

Abstract
Alzheimer’s disease (AD) is the most common dementia, and no disease-modifying therapeutic agents are currently available. BDNF/TrkB signaling is impaired in AD and is associated with prominent delta-secretase (δ-secretase, also known as asparaginyl endopeptidase or legumain) activation, which simultaneously cleaves both APP and Tau and promotes Aβ production and neurofibrillary tangles (NFT) pathologies. Here we show that the optimized δ-secretase inhibitor (#11a) or TrkB receptor agonist (CF3CN) robustly blocks δ-secretase activity separately, and their combination synergistically blunts δ-secretase, exhibiting promising therapeutic efficacy in 3xTg AD mouse model. The optimal δ-secretase inhibitor reveals demonstrable brain exposure and oral bioavailability, suppressing APP N585 and Tau N368 cleavage by δ-secretase. Strikingly, CF3CN treatment evidently escalates BDNF levels. Both #11a and CF3CN display strong in vivo PK/PD properties and ability to suppress δ-secretase activity in the brain. Orally administrated CF3CN strongly activates TrkB that triggers active Akt to phosphorylate δ-secretase T322, preventing its proteolytic activation and mitigating AD pathologies. #11a or CF3CN significantly diminishes AD pathogenesis and improves cognitive functions with the combination exhibiting the maximal effect. Thus, our data support that these derivatives are strong pharmaceutical candidates for the treatment of AD.
Keywords: CF3CN, #11a, BDNF, Delta-secretase, Alzheimer’s Disease, Neuroprotection
Graphical Abstract
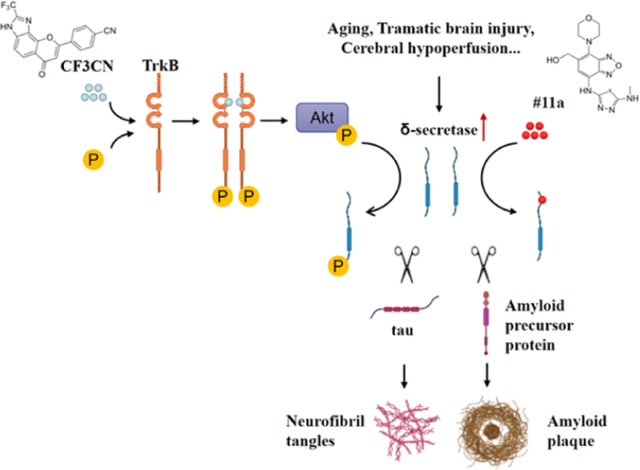
Introduction
Alzheimer’s disease (AD), the leading cause of dementia, is the most common neurodegenerative disease with aging as the major risk factor. The characteristic hallmarks include extracellular senile plaques, mainly composed of Aβ aggregates, and intraneuronal neurofibrillary tangles (NFT), principally consisting of hyperphosphorylated and truncated Tau. In addition, the pathological features involve progressive neuronal loss and chronic neuroinflammation (Hardy and Selkoe, 2002). The most prominent clinical manifestation is the gradual cognitive decline with aging. At present, drugs used for the treatment of AD only slightly delay the inevitable symptomatic progression of the disease and do not affect the main neuropathological hallmarks of the disease (Cummings et al., 2017).
AEP (asparagine endopeptidase, also called legumain) is an acidosis-activated cysteine protease that predominantly resides within endo-lysosomes (Dall and Brandstetter, 2016). Recently, we report that AEP acts as δ-secretase that simultaneously cleaves both APP N585 and Tau N368 and escalates Aβ production and Tau aggregation. Deletion of δ-secretase from AD mouse models strongly decreases senile plaque deposition and abrogates NFT pathologies, ameliorating cognitive dysfunctions (Zhang et al., 2014b; Zhang et al., 2015). Consequently, blockade of δ-secretase with its specific small-molecule inhibitor (#11) mitigates AD pathogenesis, displaying potential therapeutic efficacy in AD mouse models (Zhang et al., 2017). Mounting evidence documents that BDNF (brain-derived neurotrophic factor), a cognate ligand for TrkB receptors, is reduced in the brains of AD patients (Connor et al., 1997; Ferrer et al., 1999; Phillips et al., 1991). BDNF, a neurotrophic growth factor, regulates neuronal development, differentiation, and survival in both the peripheral nervous system and CNS (Huang and Reichardt, 2003). Notably, BDNF-provoked Akt phosphorylates δ-secretase on residue T322 and triggers its lysosomal translocation and inactivation. When BDNF levels are reduced in neurodegenerative diseases, AEP T322 phosphorylation is attenuated. Consequently, it is activated and translocates into the cytoplasm (Wang et al., 2018b). Interestingly, we found that Tau N368 fragment directly binds TrkB receptors and blocks its neurotrophic signals, inducing neuronal cell death. Knockout of BDNF or TrkB receptors provokes δ-secretase activation via reducing T322 phosphorylation and subsequent Tau N368 cleavage, inducing AD-like pathology and cognitive dysfunction (Xiang et al., 2019). Remarkably, δ-secretase is age-dependently elevated and activated in the brain, which is upregulated by the crucial transcription factor C/EBPβ (Wang et al., 2018a). Most recently, we show that deprivation of BDNF/TrkB increases inflammatory cytokines and activates the JAK2/STAT3 pathway, resulting in upregulation of C/EBPβ. This, in turn, leads to increased expression of δ-secretase, APP and Tau fragmentation by δ-secretase and neuronal loss (Wang et al., 2019). Noticeably, longitudinal Tau N368 in CSF of AD patients inversely correlates with metabolic 18F-FDG PET in human AD brains (Leuzy et al., 2019). In addition, employing a Tau PET, we show that Tau N368 is a tangle-enriched fragment and that the CSF ratio Tau368/t-Tau reflects the tangle pathology. Hence, this novel Tau biomarker could be used to improve diagnosis of AD and to facilitate the development of drug candidates targeting Tau pathology (Blennow et al., 2020).
Accumulating evidence demonstrates that BDNF displays a protective role against AD pathogenesis. BDNF increases learning and memory of demented animals (Ando et al., 2002). BDNF gene delivery has been shown to be a novel potential therapeutic in diverse models related to AD (Nagahara et al., 2009). Thus, preclinical evidence strongly supports that BDNF might be useful as a therapeutic agent for a variety of neurological disorders. However, the outcomes of several clinical trials using recombinant BDNF are disappointing (Henriques et al., 2010). Presumably, this is due to poor delivery and the short in vivo half-life of BDNF (Sakane and Pardridge, 1997; Soderquist et al., 2009). Employing a cell-based screening, we identified that 7,8-dihydroxyflavone (7,8-DHF) specifically binds to the extracellular domain of the TrkB receptor and acts as a selective TrkB agonist, which mimics the physiological actions of BDNF (Jang et al., 2010). Oral administration of 7,8-DHF activates TrkB receptors in the brains and induces BDNF-like behavioral phenotypes such as enhanced learning and memory, and anti-stress or antidepressant-like effects in rodents in a TrkB-dependent manner (Andero et al., 2011; Choi et al., 2010; Liu et al., 2010). Recently, several independent studies show that 7,8-DHF rescues memory deficits in different AD animal models. Therefore, 7,8-DHF represents an innovative orally bioactive therapeutic agent for treating AD (Devi and Ohno, 2012; Zhang et al., 2014a). To improve 7,8-DHF oral bioavailability and in vivo PK (pharmacokinetics) profiles, we developed a prodrug of 7,8-DHF, R13, with a carbamate modification of the catechol group, and found that R13 possesses much improved drug-like properties. Chronic oral administration of R13 activates TrkB signaling and prevents Aβ deposition in 5xFAD mice, inhibiting the pathological cleavage of APP and Tau by δ-secretase. Moreover, R13 reduces the loss of hippocampal synapses and ameliorates memory deficits in a dose-dependent manner (Chen et al., 2018). Utilizing medicinal chemistry and iterative organic synthesis and in vitro activity assays, we have also optimized 7,8-DHF into a very stable CF3CN synthetic derivative that displays TrkB agonism in cellular assays with an EC50 of ~26.4 nM. Chronic oral administration of CF3CN reveals promising therapeutic effects in 5xFAD mouse model (Chen et al., 2021). In the current study, we have also generated an optimized δ-secretase inhibitor using structure-based drug design, medicinal chemistry, in vitro ADMET (Absorption, Distribution, Metabolism, Excretion and Toxicity) screening and an in vivo PK triage strategy. The preclinical candidate #11a exhibits a potent inhibitory effect. Either #11a or CF3CN reveals notable therapeutic efficacy in mitigating AD pathologies, enhancing cognitive functions in 3xTg AD mice with the strongest effects observed after combination of both #11a and CF3CN. Interestingly, CF3CN robustly enhances BDNF levels in the brain and strongly phosphorylates AEP T322, antagonizing δ-secretase activity.
Materials and Methods
Mice and Reagents
3xTg mice on a C57BL/6J background were obtained from the Jackson Laboratory and were bred in a pathogen-free environment in accordance with Emory Medical School guidelines. #11a and CF3CN were firstly dissolved in DMSO with good dissolvability and then added to the 0.5% methyl cellulose solution. The 3 months old 3xTg mice was treated with daily oral injection of these compounds (7.5 mg/kg for #11a, 3 mg/kg for CF3CN) or their mixture (7.5 mg/kg #11a + 3 mg/kg CF3CN) consecutively for 3 months. Animals were equally divided into groups for each sex. To study the cognitive functions of the mice after 3 months drug treatment, Morries Water Maze and fear condition were performed. The animals were sacrificed after behaviour test. The protocol was reviewed and approved by the Emory Institutional animal care and use committee.
Anti-TrkB antibody was purchased from Biovision. Anti-Phospho-TrkB Y816 was raised against [H]-CKLQNLAKASPV-pY-LDILG-[OH] (aa806–822; EM437 and EM438) as rabbit polyclonal antibody. Anti-C/EBPβ and anti-actin were purchased from Santa Cruz Biotechnology. Anti-Akt, anti-p-Akt, anti-ERK, anti-p-ERK, anti-AEP, anti-p-C/EBPβ, anti-BDNF, anti-spinophilin, anti-PSD95, anti-Glu2, anti-synaptotagamine were purchased from Cell Signaling. Synthetic Aβ (1–42) was purchased from rPeptide. Recombinant AEP was purchased from Novoprotein. AEP substrate Z-Ala-Ala-Asn-AMC was purchased from Bachem. Human Aβ40 and Aβ42 ELISA kits, pro-inflammatory cytokines IL-1β, TNF-α, IL-6 ELISA kits were purchased from Invitrogen. The In situ Cell Death Detection Kit was purchased from Roche.
Primary Neuron Cultures
Primary rat cortical neurons were cultured as previously described. Neuron Cultured 13 days in vitro (DIV13) were treated with different dose of #11A (0, 5, 10, 50, 100, 500,1000 nM), or treated with vehicle, #11A, CF3CN or the mixture for 30 min before Aβ42 treatment. Then neurons were exposed to 20 μM pre-aggregated Aβ42 and incubated for 24 h. After Aβ42 exposure, neurons were incubated with LE28 for 5 h, then fixed in 4% formaldehyde, permeabilized, and stained with DAPI. Pictures of the Neurons were taken by fluorescence microscopy. LE28 signal intensity was scored using computer software Image J.
