 |
| Heart Atrial Appendage | 4.22E-12 | 0.006 | |
| Stomach | 4.26E-12 | 0.024 | |
| Artery Tibial | 3.55E-12 | 0.024 | |
| Lung | 3.11E-12 | 0.032 | |
| FN trabecular vBMD | Heart Left Ventricle | 3.95E-12 | <0.001 |
| Heart Atrial Appendage | 2.01E-12 | 0.007 | |
| Stomach | 2.08E-12 | 0.023 | |
| Artery Tibial | 1.74E-12 | 0.024 | |
| Lung | 1.53E-12 | 0.031 | |
| FN integral vBMD | Heart Left Ventricle | 6.93E-12 | <0.001 |
| Heart Atrial Appendage | 3.57E-12 | 0.006 | |
| Stomach | 3.60E-12 | 0.023 | |
| Artery Tibial | 2.99E-12 | 0.024 | |
| Lung | 2.63E-12 | 0.031 |
Notes: Bold tissues are associated with multiple vBMD traits at lumbar spine or femoral neck, respectively.
Subsequently, we applied MultiXcan to integrate predicted gene expression profiles from the multiple tissues relevant to specific vBMDs and identified seventy significant genes (p value < 0.001, a significance level used in earlier studies (44-46)). One gene, LYRM2, was significantly associated with vBMD at 10% false discovery rate (Table 3 and Supplementary Table 2 (30)). Some of the significant genes had been identified in the previous GWAS studies for osteoporosis, such as NME8 (47). In addition, we also found some novel putative osteoporosis-related genes, such as DNAAF2, SPAG16, M6PR, COG4, CCT8, which were not reported from GWAS or TWAS for osteoporosis-related traits. Among all the significant vBMD-related genes, nine genes, such as LYRM2, had significant associations with femoral neck (FN) trabecular vBMD and nine genes, such as ODF2L, were significantly associated with femoral neck cortical vBMD. In addition, twenty genes, for example, SLC2A10, CPEB2, achieved the statistical significance for lumbar spine (LS) trabecular vBMD. Interestingly, the genes associated with FN trabecular vBMD are distinct from those associated with LS trabecular vBMD, which are also different from the genes associated with FN cortical vBMD. These results suggest that cortical and trabecular vBMD at different skeletal sites may be under different genetic controls.
Table 3.
Most significant genes associated with vBMDs
| Ensemble ID | Gene Symbol | P | FDR P | Traits |
|---|---|---|---|---|
| ENSG00000083099 | LYRM2 | <0.00001 | 0.01 | FN trabecular vBMD |
| ENSG00000086288 | NME8 | 0.00007 | 0.52 | L2 integral vBMD |
| ENSG00000173638 | SLC19A1 | 0.00004 | 0.22 | L3 integral vBMD |
| ENSG00000197496 | SLC2A10 | 0.00008 | 0.25 | L4 trabecular vBMD |
Notes: Genes with p < 0.0001 are listed here, and the rest of significant genes (p < 0.001) are listed in Supplementary Table 2.
Functional annotation analysis of significant genes
To explore the functional importance of the significant genes for vBMDs (i.e., significant genes in Table 3 and supplementary table 2), we performed PPI analysis to identify the potential interactions of the novel genes in our results and known osteoporosis-related genes reported in previous GWAS. A significant PPI network was formed among these genes (Figure 2). Some novel genes, such as DNAAF2, SPAG16, were connected with some known osteoporosis-related genes, like NME8.
Figure 2.
