

Abstract
B-cell acute lymphoblastic leukemia (ALL) affects both pediatric and adult patients. Chemotherapy resistant tumor cells that contribute to minimal residual disease (MRD) underlie relapse and poor clinical outcomes in a sub-set of patients. Targeting mitochondrial oxidative phosphorylation (OXPHOS) in the treatment of refractory leukemic cells is a potential novel approach to sensitizing tumor cells to existing standard of care therapeutic agents. In the current study, we have expanded our previous investigation of the mitoNEET ligand NL-1 in the treatment of ALL to interrogate the functional role of the mitochondrial outer membrane protein mitoNEET in B-cell ALL. Knockout (KO) of mitoNEET (gene: CISD1) in REH leukemic cells led to changes in mitochondrial ultra-structure and function. REH cells have significantly reduced OXPHOS capacity in the KO cells coincident with reduction in electron flow and increased reactive oxygen species. In addition, we found a decrease in lipid content in KO cells, as compared to the vector control cells was observed. Lastly, the KO of mitoNEET was associated with decreased proliferation as compared to control cells when exposed to the standard of care agent cytarabine (Ara-C). Taken together, these observations suggest that mitoNEET is essential for optimal function of mitochondria in B-cell ALL and may represent a novel anti-leukemic drug target for treatment of minimal residual disease.
Keywords: cisd2, mitochondrial dysfunction, glitazones, chemoresistance
Graphical Abstract
1. Introduction
MitoNEET (gene CISD1) is an iron-sulfur containing protein located on the outer mitochondrial membrane [1]. This evolutionally conserved zinc finger protein contains a CDGSH-iron-sulfur domain [1–3] where the iron sulfur [2Fe-2S] clusters are redox active and thought to contribute to the redox and pH-sensor regulation of OXPHOS bioenergetics in mitochondria [1, 4–6]. Originally discovered as an off-target effector for the anti-diabetic glitazone drug pioglitazone, mitoNEET was found to have therapeutic benefits on β-oxidation and lipid metabolism unrelated to its peroxisome proliferator-activated receptor (PPAR)-γ activation [7–9]. MitoNEET has been identified in bioinformatic and functional studies to be associated with several cancers, including breast and lung cancer [10–12]. In breast cancer for instance, mitoNEET knockout led to a dramatic reduction in solid tumor size, and studies suggested that ligands targeting mitoNEET may modulate cancer proliferation and chemo-resistance [13, 14].
Cancer cells exhibit differential metabolism compared to normal cells [15–17], and while it is known that cancer cells are mainly glycolytic in nature (Warburg effect), alterations in mitochondrial oxidative phosphorylation (OXPHOS) can contribute to changes in chemo-resistance through mechanisms that remain to be fully elucidated [18]. Targeting mitochondrial OXPHOS in cancer cells has been suggested as a novel approach to eradicate minimal residual disease (MRD) in leukemia patients [19, 20]. MRD persists in some B-cell acute lymphoblastic leukemia (ALL) patients following completion of chemotherapy treatment, with the presence of surviving cells being a negative prognostic indicator for patients. The presence of MRD posttreatment has been attributed to leukemic cell populations residing in the bone marrow microenvironment (BMM), due to changes in cellular metabolism [21, 22]. This unique anatomical microenvironment is considered a “site of sanctuary” for leukemic cells and contributes to disease relapse and chemo-resistance through multiple metabolic pathways [21, 23].
Our previous studies of a first-in-class mitoNEET ligand, NL-1, showed activity in B-cell ALL, which prompted interest in evaluating mitochondrial bioenergetic function in the setting of mitoNEET loss, to evaluate the potential of leveraging this pathway for novel therapeutic benefit in B-cell ALL [24].
2. Material and Methods
2.1. Cell culture and materials
Human derived REH cells (ATCC #CRL-8286) were purchased and maintained in RPMI 1640 supplemented with 10% FBS and 1x streptomycin/penicillin antibiotics. MitoNEET (gene: CISD1) knockout (KO) cells were generated by Synthego (Menlo Park, CA) using the CRISPR/Cas9-based gene edition system in REH cells. The guide sequence was GAUCUCCACAUAGGGGCCAG. The PCR primers used were TTCTAGGGAGCAGGTCCTGACT (forward) and TCCTTTCCTACAAGCTCGTGGG (reverse). The control cells used throughout the study were the empty vector transduced control REH cells (vector control; VC).
2.2. GFP labeled cells
REH cells (5,000 cells/100 μl) were placed in fresh medium containing 5 μg/ml Polybrene in a 96 well plate. Firefly Luciferase-F2A-GFP Lentivirus (Fluc-F2A-GFP; Biosettia, San Diego, CA) was added (5 μl/well). The plate was spun at 1,000 x g for 60 min at room temperature then placed in 37° C, 5% CO2 incubator for 24 hrs. The next day, the media was replaced with fresh media to remove lentivirus and Polybrene. After 12 hrs, 500 μg/ml hygromycin was added for 72 hrs to select for transduced cells.
2.3. Cell proliferation studies
WST-8 assay proliferation assay. Viable cells were quantitated using the Cell Counting Kit-8 (Dojindo Molecular Technology Inc., cat#CK04) according to manufacturer’s instructions at indicated time points. Briefly, 10 μl of the assay reagent was added to each well of the 96-well plate and incubated for 2 hrs at 37 °C, after which the plates were read on a BioTek Cynergy 5 plate reader at 450 nm absorbance. MitoNEET KO cells were compared to VC cells for analysis. In addition, GFP-labeled REH proliferation studies were completed by plating fluorescent REH VC and KO cells at 50,000/well in 100 μl/well followed by reading the plate at the indicated time points using a Molecular Devices SpectraMax ID5 plate reader.
2.4. Ara-C studies
Cells were plated in 96-well plates at a density of 50,000 cells/well in 100 μl. Cells were treated with Ara-C at indicated concentrations, and cell proliferation was measured after 48 hrs of incubation with the compounds using a Cell Counting Kit-8 (Dojindo Molecular Technology Inc. Cat#CK04), which was used according to manufacturer’s instructions. Briefly, 10 μl of the assay reagent was added to each well and incubated for 2 hrs at 37 °C, after which the plates were read on a BioTek Cynergy 5 plate reader at 450 nm absorbance. Untreated cells were used as controls.
2.5. ATP studies
Cells were plated in 96-well plates at a density of 50,000 cells/well in 100 μl. ATP was quantitated using the CellTiter-Glo 2.0 Assay (Promega Cat#G9241) according to manufacturer’s instructions and read using a Molecular Devices SpectraMax ID5 plate reader.
2.6. Western blot analysis
MitoNEET VC and KO ALL cells were cultured in suspension prior to lysis in RIPA buffer, and protein concentrations were determined using the bicinchoninic acid (BCA) protein assay kit (Cat#23225 Pierce-ThermoFisher Scientific, Waltham, MA). Equal quantities of proteins were resolved on 4-20% Mini-Protean TGX Precast Protein Gels (BioRad Cat#45610994) and transferred to nitrocellulose membranes. Membranes were blocked in TBS/0.1% Tween-20 with 5% nonfat dry milk for 1 hr at RT. Blots were probed with the indicated primary antibodies overnight then rinsed with TBS/0.1% Tween-20. Blots were incubated with horseradish peroxidase-conjugated secondary antibodies for 1 hr at RT, then rinsed 3X with TBS/0.1% Tween-20. Signal was visualized using Immobilon Western Chemiluminescent HRP Substrate (Millipore Cat#WBKLS0500) on the Amersham Imager AI680. The following antibodies CISD1/MitoNEET (#83775), SOD2 (#13141), GPX1 (#3286), GPX4 (#52455) and β-actin (#8457) were purchased from Cell Signaling Technologies (Danvers, MA). Western blots are representative of at least three independent experiments.