Assessing AEP activity in Vivo
Six-month-old 3xTg mice were injected with LE28 (10 nmol in 20% DMSO/PBS, ~2 mg/kg) by tail vein, 0.5 h after oral gavage of vehicle, #11A, CF3CN or the mixture of #11A and CF3CN. Mice were anesthetized with isoflurane and then imaged at 2 h after injection using an IVIS 100 system.
AEP activity assay
Tissue homogenates or cell lysates (10 μg) were incubated in 200 μl assay buffer (20 mM citric acid, 60 mM Na2HPO4, 1 mM EDTA, 0.1% CHAPS and 1 mM DTT, pH 6.0) containing 20 μM AEP substrate. AMC released by substrate cleavage was quantified by measuring at 460 nm in a fluorescence plate reader at 37℃ for 2 h in kinetic mode for 3 min.
Western Blot analysis
The mouse brain hippocampus tissue samples or cell were lysed in lysis buffer (50 mM Tris, 40 mM NaCl, 1 mM EDTA, 0.5% Triton X-100, 1.5 mM Na3VO4, 50 mM NaF, 10 mM sodium pyrophosphate and 10mM sodium β-glycerophosphate, pH 7.0, supplemented with a cocktail of protease inhibitors), and centrifuged for 15 min at 16000 g. The supernatant was boiled in SDS loading buffer. After SDS-PAGE, the samples were transferred to a nitrocellulose membrane. The membrane was blocked with TBST containing 5% nonfat milk for 1 h, followed by the incubation with primary antibody at 4 °C overnight, and with the secondary antibody for 1 h. The membrane was developed using the enhanced chemiluminescent detection system.
Immunohistochemistry
Free-floating 30 μm-thick serial section were blocked in 1% BSA and 0.3% Triton X-100 for 30 min, followed by incubation with primary antibodies for overnight at 4 °C. After being washed with TBS, the sections were incubated with a mixture of Alexa Fluor 488-/594- and CY5-coupled secondary antibodies (Invitrogen) for detection. TUNEL staining were detected using in situ Cell death detection kits according to the manufacture’s instruction. Images were acquired through Confocal (Olympus FV 1000).
Golgi staining
Mouse brains were fixed in 4% paraformaldehyde for 24 h, and then immersed in 3% potassium bichromate for 3 days in the dark. The solution was changed each day. Then the brains were transferred into 2% silver nitrate solution and incubated for 24 h in the dark. Vibratome sections were cut at 60 μm, air dried for 10min, dehydrate through 95% and 100% ethanol, cleared in xylene and cover slipped.
Aβ40 and 42 ELISA
To detect the Aβ deposition in 3xTg mice, the mouse brains were homogenized in 8x mass of 5 M guanidine HCl/50 mM Tris-HCl (pH 8.0), and incubated at room temperature for 3 h. Then the samples were diluted with cold reaction buffer and centrifuged at 16000 g for 20 min at 4° C. The supernatant was analyzed by human Aβ40 and Aβ42 ELISA kits according to the manufacture’s instruction.
Morris Water Maze
Six-month-old 3xTg mice were trained in a round, water-filled tub (52 inches diameter) in an environment rich with extra maze cues. Each subject was given 3 trails/ day for 5 consecutive days with a 15-min intertrial interval. The maximum trial length was 60 s, and if subjects did not reach the platform in the allotted time, they were manually guided to do it. Following the 5 days task acquisition, a probe trial was presented, during which time the platform was removed. All trials were analyzed for latency and swim speed by means of MazeScan (Clever Sys, Inc.).
Contextual and Cued fear conditioning
The ability to form and retain an association between an aversive experience and environmental cues was tested with a standard fear conditioning paradigm that occurs over a period of 3 d. Mice were placed in the fear conditioning apparatus composed of Plexiglass with a metal shock grid floor and allowed to explore the enclosure for 3 min. Following this habituation period, 3 conditioned stimulus (CS)-unconditioned stimulus (US) pairings were presented with a 1 min intertrial interval. The CS was composed of a 20 s, 85-dB tone and US was composed of 2 s of a 0.5-mA footshock, which was co-terminate with each CS presentation. One minute following the last CS-US presentation, mice were returned to their home cage. On day 2, the mice were presented with a context test, during which subjects were placed in the same chamber used during conditioning on day 1 and the amount of freezing was recorded via a camera and the software provided by Coulbourn. No shocks were given during the context test. On day 3, a tone test was presented, during which time subjects were exposed to the CS in a novel compartment. Initially, animals were allowed to explore the novel context for 2 min. Then the 85-db tone was presented for 6 min and the amount of freezing behavior was recorded.
Statistical analysis
All data are expressed as Mean ± S.E.M. from three or more independent experiments, and the level of significance between two groups was assessed with Student’s t-test. For more than two groups, one-way ANOVA test was applied. A value of p< 0.05 was considered to be statistically significant.
Results
Optimization of δ-secretase inhibitor compound #11
Via high-throughput screening and co-crystallization, we obtained a lead compound #11 that acts as a specific δ-secretase allosteric inhibitor that is highly brain permeable (Zhang et al., 2017). To further explore #11 structure-activity relationship (SAR) of compound #11, we synthesized compound #2 and #3, and found the N atom in the morpholino groups (compound #2) is indispensable. Moreover, the para-position primary amine is also critical for the inhibitory activity of #11 (compound #3). Among the top validated hits, two compounds BB1 and #10 (Zhang et al., 2017) contain phenyl substituted amino-five-membered aromatic rings (thiadiazole or thiazole), which appear to be active fragments. Since the lead compound #11 is also quite small with a primary amine group on the benzene ring, accordingly, we tethered these fragments together and synthesized a series of derivatives (Figure S1). In vitro δ-secretase inhibition assay revealed that the IC50 increased from 250 nM for the lead compound 1 (#11) to 4 nM (compound #6). Substituting the thiadiazole with an electron-withdrawing group diminished the inhibitory activity (compound #7 & #8), whereas electron-donating groups escalated the effect (compound #4, #5 & #6). However, thiol groups are usually unstable and labile for oxidation or conjugation. Hence, we replaced it with a methyl-amino group, because mono-methylated group is a H-bond donor like the thiol group and is more metabolically stable than the dimethyl-amino group. Excitingly, replacing -SH in compound #6 with CH3NH-did not cripple its potency, and compound #9 exhibited an IC50 of ~5 nM (Figure 1A). However, ethyl-amino replacement reduced the IC50 to 18 nM (compound #10). Strikingly, replacing the benzene ring with pyridine in compound #9 completely suppressed the inhibitory activity (compound #11).
Figure 1. # 11a inhibits AEP activity in primary neurons.
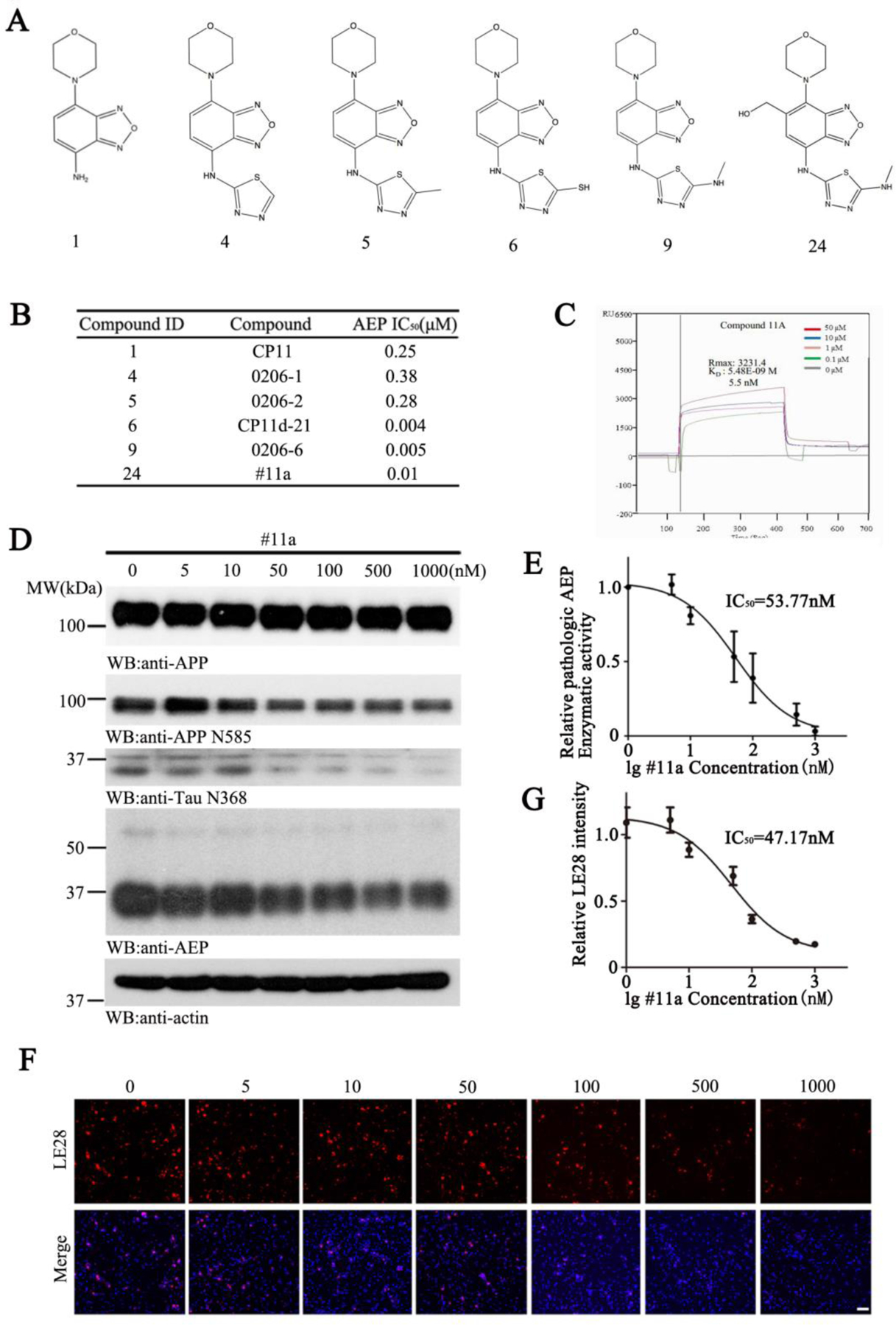
(A) Chemical Structure of the top 6 candidates.
(B) IC50 values of the compounds toward AEP.
(C) CP24 (#11a) binds to active AEP recombinant protein in Biacore assay. Active AEP recombinant protein was immobilized on Sam5 Chip.
(D) Immunoblotting analysis of AEP in vitro.
(E) Enzymatic activity of AEP was determined using an AEP fluorogenic substrate.
