

Abstract
An important function of the gut microbiome is the fermentation of non-digestible dietary fibers into short chain fatty acids (SCFAs). The three primary SCFAs: acetate, propionate, and butyrate, are key mediators of metabolism and immune cell function in the gut mucosa. We previously demonstrated that butyrate at high concentrations decreased human gut lamina propria (LP) CD4 T cell activation in response to enteric bacteria exposure in vitro. However, to date, the mechanism by which butyrate alters human gut LP CD4 T cell activation remains unknown. In this current study, we sought to better understand how exposure to SCFAs across a concentration range impacted human gut LP CD4 T cell function and activation. LP CD4 T cells were directly activated with T cell receptor (TCR) beads in vitro in the presence of a physiologic concentration range of each of the primary SCFAs. Exposure to butyrate potently inhibited CD4 T cell activation, proliferation, and cytokine (IFNγ, IL-17) production in a concentration dependent manner. Butyrate decreased the proliferation and cytokine production of T helper (Th) 1, Th17 and Th22 cells, with differences noted in the sensitivity of LP versus peripheral blood Th cells to butyrate’s effects. Higher concentrations of propionate and acetate relative to butyrate were required to inhibit CD4 T cell activation and proliferation. Butyrate directly increased the acetylation of both unstimulated and TCR-stimulated CD4 T cells, and apicidin, a Class I histone deacetylase inhibitor, phenocopied butyrate’s effects on CD4 T cell proliferation and activation. GPR43 agonism phenocopied butyrate’s effect on CD4 T cell proliferation whereas a GPR109a agonist did not. Our findings indicate that butyrate decreases in vitro human gut LP CD4 T cell activation, proliferation, and inflammatory cytokine production more potently than other SCFAs, likely through butyrate’s ability to increase histone acetylation, and potentially via signaling through GPR43. These findings have relevance in furthering our understanding of how perturbations of the gut microbiome alter local immune responses in the gut mucosa.
Keywords: SCFA, butyrate, T helper cell, T cell activation, human gut T cell, CD4
INTRODUCTION
Butyrate is a short chain fatty acid (SCFA) produced through fermentation of non-digestible dietary fibers by anaerobic bacteria in the lumen of the gastrointestinal tract. Butyrate has a myriad of effects, both locally and systemically, that are beneficial to the host {reviewed in [1, 2]}. In the gut, butyrate is a critical energy source for epithelial cells, regulates epithelial cell function and has an immunomodulatory impact on host innate and adaptive immunity [1, 2]. Butyrate functions via multiple mechanisms that include signaling through G-protein-coupled receptors (GPRs), histone deacetylase (HDAC) inhibition and modulating cellular metabolism [3–5]. Decreased abundances of gut-associated butyrate-producing bacteria (BPB) have been observed in inflammatory diseases of the gut including Inflammatory Bowel Disease (IBD) and in persons with HIV (PWH), where lower abundances of BPB genera were associated with increased levels of inflammation and immune activation [6–11]. Furthermore, the oral administration of known metabolic substrates for BPB resulted in increased abundances of colonic BPB and concomitantly decreased systemic inflammation and immune activation in PWH, indicating that increasing concentrations of butyrate in the gastrointestinal tract may alleviate inflammation [12]. Understanding the mechanisms by which butyrate contributes to gut homeostasis and how low tissue concentrations of butyrate differentially modulate host immunity may lead to novel approaches to maximize butyrate’s beneficial anti-inflammatory effects in individuals with inflammatory disorders of the gut mucosa.
To date, the role of butyrate on human CD4 T cell function has primarily been investigated using peripheral blood (PB) T cells, where butyrate was shown to impact both T regulatory and T helper (Th) cell differentiation and function. For example, in vitro exposure of human naïve CD4 T cells to IL-2, TGFβ and butyrate induced a high fraction of T regulatory cells [13]. Addition of butyrate to differentiating total CD4 T cells promoted production of IL-10, resulting in decreased frequencies of IL-17-expressing CD4 T cells [14]. Similarly, butyrate significantly reduced frequencies of Th17 cells expanded in response to in vitro exposure of PB mononuclear cells (PBMC) to gram-negative cell wall component lipopolysaccharide (LPS) [15]. Investigations into the epigenetic mechanisms that regulate expression of the Th17 transcription factor RORγt highlighted that the ability of butyrate to modulate Th17 function was dependent of the state of Th17 cell differentiation [16]. While the addition of butyrate to already differentiated Th17 cells induced RORγt expression and IL-17 secretion, exposure of naïve CD4 T cells undergoing differentiation to Th17 cells to butyrate resulted in a down-regulation of RORγt and IL-17 production [16].
In contrast to PB CD4 T cells, gut mucosal CD4 T cells are primarily resident effector memory cells that display distinct phenotypic and functional characteristics [17–19]. Given their proximity to the gut lumen and consistent exposure to higher concentrations of butyrate, these cells may be more primed to respond to butyrate compared to the CD4 T cell populations in the periphery. Thus, results of studies characterizing how human PB CD4 T cells respond to butyrate [13–16] may not directly translate to the effect of butyrate on CD4 T cells in the gut mucosal compartment. We previously demonstrated that the addition of high (but physiologically relevant) concentrations of exogenous butyrate reduced gut lamina propria (LP) CD4 T cell activation after exposure to enteric bacteria in vitro [9]; however, the mechanisms by which butyrate modulated those responses was not investigated. In this current study, we utilized an in vitro human LP mononuclear cell (LPMC) model to comprehensively evaluate the impact of a range of physiologically relevant concentrations of butyrate and other gut-associated SCFAs on LP CD4 T cell function and then probed the mechanisms by which butyrate modulated those responses.
RESULTS
In vitro exposure to exogenous butyrate decreases TCR-mediated LP CD4 T cell activation and proliferation in a concentration dependent manner.
To directly evaluate the impact of butyrate on human gut LP CD4 T cells and mitigate off target effects of butyrate on antigen presenting cells (APC) [20–23], we evaluated the ability of butyrate to modulate LP CD4 T cell activation and proliferation in the setting of T cell receptor (TCR)/CD28-mediated activation. Total LPMC were activated with or without TCR-stimulatory beads in the presence of a non-toxic, physiologically relevant concentration range of butyrate (0.0625–0.5mM). Given that concentrations of butyrate have not been reliably measured directly in human gut tissue, we used a concentration range of butyrate based on reported measured concentrations of butyrate in the gut lumen and in the portal circulation of human subjects [24, 25]. Specifically, the concentration range was based on 1) values that were less than the gut lumen but greater than the portal circulation thereby reflecting the likely concentrations that tissue LPMC would be exposed to and 2) the dose range that had limited impact on overall viability with or without TCR stimulation, in our in vitro system (Supp Fig. 1a–b).
LPMC were stimulated with or without TCR-stimulatory beads in the presence or absence of butyrate and levels of T cell proliferation and activation measured by multicolor flow cytometry after four days of culture (Supp. Fig. 2). As expected, TCR-mediated stimulation of LPMC in the absence of butyrate (i.e. 0mM) induced CD4 T cell proliferation (CFSEdim) (Fig. 1a) and increased percentages of LP CD4 T cells expressing CD25 (Fig. 1b) and co-expressing HLA-DR and CD38 (Fig. 1c). Increasing concentrations of butyrate led to a decrease in the percentage of proliferated LP CD4 T cells, with the highest concentration tested (0.5mM) completely inhibiting TCR-mediated proliferation (Fig. 1a). Butyrate also decreased the percentage of CD25+ and CD38+HLA-DR+ (Fig. 1b,c) LP CD4 T cells in a concentration dependent manner with maximal effects noted at concentrations ≥0.25mM. In the absence of TCR-mediated stimulation, butyrate did not induce proliferation (Fig. 1a) or alter the percentages of LP CD4 T cells co-expressing HLA-DR and CD38 (Fig. 1c). However, butyrate increased the expression of CD25 on unstimulated LP CD4 T cells by an average 1.4-fold at concentrations of ≥0.125mM (Fig. 1b). Butyrate has been reported to promote the differentiation of human PB and murine gut and splenic Foxp3+ T regulatory cells [13, 15, 26–28], which also express high levels of CD25 [29]. Whereas butyrate increased the expression of CD25 on LP CD4 T cells at 0.25mM, the percentages of CD25+ Foxp3-expressing LP CD4 T cells were not increased in LPMC exposed to butyrate alone (Supp Fig 3). This suggests that the increased expression of CD25 on LP CD4 T cells in response to exogenous butyrate in vitro is not due to expansion of T regulatory cells.
Fig. 1.
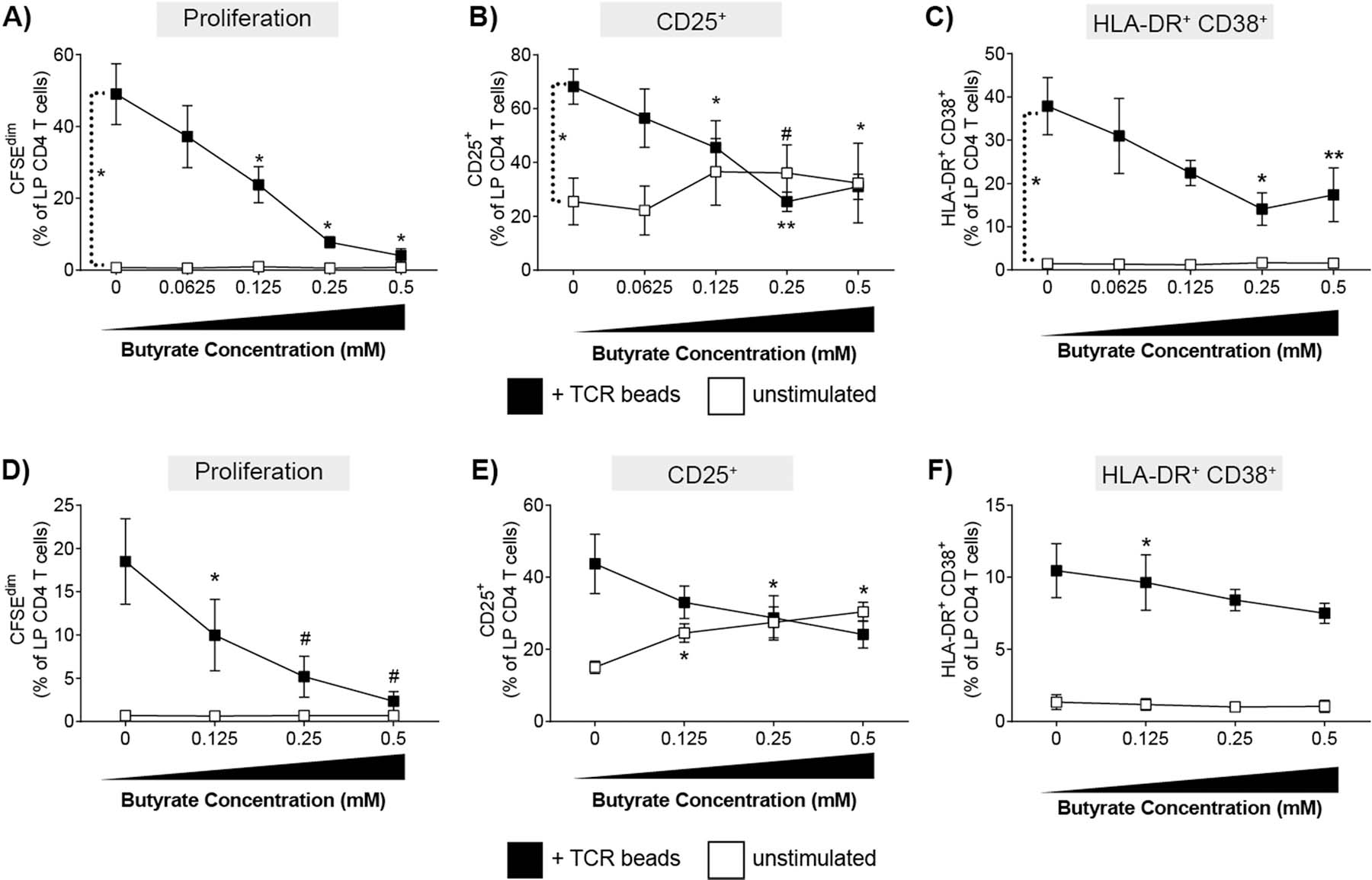
Butyrate reduces TCR-mediated proliferation and activation LP CD4 T cells in a concentration dependent manner. (A-C) Lamina propria mononuclear cells (LPMC; N = 3) or (D-F) purified LP CD4 T cells (N = 3) were pre-labelled with CFSE and cultured with or without TCR-activating beads (TCR beads) and exogenous butyrate (0.0625–0.5 mM) for four days and percentages of LP CD4 T cells that (A,D) proliferated (CFSEdim) or expressed (B, E) CD25 or (C, F) HLA-DR+CD38+ determined using multi-color flow cytometry. FM2 (Fluorescence minus two; HLA-DR and CD25) and isotype control (CD38) values have been subtracted. Values are shown as mean ± SEM. Statistical analysis: Paired t tests were conducted to determine differences in proliferation or activation between unstimulated and TCR bead stimulated conditions and between each butyrate concentration versus no butyrate conditions. *P < 0.05, *P < 0.01, #P < 0.06.
