

Abstract
Drug delivery to the brain is limited by poor penetration of pharmaceutical agents across the blood-brain barrier (BBB), within the brain parenchyma, and into specific cells of interest. Nanotechnology can overcome these barriers, but its ability to do so is dependent on nanoparticle physicochemical properties including surface chemistry. Surface chemistry can be determined by a number of factors, including by the presence of stabilizing surfactant molecules introduced during the formulation process. Nanoparticles coated with poloxamer 188 (F68), poloxamer 407 (F127), and polysorbate 80 (P80) have demonstrated uptake in BBB endothelial cells and enhanced accumulation within the brain. However, the impact of surfactants on nanoparticle fate, and specifically on brain extracellular diffusion or intracellular targeting, must be better understood to design nanotherapeutics to efficiently overcome drug delivery barriers in the brain. Here, we evaluated the effect of the biocompatible and commonly used surfactants cholic acid (CHA), F68, F127, P80, and poly(vinyl alcohol) (PVA) on poly(lactic-co-glycolic acid)-poly(ethylene glycol) (PLGA-PEG) nanoparticle transport to and within the brain. The inclusion of these surfactant molecules decreases diffusive ability through brain tissue, reflecting the surfactant’s role in encouraging cellular interaction at short length and time scales. After in vivo administration, PLGA-PEG/P80 nanoparticles demonstrated enhanced penetration across the BBB and subsequent internalization within neurons and microglia. Surfactants incorporated into the formulation of PLGA-PEG nanoparticles therefore represent an important design parameter for controlling nanoparticle fate within the brain.
Keywords: polymeric nanoparticles, brain drug delivery, blood-brain barrier, cellular uptake, diffusion
Graphical Abstract
The brain’s uniquely restrictive biological barriers, including the blood-brain barrier (BBB) and brain parenchyma, make target cells within the brain inaccessible to nearly all therapeutic molecules. While invasive delivery techniques like intraparenchymal injection can increase therapeutic accumulation in the brain, a need remains for noninvasive delivery strategies. Nanotechnology is one promising avenue for systemic delivery of neurotherapeutics since nano-sized particles can overcome transport barriers and achieve accumulation within the brain.1, 2 Recently, drug-loaded nanoparticles have demonstrated efficacy in models of glioblastoma,3 neurodegenerative disorders,4 and neonatal hypoxic-ischemia,5 among other brain injuries.6, 7
For effective therapeutic delivery in the brain, nanoparticles must be designed to have biologically advantageous properties. One important characteristic is controlled and nontoxic degradability, which impacts drug release kinetics as well as nanoparticle clearance. The biodegradable polymer poly(lactic-co-glycolic acid) (PLGA) is commonly used for nanoparticle formulation as its degradation kinetics are both well-known and tailorable.8–10 A second important nanoparticle characteristic is the ability to avoid serum protein binding and subsequent clearance by immune cells. Surface modification with poly(ethylene glycol) (PEG) has been widely used to reduce protein adsorption and limit nonspecific cellular uptake.11, 12 As a result, a PEG surface layer can confer enhanced nanoparticle diffusivity, which is especially important in the confined brain extracellular space (ECS).13 Since nanoparticle surfaces mediate interactions between the nanoparticle and biological environment, further surface functionality can be added to PLGA-PEG nanoparticles to improve cell penetration or cell-specific targeting.
One important class of molecules present at nanoparticle surfaces are surface acting agents, referred to as surfactants. During the formulation process of PLGA-PEG nanoparticles, surfactants are commonly introduced to reduce surface tension at the interface between the organic polymer solution and the aqueous phase. Although nanoparticles can be formulated without surfactants,14 these molecules promote nanoparticle formation and stabilization. Additionally, studies have shown that surfactant coatings can alter nanoparticle-cell interactions. Tween surfactants, especially Tween 80 (or polysorbate 80, P80), can specifically enhance nanoparticle accumulation in the brain.15 Pluronic surfactants, including poloxamer 188 (Pluronic® F68, F68) and poloxamer 407 (Pluronic® F127, F127), are able to inhibit P-glycoprotein efflux transporters to remain localized intracellularly.16–18 However, nearly all prior studies have evaluated surfactant effects after incubating previously-formulated nanoparticles in fresh surfactant solutions to produce a dense surface coating. Moreover, none have investigated nanoparticle diffusion past the BBB, which has dependence on surface presentation of PEG.19 We hypothesized that nanoparticles formulated with PEG and surfactants may be optimally designed for transport to and within the brain.
In the present study, we compared PLGA-PEG formulations with surfactants P80, F68, and F127 to a control formulation without surfactant in deionized (DI) water. We also studied the surfactants poly(vinyl alcohol) (PVA), the most commonly used surfactant for nanoparticle stabilization,20 and cholic acid (CHA), an anionic surfactant.21 With each formulation, we evaluated nanoparticle transport in the brain at multiple time and length scales, i.e. molecular-level diffusion as well as cellular- and whole organ-scale nanoparticle accumulation. Our results provide insight into nanoparticle design for improved penetration of biological barriers for therapeutic delivery in the brain.
Results
Surfactants and PEG enhance nanoparticle stability and diffusive ability
To isolate the role of surfactant on biodistribution, cellular uptake, and diffusive ability, we controlled for nanoparticle size and surface charge. Based on previous work, colloidally stable nanoparticles with hydrodynamic diameters below 114 nm and near-neutral zeta potential (ζ-potential) can transport efficiently to and within the brain.13, 22 The formulations used in this study had average diameters between 55–69 nm and ζ-potentials between −6.5 and −3.0 mV (Table 1). The PEG layer and surfactant molecules decrease nanoparticle aggregation and increase stability, which is indicated by low polydispersity indices (PDI < 0.20) of formulations with both PEG and surfactant. However, nanoparticles without PEG (PLGA/F127) or surfactant (PLGA-PEG/DI) had elevated PDIs of 0.20 and 0.22, respectively. We also assessed nanoparticle stability after incubation in rat serum to mimic in vivo conditions (Figure 1A). The formulation without PEG (PLGA/F127) increased 25 nm in diameter, on average, after 8 hours of incubation whereas all PEGylated formulations changed less than 10 nm in this timeframe. Using mass spectrometry, we confirmed that surfactant molecules compose a low weight percent of the PLGA-PEG/P80 (0.01%) and PLGA-PEG/CHA (0.005%) formulations (Supplementary Figure 1). Although we expect surfactant concentration to be similarly low in the other formulations, we were unable to quantify those contributions due to limitations of mass spectrometry with polydisperse polymer species.23
Table 1.
Physicochemical properties and diffusivity of PLGA-PEG nanoparticles formulated with biocompatible surfactants. Nanoparticles were characterized in terms of hydrodynamic diameter, mean surface charge (ζ-potential), and the polydispersity index (PDI) by dynamic light scattering at 25°C and pH 7.2 in 10 mM NaCl. All values are reported as mean ± standard error of the mean (SEM) (n=3). Effective diffusion coefficients in rat brain tissue (Db) were extracted at a τ = 0.8 s from nanoparticle trajectories over fifteen videos across three brain slices for each formulation, and compared to theoretical nanoparticle diffusion in artificial cerebrospinal fluid (aCSF) based on the Stokes-Einstein equation and mean particle diameter.
| Polymer/ Surfactant | Number Mean ± SEM (nm) | PDI | ζ-potential ± SEM (mV) | Ensemble Db (x10−4 μm2/s) | DaCSF/ Db | Number of Trajectories |
|---|---|---|---|---|---|---|
| PLGA/ 1% F127 | 68.2 ± 4.0 | 0.20 | −6.28 ± 0.3 | 1.02 | 94,000 | 557 |
| PLGA-PEG/ 1% P80 | 59.6 ± 1.5 | 0.08 | −3.63 ± 0.8 | 4.72 | 23,000 | 568 |
| PLGA-PEG/ 5% F68 | 65.5 ± 4.1 | 0.09 | −4.33 ± 0.4 | 5.98 | 17,000 | 569 |
| PLGA-PEG/ 1% F127 | 59.7 ± 1.3 | 0.08 | −4.06 ± 1.4 | 7.50 | 15,000 | 839 |
| PLGA-PEG/ 5% PVA | 65.3 ± 2.4 | 0.11 | −3.24 ± 0.9 | 8.48 | 12,000 | 339 |
| PLGA-PEG/ DI H2O | 55.0 ± 2.9 | 0.22 | −4.58 ± 0.6 | 12.9 | 9,000 | 590 |
| PLGA-PEG/ 3% CHA | 56.2 ± 3.0 | 0.17 | −6.11 ± 0.6 | 21.7 | 5,000 | 308 |
Figure 1.
