

Abstract
Dendritic cells (DCs) rely on glycolysis for their energy needs to induce pro-inflammatory antigen-specific immune responses. Therefore, inhibiting DC glycolysis, while presenting the self-antigen, may prevent pro-inflammatory antigen-specific immune responses. Previously we demonstrated that microparticles with alpha-ketoglutarate (aKG) in the polymer backbone (paKG MPs) were able to generate anti-inflammatory DCs by sustained delivery of the aKG metabolite, and by modulating energy metabolism of DCs. Herein, we demonstrate that paKG MPs-based delivery of a glycolytic inhibitor, PFK15, using paKG MPs induces anti-inflammatory DCs (CD86LoMHCII+) by down-regulating glycolysis, CD86, tnf and IL-6 genes, while upregulating oxidative phosphorylation (OXPHOS) and mitochondrial genes. Furthermore, paKG MPs delivering PFK15 and a self-antigen, collagen type II (bc2), in vivo, in a collagen-induced autoimmune arthritis (CIA) mouse model, normalized paw inflammation and arthritis score, by generating antigen-specific immune responses. Specifically, these formulations were able to reduce activation of DCs in draining lymph nodes and impressively generated proliferating bc2-specific anti-inflammatory regulatory T cells in joint-associated popliteal lymph nodes. These data strongly suggest that sustained glycolytic inhibition of DCs in the presence of an antigen can induce antigen-specific immunosuppressive responses, therefore, generating a technology that can be applicable for treating autoimmune diseases.
Keywords: immunoengineering, biomaterials, autoimmune diseases, drug delivery, immunometabolism
INTRODUCTION
While the etiology of several autoimmune diseases, including rheumatoid arthritis (RA) is unknown and the pathological mechanisms that drive the disease can differ between affected individuals, in all cases pro-inflammatory dendritic cells (DCs) and self-reactive effector T lymphocytes (e.g. T helper 1 (Th1) and Th17 cells) are implicated in disease pathogenesis[1,2]. Although DCs maintain adaptive immune responses by presenting antigens to T cells for their activation and differentiation, in autoimmunity, pro-inflammatory DCs develop and trigger autoimmune responses by presenting and priming effector T cells to self-antigens[3–5]. In contrast, the presentation of a self-antigen by DCs with an anti-inflammatory phenotype can lead to the expansion of antigen-specific T cells, specifically regulatory T cells (TREGs – anti-inflammatory cell)[6–8]. Studies suggest that imperfections in central and/or peripheral tolerance driven by DC-based responses, are one of the leading causes in the failure of suppressing self-reactive effector T lymphocytes, and development of autoimmunity[3,9,10]. Therefore, DCs illustrate a prime immune cell to target for autoimmune therapeutic intervention[8].
Furthermore, pro-inflammatory DCs, which have increased costimulatory CD86 expression for the activation of naïve T cells, can infiltrate inflamed autoimmune tissues or lymph nodes (LNs) to secrete pro-inflammatory cytokines (e.g. TNFα and IL-6) for the differentiation of T cells into pro-inflammatory Th1 and Th17 cells, which are implicated in the pathogenesis of autoimmune diseases[1,2,11]. Notably, pro-inflammatory immune cells, such as DCs (CD11c+CD86HiMHCII+) utilize glycolysis for cellular energy. Interestingly, increased glycolytic activity in autoimmune tissues have been associated to the progression of a number of autoimmune diseases such as RA, systemic lupus erythematosus (SLE), multiple sclerosis (MS) and vasculitis[12–15]. Pro-inflammatory DCs require a significant amount of energy when mounting an inflammatory response against an antigen (e.g. collagen in the case of RA), and therefore, utilize glycolysis for a higher rate of adenosine triphosphate (ATP) synthesis[10,16,17]. On the contrary, anti-inflammatory DCs (CD11c+CD86LoMHCII+) rely on the Krebs cycle and oxidative phosphorylation (OXPHOS) for cellular energy[10,16,18]. Therefore, shifting energy production in pro-inflammatory DCs from glycolysis to the Krebs cycle by reducing glycolysis may prevent pro-inflammatory cellular phenotypes and subsequently reduce autoimmune symptoms. Although the systemic delivery of glycolytic inhibitors may prevent pro-inflammatory phenotypes, systemic inhibition of glycolysis can lead to global and non-specific glycolytic inhibition[19,20]. Interestingly, the glycolytic enzyme 6-Phosphofructo-2-Kinase/Fructose-2,6-biphosphatase 3 (PFKFB3), which regulates the glycolytic rate in several immune cells that are responsible for the progression of RA, such as DCs and fibroblast-like synoviocytes (FLSs), represents a novel target for glycolytic inhibition[12,21]. The upregulation of the PFKFB3 enzyme has been observed and is correlated with synovial inflammation in RA by promoting the destructive behavior of RA FLSs[12]. However the systemic delivery of the transient glycolytic inhibitor, PFK15 (inhibits PFKFB3 enzyme), decreased the chemotactic migration of RA FLSs in a RA collagen-induced arthritis (CIA) mouse model[12]. Additionally, it has been demonstrated in a human rhabdomyosarcoma (RD) cancerous cell line that the modulation of glucose metabolism can be achieved by inhibiting PFKFB3 in cancer cells when delivering PFK158 (an analog of PFK15)[22]. Therefore, targeted inhibition of PFKFB3 in immune cells, such as DCs, may be beneficial in decreasing inflammation and reducing global and toxic effects, while generating antigen-specific responses. However, the effect of PFK15 on DCs has yet to be elucidated.
The metabolic reprogramming of pro-inflammatory DCs, using a transient glycolytic inhibitor may partially block glycolysis while allowing the Krebs cycle to proceed, in turn, this may induce anti-inflammatory DC phenotypes with lowering of co-stimulatory CD86 expression, while producing anti-inflammatory cytokines for the induction of CD4+ TREGs[1–3]. In fact, antigen-specific DC immunotherapies, using adoptive-transfer in mice, has shown to be an effective approach in tolerizing self-reactive antigen-specific T cells, while permitting the host’s immune system to identify other foreign antigens[23,24]. However, adoptive transfer can be a challenge to translate efficiently, economically and safely (particularly when the transferred cells originate from another individual, in turn risking the infliction of graft versus host disease)[25–28]. Thus, modulating T cell responses by modulating the individuals own DC function in vivo, may be a more viable therapeutic approach for treating autoimmune diseases. However, to prevent non-specific immune suppression and optimize the ability to deliver content to DCs, a biodegradable carrier with encapsulating abilities would be needed to deliver the glycolytic inhibitor and self-antigen[29–33]. Remarkably microparticles (MPs) can be synthesized from the immunosuppressive Krebs cycle metabolite, alpha-ketoglutarate (aKG)[34], which, as the MP degrades, aKG can fuel the Krebs cycle while simultaneously delivering its encapsulated contents within DCs[35]. It is important to note that aKG can regulate cell function, metabolism and growth. In fact, aKG controls amino acid production, functions as a reactive oxygen species (ROS) scavenger, regulates G protein function (therefore acting as a signaling molecule), and given that aKG is a homologue of glutamine, it impedes protein catabolism, enhances nitrogen retention, and can operate as an immunomodulatory molecule[36,37]. Additionally, aKG can promote collagen genesis by regulating the stability of hypoxia-inducible factor (HIF)-1 and increasing the expression of prolyl hydroxylases (PHDs)[34,36,38]. Furthermore, aKG has shown to have prophylactic benefit in murine colorectal cancer by preventing intestinal inflammation via the NF-κB pathway[39]. The metabolic function of aKG in cells and organisms creates numerous therapeutic interventions for remedying diseases. Therefore, modulating the level of the aKG metabolite within pro-inflammatory DCs may alter synovial tissue cell responses and restore homeostasis in the synovium for the treatment of RA.
Although standard immunosuppressants used to treat RA, such as, methotrexate (MTX), and sulfasalazine, have modulatory effects on immunometabolism via folate metabolism[40–43], these immunosuppressants generate non-specific and systemic immunosuppression. However, antigen-specific therapies show promise in altering antigen-specific adaptive immune responses and therefore may limit toxic global effects[44]. Moreover, subcutaneous delivery of MPs form an injection site that has shown to attract antigen presenting cells (APCs), such as DCs[45,46], for phagocytosis and for a potential subsequent modulation of the adaptive immune responses. Therefore, the delivery of MPs creates a platform for targeting immune cell types that are known to play a substantial role in autoimmunity.
Herein, we engineered immunosuppressive poly aKG (paKG) MPs to concurrently deliver the glycolytic inhibitor, PFK15, and the CIA specific antigen, bc2 (bovine collagen type II)[47], to CIA mice. It is hypothesized that as the paKG MPs degrade, the delivery of bc2 may permit DCs to present the antigen, while the delivery of PFK15 may reduce glycolysis and CD86 expression in pro-inflammatory DCs. Additionally, the intracellular delivery of PFK15 and aKG within DCs may act together to fulfill the Krebs cycle-mediated cellular energy requirements, while potentially reducing pro-inflammatory glycolysis-mediated cellular energy requirements for the induction of anti-inflammatory DCs. Furthermore, this induction of anti-inflammatory DCs may lead to an induction of suppressive antigen-specific T cell responses. This study demonstrates that the metabolic reprogramming of DCs, in the presence of an antigen, generates anti-inflammatory DC and T cell responses for the treatment of arthritis symptoms in CIA mice. Therefore, this MP technology may be applicable to treating autoimmune diseases with a similar pathogenesis to RA.
MATERIALS AND METHODS
Fabrication of polymers
paKG polymers were generated using previously reported methodologies[35]. Briefly, a 1:1 mole ratio of ketoglutaric acid and 1,10-decanediol were added in a round-bottom flask. Under vacuum, the contents were stirred at 130 °C for 2 hrs in the presence of SnCl2 as the catalyst. The synthesized polymer was then precipitated and washed in methanol. The residual methanol was then evaporated using a rotary evaporator.
paKG, paKG(PFK15), and paKG(PFK15+bc2) microparticle synthesis
Microparticles were generated using previously reported water-oil-water emulsion techniques[48–50]. Briefly, 50 mg of paKG polymers were dissolved in 1 mL of dichloromethane (DCM) (VWR, Radnor, PA). Microparticles consisting of PFK15 (Selleckchem, Houston, TX), included 1mg of PFK15 in the paKG and DCM mixture. This mixture was then combined with 10 mL of 2% polyvinyl alcohol (PVA) (Acros Organics, Fairlawn, NJ) in DIH2O and homogenized at 30,000 rpm with a handheld homogenizer for 2 min.
In order to generate microparticles that consisted of bc2, 4mg of soluble bc2 (in 250μl acetic acid) (Chondrex, Redmond, WA) was added to the paKG dissolved in DCM, and sonicated for 1 min. The emulsion thus generated was then combined with 50 mL of 1 % PVA (and stirred at 400 rpm for 3 hrs to evaporate DCM. The formed particles were centrifuged at 2000 × Gs for 5 min (Eppendorf, Hauppauge, NY). The supernatant was discarded and resuspended in DIH2O. This was repeated 3 times to remove any residual PVA. These particles were then resuspended in 5 ml of DIH2O, placed in −80 °C for a minimum of 2 hrs, and lyophilized for 48 hrs. The microparticles were stored at −20 °C and used for subsequent experiments.
Microparticle characterization
Images of the microparticle were obtained with scanning electron microscope XL30 Environmental FEG - FEI at Erying Materials Center at Arizona State University. Moreover, the size of the microparticles was determined by dynamic light scattering (Zetasizer Nano, Cambridge, UK).
PFK15 encapsulation and loading efficiency
To measure the amount of PFK15 that was loaded and encapsulated within the paKG MPs 10 mg of the particles was dissolved in 100 μL of DMSO. The absorbance of this solution was measured at 370 nm using absorbance spectrophotometer (plate reader SpectraMax M5, Molecular Devices, San Jose, CA). DMSO by itself and paKG particles not encapsulating PFK15 were used for background subtraction.
bc2 loading and encapsulation efficiency
To measure the amount of bc2 that was loaded and encapsulated within the paKG MPs 10 mg of the particles was dissolved in 500 μL of DCM. The bc2 was then extracted by adding 500 μL of PBS, vortexing vigorously, and collecting the aqueous phase. This step of extraction was repeated three times, and the PBS was combined together in an eppendorf tube. A bicinchoninic assay (BCA) assay (Pierce, Thermo Fisher Scientific, Waltham, MA) was utilized to measure the bc2 encapsulation and loading efficiency per the manufacturer’s instructions. The loading efficiency of each batch was equated to the mass of bc2 measured by absorbance per mass of MPs. The encapsulation efficiency of each batch of MPs was equivalent to the actual loading divided by theoretical bc2 loading multiplied by 100.
Release kinetics of PFK15 and degradation rate of paKG-based microparticles
Release kinetics of PFK15 was determined by incubating the MPs in 1 mL of 0.2% Tween 80 (made in 1x phosphate buffered saline (PBS – pH 7.4)). Triplicates of each sample were placed on a rotisserie rotator at 37 °C. The samples were centrifuged at 2000 × Gs for 5 min and 800 μL of the supernatant was carefully transferred to a new 1.5 mL centrifuge tube (Eppendorf, Hauppauge, NY) and stored in −80 °C until use. The displaced 800 μL in the original eppendorf tubes was replaced with 800 μL of new 0.2% Tween 80 buffer and placed on the rotisserie rotator at 37 °C. This was repeated on days 1–4, 6–8, and 10. The degradation rate of the MPs was observed by studying the release kinetics of aKG from each MP type. The release kinetics of aKG from paKG, paKG(bc2), paKG(PFK15), and paKG(PFK15+bc2) followed each of these steps, with the exemption of these MPs being incubated in 1x PBS (pH 7.4) rather than in 0.2% Tween 80 made in 1x PBS (pH 7.4).