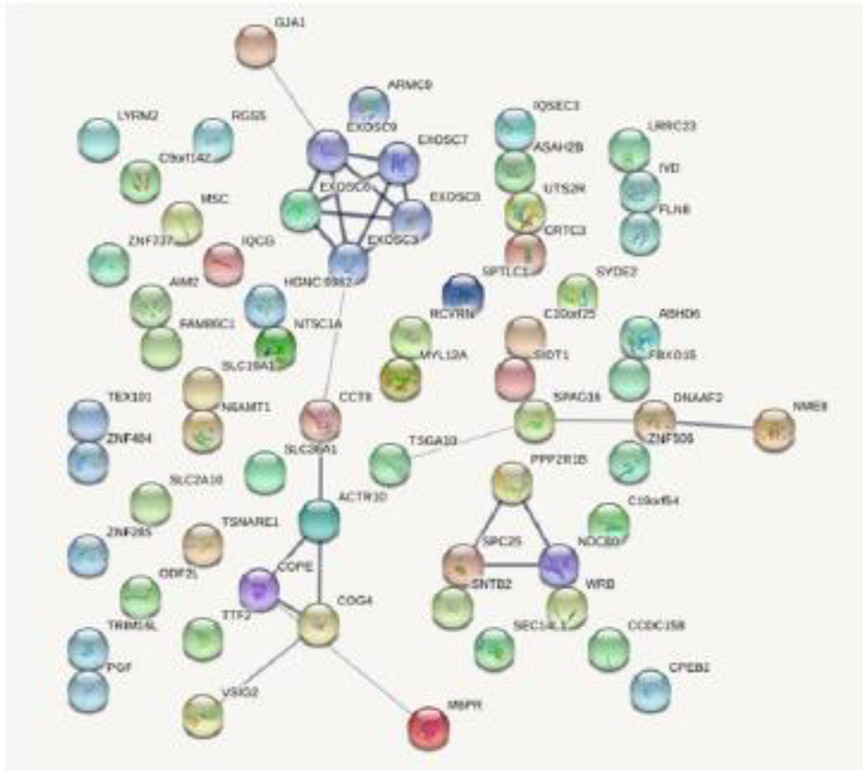
PPI network of significant genes associated with vBMD. The minimum required interaction score is set to 0.400. Line thickness indicated the strength of data support.
To further seek out the biological mechanisms underlying the observed associations between these significant genes and vBMDs, we performed the GO and KEGG pathway analysis. GO analysis demonstrated that these significant genes were relatively enriched in one biological processes (Table 4). However, KEGG analysis did not identify any pathway that is significantly enriched for the vBMD associated genes.
Table 4.
Enriched GO terms in vBMD associated genes
| Data Source | Term | Count | P | Genes |
|---|---|---|---|---|
| GO-BP | GO:0007288 ~ sperm axoneme assembly | 2 | 2.1E-2 | IQCG, SPAG16 |
BP: Biological Process.
CONCLUSIONS
In this study, we performed a novel multi-tissue based TWAS to integrate genetic component of gene expression with GWAS results in vBMD and identified a number of putative novel osteoporosis-associated gene.
Most previous genetic research for osteoporosis focused on areal BMD, but very few studies focused on separate genetic control for bone microstructures, namely, trabecular vBMD and cortical vBMD. Our results indicate that trabecular and cortical bone microstructures may be under distinct gene control. This conclusion is consistent with the fact that patients with idiopathic osteoporosis often have a predominantly cortical or trabecular bone-related problem (48). Trabecular bone is more active and likely to be affected compared with cortical bone, hence, trabecular bone is more subject to bone remodeling (49). Not only BMD, but also bone microstructure would be affected. The weak spicules of trabecular bone break are replaced by weaker bone. This explains why the wrist, hip and spine are common osteoporotic fracture sites rather than other sites, as these sites have a relatively higher trabecular bone to cortical bone ratio (50). Different genes/biological pathways may contribute to the variances in bone compositions at different skeletal sites (51,52). In accordance, we observed that different genes were associated with trabecular and cortical vBMDs. Specifically, twenty-nine genes had significant associations with trabecular vBMD while nine different genes were significantly associated with cortical vBMD. We compared our results with the previous GWAS study by Nielson et al.(8) who identified significant vBMD associations at five loci, including: 1p36.12, containing WNT4 and ZBTB40; 8q24, containing TNFRSF11B; and 13q14, containing AKAP11 and TNFSF11. However, none of the 5 genes showed significant association of predicted gene expression with vBMD in our study.
We first used stratified LD score regression to identify vBMD relevant tissues, for which vBMD heritability is enriched in regions surrounding genes with the highest specific expression in a given tissue. Several tissues, such as adrenal gland, left ventricle heart, skeletal muscle, and stomach, showed significant associations with multiple vBMD traits. Through literature mining, we found interesting evidence supporting the biological connections between these tissues and bone metabolism.