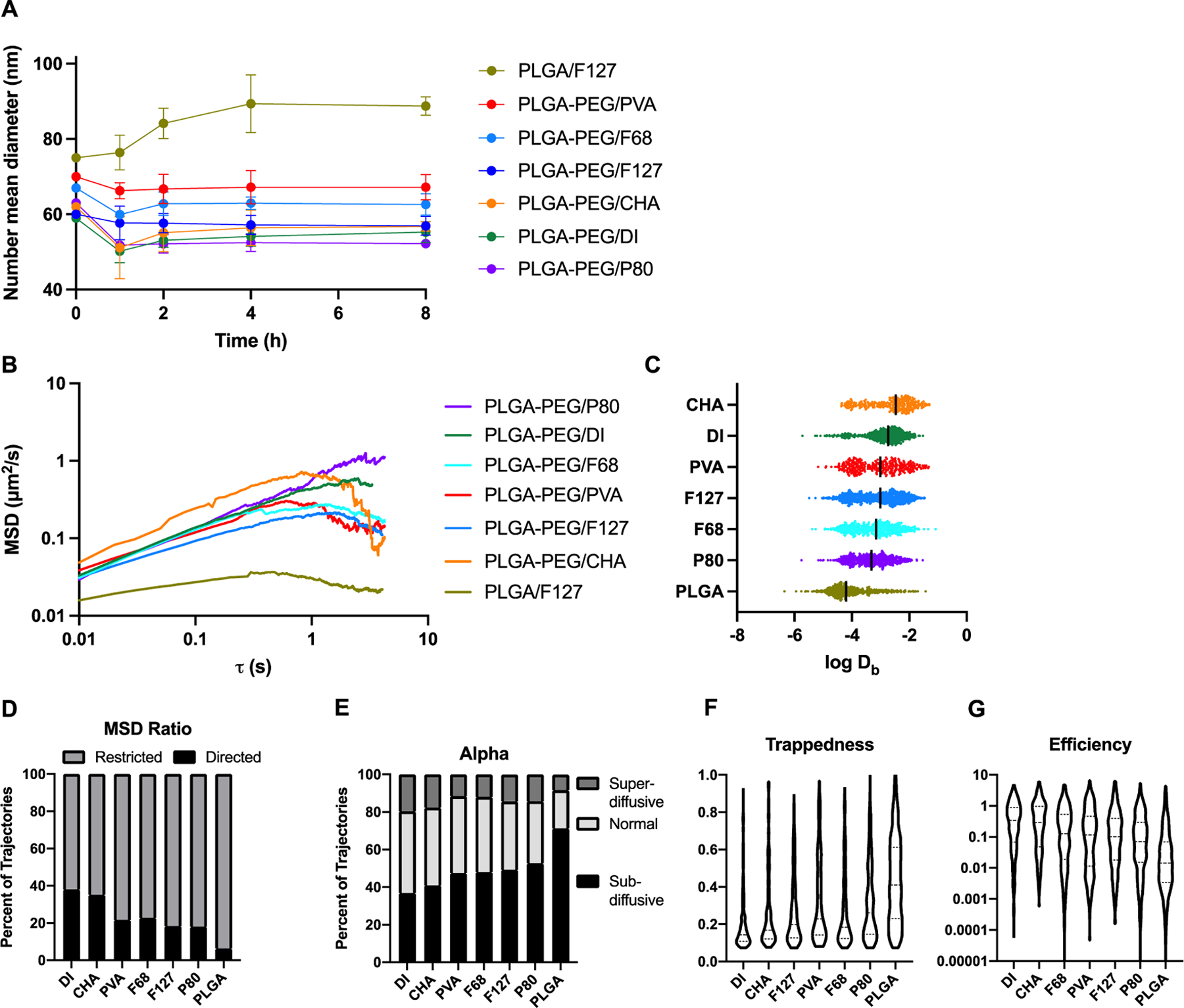
Characterization of nanoparticle stability and diffusion in brain tissue. (A) After incubation in rat serum, the PLGA/F127 formulation increased in average size, while all other formulations remained close to their original size. (B) Nanoparticle diffusion trajectories through brain tissue were analyzed to calculate ensemble-averaged mean squared displacement at time lags up to 6.5 s. (C) Log of Db at 0.8 s were extracted for each trajectory (1 dot = 1 trajectory). (D) The aspect ratio of the MSD curve for each trajectory was extracted and classified as subdiffusive (<0) or superdiffusive (>0). (E) The anomalous exponent α was extracted for each trajectory and classified as superdiffusive (>1), normal (1), or subdiffusive (<1). The (F) trappedness and (G) efficiency of each trajectory was calculated and plotted as violin plots.
To probe molecular-scale interactions in the brain microenvironment, we characterized the diffusive ability of each nanoparticle formulation in 300 μm brain slices prepared from healthy rats. At shorter length and time scales, nanoparticle transport is governed by diffusion. Each PLGA-PEG formulation demonstrates a positive slope of ensemble-averaged mean squared displacement (<MSD>) over time, while the PLGA/F127 formulation shows more stagnant growth (Figure 1B), indicating limited diffusive ability. At a time interval of 0.8 seconds, Db were extracted for each trajectory (Figure 1C). The ensemble-averaged Db for each formulation at this time scale are available in Table 1. The number of analyzed trajectories and a fold-change comparison to diffusion in artificial cerebrospinal fluid (DaCSF) are also included as summary statistics in Table 1. The Db distributions indicate significantly enhanced diffusive ability of PLGA-PEG nanoparticles with the anionic surfactant CHA (PLGA-PEG/CHA, Db=21.7x10−4 μm2/s) compared to no surfactant (PLGA-PEG/DI, Db=12.9x10−4 μm2/s, p<0.0001). However, the presence of nonionic surfactants reduced diffusive ability: compared to PLGA-PEG/DI nanoparticles, we observed 1.52-fold, 1.72-fold, 2.16-fold, and 2.73-fold slower diffusion with the PLGA-PEG/PVA, PLGA-PEG/F127, PLGA-PEG/F68, and PLGA-PEG/P80 formulations, respectively. Although the reductions were statistically significant (p<0.001 for all), the effect of surfactant incorporation was much smaller than the effect of the PEG layer. Without a PEG layer, nanoparticles appeared immobilized in the brain: PLGA-PEG/F127 nanoparticles (Db=7.50x10−4 μm2/s) exhibited 7-fold increased diffusive ability compared to PLGA/F127 nanoparticles (Db=1.02x10−4 μm2/s, p<0.0001). This result is in alignment with previous studies on the importance of a PEG layer for enhanced diffusive behavior.24
Individual trajectories were then analyzed for geometric features to distinguish between subtypes of diffusive transport. Characterization of the <MSD> curves showed that PLGA/F127 trajectories exhibited subdiffusive behavior more frequently than any other formulation, while the PLGA-PEG/DI and PLGA-PEG/CHA formulations were more likely to demonstrate superdiffusive behavior (Figure 1D). These results were closely aligned with extraction of α, the anomalous diffusion coefficient, for each nanoparticle trajectory. The plurality of PLGA/F127 trajectories were classified as subdiffusive (α<1) while most PLGA-PEG/DI and PLGA-PEG/CHA trajectories had normal (α=1) diffusive behavior (Figure 1E). Average trappedness, a geometric feature which describes the probability of the nanoparticle being trapped within a given radius, was highest for the PLGA/F127 nanoparticles and lowest for PLGA-PEG/DI nanoparticles (Figure 1F). Conversely, PLGA/F127 nanoparticles were least efficient – that is, each time step resulted in small net displacements – while PLGA-PEG/DI nanoparticles were most efficient (Figure 1G). For each geometric feature, all formulations with surfactants existed on a continuum between the immobilized PLGA/F127 nanoparticles and the diffusive PLGA-PEG/DI nanoparticles. Therefore, the results of multiple particle tracking (MPT) indicate that the inclusion of nonionic surfactants in nanoparticle formulations will increase the likelihood of cellular interaction – indicated by increasing subdiffusion, trappedness, and decreasing efficiency – at short length and time scales.