The release of PFK15 and aKG from the microparticles over the 10 day time period was quantified by performing high-performance liquid chromatography (HPLC – Agilent Infinity II 1260, Santa Clara, CA). Specifically, for PFK15 release, a Gemini 18C column, 3 μm, 4.6 × 100 mm was utilized with the gradient mobile phase of 80:20 water:methanol, at 1 mL/min flow rate for 10 mins and the absorbance was performed at 330 nm. A standard curve of PFK15 in 0.2% tween 80 was generated and the area under the curve was calculated to quantify the concentration of the PFK15 released over a set time using the ChemStation analysis software as per manufacturer’s directions. The release of aKG was quantified in a mobile phase of water. A 50 μL of injection volume was utilized in a Gemini 18C column, 3 μm, 4.6 × 100 mm (Agilent Technologies, Santa Clara, CA). A flow rate of 1.2 mL/min for 10 mins was utilized and the absorbance was determined using a UV detector at 210 nm. The area under the curve was determined to quantify the concentration of aKG using the ChemStation analysis software as per manufacturer’s directions.
Release kinetics of bc2
Bc2 was conjugated to fluorescence isothiocyanate (FITC) and the FITC-labelled paKG(bc2) and paKG(PFK15+bc2) microparticles were synthesized as described under microparticle synthesis. Specifically, 4 mg of bc2 was incubated with 10 mg of FITC in 10X PBS at 60 °C for 3 hours. Next, bc2 conjugated to FITC (bc2-FITC) was separated from unconjugated FITC by precipitating bc2-FITC in acetone. Next, the precipitate was washed in 1 mL acetone 3 times by centrifuging at 1000 × Gs, and then lyophilized to remove any water content. This lyophilized bc2-FITC was then utilized to generate MPs.
The release kinetics of bc2-FITC from the MPs followed the method of PFK15 release kinetics, with the exemption of these bc2-FITC MPs being incubated in 1x PBS (pH 7.4) rather than 0.2% Tween 80 made in 1x PBS (pH 7.4). The release of the bc2-FITC from the MPs over the 10 day time period was analyzed by measuring fluorescence of the releasates at excitation 488 nm, emission 530 nm, using a SpectraMax M2/M2e microplate reader (Molecular Devices, San Jose, CA).
Dendritic cell isolation and culture
Hematopoietic stem cells (HSC) were isolated from the bone marrow of 6–8 week-old C57BL/6j mice in accordance with the institutional animal care and use committee (IACUC) of Arizona State University for the approved protocol of 19–1712R. Immature BMDCs were obtained from the isolated HSCs using a modified 10-day protocol[51–55]. Briefly, the femur and tibia were extracted from the mice and placed in a wash media that consisted of DMEM/F-12 (1:1) with L-glutamine (VWR, Radnor, PA), 10% fetal bovine serum (Atlanta Biologics, Flowery Branch, GA) and 1% penicillin-streptomycin (VWR, Radnor, PA). The bone marrow from the femur and tibia was flushed out with 10 mL wash media and pipetted to generate a homogeneous suspension. The homogenous suspension was then centrifuged at 300 × Gs for 5 mins and the supernatant was discarded. The cell pellet was then resuspended in 3 mL of 1x red blood cell (RBC) lysis buffer for 3 mins at 4°C to lyse red blood cells. The cell suspension was then centrifuged at 300 × Gs for 5 mins and was re-suspended in DMEM/F-12 with L-glutamine (VWR, Radnor, PA), 10% fetal bovine serum, 1% sodium pyruvate (VWR, Radnor, PA), 1% non-essential amino acids (VWR, Radnor, PA), 1% penicillin–streptomycin (VWR, Radnor, PA) and 20 ng/ml recombinant mouse GM-CSF (VWR, Radnor, PA) (DC media). The cell suspension was then transferred to a tissue culture-treated T-75 flask (day 0) and incubated in 37 °C, 5% CO2 incubator. On day 2 (48 hrs later), the floating cells from the flask were collected, centrifuged, re-suspended in fresh media and seeded in 6-well low attachment plates (VWR, Radnor, PA) for 6 days. Half of the media was replenished every other day. On day 8, the cells were lifted from the low attachment plates by gently pipetting and seeding the cells on 96 well round bottom tissue culture-treated polystyrene plates for 2 additional days before treating them (any cells remaining on the 6 well low attachment plates were resuspended in 20 mM EDTA (made in 1x PBS) and incubated at 37 °C for 10 mins to be seeded on the 96 well plates). On day 10, the cells were treated with either paKG MPs (0.1mg/mL), paKG(PFK15) MPs (0.1mg/mL), paKG(PFK15+bc2) MPs (0.1mg/mL), soluble aKG (0.1mg/mL), diol (0.1mg/mL), soluble bc2 (75 μg/mL), 0.1% DMSO, or PFK15 (200 nM). In order to test how each treatment group would respond to inflammation, 1 μg/mL of LPS was also added to each condition. No treatment and LPS alone were used as a control. The yield and purity of the DCs (CD11c, MHCII and CD86) were determined with immunofluorescence staining and flow cytometry.
Confocal microscopy
The modified 10-day protocol was followed to acquire mature BMDCs from HSCs[51–55]. On day 10, 100,000 cells were seeded on a glass slide within 24 well plates and were incubated for 24 hrs in 37°C. The cells were then treated with fluorescently labeled rhodamine-paKG MPs and the nucleus was stained with DAPI. Samples were imaged with a Nikon C2 laser scanning confocal microscope using a 60x, oil-immersion lens with numerical aperture of 1.4. DAPI and fluorescently labeled rhodamine-paKG MPs were excited with 405 nm and 561 nm lasers respectively, coupled with appropriate blue and red channel emission detection. Fluorescent channels were scanned sequentially and transmitted light from the 561 nm laser was used for differential interference contrast (DIC). Image dimensions were 1024 × 1024 pixels scanned with a digital zoom of 2x. Z-stacks were created in the same manner with a step size of 0.25 μm between optical slices. Cells treated with rhodamine-paKG MPs and untreated cells were used as negative imaging controls to identify the signal of interest. Laser intensity and detector gain were adjusted to eliminate background or autofluorescence and avoid pixel saturation. The Nikon software, Elements was used to adjust the intensity scale, create orthogonal views, and convert images to 8-bit TIFF format.
Mixed lymphocyte reactions
Spleen of 6–8-week-old BALB/c mice were isolated for mixed lymphocyte reaction (MLR) studies. Cells were extracted from the spleen by applying firm pressure from a pestle against a cell strainer. The effluent was centrifuged at 300 × Gs for 5 min. The supernatant was discarded and the splenocytes were then resuspended in 3 mL of 1x RBC lysis buffer for 3 min at 4 °C. The cells were then centrifuged at 300 × Gs for 5 min and the pellet was resuspended in DMEM/F-12 with L-glutamine (VWR, Radnor, PA), 10% fetal bovine serum, 1% sodium pyruvate (VWR, Radnor, PA), 1% non-essential amino acids (VWR, Radnor, PA), and 1% penicillin–streptomycin (VWR, Radnor, PA). After resuspension, the splenocytes were used in MLR and seeded with BMDCs from DBA/1j mice on day 10 of the BMDC isolation protocol[51–55], shortly after the treatment of the cells. The splenocyte to BMDC ratio was 20:1 for 48–72 hrs. No treatment was used as a control.
Metabolomic LC-MS studies
Bone marrow from the femur and tibia of C57BL/6j mice were isolated to derive BMDCs[51–55]. The cells were cultured in 6 well plates at 1 million cells per well. A concentration of 50 μg/well of paKG MPs, or paKG(PFK15) MPs were added to the wells, and no treatment was used as a control. Following 24 hrs of culture, the supernatant was discarded, and the cells were washed with 2 mL of 37 °C 1x PBS. Immediately following the PBS wash, 1 mL of 80:20 methanol:H2O (−80 °C) was added into the plates. The plates were then kept on dry ice to assist in metabolite extraction. The cells were then scraped with a cell scraper (VWR, Radnor, PA), and placed into eppendorf tubes. The samples were centrifuged at 16,000 × Gs for 5 min at 4 °C (Eppendorf, Hauppauge, NY, USA). The supernatant was transferred to a vial and completely dried using a speedvac concentrator. The pellets were used to quantify the total protein using Nanodrop 2000 (ThermoFisher Scientific, Waltham, MA, USA).
LC-MS/MS methods were performed using previous reported methodologies.[56–63] Agilent 1290 UPLC-6490 QQQ-MS (Santa Clara, CA) system was used to perform LC-MS. A total of 10 μL of the processed samples were injected twice. The injection of the processed samples was analyzed using the negative and positive ionization modes. Both chromatographic separations were performed in hydrophilic interaction chromatography (HILIC) mode on a Waters XBridge BEH Amide column (150 × 2.1 mm, 2.5 μm particle size, Waters Corporation, Milford, MA). The following settings were utilized in these studies: 0.3 mL/min for the flow rate, 4 °C for the auto-sampler temperature, and 40 °C for the column compartment. The mobile phase consisted of Solvents A - 10 mM ammonium acetate, 10 mM ammonium hydroxide in 95% H2O/5% ACN and Solvent B - 10 mM ammonium acetate, 10 mM ammonium hydroxide in 95% acetonitrile (ACN)/5% H2O. Following the initial 1 min isocratic elution of 90% of solvent B, the amount of Solvent B decreased by 40% at t=11 min. The make-up of Solvent B was consistently at 40% for 4 min (t=15 min) and then steadily increased to 90% for the next injection.
The mass spectrometer also has an electrospray ionization (ESI) source. Multiple-reaction-monitoring (MRM) mode was used for targeted data acquisition with a total of ~320 MRM transitions between negative and positive modes. Agilent Masshunter Workstation software (Santa Clara, CA) controlled the LC-MS system, and Agilent MassHunter Quantitative Data Analysis (Santa Clara, CA) was used to integrate the extracted MRM peaks.
Seahorse assay
The extracellular acidification rate (ECAR) and the oxygen consumption rate (OCR) of BMDCs were determined with the Seahorse Extracellular Flux XF-96) analyzer (Seahorse Bioscience, North Billerica, MA) as previously described[64]. Briefly, cells were seeded at 50,000 cells per well in Seahorse XF-96 plates and treated with 10 μg/well of paKG MPs, paKG(PFK15) MPs, or soluble aKG with or without 1 μg/mL of LPS. These conditions were cultured for 24 hrs prior to isolating the intracellular metabolites. The media was then removed and replaced with DMEM in the absence of glucose. Injections of D-glucose (10 mM), oligomycin (1 mM), and 2-deoxyglucose (100 mmol/L) were then performed. Glycolysis was quantified by the ECAR after the injection of D-glucose and the maximum glycolytic capacity was a measurement of the ECAR after the injection of oligomycin. The measurement of ECAR after the injection of 2-deoxyglucose represented non-glycolytic activity. There were 10 technical replicates of the samples.
For analysis, the paKG MPs and paKG(PFK15) MPs were normalized to protein amount and compared to the no treatment control.
Flow cytometry
All immunofluorescence antibodies were purchased and used as is (BD biosciences, Tonbo Biosciences, Biolegend, Thermo Scientific, Invitrogen). The 0.1% flow staining buffer was prepared by mixing 0.1% bovine serum albumin (VWR, Radnor, PA), 2mM Na2EDTA (VWR, Radnor, PA) and 0.01% NaN3(VWR, Radnor, PA) in DIH2O. Fixable dye eF780 (ThermoFisher Scientific, Waltham, MA, USA) was used for live/dead staining. Flow cytometry was completed by following the manufacturer’s guidance of the Attune NXT Flow cytometer (ThermoFisher Scientific, Waltham, MA, USA) in the Arizona State University’s flow cytometry core.
RNA Seq
BMDC’s were derived using the modified 10-day protocol[51–55] and were treated with paKG-based MPs on day 10. RNA was extracted using an RNA purification kit (Invitrogen, Thermo Fisher Scientific, Waltham, MA). RNA-seq reads for each sample were quality checked using FastQC v0.11.8 and aligned to the mouse genome build 38_89 (Ensembl_GRCm38_89) top level assembly using STAR v2.5.3a. A series of quality control metrics were generated on the STAR outputs. Stringtie-1.3.6 was used to report read counts, FPKM values (Fragments Per Kilobase of transcript per Million mapped reads) and TPM (Transcripts Per Million). Differential expression (DE) analysis was performed with EdgeR package from Bioconductor v3.2 in R 3.6.2. Multi-dimensional scaling (MDS) plot was drawn by plotMDS, in which distances correspond to leading log-fold-changes between samples. EdgeR applied an overdispersed Poisson model to account for variance among biological replicates. Empirical bayes tagwise dispersions are also estimated to moderate the overdispersion across transcripts. Then a negative bionomial generalized log-linear model was fit to the read counts for each gene for all comparison pairs. For each pairwise comparison, genes with false discovery rate (FDR) <0.05 were considered significant and log2-fold changes of expression between conditions (logFC) were reported. FDR was calculated following Benjamini & Hochberg (1995) procedure, the expected proportion of false discoveries amongst the rejected hypotheses. The false discovery rate is a less stringent condition than the family-wise error rate, so these methods are more powerful than the others.
Biodistribution studies of microparticles
paKG MPs were crosslinked with coumarin fluorescent dye (highly hydrophobic dye) and were injected subcutaneously in C57BL/6j (n=2) mice to observe the DC and macrophage infiltration in the injection site and the trafficking of the particles to the LNs. Mice were injected with 1 mg of paKG-coumarin particles near the hind legs. After 24 hrs, mice were sacrificed and the draining ING LNs, control cLNs, and injection sites were isolated and stained for CD11c, CD11b, and F4/80. Antibody staining was analyzed with flow cytometry.
CIA induction
The IACUC of Arizona State University approved animal studies regarding rheumatoid arthritis protocol number: 19–1712R. Eight to ten week old male DBA/1j (Jackson Laboratories) mice were injected with complete Freund’s adjuvant (CFA) + bc2 emulsion on day 0, and on day 21 booster. incomplete Freund’s adjuvant (IFA) + bc2 was injected one-third of the way down the tail. Mice were placed in a restrainer for CFA and IFA injections and tails were sanitized with alcohol swabs prior to the injections. Additionally, mice received an intraperitoneal injection of LPS on day 28 to synchronize and trigger the onset of the disease.