(F) Microscopy images of neurons exposed to different concentration of #11a, followed by incubation with LE28. Scale bar, 50μm.
(G) Quantification of LE28 fluorescent intensity in primary neurons.
Usually, benzene-diamine is metabolically unstable and easily modified by glutathione (GSH) conjugation in the liver. As expected, an in vitro active metabolite trapping assay demonstrated that compound #9 was covalently modified by GSH in liver microsomes (Figure S2). To alleviate this potential liver toxicity, we designed derivatives based on the co-crystal structure of #11 and active AEP (Figure S3). According to the docking model generated by compound #9 and AEP, there was no hydrogen bonding between the backbone pharmacophore and active AEP protein. The major stabilizing forces were hydrophobic interactions between #9 and His148, Tyr217 and Tyr228. Arg44 was observed to be close to the benzene ring in the benzoxadiazole of the compound. Compound #24–27 were designed to introduce various substituents in the X position that would generate possible hydrogen bonding with Arg44. As expected, compound #24 (#11a) exhibited the anticipated activity with an IC50 of ~10 nM (Figure 1B). Blocking the benzene ring with substitutive groups in compound #24–30, however, reduced their potencies as compared to compound #9. A BIACORE in vitro binding assay with purified recombinant active δ-secretase and compound #11a showed the binding affinity Kd was ~5.5 nM (Figure 1C). Compared to compound #9, #11a was stable in a reactive metabolite trapping assay without GSH conjugation (Figure S2). The structure of AEP shares similarities with the caspase family of cysteine proteases that are frequently activated in various apoptotic cells. Importantly, an in vitro inhibition assay revealed that #11a (compound #24) inhibited caspase-3 with an IC50 of ~30.2 µM, conferring 3020-fold greater selectivity for AEP (Table. S1). To assess whether the optimized #11a blocks δ-secretase in primary neurons, we incubated different concentrations of #11a in the presence of aggregated Aβ for 24 h. Immunoblotting revealed that #11a dose-dependently attenuated APP N585 and Tau N368 fragmentation by active AEP. Noticeably, AEP auto-activation via proteolytic cleavage was also gradually blocked as #11a doses progressively escalated (Figure 1D). The enzymatic activity of AEP as measured in vitro with a fluorogenic substrate displayed an IC50 of ~53.8 nM (Figure 1E). Alternatively, we also employed a specific fluorescent activity-based probe LE28 that covalently binds with active AEP (Edgington-Mitchell et al., 2016), and found that the IC50 was ~47.2 nM (Figure 1F & G). Hence, #11a is an optimized and stable lead compound with potent inhibitory effect against AEP.
In vivo PK profiles and oral bioavailability of optimal derivative #11a
To assess the druggability of #11a, we conducted a panel of in vitro ADMET assays. Biological/chemical stability assays revealed that this compound was very stable in PBS and human/mouse fluids with a half-life >469 min (Table. S2). A hepatocyte stability assay showed that it was very stable in human or rat hepatocytes with a half-life > 340 min and 480 min, respectively (Table. S3). A plasma protein binding assay revealed that the unbound fraction of #11a was around 43.3% and 26.9% in human and mouse plasma, respectively (Table. S4). Caco-2 and MDCK-MDR permeability assays demonstrated that the efflux ratios were 3.59 and 37.2, respectively (Table. S5 & 6), indicating that it might not be an optimal drug for cell or tissue permeability. PAMPA-BBB permeability assays revealed that the Pe (x 10−6 cm/s) was around 1.6, suggesting that it can penetrate the brain blood barrier. To ascertain this this compound without safety issue, we conducted a hERG channel inhibition assay and found no concentration-dependent cardiac K+ ion channel inhibition (Table. S7).
To investigate the in vivo PK profile of #11a, we administered 2 mg/kg i.v. or 5 mg/kg P.O. to 2-month-old ICR mice, followed by monitoring drug concentrations in the plasma and brain at different time points by LC/MS. The plasma half-life t1/2 was ~1.23 h with tmax at ~2.0 h and a Clearance rate CL 885.7 ml/min. The plasma Cmax was 59.4 ng/ml and the volume of distribution Vz was ~18.0 (L/kg), fitting with its lipophilicity, and it was not tightly bound to plasma proteins. The oral bioavailability (F) was ~129.8% based on the calculated AUClast, in agreement with its stability and good absorption. The drug was detectable in the brain at 2–4 h after oral administration, supporting that it is brain permeable (Figure 2). The summary of the in vitro ADMET and chemical and physical characteristics and in vivo PK features is included in Table. S8.
Figure 2. CP24 (#11a) concentrations in the brain and the plasma after oral administration or IV injection of CP24 (#11a).
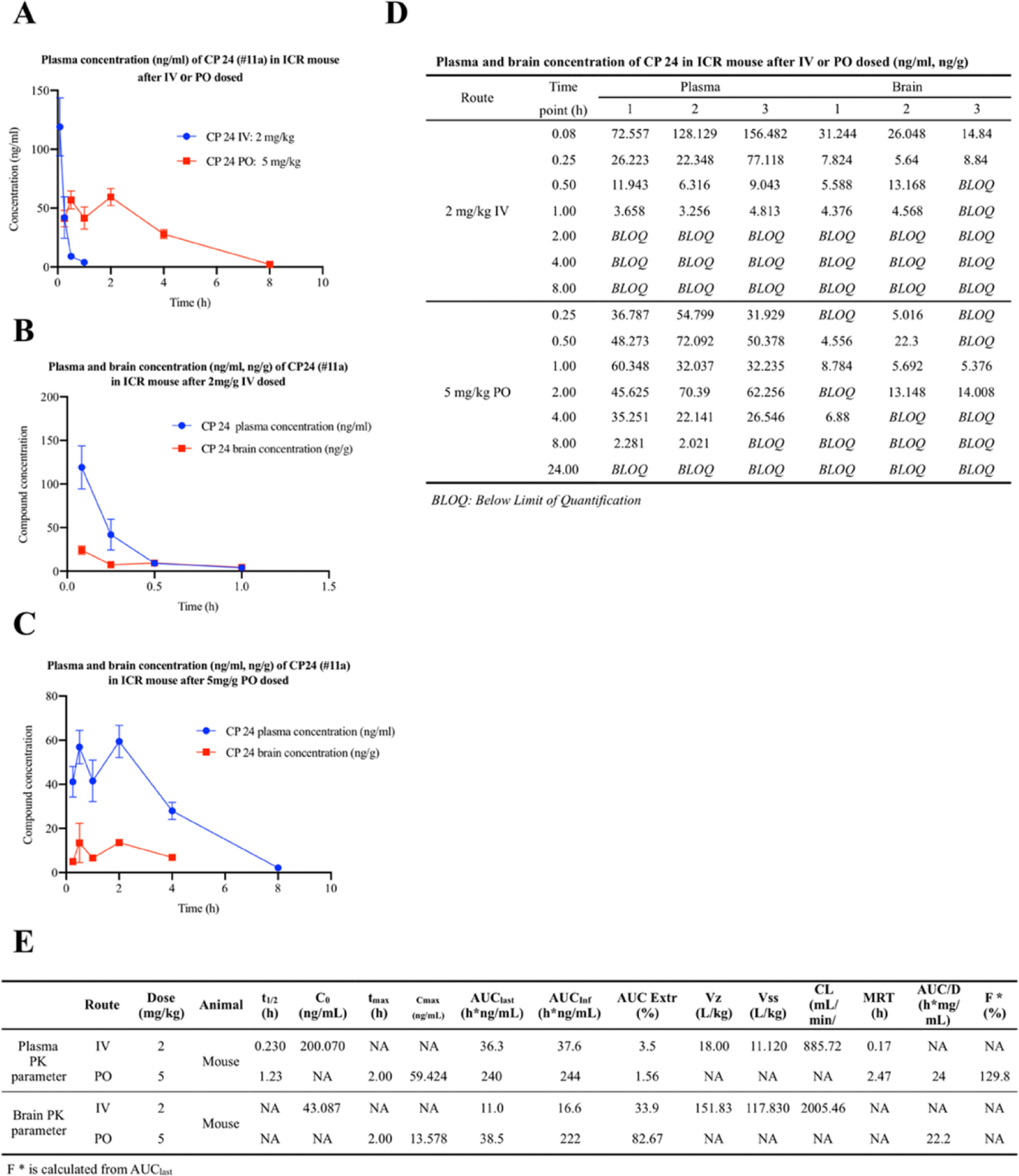
(A) Three ICR mice were given 5 mg/kg of CP24 (#11a), which was dissolved in DMSO and resuspended in 5% DMSO 95% methylcellulose (0.5%, wt/vol). Three ICR mice were given 2 mg/kg of CP24 (#11a), which was dissolved in PBS. At indicated time points, blood samples were collected from all three mice. At indicated time points, blood samples were collected from all three mice. CP24 (#11a) was quantitatively analyzed by LC-MS/MS.
(B) Nine ICR mice were given 2 mg/kg of CP24 (#11a), which was dissolved in PBS. At each indicated time points, three mice per group were sacrificed and serum and brain samples were collected. CP24 (#11a) was quantitatively analyzed by LC-MS/MS.
(C) Nine ICR mice were given 5 mg/kg of CP24 (#11a), which was dissolved in DMSO and resuspended in 5% DMSO 95% methylcellulose (0.5%, wt/vol). At each indicated time points, three mice per group were sacrificed and serum and brain samples were collected. CP24 (#11a) was quantitatively analyzed by LC-MS/MS.
(D) Plasma and brain concentrations of CP 24 (#11a) in ICR mouse after IV or PO dosed.
(E) CP24 (#11a) in vivo PK profiles.
#11a, CF3CN and their combination strongly inhibit AEP activity in primary neurons
CF3CN is a newly optimized synthetic 7,8-DHF derivative that exhibits stronger TrkB binding affinity (kd ~ 80.2 nM) and more potent agonistic activity (26.4 nM) than previously available compounds (Chen et al., 2021). We have recently reported that BDNF/TrkB signaling triggers δ-secretase T322 phosphorylation by active Akt, suppressing its activation (Wang et al., 2018b). To explore whether a combination of both TrkB agonist and δ-secretase inhibitor exhibits a synergistic effect in blocking the protease activity, we pre-treated primary neuronal cultures with #11a (10 nM), CF3CN (10 nM) or both drugs together, followed by stimulation with 2 µM aggregated Aβ for 24 h. Immunoblotting showed that p-TrkB was activated by CF3CN, and the mixture further escalated p-TrkB signals. Consequently, the downstream effectors p-Akt and p-ERK oscillated with the upstream p-TrkB activities. As a result, p-AEP T322 correlated with p-Akt levels, leading to repression of δ-secretase proteolytic activation. Accordingly, the downstream substrate APP N585 and Tau N368 fragmentation tightly mirrored the upstream protease activities (Figure 3A & B, p-TrkB/TrkB: F=92.25, p<0.0001; p-Akt/Akt: F=69.28, p<0.0001; p-ERK/ERK: F=39.26, p<0.0001). Use of a fluorogenic substrate revealed that #11a significantly inhibits the proteolytic activity of δ-secretase, as did CF3CN. The maximal inhibitory effect occurred in the presence of their combination (Figure 3C, F=126.2, p<0.0001). To further validate this synergistic effect, we used the activity-based probe LE28 to measure residual protease activity in primary neuronal cultures treated with these drugs and observed similar effects (Figure 3D & E, F=102.4, p<0.0001). Therefore, our findings support that either #11a or TrkB agonist CF3CN robustly antagonizes δ-secretase activation in primary neurons, and their mixture exhibits the synergistic effect in inhibiting δ-secretase.