Ex vivo assessments do not reveal surfactant-mediated differences in cell uptake or toxicity
Given the observed differences in nanoparticle diffusion after ex vivo nanoparticle application, we investigated whether the organotypic brain slice platform would reveal surfactant-mediated changes in uptake by brain cells. We assessed nanoparticle uptake into neurons and microglia within 4 h of exposure by confocal imaging (Supplementary Figure 2A) and quantitation by flow cytometry of microglia (Figure 2A). The results showed uptake of PLGA-PEG/DI nanoparticles in 25.2% of microglia, which decreased to 11.0%, 13.2%, 8.6%, 10.1%, 12.1%, and 12.9% for PLGA-PEG/CHA, F68, F127, P80, PVA, and PLGA/F127, respectively. No significant differences exist between nanoparticles formulated with surfactant. We investigated cytotoxicity as a potential confounding factor in nanoparticle uptake studies, since cells exposed to a high surfactant dose would undergo cell death25 and therefore not be counted in flow cytometry analysis. Using propidium iodide staining (Figure 2B) and lactate dehydrogenase release (Supplementary Figure 2B) as two measures of cell death after four hours of nanoparticle incubation, all samples were within a normal 5–25% cytotoxicity range.
Figure 2.
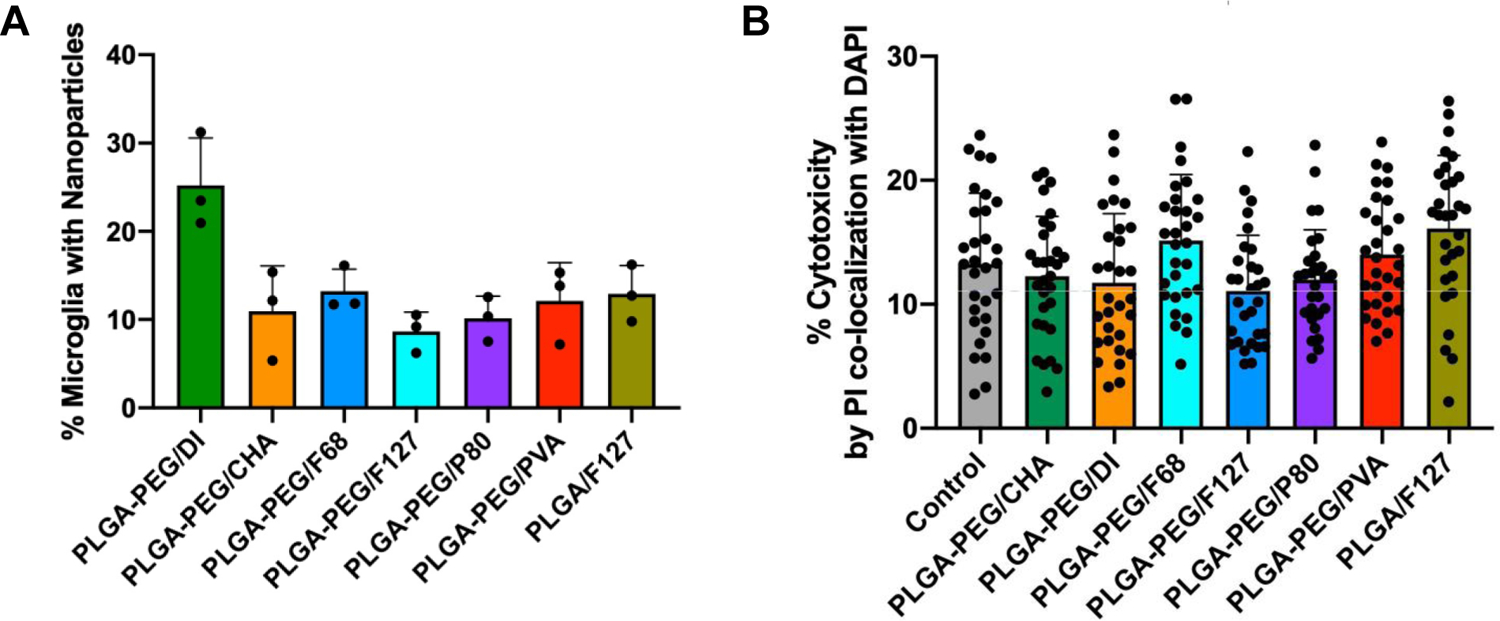
Assessment of surfactant effects on nanoparticle transport in organotypic brain slices. (A) Flow cytometry analysis indicate that all formulations with surfactant demonstrated similar levels of microglial uptake within 4 h. PLGA-PEG/DI achieved elevated levels of uptake. (B) Propidium iodide (PI)-positive cell counts, as a proportion of total cells, demonstrate no significant differences in cytotoxicity across all treatment conditions.
Nonionic surfactants enhance BBB permeation and accumulation in the brain
Nanoparticle fate in the brain is dependent on favorable circulation kinetics, biodistribution, and transport across the BBB, which must be assessed in vivo. We administered each PLGA-PEG formulation in healthy rat pups by intravenous tail vein injection. Four hours after administration, only PLGA-PEG/P80 nanoparticles were able to extravasate across the healthy BBB and uptake in neurons and microglia (Figure 3). To quantify nanoparticle penetration across the BBB, we used a capillary depletion technique on homogenized brain tissue which separated brain capillaries from the parenchyma.26 Results are presented as the percent of injected dose (%ID) per gram brain parenchyma or brain capillary (Figure 3A). Only PLGA-PEG/P80 nanoparticles achieved a significantly higher concentration in the brain parenchyma compared to brain capillaries (17.8 vs 7.4 %ID per g tissue, p=0.028). However, all nonionic surfactants improved BBB permeation compared to the no-surfactant control: without a surfactant, nanoparticle concentration in brain capillaries was 14.9-fold higher than in the parenchyma; F68, F127, P80, and PVA reduced this value to 3.2-, 0.9-, 0.4-, and 1.2-fold, respectively. Interestingly, the formulation with anionic CHA demonstrated nearly no accumulation in either the brain capillaries or parenchyma.
Figure 3.