CIA measurements and treatment studies
Baseline weights, paw thickness measurements and paw photos (for arthritic scores) were obtained. Weights were captured 1–3 times a week to assure mice were within a healthy weight range. Paw thickness measurements and paw photos were collected 1–3 times a week starting from day 21 to the end of study. Paw thickness measurements were performed using Vernier calipers. Paw photos were obtained for arthritic scores with a scale of 0 to 6 where a score of 3 or higher represented moderate or severe arthritis. CIA treatment mice were subcutaneously injected with 0.5 mg of paKG(PFK15+bc2) MPs, paKG(PFK15) MPs, paKG, or 1x PBS (50 μL) near the right and left hind leg on day 35 and day 40. The ING LNs, pLNs, cLNs, injection site and spleen were harvested to analyze T cell responses, ex vivo recall reactions, and DC and macrophage responses.
Recall reaction ex vivo culture
Recall reactions were performed on cells isolated from the cLN and spleen. Plates were coated with anti-CD3 (1 μg/mL) and either incubated in 4°C overnight or, for 2 hrs at 37 °C. Anti-CD3 was then removed with a multichannel pipettor and rinsed with 1x PBS twice. After the removal of PBS, cells from the extracted organs were isolated, incubated for 3 mins at room temperature in 1x RBC lysis buffer, and approximately 106 cells/mL were seeded. Anti-CD28 (2 μg/mL) and IL-2 (10 ng/mL) were added to stimulate the seeded cells. Cells were cultured for 48–72 hrs prior to antibody staining and flow cytometry.
Statistical Analysis
Data are expressed as mean ± standard error. Comparison between two groups was performed using Student’s t-test (Microsoft, Excel). Comparisons between multiple treatment groups were performed using one-way ANOVA, followed by Bonferroni multiple comparisons, and p-values of 0.05 was considered statistically significant (GraphPad Prism Software 6.0, San Diego, CA)
RESULTS
PFK15 reduces glycolysis and reduces co-stimulatory CD86 expression in bone marrow-derived DCs in vitro
In this study, the ability of PFK15 to reduce glycolysis in bone marrow-derived DCs (BMDCs) in the presence or absence of lipopolysaccharide (LPS, mimicking inflammation in vitro) was tested (Figure 1a). To assess this, the extracellular acidification rate (ECAR, representative of glycolysis) of BMDCs was determined by extracellular flux assays. BMDCs treated with LPS only and no treatment were used as controls. It was observed that, in the presence of LPS, PFK15 significantly lowered glycolysis (ECAR) in BMDCs as compared to LPS alone. Interestingly, ECAR in BMDCs treated with PFK15 alone was not significantly different than no treatment (Figure 1b). These data show that, in the presence of LPS, PFK15 reduces glycolysis in BMDCs, whereas, in the absence of LPS, PFK15 does not affect BMDC glycolysis.
Figure 1: PFK15 reduces glycolysis in BMDCs and prevents their activation in vitro.
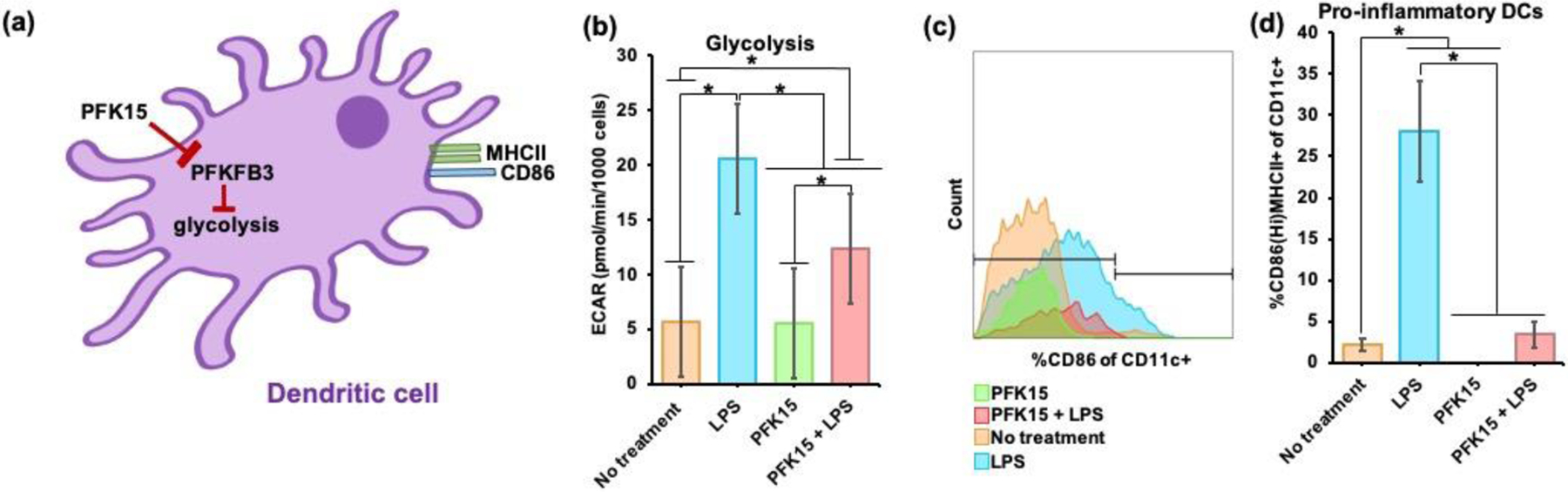
(a) Schematic of soluble PFK15 reprograms glycolysis in BMDCs with PFKFB3 inhibition. (b) In the presence of LPS, PFK15 significantly lowered the glycolysis (extracellular acidification rate - ECAR) in BMDCs (n = 30–36, avg ± SEM, * - p<0.05). (c) Flow cytometry demonstrates that PFK15 is capable of decreasing CD86 expression in BMDCs. (d) in the presence of LPS, PFK15 significantly decreased CD86HiMHCII+ BMDC populations as compared to LPS, respectively (n = 5–11, avg ± SEM, * - p<0.05).
Next, using flow cytometry, it was assessed if the changes in BMDC glycolytic rates can affect surface co-stimulatory CD86 expression in BMDCs. In this study, pro-inflammatory DCs were defined as the frequency of CD86HiMHCII+ of CD11c+ cells and anti-inflammatory DCs were defined as CD86LoMHCII+ of CD11c+ cells. It was observed that PFK15, with and without LPS, reduced CD86 expression in vitro in CD11c+ BMDCs when compared to LPS and no treatment groups (Figure 1c, Figure S1a, Table 1). It was also observed that PFK15, in the presence of LPS, significantly decreased the frequency of CD86HiMHCII+ of CD11c+ BMDCs by approximately 8-fold when compared to LPS alone treatment. Similarly, in the absence of LPS, PFK15 also significantly decreased the frequency of the pro-inflammatory BMDC populations as compared to no treatment (Figure 1d). Interestingly, PFK15, with and without LPS, did not significantly impact anti-inflammatory BMDC populations as compared to LPS and no treatment, respectively (Figure S1b). Overall, these data suggest that PFK15 can suppress CD86 expression in BMDCs in vitro and can also reduce glycolysis in BMDCs in the presence of LPS.
Table 1.
The table below represents the antibodies and protein transport inhibitors that were utilized in these studies.
| S. No. | Target | Fluorophore | Company | Catalog # | Clone |
|---|---|---|---|---|---|
| 1 | CD4 | BB700 | BD Biosciences | 566407 | RM4–5 |
| 2 | CD8 | APC-R700 | BD Biosciences | 564983 | 53–6.7 |
| 3 | CD25 | PECy7 | BD Biosciences | 552880 | PC61 |
| 4 | Tbet | BV785 | BioLegend | 644835 | 4B10 |
| 5 | FoxP3 | eF450 | Invitrogen | 48-5773-82 | FJK-16s |
| 6 | RORγT | BV650 | BD Biosciences | 564722 | Q31-378 |
| 7 | Ki67 | FITC | Invitrogen | 11-5698-82 | SolA15 |
| 8 | GATA3 | BV711 | BD Biosciences | 565449 | L50–823 |
| 9 | CD279 | PE | BD Biosciences | 551892 | J43 |
| 10 | CD44 | BV480 | BD Biosciences | 566200 | IM7 |
| 11 | CD62L | SB600 | Invitrogen | 63-0621-80 | MEL-14 |
| 12 | MHC-Tetramer: - I-A(q) bovine collagenII 271–285 GEPGIAGFKGEQGPK | APC | NIH Tetramer Core Facility | NA | - |
| 13 | CD11c | PE | BioLegend | 117308 | N418 |
| 14 | CD86 | SB600 | ThermoFisher Scientific | 63-0862-82 | GL1 |
| 15 | MHC | APC | BioLegend | 107614 | M5/114.15.2 |
| 16 | CD11b | FITC | Tonbo | 35–0112-U500 | XMG1.2 |
| 17 | F4/80 | BV702 | Invitrogen | 67-4801-08 | BM8 |
PFK15 is encapsulated in paKG-based MPs to simultaneously deliver aKG and PFK15 to DCs
In order for PFK15 and aKG to act on the same DC, PFK15 and aKG would need to be delivered together. Our prior work demonstrated, in vitro, that paKG MPs, can associate with DCs and release aKG in a sustained manner as the MP degrades via hydrolysis of the ester bonds [35,65]. Importantly, this technology can be designed to encapsulate desired materials for delivery[35]. Therefore, encapsulating PFK15, to inhibit glycolysis, within the paKG MPs, for the simultaneous delivery of aKG, may allow for Krebs cycle-mediated cellular energy and OXPHOS in DCs. This approach may potentially inhibit pro-inflammatory DC function and favor anti-inflammatory DC phenotypes. To test this, the hydrophobic PFK15 molecule was encapsulated in paKG MPs (termed paKG(PFK15) MPs) using water-in-oil emulsion techniques. The morphology and heterogeneity of paKG and paKG(PFK15) MPs were confirmed by scanning electron microscopy (SEM). MPs displayed spherical shapes with a smooth surface morphology (Figure 2a). The average diameter of the paKG MPs was 2.3 ± 0.3 μm and paKG(PFK15) MPs was 1 ± 0.1 μm, as determined by dynamic light scattering (DLS) (Figure S2a). The percentage encapsulation efficiency of PFK15 within paKG MPs was 68% and loading was 17 ± 4 μg/mg of paKG MPs. The cumulative release kinetics of PFK15 from paKG(PFK15) MPs demonstrated a release of 17.7 ± 5.7 μg/mg of paKG MPs by day 10 (Figure S2b).
Figure 2: paKG(PFK15) MPs modulate the metabolic profile of BMDCs in vitro.
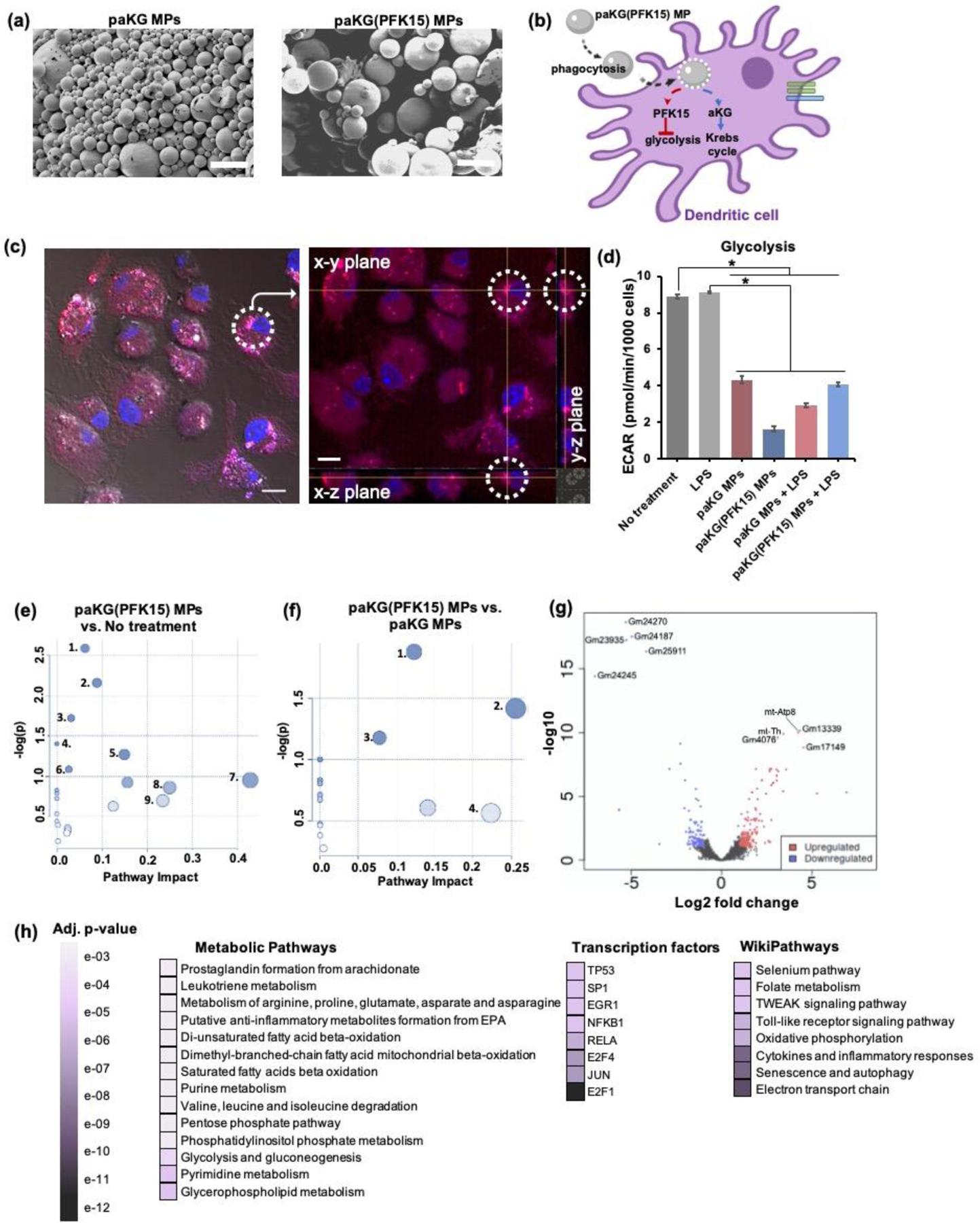
(a) SEM of paKG MPs and paKG(PFK15) MPs demonstrate spherical and heterogenous MPs (scale bar = 10 μm, magnification 2000x). (b) Schematic of paKG(PFK15) MPs modulating BMDC metabolism. (c) BMDCs phagocytose paKG MPs (paKG MP – red, nucleus – blue; gray = DIC filter, scale bar = 10 μm). The dashed circle and crossbar (yellow lines meet), in the right image, shows the same particle in different planes (x,y,z). (d) In the presence of LPS, paKG(PFK15) MPs significantly reduced the glycolysis (ECAR) in BMDCs as compared to LPS alone (n = 15–18, avg ± SEM, * - p<0.05.) (e, f) Metabolomics of BDMCs, (e) paKG(PFK15) MPs significantly modulate 28 intracellular metabolite levels and their respective signaling pathways, significantly as compared to no treatment. paKG(PFK15) MPs vs. no treatment: 1 – Citrate cycle; 2 – Alanine, asparate and glutamate metabolism; 3 – Butanoate metabolism; 4 – Pyruvate metabolism; 5 – Glycolysis/gluconeogenesis; 6 – Cysteine and methionine metabolism; 7 – Taurine and hypotaurine metabolism; 8 – Ascorbate and alderate metabolism; 9 – Nicotinate and nicotinamide metabolism, (f) paKG(PFK15) MPs significantly modulated 34 intracellular metabolite levels significantly as compared to paKG MPs. paKG(PFK15) MPs vs. paKG MPs: 1 – Histidine metabolism; 2 – Glutathione metabolism; 3 – Arginine and proline metabolism; 4 – Alanine, asparate and glutamate metabolism. Pathway impact – number of metabolites modified significantly in a pathway; -log(p) – level of modulation (n = 3, p<0.05). (g, h) RNA-seq of BMDCs, (g) a volcano plot of RNA-seq results display the fold changes (log2 fold change) and the false discovery rate (FDR)-adjusted p values (-log10 (FDR)) in genes that were upregulated (red) and downregulated (blue) when comparing paKG(PFK15) MP treated BMDCs to no treatment, (h) a gene enrichment map is represented by the statistical significance (FDR-adjusted p value) of enriched pathways. The darker purple colors represent higher significant differences between paKG(PFK15) MPs and no treatment (n = 3).