Figure 3. #11a, CF3CN and their mixture inhibit AEP in primary neurons.
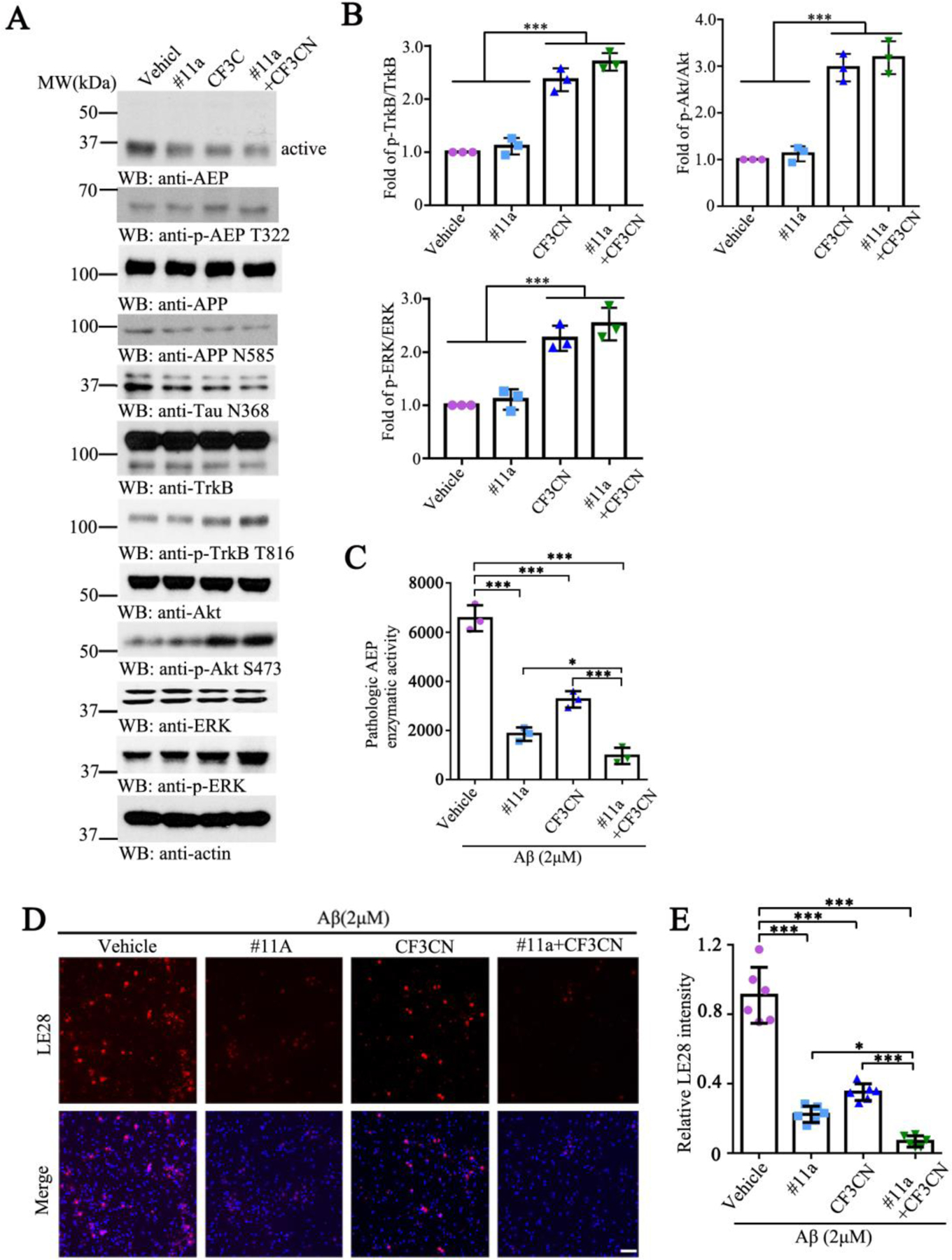
(A) Immunoblotting analysis of AEP, the p-TrkB and its downstream in vitro.
(B) Quantitative analysis of p-TrkB/TrkB, p-Akt/Akt and p-ERK/ERK in primary neurons. (n=3, one-way ANOVA)
(C) Enzymatic activity of AEP was determined using a fluorogenic substrate. (n=3, one-way ANOVA)
(D) Microscopy images of neurons exposed to Vehicle, #11a, CF3CN and their mixture, followed by incubation with LE28. Scale bar, 50 μm.
(E) Quantification of LE28 fluorescent intensity in primary neurons. (n=6, one-way ANOVA)
(Data represent the meauns ± SEM; *P < 0.05, **P < 0.01, ***P< 0.001).
#11a, CF3CN and their combination potently block δ-secretase in the brain in 3xTg mice
To assess whether orally administrated #11a or CF3CN inhibits δ-secretase in the mouse brains, we administered these compounds to 3-month-old 3xTg mice daily by oral gavage for 3 months (7.5 mg/kg for #11a, 3 mg/kg for CF3CN or their combination). At the end of the treatment, we monitored both APP and Tau proteolytic fragmentation in the brain via immunoblotting and found that APP N373 and N585 fragmentation by δ-secretase were prominently decreased by #11a or CF3CN. The maximal effect was observed in the samples treated with the combination of both #11a and CF3CN. Tau cleavage at N368 also exhibited a similar pattern. As observed in primary neuronal cultures, p-TrkB was strongly activated by CF3CN and the mixture elicited a stronger effect on p-TrkB. The downstream activation of Akt and ERK tightly coupled with the upstream p-TrkB activities. Again, p-AEP T322 levels closely correlated with p-Akt signals. As a consequence, δ-secretase proteolytic activation was strongly inhibited by #11a and the mixture (Figure 4A & B, p-TrkB/TrkB: F=6.866, p=0.0133; p-Akt/Akt: F=16.56, p=0.0009; p-ERK/ERK: F=12.82, p=0.002). D-secretase enzymatic assay with the brain lysates demonstrated the same inhibitory effect as the primary neuronal cultures (Figure 4C, F=68.47, p<0.0001).
Figure 4. #11a, CF3CN and their mixture block AEP in the brain of 3xTg mice.
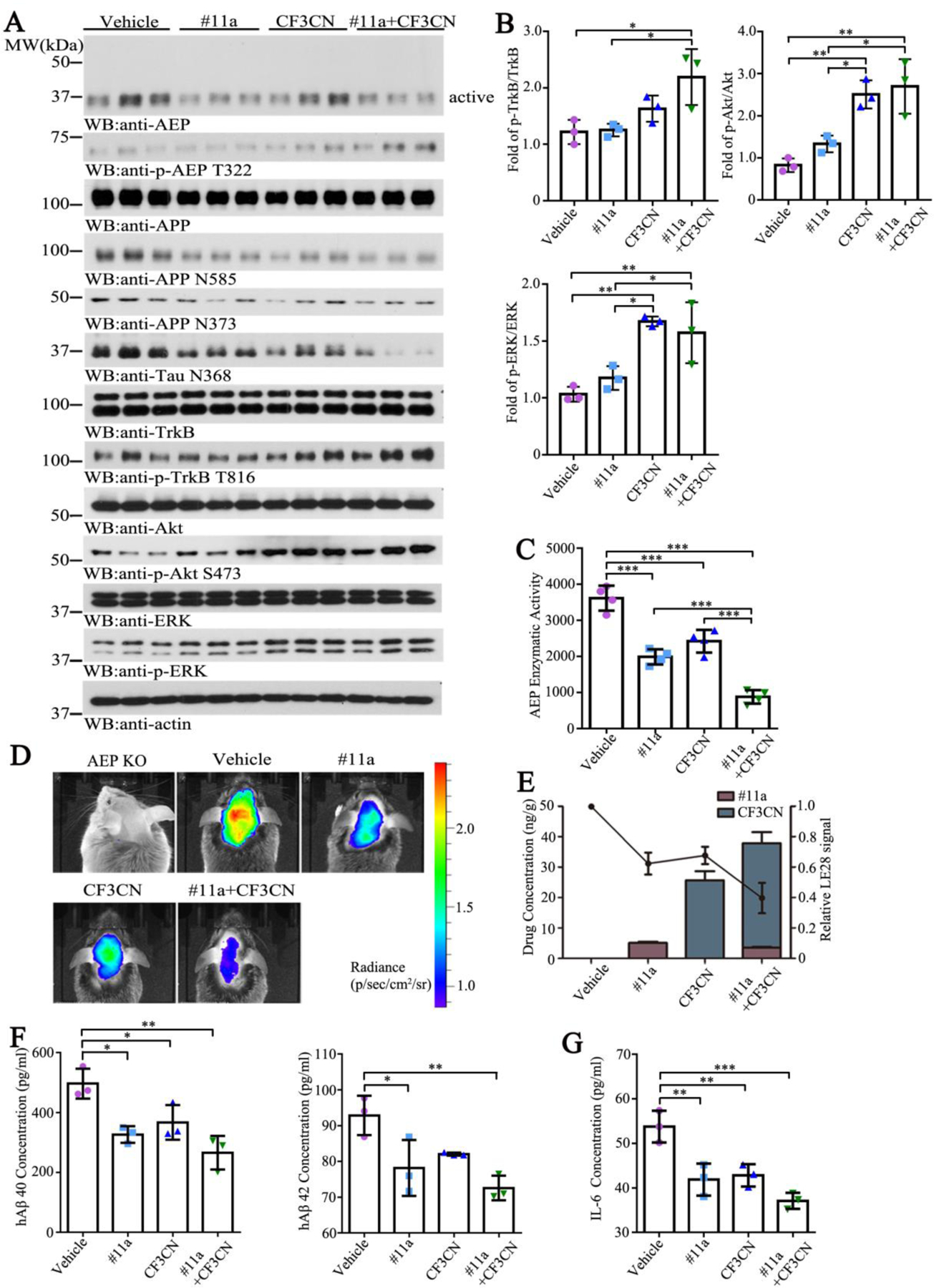
(A) Immunoblotting analysis of AEP, the p-TrkB and their downstream in vitro.
(B) Quantitative analysis of p-TrkB/TrkB, p-Akt/Akt and p-ERK/ERK in the brain in 3xTg mice. (n=3, one-way ANOVA)
(C) Enzymatic activity of AEP was determined using a fluorogenic substrate. (n=3, one-way ANOVA) (D) Imaging of 3xTg mice by using IVIS 100 after injection with LE28.