Exposure of purified LP CD4 T cells to exogenous butyrate decreased TCR-mediated LP CD4 T cell activation and proliferation in a concentration dependent manner.
To determine if butyrate decreased proliferation and activation by acting directly on LP CD4 T cells, purified LP CD4 T cells were exposed to the same non-toxic doses of butyrate (Supp Fig 1c) in the presence and absence of TCR stimulation (Fig. 1d–f). In the absence of butyrate, TCR bead stimulation of purified LP CD4 T cells induced proliferation and activation (Fig 1d–f) although to a lesser degree than observed with total LPMC (Fig. 1a–c), suggesting optimal stimulation of LP CD4 T cells in response to TCR-mediated stimulation requires the presence of other cells. Similar to total LPMC cultures, butyrate significantly reduced LP CD4 T cell proliferation and CD25 expression in a concentration dependent manner when purified CD4 T cells were stimulated with TCR stimulatory beads (Fig 1d,e). Conversely, butyrate had a much less pronounced inhibitory effect on CD38+HLA-DR+ LP CD4 T cells (Fig. 1f) suggesting butyrate may act on other LPMC that then directly or indirectly modulate CD38 and HLA-DR co-expression by LP CD4 T cells. In the absence of TCR stimulation, the addition of butyrate directly increased the expression of CD25 on LP CD4 T cells (Fig. 1e) similar to the effect noted in total LPMC cultures (Fig1b), indicating that exogenous butyrate may directly regulate the expression of CD25 on human gut CD4 T cells.
Butyrate decreased LP Th cell proliferation and cytokine production.
Th1, Th17 and Th22 cells in the intestinal LP are critical in maintaining gut barrier homeostasis and are key mediators of mucosal immunity [30, 31]. Therefore, the impact of butyrate on the proliferative capacity of LP Th1 (IFNγ+IL-17-), Th17 (IL-17+IFNγ±) and Th22 (IL-22+IL-17-IFNγ-) subsets was assessed by multi-color flow cytometry (Supp Fig 4). TCR bead stimulation (in the absence of butyrate) drove significant increases in net proliferation (TCR-stimulated minus unstimulated) of all Th subsets, although to differing degrees with Th1 (mean proliferation: 53.3±5.0% [SEM]) and Th22 (55.8±5.1%) cells proliferating to a greater degree than Th17 cells (35.7±7.4%) (Fig. 2a–c). Butyrate significantly decreased the net proliferation of all Th subsets in a concentration dependent manner irrespective of the level of Th proliferation induced by T cell activating beads (Fig 2a–c). Th1 and Th22 cells responded similarly to butyrate, with comparable percent decreases in proliferation at each concentration of butyrate tested (Fig 2a,b). Th17 cells also decreased in a concentration dependent manner, although, in comparison to Th1 and Th22 cells, the magnitude of the average percent decrease was higher at the lower concentrations of butyrate (Fig. 2c).
Fig. 2.
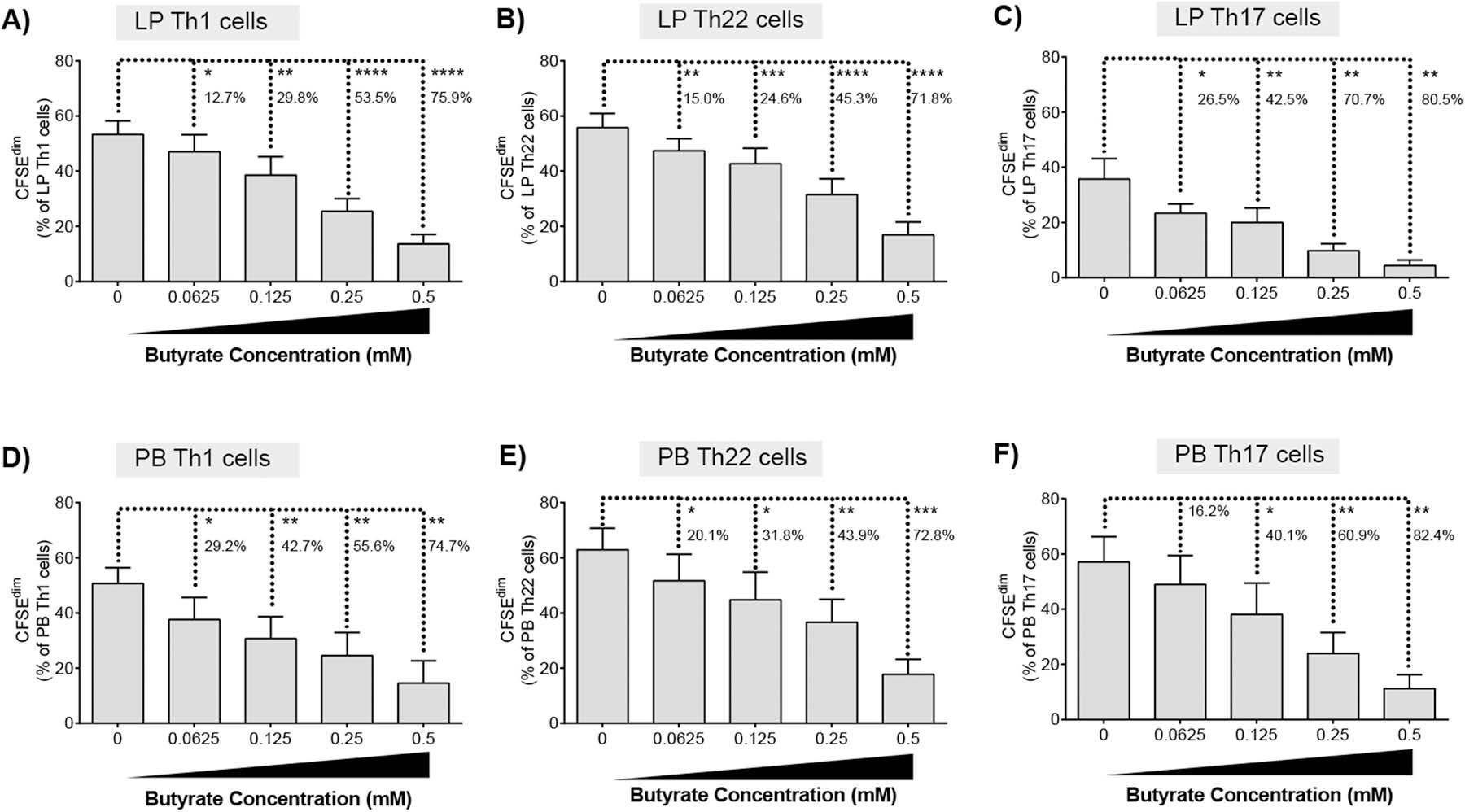
Butyrate decreases proliferation of LP and PB T helper cells. CFSE-labelled (A) LPMC (N = 6) or (B) PBMC (N = 6) were cultured with TCR-activating beads and exogenous butyrate (0.0625–0.5 mM) for four days with mitogenic stimulation during the last 4 h of culture. Percentages of proliferating (A, D) Th1, (B, E) Th22 or (C, F) Th17 cells were determined using multi-color flow cytometry. Isotype control values have been subtracted. Values are shown as mean ± SEM of net proliferation (TCR-stimulated minus unstimulated). Statistical analysis: Paired t tests were conducted to determine differences in Th subset proliferation between butyrate concentrations versus no butyrate conditions. Percentages represent the average percentage decrease in Th cell proliferation versus no butyrate. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
To determine if butyrate-mediated decrease in LP Th cells proliferation similarly reduced cytokine production, levels of secreted IL-17 and IFNγ were measured in culture supernatants (Supp Fig 5). In the presence of TCR stimulation, butyrate significantly decreased the production of IL-17 by purified CD4 T cells in a dose dependent manner (Supp Fig 5a). Butyrate maximally decreased the production of IFNγ at a concentration of 0.25mM but had variable activity at lower doses (Supp Fig 5b). Butyrate had no observable impact on IL-17 or IFNγ production in the absence of TCR stimulation.
The impact of butyrate on proliferation of PB Th subsets was next tested to investigate if the concentration dependent decrease in LP Th cell proliferation in response to butyrate was intrinsic to gut Th cells. Following exposure of PBMC to TCR-stimulatory beads, PB Th1, Th22 and Th17 cells all proliferated to similar degrees in the absence of butyrate (Th1: (53.3±5.0%; Th22 62.9±7.8%; Th17: 57.1±9.2%) (Fig. 2d–f) contrasting to the differential proliferative responses observed for LP Th cells (Fig. 2a–c). Butyrate inhibited PB Th1, Th22 and Th17 cell proliferation in a concentration dependent manner as observed for LP Th cells. PB Th1 cells had the greatest decrease (29.2%) in proliferation at the lowest concentration of butyrate tested (Fig. 2d) relative to Th22 (20.1%) (Fig. 5e) and Th17 (16.2%) (Fig. 2f) populations. Differences in the response of the Th subsets to the lowest butyrate concentration (0.0625mM) appeared to be tissue site dependent. Exposure of LP Th1 to low dose butyrate decreased proliferation by 12.7% (Fig. 2a) but decreased PB Th1 proliferation by 29.2% (Fig. 2d). Butyrate decreased LP Th17 proliferation by 26.5% (Fig. 2c) versus a 16.2% decrease for PB Th17 cells (Fig. 2f) at the lowest concentration tested (0.0625 mM).
Fig. 5.