Furthermore, paKG MP endotoxin levels were determined to be <0.01 EU/mL, therefore, suggesting minimal effect on DC activation[35]. Overall, these data suggest that paKG MPs may be a feasible approach to simultaneously deliver PFK15 and aKG.
BMDCs phagocytose paKG-based MPs in vitro
In order for paKG(PFK15) MPs to modulate DC function, paKG MPs need to be phagocytosed by DCs to deliver PFK15 and aKG intracellularly (Figure 2b). Confocal imaging demonstrated that paKG MPs encapsulating rhodamine 6G, a fluorescent dye, can be internalized by BMDCs as observed by images containing magenta (paKG(rhodamine 6G) MPs) and blue (nucleus of BMDCs) (Figure 2c). Additionally, live/dead viability stains, as observed by flow cytometry, confirmed that the 0.01 mg/mL concentration of paKG-based MPs that were added to the BMDC cultures were non-toxic[35]. In all, these data demonstrate that BMDCs can phagocytose paKG-based MPs in vitro.
paKG(PFK15) MPs reduce BMDC glycolysis and alter metabolic pathways
Since metabolites can control DC function[18], and since it was observed that PFK15 alone can modify the glycolytic rate and activation of pro-inflammatory BMDCs, the effect of delivering PFK15 in paKG MPs on the intracellular metabolic profile of BMDCs needs to be investigated. To test this, extracellular flux assays and metabolomic studies were performed. BMDCs with LPS and no treatment were used as controls. It was observed that, paKG(PFK15) MPs, significantly decreased glycolysis (ECAR) in BMDCs treated with LPS by approximately 2-fold, as compared to LPS alone. Similarly, paKG(PFK15) MPs, without LPS treatment, also significantly decreased glycolysis by approximately 5-fold, as compared to no treatment (Figure 2d). Together, these data demonstrate that PFK15 encapsulated in paKG MPs can reduce glycolysis in BMDCs.
Next, the ability of paKG(PFK15) MPs to impact metabolite accumulation and metabolic pathways, other than glycolysis, in BMDCs was investigated. Using liquid chromatography targeted mass spectrometry (LC-MS/MS), it was determined that the delivery of PFK15 from paKG MPs significantly affected 28 metabolites out of the 299 intracellular metabolites tested, as compared to no treatment. Additionally, 34 out of the 299 intracellular metabolites tested were affected when comparing paKG(PFK15) MPs to paKG MPs. The Kyoto Encyclopedia of Genes and Genomes (KEGG) database and metabolite intensities were utilized to identify the metabolic pathways that were the most impacted and an interactive visualization framework was constructed to visualize the relevant pathways. The metabolic pathways that had an impact value > 0.2 or −log (p) > 1.0 were determined as the pathways most significantly affected. When comparing paKG(PFK15) MPs to no treatment, 9 metabolic pathways in BMDCs were significantly impacted with a ~2–5 fold alteration in 28 metabolites within amino acid metabolism, carbohydrate metabolism, ascorbate and alderate metabolism, and pathways involved in metabolism of cofactors and vitamins (Figure 2e). Furthermore, when comparing paKG(PFK15) MPs to paKG MPs, 34 metabolites were significantly impacted in BMDC metabolic pathways involved in amino acid metabolism such as histidine metabolism; glutathione metabolism, arginine and proline metabolism; alanine and aspartate and glutamate metabolism (Figure 2f). In all, these data demonstrate that paKG(PFK15) MPs modulate BMDC metabolism by directly changing the intracellular metabolite profile.
paKG(PFK15) MPs alter genes encoding pro- and anti- inflammatory responses in BMDCs
In order to understand the relation between the phenotypic changes observed in BMDCs to cellular signaling pathways, RNA-seq on treated BMDCs was performed. When comparing paKG(PFK15) MPs vs. no treatment, volcano plots demonstrated an upregulation in protein coding (mt-Atp8; log2 fold change: 4), Mt-tRNA (mt-Th; log2 fold change: 3), and pseudogenes that are unprocessed (Gm13339; log2 fold change: 4) and processed (Gm4076 (log2 fold change: 3) and Gm17149 (log2 fold change: 4)). Furthermore, these volcano plots highlighted an upregulation and enrichment of the electron transport chain (ETC) (adjusted (adj.) p-value: 3.6e-15), oxidative phosphorylation (adj. p-value: 1.1e-10), and mRNA processing (adj. p-value: 5.6e-05) when comparing paKG(PFK15) MP treated BMDCs to no treatment (Figure 2g). Moreover, pathways such as the metabolic pathway, transcription factor pathways and wikipathways demonstrated an inverse relation in gene expression when comparing paKG(PFK15) MP treated BMDCs to no treatment. These gene sets within paKG(PFK15) MP treated BMDCs, were associated with immune response pathways that are involved in increasing anti-inflammatory metabolite formation and fatty acid oxidation, and decreasing pro-inflammatory associated genes related to glycolytic processes, toll-like receptor signaling and cytokines and inflammatory responses. An upregulation of genes that encoded ETC, TGFβ signaling pathway, and glutathione metabolism in paKG(PFK15) MPs were of particular relevance. Furthermore, BMDCs treated with paKG(PFK15) MPs demonstrated a downregulation in genes related to pro-inflammatory responses, such as genes encoding Tnf, pfkfb3, HIF1α and several genes associated with the IL-6 signaling pathway (Figure 2h). In all, RNA-seq data demonstrated that paKG(PFK15) MPs modulate immune responses in BMDC’s by altering the expression of genes that are involved in pro- and anti-inflammatory immune processes.
CIA-associated antigen, bc2, is encapsulated within paKG(PFK15) MPs to reduce bc2-driven inflammation in the paws of CIA mice
The CIA-associated antigen, bc2, was encapsulated within paKG(PFK15) MPs (termed paKG(PFK15+bc2) MPs) to generate antigen-specific immune suppression via glycolytic inhibition for the treatment of RA in CIA mice. Bc2 is the antigen that triggers RA symptoms in the CIA mouse model[47], thus, retraining the immune system to tolerate bc2 may reinstate immune tolerance in CIA mice. This may be achieved in vivo by delivering bc2 to anti-inflammatory DCs that sustain energy from the Krebs cycle (attained by delivering aKG) and, concurrently, have a reduced energy reliance on glycolysis (attained by delivering PFK15). Therefore, PFK15+bc2 was encapsulated in paKG MPs using water-oil-water emulsion techniques, for the potential simultaneous delivery of aKG, PFK15, and bc2 to the same DC as the MP degrades. paKG MPs encapsulating bc2 (termed paKG(bc2) MPs) were synthesized, using water-oil-water emulsion techniques, and utilized as a control.
SEM analysis demonstrated that paKG(bc2) and paKG(PFK15+bc2) MPs were more porous than the paKG and paKG(PFK15) MPs (Figure S2c, Figure 3a). DLS determined the average diameter of the paKG(bc2) MPs to be 1.0 ± 0.1 μm and the paKG(PFK15+bc2) MPs to be 1.5 ± 0.2 μm (Figure S2a, Figure 3b). The percentage encapsulation efficiency of bc2 was 54.9% and the percentage encapsulation efficiency of PFK15 was 47.4%. Additionally, the loading of bc2 was 13.7 ± 36.7 μg/mg of MPs and the loading of PFK15 was 11.8 ± 10.9 μg/mg of paKG MPs. The cumulative release kinetics of PFK15 from paKG(PFK15+bc2) MPs demonstrated a release of 29.7 ± 3.0 μg/mg of paKG MPs by day 10 (not significantly different than PFK15 release from paKG(PFK15) MPs - Figure S2b) and release kinetics of the FITC-labelled bc2 from paKG(bc2) and paKG(PFK15+bc2) MPs demonstrated a sustained cumulative release of bc2 over a 10 day time period (no significant differences between the release of the FITC-labelled bc2 from the paKG(bc2) and paKG(PFK15+bc2) MPs - Figure S2d). Furthermore, each of these paKG based MPs demonstrated a constant degradation rate, as observed by the release of aKG over a 10 day time period. paKG(PFK15) MP degradation was significantly different than paKG(PFK15+bc2) degradation from days 1–10 and paKG(bc2) MP degradation was significantly different than paKG(PFK15+bc2) MPs on day 8. Significance was compared against paKG(PFK15+bc2) MPs (Figure S2e).
Figure 3: paKG(PFK15+bc2) MPs reduce symptoms of RA in vivo in a CIA mouse model.
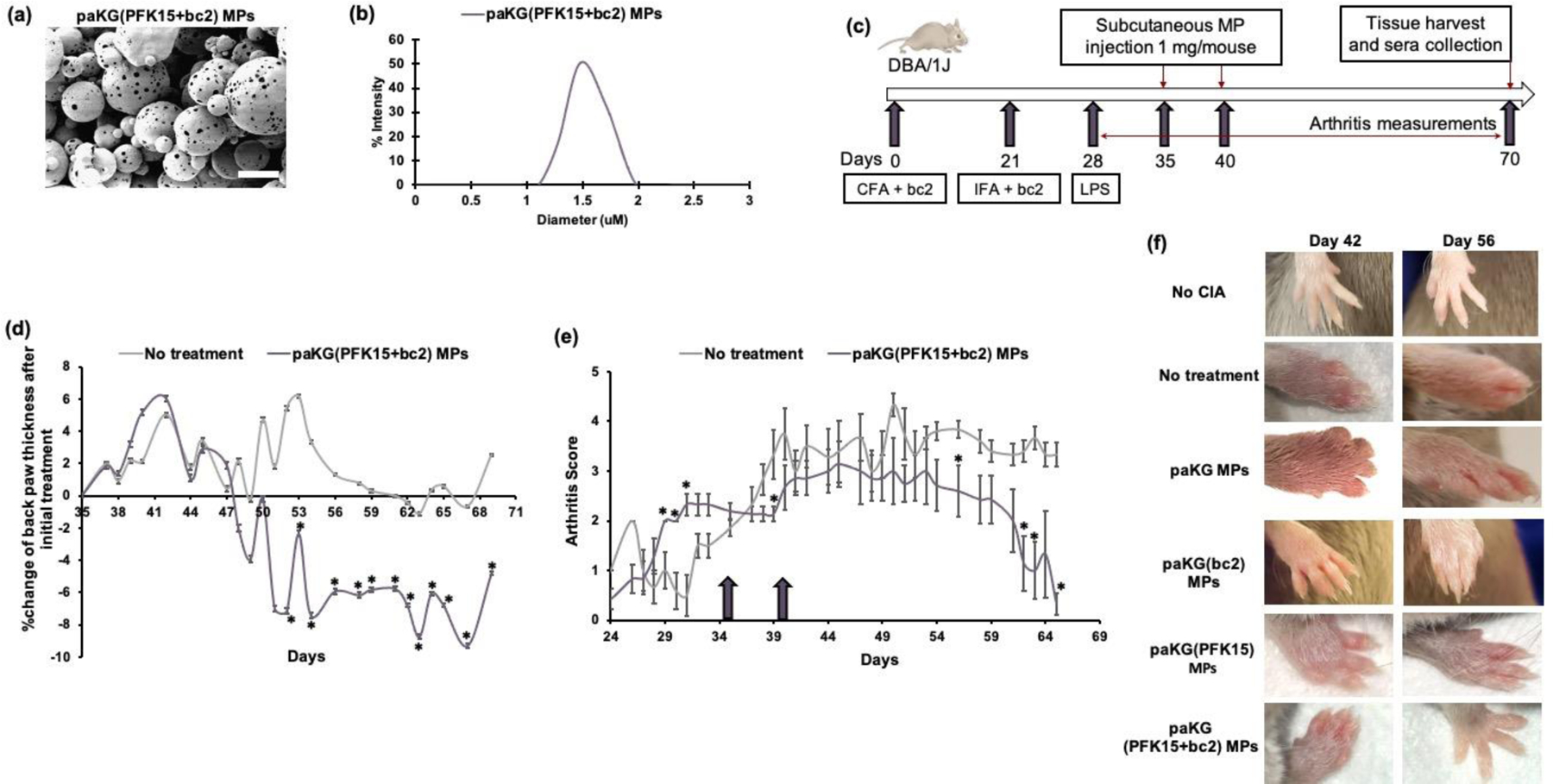
(a) SEM of paKG(PFK15+bc2) MPs demonstrate porous, spherical and heterogenous MPs (scale bar = 10 μm, magnification 2000x). (b) The average diameter of paKG(PFK15+bc2) MPs was found to be 1.5 ± 0.2 μm. (c) Schema of in vivo CIA studies. (d) Average of the normalized percent change in back paw thickness after the first dose of paKG(PFK15+bc2) MPs as compared to no treatment mice (n = 9–11, avg ± SEM, * - p<0.05). (e) Average of the arthritic back paw scores. Arrows signify treatment days on days 35 and 40 (n = 6–9, avg ± SEM * - p<0.05). (f) Representative front paw images of control mice and paKG(PFK15+bc2) MP treated mice on days 42 and 56.