(E) Relationship between quantified LE28 signal intensity and drug concentrations in 3xTg mice brain. (n=3, one-way ANOVA)
(F) hAβ 40 and hAβ 42 levels were determined by ELISA. Both of hAβ 40 and hAβ 42 concentrations were reduced by #11a treatment. (n=3, one-way ANOVA)
(G) pro-inflammatory cytokines were determined by ELISA. The level of IL-6 was diminished by #11a and CF3CN treatments. (n=3, one-way ANOVA)
(Data are shown as mean ± SEM. *p<0.05, **p<0.01, ***p<0.001).
To gain the insight into the in vivo PK/PD relationship, we treated 6-month-old 3xTg mice with the indicated compounds by oral gavage. After 2 h, we monitored δ-secretase activity in the brains by performing the in vivo imaging with the activity-based probe LE28. As a negative control, there was no fluorescent signal in 3xTg/AEP-deficient brains. The strongest δ-secretase activity was observed in 3xTg mice, and this was substantially reduced by #11a or CF3CN. Again, the maximal inhibitory effect occurred with the combination treatment (Figure 4D). At the same time, we also sacrificed another group of mice 2 h after oral gavage and prepared the brain and plasma samples for quantitative analysis of the concentrations of #11a and CF3CN (Table. S9). The relationship between δ-secretase activities and drug concentrations in the brain were quantified in Figure 4E. The in vivo PK/PD curve was in good agreement with δ-secretase activity measured in the brain lysates in Fig 4C. Quantification of human Aβ40 and 42 in the brains showed that both peptides were significantly reduced by #11a and the mixture of both #11a and CF3CN. However, CF3CN only trended towards Aβ reduction but the differences were not statistically significant (Figure 4F, hAβ40: F=11.73, p=0.0027; hAβ42: F=8.512, p=0.0072). Since AD is associated with chronic neuro-inflammation, accordingly, we examined pro-inflammatory cytokine IL-6 levels in the brain and found that it was significantly diminished by both #11a and CF3CN with the combination exhibiting the strongest effect (Figure 4G, F=16.92, p=0.0008).
#11a, CF3CN and their combination robustly diminish AD pathogenesis in 3xTg mice
To further analyze the effects of the drugs on AD pathogenesis, we conducted immunofluorescent (IF) co-staining on the brain sections with various antibodies. In alignment with IB findings in the brain lysates, we found that the C-terminal Aβ-containing APP C586 fragments, resulting from APP N585 cleavage by δ-secretase, echoed the intensity of upstream active protease (Figure 5A & B, AEP: F=36.75, p<0.0001; APP C586: F=38.63, p<0.0001). Consequently, Aβ levels were evidently reduced by #11a or CF3CN and their combination exhibited the strongest effect. Thioflavin S (ThS) co-staining revealed that some of extracellular Aβ were deposited into the aggregates (Figure 5C & D, Th S: F=82.1, p<0.0001; Aβ: F=64.76, p<0.0001). Next, we also co-stained Tau N368 and p-Tau AT8 and found that neuronal Tau N368 activities were prominently decreased in the drug-treated groups as compared to vehicle control and the maximal inhibitory effect was found in the samples treated with the mixture (Figure 5E & F, AT8: F=33.53, p<0.0001; Tau N368: F=71.46, p<0.0001). The hippocampal Tau pathologies were further validated by the Silver staining, which manifested the similar patterns as those exhibited by the IF Tau N368/AT8 co-staining (Figure 5G), suggesting that Tau N368 fragment produced by δ-secretase cleavage was hyperphosphorylated and aggregated into NFTs in the hippocampus. Hence, δ-secretase inhibitor #11a and TrkB agonist CF3CN both mitigate AD pathogenesis and their combination exhibits a stronger therapeutic effect.
Figure 5. #11a, CF3CN and their mixture diminish AD pathologies in3xTg mice.
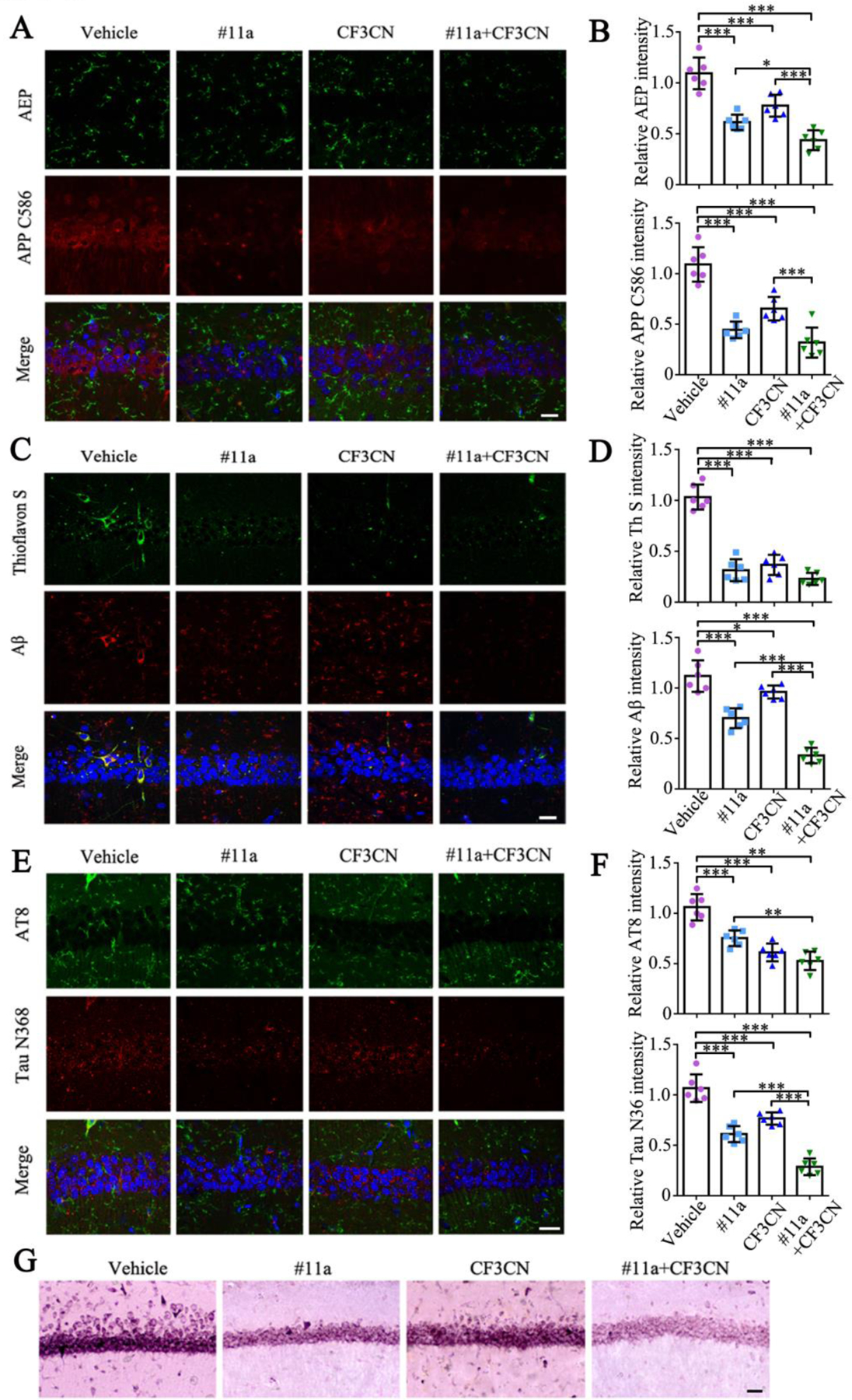
(A-B) Immunofluorescence staining of AEP and APP C586 in 3xTg mice brain sections. Scale bar, 20μm. (n=6, one-way ANOVA)
(C-D) Immunofluorescence staining of Aβ and Thioflavin S in 3xTg mice brain sections. Scale bar, 20μm. (n=6, one-way ANOVA)
(E-F) Immunofluorescence staining of AT8 and TauN368 in 3xTg mice brain sections. Scale bar, 20μm. (n=6, one-way ANOVA)
(G) Silver staining of 3xTg mice brain sections. Scale bar, 50μm.
(Data represent the means ± SEM; *P < 0.05, **P < 0.01, ***P< 0.001).
CF3CN strongly activates TrkB receptors and elevates BDNF in the brain, decreasing the synaptic loss.
CF3CN mimics the biological actions of BDNF and activates p-TrkB signaling. Previous studies show that BDNF induces its own mRNA production in primary neuronal cultures. Moreover, newly synthesized BDNF induced by the self-amplification system contributes to the synaptic maturation and function (Nakajima et al., 2015). To investigate whether CF3CN induces endogenous BDNF expression, we conducted IF co-staining on the brain sections from 3xTg mice chronically treated with various compounds. P-TrkB/TrkB co-staining revealed that CF3CN and #11a/CF3CN groups robustly provoked p-TrkB activation in the hippocampal neurons, whereas vehicle control or #11a failed (Figure 6A & B, TrkB: F=6.203, p=0.0037; p-TrkB: F=135.5, p<0.0001). As expected, anti-BDNF staining displayed prominent BDNF signals in the brains where p-TrkB was activated by CF3CN (Figure 6C & D, F=71.92, p<0.0001). Quantification of BDNF by ELISA revealed that BDNF levels were significantly increased by CF3CN and further escalated by the combination of #11a + CF3CN (Figure 6E, F=19.07, p=0.0005). Quantitative RT-PCR (qRT-PCR) analysis showed that BDNF mRNA levels were strongly increased in the brains where TrkB receptors were phosphorylated and activated, underscoring that CF3CN induces BDNF production via stimulation of TrkB neurotrophic signaling (Figure S4A, F=36.77, p<0.0001). Our recent finding shows that BDNF and C/EBPβ inversely regulate each other in AD pathogenesis (Wang et al., 2019). In consequence, we monitored C/EBPβ status by immunoblotting and found that both p-C/EBPβ T235, a biomarker for its activation, and its total levels were greatly diminished in the brains treated with CF3CN or #11a+ CF3CN. By contrast, BDNF concentrations were inversely escalated in these samples (Figure 6H). Thus, CF3CN activates p-TrkB and elevates endogenous BDNF expression in the brains of 3xTg mice, associated with potent suppression of C/EBPβ.
Figure 6. CF3CN activates TrkB receptors and elevates BDNF in the brain, decreasing the synaptic loss.