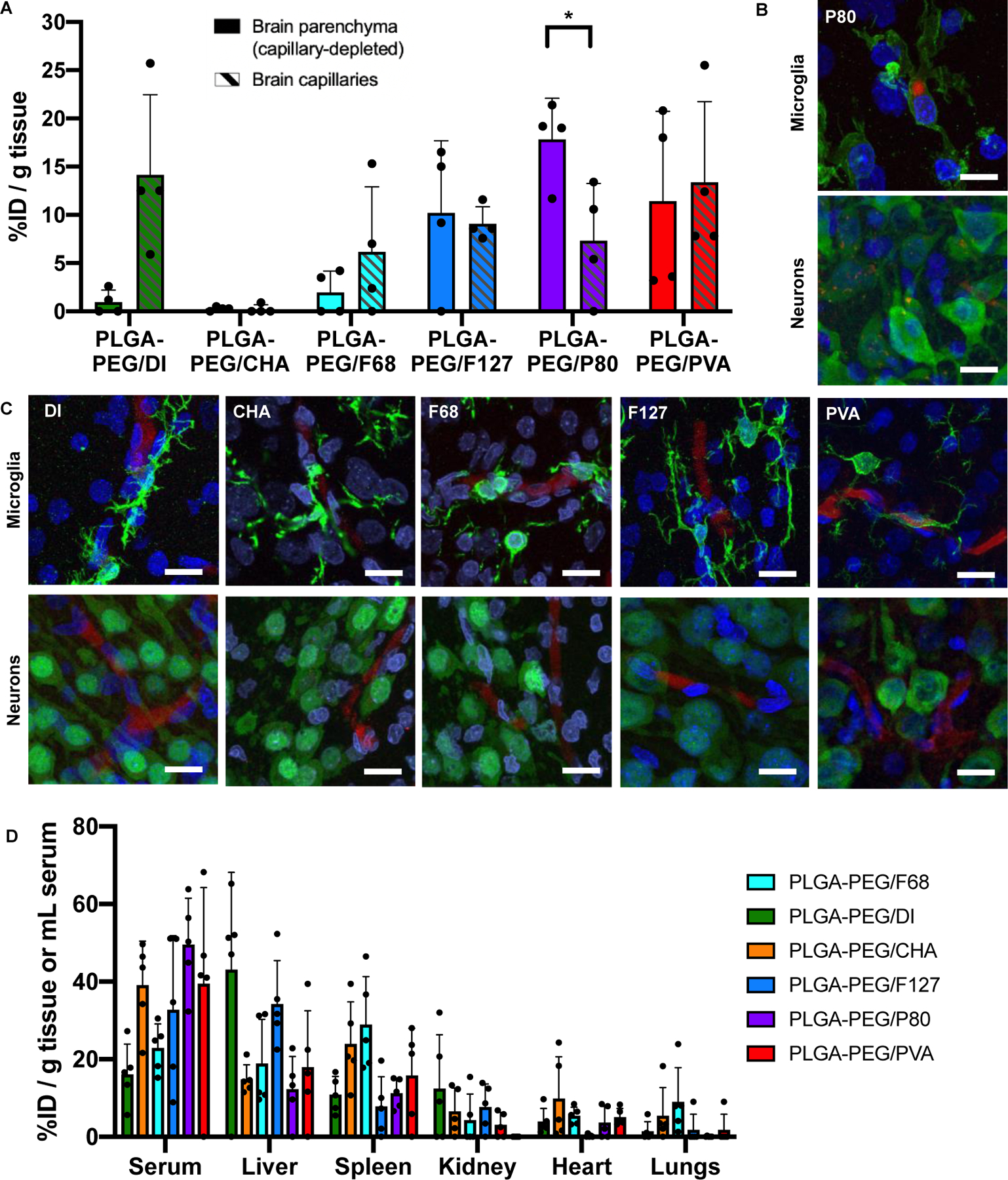
Distribution of biodegradable, PEGylated nanoparticles (red) in the brain and major organs at t=4h. (A) PLGA-PEG/P80 nanoparticles, unlike all other formulations, exhibit significantly higher accumulation (p=0.0280) in the brain parenchyma (left bars, solid fill) compared to brain capillaries (right bars, hashed). (B) PLGA-PEG/P80 nanoparticles can internalize within some microglia (green, top) and neurons (green, bottom) in the brain parenchyma. (C) PLGA-PEG/DI, PLGA-PEG/CHA, PLGA-PEG/F68, PLGA-PEG/F127, and PLGA-PEG/PVA nanoparticles do not exhibit patterns of microglial (top row) or neuronal (bottom row) uptake, and instead appear associated within the vasculature. (D) Nanoparticles demonstrate accumulation in the serum, liver, and spleen, with minimal signal from the kidney, heart, and lungs. (A, D): Each dot represents one pup for a total of n=4 (brain) or n=5 (major organs). (B-C): All cell nuclei (blue) are stained with DAPI and all scale bars represent 20 μm.
We then sought to understand the fate of nanoparticles once within the brain parenchyma. Using confocal imaging, we found evidence of PLGA-PEG/P80 uptake in microglia and neurons within the hippocampus (Figure 3B). In contrast, PLGA-PEG nanoparticles formulated without surfactant or with CHA, F68, F127, or PVA showed greater association with blood vessel structures (Figure 3C). Staining of tight junction protein zona occludens 1 (ZO-1) confirmed these regions as brain capillaries (Supplemental Figure 3A). The accumulation of nanoparticles at the healthy BBB persists at least for 24 hours: patterns of PLGA-PEG/PVA nanoparticle localization in brain capillaries appear consistent at both 4 hours and 24 hours after administration, with no visible additional neuronal or microglial uptake of nanoparticles (Supplemental Figure 3B–D). Comparison with the PLGA/F127 formulation after 24 hours indicates that the absence of a PEG layer does not enable increased BBB transcytosis or cellular uptake (Supplemental Figure 3C–D). Finally, analysis of nanoparticle biodistribution across serum and major organs supports that a large fraction of nanoparticles are still in circulation at 4 h. The liver and spleen also demonstrate nanoparticle accumulation, while more limited uptake was found in the kidney, heart, and lungs.
Polysorbate 80 is surface-associated and influences serum protein adsorption
Surfactants, including P80, may achieve favorable nanoparticle fate in the brain by facilitating plasma protein adsorption or desorption to the nanoparticle surface as the “protein corona” biolayer develops.27 However, this phenomenon is poorly studied for nanoparticles with both PEG and surfactants. We first used time-of-flight secondary ion mass spectrometry to establish that P80 is present on the nanoparticle surface. Positive and negative ion control spectra were taken from PLGA-PEG/DI nanoparticles and the P80 surfactant to determine unique peaks for each material. These peaks were then used to create a peak ratio A/(A+B) where A = sum of all P80 peaks and B = sum of all nanoparticle peaks. Figures 4A and 4B show the positive and negative ion peak ratios respectively from the ToF-SIMS data. Supplementary Table 1 shows the proposed chemical identifications of the selected peaks for each material. As seen in Figures 4A and 4B, the PLGA-PEG/P80 nanoparticles show a higher relative intensity of the P80 peaks relative to the PLGA-PEG/DI nanoparticles (p=0.001 and p<0.0001 for the positive and negative ion peaks, respectively) confirming the presence of P80. We next incubated PLGA-PEG/DI and PLGA-PEG/P80 nanoparticles in rat plasma for four hours at 37°C and subsequently quantified the amount of plasma proteins adsorbed to the nanoparticles (Figure 4C). Our results show significantly increased levels of protein adsorption in the PLGA-PEG/P80 (p=0.0382) compared to nanoparticles without surfactant. We further found evidence that elevated serum protein adsorption alters the nanoparticle surface charge: after the serum incubation, a significant negative shift in ζ-potential was observed for PLGA-PEG/P80 nanoparticles (−3.6 to −11.2 mV, p=0.0002) but not for PLGA-PEG/DI nanoparticles (Figure 4D). This shift was not associated with a change in nanoparticle stability or polydispersity (Supplemental Figure 4A) and is larger than the shift experienced by any other surfactant-formulated nanoparticle (Supplemental Figure 4B). For comparison, quantification of serum protein adsorption on the other surfactant-formulated nanoparticles is presented in Supplemental Figure 5.
Figure 4.
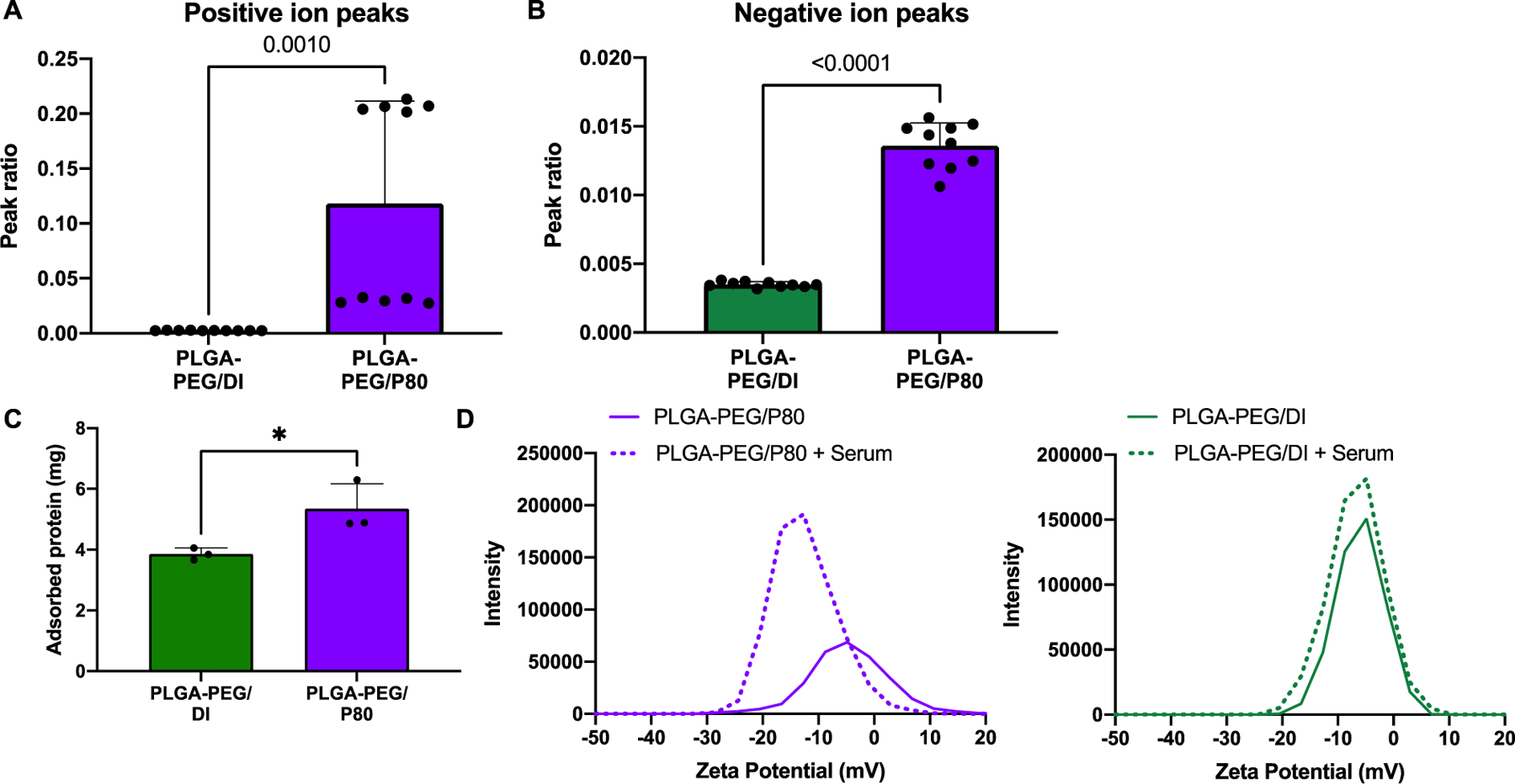
PLGA-PEG/P80 surface analysis and serum protein adsorption. Positive ion (A) and negative ion (B) peak ratio from PDG-PEG/DI and PLG-PED/P80 ToF-SIMS data. (C) Compared to the PLGA-PEG/DI control, PLGA-PEG/P80 nanoparticles exhibited increased protein adsorption (p=0.0382). (D) PLGA-PEG/P80 demonstrated a negative shift in ζ-potential after plasma incubation, which was not observed with the PLGA-PEG/DI control.