To determine if the metabolic inhibition in the presence of the bc2 antigen can reverse RA symptoms in vivo, the CIA model in DBA/1j mice was utilized. The severity of the arthritis was determined by an arthritic score on a scale of 0 to 6. A score of 3 or higher signified moderate arthritis. Mice were administered 0.5 mg of either empty paKG MPs, paKG(bc2) MPs, paKG(PFK15) MPs, or paKG(PFK15+bc2) MPs subcutaneously, near each hind leg on day 35 (an average arthritic score of 3) and day 40 (Figure 3c). Injections of phosphate buffered saline (PBS) were used as a no treatment positive control and mice with no CIA were used as a negative control. Since the treatments were administered near the two hind legs, the back paws of the mice were denoted as the local physiological measurements and were observed to determine the local effects of the disease and treatment. The front paws of the mice were observed to determine the systemic effects of the disease and treatment. It was determined that the percent change of the back paw thickness, after the day 35 treatment of paKG(PFK15+bc2) MPs was significantly lower than no treatment on days 52 till the end of the study, day 69 (Figure 3d). Moreover, when comparing arthritic scores, it was observed that, after the day 35 treatment, the paKG(PFK15+bc2) MP treated mice also had significantly lower arthritic scores than untreated mice on days 39, 56, 62, 63 and the end of study measurement, day 65 (day 65 back paw score was 0.3) (Figure 3e). Notably, the decrease in back paw thickness of paKG(PFK15+bc2) MP treated mice was highest when compared to all the paKG-based MP control groups, except for paKG(PFK15) MP treated mice (Figure S3). Whereas the decrease in back paw arthritic scores were less in all control paKG-based MP treatment groups as compared to paKG(PFK15+bc2) MP treated mice (Figure S4). Moreover, it was observed that the front paws of mice treated with paKG(PFK15+bc2) MPs on day 56 demonstrated decreased swelling as compared to no treatment, and paKG MP, paKG(bc2) MP, and paKG(PFK15) MP treated mice (Figure 3f), suggesting a systemic response within 3 weeks of the first treatment. These data suggest that local injections of paKG(PFK15+bc2) MPs are a viable strategy to systemically treat RA. Furthermore, the reduction in paw swelling, in the paKG(PFK15+bc2) MP treatment group, indicates that the delivery of PFK15+bc2 via paKG MPs may induce antigen-specific immune suppression via glycolytic inhibition in DCs in a CIA mouse model.
DCs and macrophages infiltrate MP injection site and traffic MPs to LNs within 24 hrs
The modulation of physiological measurements in paKG(PFK15+bc2) MP treated mice in a CIA mouse model may indicate that the DCs are interacting with the paKG(PFK15+bc2) MPs for the generation of antigen-specific immune suppression. To test if DCs are indeed phagocytosing the MPs in vivo, paKG MPs were crosslinked with a coumarin (blue) dye and were subcutaneously injected near each hind leg of healthy C57BL/6j mice. Mice were sacrificed 24 hrs after the treatment injections and the injection sites, draining LNs (dLNs) (inguinal LNs (ING LNs) – located in the abdominal region) and control cervical LNs (cLNs – located in the neck) were harvested. As observed by flow cytometry, CD11c+ DCs and CD11b+F4/80+ macrophages invaded the site of injection and phagocytosed paKG MPs within the injections site, as observed by coumarin+CD11c+ (graphically represented by dark blue) and coumarin+CD11b+F4/80+ cell populations (graphically represented by light blue) (Figure S5a, Table 1). Moreover, the DCs and macrophages carried the particles to the ING dLNs, where an increased amount of coumarin+ DCs and coumarin+ macrophages were observed in the ING dLNs as compared to the control cLNs (Figure S5b, Table 1). Overall, these data suggest that the local subcutaneous injections of MPs in vivo, caused CD11c+ cells to infiltrate the injection site and traffic these MPs to the LNs for a systemic modulation of DC responses.
paKG(PFK15+bc2) MPs increase the frequency of CD86LoMHCII+ BMDC populations in vitro
Since DCs indeed phagocytose paKG-based MPs in vivo, it was hypothesized that the observed reduction in paw swelling in the CIA mice treated with paKG(PFK15+bc2) MPs, may be due to the paKG(PFK15+bc2) MPs modulating DC function (Figure 4a). Therefore, the effect of paKG(PFK15+bc2) MPs on cultured BMDCs, in the form of altered CD86 expression in CD11c+ cells, was tested in vitro. BMDC cultures were treated with the paKG(PFK15+bc2) MPs, and no treatment, paKG MPs, paKG(bc2) MPs, paKG(PFK15) MPs and LPS conditions were used as controls. It was observed that paKG MPs, paKG(bc2) MPs, paKG(PFK15) MPs and paKG(PFK15+bc2) MPs with LPS reduced CD86 expression in CD11c+ cells when compared to LPS alone condition (Figure 4b, 4c, 4d and 4e separated into four graphs for ease of visualization. The no treatment and LPS group remained consistent in each of the four graphs.). Specifically, paKG(PFK15+bc2) MPs, with LPS, significantly decreased CD86 expression in CD11c+ cells by approximately 9-fold when compared to LPS alone (Figure 4f). Moreover, BMDCs treated with paKG(PFK15+bc2) MPs, significantly increased the frequency of CD86LoMHCII+ BMDCs when compared to no treatment (Figure 4g). Furthermore, all paKG-based MPs, in the presences of LPS, demonstrated significantly lower CD86HiMHCII+ BMDC frequencies as compared to LPS alone (Figure 4h). In all, these data suggest that the delivery of PFK15+bc2 via paKG MPs can decreased CD86 expression for an increased frequency of CD86LoMHCII+ BMDC populations in vitro.
Figure 4: paKG(PFK15+bc2) MPs alter DC function in vitro and in vivo in a CIA mouse model.
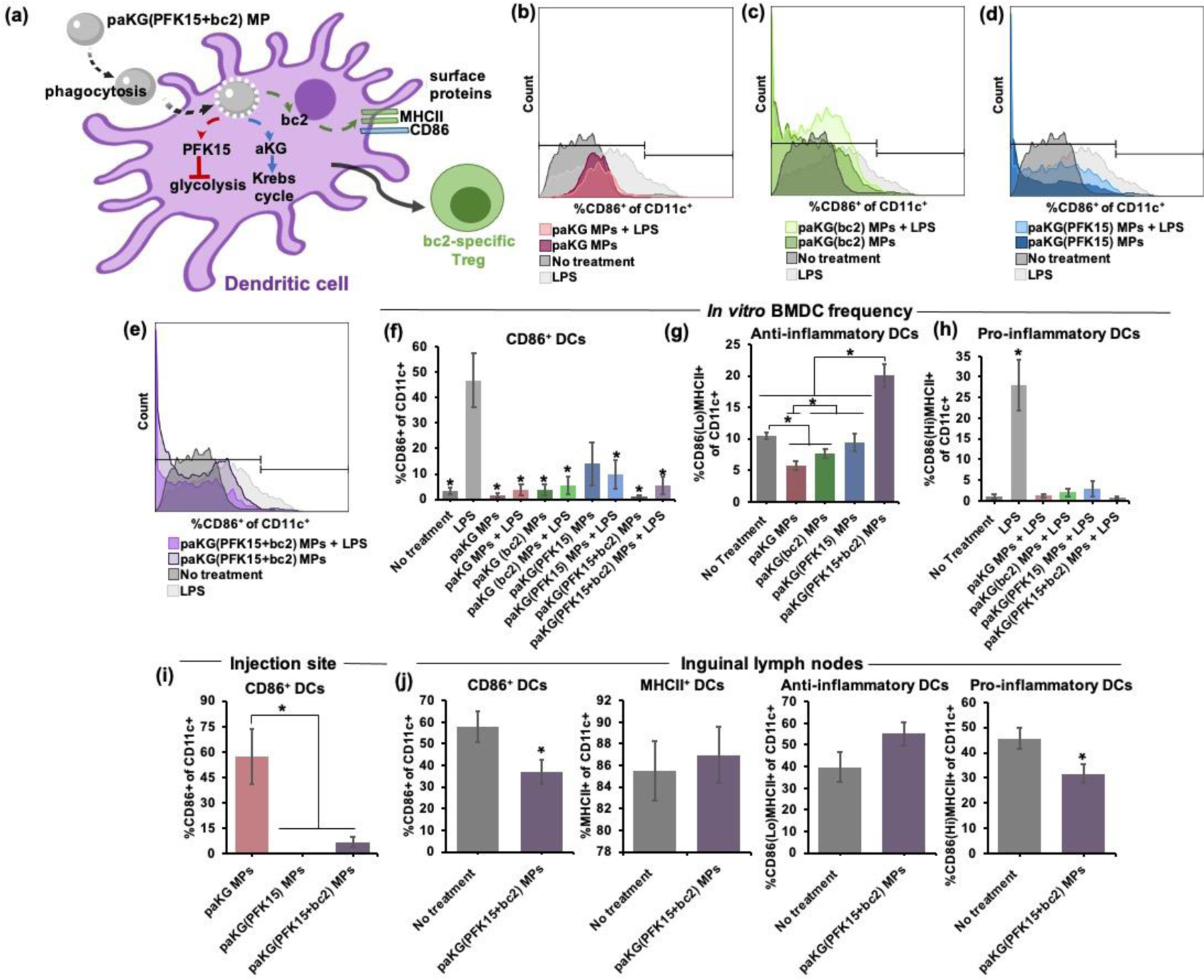
(a) Schematic of paKG(PFK15+bc2) MPs reprogramming DC surface proteins for the modulation of adaptive immune responses. (b-e) Histogram plots of CD11c+CD86+ frequencies demonstrate that paKG-based MPs in the presence LPS have lower CD86 expression than LPS alone, (b) paKG MPs with and without LPS, (c) paKG(bc2) MPs with and without LPS, (d) paKG(PFK15) MPs with and without LPS, (e) paKG(PFK15+bc2) MPs with and without LPS. (f) In the presence of LPS, paKG-based MPs significantly decreased the frequency of CD11c+CD86+ cells in vitro as compared to LPS (n = 5–11, avg ± SEM, *- p<0.05). (g) paKG(PFK15+bc2) MPs significantly increased the percentage of CD86LoMHCII+ of CD11c+ cells in vitro as compared to no treatment and all other paKG-based MPs (n = 6–12, avg ± SEM, *- p<0.05). (h) All paKG-based MPs, in the presence of LPS, have a significantly lower frequency of CD86HiMHCII+ of CD11c+ cells in vitro as compared to LPS (n = 5–11, avg ± SEM, *- p<0.05). (i) paKG MPs encapsulating PFK15, reduce CD11c+CD86+ frequency within the MP injection sites of CIA mice (n = 4–8, avg ± SEM, *- p<0.05). (j) In vivo CD11c+ frequencies within the inguinal lymph nodes of CIA mice (n = 5–7, avg ± SEM, *- p<0.05).