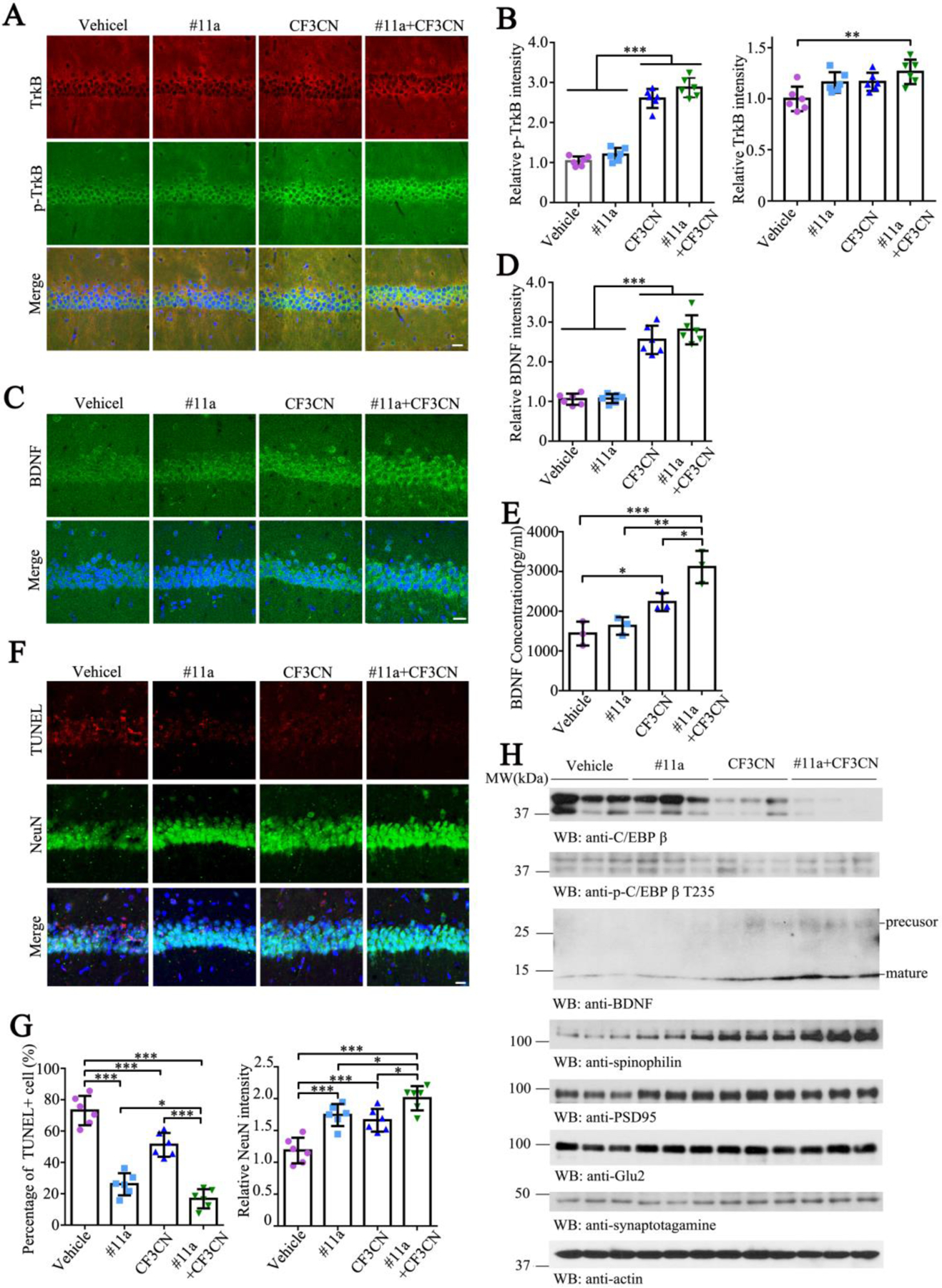
(A-B) Immunofluorescence staining of TrkB and p-TrkB in 3xTg mice brain sections. Scale bar, 20 μm. (n=6, one-way ANOVA)
(C-D) Immunofluorescence staining of BDNF in 3xTg mice brain. Scale bar, 20 μm. (n=6, one-way ANOVA)
(E) BDNF levels were determined by ELISA. (n=3, one-way ANOVA)
(F-G) Immunofluorescence staining of NeuN and TUNEL in 3xTg mice brain sections. Scale bar, 20 μm. (n=6, one-way ANOVA)
(H) Immunoblotting analysis of synaptic markers in 3xTg mice brain lysates.
(Data represent the means ± SEM; *P < 0.05, **P < 0.01, ***P< 0.001).
One of major roles of BDNF/TrkB neurotrophic signaling is to promote neuronal survival. Accordingly, we performed NeuN and TUNEL co-staining and found that the apoptosis in the hippocampal neurons in the vehicle group was greatly suppressed after treatment with #11a or CF3CN. Again, the combination of #11a and CF3CN exhibited the strongest anti-apoptotic activity. In alignment with these observations, we found that NeuN signals were conversely correlated with TUNEL activities in the hippocampus (Figure 6F & G, TUNEL: F=66.89, p<0.0001; NeuN: F=20.24, p<0.0001), indicating that δ-secretase inhibitor #11a or TrkB agonist CF3CN robustly inhibits neuronal apoptosis and enhances neuronal survival, and their mixture synergistically augments the pro-survival effect. In addition to neuronal survival, BDNF/TrkB neurotrophic signaling also mediates the synaptic plasticity. Subsequently, we performed immunoblotting and found that the synaptic proteins including spinophilin, PSD-95, synaptotagamine and GluR2 were all increased in the brains treated with CF3CN as compared to the vehicle control. Though #11a also augmented these synaptic proteins, the activity was not as robust as the TrkB agonist, though the maximal effect was observed after combination treatment (Figure 6H). Consistent with these observations, Golgi staining for the dendritic spines also revealed similar patterns (Figure S4B & C, F=10.19, p=0.0003).
#11a, CF3CN and their mixture significantly improve the learning and memory in 3xTg mice
Next, we examined effects on spatial memory using Morris Water Maze (MWM) assay. During the training period of 5 days, mice showed reduced latencies and distances to find the hidden platform. As expected, 3xTg mice treated with #11a or CF3CN showed shorter latency periods than vehicle control group. Again, groups treated with a combination of #11a + CF3CN exhibited the shortest latency, representing the most robust therapeutic effects on restoration of spatial memory (Figure 7A & B, F=5.404, p=0.0069). All of the groups exhibited comparable swim speeds, indicating that the compounds do not affect the motor function of the animals (Figure 7D, F=0.7195, p=0.5520). In the probe test, the percentage of time spent in quadrant from which the hidden platform was removed was significantly longer for #11a or CF3CN than the vehicle control, indicating better memory recall. Again, the combination group exhibited the most pronounced effect (Figure 7C, F=17.98, p<0.0001). Hence, inhibition of δ-secretase with an optimized compound #11a or activation of TrkB receptors with an improved agonist CF3CN in 3xTg mice restores learning and memory capabilities. Combination treatment with both agents exhibits synergistic therapeutic efficacy. Chronic treatment with these compounds revealed no demonstrable toxicity as revealed by the CBC (complete blood chemistry) analysis (Figure S5A & B, WBC: F=1.122, p<0.3959; RBC: F=2.69, p=0.1170; ALT: F=0.6647, p=0.5968; BUN: F=3.769, p=0.0592) and pathological examination of each organ from 3xTg mice (Figure S5C). Therefore, these compounds are safe for chronic oral administration in mice.
Figure 7. #11a, CF3CN and their mixture improve the learning and memory in 3xTg mice.
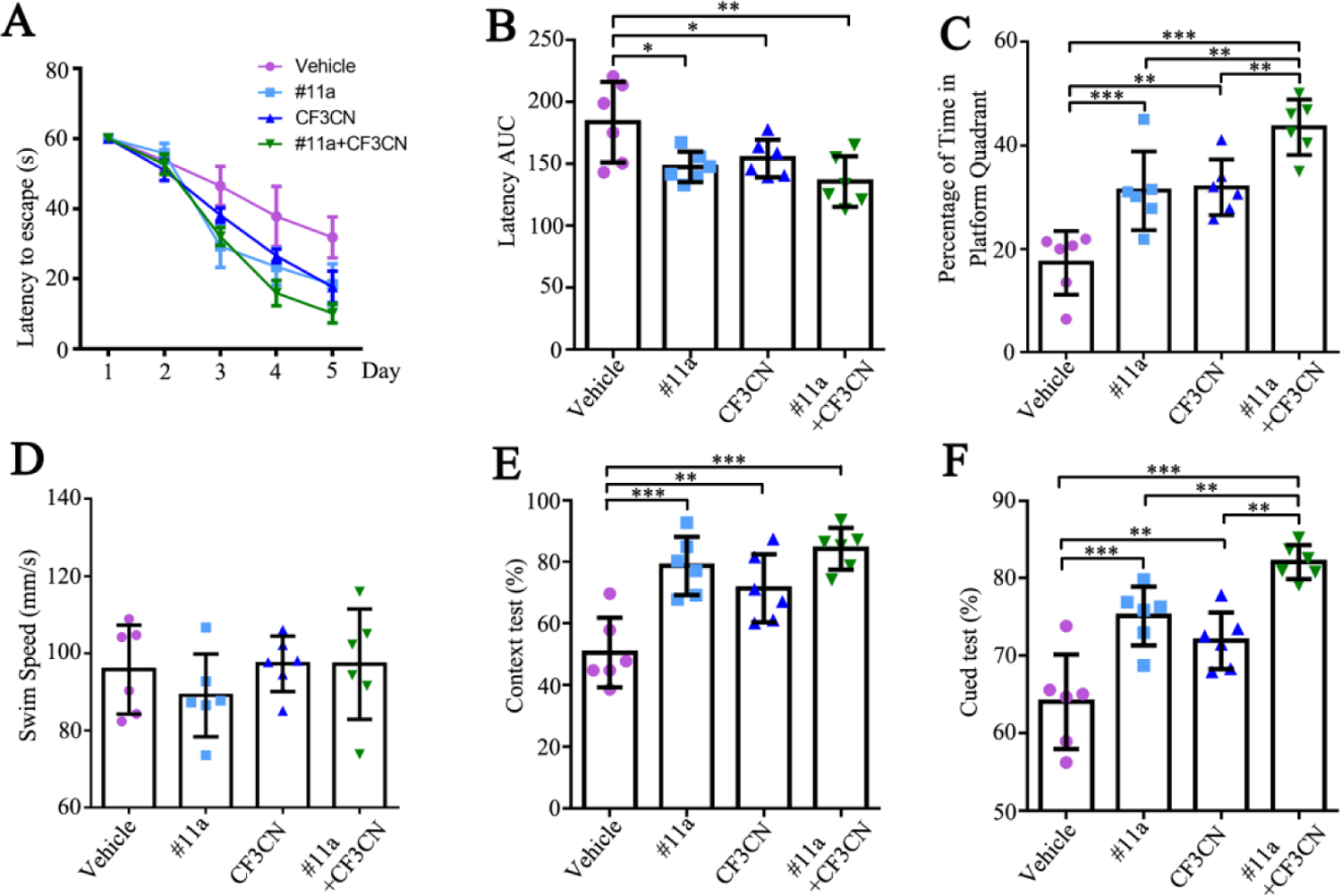
(A-B) Latency to mount the submerged platform in Morris Water Maze (MWM) assay. The area under curve of latency (latency AUC) were less in #11a and CF3CN treatment group. (n=6, one-way ANOVA)
(C) Percentage of time in platform quadrant in Morris Water Maze (MWM) assay. The percentage of time spent in platform quadrant was longer in #11a and CF3CN group. (n=6, one-way ANOVA)
(D) Swim speed in Morris Water Maze (MWM) assay. (n=6, one-way ANOVA)
(E-F) Fear Conditioning tests. (n=6, one-way ANOVA)
(Data represent the means ± SEM; *P < 0.05, **P < 0.01, ***P< 0.001).