Adrenal Gland
The adrenal gland is composed of the outer cortex and the inner medulla. Glucocorticoids are secreted by the middle region of the adrenal cortex (53). Excess glucocorticoids can cause detrimental effects on bone physiology, which leads to osteoporosis and bone fracture. The cellular mechanisms by which glucocorticoids affects bone are complicated with a range of direct and indirect effects on the multiple cell types present in bone and on other tissues important in fracture protection (54). The most important effects among them appear to be direct actions of glucocorticoids on osteoblasts to reduce their activities and cause their apoptosis and the stimulatory action on the activity of osteoclasts (55,56).
Left Ventricle Heart
In a mouse model of postinfarction heart failure, bone marrow plasma levels of RANKL were significantly (p = 0.03) increased in mice with severely impaired left-ventricular function (57). The alterations in bone marrow plasma levels of RANKL in chronic postinfarction mice were consistent with similar findings observed in patients with chronic heart failure (CHF). Studies have shown that bone marrow plasma from patients with CHF stimulated the formation of osteoclasts, and this effect could be blocked by RANKL antibodies (57). Additionally, studies have provided evidence that increasing RANKL expression by various stress conditions could stimulate osteoclast differentiation (58). These results suggested a direct pathophysiological pathway linking the function of the left ventricle heart with bone remodeling.
Skeletal Muscle
Results from basic and clinical research of bone and muscle suggested close interactions between bone and skeletal muscles via local and humoral signaling pathways in addition to their musculoskeletal functions (59). In fact, age-related muscle loss may coexist with osteoporosis, establishing a vicious cycle between dysfunctional muscle and bone (60). Meanwhile, endocrine factors influence muscle and bone through protein catabolism. For example, active vitamin D can increase osteoglycin level, which leads to rescuing the decrease in myotubular differentiation and suppressing osteoblast differentiation through muscle-derived soluble factors (61). Insulin-like growth factor binding proteins are secreted from muscle. Insulin-like growth factor binding protein 2 (IGFBP-2), which is a member of insulin-like growth factor binding protein, facilitates the differentiation of osteoblasts through an interaction of the heparin binding domain-1 with receptor tyrosine phosphatase β (62). Other factors, such as inflammatory, nutritional states, etc., can also affect the interaction of bone with skeletal muscle from different points of view.
By integrating information available across multiple vBMD relevant tissues, we identified seventy genes associated with vBMDs at various skeletal sites. Among them, many genes, such as NME8, LYRM2, and RGS5 have previously been reported to have a connection with BMD/osteoporosis.
NME8 -- NME/NM23 Family Member 8, has an extremely high expression specifically in bone tissue (63). Estrada et al. discovered rs10226308, a SNP mapped with NME8, was significantly associated with lumbar spine BMD (p = 6 × 10−13) (47). However, the functional roles of NME8 in bone metabolism are currently unknown.
LYRM2 -- LYR motif containing 2, is the one of the top ten most significantly upregulated differentially expressed genes in trabecular osteocytes between osteoporosis and normal controls (64,65). In a previous study, LYRM2 was found to be significantly differentially expressed in peripheral blood monocytes between osteoporosis patients and normal controls (65). However, the functional roles of LYRM2 associated with bone metabolism are still unknown and warrants further investigation.
RGS5 -- Regulator of G Protein Signaling 5, which is a GTPase activating protein, was found to have overexpression in parathyroid tumors associated with primary hyperparathyroidism (PHPT) and previous research showed that the calcium-sensing receptor (CASR) signaling could be inhibited by RGS5. Given the characteristics of the bone in PHPT patients, namely, preserved or increased trabecular bone and cortical bone loss (66), Balenga et al. assessed the microstructure of trabecular bone in mice femurs and discovered that the significant increase of trabecular bone volume correlated with parathyroid hormone (PTH) levels regulated by RGS5 parathyroid-specific overexpression (67). In our study, RGS5 was also found to have a significant association with femoral neck cortical BMD, which was also worth further validation.
In addition, we identified a number of novel putative vBMD associated genes, such as DNAAF2, SPAG16, M6PR, COG4, CCT8. Many of these novel genes formed significant PPI network with known osteoporosis-related genes, highlighting their functional potentials on regulating osteoporosis risk. Interestingly, based on GeneCards (https://www.genecards.org/), some of these novel genes (e.g., DNAAF2, SPAG16) exhibit higher expression levels in bone marrow compared with other tissues, suggesting that these novel genes may be associated with bone metabolism in some degree.