The combinatorial delivery of paKG and PFK15 decreases CD86 expression of CD11c+ cells in the injection sites of CIA mice
Since in vitro BMDC studies demonstrated that the combinatorial delivery of paKG and PFK15 can decrease CD86 expression as well as suppress pro-inflammatory gene expression, it was hypothesized that the simultaneous delivery of paKG and PFK15 can also lead to a modulation in the expression of CD86 in CD11c+ cells in vivo in CIA mice. CIA induced mice treated with paKG(PFK15+bc2) MPs were euthanized on day 70 and flow cytometry was performed on the treatment injection sites and the ING LNs. The LNs are crucial secondary lymphoid organs where DCs and T cells engage for the initiation of adaptive cell immunity[66]. Therefore, if DCs are modulated in the LNs then this may lead to an eventual modulation in T cell responses as well. However, for this to occur, DCs would need to first infiltrate the MP injection sites and traffic the MPs to the LNs. Therefore, the treatment injection sites were examined to assess if CD11c+ or F4/80+ (macrophage marker) cells infiltrate the injection sites in CIA mice. Since macrophages are professional phagocytes, they were utilized for comparison. It was observed that mice treated with paKG-based MPs had an infiltration of CD11c+ and F4/80+ cells (Figure S6). Furthermore, the injection sites of mice treated with an MP formulation consisting of both paKG and PFK15, demonstrated significantly lower percentages of CD86+ expression in CD11c+ cells as compared to the injection site of mice treated with empty paKG MPs (Figure 4i, Figure S1). These data suggest that CD11c+ and F4/80+ cells infiltrate the injection sites of paKG-based MPs in CIA mice, and the delivery of PFK15, via paKG MPs, drives the reduction in CD11c+CD86+ expression at the injection sites.
paKG(PFK15+bc2) MPs decrease CD86 expression of CD11c+ cells in the ING LNs of CIA mice
Since biodistribution studies determined that CD11c+ cells infiltrate the injection sites, and that these cells can transport the particles to the LNs, we next tested the ability of these paKG-based MPs to modulate CD86 expression in the local ING LNs of CIA mice. It was determined that the ING LNs of mice treated with the paKG-based MPs had significantly lower CD11c+CD86+ expression as compared to the ING LNs of the no treatment control. Notably, MHCII expression in CD11c+ cells in the paKG(PFK15+bc2) MP treatment group was not significantly different from no treatment. However, the ING LNs of the paKG(PFK15+bc2) MP treatment group had an increase in CD86LoMHCII+ of CD11c+ DC populations and a significant decrease in CD86HiMHCII+ of CD11c+ populations when compared to the no treatment ING LNs (Figure 4j, Figure S7). Overall, these data indicate that the paKG(PFK15+bc2) MPs decrease CD86 expression of CD11c+ cells that infiltrate the injection sites, and given the biodistribution data, these cells then traffic the paKG(PFK15+bc2) MPs to the LNs, where a decrease in CD86 expression in CD11c+ cells is also observed.
paKG MPs delivering PFK15 and bc2 induced antigen-specific T cell responses in local and systemic sites of CIA mice
Thus far, paKG(PFK15+bc2) MPs have shown to decrease CD86HiMCHII+ of CD11c+ populations in vitro and in vivo, and since DCs play a central role in initiating and modulating T cell responses, the ability of paKG(PFK15+bc2) MPs modulating T cell responses was assessed in vivo in a CIA mouse model. After mice were euthanized, the popliteal LNs (pLNs – near hind legs) and spleen were extracted and analyzed for different T cell phenotypes using flow cytometry. The pLNs are localized near the knee joint and therefore were denoted as the local lymphatic measurement of the knee. The spleen and cLNs represented the systemic measurements of the treatment. Notably, the draining pLNs of paKG(PFK15+bc2) MP treated mice had significantly increased proliferating TREG populations as compared to no treatment. Additionally, mice receiving paKG(PFK15+bc2) MPs, when compared to no treatment, had significantly decreased TH1, TC17, activated CD8+Tbet+RORγT+, and proliferating and activated CD8+Foxp3+RORγT+ (~8-fold) populations in the pLNs. Importantly, paKG(PFK15+bc2) MP treatment groups showed significantly higher percentages of proliferating antigen-specific TREG populations (~2-fold) and significantly decreased percentages of activated antigen-specific TH17 cells in the pLNs when compared to no treatment (Figure 5a, Figure S8, Figure S9a, Table 1). These data indicate that CIA mice treated with paKG(PFK15+bc2) MPs can lead to an increase in anti-inflammatory responses and a decrease in inflammatory T cell responses in local sites. Importantly, paKG(PFK15+bc2) MP treated mice can generate antigen-specific immune suppression in the draining pLNs. Furthermore, when assessing the systemic T cell effects of paKG(PFK15+bc2) MPs in the spleen, it was observed that paKG(PFK15+bc2) MP treated mice had significantly decreased inflammatory T cell populations of activated TH1, activated TH17 and proliferating CD4+Tbet+RORγT+ populations as compared to the no treatment control (Figure S9b). Interestingly, there is a decreased frequency of anti-inflammatory T cell responses in both the pLNs and spleen of the paKG and paKG(bc2) MP treated mice as compared to the paKG(PFK15) and paKG(PFK15+bc2) MP treatment groups. These findings may suggest that the combinatorial delivery of both paKG and PFK15 are required to elicit more potent anti-inflammatory T cell responses (Figure S9a,b). Overall, these data strongly suggest that the delivery of PFK15+bc2 via paKG MPs can generate immunosuppressive antigen-specific T cell responses in vivo in local sites. Specifically, the T cell phenotype of CIA mice treated with paKG(PFK15+bc2) MPs appears to be skewed toward bc2-specific anti-inflammatory responses as observed by an up-regulation of bc2-specific TREG populations and down regulation of pro-inflammatory and activated bc2-specific TH17 responses in the pLNs, which may potentially be due to the modulation of DC responses in the LNs.
Figure 5: paKG(PFK15+bc2) MPs alter T cell responses in vivo in a CIA mouse model.
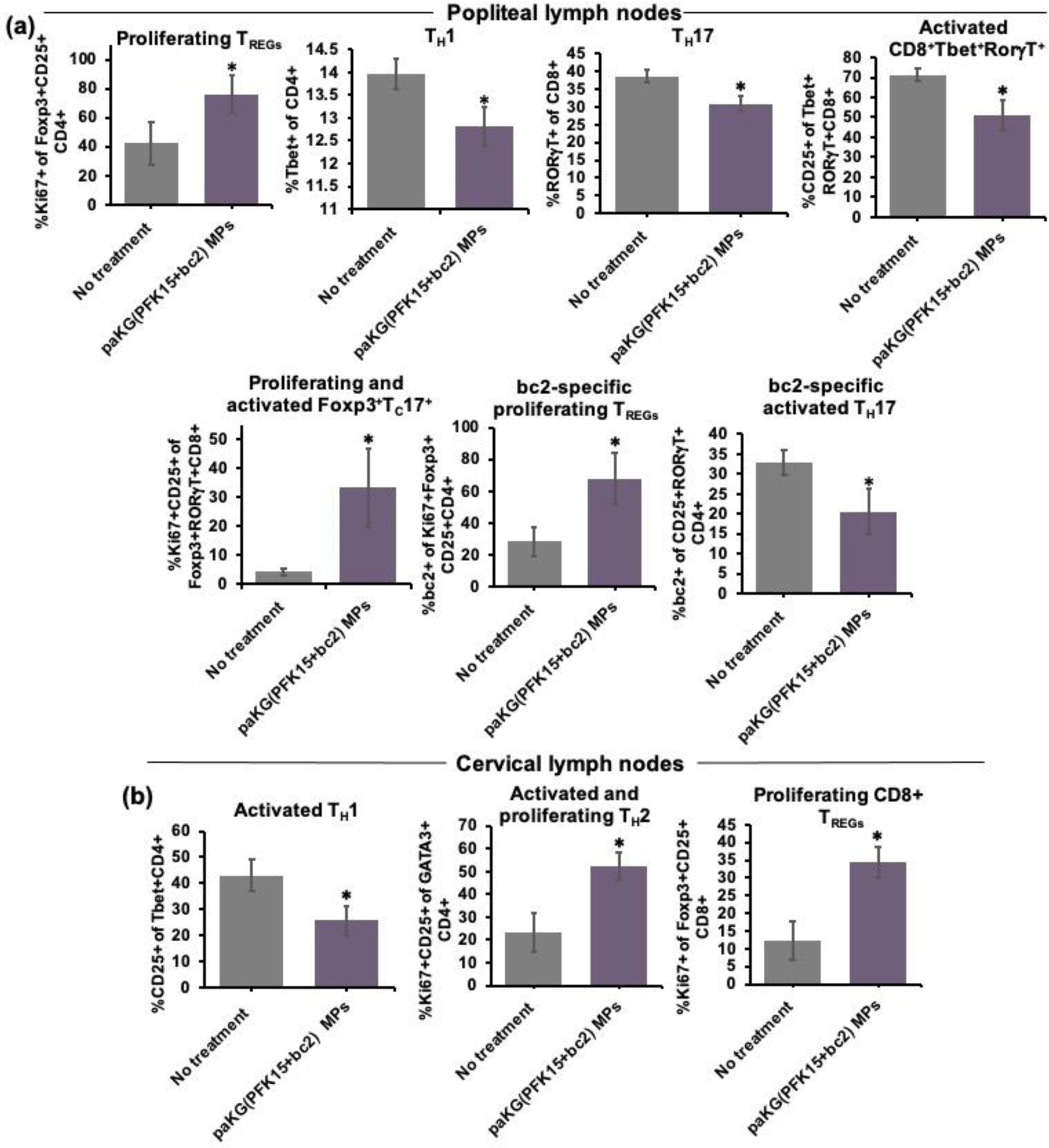
(a)In vivo T cell frequencies in the popliteal lymph nodes of CIA mice (n = 3–4; avg ± SEM, *- p<0.05). (b) Ex vivo T cell frequencies within the cervical lymph nodes of CIA mice, in the presence of the bc2 antigen (n = 10–14; avg ± SEM, *- p<0.05).
In order to confirm if the observed antigen-specific T cell responses in the pLNs of the CIA mouse model were also occurring systemically, ex vivo recall reactions were performed on the non-draining cLNs. After CIA mice were euthanized on day 70, cells from the non-draining cLNs were isolated from paKG(PFK15+bc2) MP treated mice and the control no treatment mice, and these cells were then restimulated with the bc2 antigen or the control, bovine serum albumin (BSA) protein. A heatmap was utilized to qualitatively depict if T cells exposed to bc2, proliferated differentially as compared to T cells exposed to BSA (scale of fold change in proliferation is shown - Figure S10). Overall, a lower frequency in pro-inflammatory CD4+ T cells was observed in paKG(PFK15+bc2) MP treated mice as compared to no treatment (Figure S10). Specifically, it was observed that the frequency of activated TH1 cells was significantly decreased in mice treated with paKG(PFK15+bc2) MPs as compared to the no treatment control. Additionally, activated TH2 cells (~2-fold) and proliferating Foxp3+CD25+CD8+ (~3-fold) were significantly higher in the paKG(PFK15+bc2) MP treatment group, however, this difference was not observed in the no treatment mice (Figure 5b, Table 1). Importantly, the paKG(PFK15+bc2) MP treatment group displayed lower pro-inflammatory bc2-specific CD4+ T cell populations as compared to no treatment (Figure S10). Taken together, these data strongly suggest that T cells in the paKG(PFK15+bc2) treatment group are capable of suppressing pro-inflammatory antigen-specific T cell responses in systemic sites when being reintroduced to the self-antigen weeks after the initial treatment. Importantly, this data suggests that the shift toward immunosuppressive responses may potentially be due to the generation of proliferating antigen-specific TREGs in the pLNs.
DISCUSSION
Current clinically approved therapies for autoimmune diseases such as RA, SLE, and Hashimoto’s disease do not induce extended periods of drug-free remission or restoration of immune tolerance to self-antigens[67]. The findings in this study demonstrates that the glycolytic suppression of DCs in the presence of the self-antigen can lead to the generation of suppressive antigen-specific immune responses for the treatment of arthritic symptoms in CIA mice. The glycolytic energy pathway is utilized to generate ATP by DCs and may determine if DCs will display pro-inflammatory phenotypes[10,17,68]. Although studies have demonstrated that glycolytic inhibitors can modulate glucose metabolism[19,69–71], the systemic delivery of inhibitors can lead to global and non-specific glycolytic inhibition[19]. Thus, this present work encapsulated a glycolytic inhibitor, PFK15, within paKG MPs to control local and/or intracellular delivery to phagocytes such as DCs for a shift in glycolytic energy reliance toward the Krebs cycle[35]. Although, delivery of biomolecules to DCs have been achieved in vivo using polymeric particles such as poly lactic-co-glycolic acid (PLGA) MPs, these particles are utilized as inert delivery vehicles, and may not participate actively in modulating DC responses[32,72–75]. However, the paKG MPs themselves are immunosuppressive[35] and when coupling this technology with PFK15, a shift in glycolytic energy reliance toward the Krebs cycle within DCs occurred, as observed by an alteration in DC metabolism and gene expression.
Furthermore, the co-delivery of PFK15 and the CIA associated antigen, bc2, within paKG MPs to the same DC may have led to the subsequent induction of suppressive antigen-specific immune responses observed within CIA mice. Co-delivering encapsulated content, specifically an antigen and oligonucleotides within PLGA MPs to BMDCs, has previously been reported as a more potent response as compared to delivering the antigen or oligonucleotide alone within PLGA MPs to BMDCs[76]. This potent BMDC response may stimulate from the same DC being delivered the same encapsulated MP content[76]. Therefore, PFK15+bc2 was co-encapsulated within paKG MPs to increase the likelihood of co-delivery to the same DC. As observed by CD86 expression, paKG(PFK15+bc2) MPs were able to decrease pro-inflammatory DC populations in vitro and in vivo, as well as increase the citrate cycle (Krebs cycle – as observed by metabolomics analyses), OXPHOS, and the ETC while reducing glycolysis (ECAR) and genes encoding pro-inflammatory responses within BMDCs in vitro. Since DCs are efficient APCs[77–79], this data suggested that the paKG(PFK15+bc2) MPs may induce suppressive antigen-specific immune responses, however, the generation of antigen-specific TREGs in vivo can be difficult to achieve and such therapies are currently not available in clinic[80]. Nonetheless, this present work demonstrated an increased frequency of proliferating antigen-specific TREGs in the local draining lymph nodes of the joints (pLNs) of paKG(PFK15+bc2) MP treated mice, which may have occurred as a result of altering DC glucose metabolism. Although restricting glucose from DCs, and introducing T cells in the presence of glucose to DCs has previously shown to induce pro-inflammatory T cell responses[81], this present study indicates that if glucose is consistently restricted then DCs can induce anti-inflammatory T cell responses. Importantly, this present work suggests that the modulation of the local (back paw thickness and scores) and systemic (front paw images) physiological measurements may have occurred as a result of the increase in proliferating antigen-specific TREGs, in local sites, due to the delivery of paKG(PFK15+bc2) MPs. Additionally, this study suggests that paKG(PFK15+bc2) MPs can generate extended immunosuppressive T cell responses weeks after treatment when being reintroduced to the self-antigen. Specifically, ex vivo T cell studies demonstrated a systemic decrease in pro-inflammatory bc2-specific CD4+ T cell populations of mice treated with paKG(PFK15+bc2) MPs when being reintroduced to the self-antigen.