Discussion
BDNF/TrkB signaling acts as an important regulator of amyloidogenic processing. For example, Aβ production is reduced by BDNF in primary neuronal cultures (Rohe et al., 2009), but it is increased by BDNF deprivation (Matrone et al., 2008). This inverse relationship also occurs between pathologic Tau containing NFT and BDNF in neurons (Murer et al., 1999). Clearly, BDNF possesses a protective role against AD pathogenesis. The beneficial effect of BDNF has been reported in animals with cognitive deficits (Ando et al., 2002), and studies in AD models show that BDNF has a neuroprotective effect against β-amyloid toxicity. Activating BDNF/TrkB signaling via the agonists or synaptogenic molecular cascades such as the PKC-BDNF pathway have been proposed to ameliorate the synaptic loss and promote neuro-regeneration as potential anti-AD therapeutic strategies (Sun and Alkon, 2019). In the current work, we report an optimized δ-secretase inhibitor #11a, which is optimized via structure-based drug design and reiterative organic synthesis, δ-secretase inhibitory assays and in vitro ADMET. The optimal lead compound #11a possesses optimal oral bioavailability and in vivo PK profiles (Figure 2). Notably, it displays tight in vivo PK/PD relationship and strongly blocks δ-secretase activity in the brain upon oral administration. Interestingly, CF3CN also antagonizes δ-secretase protease activity in the brain via activation of the TrkB/Akt pathway, which robustly phosphorylates δ-secretase on T322 and suppresses its proteolytic cleavage and activation. Consequently, APP N585 and Tau N368 fragmentation by δ-secretase were predominantly inhibited, attenuating Aβ production and Tau hyperphosphorylation. In alignment with the mitigated biochemical events, ThS-positive Aβ aggregates and Tau pathology validated with Silver staining and neuronal loss manifested with NeuN/TUNEL staining in the hippocampus are all significantly alleviated with treatment either #11a or CF3CN. The maximal effect occurs with their combination. Accordingly, the dendritic spines and synaptic degeneration in the brains of 3xTg mice are substantially inhibited by δ-secretase inhibitor #11a or TrkB agonist CF3CN, and their combination elicits the maximal effect. To support these observations, immunoblotting analysis also shows that synaptic structural proteins such as PSD-95, synaptagmine, spinophilin, etc. are evidently augmented, fitting with BDNF/TrkB neurotrophic activities in synaptic plasticity (Figure 6H).
In addition to direct activation of TrkB receptors as an agonist, CF3CN also strongly enhances BDNF levels in the brain. Hence, CF3CN triggers TrkB activation via both its own TrkB receptor agonistic activity and elevating endogenous BDNF secretion in the brain. As a result, either #11a or CF3CN significantly attenuates the cognitive deficits in 3xTg mice with their combination displaying the maximal effect. It remains unclear how CF3CN induces BDNF production in the brain. Presumably, CF3CN activates p-TrkB that phosphorylates CREB, a well-characterized BDNF transcription factor. Recently, it has been reported that BDNF induces its own mRNA production in primary neuronal cultures. Moreover, newly synthesized BDNF induced by the self-amplification system contributes to the synaptic maturation and function (Nakajima et al., 2015). Moreover, CREB family transcription factors are required for the early induction of all the major BDNF transcripts, whereas CREB directly binds only to BDNF promoter IV, is phosphorylated in response to BDNF/TrkB signaling, and activates transcription from BDNF promoter IV by recruiting CBP (Esvald et al., 2020). In consequence, qRT-PCR analysis shows that BDNF mRNA levels in the brains are strongly augmented upon CF3CN treatment (Figure S4A).
CF3CN mitigates AD pathologies not only via activation of the BDNF/TrkB signaling pathway but also via suppression of δ-secretase through phosphorylation by TrkB-activated Akt. Since both CF3CN and #11a affect the activity of δ-secretase, the crucial age-dependent protease that simultaneously cleaves both APP and Tau in AD pathogenesis, the combination of these two compounds exhibits the greatest activity in attenuating AD pathology, resulting in the most prominent alleviation of cognitive defects. The synergistic effect might result from the lowest level of Tau N368, which binds the kinase domain on TrkB receptors and blocks its neurotrophic effect (Xiang et al., 2019). Subsequently, higher p-TrkB/p-Akt activities trigger stronger p-AEP T322 signals, robustly blocking its proteolytic activation (Figure 3A and Fig. 4A). The target engagement in AD mouse brain is validated with in vivo δ-secretase activity assay in 3xTg mice using fluorescent activity-based probe LE28 (Figure 3D & E). This observation is further confirmed with a fluorogenic substrate for δ-secretase in brain lysates (Figure 3C). Consistent with reduced δ-secretase activity, APP N373/585 and Tau N368 truncation by δ-secretase were evidently attenuated. Aβ quantification mirrors δ-secretase enzymatic activity (Figure 4F). These biochemical processes are validated by immunofluorescent co-staining on the brain sections. For instance, the fluorescent staining of Aβ-bearing fragment APP C586, a C-terminal fragment from APP N585 cleavage by δ-secretase, reflected AEP intensities. Subsequently, aggregated Aβ signals correlate with ThS staining activities. In alignment with these findings, Tau N368 activities also oscillate with AT8 signals, in agreement with Silver staining in the hippocampus (Figure 5).
Most recently, we report that BDNF/TrkB reduction increases inflammatory cytokines and activates the JAK2/STAT3 pathway, resulting in the upregulation of C/EBPβ/δ-secretase pathway, which mediates both APP and Tau fragmentation by δ-secretase and neuronal loss (Wang et al., 2019). In this work, we find that activation of BDNF/TrkB signaling strongly decreases C/EBPβ, which is inversely coupled to escalation of BDNF in the brains using the immunoblotting analysis (Figure 6H). One of AD pathological features is chronic neuro-inflammation, most likely induced by the formation of Aβ deposits. Notably, Aβ and IL-6 additively augments C/EBPβ activity (Strohmeyer et al., 2014). Previous studies show that C/EBPβ promotes the production of inflammatory mediators, and C/EBP family members are themselves induced by the classical pro-inflammatory triad of IL-1β, IL-6, and tumor necrosis factor alpha (TNF-α) (Poli, 1998; Wedel and Ziegler-Heitbrock, 1995), all of which are highly elevated in the AD brain (Akiyama et al., 2000). As expected, quantification of IL-6 in the brains after drug treatment reveals that activation of BDNF/TrkB pathway robustly represses IL-6, fitting with C/EBPβ reduction (Figure 4G).
Despite tremendous effort and billions of invested dollars, only five medical treatments have been approved for AD, which mainly act to control symptoms rather than alter the course of the disease. Reduced acetylcholine neurotransmission due to loss of neurons in the basal forebrain and depletion of choline acetyltransferase are observed in AD pathology. Hence, currently, there are two types of medication to treat AD: cholinesterase inhibitors and NMDA antagonist. The first category of inhibitors blocks the activity of acetylcholine transferase, thereby increasing acetylcholine levels in the CNS to restore cognitive functions. The second inhibits the over-activation of NMDA-induced glutamate excitotoxicity. These drugs can only partially alleviate the symptoms. Our discovery that δ-secretase simultaneously modulates both APP and Tau pathways and promotes AD pathogenesis provides an unprecedented drug target. Based on our previous co-crystal structure between #11 and active δ-secretase, we have designed and synthesized a series of derivatives that demonstrate gradually enhanced potency against δ-secretase with improved PK profiles (Figure 1 & 2). The optimal #11a exhibits optimal oral bioavailability, tight in vivo PK/PD relationship and acceptable brain exposure, suggesting that this compound and CF3CN are good preclinical candidates with reasonable drug-like properties that warrant further development to treat this devastating neurodegenerative disease.
Supplementary Material
Supplementary Figure 1: Chemical structures of AEP inhibitor derivatives.
Supplementary Figure 2: in vitro active metabolite trapping assay of CP9 and CP24 (#11a).
Supplementary Figure 3: Drug design and optimization of AEP inhibitor compound 9.
Supplementary Figure 4: #11a, CF3CN and their mixture increase BDNF mRNA expression and prevent synaptic loss in the hippocampus of 3xTg mice
Supplementary Figure 5: 12 weeks treatments with #11a, CF3CN or two compounds combination demonstrate no toxic effect on mice tissues or the blood in 3xTg mice.
Table S1: Inhibitory efficacy of compounds on AEP and Casepase-3.
Table S2: Biological and chemical stability assay.
Table S3: Hepatocyte stability assay.
Table S4: Plasma protein binding assay.
Table S5: Caco-2 permeability assay.
Table S6: MDCK-MDR1 permeability assay.
Table S7: hERG channel inhibition assay.
Table S8: CP24 (#11a) pharmacological property Profile.
Table S9: Drug concentrations of #11a and CF3CN in the plasma and the brain after oral administration.
Highlights.
Optimized δ-secretase inhibitor (#11a) and TrkB receptor agonist (CF3CN) combination synergistically blunts δ-secretase.
CF3CN strongly activates p-TrkB that triggers active Akt to phosphorylate δ-secretase T322, preventing its proteolytic activation and mitigating AD pathologies.
#11a or CF3CN significantly diminishes AD pathogenesis and improves cognitive functions with the combination exhibiting the maximal effect.
ACKNOWLEDGEMENTS
This work was supported by grants from NIH grant (RF1, AG051538) to K. Y. The authors are thankful for Dr. Arthur W. English at Cell Biology Department at Emory University for critical proof reading the manuscript and providing a lot of valuable advice. This study was supported in part by the Rodent Behavioral Core (RBC), which is subsidized by the Emory University School of Medicine and is one of the Emory Integrated Core Facilities. Additional support was provided by the Viral Vector Core of the Emory Neuroscience NINDS Core Facilities (P30NS055077). Further support was provided by the Georgia Clinical & Translational Science Alliance of the National Institutes of Health under Award Number UL1TR002378.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURE
K.Y. is a shareholder of Wuhan Yuanzheng Pharmaceuticals, Inc. and Shanghai Braegen Pharmaceuticals, Inc. The authors declare of no competing financial interest.