2.7. Microscopy
For the MitoTracker and mitoNEET co-localization study, cells were stained with 100 nM MitoTracker Deep Red cat #M22426 for 30 min at 37 ° C. Cells were washed with 1X PBS then fixed in 4% PFA for 15 min at 37 ° C. Cells were washed with 1X PBS then incubated with 0.4% Triton X-100 in PBS for 10 min at RT. Cells were washed 1X in PBS before adding mitoNEET antibody (Cell signaling Technologies cat #83775), 1 μl diluted in 200 μl of PBS + 5% BSA, for 1hr at RT. The cells were washed with 1X PBS before adding donkey anti-rabbit AF555 diluted 1:400 for 1 hr at RT. Cells were then washed 2 times in PBS before slides were made and Prolong with DAPI was used prior to adding a coverslip. The SIM (Structured Illumination Microscopy) images were acquired using a Nikon A1R w/ N-SIM E microscope, using the 100X/1.49 SR objective. For the TOM20 and mitoNEET co-localization study, cells were counted, washed with 1X PBS, then fixed in 4% PFA for 15 min at 37 ° C. Cells were washed with 1X PBS then incubated with 0.4% Triton X-100 in PBS for 10 min at RT. Cells were washed 3X with 1X PBS then incubated for 1 hr at RT with 5% goat serum in 1X PBS. Blocking buffer was removed then 1:200 dilution of TOM20 Coralite 488-conjugated polyclonal antibody (Proteintech cat#CL488-11802) added for 1.5 hr at RT. They were washed 3X in 1XPBS before adding mitoNEET antibody (Cell Signaling Technologies cat #83775), 1 μl diluted in 200 μl of PBS + 5% BSA, for 1hr at RT. The cells were washed with 1X PBS before adding donkey anti-rabbit AF555 diluted 1:400 for 1 hr at RT. They were then washed 2X in 1X PBS before slides were made and Prolong with DAPI was used prior to adding a coverslip. The images were taken using the 100X/1.49 SR objective on the Nikon A1R confocal. For the Nile Red assay, following isolation of REH cells, 4 million cells were re-suspended in PBS containing 2 μM of Nile Red (Invitrogen, Carlsbad, CA) for 10 min. Following incubation, the cells were rinsed with PBS, re-suspended in PBS, and cytospun on slides. The Nile Red images were taken on the Nikon A1R confocal microscope using the 60X/1.4 objective.
2.8. Metabolite characterization
To determine levels of glutathione (GSH) and GSSH, a luciferase-based GSH/GSSG assay from Promega (cat# V6611) was used, according to the manufacturer’s protocol. NADP/NADPH levels were measured using a luciferase-based kit from Promega (cat# G9081) according to the manufacturer’s protocol. Cells were seeded at 50,000/well in 100 μl/well in 96-well white plates with an opaque bottom. Luminescent readings were completed using a Cytation 5 multi-modal plate reader (BioTek).
2.9. Electron microscopy
A cell suspension was fixed in glutaraldehyde and paraformaldehyde solution for 1 hr before being pelleted by centrifugation. Pellets were enrobed in agarose and then stained with osmium tetroxide (OsO4) solution. The agarose pieces containing cells were then gradually dehydrated, infiltrated, and embedded in EPON resin. Ultra-thin sections (70-90 nm) were collected onto copper grids and further stained with uranyl acetate and lead citrate. Sections were imaged under TEM at 80KV to visualize mitochondrial ultrastructure.
2.10. Cellular respiration
A MitoStress Test assay, utilizing the Seahorse XFe96 Bioanalyzer (Agilent Technologies, Santa Clara, CA) was used to assess the impact of mitoNEET on cellular respiration. One hour prior to the start of the assay, MitoNEET KO and VC REH cells, suspended in Mito-Stress assay media containing glucose, L-glutamine and sodium pyruvate, were seeded into Seahorse 96-well XF cell culture microplates coated with Cell-Tak Cell and Tissue Adhesive (Corning Inc., Glendale, AZ) at a density of 100,000 cells/well. Cell-Tak solution contained a ratio of 289 μl of 0.1 M sodium bicarbonate, 1 μl of 5N NaOH, and 10 μl of Cell-Tak, with 10 μl added to wells, then discarded after 20 mins by 2 washes of 100 μl of water. For the Glycolysis Stress Test, media containing 49.5 mL of sterile XF DMEM Medium, pH 7.4 with 500 μl of Seahorse XF 200 mM glutamine was added to the cells. The 96-well plate was centrifuged at 200 x g for one minute and placed in a non-CO2 incubator for 1 hr prior to beginning the experiment. The Seahorse XFe96 measured oxygen consumption rate (OCR) under specific conditions using sequential addition of oligomycin, carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP), antimycin-A and rotenone for the MitoStress Test. For the Glycolysis Stress test, glucose, oligomycin and 2-deoxygclucose were added as per kit instructions.
2.11. Electron flow assay
An Electron Flow Assay utilizing the Seahorse XFe96 Bioanalyzer (Agilent Technologies, Santa Clara, CA) was used to assess the impact of mitoNEET on activities of ETC complexes in permeabilized cells. REH VC and mitoNEET KO cells were seeded into Seahorse 96-well XF cell culture microplates coated with Cell-Tak Cell and Tissue Adhesive (Corning Inc., Glendale, AZ) at a density of 300,000 cells/well 1 hr before the assay was performed. The plate was centrifuged at 200 x g for 60 sec and placed in a non-CO2 incubator for 1 hr prior to the start of the experiment. The XFe96 Bioanalyzer was calibrated with the following injections added to their respective injection ports: Port A: rotenone (2 μM); Port B: succinate (10 mM); Port C: antimycin-A (4 μM) and Port D: ascorbate (10 mM) + cytochrome c electron donor N,N,N′,N′-tetramethyl-p-phenylenediamine (TMPD; 100 μM). Pyruvate (10 mM), malate (1 mM), FCCP (4 μM) and Seahorse XF Plasma Membrane Permeabilizer (2 nM) were added to the XFe96 cell plate immediately before beginning the assay. Two mix-wait-measure cycles consisting of 30 sec mix, 30 sec wait, and 2 min measure were performed before the first injection and following each injection. ETC complex I activity was calculated as the complex I substrate (pyruvate + malate)-driven oxygen consumption sensitive to rotenone inhibition. ETC complex II/III activity calculates as the complex II/III substrate (succinate)-driven oxygen consumption sensitive to antimycin-A inhibition.
2.12. Superoxide assay with dihydroethidium bromide (DHE)
Cells were plated in 96-well V-bottom plate at a density of 100,000 cells/well in 100 μl. DHE was quantitated using a superoxide ROS Detection Cell-Based Assay Kit (Cayman Chemical Cat#601290) according to manufacturer’s instructions and read using a Molecular Devices SpectraMax ID5 plate reader.
3. Results and Discussion
MitoNEET is a redox active [2Fe-2S] cluster-containing protein which has been shown to regulate the oxidative capacity of mitochondria and cellular iron homeostasis [25]. Bioinformatic analyses and studies of cancers including breast, lung, and prostate cancer have reported that mitoNEET over-expression may contribute to tumor growth, and the knockdown or knockout of this protein reduces cell proliferation and/or tumor size, demonstrating a potential role for mitoNEET in cancer growth [10, 12, 14, 26]. MitoNEET-overexpression has also been shown to attenuate the effects of oxidative stress in cardiomyocytes and other cell types [27–30]. We determined that mitoNEET is expressed in primary patient samples isolated during relapse of B-ALL (Supplemental Fig. 1). We also recently showed that the mitoNEET ligand, NL-1, exhibited anti-proliferative activity in human REH cells, prompting our investigation into the role of mitoNEET in mitochondrial function in REH leukemic cells [24]. The need for novel approaches to treating MRD in ALL and potential role of mitoNEET in tumor progression, led us to postulate that mitoNEET ligands may provide benefit in ALL treatment by influencing mitochondrial metabolism.