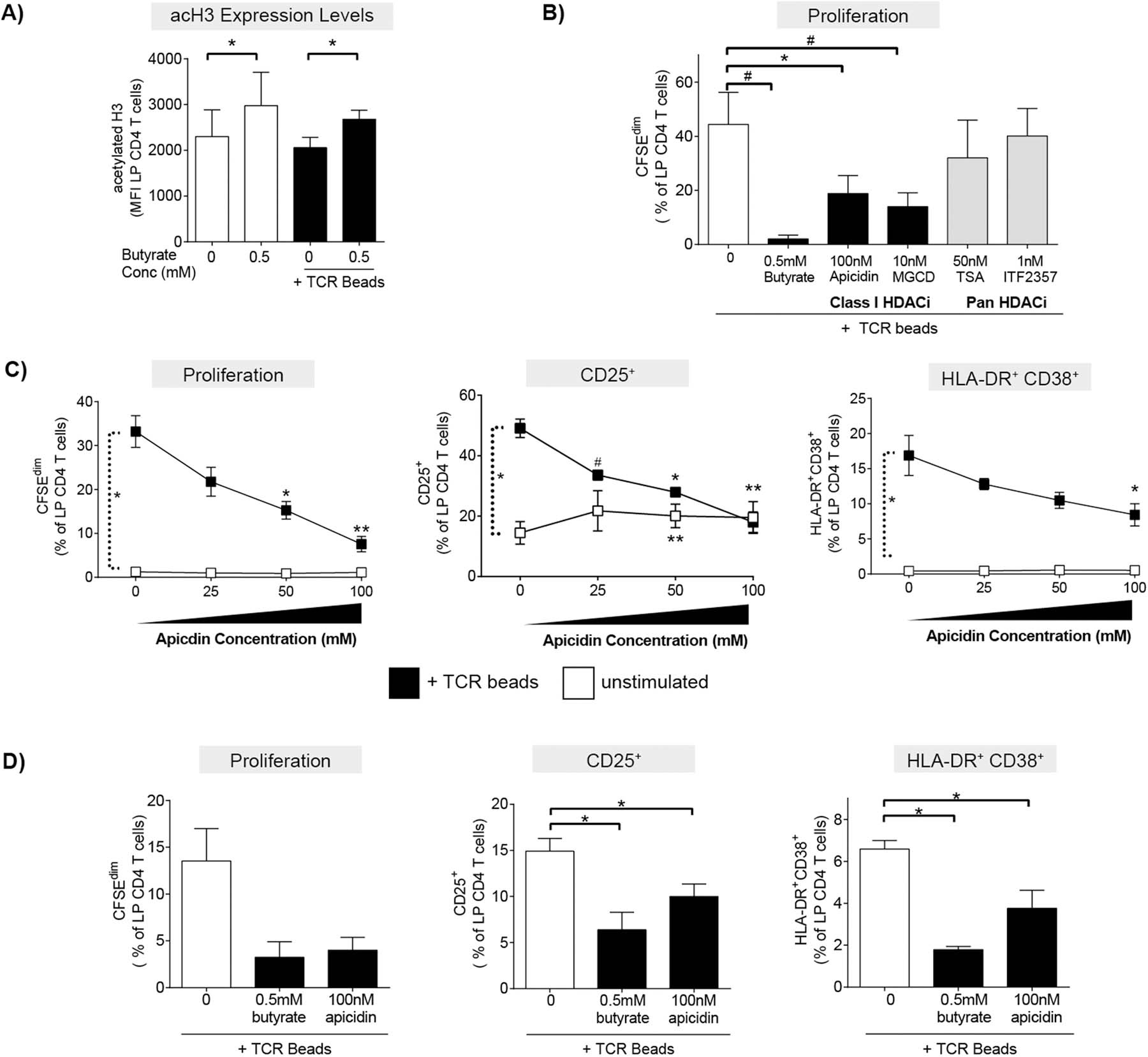
HDAC inhibitors phenocopy butyrate’s ability to decrease LP CD4 T cell proliferation and activation. (A) Levels of acetylation of histone 3 lysine 9 (H3K9) were measured in LP CD4 T cells by multi-color flow cytometry 24hrs after culture of LPMC (N = 4) in the presence or absence of exogenous butyrate (0.5 mM) and TCR-activating beads (TCR beads). Values are shown as the mean fluorescence intensity (MFI) of acetylation of H3K9 (acetylated H3) (mean ± SEM). Statistical analysis: Paired t-tests were conducted to determine differences in acetylation of H3K9 levels in LP CD4 T cells in the presence or absence of butyrate in unstimulated or TCR bead-stimulated conditions. *P < 0.05. (B) CFSE-labelled LPMC (N = 3) were exposed to TCR beads and Class I specific HDAC inhibitors (HDACi’s; apicidin and MGCD) or pan HDACi’s (TSA and ITF2357) and levels of LP CD4 T cell proliferation (CFSEdim) determined at 4 days using multi-color flow cytometry. Values are shown as mean ± SEM. Statistical analysis Paired t tests were conducted to determine differences in proliferation between TCR bead-stimulated LPMC in the absence of butyrate (open bar) and in the presence of butyrate or HDAC inhibitors (filled bars). *P < 0.05, #P ≤ 0.07. (C) LPMC (N = 3) or (D) purified LP CD4 T cells (N = 3) were cultured with or without TCR beads and Apicidin (25‒100 nM) for four days and percentages of LP CD4 T cells that proliferated (CFSEdim) or expressed CD25 or HLA-DR+CD38+ determined using multi-color flow cytometry. FM2 (Fluorescence minus two; HLA-DR and CD25) and isotype control (CD38) values have been subtracted. Values are shown as mean ± SEM. Statistical analysis: Paired t tests were conducted to determine differences in proliferation or activation between no stimulated and TCR bead-stimulated conditions and between butyrate or apicidin concentrations versus no butyrate/apicidin conditions in the presence or absence (C only) of TCR beads. *P < 0.05, **P < 0.01, #P ≤ 0.07.
The timing of butyrate exposure relative to TCR stimulation differentially impacted the inhibition of LP CD4 T cell proliferation.
To determine if the timing of butyrate exposure relative to TCR signaling was critical in the ability of butyrate to modulate LP CD4 T cell proliferation, butyrate was added at various times before (pre-treatment) and after (post-treatment) the addition of the TCR-stimulatory beads to total LPMC cultures (Fig. 3a,b). For these assays, butyrate was added at 0.5 mM, the concentration at which the maximum effect on LP T cell proliferation was observed (Fig. 1a). Exposure of LPMC to butyrate for ≤18hrs before TCR-mediated stimulation did not further reduce LP CD4 T cell proliferation beyond what was observed when butyrate was added simultaneously (0hrs) with TCR stimulatory beads (Fig 3a). For the post-treatment assays, the addition of butyrate 6hrs after TCR-induced stimulation decreased proliferation to similar levels as those when butyrate was added simultaneously with TCR stimulation (Fig. 3b). However, the addition of butyrate at or after 18hrs of TCR stimulation limited butyrate inhibition of LP CD4 T cell proliferation, although the levels of proliferation were still lower than in the absence of butyrate (Fig 3b).
Fig. 3.
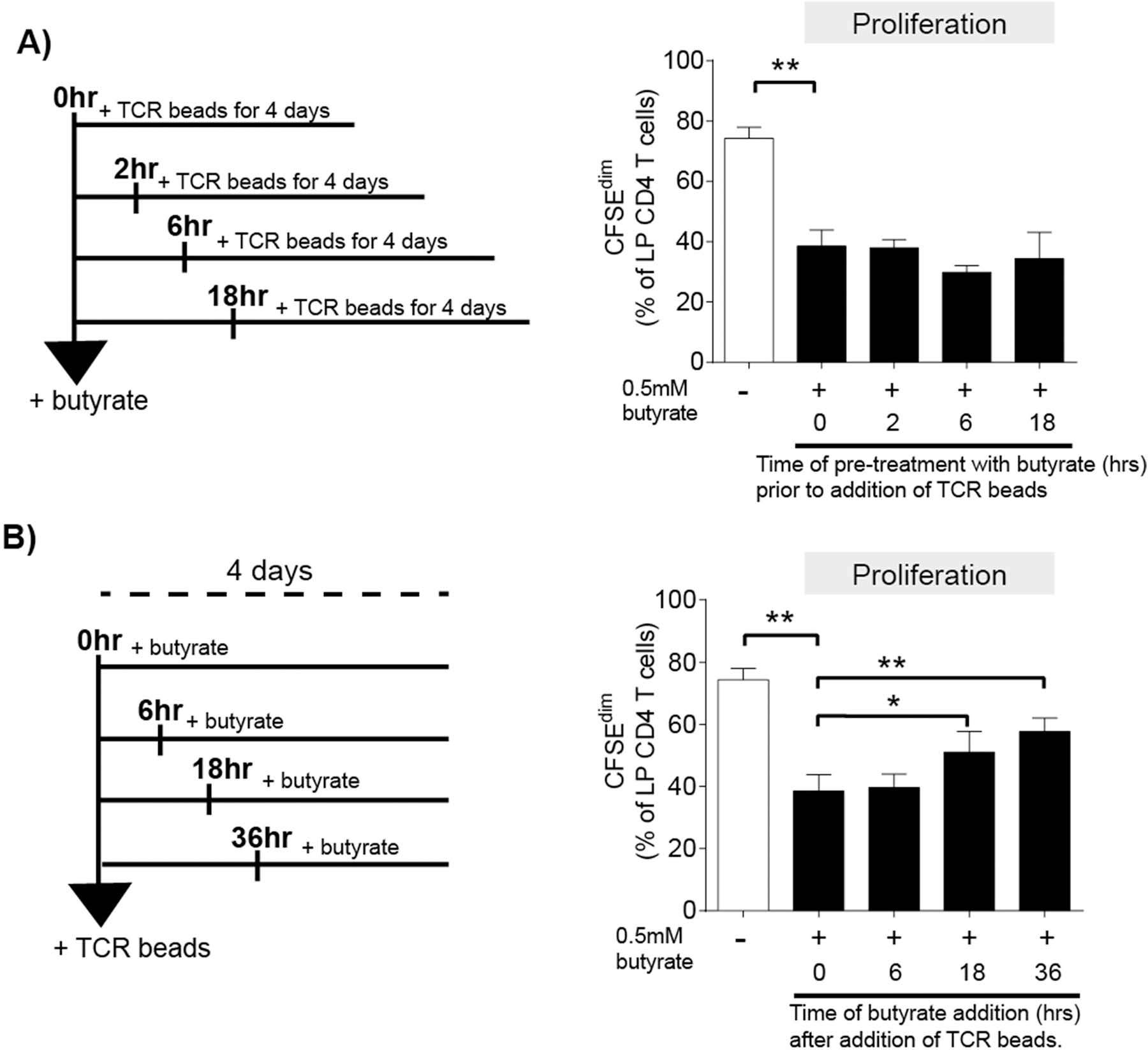
Inhibition of LP CD4 T cell proliferation is dependent of the timing of butyrate exposure relative to TCR stimulation. LPMC (N = 3) were pre-labelled with CFSE and either (A) pre-treated with exogenous butyrate (0.5 mM) for 0, 2, 6 and 18 h before the addition of TCR-activating beads (TCR beads) or (B) exposed to exogenous butyrate 0, 6, 18 or 36 h after the addition of TCR beads. Percentages of proliferated (CFSEdim) LP CD4 T cells was measured 4 days after the addition of TCR beads using multi-color flow cytometry. Values are shown as mean ± SEM. Statistical analysis: Paired t tests were conducted to determine differences in proliferation between TCR bead-stimulated LPMC in the absence of butyrate (open bar) versus the simultaneous addition of butyrate (filled bar; 0hrs) as well as comparisons between 0hrs butyrate and each time point either pre- or post-treatment. *P < 0.05, **P < 0.01.