In light of the novel findings of vBMD associated tissues and genes, our study also has a few limitations. First, we were unable to include bone tissue/cells (e.g., osteoblast, osteoclast) in our TWAS analysis. This is because there is no gene expression data of bone tissue/cells in the GTEx reference panel used in this study. In addition, though there are a few published osteoblast/osteoclast eQTL (expression quantitative trait locus) studies (68,69), these studies did not release the individual-level genotype and transcriptome data for data sharing, and thus we were unable to leverage these studies for the TWAS analysis. Second, the sample sizes of GTEx reference panels were relatively small at present, and thus some gene expression levels may not be accurately predicted (70). Third, TWAS tend to identify multiple significant genes per locus, and many of them may not be causal due to confounding through linkage disequilibrium among SNPs. There may exist a concern of collinearity(71). Fourth, the GWAS cohorts for vBMD are relatively small compared to many other osteoporosis-related traits, which may limit the statistical power of this study to identify osteoporosis-related genes. Future studies are needed to expand the sample sizes of both gene expression reference panel and GWAS cohorts, to develop new methods to overcome the confounding problem of linkage disequilibrium among SNPs, as well as to acquire gene expression data in primary human bone tissues/cells.
In summary, we performed the first multi-tissue TWAS for vBMD and identified a number of novel vBMD associated genes in several tissues that are significantly relevant to vBMD. Our findings highlighted the power of integrating tissue-specific gene expression data with GWAS statistics to reveal novel insights into the potential pathogenesis mechanisms of osteoporosis.
Supplementary Material
Highlights.
We performed the first TWAS for volumetric BMD across multiple tissues.
We identified 70 significant genes associated with vBMD.
NME8 and LYRM2 have been revalidated to have a connection with BMD/osteoporosi.
DNAAF2 and SPAG16 were identified to be putative vBMD associated genes.
ACKNOWLEDGMENT
This work was benefited by the support of grants from the National Institutes of Health [R01MH107354, R01MH104680, R01GM109068, R01AR069055, U19AG055373, R01DK115679, R01AG061917].
Framingham: The study was supported by grants from the US National Institute for Arthritis, Musculoskeletal and Skin Diseases and National Institute on Aging (R01AR41398 and R01AR061162 to DPK; R01AR050066 to DK). The Framingham Heart Study of the National Heart, Lung, and Blood Institute of the National Institutes of Health and Boston University School of Medicine were supported by the National Heart, Lung, and Blood Institute's Framingham Heart Study (N01-HC-25195) and its contract with Affymetrix, Inc. for genotyping services (N02-HL-6-4278). Analyses reflect intellectual input and resource development from the Framingham Heart Study investigators who participated in the SNP Health Association Resource (SHARe) project. A portion of this research was conducted using the Linux Cluster for Genetic Analysis (LinGA-II) funded by the Robert Dawson Evans Endowment of the Department of Medicine at Boston University School of Medicine and Boston Medical Center.
Osteoporotic Fractures in Men (MrOS)-USA: The Osteoporotic Fractures in The Osteoporotic Fractures in Men (MrOS) Study was funded by National Institutes of Health. The following institutes provide support: the National Institute on Aging (NIA), the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), the National Center for Advancing Translational Sciences (NCATS), and NIH Roadmap for Medical Research under the following grant numbers: U01AG027810, U01AG042124, U01AG042139, U01AG042140, U01AG042143, U01AG042145, U01AG042168, U01AR066160, and UL1TR000128. The National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) provides funding for the MrOS ancillary study ‘GWAS in MrOS and SOF’ under the grant number RC2AR058973. CMN is supported by NIAMS K01AR062655.