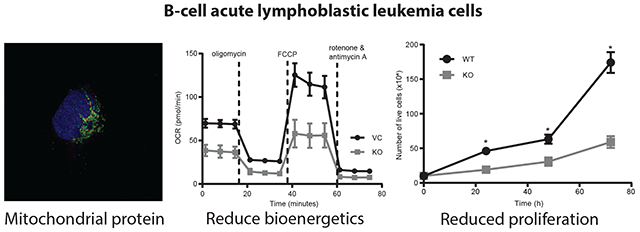
To evaluate the role of mitoNEET in leukemic cells, we generated a mitoNEET knockout (KO) cell-line in REH B-cell ALL cells using CRISPR-Cas 9 methodology (Fig. 1A). Since mitoNEET has previously been shown to localize on the outer mitochondrial membrane of mitochondria [5, 6, 31], we confirmed through co-staining with MitoTracker that mitoNEET is localized with the mitochondria in the VC REH cells, and mitoNEET staining is lost in the mitoNEET KO cells (Fig. 1B) consistent with previously published studies [5, 6]. As an additional orthogonal evaluation of mitoNEET localization, mitoNEET was shown to localize with the translocase of the outer membrane protein (TOM20), a well-known mitochondrial marker (Supplemental Fig. 2). To visualize the mitochondrial ultra-structure in more detail, TEM imaging was employed and demonstrated that the mitochondrial ultrastructure was altered in mitoNEET KO cells (Fig. 1C). Ultra-structural alterations in mitochondria where mitoNEET was knocked-out, have also been noted by other groups, with noticeable decrease in the complexity of the cristae structure located in mitoNEET KO cellular mitochondria [1, 32]. Since mitochondrial function is closely tied to the size and shape of the mitochondria [33], we expected the mitoNEET KO mitochondria to have diminished functional capacity, specifically the bioenergetic OXPHOS capacity of the leukemia cells [32].
Fig. 1.
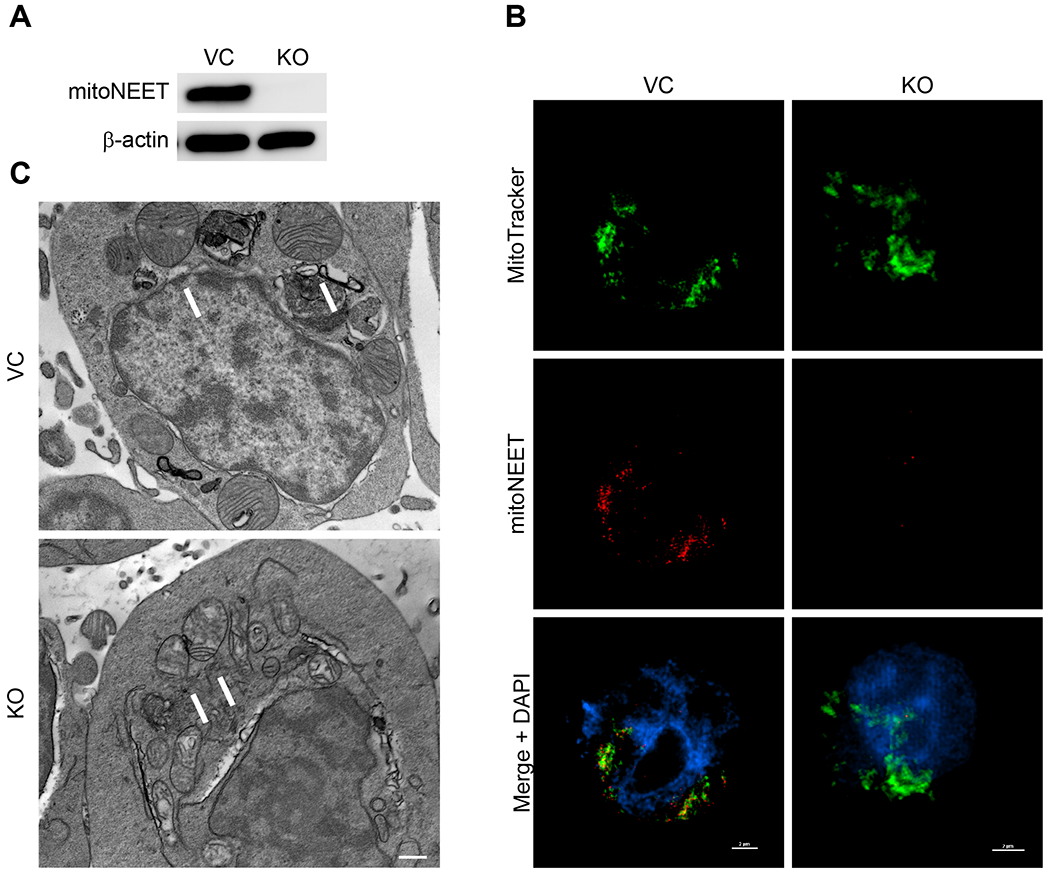
A) The mitochondrial protein mitoNEET was knocked out (KO) of REH cells using CRISPR/Cas9 for comparison to vector control cells (VC) and the protein expression determined with Western Blot. Beta-actin was used as the loading control (N = 3). B) Cellular localization of mitoNEET in REH cells. MitoNEET (red) is located on mitochondria, which are stained with MitoTracker Deep Red (green). Co-localization is observed from the yellow/orange coloring in the overlay in the VC cells. MitoNEET KO cells do not show localized staining in the mitochondria. Scale bar = 2 μm. C) TEM micrographs of REH cells. The KO cells are characterized by structural changes in the size and shape of the cristae in mitochondria, which are indicated by arrows, compared to the VC. Scale bar = 500 nm.
Based on previous studies showing mitoNEET regulates mitochondrial bioenergetic function [1, 29, 34], we utilized the Seahorse Bioanalyzer to characterize mitochondrial function including OXPHOS (measured by the oxygen consumption rate; OCR) and glycolysis (measured by the extracellular acidification rate; ECAR). MitoNEET knockout in REH cells was associated with reduction in overall mitochondrial function, including diminished non-mitochondrial oxygen consumption, basal respiration, maximal respiration, proton leak, ATP production, and spare respiratory capacity (Fig. 2A–F). Fig. 2G and 2H show an overall reduction in OCR and a nonsignificant decrease in ECAR. When the cells were analyzed using the Glycolysis Stress test, no significant differences were found between the KO and VC cells (Fig. 2I), even though the total ATP content in the KO cells was significantly reduced (Fig. 2J) [19, 35, 36]. These findings are consistent with earlier work focused on cardiac and neuronal cells showing the loss of mitoNEET in mitochondria greatly affected mitochondrial bioenergetic (OXPHOS) function, although loss of mitoNEET did not result in a complete shutdown of the electron transport chain (ETC) function [30, 34]. As a final evaluation of mitochondrial function, we assessed electron flow rates through the ETC (Fig. 3A and 3B). In comparison to the VC cells, there was a significant decrease in electron flow rates in the KO cells. Complex I (Fig. 3C), as well as the Complex II/III activity (Fig. 3D) was reduced, providing additional evidence that a loss of mitoNEET in leukemia cells impairs the OXPHOS capacity function of mitochondria.
Fig. 2.
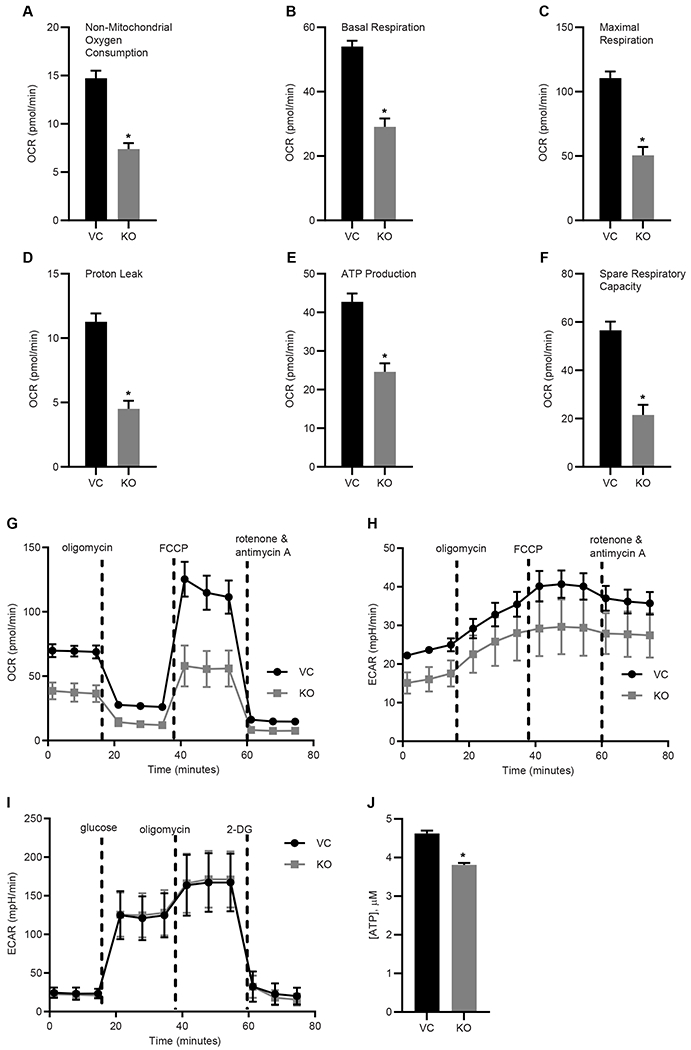
Mitochondrial function in REH leukemia cells is disrupted. A mitochondrial stress test kit with the Seahorse bioanalyzer shows reduction in several key aspects of mitochondrial function including: A) non-mitochondrial oxygen consumption, B) basal respiration, C) maximal respiration, D) proton leak, E) ATP production, F) spare respiratory capacity; G) oxygen consumption (OCR) graphs of mitochondrial function. H) The extrasellar acidification rate (ECAR) graph, and I) Glycolysis stress test (no significant difference). J) ATP content (μM) is reduced in the mitoNEET KO cells. Data are shown average ± SD, where N = 8. *P<0.05.