Higher concentrations of propionate and acetate relative to butyrate were required to inhibit LP CD4 T cell activation and proliferation.
Acetate and propionate are the other primary SCFAs generated in the gut by the metabolic activity of commensal bacteria, with acetate present at the highest concentrations and propionate and butyrate present in similar amounts [24, 25]. To determine if butyrate’s ability to decrease TCR-mediated LP CD4 T proliferation and activation were shared by these other SCFAs, total LPMC were activated with TCR-stimulating beads in the presence of varying physiologic and non-toxic (Supp Fig 1d) concentrations of acetate and propionate. Similar to butyrate, these concentrations were determined using values reflective of human gut lumen and portal circulation[24, 25] concentrations and those that were non-toxic in our assays (Supp Fig 1).
The addition of acetate decreased TCR-stimulated LP CD4 T cell proliferation and CD25 expression on LP CD4 T cells in a concentration dependent manner, albeit requiring higher concentrations than butyrate to achieve similar inhibition (Fig 4 a,b). Acetate decreased the percentage of HLA-DR+CD38+ LP CD4 T cells only at the highest concentration tested (10mM) (Fig 4c). Propionate decreased LP CD4 T cell proliferation and CD25 expression at approximately twice the concentrations where butyrate demonstrated maximal activity (Fig 4 d,e). Much like acetate, propionate decreased the percentage of HLA-DR+CD38+ LP CD4 T cells only at the highest concentration tested (1mM) (Fig. 4f). Unlike butyrate, acetate and propionate had no impact on the expression of CD25 on LP CD4 T cells in the absence of TCR stimulation (data not shown), suggesting the effect of driving low levels of CD25 expression on resting LP CD4 T cells is specific to butyrate.
Fig. 4.
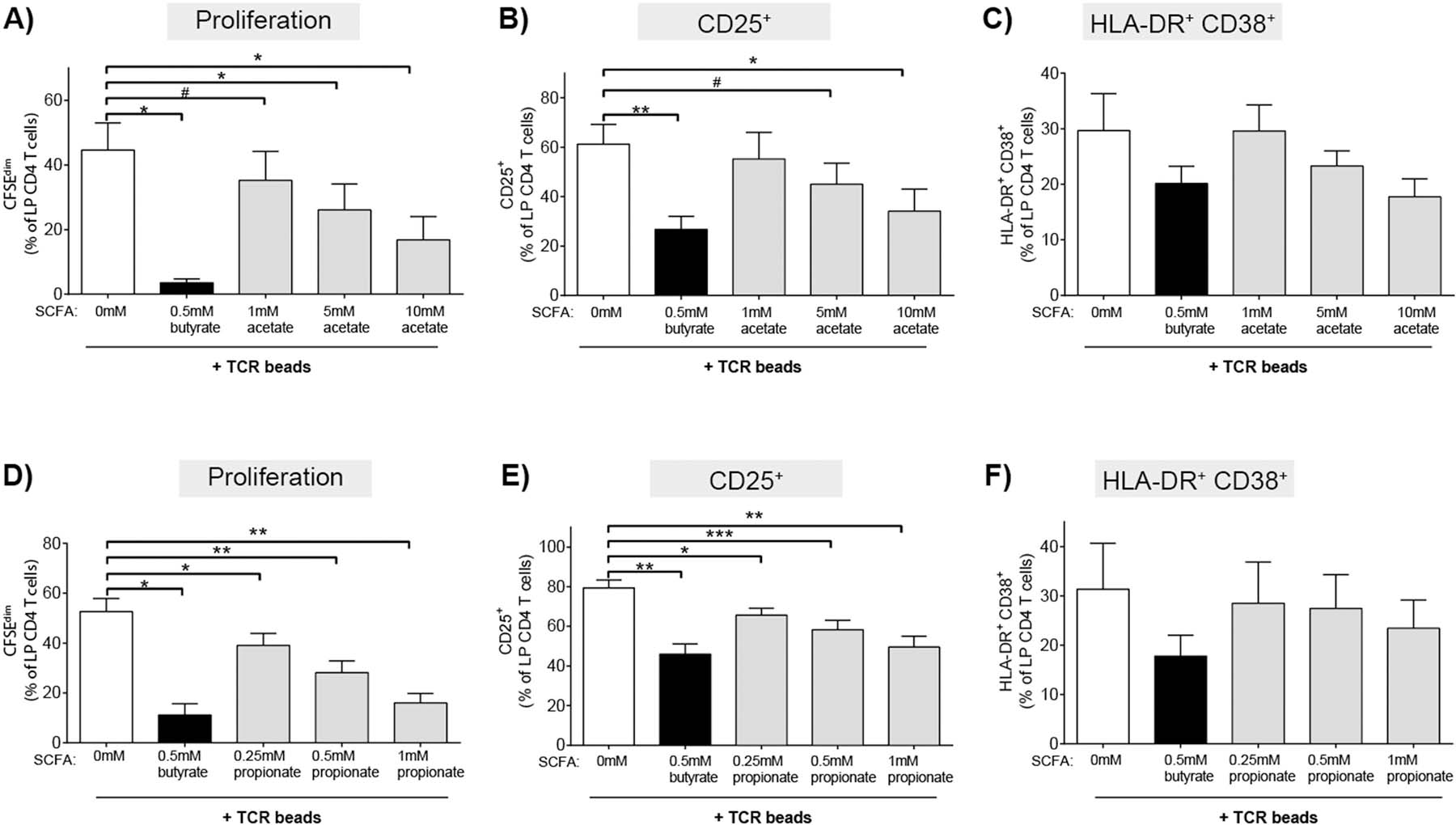
Higher concentrations of acetate and propionate relative to butyrate are required to inhibit LP CD4 T cell proliferation and activation. LPMC were pre-labelled with CFSE and cultured with TCR-activating beads (TCR beads) and exogenous butyrate (0.5 mM) or (A-C) acetate (1‒10 mM; N = 3) or (D-F) propionate (0.25‒10 mM; N = 4) for four days and percentages of LP CD4 T cells that (A,D) proliferated (CFSEdim) or expressed (B, E) CD25 or (C, F) HLA-DR+CD38+ determined using multi-color flow cytometry. FM2 (Fluorescence minus two; HLA-DR and CD25) and isotype control (CD38) values have been subtracted. Values are shown as mean ± SEM of net proliferation or activation (TCR-stimulated minus unstimulated). Statistical analysis: Paired t tests were conducted to determine differences in proliferation or activation between each SCFA concentration (filled bars) versus no SCFA condition (open bar). *P < 0.05, **P < 0.01, ***p < 0.01, #P ≤ 0.07.
Butyrate increased histone acetylation in LP CD4 T cells
Butyrate is a known class I and II HDAC inhibitor (HDACi) [5, 32], with limited studies demonstrating butyrate’s HDACi activity in human T cells [33–35]. To probe potential mechanisms by which butyrate decreased human LP CD4 T cell proliferation and activation, levels of acetylation of histone 3 lysine 9 (acH3K9), an indirect indicator of HDAC inhibition [36], were measured in LP CD4 T cells by multicolor flow cytometry after short term (24hr) culture (Supp Fig 6a). The presence of acH3K9 in the majority of cells irrespective of stimulation or butyrate exposure was to be expected, as these cells were viable and have active genomes with acetylated histones. Given that nearly all viable cells were positive for acH3K9, we measured overall expression levels by mean fluorescence intensity (MFI) to extrapolate the impact of butyrate on histone acetylation. Exposure of unstimulated LPMC to butyrate (0.5mM) significantly increased histone acetylation in LP CD4 T cells relative to controls without butyrate (Fig 5a). The butyrate-induced shifts in MFI were in keeping with previously published studies where the addition of acetate and TSA to human PBMC similarly increased overall MFI of acH3K9[35]. To further confirm that butyrate alters HDAC activity in human LP CD4 T cells, Trichostatin A (TSA), a known pan HDACi, also increased acH3K9 levels in resting LP CD4 T cells (Supp Fig 6b). Butyrate’s ability to increase histone acetylation in LP CD4 T cells in the absence of TCR-stimulation was confirmed by western blot (Supp Fig 6c). When LPMC were activated with TCR-stimulating beads, the effect of butyrate to increase acH3K9 expression in TCR-activated LP CD4 T cells was similar to that observed without TCR stimulation (Fig 5a), indicating that butyrate is the primary driver of the observed increase in acH3K9.
Class I HDAC inhibitors phenocopied butyrate’s inhibition of LP CD4 T cell activation and proliferation
To further confirm HDAC inhibition was a mechanism by which butyrate altered LP CD4 T cell function, the ability of a panel of known class I specific HDACi’s (apicidin and MGCD) and pan HDACi’s (TSA and ITF2357) to phenocopy butyrate’s effect on LP CD4 T cell proliferation was next investigated. Initial studies were carried out to determine the maximal concentration of each HDACi that did not induce toxicity (data not shown), and these maximal concentrations were used in subsequent assays. The addition of apicidin and MGCD (class I HDACi’s) to TCR-stimulated LPMC decreased LP CD4 T cell proliferation to a greater degree than the addition of either pan HDACi (TSA and ITF2357) (Fig 5b). A concentration-dependent effect of apicidin on TCR-mediated LP CD4 T cell proliferation and activation was next tested. Apicidin decreased LP CD4 T cell proliferation, CD25 expression and co-expression of HLA-DR and CD38 on LP CD4 T cells in a concentration dependent manner (Fig 5c). In the absence of TCR stimulation, apicidin had little impact on LP CD4 T cell proliferation or percentages of HLA-DR+ CD38+ LP CD4 T cells (Fig. 5c). However, similar to the observations of CD25 expression in the presence of butyrate, the percentage of CD25+ LP CD4 T cells increased on average 1.5-fold at all concentrations of apicidin tested (Fig 5c), indicating that the expression of CD25, in the absence of TCR stimulation, may be related to HDAC activity.
To determine if apicidin also acted directly on LP CD4 T cells to decrease T cell proliferation and activation as we had observed for butyrate (Fig. 1d), the maximal concentration of apicidin (100 nM) was added to purified LP CD4 T cells in the presence of TCR-stimulatory beads (Fig. 5d). Apicidin decreased LP CD4 T cell proliferation and the percentage of LP CD4 T cells that expressed CD25 (Fig 5d). While TCR stimulation of purified CD4 T cells induced small increases in the frequency of HLA-DR+CD38+ positive LP CD4 T cells, apicidin decreased expression by 43% (Fig. 5d).
Known butyrate receptors, GPR109a and GPR43, were minimally expressed on LP CD4 T cells.
Butyrate is known to signal through GPRs (GPR41, GPR43 and GPR109a) on the cell surface [3, 37–40], but the expression of these receptors and their functional importance on human gut CD4 T cells has yet to be clearly established. While butyrate can signal through all three of these receptors, we chose to focus our studies on GPR43 and GPR109a due to the established importance of butyrate signaling through these receptors in the modulation of immune responses [38, 41–44]. We determined the extent to which LP CD4 T cells expressed these receptors by staining for surface expression of GPR43 and GPR109a on LP CD4 T cells and analyzing by multi-color flow cytometry directly ex vivo (baseline) and after TCR bead stimulation (4 days) (Supp Fig 7a). Low percentages of LP CD4 T cells expressed GPR43 (1.0% ± 0.6%) or GPR109a (0.5% ± 0.01%) at baseline (Supp Fig 7b). Myeloid dendritic cells (mDC) and macrophages are known to express GPR43 and GPR109a[38, 41, 45]. Therefore, to confirm the ability of the antibodies to adequately detect surface expression of these GPRs, we evaluated GPR43 and GPR109a expression on CD3- cells, which would be enriched for both mDC and macrophages[46–49], within the same LPMC samples (Supp Fig. 7c). On average, 24.17% ± 1.12% of LP CD3- cells expressed GPR43; however, GPR109a expression remained low (4% ± 1.15%). Given the relatively low expression of GPR109a on the CD3- population, we specifically investigated the expression on intraepithelial mononuclear cells (IEMC), a population of cells previously described to have higher levels of expression[50]. Intraepithelial CD3-CD19-HLA-DR+ myeloid cells expressed GPR109a (20.9% ± 2.5%) (Supp Fig. 8), confirming that the low expression detected on LP CD4 T cells was not due to technical problems with the antibody.