Footnotes
Declarations of interest: none
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCE
- 1.NIH Consensus Development Panel on Osteoporosis Prevention, Diagnosis, and Therapy, March 7–29, 2000: highlights of the conference. South Med J. 2001;94(6):569–573. [PubMed] [Google Scholar]
- 2.Burge R, Dawson-Hughes B, Solomon DH, Wong JB, King A, Tosteson A. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005–2025. J Bone Miner Res. 2007;22(3):465–475. [DOI] [PubMed] [Google Scholar]
- 3.Anderson DE DS, Allaire BT, et al. . The associations between QCT-based vertebral bone measurements and prevalent vertebral fractures depend on the spinal locations of both bone measurement and fracture. Osteoporos Int. 2014;25(2):559–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Langsetmo L, Peters KW, Burghardt AJ, et al. Volumetric Bone Mineral Density and Failure Load of Distal Limbs Predict Incident Clinical Fracture Independent HR-pQCT BMD and Failure Load Predicts Incident Clinical Fracture of FRAX and Clinical Risk Factors Among Older Men. J Bone Miner Res. 2018;33(7):1302–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li N, Li XM, Xu L, Sun WJ, Cheng XG, Tian W. Comparison of QCT and DXA: Osteoporosis Detection Rates in Postmenopausal Women. Int J Endocrinol. 2013; 2013:895474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Orwoll ES, Oviatt SK, Mann T. The impact of osteophytic and vascular calcifications on vertebral mineral density measurements in men. J Clin Endocrinol Metab. 1990;70(4):1202–1207. [DOI] [PubMed] [Google Scholar]
- 7.Yang TL, Shen H, Liu A, et al. A road map for understanding molecular and genetic determinants of osteoporosis. Nat Rev Endocrinol. 2020;16(2):91–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nielson CM, Liu CT, Smith AV, et al. Novel Genetic Variants Associated With Increased Vertebral Volumetric BMD, Reduced Vertebral Fracture Risk, and Increased Expression of SLC1A3 and EPHB2. J Bone Miner Res. 2016;31(12):2085–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lappalainen T, Sammeth M, Friedlander MR, et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013;501(7468):506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang X, Joehanes R, Chen BH, et al. Identification of common genetic variants controlling transcript isoform variation in human whole blood. Nat Genet. 2015;47(4):345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Westra HJ, Peters MJ, Esko T, et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet. 2013;45(10):1238–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Albert FW, Kruglyak L. The role of regulatory variation in complex traits and disease. Nat Rev Genet. 2015;16(4):197–212. [DOI] [PubMed] [Google Scholar]
- 13.Consortium GT. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45(6):580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu L, Shi W, Long J, et al. A transcriptome-wide association study of 229,000 women identifies new candidate susceptibility genes for breast cancer. Nat Genet. 2018;50(7):968–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barbeira AN, Pividori M, Zheng J, Wheeler HE, Nicolae DL, Im HK. Integrating predicted transcriptome from multiple tissues improves association detection. PLoS Genet. 2019;15(1):e1007889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma M, Huang DG, Liang X, et al. Integrating transcriptome-wide association study and mRNA expression profiling identifies novel genes associated with bone mineral density. Osteoporos Int. 2019;30(7):1521–1528. [DOI] [PubMed] [Google Scholar]
- 17.Gamazon ER, Wheeler HE, Shah KP, et al. A gene-based association method for mapping traits using reference transcriptome data. Nat Genet. 2015;47(9):1091–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Du Y, Li P, Wen Y, et al. Evaluating the Correlations Between Osteoporosis and Lifestyle-Related Factors Using Transcriptome-Wide Association Study. Calcif Tissue Int. 2020;106(3):256–263. [DOI] [PubMed] [Google Scholar]
- 19.Liu Y, Shen H, Greenbaum J, et al. Gene Expression and RNA Splicing Imputation Identifies Novel Candidate Genes Associated with Osteoporosis. J Clin Endocrinol Metab. 2020; 105(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Samelson EJ, Christiansen BA, Demissie S, et al. QCT measures of bone strength at the thoracic and lumbar spine: the Framingham Study. J Bone Miner Res. 2012;27(3):654–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoffmann U, Massaro JM, Fox CS, Manders E, O'Donnell CJ. Defining normal distributions of coronary artery calcium in women and men (from the Framingham Heart Study). Am J Cardiol. 2008; 102(9): 1136–1141, 1141 e1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cupples LA, Arruda HT, Benjamin EJ, et al. The Framingham Heart Study 100K SNP genome-wide association study resource: overview of 17 phenotype working group reports. BMC Med Genet. 2007;8Suppl 1:S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blank JB, Cawthon PM, Carrion-Petersen ML, et al. Overview of recruitment for the osteoporotic fractures in men study (MrOS). Contemp Clin Trials. 2005;26(5):557–568. [DOI] [PubMed] [Google Scholar]