Fig. 3.
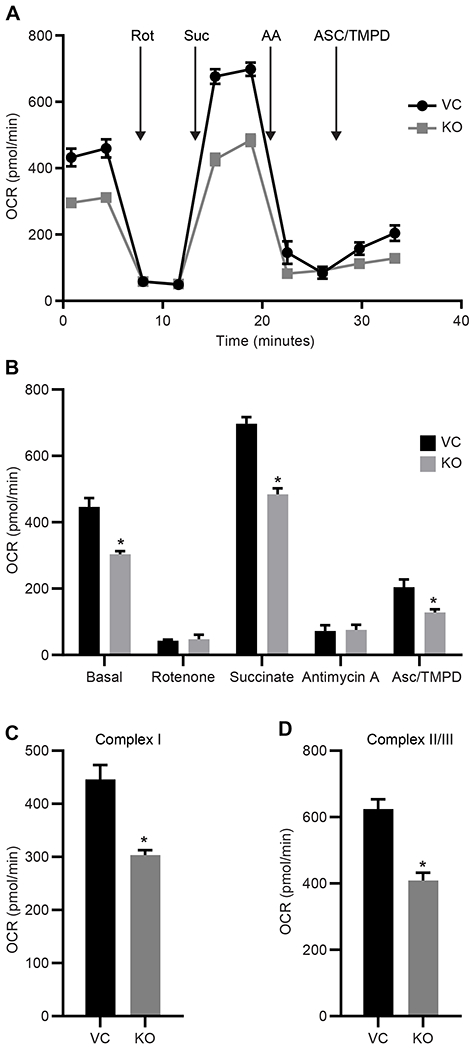
Mitochondrial electron transport chain (ETC) function is diminished in mitoNEET KO REH cells. A) OCR graph showing substrate and inhibitor addition, B) averaged OCR response, C) Complex I Activity from the ETC data and D) Complex II/III activity. Data are shown as the average ± SD, where N = 8. *P<0.05. Abbreviations: rotenone (rot), succinate (sue), antimycin A (AA), ascorbate (asc), and N,N,N′,N′-tetramethyl-p-phenylenediamine (TMPD).
The mitochondrial OXPHOS and electron flow have previously been shown to be influenced by a loss of mitoNEET [37], with overexpression of mitoNEET leading to increased mitochondrial OXPHOS [38]. These changes in mitochondrial function in REH cells prompted us to evaluate other parameters of mitochondrial function including reactive oxygen species (ROS) production and mitochondrial membrane potential. In REH cells, the KO of mitoNEET led to increased levels of hydrogen-peroxide ROS production measured by the Amplex Red assay kit (Fig. 4A), and a decrease in the mitochondrial membrane potential (Fig. 4B), with the latter being a contributor of mitochondrially-derived ROS [39]. We found that the basal level of superoxide was significantly increased in the KO cells as measured by the superoxide indicator dye DHE, and as expected, antimycin A, an inhibitor of the ETC complex III, induced a significant increase in superoxide load in the KO cells as compared to VC cells (Fig. 4C). The antioxidant N-acetyl-cysteine was able to significantly reduce the superoxide levels in both the KO and VC cells. Combined with the results from the Mito-Stress and electron flow assays, these findings support a potential role for mitoNEET in the regulation of mitochondrial production of ATP under normal physiological parameters in concert with low production of noxious radicals. The absence of mitoNEET in leukemia cells leads to mitochondrial dysfunction and identifies a potential vulnerability that could be manipulated during treatment.
Fig. 4.
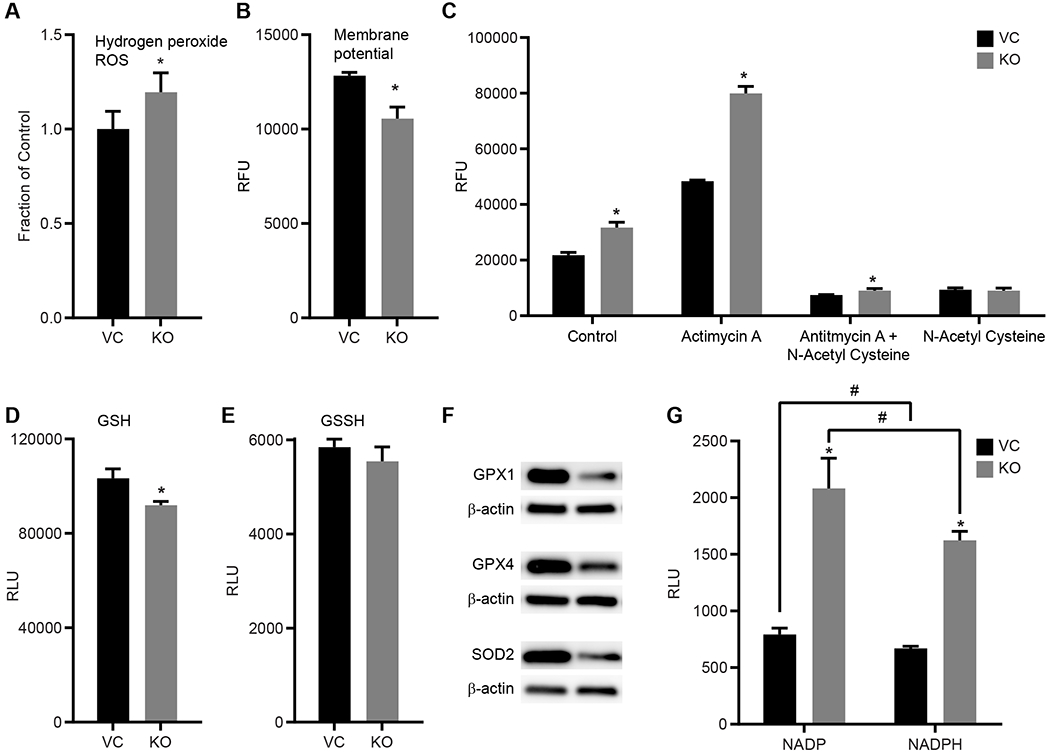
MitoNEET KO leads to mitochondrial dysfunction in REH cells. A) hydrogen peroxide ROS levels are increased in KO cells compared to VC; B) mitochondrial membrane potential is decreased in KO cells compared to VC; C) DHE detection of superoxide indicates increased levels in the KO cells compared to VC. The positive control antimycin A increased superoxide in both the VC and KO cells. The antioxidant N-acetyl cysteine (NAC) decreased both endogenous as well as antimycin A-induced superoxide production. Data are shown as the average ± SD, where N = 3-4. *P<0.05. D) Thiol content in REH cells with levels of reduced (GSH) and E) oxidized (GSSH) glutathione were measured in the KO cells compared to the VC. The loss of mitoNEET was correlated with a decrease in thiol content. F) Western Blot analysis of glutathione peroxidase (GPX) 1 and 4 as well as superoxide dismutase 2 (SOD2) showed a decrease in protein content in the mitoNEET KO cells compared to the VC. Equal protein loading per lane verified with β-actin staining. Data are shown average ± SD, where N = 8. *P<0.05. G) Metabolites NADP and NADPH in REH cells show significant increase in mitoNEET KO cells compared to the controls (VC). RLU average ± SD, where N = 8. *P<0.05.