Discussion
In this study, we have evaluated the influence of surfactant on nanoparticle biodistribution, transport within the brain, and cellular fate. The results suggest that surfactant molecules increase interactions between nanoparticles and the brain microenvironment. This was demonstrated by improved BBB penetration, decreased diffusive ability through the brain ECS, and increased uptake within neurons and microglia for formulations with surfactant compared to a non-surfactant control. Importantly, surfactants were able to alter nanoparticle behavior solely after incorporation into the formulation process, without any additional incubation steps for surface coating or adsorption performed in previous studies. This suggests that complete surface coverage is not necessary to achieve surfactant-mediated interactions with components of the brain microenvironment.
One important surfactant-mediated interaction confirmed in this study was that between P80 and cells of the BBB. PLGA-PEG/P80 nanoparticles, unlike the other formulations administered in vivo, extravasated beyond brain capillary cells and localized within neurons and microglia in the healthy brain. Previous studies support the ability of a P80 surface coating to enhance nanoparticle accumulation in the brain.28–30 The mechanism of this enhanced transport has been attributed to receptor-mediated transcytosis: P80 promotes the adsorption of apolipoprotein B and E, which in turn bind to low-density lipoprotein receptors (LDL-Rs) on brain endothelial cells.16 A similar phenomenon has been described with F68-coated PLGA formulations,31 and several previous studies have concluded that both F68- and P80-coated PLGA nanoparticles enhance drug penetration in the brain.32, 33 In comparison to these studies, which used fluorescence imaging to qualitatively show BBB penetration, our study used capillary depletion to quantify fluorescence signal in capillary-rich and capillary-depleted brain fractions. This method is robust for bright fluorophores like AlexaFluor but is sensitive to in vivo processing parameters including perfusion quality, similar to other methods for determining BBB permeability.34 Our results suggest that surfactant-mediated BBB penetration can still occur with low surfactant amounts (i.e. without specific coating steps) and despite nanoparticle PEGylation.
Additionally, the subsequent cellular internalization of PLGA and PLGA-PEG nanoparticles had not previously been characterized, despite the importance of intracellular drug delivery for many neurological disease targets. Neuronal- or microglial-specific delivery of nanotherapeutics enables drugs to bypass transporters which may be involved in pathological processes,35 and instead provide direct intracellular therapeutic effects with limited extracellular consequences. Neurons and microglia utilize different transport pathways for nanoparticle internalization: neuronal uptake largely occurs through clathrin-mediated endocytosis,36 but microglia can additionally leverage phagocytosis and macropinocytosis pathways associated with macrophage cells.37 While the effect of PEGylation on decreasing phagocytic uptake is well-known, studies on the effect of surfactant have only involved non-PEGylated formulations.30, 32, 33 We attempted to evaluate cellular internalization using confocal imaging and flow cytometry in an ex vivo model, but no significant differences between surfactant formulations were observed. Using the in vivo model, however, we observed PLGA-PEG/P80 localization within neurons and microglia. This result aligns with one previous study demonstrating neuronal uptake of P80-coated human serum albumin nanoparticles38 and motivates further investigation of the mechanism driving nanoparticle fate beyond BBB penetration.
The formation of a protein corona at the nanoparticle surface is one important driver of biological behavior. We demonstrate that, even on well-PEGylated nanoparticles, surfactant choice influences the extent of plasma protein adsorption. Based on previous studies evaluating the protein corona of biodegradable, polymeric nanoparticles, the major adsorbed proteins are likely albumin, fibrinogen, immunoglobulin G (IgG) and its light chains, and the apolipoproteins ApoA-I and ApoE.39 However, the exact composition of the protein corona likely differs between our different surfactant formulations and is known to change upon transport across the BBB.27 Our results suggest that differences in the in vivo fate of biodegradable nanoparticles reflect differences in the protein corona, given that formulations with enhanced brain uptake (PLGA-PEG/F127, P80, and PVA) presented higher levels of adsorbed protein compared to those that had low brain uptake (PLGA-PEG/CHA and F68). The in-depth characterization of surfactant effects on nanoparticle protein coronas, especially considering dynamic changes as the nanoparticle transports through various brain compartments, is an important area for future work.
Even at very short scales of length and time, surfactants still impart influence on nanoparticle transport in the brain. From multiple particle tracking analysis, we showed that the presence of nonionic surfactant molecules hinders PLGA-PEG nanoparticle diffusion compared to control nanoparticles without surfactant. The heterogeneous surface produced by the incorporation of nonionic surfactant molecules within a PEG layer may increase viscous drag in the brain extracellular space, slowing diffusive behavior. In contrast, the inclusion of an anionic surfactant like CHA increased diffusive behavior. We speculate that the small size of CHA likely minimizes interference with surface PEG chains, allowing more efficient PEG coverage and greater inert behavior of the nanoparticle. Additionally, individual CHA molecules may contribute to enhanced diffusion due to electrostatic repulsion effects with negatively charged extracellular matrix (ECM) proteins.40 These results are further evidence that surfactant molecules increase interactions between the nanoparticle and its biological environment, which aligns with a previous study on the hindrance effect of PVA on nanoparticle diffusion through cervico-vaginal mucus.20 In comparison, nanoparticles without a PEG layer were completely immobilized in brain tissue. Not only was the average diffusive ability of a PLGA nanoparticle more than 7-fold slower than its PEGylated counterpart, but extracted trajectory features also described nanoparticles which were more subdiffusive, constrained, trapped, and less efficient than any other formulation. Only one previous study has reported these trajectory features for nanoparticles diffusing within a biological environment; gold nanoparticles exhibited both super- and sub-diffusion within fibroblasts in vitro.41 As quantifying and classifying nanoparticle diffusion becomes more widespread in the drug delivery literature, it will be important to create further distinctions between specific nanoparticle transport modes, such as superdiffusion by an ATP-driven transport process versus intracellular flow gradients. Our results suggest that the incorporation of surfactants into PLGA-PEG formulations results in nanoparticles that balance diffusive ability and cellular interaction.