TCR-bead stimulation of LPMC in the absence of butyrate significantly increased the percentage of GPR43 expressing LP CD4 T cells, although these percentages remained low (<5% of total LP CD4 T cells) and butyrate had no impact on TCR-induced GPR43 expression (Supp Fig 7d). Similar to GPR43, GPR109a expression increased after the addition of TCR stimulating beads, although this increase was not significant, and the overall frequency of positive LP CD4 T cells remained low (< 1.0%) (Supp Fig 7d). Percentages of GPR109a+ LP CD4 T cells were significantly decreased in both TCR-stimulated and unstimulated conditions by the addition of butyrate (Supp Fig 7d).
GPR43 signaling reduces LP CD4 T cell proliferation, despite low expression of GPR43 on LP CD4 T cells.
To determine if relatively low expression of GPR43 and GPR109a on LP CD4 T cells precluded a role for these receptors in butyrate’s effect on LP T cell function, we exposed purified LP CD4 T cells to non-toxic doses of GPR43 (4-CMTB) and GPR109a (MK 1903) agonists and TCR-stimulatory beads and measured levels of LP CD4 T cell proliferation 4 days later (Fig 6; Supplementary Figure 8b). These investigations were performed using purified CD4 T cells to confirm that alterations in T cell function were a direct effect on LP CD4 T cells and not mediated via other LP cells. Furthermore, we focused on proliferation of LP CD4 T cells due to the more robust effect of butyrate on this T cell function (Fig. 1a) versus the more subtle changes in T cell activation phenotype assessed by HLA-DR and CD38 co-expression (Fig. 1f). Initial studies confirmed activity of these agonists with both GPR43 and GPR109a agonists similarly altering TNFα production by PBMC stimulated with Escherichia coli lysates (data not shown), as previously reported for GPR43 agonists[44]. Surprisingly, GPR43 agonism decreased TCR-induced LP CD4 T cell proliferation in a dose dependent manner, with 10μm and 100μm reducing proliferation similar to butyrate. However, the GPR109a agonist did not impact T cell proliferation at any dose tested.
Fig. 6.
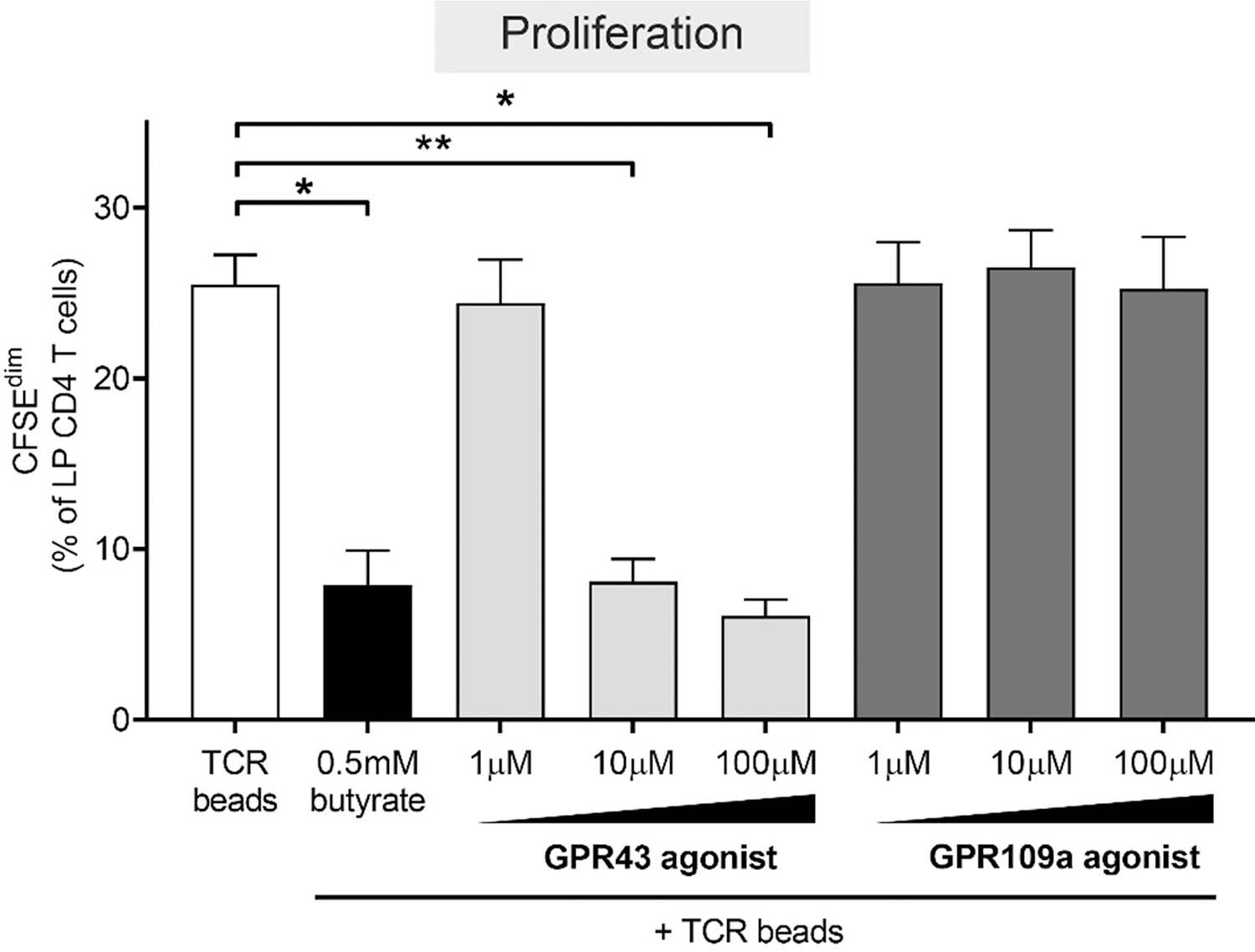
GPR43 agonist phenocopies butyrate’s ability to decrease LP CD4 T cell proliferation. CFSE-labelled purified LP CD4 T cells (N = 4) were cultured with TCR beads, with or without butyrate (0.5 mM) or GPR agonists (1‒100 μM; N = 3 for 1 μM and 100 μM) for four days and percentages of LP CD4 T cells that proliferated (CFSEdim) determined using multi-color flow cytometry. Values are shown as mean ± SEM of net proliferation (TCR-stimulated minus unstimulated). Statistical analysis: Paired t tests were conducted to determine differences in proliferation between butyrate or GPR agonist concentrations versus no butyrate or GPR agonist conditions. *P < 0.05, **P < 0.01.
Discussion
Low abundances of bacterial taxa containing BPB have been reported as a feature of dysbiosis in human inflammatory disorders of the gut mucosa and associated with inflammation and immune activation [6, 7, 9, 12, 51]. Furthermore, low concentrations of stool butyrate are associated with inflammatory disorders of the gut mucosa [12, 52, 53]. However, studies extensively probing how lower tissue concentrations of butyrate may impact local gut immune cells, particularly LP CD4 T cells, which are the major constituents of human gut mucosa and play important roles in gut homeostasis [54], are limited. Therefore, we utilized an in vitro human LPMC model to investigate human gut LP CD4 T cell responses to concentrations of butyrate broadly reflecting “low” and “high” physiologic butyrate tissue concentrations, and the mechanisms by which butyrate drove these responses. T cells were stimulated in an antigen-independent manner via TCR/CD28 bead stimulation in part to induce robust CD4 T cell activation, and in part to avoid off target effects of butyrate on antigen-presenting cells [55] and thereby directly observe butyrate’s ability to impact LP CD4 T cell function.
In these studies, concurrent addition of high doses of butyrate directly decreased TCR-mediated LP CD4 T cell activation and proliferation, including Th1, Th17 and Th22 proliferation, but lower doses had a more limited impact. A previous study also noted dose-dependent effects of butyrate such that lower concentrations (0.25mM) of butyrate promoted murine CD4 T regulatory cell differentiation and higher concentrations (1mM) induced T-bet expression and IFNγ production in all T cell subsets [56]. Although LP Th17 cells had the lowest average proliferation in response to TCR stimulation, this Th subset appeared to have increased sensitivity to butyrate’s anti-proliferative effect as demonstrated by potent inhibition of IL-17 production and Th17 cell proliferation. This was not reflected in PB Th17 responses in the presence of butyrate, potentially highlighting the gut-specificity of butyrate’s actions. The varied sensitivity of CD4 T cells to butyrate could be attributed to differences in expression of GPRs or in the transport of butyrate into the cell. CD147, a surface protein that is important in the transport of butyrate through MCT-1, is known to be variably expressed on human CD4 T cells in the periphery[57–59]. Although we did not quantify the expression of CD147 on LP Th17 cells, it is possible that the associated greater ability for butyrate to be transported into LP Th17 cells, through higher expression of CD147, may explain their increased sensitivity to butyrate and/or account for differences in butyrate’s ability to inhibit LP Th17 proliferation at lower concentrations. Furthermore, differential expression of GPR43 may additionally explain the differences in sensitivity to butyrate in LP Th cells. We have previously shown human LP Th17 cells had the greatest proliferative capacity in response to in vitro exposure to enteric bacteria [60], and increased fractions of gut-resident bacteria-reactive Th17 cells have been noted in in patients with IBD [61]. Given that IBD is associated with lower abundance of BPB, it will be important to further investigate if our current observations have broad applicability to bacteria-reactive Th cells, particularly Th17 cells, and provide a potential link between decreased tissue butyrate levels and IBD-associated overt Th17 cell inflammation.
Mechanistic assays presented in this study advocate that butyrate alters human LP CD4 T cell activation and proliferation through HDAC inhibition as evidenced by 1) an increase in histone acetylation in LP CD4 T cells on exposure of LPMC to butyrate and 2) the ability of apicidin, a known class I HDACi, to phenocopy butyrate’s inhibition of activation and proliferation of purified LP CD4 T cells. These observations are in keeping with several studies demonstrating butyrate’s ability to function as an HDACi in murine splenic and human PB CD4 T cells [16, 35, 37, 62]. Interestingly, although we noted low frequencies of GPR43+ LP CD4 T cells, the direct agonism of this receptor phenocopied butyrate’s impact on LP CD4 T cell proliferation. Conversely, direct agonism of the GPR109a receptor, had no observable impact. These observations may explain the ability of butyrate, propionate and acetate to similarly decrease LP CD4 T cell activation, since these SCFAs all signal through GPR43 and not GPR109a, a GPR specific to butyrate. A role for GPR43 signaling has also been implicated in the negative regulation of murine mucosal CD4 T cells[14]. Additional studies are required to extensively address the role of GPR43 signaling in the modulation of LP CD4 T cell function by butyrate, including determining whether butyrate inhibits HDAC in a GPR43 dependent manner, as was observed with propionate treatment of murine colonic T regulatory cells where enhanced histone acetylation was dependent of GPR43 expression[28]. Furthermore, although the presented data largely supports roles for HDACi and possibly GPR43 signaling, the role of other butyrate related pathways such as alterations in cellular metabolism [63] and PPARγ [64–66] signaling cannot be ruled out.