- 24.Orwoll E, Blank JB, Barrett-Connor E, et al. Design and baseline characteristics of the osteoporotic fractures in men (MrOS) study--a large observational study of the determinants of fracture in older men. Contemp Clin Trials. 2005;26(5):569–585. [DOI] [PubMed] [Google Scholar]
- 25.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38(8):904–909. [DOI] [PubMed] [Google Scholar]
- 27.Das S, Forer L, Schonherr S, et al. Next-generation genotype imputation service and methods. Nat Genet. 2016;48(10):1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCarthy S, Das S, Kretzschmar W, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016;48(10):1279–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Finucane HK, Reshef YA, Anttila V, et al. Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat Genet. 2018;50(4):621–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu A, Liu Y, Su K-J, et al. Supplementary Tables. Figshare Deposited 18 December 2020. 10.6084/m9.figshare.13428416.v1. [DOI] [Google Scholar]
- 31.Finucane HK ea. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat Genet. 2015;47:1228–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Finucane HK, Bulik-Sullivan B, Gusev A, et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat Genet. 2015;47(11):1228–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wagner GP, Kin K, Lynch VJ. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012;131(4):281–285. [DOI] [PubMed] [Google Scholar]
- 34.https://alkesgroup.broadinstitute.org/LDSCORE/. Accessed.
- 35.Bulik-Sullivan B, Finucane HK, Anttila V, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015;47(11):1236–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bulik-Sullivan BK, Loh PR, Finucane HK, et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet. 2015;47(3):291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zou H, Hastie T. Regularization and variable selection via the elastic net (vol B 67, pg 301, 2005). J R Stat Soc B. 2005;67:768–768. [Google Scholar]
- 38.Gamazon ER, Wheeler HE, Shah KP, et al. A gene-based association method for mapping traits using reference transcriptome data. Nature Genetics. 2015;47:1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barbeira AN, Dickinson SP, Bonazzola R, et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat Commun. 2018;9(1):1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4(1):44–57. [DOI] [PubMed] [Google Scholar]
- 43.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheng B, Liang X, Wen Y, et al. Integrative analysis of transcriptome-wide association study data and messenger RNA expression profiles identified candidate genes and pathways for inflammatory bowel disease. J Cell Biochem. 2019;120(9):14831–14837. [DOI] [PubMed] [Google Scholar]
- 45.Leonenko G, Sims R, Shoai M, et al. Polygenic risk and hazard scores for Alzheimer's disease prediction. Ann Clin Transl Neurol. 2019;6(3):456–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schwantes-An TH, Darlay R, Mathurin P, et al. Genome-wide association study and meta-analysis on alcohol-related liver cirrhosis identifies novel genetic risk factors. Hepatology. 2020. [DOI] [PubMed] [Google Scholar]
- 47.Estrada K, Styrkarsdottir U, Evangelou E, et al. Genome-wide meta-analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nat Genet. 2012;44(5):491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Laroche M Pattern of bone mineral density in idiopathic male osteoporosis. Rheumatol Int. 2012;32(10):3093–3096. [DOI] [PubMed] [Google Scholar]
- 49.Shah VN, Sippl R, Joshee P, et al. Trabecular bone quality is lower in adults with type 1 diabetes and is negatively associated with insulin resistance. Osteoporos Int. 2018;29(3):733–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Osteoporosis. https://en.wikipedia.org/wiki/Osteoporosis. Accessed.
- 51.Kemp JP, Medina-Gomez C, Estrada K, et al. Phenotypic dissection of bone mineral density reveals skeletal site specificity and facilitates the identification of novel loci in the genetic regulation of bone mass attainment. PLoS Genet. 2014;10(6):e1004423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paternoster L, Lorentzon M, Lehtimaki T, et al. Genetic determinants of trabecular and cortical volumetric bone mineral densities and bone microstructure. PLoS Genet. 2013;9(2):e1003247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adrenal Gland. https://training.seer.cancer.gov/anatomy/endocrine/glands/adrenal.html. Accessed.