One limitation of this study was the inability to characterize the amount distribution, or orientation of surfactant at the nanoparticle surface. While mass spectrometry determined that surfactant contributions to the PLGA-PEG/CHA and PLGA-PEG/P80 were small (less than 0.01% by weight), it could not be used for polydisperse polymeric surfactants and did not provide any information on the spatial distribution of these molecules. ToF-SIMS determined that P80 surfactant molecules were present at the nanoparticle surface but could not quantify the extent of surface coverage or distribution throughout the nanoparticle. Determination of these additional parameters would provide additional insight to the minimum effective dose of surfactant required to achieve biological effects. However, these measurements have not previously been done for biodegradable polymeric nanoparticles and are challenging due to chemical similarities between surfactant molecules and the PLGA-PEG polymer itself. Development of this technique would enable the formulation of nanoparticles with varying degrees of surface presentation of surfactant, which is likely a key determinant of nanoparticle behavior in the brain. Additionally, the exact role of surfactant length and hydrophobicity may be elucidated by focusing investigations to certain classes of surfactants, like the Pluronics® or Tweens. Such effects have been preliminarily identified for example on in vitro macrophage uptake,42 but characterization in the unique brain microenvironment is currently lacking. The continued study of the biological roles of surfactant molecules elucidates the nuance of the role of surfactants in nano-neurotherapeutic formulation and can inform the design of nanoparticles for effective transport into and within the brain.
Conclusion
We have shown that the incorporation of surfactant molecules in PLGA-PEG formulations enhances nanoparticle interaction within the brain microenvironment. Nonionic surfactants mediate enhanced BBB penetration after intravenous administration: PLGA-PEG/F68, PLGA-PEG/F127, PLGA-PEG/PVA, and PLGA-PEG/P80 nanoparticles exhibit 2– 11-, 12-, and 19-fold greater uptake in the brain parenchyma, respectively, compared to PLGA-PEG nanoparticles without surfactant. We observed hindered diffusive transport of nanoparticles containing surfactants, demonstrating the role of surfactants in increasing short-term interactions with cells. Charged surfactants, such as the anionic CHA, enable faster overall nanoparticle diffusion likely due to electrostatic interactions with brain ECM components. Overall, the presence of a surfactant influences the ability of nanoparticles to overcome biological barriers in the brain. Additional studies on key nanoparticle design parameters, including surfactant spatial distribution and surface coverage for effective delivery to the brain, will enable future development, implementation, and clinical translation for polymer nanoparticles in treating neurological disorders.
Materials and Methods
Materials
PLGA45k (50:50)-mPEG5k (PLGA-PEG) or PLGA45k (50:50) (PLGA) polymers were purchased from Akina PolySciTech. AlexaFluor 555 (AF555) and 647 (AF647) NHS Ester for polymer labeling were purchased from ThermoFisher. Cholic acid (bile salts), Pluronic® F68, Pluronic® F127, P80, and PVA (27 kDa MW) were purchased from Sigma. Solvents for nanoparticle formulation, including acetone and 1x phosphate buffered saline (PBS), were used as received. Capillary depletion buffer (CDB) was made with 10 mM (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) (HEPES), 141 mM sodium chloride (NaCl), 4 mM potassium chloride, 2.8 mM calcium chloride, 1 mM magnesium sulfate, 1 mM monosodium phosphate, and 10 mM glucose (Sigma). CDB with dextran (CDB-D) was made by dissolving 2.6 g dextran (67,300 Da MW, Sigma) in 7.4 mL CDB. Slice culture media (SCM) was made with 50% minimum essential media, 45% Hank’s Balanced Salt Solution (HBSS) with calcium and magnesium, 5% horse serum, and 1% glutamine and penicillin-streptomycin each (Gibco). FACS media was formulated with 90% 1xHBSS without calcium or magnesium, 10% fetal bovine serum, and 1% 1M HEPES. Percoll solution for FACS was purchased from Sigma.
Nanoparticle formulation
Nanoparticles were prepared by nanoprecipitation. PLGA-PEG or PLGA was dissolved in acetone at a concentration of 20 mg/mL. The polymer solution (organic phase) was then added dropwise into 25 mL of one of the following aqueous solutions: 1% P80, 5% PVA, 1% F127, 5% F68, 3% CHA, or no surfactant in deionized water. Nanoparticles formed spontaneously and were stirred for 3 h at 700 rpm to remove the organic solvent. Nanoparticles were collected and washed twice by ultracentrifugation with deionized water at 100,000xg for 25 min. Finally, the nanoparticles were resuspended in 1 mL deionized water or in sterile PBS for animal experiments. Nanoparticles were used immediately. For fluorescently-labeled nanoparticles, the same nanoparticle formulation procedure was used with PLGA and PLGA-PEG and conjugation of AF555 or AF647 was achieved by attachment to the free COOH on the PLGA backbone, as described previously.13
Nanoparticle characterization by dynamic light scattering and mass spectrometry
The particle size and PDI of PLGA-PEG nanoparticles formulated in CHA, (PLGA-PEG/CHA), F127 (PLGA-PEG/F127), F68 (PLGA-PEG/F68), P80 (PLGA-PEG/P80), PVA (PLGA-PEG/PVA), or no surfactant (PLGA-PEG/DI) and PLGA nanoparticles formulated in F127 (PLGA/F127) were measured by dynamic light scattering. The ζ-potential was determined using a zeta potential analyzer (NanoSizer Zeta Series, Malvern Instruments, Malvern, UK). Samples were diluted to appropriate concentrations to obtain accurate measurements in 10 mM NaCl at room temperature, pH 7.4, as described previously.21 For the serum stability assay, nanoparticles were incubated in rat serum at 37°C and aliquots were removed at 1, 2, 4, 8, and 24h for DLS characterization.
To quantify the amount of surfactant present in each formulation, standard solutions of each surfactant were injected into a triple quadrupole LC-MS/MS system (AB Sciex 5600 QTOF) equipped with a Waters BEH column (50 mm, 2.1x150 mm). Surfactant was eluted using two mobile phases, HPLC grade water and acetonitrile at 0.3 mL/min and identified on the chromatogram by molecular weight. For surfactants P80 and CHA, where a sample peak was identified, a calibration curve from standard solutions was created and used to determine the amount of surfactant present in PLGA-PEG/P80 and PLGA-PEG/CHA, respectively (Supplementary Figure 1A). For surfactants F68, F127, and PVA, no sample peak could be identified (Supplementary Figure 1B).
Animal experiments and ethics statement
This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All of the animals were handled according to approved Institutional Animal Care and Use Committee (IACUC) protocols (#4383–01 and #4383–02) of the University of Washington, Seattle, WA, USA. The University of Washington has an approved Animal Welfare Assurance (#A3464–01) on file with the NIH Office of Laboratory Animal Welfare (OLAW), is registered with the United States Department of Agriculture (USDA, certificate #91-R-0001), and is accredited by AAALAC International. Every effort was made to minimize suffering. Time-mated pregnant female Sprague–Dawley rats (virus antibody-free CD® (SD) IGS, Charles River Laboratories, Raleigh, NC, USA) were purchased and arrived on estrous (E) day 17. Dams were housed individually and allowed to acclimate to their environment for a minimum of 3 days prior to delivering. The day of birth was defined as postnatal (P) day 0. Litters containing both sexes were cross-fostered and culled to 12 animals early after birth. Before and after the experiment, each dam and her pups were housed under standard conditions with an automatic 12 h light/dark cycle, a temperature range of 20–26°C, and access to standard chow and autoclaved tap water ad libitum. The pups were checked for health daily.
Cell uptake and death in organotypic brain slices
In cultured brain slices, cytotoxicity of surfactant-formulated nanoparticles was determined by lactate dehydrogenase (LDH) assay and propidium iodide (PI) staining. Brain slices were collected from P9 pups and left to rest overnight in the incubator (maintained at 37°C, with constant humidity and 95% air and 5% CO2).43 Each nanoparticle formulation was diluted to 1 mg/mL in SCM and 100 μL of the diluted nanoparticles was added on top of each slice. The media was collected 4 h after treatment start time for LDH analysis, and fresh media containing 5 μg/ml PI was added for 1 hour. The slices were then fixed, stained with DAPI, and imaged using a Nikon A1R with a 40x objective. For every slice, five images were acquired from each brain region of interest (cortex and thalamus). Image acquisition settings were consistent for all images. For each image, DAPI+ cells (total cells) and PI+ cells (dead cells) were counted manually in ImageJ (NIH) after applying an Otsu threshold and fluorescent cutoff to aid in visualization. The PI+/DAPI+ cell ratio was expressed as the percentage of dead cells in an individual image.
For LDH cytotoxicity analysis, media samples were thawed to room temperature and LDH assays (Cayman Chemical) were conducted according to the manufacturer’s protocol. 100 μL of the sample was added to 100 μL of LDH reaction buffer in triplicate to 96-well plates on ice and the plates were gently shaken in a 37°C incubator. After 30 min, the plates were returned to the ice and then measured by UV-Vis (SpectraMax M5, Molecular Devices) for absorbance at 490 nm. Percent cytotoxicity was calculated as the sample absorbance normalized to the 4 h absorbance of the Triton-X condition x100%.