Although our study primarily focused on butyrate’s ability to modulate LP CD4 T cell function, we also investigated functional responses of LP CD4 T cells after exposure to acetate and propionate, the other primary SCFAs found in the human colon [1, 4–7]. Both acetate and propionate significantly decreased LP CD4 T cell activation and proliferation, although higher concentrations than butyrate were needed to achieve a similar effect. This concentration dependent difference in effects on LP T cell function may reflect the ability of these SCFAs to inhibit class I HDAC activity since all three SCFAs have been reported to inhibit this class of HDACs, with butyrate generally considered to be the most potent [5, 35, 40, 67]. Our observations also suggest that there may be redundancies in the ability of SCFAs to modulate LP CD4 T cell responses in the gut mucosa such that lower concentrations of one SCFA may have a limited impact on overall LP CD4 T cell activation if higher concentrations of another are present.
A noteworthy observation made in these studies is that the anti-proliferative effect of butyrate on LP CD4 T cells required butyrate to be present before or early after TCR stimulation, and that addition of butyrate late into the T cell response had a diminished anti-proliferative effect. The temporal importance of the ability of butyrate to alter T cell proliferation may be due to the mechanism by which butyrate alters this T cell response. For example, if butyrate-mediated HDAC inhibition is responsible for the decreased proliferation as our model suggests, it must be present early in the events of T cell activation to alter the expression of genes involved in the cell cycle progression (e.g., cyclins [68]). Importantly, these observations have clinical implications and may explain the inconsistent results and often limited impact of butyrate-based therapies administered to individuals with IBD with active disease [49, 69–71] that is characterized by marked LP T cell activation. It is possible that butyrate interventions would be more efficacious if given to individuals who were currently in milder disease states with limited T cell activation rather than those with more overt T cell activation. Similarly, if butyrate concentrations are increased for an extended period, the activation of newly recruited CD4 T cells would likely be dampened and thus act to limit the severity of the disease. To translate these in vitro findings into a clinical setting, future studies in which butyrate is administered during active inflammation in the gut mucosa should characterize butyrate’s effect on T cell activation and cytokine production.
Conclusions
Our study demonstrates that butyrate decreases TCR-driven gut human LP CD4 T cell activation, proliferation, and cytokine production over a wide physiologically relevant concentration range in vitro. Moreover, lower physiologic concentrations of butyrate in the gut mucosa may selectively regulate Th subset activation. We postulate that in inflammatory disorders of the gut mucosa, such as IBD and HIV-1, low abundances of BPB seen with dysbiosis and associated lower concentrations of tissue butyrate may be a contributing factor to high levels of LP CD4 T cell activation and production of inflammatory cytokines. It is possible that this scenario would have the greatest effect on T cells that are newly recruited into the gut mucosa and their subsequent unchecked activation in response to translocating enteric bacteria [9, 51, 72, 73]. Ultimately, these findings suggest that modulating butyrate concentrations either in the tissue or gut lumen could have beneficial anti-inflammatory effects in individuals with inflammatory disorders of the gut mucosa and minimize disease progression [12].
METHODS
Human intestinal LPMC and IEMC isolation.
Macroscopically normal human jejunal tissue was obtained from patients undergoing elective abdominal surgery and would have been otherwise discarded, was used in this study. Tissue samples from individuals with a clinical history of recent chemotherapy (less than eight weeks), hematologic malignancy, radiation, HIV-1 infection, immune-suppressing drug use or inflammatory bowel conditions were excluded from this study. Patients gave informed consent to allow unrestricted use of tissue specimens in research and all samples were de-identified to laboratory staff with only age, sex, the reason for surgery and relevant treatment status information available. The use of these samples for research purposes was reviewed by the Colorado Multiple Institutional Review Board (COMIRB) at the University of Colorado Anschutz Medical Campus and met criteria defined by their policies formed in accordance with regulations outlined by the FDA and OHRP, to be deemed “Not Human Research”. Though exempt research is not covered by federal regulations, this research was conducted in accordance with the ethical guidelines outlined in the Belmont Report and the University of Colorado policies on responsible conduct of research. LPMC were isolated from tissue samples as previously detailed [9, 72, 74–77]. Briefly, tissue (muscle and fat removed) was treated with multiple rounds of EDTA to disassociate epithelial and intra-epithelial mononuclear cells (IEMC) which were subsequently cryopreserved. The remaining tissue underwent collagenase-digestion to isolate LP cells and single cell suspensions cryopreserved. The majority of jejunal LPMC are CD3+ T cells. CD4 T cells constitute, on average, 65 ± 1.5% of viable LP leucocytes with smaller percentages of CD8 T cells, B cells and innate immune cells present, as previously detailed[78].
PBMC isolation
PBMC were isolated from whole blood of healthy donors (N=6) who voluntarily gave written informed consent. Whole blood collection was approved by COMIRB. Blood was drawn in vacutainer tubes containing sodium heparin. PBMC were isolated using Ficoll density gradient centrifugation as previously detailed [79–84] and cryopreserved for long term storage in liquid nitrogen.
LPMC culture model.
LPMC were counted and plated at 1x106 LPMC/mL in 48 well culture plates using complete culture media (RPMI + 10% human AB serum + 1% penicillin/streptomycin/glutamine + 500µg/ml Zosyn (piperacillin/tazobactam) and incubated at 37°C with 5% CO2 and 95% humidity for 24 hrs. to four days as indicated in figure legends. Overall viability of LPMC before culture was on average greater than 78%. T cell activating beads (anti-CD3/CD2/CD28; Miltenyi) were added to culture conditions in a 1:25 (bead: LPMC) ratio. For assays using purified CD4 T cells, LP CD4 T cells were isolated using the EasySep immunomagnetic negative selection kit (Stemcell Technologies) according to the manufacturer’s protocol. On average LP CD4 T cells were enriched to 97.1% of viable leucocytes (n=9).
SCFA assay conditions:
Unless otherwise noted, LPMC were cultured with SCFAs added concurrently with TCR activating beads. Butyric acid, glacial acetic acid and propionic acid were diluted in culture media to working stocks (10–20 mM), raised to a physiologic pH (7.5) using NaOH, filtered and serially diluted to appropriate concentrations at the time of assay set up (SCFA concentrations; Acetate 0.25–10 mM, Propionate 0.25–2 mM, Butyrate 0.0625–0.5 mM). For studies examining the impact of butyrate pre-treatment on LP CD4 cell proliferation. LPMC were plated and exposed to butyrate (butyric acid) for 0, 2, 6 and 8 hours before exposure to TCR activating beads. The conditions examining the impact of butyrate addition after TCR activating bead exposure, butyrate was added at 0, 6, 18 and 36 hours after TCR bead exposure.
Measurement of LP CD4 T cell activation:
After four days of culture with or without butyrate and with or without TCR stimulation, culture conditions were harvested and analyzed by multi-color flow cytometry. Detailed gating strategies to identify CD4 T cells are outlined in supplemental figures (Supplemental Figs 2). Briefly, to enumerate populations of interest, viable LP CD4 T cells were identified (Zombie dye negative) within a total lymphocyte gate based on forward and side scatter and doublets were excluded using forward-scatter-height versus forward-scatter-width. CD4 T cells were identified as CD3+ (PerCpCy5.5, TONBO, Clone: OKT3), CD4+ (V450, TONBO, Clone: RPA-T4) and CD8− (APC, TONBO, Clone: RPA-T8) cells. LP CD4 T cell activation was determined by enumerating the percentage of CD4 T cells that co-expressed CD38 (AF700, eBiosciences, Clone: HIT2) and HLA-DR (APC-Cy7, BioLegend, Clone: L243) as well as the percentage of cells that expressed CD25 (PE, BD, Clone: M-A251). For final reported values, isotype and FMO control values were subtracted, and data displayed as the net value.
LP CD4 T cell proliferation:
In experiments enumerating proliferation of LP T cells, LPMC or purified LP CD4 T cells were labeled with 1 μM CellTrace CFSE (Invitrogen) per manufacturer’s instructions before setting up culture conditions. CFSE-labeled cells were exposed to various concentrations of SCFA as mentioned above. LPMCs were cultured with or without TCR activating beads for four days at 37°C, 5% CO2 and 95% humidity. Cultures were collected, and CD4 T cell proliferation (CFSEdim) was determined by multi-color flow cytometry. Proliferating CD4 T cells were enumerated as the percentage of CFSEdim CD4 T cells with the CFSE gate established on the unstimulated control condition.
Measurement of LP CD4 T regulatory cell frequency:
LPMC were cultured for 4 days in the presence or absence of butyrate (0.25 and 0.5 mM). After four days of culture, cells were stained using FOXP3 Fix/Perm Buffer Set (Biolegend) with the following antibodies: CD45 (PerCpCy5.5, eBiosciences, Clone: 2d1), CD3 (PE-Dazzle, Biolegend, Clone: UCHT1), CD4 (APC, TONBO, Clone: RPA-T4),CD8 (PE-Cy7, TONBO, Clone: RPA-T8), CD25 (PE, BD, Clone: M-A251) and FOXP3 (AF488, eBiosciences, Clone: PCH101) and rat IgG2a (eBiosciences).
LP and PB CD4 T helper cell proliferation:
LPMC or PBMC (1x106 cells/ml) were pre-labeled with CFSE and after 4 days of culture, cells were stimulated with a final concentration of 100ng/ml of phorbol myristate acetate (100ng/ml; Sigma-Aldrich) and Ionomycin (100μg/ml; Sigma-Aldrich) in the presence of 1 μg/ml Brefeldin A (Golgi Plug; BD Biosciences) for 4 h at 37°C in 5% CO2. LPMC were collected and measurement of proliferating Th cells determined using multi-color flow cytometry. LP CD4 T cells were identified using the following antibodies: CD3 (PerCpCy5.5, TONBO, Clone: OKT3), CD4 (APC, Biolegend, Clone: RPA-T4) and CD8 (PE-Dazzle, TONBO, Clone: RPA-T8) and viability dye. Cells were fixed using Medium A (Invitrogen, Carlsbad, CA) and permeabilized using Medium B (Invitrogen). Intracellular cytokine staining was then used to characterize Th subsets[85, 86]. The following antibodies were used IL-17 (V450, eBiosciences, CLONE: N49–653), IFNγ (AF700 BD, clone: B27), and IL-22 (PE-Cy7, Biolegend, clone:22URTI) Matched isotypes were used to determine positive staining and CFSE labeling to determine proliferation. Th subsets were characterized as follows: Th17 were any LP CD4 T cell producing IL-17, Th1 were LP CD4 T cells that produced IFNγ and could also produce IL-22, Th22 were LP CD4 T cells that only produced IL-22.
Quantification of secreted cytokine production:
Levels of IL-17 and IFNγ in LPMC culture supernatants were quantified using ELISAs (ThermoFisher). Assays were carried out according as per manufacturers protocols (Analytical sensitivity: IL-17 4pg/ml, IFNγ 4pg/ml).