The [2Fe-2S] clusters of mitoNEET are redox active, similar to other Fe-S containing proteins [40, 41], and supports the maintenance of free thiol content of cells including the antioxidant glutathione [42]. Electron paramagnetic resonance (EPR) studies further show that mitoNEET interacts with cytosolic metabolites such as glutathione, and NADPH/NADH [43]. Since these metabolites are essential for supporting the redox reactions in cellular processes, we evaluated their levels in the mitoNEET KO cells. Fig. 4D shows that the level of glutathione in the KO cells was dramatically reduced when compared to the VC cells, while no difference was observed in the levels of the oxidized GSSH (Fig. 4E). We also found that the protein levels of glutathione peroxidase (GPX) 1 and GPX4 were reduced in the KO cells, as well as the manganese-dependent superoxide dismutase (Mn-SOD; SOD2) (Fig. 4F). GPX1 is mainly located in the cytosol of most cell types, while GPX4 (also known as phospholipid hydroperoxide) is located in the mitochondrial and cytosolic spaces, and plays a role in lipid metabolism [39, 44]. The observed reduction of protein levels might be due, in part, to the higher load of ROS produced by the cells and decreased interaction with mitoNEET [42], and may also play a role in ferroptosis and iron dysregulation pathways [11, 45, 46]. Additionally, previous studies have indicated that some leukemias have increased levels of NADPH, which plays an important role in redox mechanisms [47, 48], and may explain the resulting increases in ROS detected. NADH+ and NADPH levels are dramatically increased in the KO cells (Fig. 4G), suggesting that reduced mitoNEET expression impacts the metabolism of NADH/NADPH and the metabolic signaling of these factors.
It was previously suggested that mitoNEET may play a role in lipid metabolism since patients taking pioglitazone had decreased free fatty acid plasma levels, while overexpression of mitoNEET in an obese mouse model led to significant improvements in lipid metabolism [7, 29, 30, 49]. Furthermore, the glitazone derivative PNU-91325 increased fatty acid synthesis in liver HepG2 cells [9]. Recently, it has been suggested that metabolic dysregulation may contribute to MRD, and taken together with the mitoNEET-associated effect on lipids alterations, we assessed the level of lipids in the leukemia KO and VC cells using Nile Red lipid staining [50]. As demonstrated in Fig. 5A, loss of mitoNEET led to a significant decrease in lipid levels in the cells. In order to explore this relationship between mitoNEET and fatty acids [9, 29], we evaluated the levels of free fatty acids via a triglyceride assay, but did not find any significant differences between the KO and the VC cells (Fig. 5B). Lastly, evaluation of the fatty acid metabolism utilizing the Seahorse BioFlux Analyzer evaluated the effect of mitoNEET KO on β-oxidation in the REH cells (Fig. 5C). As part of the assay, etomoxir C, an irreversible inhibitor (due to the covalent binding epoxide moiety) to the carnitine palmitoyltransferase-1, was used, as it prevents the formation of the acyl carnitines which is necessary for the production of ATP from fatty acid oxidation [51]. We found that the KO cells had lower levels of fatty acid oxidation when treated with etomoxir C, but this decrease was not statistically significant. Future studies will evaluate the β-oxidation pathways in ALL, especially those involved with Co-A derivatives, as fatty acids may play a role in mitochondrial autophagy [9, 29, 52].
Fig. 5.
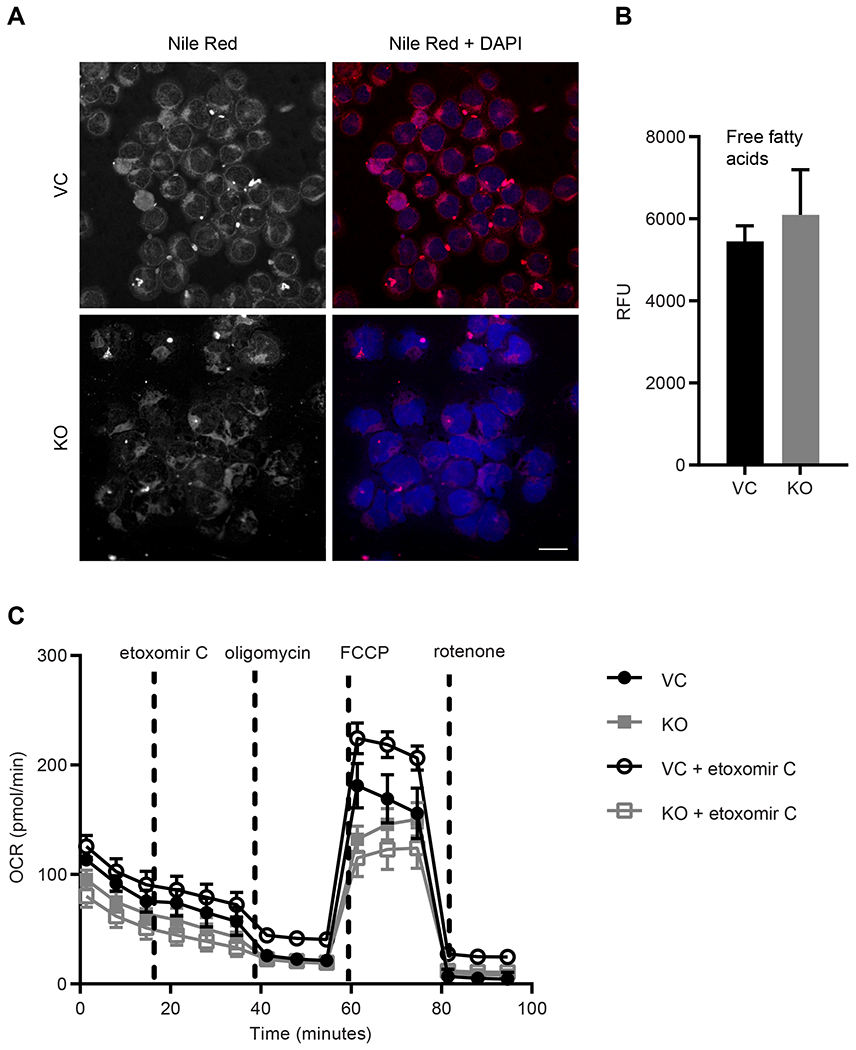
mitoNEET KO leads to a decrease in cellular lipid levels. A) Nile Red staining shows lipid levels and DAPI nuclear staining, scale bar = 20 μm; B) free fatty acid (FFA) levels are unchanged with mitoNEET KO, C) β-oxidation assay with the Seahorse BioFlux Analyzer show that OCR is reduced in mitoNEET KO cells, average ± SD, where N = 8.
Mitochondrial metabolism in cancer cells has been suggested as a potential novel target for anticancer drug development [38]. MitoNEET has been suggested to play a role in cancer cell proliferation, where previous studies in breast cancer showed that elevated levels of mitoNEET led to increased cell proliferation [14, 53]. We evaluated the effect mitoNEET KO on REH cell proliferation and found that KO cells showed reduced proliferation (Fig. 6 and Supplemental Fig. 3), and a similar effect on cell proliferation was seen in our previously published studies showing that NL-1, a mitoNEET ligand, reduced cell proliferation in a B-cell ALL cancer model [24]. We next evaluated the effect of mitoNEET KO, in combination with the anti-cancer drug cytarabine (Ara-C), a common treatment of leukemia. Ara-C is an antimetabolic agent which interferes with the normal synthesis of DNA [54]. We found that treatment with Ara-C led to a significant decrease in cell proliferation in KO cells compared to the VC cells (Fig. 7), suggesting that the increased ROS-based stress on the cell with mitoNEET KO led to increase susceptibly of the cells to Ara-C. Since mitochondrial metabolism is needed for tumor growth to support proliferation [38], several groups have suggested that targeting mitochondria might be a useful strategy in cancer therapy. However, this strategy has been explored predominantly in solid tumors with insight into hematological cancers remaining less explored [55, 56]. By exploiting this metabolic vulnerability in leukemias, future combination therapies, including therapies targeting mitochondria (e.g. metformin [38]) or specifically mitoNEET, might provide additional benefit to patients.