Flow cytometry
Flow cytometry was used to quantify the proportion of microglia with nanoparticle uptake. Fresh brain slices (3 slices per sample and n=3 samples per condition) were prepared as described previously and immediately incubated with a nanoparticle condition: 100 μL of PLGA-PEG/DI, PLGA-PEG/CHA, PLGA-PEG/F68, PLGA-PEG/F127, PLGA-PEG/P80, PLGA-PEG/PVA, or PLGA/F127, or no nanoparticles. After 4 h, slices from each experimental group were placed in 1 mL Accutase. Samples were gently shaken on ice for 30 min and then carefully pipetted to ensure tissue was fully homogenized. The sample was then transferred to a new tube through a top filter (Pierce Tissue Strainers) until all the homogenate was filtered. During this process, HBSS and 25 mM HEPES were added to dilute homogenate to a final volume of 10 mL. The tube was spun down at 600xg at 4°C for 5 min to pellet cells, then aspirated and the supernatant discarded. 100% FBS was then added to resuspend the cell pellet. Percoll Solution (final concentration 33%, GE Healthcare) was added to the cell suspension and mixed well, and then FACS media was added to the suspension. The cell suspension was centrifuged for 15 min at 800xg and 4°C, and then the supernatant was aspirated, leaving the cell pellet at the bottom. To wash excess Percoll Solution, the pellet was resuspended in FACS media and centrifuged for 10 min at 600xg and 4°C, and again the supernatant was removed. The final pellet was resuspended in FACS media for staining. Fc block (BD Biosciences) was added to the FACS media cell suspension and incubated for 5 min on ice, and then cells were stained with DAPI (1:10,000) and FITC CD11b (1:200). Appropriate controls for CD11b gating were done with an aliquot of the control sample. The cells were stained with the above stains for 15 min and washed 3 times with FACS media for 4 min at 1000xg and 4°C. The BD LSRII (BD Biosciences) machine recorded cells in each sample with fluorescence in the DAPI, CD11b, and AF555 channels until 100,000 events (live cells) were reached. Analysis of the cytometry data was performed in FCS Express 7 Research and representative data is shown in Supplemental Figure 6.
Biodistribution, capillary depletion, and nanoparticle quantification
AF647-labeled PLGA and PLGA-PEG nanoparticles were administered via tail vein (150 mg/kg) in P9 pups (n=5). We chose P9 because P7–10 rats are commonly used in models of neonatal brain injury, where nanotherapeutic development is critically needed. Although the misconception persists that the BBB is immature or incompletely formed in neonates,44 several studies in a number of species have demonstrated that tight junctions between brain endothelial cells are functionally effective as soon as the first blood vessels penetrate the parenchyma in the developing brain,45, 46 including in rats.47 Four hours after nanoparticle administration, the pup was sacrificed and the animal was perfused with 20 mL 1xPBS. Capillary depletion was conducted on freshly extracted brains (n=4) according to the protocol described by Banks et al..26 Briefly, brains were homogenized in 0.8 mL CDB and then mixed with 1.6 mL CDB-D on ice. The homogenate was centrifuged at 5400xg for 15 min at 4°C. The middle, clear layer was separated as the capillary-depleted brain fraction and the bottom, red pellet was resuspended in 0.3 mL PBS as the capillary-rich brain fraction. All other organs were homogenized in PBS at a 1 g/mL concentration and centrifuged at 10,000xg for 10 min at 4°C to remove cellular debris.
Nanoparticle concentration in the capillary-rich and capillary-depleted brain fractions and major organs were determined by measurement of sample fluorescence intensities (excitation 625 nm/ emission 665 nm) using UV-Vis spectroscopy. Separate calibration curves were created for each tissue fraction and nanoparticle formulation combination (Supplementary Figure 7). The same nanoparticle batch was used for injection and calibration curves. The analysis was conducted by first subtracting blank fluorescence values for tissue from a control animal. All calculated nanoparticle concentrations were normalized by injected dose (ID) and then weight to find % ID per mg tissue.
Immunohistochemistry for microglial and neuronal co-localization
Nanoparticle co-localization after in vivo administration was evaluated for each formulation group by placing freshly extracted brains in a formalin-to-30% sucrose gradient and then sectioning on a Leica cryostat into 30 μm sections. For microglia, a primary antibody solution (1:250 rabbit anti-Iba1, Wako) was prepared in 1xPBS containing 1% Triton-X (Sigma) and 3% normal goat serum (Sigma) and was added to tissue sections for 4 h in a humidified chamber at room temperature. Sections were washed twice in 1xPBS. A secondary antibody solution was prepared in 1xPBS and 1% Triton-X and added to tissue sections for 2 h. For neurons, a pre-conjugated antibody solution (1:500 anti-NeuN AlexaFluor 488, Abcam) was prepared in 1xPBS containing 1% Triton-X (Sigma) and added to tissue sections for 6 h in a humidified chamber at room temperature. Sections were washed twice in 1xPBS and then stained with 1:10,000 DAPI for 10 min (Invitrogen). Slides were washed and dried for 30 min in the dark. Mounting medium (Dako, Agilent Technologies, Santa Clara, CA) was added to each slide and a glass coverslip placed on top. Slides were stored at 4°C until imaged on an A1 confocal microscope (Nikon Instruments) and at 20°C for long-term storage.
Cellular co-localization was also evaluated in ex vivo brain tissue. Brain slices were collected from P9 pups and 100 μL of surfactant-formulated PLGA or PLGA-PEG nanoparticles (1 mg/mL) was immediately added on top of each slice. After 4 h, slices were washed with 1xPBS, fixed with 10% formalin for 1 h, and then washed again. For microglial staining, a primary antibody solution (1:250 rabbit anti-Iba1, Wako) was prepared in 1xPBS containing 3% Triton-X (Sigma) and 6% normal goat serum (Sigma). For neuronal staining, the antibody solution (1:500 anti-NeuN Alexa Fluor 488, Abcam) was prepared in 1xPBS containing 3% Triton-X. Antibody solutions were added to the slices for 6 h at room temperature and then washed twice. For microglia, a secondary antibody solution (1:500 goat anti-rabbit Alexa Fluor 488, Abcam) was prepared in 1xPBS containing 3% Triton-X and subsequently was added to the slices at room temperature for 2 h. After washing twice, all slices were stained with 1:10,000 DAPI (Invitrogen) for 15 min and stored in 1xPBS at 4°C until imaged on an A1 confocal microscope.
Multiple Particle Tracking (MPT) in organotypic brain slices
Fresh brain slices from P9 pups were prepared as described previously24, 48 and used for MPT analysis to evaluate diffusive ability of surfactant-formulated PLGA-PEG nanoparticles in the living brain. Slices were transferred to 35 mm glass bottom imaging disks and 2 μL of AF555-labeled nanoparticles were injected directly into brain tissue. Visualization of the nanoparticles was accomplished with the excitation/emission spectra specific to AF555. Five 6.5 s videos were collected per slice at 10 Hz and 40x magnification via fluorescent microscopy with a cMOS camera (Hamamatsu Photonics Corporation, Bridgewater, NJ). Trajectories from each video were segmented and recorded with respect to x-position (x), y-position (y), and time step (t) using the TrackMate ImageJ plugin.