Studies to understand butyrate’s mechanism of action
Quantification of histone H3 acetylation:
To determine Histone H3 lysine 9 (H3K9) acetylation levels by flow cytometry, LPMC were cultured with or without butyrate (0.5 mM) and TCR activating beads and harvested after 24 hours of culture. LP CD4 T cells were identified using the following antibodies: CD3 (PerCpCy5.5, TONBO, Clone: OKT3), CD4 (APC, Biolegend, Clone: RPA-T4) and CD8 (PE-Dazzle, TONBO, Clone: RPA-T8) and viability dye. Cells were next fixed using Medium A and permeabilized using Medium B (Invitrogen) and stained according to the protocol outlined in [87] for acH3K9 (Millipore) with a Donkey anti-rabbit IgG secondary antibody used (AF488, Biolegend) to determine acH3 expression. Expression levels of acH3 were determined by LP CD4 T cell mean fluorescence intensity (MFI) with control secondary antibody only values removed.
H3K9 protein levels were also assessed using western blot. LP CD4 T cells were purified as detailed above and cultured in the presence of butyrate at 0.125 and 0.5 mM for 24 hours. Protein was extracted using Tris lysis buffer in the presence of a protease inhibitor (Roche). Total protein was quantified for normalization using a Pierce BCA protein assay kit and lysates were solubilized in Laemmli Sample Buffer (Bio-Rad) and were loaded into a 4–20% gradient Criterion Gel (Bio-Rad) and then subsequently transferred to PVDF membranes (Invitrogen). The following antibodies were used: anti–β-actin (conc: 1:50,000, Abcam) and anti-acH3K9 (conc: 1:10,000, Rabbit Polyclonal, Millipore), membranes were blocked with 5% skim milk for 2 hrs. Peroxidase goat anti-rabbit IgG and peroxidase goat anti-mouse IgG were used according to manufactures recommendations. Blots were visualized using a Chemidoc XRS+ system (Bio-Rad), and expression levels were quantified using ImageJ software[88].
Small molecule HDACi assay:
Trichostatin A, Apicidin, MGCD, and ITF2357 were diluted to working stocks of 1000 nM in culture media and serially diluted at the time of assay set up to concentrations indicated in figure legends. Apicidin, MGCD, and ITF2357 were kindly provided by Dr. Timothy McKinsey (University of Colorado Anschutz Medical Campus). The impact of HDACi on TCR-mediated LP CD4 T cell activation and proliferation was determined by examination of changes in LP CD4 T cell expression of activation markers (HLA-DR+CD38+ and CD25+) and Proliferation (CFSEdim).
GPR expression:
GPR43 and GPR109a expression levels on LP CD4 T cells were determined ex vivo and after four days of culture. LP CD4 T cells were cultured in the presence or absence of TCR activation and butyrate (0.5mM). LP CD4 T cell flow staining was determined by CD45 (PerCp-Cy5.5, eBiosciences, Clone: 2d1), CD3 (PE-Dazzle, Biolegend, Clone UCHT1), CD4 (VF450, TONBO, Clone: RPA-T4), CD8 (PE-Dazzle, TONBO, Clone: RPA-T8) and viability dye. Cells were also stained for GPR109a (PE, R&D, clone: 245106) and GPR43 (AF647, Bioss Antibodies, clone: polyclonal) expression. GPR109a expression was also measured on IEMC using the following panel: CD45 (PerCp-Cy5.5; Invitrogen, Clone: 2D1), CD3 (PE-Dazzle, Biolegend, Clone UCHT1), CD19 (PECy5, BD Biosciences, Clone HIB19, HLA-DR (APC-Cy7, BioLegend, Clone: L243) and viability dye. Isotype controls were used to determine positive staining.
GPR43 and GPR109a agonist studies:
MK1903 (GPR109a agonist, Tocris Bioscience)) and 4-CMTB (GPR43 agonist, Tocris Bioscience)) were reconstituted in DMSO per the manufacturer’s instructions and working stocks (100mM) stored at 4 degrees C. Working stocks were serially diluted in culture media and added to CFSE-labeled purified LP CD4 T cells and proliferation in response to TCR-bead stimulation measured after 4 days as detailed above.
Flow cytometry Acquisition and Analysis
All flow cytometry data were acquired on an LSRII flow cytometer (BD Biosciences) and analyzed using FlowJo version 10. Cytometer Setup and Tracking feature using BD FACSDIVA software (V6.1) and routine quality control were performed daily as previously described [72].
Statistical analysis
Statistical analysis and graphs were generated using GraphPad Prism version 6 for Windows. Parametric two-tailed t-tests were performed to examine differences between groups of matched paired data as described in the figure legends. All assays had a minimum of N=3 samples and significance levels were set to p<0.05.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the National Institutes of Health R01 AI108404 (to C. W.) and the Colorado Clinical and Translational Sciences Institute Grant TL1 TR002533 (to J.K.). We thank Dr. Timothy McKinsey (University of Colorado Anschutz Medical Campus) for providing Apicidin, MGCD, TSA and ITF2357. We thank the University of Colorado Cancer Center for cell flow sorting services (P30CA046934). We would also like to thank Moriah Castleman, Allison Christians, Jay Liu, and Sabrina Nesladek for assistance with tissue processing.
This work was funding by National Institutes of Health Grant Number R01 AI108404 (CW) and CCTSI TL1 (JK)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
ADDITIONAL INFORMATION
The authors declare no competing interests.
Disclosure: The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
DATA AVAILABILITY
The data generated during this study are available from the corresponding author on request.
REFERENCES
- 1.Parada Venegas D, et al. , Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front Immunol, 2019. 10: p. 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kayama H, Okumura R, and Takeda K, Interaction Between the Microbiota, Epithelia, and Immune Cells in the Intestine. Annu Rev Immunol, 2020. 38: p. 23–48. [DOI] [PubMed] [Google Scholar]
- 3.Kim MH, et al. , Short-chain fatty acids activate GPR41 and GPR43 on intestinal epithelial cells to promote inflammatory responses in mice. Gastroenterology, 2013. 145(2): p. 396–406e1–10. [DOI] [PubMed] [Google Scholar]
- 4.Hamer HM, et al. , Review article: the role of butyrate on colonic function. Aliment Pharmacol Ther, 2008. 27(2): p. 104–19. [DOI] [PubMed] [Google Scholar]
- 5.Davie JR, Inhibition of histone deacetylase activity by butyrate. J Nutr, 2003. 133(7 Suppl): p. 2485S–2493S. [DOI] [PubMed] [Google Scholar]
- 6.McHardy IH, et al. , HIV Infection is associated with compositional and functional shifts in the rectal mucosal microbiota. Microbiome, 2013. 1(1): p. 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mutlu EA, et al. , A compositional look at the human gastrointestinal microbiome and immune activation parameters in HIV infected subjects. PLoS Pathog, 2014. 10(2): p. e1003829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun Y, et al. , Fecal bacterial microbiome diversity in chronic HIV-infected patients in China. Emerg Microbes Infect, 2016. 5: p. e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dillon SM, et al. , Low abundance of colonic butyrate-producing bacteria in HIV infection is associated with microbial translocation and immune activation. AIDS, 2017. 31(4): p. 511–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Machiels K, et al. , A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut, 2014. 63(8): p. 1275–83. [DOI] [PubMed] [Google Scholar]
- 11.Takahashi K, et al. , Reduced Abundance of Butyrate-Producing Bacteria Species in the Fecal Microbial Community in Crohn’s Disease. Digestion, 2016. 93(1): p. 59–65. [DOI] [PubMed] [Google Scholar]
- 12.Serrano-Villar S, et al. , The effects of prebiotics on microbial dysbiosis, butyrate production and immunity in HIV-infected subjects. Mucosal Immunol, 2017. 10(5): p. 1279–1293. [DOI] [PubMed] [Google Scholar]
- 13.Schmidt A, et al. , Comparative Analysis of Protocols to Induce Human CD4+Foxp3+ Regulatory T Cells by Combinations of IL-2, TGF-beta, Retinoic Acid, Rapamycin and Butyrate. PLoS One, 2016. 11(2): p. e0148474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun M, et al. , Microbiota-derived short-chain fatty acids promote Th1 cell IL-10 production to maintain intestinal homeostasis. Nat Commun, 2018. 9(1): p. 3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Asarat M, et al. , Short-Chain Fatty Acids Regulate Cytokines and Th17/Treg Cells in Human Peripheral Blood Mononuclear Cells in vitro. Immunol Invest, 2016. 45(3): p. 205–22. [DOI] [PubMed] [Google Scholar]
- 16.Salkowska A, et al. , Differentiation stage-specific effect of histone deacetylase inhibitors on the expression of RORgammaT in human lymphocytes. J Leukoc Biol, 2017. 102(6): p. 1487–1495. [DOI] [PubMed] [Google Scholar]
- 17.Booth JS, et al. , Characterization and functional properties of gastric tissue-resident memory T cells from children, adults, and the elderly. Front Immunol, 2014. 5: p. 294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumar BV, et al. , Human Tissue-Resident Memory T Cells Are Defined by Core Transcriptional and Functional Signatures in Lymphoid and Mucosal Sites. Cell Rep, 2017. 20(12): p. 2921–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schluns KS and Klonowski KD, Protecting the borders: tissue-resident memory T cells on the front line. Front Immunol, 2015. 6: p. 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nastasi C, et al. , The effect of short-chain fatty acids on human monocyte-derived dendritic cells. Sci Rep, 2015. 5: p. 16148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nascimento CR, et al. , The short chain fatty acid sodium butyrate regulates the induction of CD1a in developing dendritic cells. Immunobiology, 2011. 216(3): p. 275–84. [DOI] [PubMed] [Google Scholar]
- 22.Kaisar MMM, et al. , Butyrate Conditions Human Dendritic Cells to Prime Type 1 Regulatory T Cells via both Histone Deacetylase Inhibition and G Protein-Coupled Receptor 109A Signaling. Front Immunol, 2017. 8: p. 1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berndt BE, et al. , Butyrate increases IL-23 production by stimulated dendritic cells. Am J Physiol Gastrointest Liver Physiol, 2012. 303(12): p. G1384–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cummings JH, et al. , Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut, 1987. 28(10): p. 1221–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bloemen JG, et al. , Short chain fatty acids exchange across the gut and liver in humans measured at surgery. Clin Nutr, 2009. 28(6): p. 657–61. [DOI] [PubMed] [Google Scholar]
- 26.Arpaia N, et al. , Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature, 2013. 504(7480): p. 451–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Furusawa Y, et al. , Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature, 2013. 504(7480): p. 446–50. [DOI] [PubMed] [Google Scholar]
- 28.Smith PM, et al. , The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science, 2013. 341(6145): p. 569–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baecher-Allan C, Viglietta V, and Hafler DA, Human CD4+CD25+ regulatory T cells. Semin Immunol, 2004. 16(2): p. 89–98. [DOI] [PubMed] [Google Scholar]
- 30.Guglani L and Khader SA, Th17 cytokines in mucosal immunity and inflammation. Curr Opin HIV AIDS, 2010. 5(2): p. 120–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shale M, Schiering C, and Powrie F, CD4(+) T-cell subsets in intestinal inflammation. Immunol Rev, 2013. 252(1): p. 164–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rada-Iglesias A, et al. , Butyrate mediates decrease of histone acetylation centered on transcription start sites and down-regulation of associated genes. Genome Res, 2007. 17(6): p. 708–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luu M, et al. , Regulation of the effector function of CD8(+) T cells by gut microbiota-derived metabolite butyrate. Sci Rep, 2018. 8(1): p. 14430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fontenelle B and Gilbert KM, n-Butyrate anergized effector CD4+ T cells independent of regulatory T cell generation or activity. Scand J Immunol, 2012. 76(5): p. 457–63. [DOI] [PubMed] [Google Scholar]