References:
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O’Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T, 2000. Inflammation and Alzheimer’s disease. Neurobiol Aging 21, 383–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andero R, Heldt SA, Ye K, Liu X, Armario A, Ressler KJ, 2011. Effect of 7,8-dihydroxyflavone, a small-molecule TrkB agonist, on emotional learning. Am J Psychiatry 168, 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando S, Kobayashi S, Waki H, Kon K, Fukui F, Tadenuma T, Iwamoto M, Takeda Y, Izumiyama N, Watanabe K, Nakamura H, 2002. Animal model of dementia induced by entorhinal synaptic damage and partial restoration of cognitive deficits by BDNF and carnitine. J Neurosci Res 70, 519–527. [DOI] [PubMed] [Google Scholar]
- Blennow K, Chen C, Cicognola C, Wildsmith KR, Manser PT, Bohorquez SMS, Zhang Z, Xie B, Peng J, Hansson O, Kvartsberg H, Portelius E, Zetterberg H, Lashley T, Brinkmalm G, Kerchner GA, Weimer RM, Ye K, Hoglund K, 2020. Cerebrospinal fluid tau fragment correlates with tau PET: a candidate biomarker for tangle pathology. Brain 143, 650–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Ahn EH, Liu X, Wang ZH, Luo S, Liao J, Ye K, 2021. Optimized TrkB Agonist Ameliorates Alzheimer’s Disease Pathologies and Improves Cognitive Functions via Inhibiting Delta-Secretase. ACS Chem Neurosci [DOI] [PMC free article] [PubMed]
- Chen C, Wang Z, Zhang Z, Liu X, Kang SS, Zhang Y, Ye K, 2018. The prodrug of 7,8-dihydroxyflavone development and therapeutic efficacy for treating Alzheimer’s disease. Proc Natl Acad Sci U S A 115, 578–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DC, Maguschak KA, Ye K, Jang SW, Myers KM, Ressler KJ, 2010. Prelimbic cortical BDNF is required for memory of learned fear but not extinction or innate fear. Proc Natl Acad Sci U S A 107, 2675–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor B, Young D, Yan Q, Faull RL, Synek B, Dragunow M, 1997. Brain-derived neurotrophic factor is reduced in Alzheimer’s disease. Brain Res Mol Brain Res 49, 71–81. [DOI] [PubMed] [Google Scholar]
- Cummings J, Lee G, Mortsdorf T, Ritter A, Zhong K, 2017. Alzheimer’s disease drug development pipeline: 2017. Alzheimers Dement (N Y) 3, 367–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dall E, Brandstetter H, 2016. Structure and function of legumain in health and disease. Biochimie 122, 126–150. [DOI] [PubMed] [Google Scholar]
- Devi L, Ohno M, 2012. 7,8-dihydroxyflavone, a small-molecule TrkB agonist, reverses memory deficits and BACE1 elevation in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 37, 434–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgington-Mitchell LE, Wartmann T, Fleming AK, Gocheva V, van der Linden WA, Withana NP, Verdoes M, Aurelio L, Edgington-Mitchell D, Lieu T, Parker BS, Graham B, Reinheckel T, Furness JB, Joyce JA, Storz P, Halangk W, Bogyo M, Bunnett NW, 2016. Legumain is activated in macrophages during pancreatitis. Am J Physiol Gastrointest Liver Physiol 311, G548–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esvald EE, Tuvikene J, Sirp A, Patil S, Bramham CR, Timmusk T, 2020. CREB Family Transcription Factors Are Major Mediators of BDNF Transcriptional Autoregulation in Cortical Neurons. J Neurosci 40, 1405–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Marin C, Rey MJ, Ribalta T, Goutan E, Blanco R, Tolosa E, Marti E, 1999. BDNF and full-length and truncated TrkB expression in Alzheimer disease. Implications in therapeutic strategies. J Neuropathol Exp Neurol 58, 729–739. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ, 2002. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. [DOI] [PubMed] [Google Scholar]
- Henriques A, Pitzer C, Schneider A, 2010. Neurotrophic growth factors for the treatment of amyotrophic lateral sclerosis: where do we stand? Front Neurosci 4, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF, 2003. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem 72, 609–642. [DOI] [PubMed] [Google Scholar]
- Jang SW, Liu X, Yepes M, Shepherd KR, Miller GW, Liu Y, Wilson WD, Xiao G, Blanchi B, Sun YE, Ye K, 2010. A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc Natl Acad Sci U S A 107, 2687–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuzy A, Cicognola C, Chiotis K, Saint-Aubert L, Lemoine L, Andreasen N, Zetterberg H, Ye K, Blennow K, Höglund K, Nordberg A, 2019. Longitudinal tau and metabolic PET imaging in relation to novel CSF tau measures in Alzheimer’s disease. Eur J Nucl Med Mol Imaging 46, 1152–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Chan CB, Jang SW, Pradoldej S, Huang J, He K, Phun LH, France S, Xiao G, Jia Y, Luo HR, Ye K, 2010. A synthetic 7,8-dihydroxyflavone derivative promotes neurogenesis and exhibits potent antidepressant effect. J Med Chem 53, 8274–8286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matrone C, Ciotti MT, Mercanti D, Marolda R, Calissano P, 2008. NGF and BDNF signaling control amyloidogenic route and Abeta production in hippocampal neurons. Proc Natl Acad Sci U S A 105, 13139–13144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murer MG, Boissiere F, Yan Q, Hunot S, Villares J, Faucheux B, Agid Y, Hirsch E, Raisman-Vozari R, 1999. An immunohistochemical study of the distribution of brain-derived neurotrophic factor in the adult human brain, with particular reference to Alzheimer’s disease. Neuroscience 88, 1015–1032. [DOI] [PubMed] [Google Scholar]
- Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM, Wang L, Blesch A, Kim A, Conner JM, Rockenstein E, Chao MV, Koo EH, Geschwind D, Masliah E, Chiba AA, Tuszynski MH, 2009. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat Med 15, 331–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima S, Numakawa T, Adachi N, Ooshima Y, Odaka H, Yoshimura A, Kunugi H, 2015. Self-amplified BDNF transcription is a regulatory system for synaptic maturation in cultured cortical neurons. Neurochem Int 91, 55–61. [DOI] [PubMed] [Google Scholar]
- Phillips HS, Hains JM, Armanini M, Laramee GR, Johnson SA, Winslow JW, 1991. BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer’s disease. Neuron 7, 695–702. [DOI] [PubMed] [Google Scholar]
- Poli V, 1998. The role of C/EBP isoforms in the control of inflammatory and native immunity functions. J Biol Chem 273, 29279–29282. [DOI] [PubMed] [Google Scholar]
- Rohe M, Synowitz M, Glass R, Paul SM, Nykjaer A, Willnow TE, 2009. Brain-derived neurotrophic factor reduces amyloidogenic processing through control of SORLA gene expression. J Neurosci 29, 15472–15478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakane T, Pardridge WM, 1997. Carboxyl-directed pegylation of brain-derived neurotrophic factor markedly reduces systemic clearance with minimal loss of biologic activity. Pharm Res 14, 1085–1091. [DOI] [PubMed] [Google Scholar]
- Soderquist RG, Milligan ED, Sloane EM, Harrison JA, Douvas KK, Potter JM, Hughes TS, Chavez RA, Johnson K, Watkins LR, Mahoney MJ, 2009. PEGylation of brain-derived neurotrophic factor for preserved biological activity and enhanced spinal cord distribution. J Biomed Mater Res A 91, 719–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohmeyer R, Shelton J, Lougheed C, Breitkopf T, 2014. CCAAT-enhancer binding protein-beta expression and elevation in Alzheimer’s disease and microglial cell cultures. PLoS One 9, e86617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun MK, Alkon DL, 2019. Neuro-regeneration Therapeutic for Alzheimer’s Dementia: Perspectives on Neurotrophic Activity. Trends Pharmacol Sci 40, 655–668. [DOI] [PubMed] [Google Scholar]
- Wang ZH, Gong K, Liu X, Zhang Z, Sun X, Wei ZZ, Yu SP, Manfredsson FP, Sandoval IM, Johnson PF, Jia J, Wang JZ, Ye K, 2018a. C/EBPbeta regulates delta-secretase expression and mediates pathogenesis in mouse models of Alzheimer’s disease. Nat Commun 9, 1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZH, Wu W, Kang SS, Liu X, Wu Z, Peng J, Yu SP, Manfredsson FP, Sandoval IM, Liu X, Wang JZ, Ye K, 2018b. BDNF inhibits neurodegenerative disease-associated asparaginyl endopeptidase activity via phosphorylation by AKT. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZH, Xiang J, Liu X, Yu SP, Manfredsson FP, Sandoval IM, Wu S, Wang JZ, Ye K, 2019. Deficiency in BDNF/TrkB Neurotrophic Activity Stimulates delta-Secretase by Upregulating C/EBPbeta in Alzheimer’s Disease. Cell Rep 28, 655–669e655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedel A, Ziegler-Heitbrock HW, 1995. The C/EBP family of transcription factors. Immunobiology 193, 171–185. [DOI] [PubMed] [Google Scholar]
- Xiang J, Wang ZH, Ahn EH, Liu X, Yu SP, Manfredsson FP, Sandoval IM, Ju G, Wu S, Ye K, 2019. Delta-secretase-cleaved Tau antagonizes TrkB neurotrophic signalings, mediating Alzheimer’s disease pathologies. Proc Natl Acad Sci U S A 116, 9094–9102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Liu X, Schroeder JP, Chan CB, Song M, Yu SP, Weinshenker D, Ye K, 2014a. 7,8-dihydroxyflavone prevents synaptic loss and memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 39, 638–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Obianyo O, Dall E, Du Y, Fu H, Liu X, Kang SS, Song M, Yu SP, Cabrele C, Schubert M, Li X, Wang JZ, Brandstetter H, Ye K, 2017. Inhibition of delta-secretase improves cognitive functions in mouse models of Alzheimer’s disease. Nat Commun 8, 14740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Song M, Liu X, Kang SS, Kwon I-S, Duong DM, Seyfried NT, Hu WT, Liu Z, Wang J-Z, Cheng L, Sun YE, Yu SP, Levey AI, Ye K, 2014b. Cleavage of tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer’s disease. Nature Medicine 20, 1254–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Song M, Liu X, Su Kang S, Duong DM, Seyfried NT, Cao X, Cheng L, Sun YE, Ping Yu S, Jia J, Levey AI, Ye K, 2015. Delta-secretase cleaves amyloid precursor protein and regulates the pathogenesis in Alzheimer’s disease. Nat Commun 6, 8762. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Chemical structures of AEP inhibitor derivatives.
Supplementary Figure 2: in vitro active metabolite trapping assay of CP9 and CP24 (#11a).
Supplementary Figure 3: Drug design and optimization of AEP inhibitor compound 9.
Supplementary Figure 4: #11a, CF3CN and their mixture increase BDNF mRNA expression and prevent synaptic loss in the hippocampus of 3xTg mice
Supplementary Figure 5: 12 weeks treatments with #11a, CF3CN or two compounds combination demonstrate no toxic effect on mice tissues or the blood in 3xTg mice.
Table S1: Inhibitory efficacy of compounds on AEP and Casepase-3.
Table S2: Biological and chemical stability assay.
Table S3: Hepatocyte stability assay.
Table S4: Plasma protein binding assay.
Table S5: Caco-2 permeability assay.
Table S6: MDCK-MDR1 permeability assay.
Table S7: hERG channel inhibition assay.
Table S8: CP24 (#11a) pharmacological property Profile.
Table S9: Drug concentrations of #11a and CF3CN in the plasma and the brain after oral administration.