Fig. 6.
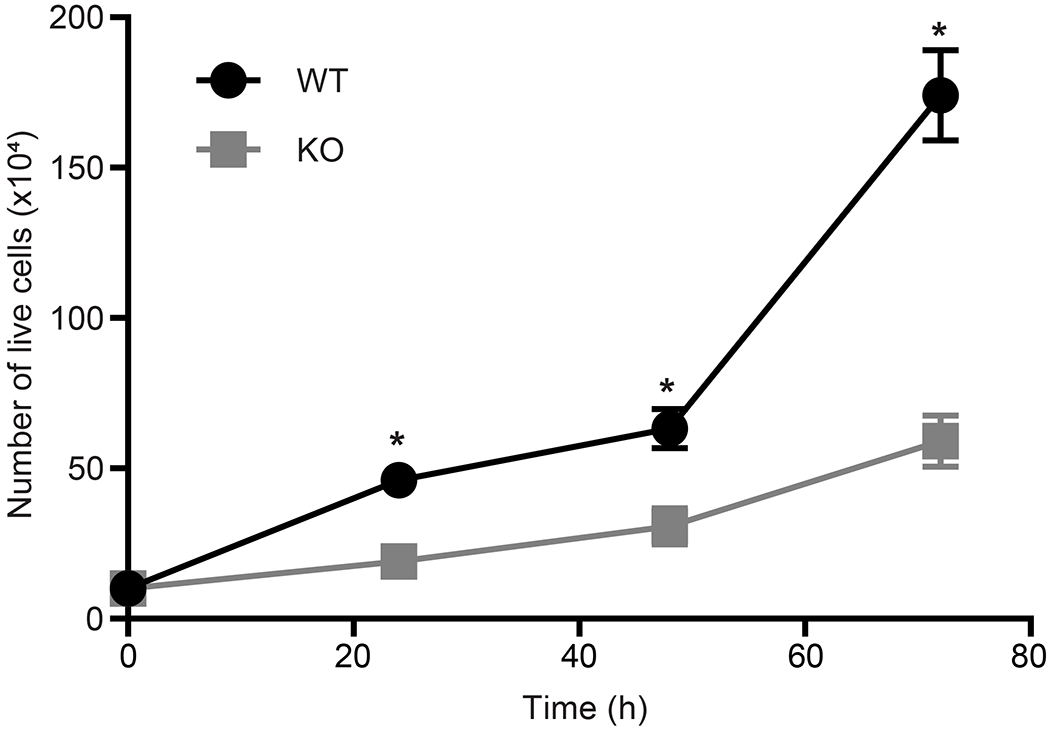
MitoNEET KO results in decreased proliferation of REH cells compared to control cells (VC). Data are shown average ± SD, where N = 4. *P<0.05.
Fig. 7.
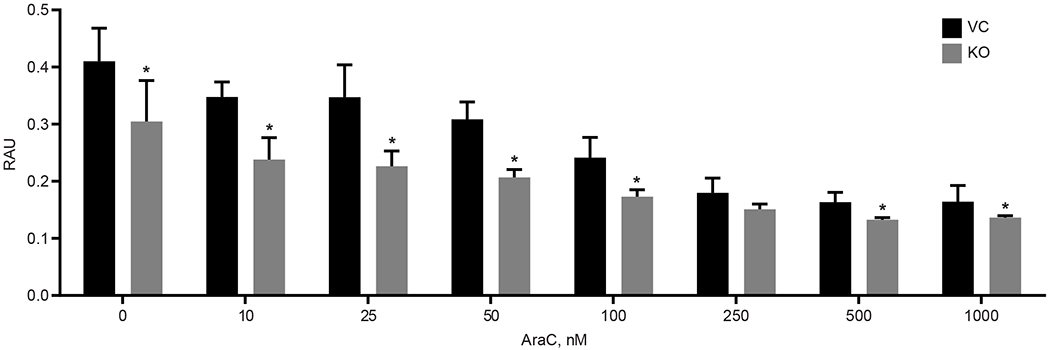
MitoNEET KO in REH cells increases sensitivity to the ALL anti-cancer drug Ara-C. Proliferation was significantly decreased in the KO cells compared to the VC. Data are shown average ± SD, where N = 4. *P<0.05.
In conclusion, we have characterized the effects of mitoNEET KO on mitochondrial bioenergetic function in human REH leukemic cells. Loss of mitoNEET dramatically affected mitochondrial bioenergetic function, decreased cellular OXPHOS, including dysregulated ROS generation, and decreased tumor cell proliferation. Based on these findings, targeting mitochondrial metabolism in B-ALL cancer cells through interruption of mitoNEET function provides a possible novel approach to of the treatment refractory MRD. Future studies will interrogate the potential of targeting mitochondrial function to sensitize treatment-refractory ALL to currently available chemotherapeutic agents.
Supplementary Material
Supplemental Fig. 1. MitoNEET protein levels in primary cancer specimens from two B-cell ALL patients. Equal protein loading per lane verified with β-actin staining.
Supplemental Fig. 2. Co-localization of mitoNEET (red) with TOM20 (green), a mitochondrial marker, with yellow areas showing overlap in REH cells. DAPI was used for nuclear staining. Scale bar = 2 μm.
Supplemental Fig. 3. Cell proliferation in the REH KO and VC cells was determined by measuring the fluorescence in GFP-expressing cells. *P<0.05, where N = 3.
Highlights.
MitoNEET is a mitochondrial redox-active [2Fe-2S] cluster protein
Knockout of mitoNEET in B-cell acute lymphoblastic leukemia affects bioenergetics
KO cells have reduced oxidative phosphorylation and increased ROS production
KO cells show reduced lipid content with reduction in cell proliferation
KO cells exhibit increased sensitivity to cytarabine (Aca-C)
Acknowledgements
This work was supported by the National Institutes of Health [Grants U54GM104942, P20GM121322, P20RR016440, P20GM103434, P20GM103488, P20GM103434, S10OD016165, and P20GM109098,] and the Alexander B. Osborn Hematopoietic Malignancy and Transplantation Endowed Professorship (LFG), the National Heart, Lung and Blood Institute Grant HL-128485 (JMH), and the Community Foundation for the Ohio Valley Whipkey Trust (JMH). We acknowledge the use of the WVU SRF Electron Microscopy Facilities with Dr. Marcela Redigolo and UMD Electron Microscopy Core Imaging Facility with Dr. Ru-Ching Hsia. Imaging experiments were performed in the WVU Imaging Facilities which have been supported by the WVU Cancer Institute, the WVU HSC Office of Research and Graduate Education, and NIH grants [P20RR016440, P30GM103488, P20GM121322, P20GM103434, and U54GM104942].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interest
The authors declare no conflict of interest.