- 35.Bolduc JF, et al. , Epigenetic Metabolite Acetate Inhibits Class I/II Histone Deacetylases, Promotes Histone Acetylation, and Increases HIV-1 Integration in CD4(+) T Cells. J Virol, 2017. 91(16). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thiagalingam S, et al. , Histone deacetylases: unique players in shaping the epigenetic histone code. Ann N Y Acad Sci, 2003. 983: p. 84–100. [DOI] [PubMed] [Google Scholar]
- 37.Park J, et al. , Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR-S6K pathway. Mucosal Immunol, 2015. 8(1): p. 80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Singh N, et al. , Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity, 2014. 40(1): p. 128–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ganapathy V, et al. , Transporters and receptors for short-chain fatty acids as the molecular link between colonic bacteria and the host. Curr Opin Pharmacol, 2013. 13(6): p. 869–74. [DOI] [PubMed] [Google Scholar]
- 40.Aoyama M, Kotani J, and Usami M, Butyrate and propionate induced activated or non-activated neutrophil apoptosis via HDAC inhibitor activity but without activating GPR-41/GPR-43 pathways. Nutrition, 2010. 26(6): p. 653–61. [DOI] [PubMed] [Google Scholar]
- 41.Zandi-Nejad K, et al. , The role of HCA2 (GPR109A) in regulating macrophage function. FASEB J, 2013. 27(11): p. 4366–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thangaraju M, et al. , GPR109A is a G-protein-coupled receptor for the bacterial fermentation product butyrate and functions as a tumor suppressor in colon. Cancer Res, 2009. 69(7): p. 2826–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakajima A, et al. , The short chain fatty acid receptor GPR43 regulates inflammatory signals in adipose tissue M2-type macrophages. PLoS One, 2017. 12(7): p. e0179696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.D’Souza WN, et al. , Differing roles for short chain fatty acids and GPR43 agonism in the regulation of intestinal barrier function and immune responses. PLoS One, 2017. 12(7): p. e0180190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ji J, et al. , Microbial metabolite butyrate facilitates M2 macrophage polarization and function. Sci Rep, 2016. 6: p. 24838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Howe R, et al. , Evidence for dendritic cell-dependent CD4(+) T helper-1 type responses to commensal bacteria in normal human intestinal lamina propria. Clin Immunol, 2009. 131(2): p. 317–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ahluwalia B, Magnusson MK, and Ohman L, Mucosal immune system of the gastrointestinal tract: maintaining balance between the good and the bad. Scand J Gastroenterol, 2017. 52(11): p. 1185–1193. [DOI] [PubMed] [Google Scholar]
- 48.Panea C, et al. , Intestinal Monocyte-Derived Macrophages Control Commensal-Specific Th17 Responses. Cell Rep, 2015. 12(8): p. 1314–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vieira EL, et al. , Oral administration of sodium butyrate attenuates inflammation and mucosal lesion in experimental acute ulcerative colitis. J Nutr Biochem, 2012. 23(5): p. 430–6. [DOI] [PubMed] [Google Scholar]
- 50.Li Z, McCafferty KJ, and Judd RL, Role of HCA2 in Regulating Intestinal Homeostasis and Suppressing Colon Carcinogenesis. Front Immunol, 2021. 12: p. 606384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dillon SM, et al. , An altered intestinal mucosal microbiome in HIV-1 infection is associated with mucosal and systemic immune activation and endotoxemia. Mucosal Immunol, 2014. 7(4): p. 983–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhuang X, et al. , Systematic Review and Meta-analysis: Short-Chain Fatty Acid Characterization in Patients With Inflammatory Bowel Disease. Inflamm Bowel Dis, 2019. 25(11): p. 1751–1763. [DOI] [PubMed] [Google Scholar]
- 53.Zhang M, et al. , Butyrate inhibits interleukin-17 and generates Tregs to ameliorate colorectal colitis in rats. BMC Gastroenterol, 2016. 16(1): p. 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Wijk F and Cheroutre H, Mucosal T cells in gut homeostasis and inflammation. Expert Rev Clin Immunol, 2010. 6(4): p. 559–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cushing K, Alvarado DM, and Ciorba MA, Butyrate and Mucosal Inflammation: New Scientific Evidence Supports Clinical Observation. Clin Transl Gastroenterol, 2015. 6: p. e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kespohl M, et al. , The Microbial Metabolite Butyrate Induces Expression of Th1-Associated Factors in CD4(+) T Cells. Front Immunol, 2017. 8: p. 1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lecona E, et al. , Kinetic analysis of butyrate transport in human colon adenocarcinoma cells reveals two different carrier-mediated mechanisms. Biochem J, 2008. 409(1): p. 311–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang H, et al. , CD147 modulates the differentiation of T-helper 17 cells in patients with rheumatoid arthritis. APMIS, 2017. 125(1): p. 24–31. [DOI] [PubMed] [Google Scholar]
- 59.Miao J, et al. , CD147 Expressed on Memory CD4(+) T Cells Limits Th17 Responses in Patients With Rheumatoid Arthritis. Front Immunol, 2020. 11: p. 545980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dillon SM, et al. , Age-related alterations in human gut CD4 T cell phenotype, T helper cell frequencies, and functional responses to enteric bacteria. J Leukoc Biol, 2020. 107(1): p. 119–132. [DOI] [PubMed] [Google Scholar]
- 61.Hegazy AN, et al. , Circulating and Tissue-Resident CD4(+) T Cells With Reactivity to Intestinal Microbiota Are Abundant in Healthy Individuals and Function Is Altered During Inflammation. Gastroenterology, 2017. 153(5): p. 1320–1337e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cao T, et al. , The epigenetic modification during the induction of Foxp3 with sodium butyrate. Immunopharmacol Immunotoxicol, 2018: p. 1–10. [DOI] [PubMed]
- 63.Luu M and Visekruna A, Short-chain fatty acids: Bacterial messengers modulating the immunometabolism of T cells. Eur J Immunol, 2019. 49(6): p. 842–848. [DOI] [PubMed] [Google Scholar]
- 64.Wachtershauser A, Loitsch SM, and Stein J, PPAR-gamma is selectively upregulated in Caco-2 cells by butyrate. Biochem Biophys Res Commun, 2000. 272(2): p. 380–5. [DOI] [PubMed] [Google Scholar]
- 65.Korecka A, et al. , ANGPTL4 expression induced by butyrate and rosiglitazone in human intestinal epithelial cells utilizes independent pathways. Am J Physiol Gastrointest Liver Physiol, 2013. 304(11): p. G1025–37. [DOI] [PubMed] [Google Scholar]
- 66.Byndloss MX, et al. , Microbiota-activated PPAR-gamma signaling inhibits dysbiotic Enterobacteriaceae expansion. Science, 2017. 357(6351): p. 570–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Das B, et al. , Short chain fatty acids potently induce latent HIV-1 in T-cells by activating P-TEFb and multiple histone modifications . Virology, 2015. 474: p. 65–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Siavoshian S, et al. , Butyrate stimulates cyclin D and p21 and inhibits cyclin-dependent kinase 2 expression in HT-29 colonic epithelial cells. Biochem Biophys Res Commun, 1997. 232(1): p. 169–72. [DOI] [PubMed] [Google Scholar]
- 69.Eeckhaut V, et al. , Butyricicoccus pullicaecorum in inflammatory bowel disease. Gut, 2013. 62(12): p. 1745–52. [DOI] [PubMed] [Google Scholar]
- 70.Steinhart AH, et al. , Treatment of left-sided ulcerative colitis with butyrate enemas: a controlled trial. Aliment Pharmacol Ther, 1996. 10(5): p. 729–36. [DOI] [PubMed] [Google Scholar]
- 71.Scheppach W, et al. , Effect of butyrate enemas on the colonic mucosa in distal ulcerative colitis. Gastroenterology, 1992. 103(1): p. 51–6. [DOI] [PubMed] [Google Scholar]
- 72.Dillon SM, et al. , HIV-1 infection of human intestinal lamina propria CD4+ T cells in vitro is enhanced by exposure to commensal Escherichia coli. J Immunol, 2012. 189(2): p. 885–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dinh DM, et al. , Intestinal microbiota, microbial translocation, and systemic inflammation in chronic HIV infection. J Infect Dis, 2015. 211(1): p. 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dillon SM, et al. , Enhancement of HIV-1 infection and intestinal CD4+ T cell depletion ex vivo by gut microbes altered during chronic HIV-1 infection. Retrovirology, 2016. 13: p. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dillon SM, et al. , Gut dendritic cell activation links an altered colonic microbiome to mucosal and systemic T-cell activation in untreated HIV-1 infection. Mucosal Immunol, 2016. 9(1): p. 24–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Harper MS, et al. , Interferon-alpha Subtypes in an Ex Vivo Model of Acute HIV-1 Infection: Expression, Potency and Effector Mechanisms. PLoS Pathog, 2015. 11(11): p. e1005254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yoder AC, et al. , The transcriptome of HIV-1 infected intestinal CD4+ T cells exposed to enteric bacteria. PLoS Pathog, 2017. 13(2): p. e1006226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dillon SM, et al. , Quantifying HIV-1-Mediated Gut CD4(+) T Cell Deathin the Lamina Propria Aggregate Culture (LPAC) Model. Bio Protoc, 2020. 10(2): p. e3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Steele AK, et al. , Contribution of intestinal barrier damage, microbial translocation and HIV-1 infection status to an inflammaging signature. PLoS One, 2014. 9(5): p. e97171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dillon SM, et al. , The natural killer cell interferon-gamma response to bacteria is diminished in untreated HIV-1 infection and defects persist despite viral suppression. J Acquir Immune Defic Syndr, 2014. 65(3): p. 259–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Manuzak JA, et al. , Increased Escherichia coli-induced interleukin-23 production by CD16+ monocytes correlates with systemic immune activation in untreated HIV-1-infected individuals. J Virol, 2013. 87(24): p. 13252–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Manuzak J, Dillon S, and Wilson C, Differential interleukin-10 (IL-10) and IL-23 production by human blood monocytes and dendritic cells in response to commensal enteric bacteria. Clin Vaccine Immunol, 2012. 19(8): p. 1207–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dillon SM, et al. , Blood myeloid dendritic cells from HIV-1-infected individuals display a proapoptotic profile characterized by decreased Bcl-2 levels and by caspase-3+ frequencies that are associated with levels of plasma viremia and T cell activation in an exploratory study. J Virol, 2011. 85(1): p. 397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dillon SM, et al. , Plasmacytoid and myeloid dendritic cells with a partial activation phenotype accumulate in lymphoid tissue during asymptomatic chronic HIV-1 infection. J Acquir Immune Defic Syndr, 2008. 48(1): p. 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wacleche VS, et al. , New insights into the heterogeneity of Th17 subsets contributing to HIV-1 persistence during antiretroviral therapy. Retrovirology, 2016. 13(1): p. 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Eyerich S, et al. , Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J Clin Invest, 2009. 119(12): p. 3573–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rigby L, et al. , Methods for the analysis of histone H3 and H4 acetylation in blood. Epigenetics, 2012. 7(8): p. 875–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schneider CA, Rasband WS, and Eliceiri KW, NIH Image to ImageJ: 25 years of image analysis. Nat Methods, 2012. 9(7): p. 671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated during this study are available from the corresponding author on request.