Furthermore, the size of the MPs also may affect the resolution rate of RA symptoms in mice based on the higher number or faster generation of antigen-specific Tregs or decrease in antigen-specific Th17 cells. Importantly, the size of the MPs can be decreased by increasing the homogenizing speed, and this may increase the release kinetics of PFK15, aKG and bc2 protein from the particles, as the surface area will increase with decreasing particle sizes. This optimization of particle size might generate an optimal condition for in vivo treatment of RA. Interestingly, from our results it was observed that all the particles released aKG at a linear rate, and for the first 10 days, except paKG(PFK15) degraded at a similar rate. The paKG(PFK15) might have delayed degradation potentially due to the increased hydrophobicity of the particle in their core, which might prevent hydrolytic degradation afforded in particles that contained bc2. Therefore, this modification in the degradation rate and release kinetics of PFK15 from paKG MPs may play a role in response to RA symptoms in mice.
Overall, this study demonstrates that the metabolic reprogramming of pro-inflammatory DCs in the presence of a self-antigen may be a potential approach to restructuring the patient’s own immune system to mount a response against mechanisms that promote the manifestation of autoimmunity. The decrease in CD86 and Tnf expression within DCs can be useful in treating additional autoimmune diseases that have a similar pathogenesis to RA. However, to assess if delivering PFK15 and the relevant self-antigen(s) within paKG MPs could be applicable to other autoimmune disease models with a similar pathogeny, autoimmune mouse models of SLE and MS for example, would need to be tested. Promisingly, the reduction in glycolysis from the combinatorial delivery of PFK15 and aKG did demonstrate a pro-inflammatory energy reliance shift of glycolysis toward the anti-inflammatory energy reliance of the Krebs cycle in BMDCs. Therefore, this platform may be applicable to treating other autoimmune mouse models with various self-antigens for the induction of anti-inflammatory DCs and the potential subsequent induction of suppressive antigen-specific T cell responses. This shift in pro-inflammatory to anti-inflammatory immune responses within an inflammatory environment, using the paKG(PFK15+self-antigen) MP technology, is highly desirable when treating autoimmunity, especially when taking note that a pro-inflammatory environment triumphs in inflammatory autoimmune diseases.
CONCLUSION
The complexity of autoimmune diseases has shifted the scientific field toward developing individualized treatments. Therefore, progress in the field of antigen-specific immunotherapy is essential and opens pathways for personalized medicine to ensure better patient care and to fulfill the unmet clinical need of standard immunosuppressants. We engineered an anti-inflammatory metabolite-based delivery system for the generation of anti-inflammatory DCs and antigen-specific T cell responses, which does not utilize any growth factors or cytokines and thus may have higher translational potential. This is the first report, to the best of our knowledge, which demonstrates that antigen-specific responses can be generated by modulating metabolism of DCs in vivo in the presence of MPs containing a glycolytic inhibitor and self-antigen. Notably, this platform technology of glycolysis inhibition in the presence of a self-antigen provides a general strategy to generate antigen-specific immune suppression and can be applied to autoimmune diseases where the self-autoantigens are known.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to acknowledge the Flow Cytometry Core, the Regenerative Medicine Imaging Facility, the KED Genomics Core, the FEI at Erying Materials Center, the Advanced Light Microscopy Facilities, and the Department of Animal Care and Technologies at Arizona State University. Additionally, the authors would like to thank Dr. Seo, School of Molecular Sciences, Arizona State University for providing access to dynamic light scatter.
Funding:
The authors would also like to acknowledge the startup funds provided by Arizona State University to Abhinav P. Acharya for the completion of this study, NIH 1R01AI155907-01 and NIH R01AR078343.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests: Abhinav P. Acharya is affiliated with a start-up company, Immunometabolix, LLC. There are no other conflicts to declare.
Declaration of interests
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
Abhinav P. Acharya reports financial support was provided by National Institutes of Health. Abhinav P. Acharya has patent pending with Immunometabolix, LLC. N/A
Data and materials availability:
All data needed to evaluate the conclusions are present in the manuscript and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The raw data required to reproduce these findings are available upon request from the authors. The processed data required to reproduce these findings are available upon request from the authors.
REFERENCES
- [1].VanderBorght A, Geusens P, Raus J, Stinissen P, The autoimmune pathogenesis of rheumatoid arthritis: Role of autoreactive T cells and new immunotherapies, Semin. Arthritis Rheum (2001). 10.1053/sarh.2001.27736. [DOI] [PubMed] [Google Scholar]
- [2].Jensen SS, Gad M, Differential induction of inflammatory cytokines by dendritic cells treated with novel TLR-agonist and cytokine based cocktails: Targeting dendritic cells in autoimmunity, J. Inflamm (2010). 10.1186/1476-9255-7-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Audiger C, Rahman MJ, Yun TJ, Tarbell KV, Lesage S, The Importance of Dendritic Cells in Maintaining Immune Tolerance, J. Immunol (2017). 10.4049/jimmunol.1601629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Iberg CA, Jones A, Hawiger D, Dendritic Cells As Inducers of Peripheral Tolerance, Trends Immunol. (2017). 10.1016/j.it.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Durai V, Murphy KM, Functions of Murine Dendritic Cells, Immunity. 45 (2016). 10.1016/j.immuni.2016.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Maldonado RA, von Andrian UH, How Tolerogenic Dendritic Cells Induce Regulatory T Cells, in: Adv. Immunol, 2010. 10.1016/B978-0-12-380995-7.00004-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Raker VK, Domogalla MP, Steinbrink K, Tolerogenic dendritic cells for regulatory T cell induction in man, Front. Immunol 6 (2015). 10.3389/fimmu.2015.00569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Khan S, Greenberg JD, Bhardwaj N, Dendritic cells as targets for therapy in rheumatoid arthritis, Nat. Rev. Rheumatol (2009). 10.1038/nrrheum.2009.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Coutant F, Miossec P, Altered dendritic cell functions in autoimmune diseases: Distinct and overlapping profiles, Nat. Rev. Rheumatol (2016). 10.1038/nrrheum.2016.147. [DOI] [PubMed] [Google Scholar]
- [10].Pearce EL, Pearce EJ, Metabolic pathways in immune cell activation and quiescence., Immunity. (2013). 10.1016/j.immuni.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Nish SA, Schenten D, Wunderlich T, Pope SD, Gao Y, Hoshi N, Yu S, Yan X, Lee HK, Pasman L, Brodsky I, Yordy B, Zhao H, Bruning J, Medzhitov R, T cell-intrinsic role of IL-6 signaling in primary and memory responses, Elife. 2014 (2014). 10.7554/eLife.01949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zou Y, Zeng S, Huang M, Qiu Q, Xiao Y, Shi M, Zhan Z, Liang L, Yang X, Xu H, Inhibition of 6-phosphofructo-2-kinase suppresses fibroblast-like synoviocytes-mediated synovial inflammation and joint destruction in rheumatoid arthritis, Br. J. Pharmacol (2017). 10.1111/bph.13762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kaushik DK, Bhattacharya A, Mirzaei R, Rawji KS, Ahn Y, Rho JM, Yong VW, Enhanced glycolytic metabolism supports transmigration of brain-infiltrating macrophages in multiple sclerosis, J. Clin. Invest (2019). 10.1172/JCI124012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Liu R-T, Zhang M, Yang C-L, Zhang P, Zhang N, Du T, Ge M-R, Yue L-T, Li X-L, Li H, Duan R-S, Enhanced glycolysis contributes to the pathogenesis of experimental autoimmune neuritis, J. Neuroinflammation 15 (2018). 10.1186/s12974-018-1095-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Okazaki T, Shinagawa S, Mikage H, Vasculitis syndrome-diagnosis and therapy, J. Gen. Fam. Med (2017). 10.1002/jgf2.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sun L, Fu J, Zhou Y, Metabolism controls the balance of Th17/T-regulatory cells, Front. Immunol (2017). 10.3389/fimmu.2017.01632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].O’Neill LAJ, Pearce EJ, Immunometabolism governs dendritic cell and macrophage function, J. Exp. Med (2016). 10.1084/jem.20151570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ryan DG, O’Neill LAJ, Krebs cycle rewired for macrophage and dendritic cell effector functions, FEBS Lett. (2017). 10.1002/1873-3468.12744. [DOI] [PubMed] [Google Scholar]
- [19].Abboud G, Choi SC, Kanda N, Zeumer-Spataro L, Roopenian DC, Morel L, Inhibition of glycolysis reduces disease severity in an autoimmune model of rheumatoid arthritis, Front. Immunol (2018). 10.3389/fimmu.2018.01973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Seki SM, Gaultier A, Exploring non-metabolic functions of glycolytic enzymes in immunity, Front. Immunol 8 (2017). 10.3389/fimmu.2017.01549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yin Z, Bai L, Li W, Zeng T, Tian H, Cui J, Targeting T cell metabolism in the tumor microenvironment: an anti-cancer therapeutic strategy, J. Exp. Clin. Cancer Res (2019). 10.1186/s13046-019-1409-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wang C, Qu J, Yan S, Gao Q, Hao S, Zhou D, PFK15, a PFKFB3 antagonist, inhibits autophagy and proliferation in rhabdomyosarcoma cells, Int. J. Mol. Med (2018). 10.3892/ijmm.2018.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Miller SD, Turley DM, Podojil JR, Antigen-specific tolerance strategies for the prevention and treatment of autoimmune disease, Nat. Rev. Immunol (2007). 10.1038/nri2153. [DOI] [PubMed] [Google Scholar]
- [24].Theil A, Tuve S, Oelschlägel U, Maiwald A, Döhler D, Oßmann D, Zenkel A, Wilhelm C, Middeke JM, Shayegi N, Trautmann-Grill K, von Bonin M, Platzbecker U, Ehninger G, Bonifacio E, Bornhäuser M, Adoptive transfer of allogeneic regulatory T cells into patients with chronic graft-versus-host disease, Cytotherapy. 17 (2015). 10.1016/j.jcyt.2014.11.005. [DOI] [PubMed] [Google Scholar]
- [25].McDonald-Hyman C, Flynn R, Panoskaltsis-Mortari A, Peterson N, MacDonald KPA, Hill GR, Luznik L, Serody JS, Murphy WJ, Maillard I, Munn DH, Turka LA, Koreth J, Cutler CS, Soiffer RJ, Antin JH, Ritz J, Blazar BR, Therapeutic regulatory T-cell adoptive transfer ameliorates established murine chronic GVHD in a CXCR5-dependent manner, Blood. 128 (2016). 10.1182/blood-2016-05-715896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Rapoport AP, Stadtmauer EA, Aqui N, Vogl D, Chew A, Bin Fang H, Janofsky S, Yager K, Veloso E, Zheng Z, Milliron T, Westphal S, Cotte J, Huynh H, Cannon A, Yanovich S, Akpek G, Tan M, Virts K, Ruehle K, Harris C, Philip S, Vonderheide RH, Levine BL, June CH, Rapid immune recovery and graft-versus-host disease - Like engraftment syndrome following adoptive transfer of costimulated autologous T cells, Clin. Cancer Res 15 (2009). 10.1158/1078-0432.CCR-09-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Tricot G, Vesole DH, Jagannath S, Hilton J, Munshi N, Barlogie B, Graft-versus-myeloma effect: Proof of principle, Blood. 87 (1996). 10.1182/blood.v87.3.1196.bloodjournal8731196. [DOI] [PubMed] [Google Scholar]
- [28].Lokhorst HM, Wu K, Verdonck LF, Laterveer LL, Van De Donk NWCJ, Van Oers MHJ, Cornelissen JJ, Schattenberg AV, The occurrence of graft-versus-host disease is the major predictive factor for response to donor lymphocyte infusions in multiple myeloma, Blood. 103 (2004). 10.1182/blood-2003-11-3862. [DOI] [PubMed] [Google Scholar]
- [29].Siepmann J, Siepmann F, Microparticles used as drug delivery systems, Prog. Colloid Polym. Sci 133 (2006). 10.1007/2882_053. [DOI] [Google Scholar]
- [30].Nidhi, Rashid M, Kaur V, Hallan SS, Sharma S, Mishra N, Microparticles as controlled drug delivery carrier for the treatment of ulcerative colitis: A brief review, Saudi Pharm. J 24 (2016). 10.1016/j.jsps.2014.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Abdellatif AA, Microparticles Formulation as a Targeting Drug Delivery System, J. Nanomedicine Res 6 (2017). 10.15406/jnmr.2017.06.00151. [DOI] [Google Scholar]
- [32].Acharya AP, Clare-Salzler MJ, Keselowsky BG, A high-throughput microparticle microarray platform for dendritic cell-targeting vaccines, Biomaterials. (2009). 10.1016/j.biomaterials.2009.04.032. [DOI] [PubMed] [Google Scholar]