References
- [1].Wiley SE, Murphy AN, Ross SA, van der Geer P, Dixon JE, MitoNEET is an iron-containing outer mitochondrial membrane protein that regulates oxidative capacity, Proc Natl Acad Sci U S A 104(13) (2007) 5318–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Karmi O, Marjault HB, Pesce L, Carloni P, Onuchic JN, Jennings PA, Mittler R, Nechushtai R, The unique fold and lability of the [2Fe-2S] clusters of NEET proteins mediate their key functions in health and disease, J Biol Inorg Chem 23(4) (2018) 599–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Inupakutika MA, Sengupta S, Nechushtai R, Jennings PA, Onuchic JN, Azad RK, Padilla P, Mittler R, Phylogenetic analysis of eukaryotic NEET proteins uncovers a link between a key gene duplication event and the evolution of vertebrates, Sci Rep 7 (2017) 42571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Geldenhuys WJ, Leeper TC, Carroll RT, mitoNEET as a novel drug target for mitochondrial dysfunction, Drug Discov Today 19(10) (2014) 1601–6. [DOI] [PubMed] [Google Scholar]
- [5].Paddock ML, Wiley SE, Axelrod HL, Cohen AE, Roy M, Abresch EC, Capraro D, Murphy AN, Nechushtai R, Dixon JE, Jennings PA, MitoNEET is a uniquely folded 2Fe 2S outer mitochondrial membrane protein stabilized by pioglitazone, Proc Natl Acad Sci U S A 104(36) (2007) 14342–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wiley SE, Paddock ML, Abresch EC, Gross L, van der Geer P, Nechushtai R, Murphy AN, Jennings PA, Dixon JE, The outer mitochondrial membrane protein mitoNEET contains a novel redox-active 2Fe-2S cluster, J Biol Chem 282(33) (2007) 23745–9. [DOI] [PubMed] [Google Scholar]
- [7].Colca JR, McDonald WG, Waldon DJ, Leone JW, Lull JM, Bannow CA, Lund ET, Mathews WR, Identification of a novel mitochondrial protein (“mitoNEET”) cross-linked specifically by a thiazolidinedione photoprobe, Am J Physiol Endocrinol Metab 286(2) (2004) E252–60. [DOI] [PubMed] [Google Scholar]
- [8].Divakaruni AS, Wiley SE, Rogers GW, Andreyev AY, Petrosyan S, Loviscach M, Wall EA, Yadava N, Heuck AP, Ferrick DA, Henry RR, McDonald WG, Colca JR, Simon MI, Ciaraldi TP, Murphy AN, Thiazolidinediones are acute, specific inhibitors of the mitochondrial pyruvate carrier, Proc Natl Acad Sci U S A 110(14) (2013) 5422–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Harrigan GG, Colca J, Szalma S, Boros LG, PNU-91325 increases fatty acid synthesis from glucose and mitochondrial long chain fatty acid degradation: a comparative tracer-based metabolomics study with rosiglitazone and pioglitazone in HepG2 cells, Metabolomics 2(1) (2006) 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zhang A, Yang J, Ma C, Li F, Luo H, Development and Validation of a Robust Ferroptosis-Related Prognostic Signature in Lung Adenocarcinoma, Front Cell Dev Biol 9 (2021) 616271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wang D, Wei G, Ma J, Cheng S, Jia L, Song X, Zhang M, Ju M, Wang L, Zhao L, Xin S, Identification of the prognostic value of ferroptosis-related gene signature in breast cancer patients, BMC Cancer 21(1) (2021) 645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wang S, Wu C, Ma D, Hu Q, Identification of a ferroptosis-related gene signature (FRGS) for predicting clinical outcome in lung adenocarcinoma, PeerJ 9 (2021) e11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Natarajan A, Ramachandran B, Gopisetty G, Jayavelu S, Sundersingh S, Rajkumar T, Pioglitazone modulates doxorubicin resistance in a in vivo model of drug resistant osteosarcoma xenograft, Naunyn Schmiedebergs Arch Pharmacol (2020). [DOI] [PubMed] [Google Scholar]
- [14].Sohn YS, Tamir S, Song L, Michaeli D, Matouk I, Conlan AR, Harir Y, Holt SH, Shulaev V, Paddock ML, Hochberg A, Cabanchick IZ, Onuchic JN, Jennings PA, Nechushtai R, Mittler R, NAF-1 and mitoNEET are central to human breast cancer proliferation by maintaining mitochondrial homeostasis and promoting tumor growth, Proc Natl Acad Sci U S A 110(36) (2013) 14676–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kuntz EM, Baquero P, Michie AM, Dunn K, Tardito S, Holyoake TL, Helgason GV, Gottlieb E, Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells, Nat Med 23(10) (2017) 1234–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Guaragnella N, Giannattasio S, Moro L, Mitochondrial dysfunction in cancer chemoresistance, Biochem Pharmacol 92(1) (2014) 62–72. [DOI] [PubMed] [Google Scholar]
- [17].Guerra F, Arbini AA, Moro L, Mitochondria and cancer chemoresistance, Biochim Biophys Acta Bioenerg 1858(8) (2017) 686–699. [DOI] [PubMed] [Google Scholar]
- [18].Weiner-Gorzel K, Murphy M, Mitochondrial dynamics, a new therapeutic target for Triple Negative Breast Cancer, Biochim Biophys Acta Rev Cancer 1875(2) (2021) 188518. [DOI] [PubMed] [Google Scholar]
- [19].Nair RR, Piktel D, Hathaway QA, Rellick SL, Thomas P, Saralkar P, Martin KH, Geldenhuys WJ, Hollander JM, Gibson LF, Pyrvinium Pamoate Use in a B cell Acute Lymphoblastic Leukemia Model of the Bone Tumor Microenvironment, Pharm Res 37(3) (2020) 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Baffy G, Mitochondrial uncoupling in cancer cells: Liabilities and opportunities, Biochim Biophys Acta Bioenerg 1858(8) (2017) 655–664. [DOI] [PubMed] [Google Scholar]
- [21].Rellick SL, Hu G, Piktel D, Martin KH, Geldenhuys WJ, Nair RR, Gibson LF, Coculture model of B-cell acute lymphoblastic leukemia recapitulates a transcription signature of chemotherapy-refractory minimal residual disease, Sci Rep 11(1) (2021) 15840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Slone WL, Moses BS, Evans R, Piktel D, Martin KH, Petros W, Craig M, Gibson LF, Modeling Chemotherapy Resistant Leukemia In Vitro, J Vis Exp (108) (2016) e53645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Karantanou C, Godavarthy PS, Krause DS, Targeting the bone marrow microenvironment in acute leukemia, Leuk Lymphoma 59(11) (2018) 2535–2545. [DOI] [PubMed] [Google Scholar]
- [24].Geldenhuys WJ, Nair RR, Piktel D, Martin KH, Gibson LF, The MitoNEET Ligand NL-1 Mediates Antileukemic Activity in Drug-Resistant B-Cell Acute Lymphoblastic Leukemia, J Pharmacol Exp Ther 370(1) (2019) 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lipper CH, Stofleth JT, Bai F, Sohn YS, Roy S, Mittler R, Nechushtai R, Onuchic JN, Jennings PA, Redox-dependent gating of VDAC by mitoNEET, Proc Natl Acad Sci U S A 116(40) (2019) 19924–19929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Mittler R, Darash-Yahana M, Sohn YS, Bai F, Song L, Cabantchik IZ, Jennings PA, Onuchic JN, Nechushtai R, NEET Proteins: A New Link Between Iron Metabolism, Reactive Oxygen Species, and Cancer, Antioxid Redox Signal 30(8) (2019) 1083–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Habener A, Chowdhury A, Echtermeyer F, Lichtinghagen R, Theilmeier G, Herzog C, MitoNEET Protects HL-1 Cardiomyocytes from Oxidative Stress Mediated Apoptosis in an In Vitro Model of Hypoxia and Reoxygenation, PLoS One 11(5) (2016) e0156054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Joffin N, Paschoal VA, Gliniak CM, Crewe C, Elnwasany A, Szweda LI, Zhang Q, Hepler C, Kusminski CM, Gordillo R, Oh DY, Gupta RK, Scherer PE, Mitochondrial metabolism is a key regulator of the fibro-inflammatory and adipogenic stromal subpopulations in white adipose tissue, Cell Stem Cell 28(4) (2021) 702–717 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kusminski CM, Chen S, Ye R, Sun K, Wang QA, Spurgin SB, Sanders PE, Brozinick JT, Geldenhuys WJ, Li WH, Unger RH, Scherer PE, MitoNEET-Parkin Effects in Pancreatic alpha- and beta-Cells, Cellular Survival, and Intrainsular Cross Talk, Diabetes 65(6) (2016) 1534–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kusminski CM, Holland WL, Sun K, Park J, Spurgin SB, Lin Y, Askew GR, Simcox JA, McClain DA, Li C, Scherer PE, MitoNEET-driven alterations in adipocyte mitochondrial activity reveal a crucial adaptive process that preserves insulin sensitivity in obesity, Nat Med 18(10) (2012) 1539–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wiley SE, Rardin MJ, Dixon JE, Chapter 13 Localization and function of the 2Fe-2S outer mitochondrial membrane protein mitoNEET, Methods Enzymol 456 (2009) 233–46. [DOI] [PubMed] [Google Scholar]