- 54.Hardy R, Cooper MS. Adrenal gland and bone. Arch Biochem Biophys. 2010;503(1):137–145. [DOI] [PubMed] [Google Scholar]
- 55.Hofbauer LC, Rauner M. Minireview: live and let die: molecular effects of glucocorticoids on bone cells. Mol Endocrinol. 2009;23(10):1525–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hardy R, Cooper MS. Bone loss in inflammatory disorders. J Endocrinol. 2009;201(3):309–320. [DOI] [PubMed] [Google Scholar]
- 57.Leistner DM, Seeger FH, Fischer A, et al. Elevated levels of the mediator of catabolic bone remodeling RANKL in the bone marrow environment link chronic heart failure with osteoporosis. Circ Heart Fail. 2012;5(6):769–777. [DOI] [PubMed] [Google Scholar]
- 58.Elefteriou F, Ahn JD, Takeda S, et al. Leptin regulation of bone resorption by the sympathetic nervous system and CART. Nature. 2005;434(7032):514–520. [DOI] [PubMed] [Google Scholar]
- 59.Naoyuki Kawao HK. Interactions Between Muscle Tissues and Bone Metabolism. Journal of Cellular Biochemistry. 2014;116(5):687–695. [DOI] [PubMed] [Google Scholar]
- 60.Gilsanz V, Wren TA, Sanchez M, Dorey F, Judex S, Rubin C. Low-level, high-frequency mechanical signals enhance musculoskeletal development of young women with low BMD. J Bone Miner Res. 2006;21(9):1464–1474. [DOI] [PubMed] [Google Scholar]
- 61.Tanaka K, Kanazawa I, Yamaguchi T, Yano S, Kaji H, Sugimoto T. Active vitamin D possesses beneficial effects on the interaction between muscle and bone. Biochem Biophys Res Commun. 2014;450(1):482–487. [DOI] [PubMed] [Google Scholar]
- 62.Xi G, Wai C, DeMambro V, Rosen CJ, Clemmons DR. IGFBP-2 directly stimulates osteoblast differentiation. J Bone Miner Res. 2014;29(11):2427–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Leslie WD, Lix LM, Johansson H, Oden A, McCloskey E, Kanis JA. Spine-hip discordance and fracture risk assessment: a physician-friendly FRAX enhancement. Osteoporos Int. 2011;22(3):839–847. [DOI] [PubMed] [Google Scholar]
- 64.Elad Wasserman DW, Gisela Kuhn, Malka Attar-Namdar, Ralph Müller, Itai Bab. Differentially load-regulated gene expression in mouse trabecular osteocytes Bone. 2013;53(1):14–23. [DOI] [PubMed] [Google Scholar]
- 65.Li JJ, Wang BQ, Fei Q, Yang Y, Li D. Identification of candidate genes in osteoporosis by integrated microarray analysis. Bone Joint Res. 2016;5(12):594–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Silverberg SJ, Shane E, de la Cruz L, et al. Skeletal disease in primary hyperparathyroidism. J Bone Miner Res. 1989;4(3):283–291. [DOI] [PubMed] [Google Scholar]
- 67.Balenga N, Koh J, Azimzadeh P, et al. Parathyroid-Targeted Overexpression of Regulator of G-Protein Signaling 5 (RGS5) Causes Hyperparathyroidism in Transgenic Mice. J Bone Miner Res. 2019;34(5):955–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mullin BH, Zhu K, Xu J, et al. Expression Quantitative Trait Locus Study of Bone Mineral Density GWAS Variants in Human Osteoclasts. J Bone Miner Res. 2018;33(6):1044–1051. [DOI] [PubMed] [Google Scholar]
- 69.Grundberg E, Kwan T, Ge B, et al. Population genomics in a disease targeted primary cell model. Genome Res. 2009;19(11):1942–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wainberg M, Sinnott-Armstrong N, Mancuso N, et al. Opportunities and challenges for transcriptome-wide association studies. Nat Genet. 2019;51(4):592–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wu C, Pan W. A powerful fine-mapping method for transcriptome-wide association studies. Hum Genet. 2020;139(2):199–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.