- [33].Fisher JD, Acharya AP, Little SR, Micro and nanoparticle drug delivery systems for preventing allotransplant rejection, Clin. Immunol 160 (2015) 24–35. 10.1016/j.clim.2015.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wu N, Yang M, Gaur U, Xu H, Yao Y, Li D, Alpha-ketoglutarate: Physiological functions and applications, Biomol. Ther (2016). 10.4062/biomolther.2015.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Mangal JL, Inamdar S, Yang Y, Shi X, Wankhede M, Gu H, Rege K, Green MD, Curtis M, Acharya A, Metabolite releasing polymers control dendritic cell function by modulating their energy metabolism, J. Mater. Chem. B (2020). 10.1039/d0tb00790k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zdzisińska B, Żurek A, Kandefer-Szerszeń M, Alpha-Ketoglutarate as a Molecule with Pleiotropic Activity: Well-Known and Novel Possibilities of Therapeutic Use, Arch. Immunol. Ther. Exp. (Warsz) (2017). 10.1007/s00005-016-0406-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Liu G, Huang Y, Zhai L, Impact of Nutritional and Environmental Factors on Inflammation, Oxidative Stress, and the Microbiome, Biomed Res. Int (2018). 10.1155/2018/5606845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Tennant DA, Frezza C, MacKenzie ED, Nguyen QD, Zheng L, Selak MA, Roberts DL, Dive C, Watson DG, Aboagye EO, Gottlieb E, Reactivating HIF prolyl hydroxylases under hypoxia results in metabolic catastrophe and cell death, Oncogene. 28 (2009). 10.1038/onc.2009.250. [DOI] [PubMed] [Google Scholar]
- [39].Li S, Fu C, Zhao Y, He J, Tong Q, Intervention with α-ketoglutarate ameliorates colitis-related colorectal carcinoma via modulation of the gut microbiome, Biomed Res. Int (2019). 10.1155/2019/8020785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chimenti MS, Triggianese P, Conigliaro P, Candi E, Melino G, Perricone R, The interplay between inflammation and metabolism in rheumatoid arthritis, Cell Death Dis. (2015). 10.1038/cddis.2015.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Baum CL, Selhub J, Rosenberg IH, Antifolate actions of sulfasalazine on intact lymphocytes, J. Lab. Clin. Med (1981). 10.5555/uri:pii:0022214381902006. [DOI] [PubMed] [Google Scholar]
- [42].Cutolo M, Sulli A, Pizzorni C, Seriolo B, STRAUB RH, Anti-inflammatory mechanisms of methotrexate in rheumatoid arthritis, Ann. Rheum. Dis (2001). 10.1136/ard.60.8.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Spurlock CF, Olsen NJ, Aune TM, Will Understanding Methotrexate Modes of Action Teach us About Rheumatoid Arthritis?, in: Autoimmun. - Pathog. Clin. Asp. Ther. Specif. Autoimmune Dis, 2015. 10.5772/59901. [DOI] [Google Scholar]
- [44].Page A, Fusil F, Cosset FL, Antigen-specific tolerance approach for rheumatoid arthritis: Past, present and future, Jt. Bone Spine 88 (2021). 10.1016/j.jbspin.2021.105164. [DOI] [PubMed] [Google Scholar]
- [45].Liu J, Kjeken R, Mathiesen I, Barouch DH, Recruitment of Antigen-Presenting Cells to the Site of Inoculation and Augmentation of Human Immunodeficiency Virus Type 1 DNA Vaccine Immunogenicity by In Vivo Electroporation, J. Virol 82 (2008). 10.1128/jvi.02564-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Genito CJ, Batty CJ, Bachelder EM, Ainslie KM, Considerations for Size, Surface Charge, Polymer Degradation, Co-Delivery, and Manufacturability in the Development of Polymeric Particle Vaccines for Infectious Diseases, Adv. NanoBiomed Res 1 (2021). 10.1002/anbr.202000041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Myers LK, Rosloniec EF, Cremer MA, Kang AH, Minireview: Collagen-induced arthritis, an animal model of autoimmunity, Life Sci. (1997). 10.1016/S0024-3205(97)00480-3. [DOI] [PubMed] [Google Scholar]
- [48].Ratay ML, Glowacki AJ, Balmert SC, Acharya AP, Polat J, Andrews LP, Fedorchak MV, Schuman JS, Vignali DAA, Little SR, Treg-recruiting microspheres prevent inflammation in a murine model of dry eye disease, J. Control. Release (2017). 10.1016/j.jconrel.2017.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Acharya AP, Lewis JS, Keselowsky BG, Combinatorial co-encapsulation of hydrophobic molecules in poly(lactide-co-glycolide) microparticles, Biomaterials. (2013). 10.1016/j.biomaterials.2013.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ratay ML, Balmert SC, Acharya AP, Greene AC, Meyyappan T, Little SR, TRI Microspheres prevent key signs of dry eye disease in a murine, inflammatory model, Sci. Rep 7 (2017). 10.1038/s41598-017-17869-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Acharya AP, Sinha M, Ratay ML, Ding X, Balmert SC, Workman CJ, Wang Y, Vignali DAA, Little SR, Localized Multi-Component Delivery Platform Generates Local and Systemic Anti-Tumor Immunity, Adv. Funct. Mater 27 (2017). 10.1002/adfm.201604366. [DOI] [Google Scholar]
- [52].Acharya AP, Dolgova NV, Xia CQ, Clare-Salzler MJ, Keselowsky BG, Adhesive substrates modulate the activation and stimulatory capacity of non-obese diabetic mouse-derived dendritic cells, Acta Biomater. (2011). 10.1016/j.actbio.2010.08.026. [DOI] [PubMed] [Google Scholar]
- [53].Acharya AP, Dolgova NV, Clare-Salzler MJ, Keselowsky BG, Adhesive substrate-modulation of adaptive immune responses, Biomaterials. (2008). 10.1016/j.biomaterials.2008.08.040. [DOI] [PubMed] [Google Scholar]
- [54].Lewis JS, Dolgova NV, Chancellor TJ, Acharya AP, Karpiak JV, Lele TP, Keselowsky BG, The effect of cyclic mechanical strain on activation of dendritic cells cultured on adhesive substrates, Biomaterials. 34 (2013). 10.1016/j.biomaterials.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Lewis JS, Zaveri TD, Crooks CP, Keselowsky BG, Microparticle surface modifications targeting dendritic cells for non-activating applications, Biomaterials. (2012). 10.1016/j.biomaterials.2012.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Jasbi P, Mitchell NM, Shi X, Grys TE, Wei Y, Liu L, Lake DF, Gu H, Coccidioidomycosis Detection Using Targeted Plasma and Urine Metabolic Profiling, J. Proteome Res (2019). 10.1021/acs.jproteome.9b00100. [DOI] [PubMed] [Google Scholar]
- [57].Parent BA, Seaton M, Sood RF, Gu H, Djukovic D, Raftery D, O’Keefe GE, Use of metabolomics to trend recovery and therapy after injury in critically ill trauma patients, JAMA Surg. (2016). 10.1001/jamasurg.2016.0853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Eghlimi R, Shi X, Hrovat J, Xi B, Gu H, Triple Negative Breast Cancer Detection Using LC-MS/MS Lipidomic Profiling, J. Proteome Res (2020). 10.1021/acs.jproteome.0c00038. [DOI] [PubMed] [Google Scholar]
- [59].He H, Shi X, Lawrence A, Hrovat J, Turner C, Cui JY, Gu H, 2,2′,4,4′-tetrabromodiphenyl ether (BDE-47) induces wide metabolic changes including attenuated mitochondrial function and enhanced glycolysis in PC12 cells, Ecotoxicol. Environ. Saf (2020). 10.1016/j.ecoenv.2020.110849. [DOI] [PubMed] [Google Scholar]
- [60].Shi X, Wang S, Jasbi P, Turner C, Hrovat J, Wei Y, Liu J, Gu H, Database-Assisted Globally Optimized Targeted Mass Spectrometry (dGOT-MS): Broad and Reliable Metabolomics Analysis with Enhanced Identification, Anal. Chem (2019). 10.1021/acs.analchem.9b03107. [DOI] [PubMed] [Google Scholar]
- [61].Jasbi P, Baker O, Shi X, Gonzalez L, Wang S, Anderson S, Xi B, Gu H, Johnston C, Daily Red Wine Vinegar Ingestion for Eight Weeks Improves Glucose Homeostasis and Affects the Metabolome but does not Reduce Adiposity in Adults, Food Funct. (2019). 10.1039/c9fo01082c. [DOI] [PubMed] [Google Scholar]
- [62].Jasbi P, Wang D, Cheng SL, Fei Q, Cui JY, Liu L, Wei Y, Raftery D, Gu H, Breast cancer detection using targeted plasma metabolomics, J. Chromatogr. B Anal. Technol. Biomed. Life Sci (2019). 10.1016/j.jchromb.2018.11.029. [DOI] [PubMed] [Google Scholar]
- [63].Li CY, Dempsey JL, Wang D, Lee SW, Weigel KM, Fei Q, Bhatt DK, Prasad B, Raftery D, Gu H, Cui JY, PBDEs altered gut microbiome and bile acid homeostasis in male C57BL/6 mice, Drug Metab. Dispos (2018). 10.1124/dmd.118.081547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Curtis M, Kenny HA, Ashcroft B, Mukherjee A, Johnson A, Zhang Y, Helou Y, Batlle R, Liu X, Gutierrez N, Gao X, Yamada SD, Lastra R, Montag A, Ahsan N, Locasale JW, Salomon AR, Nebreda AR, Lengyel E, Fibroblasts Mobilize Tumor Cell Glycogen to Promote Proliferation and Metastasis, Cell Metab. (2019). 10.1016/j.cmet.2018.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Engineer C, Parikh J, Raval A, Review on hydrolytic degradation behavior of biodegradable polymers from controlled drug delivery system, Trends Biomater. Artif. Organs 25 (2011). [Google Scholar]
- [66].Bousso P, T-cell activation by dendritic cells in the lymph node: Lessons from the movies, Nat. Rev. Immunol (2008). 10.1038/nri2379. [DOI] [PubMed] [Google Scholar]
- [67].Chandrashekara S, The treatment strategies of autoimmune disease may need a different approach from conventional protocol: A review, Indian J. Pharmacol 44 (2012). 10.4103/0253-7613.103235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Pearce EJ, Everts B, Dendritic cell metabolism, Nat. Rev. Immunol (2015). 10.1038/nri3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Okano T, Saegusa J, Nishimura K, Takahashi S, Sendo S, Ueda Y, Morinobu A, 3-bromopyruvate ameliorate autoimmune arthritis by modulating Th17/Treg cell differentiation and suppressing dendritic cell activation, Sci. Rep (2017). 10.1038/srep42412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Redman R, Pohlmann P, Kurman M, Tapolsky GH, Chesney J, Abstract CT206: PFK-158, first-in-man and first-in-class inhibitor of PFKFB3/glycolysis: A phase I, dose escalation, multi-center study in patients with advanced solid malignancies, in: 2015. 10.1158/1538-7445.am2015-ct206. [DOI]
- [71].Xintaropoulou C, Ward C, Wise A, Marston H, Turnbull A, Langdon SP, A comparative analysis of inhibitors of the glycolysis pathway in breast and ovarian cancer cell line models, Oncotarget. (2015). 10.18632/oncotarget.4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Lewis JS, Roche C, Zhang Y, Brusko TM, Wasserfall CH, Atkinson M, Clare-Salzler MJ, Keselowsky BG, Combinatorial delivery of immunosuppressive factors to dendritic cells using dual-sized microspheres, J. Mater. Chem. B (2014). 10.1039/c3tb21460e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Lewis JS, Stewart JM, Marshall GP, Carstens MR, Zhang Y, V Dolgova N, Xia C, Brusko TM, Wasserfall CH, Clare-Salzler MJ, Atkinson MA, Keselowsky BG, Dual-Sized Microparticle System for Generating Suppressive Dendritic Cells Prevents and Reverses Type 1 Diabetes in the Nonobese Diabetic Mouse Model, ACS Biomater. Sci. Eng (2019). 10.1021/acsbiomaterials.9b00332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Wischke C, Zimmermann J, Wessinger B, Schendler A, Borchert HH, Peters JH, Nesselhut T, Lorenzen DR, Poly(I:C) coated PLGA microparticles induce dendritic cell maturation, Int. J. Pharm (2009). 10.1016/j.ijpharm.2008.08.039. [DOI] [PubMed] [Google Scholar]
- [75].Yoshida M, Babensee JE, Molecular aspects of microparticle phagocytosis by dendritic cells, J. Biomater. Sci. Polym. Ed (2006). 10.1163/156856206777996844. [DOI] [PubMed] [Google Scholar]
- [76].Román BS, Irache JM, Gómez S, Tsapis N, Gamazo C, Espuelas MS, Co-encapsulation of an antigen and CpG oligonucleotides into PLGA microparticles by TROMS technology, Eur. J. Pharm. Biopharm (2008). 10.1016/j.ejpb.2008.03.015. [DOI] [PubMed] [Google Scholar]
- [77].Patente TA, Pinho MP, Oliveira AA, Evangelista GCM, Bergami-Santos PC, Barbuto JAM, Human dendritic cells: Their heterogeneity and clinical application potential in cancer immunotherapy, Front. Immunol (2019). 10.3389/fimmu.2018.03176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Corinti S, Medaglini D, Prezzi C, Cavani A, Pozzi G, Girolomoni G, Human dendritic cells are superior to B cells at presenting a major histocompatibility complex class II-restricted heterologous antigen expressed on recombinant Streptococcus gordonii, Infect. Immun 68 (2000). 10.1128/IAI.68.4.1879-1883.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Chaperot L, Chokri M, Jacob MC, Drillat P, Garban F, Egelhofer H, Molens JP, Sotto JJ, Bensa JC, Plumas J, Differentiation of antigen-presenting cells (dendritic cells and macrophages) for therapeutic application in patients with lymphoma, Leukemia. 14 (2000). 10.1038/sj.leu.2401888. [DOI] [PubMed] [Google Scholar]
- [80].Clemente-Casares X, Blanco J, Ambalavanan P, Yamanouchi J, Singha S, Fandos C, Tsai S, Wang J, Garabatos N, Izquierdo C, Agrawal S, Keough MB, Yong VW, James E, Moore A, Yang Y, Stratmann T, Serra P, Santamaria P, Expanding antigen-specific regulatory networks to treat autoimmunity, Nature. (2016). 10.1038/nature16962. [DOI] [PubMed] [Google Scholar]
- [81].Lawless SJ, Kedia-Mehta N, Walls JF, McGarrigle R, Convery O, Sinclair LV, Navarro MN, Murray J, Finlay DK, Glucose represses dendritic cell-induced T cell responses, Nat. Commun (2017). 10.1038/ncomms15620. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions are present in the manuscript and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The raw data required to reproduce these findings are available upon request from the authors. The processed data required to reproduce these findings are available upon request from the authors.