- [32].Baker N, Patel J, Khacho M, Linking mitochondrial dynamics, cristae remodeling and supercomplex formation: How mitochondrial structure can regulate bioenergetics, Mitochondrion 49 (2019) 259–268. [DOI] [PubMed] [Google Scholar]
- [33].Berman SB, Pineda FJ, Hardwick JM, Mitochondrial fission and fusion dynamics: the long and short of it, Cell Death Differ 15(7) (2008) 1147–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Geldenhuys WJ, Benkovic SA, Lin L, Yonutas HM, Crish SD, Sullivan PG, Darvesh AS, Brown CM, Richardson JR, MitoNEET (CISD1) Knockout Mice Show Signs of Striatal Mitochondrial Dysfunction and a Parkinson’s Disease Phenotype, ACS Chem Neurosci 8(12) (2017) 2759–2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Samudio I, Fiegl M, McQueen T, Clise-Dwyer K, Andreeff M, The warburg effect in leukemia-stroma cocultures is mediated by mitochondrial uncoupling associated with uncoupling protein 2 activation, Cancer Res 68(13) (2008) 5198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wander P, Arentsen-Peters S, Pinhanos SS, Koopmans B, Dolman MEM, Ariese R, Bos FL, Castro PG, Jones L, Schneider P, Navarro MG, Molenaar JJ, Rios AC, Zwaan CM, Stam RW, High-throughput drug screening reveals Pyrvinium pamoate as effective candidate against pediatric MLL-rearranged acute myeloid leukemia, Transl Oncol 14(5) (2021) 101048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wang Y, Landry AP, Ding H, The mitochondrial outer membrane protein mitoNEET is a redox enzyme catalyzing electron transfer from FMNH2 to oxygen or ubiquinone, J Biol Chem 292(24) (2017) 10061–10067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Salem AF, Whitaker-Menezes D, Howell A, Sotgia F, Lisanti MP, Mitochondrial biogenesis in epithelial cancer cells promotes breast cancer tumor growth and confers autophagy resistance, Cell Cycle 11(22) (2012) 4174–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Suski JM, Lebiedzinska M, Bonora M, Pinton P, Duszynski J, Wieckowski MR, Relation between mitochondrial membrane potential and ROS formation, Methods Mol Biol 810 (2012) 183–205. [DOI] [PubMed] [Google Scholar]
- [40].Mena NP, Bulteau AL, Salazar J, Hirsch EC, Nunez MT, Effect of mitochondrial complex I inhibition on Fe-S cluster protein activity, Biochem Biophys Res Commun 409(2) (2011) 241–6. [DOI] [PubMed] [Google Scholar]
- [41].Orme-Johnson WH, Beinert H, On the formation of the superoxide anion radical during the reaction of reduced iron-sulfur proteins with oxygen, Biochem Biophys Res Commun 36(6) (1969) 905–11. [DOI] [PubMed] [Google Scholar]
- [42].Landry AP, Ding H, Redox control of human mitochondrial outer membrane protein MitoNEET [2Fe-2S] clusters by biological thiols and hydrogen peroxide, J Biol Chem 289(7) (2014) 4307–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Landry AP, Wang Y, Cheng Z, Crochet RB, Lee YH, Ding H, Flavin nucleotides act as electron shuttles mediating reduction of the [2Fe-2S] clusters in mitochondrial outer membrane protein mitoNEET, Free Radic Biol Med 102 (2017) 240–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kim MJ, Yun GJ, Kim SE, Metabolic Regulation of Ferroptosis in Cancer, Biology (Basel) 10(2) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Homma T, Kobayashi S, Fujii J, Cysteine preservation confers resistance to glutathione-depleted cells against ferroptosis via CDGSH iron sulphur domain-containing proteins (CISDs), Free Radic Res 54(6) (2020) 397–407. [DOI] [PubMed] [Google Scholar]
- [46].Wu ZH, Tang Y, Yu H, Li HD, The role of ferroptosis in breast cancer patients: a comprehensive analysis, Cell Death Discov 7(1) (2021) 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Bhanot H, Weisberg EL, Reddy MM, Nonami A, Neuberg D, Stone RM, Podar K, Salgia R, Griffin JD, Sattler M, Acute myeloid leukemia cells require 6-phosphogluconate dehydrogenase for cell growth and NADPH-dependent metabolic reprogramming, Oncotarget 8(40) (2017) 67639–67650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Sanchez-Sanchez B, Gutierrez-Herrero S, Lopez-Ruano G, Prieto-Bermejo R, Romo-Gonzalez M, Llanillo M, Pandiella A, Guerrero C, Miguel JF, Sanchez-Guijo F, Del Canizo C, Hernandez-Hernandez A, NADPH oxidases as therapeutic targets in chronic myelogenous leukemia, Clin Cancer Res 20(15) (2014) 4014–25. [DOI] [PubMed] [Google Scholar]
- [49].Serlie MJ, Allick G, Groener JE, Ackermans MT, Heijligenberg R, Voermans BC, Aerts JM, Meijer AJ, Sauerwein HP, Chronic treatment with pioglitazone does not protect obese patients with diabetes mellitus type II from free fatty acid-induced insulin resistance, J Clin Endocrinol Metab 92(1) (2007) 166–71. [DOI] [PubMed] [Google Scholar]
- [50].Al-Saffar NM, Titley JC, Robertson D, Clarke PA, Jackson LE, Leach MO, Ronen SM, Apoptosis is associated with triacylglycerol accumulation in Jurkat T-cells, Br J Cancer 86(6) (2002) 963–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Weis BC, Cowan AT, Brown N, Foster DW, McGarry JD, Use of a selective inhibitor of liver carnitine palmitoyltransferase I (CPT I) allows quantification of its contribution to total CPT I activity in rat heart. Evidence that the dominant cardiac CPT I isoform is identical to the skeletal muscle enzyme, J Biol Chem 269(42) (1994) 26443–8. [PubMed] [Google Scholar]
- [52].Bosc C, Broin N, Fanjul M, Saland E, Farge T, Courdy C, Batut A, Masoud R, Larrue C, Skuli S, Espagnolle N, Pages JC, Carrier A, Bost F, Bertrand-Michel J, Tamburini J, Recher C, Bertoli S, Mansat-De Mas V, Manenti S, Sarry JE, Joffre C, Autophagy regulates fatty acid availability for oxidative phosphorylation through mitochondria-endoplasmic reticulum contact sites, Nat Commun 11(1) (2020) 4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Bai F, Morcos F, Sohn YS, Darash-Yahana M, Rezende CO, Lipper CH, Paddock ML, Song L, Luo Y, Holt SH, Tamir S, Theodorakis EA, Jennings PA, Onuchic JN, Mittler R, Nechushtai R, The Fe-S cluster-containing NEET proteins mitoNEET and NAF-1 as chemotherapeutic targets in breast cancer, Proc Natl Acad Sci U S A 112(12) (2015) 3698–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Fajardo-Orduna GR, Ledesma-Martinez E, Aguiniga-Sanchez I, Mora-Garcia ML, Weiss-Steider B, Santiago-Osorio E, Inhibitors of Chemoresistance Pathways in Combination with Ara-C to Overcome Multidrug Resistance in AML. A Mini Review, Int J Mol Sci 22(9) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Vasan K, Werner M, Chandel NS, Mitochondrial Metabolism as a Target for Cancer Therapy, Cell Metab 32(3) (2020) 341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Farhadi P, Yarani R, Dokaneheifard S, Mansouri K, The emerging role of targeting cancer metabolism for cancer therapy, Tumour Biol 42(10) (2020) 1010428320965284. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. 1. MitoNEET protein levels in primary cancer specimens from two B-cell ALL patients. Equal protein loading per lane verified with β-actin staining.
Supplemental Fig. 2. Co-localization of mitoNEET (red) with TOM20 (green), a mitochondrial marker, with yellow areas showing overlap in REH cells. DAPI was used for nuclear staining. Scale bar = 2 μm.
Supplemental Fig. 3. Cell proliferation in the REH KO and VC cells was determined by measuring the fluorescence in GFP-expressing cells. *P<0.05, where N = 3.