Selected trajectory features, described below, were extracted.41 First, geometrically-averaged precision-weighted MSDs were calculated for each trajectory and timestep using the equation:
where r2 indicates the MSD determined at each step, n, for a total number of steps, N, with 3D position coordinates x(x,y,t). Then, diffusion coefficients, D, and the anomalous diffusion coefficient (Deff), alpha, were determined by fitting MSD curves to the function:
Alpha values of 1 indicate normal diffusive behavior, while values below 1 indicate subdiffusion and values above 1 indicate superdiffusion. The MSD ratio, which characterizes the shape of the MSD curve, is defined by:
where n1 and n2 represent the first and last frames of the trajectory, respectively. Ratios below 0 indicate restricted diffusion (subdiffusion) while ratios above 0 indicate superdiffusion. Trajectory efficiency (E), a measure of the nanoparticle’s net displacement compared to the sum of its step lengths, was calculated by the equation:
The fractal path dimension (Df), which can distinguish between confined and random walk trajectories, was calculated from:
where d is the largest distance between any two positions and L is the sum of all step lengths. Fractal dimension values of 2 indicate random walk trajectories and values above 2 indicate confined diffusion. Finally, trappedness (pt), the probability that a particle with diffusion coefficient D and traced for a period of time N∆t is trapped into a region r0, is given by:
All calculations were done in Python using a package available on GitHub.49
Time-of-Flight secondary ion mass spectrometry
ToF-SIMS spectra were acquired on a IONTOF ToF-SIMS 5 spectrometer using a 25 keV Bi3+ cluster ion source in the pulsed mode. Spectra were acquired for both positive and negative secondary ions over a mass range of m/z = 0 to 800. The ion source was operated at a current of 0.2 pA. Secondary ions of a given polarity were extracted and detected using a reflectron time-of-flight mass analyzer. Spectra were acquired using an analysis area of 100 micron x 100 micron. Positive ion spectra were calibrated using the CH3+, C2H3+, and C3H5+ peaks. The negative ion spectra were calibrated using the CH−, OH−, C2H−, and C4H− peaks. Calibration errors were kept below 25 ppm. Mass resolution (m/Δm) for a typical spectrum was between 5000 to 5600 for m/z = 27 (pos) and between 4000 to 6500 for m/z = 25 (neg). PLA-PEG/DI nanoparticle, surfactant and PLA-PEG/surfactant samples were drop cast on cleaned silicon wafers. 5 positive and 5 negative ion spectra were collected from random positions on each sample. Sample preparation and data acquisition was repeated on two separate dates for a total of 10 positive and 10 negative ion spectra per sample type. The positive and negative ion data were analyzed separately to generate a peak list across all spots on all samples. The peak area tables were imported into the NBToolbox spectragui (https://www.nb.uw.edu/mvsa/nbtoolbox) and used to create a peak ratio A/(A+B) where A = sum of all P80 peaks and B = sum of all nanoparticle peaks.
Plasma protein adsorption study
Plasma was collected from P9 rat pups by collecting blood into a heparin-coated tube and then centrifuging out cells at 2000xg for 10 min. 100 μL of each PLGA and PLGA-PEG nanoparticles were mixed well with 900 μL plasma and then left in a 37°C incubator. 4 h later, nanoparticles were pelleted by centrifugation at 100,000xg for 25 min to remove non-adsorbed protein. A small volume of the resuspended nanoparticles was used for dynamic light scattering characterization, as described previously. The samples were also tested for protein concentration with the Pierce BCA Protein Assay Kit (ThermoFisher). Following the manufacturer’s instructions, 25 μL of each sample was added to a 96-well plate in triplicate on ice. After addition of 200 μL BCA assay working reagent (50:1 reagent A:B), the plate was gently shaken at 37 °C for 30 min. After 30 min, absorbance of each well was measured at 562 nm on a SpectraMax M5 UV–Vis Spectrophotometer (Molecular Devices).
Statistical analysis
Statistical analysis of in vivo biodistribution and FACS microglial uptake results were made using an unpaired t-test, assuming normality. Statistical analysis of MPT data distributions was conducted using a two-tailed Mann-Whitney U-test. All analysis was performed using GraphPad version 7 (Prism, San Diego, California). A P‐value <0.05 was considered statistically significant.
Supplementary Material
Acknowledgements
This work was supported by the National Institutes of Health Predoctoral Individual National Research Service Award #F31HD095572–03, National Institute of General Medical Sciences Grant #R35GM124677, and National Science Foundation Grant #1703438. The authors would also like to thank the UW Department of Chemical Engineering Bindra Lab for access to the Malvern Zetasizer, and the UW Department of Medicinal Chemistry Mass Spectrometry Center and Research Scientist Scott Edgar for data collection and analysis. Part of this work was conducted at the Molecular Analysis Facility, a National Nanotechnology Coordinated Infrastructure (NNCI) site at the University of Washington, which is supported in part by funds from the National Science Foundation (awards NNCI-2025489, NNCI-1542101), the Molecular Engineering & Sciences Institute, and the Clean Energy Institute. D. G and L. G. gratefully acknowledge support from NIH grant EB-002027 during preparation of this manuscript as well as for some the data presented in it.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Disclosure
The authors declare no competing financial interest.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data availability:
Data is available upon request.
References
- 1.Wong HL; Wu XY; Bendayan R, Nanotechnological advances for the delivery of CNS therapeutics. Adv Drug Deliv Rev 2012, 64 (7), 686–700. [DOI] [PubMed] [Google Scholar]
- 2.Tosi G; Costantino L; Ruozi B; Forni F; Vandelli MA, Polymeric nanoparticles for the drug delivery to the central nervous system. Expert Opin Drug Deliv 2008, 5 (2), 155–74. [DOI] [PubMed] [Google Scholar]
- 3.Wohlfart S; Khalansky AS; Gelperina S; Maksimenko O; Bernreuther C; Glatzel M; Kreuter J, Efficient chemotherapy of rat glioblastoma using doxorubicin-loaded PLGA nanoparticles with different stabilizers. PLoS One 2011, 6 (5), e19121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu Z; Gao X; Kang T; Jiang M; Miao D; Gu G; Hu Q; Song Q; Yao L; Tu Y; Chen H; Jiang X; Chen J, B6 peptide-modified PEG-PLA nanoparticles for enhanced brain delivery of neuroprotective peptide. Bioconjug Chem 2013, 24 (6), 997–1007. [DOI] [PubMed] [Google Scholar]
- 5.Joseph A; Wood T; Chen CC; Corry K; Snyder JM; Juul SE; Parikh P; Nance E, Curcumin-loaded polymeric nanoparticles for neuro-protection in neonatal rats with hypoxic-ischemic encephalopathy. Nano Res 2018, 11 (10), 5670–5688. [Google Scholar]
- 6.Godinho BMDC; Ogier JR; Darcy R; Driscoll CM; Cryan JF, Self-assembling Modified beta-Cyclodextrin Nanoparticles as Neuronal siRNA Delivery Vectors: Focus on Huntington’s Disease. Mol Pharmaceut 2013, 10 (2), 640–649. [DOI] [PubMed] [Google Scholar]
- 7.Khalin I; Alyautdin R; Wong TW; Gnanou J; Kocherga G; Kreuter J, Brain-derived neurotrophic factor delivered to the brain using poly (lactide-co-glycolide) nanoparticles improves neurological and cognitive outcome in mice with traumatic brain injury. Drug Deliv 2016, 23 (9), 3520–3528. [DOI] [PubMed] [Google Scholar]
- 8.Park TG, Degradation of Poly(D,L-Lactic Acid) Microspheres - Effect of Molecular-Weight. J Control Release 1994, 30 (2), 161–173. [Google Scholar]
- 9.Park TG, Degradation of Poly(Lactic-Co-Glycolic Acid) Microspheres - Effect of Copolymer Composition. Biomaterials 1995, 16 (15), 1123–1130. [DOI] [PubMed] [Google Scholar]
- 10.Anderson JM; Shive MS, Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv Drug Deliver Rev 2012, 64, 72–82. [DOI] [PubMed] [Google Scholar]
- 11.Owens DE 3rd; Peppas NA, Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm 2006, 307 (1), 93–102. [DOI] [PubMed] [Google Scholar]
- 12.Suk JS; Xu Q; Kim N; Hanes J; Ensign LM, PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv Drug Deliv Rev 2016, 99 (Pt A), 28–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nance EA; Woodworth GF; Sailor KA; Shih TY; Xu Q; Swaminathan G; Xiang D; Eberhart C; Hanes J, A dense poly(ethylene glycol) coating improves penetration of large polymeric nanoparticles within brain tissue. Sci Transl Med 2012, 4 (149), 149ra119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lepeltier E; Bourgaux C; Couvreur P, Nanoprecipitation and the “Ouzo effect”: Application to drug delivery devices. Adv Drug Deliv Rev 2014, 71, 86–97. [DOI] [PubMed] [Google Scholar]
- 15.Kreuter J; Shamenkov D; Petrov V; Ramge P; Cychutek K; Koch-Brandt C; Alyautdin R, Apolipoprotein-mediated transport of nanoparticle-bound drugs across the blood-brain barrier. J Drug Target 2002, 10 (4), 317–325. [DOI] [PubMed] [Google Scholar]
- 16.Kreuter J, Influence of the surface properties on nanoparticle-mediated transport of drugs to the brain. J Nanosci Nanotechno 2004, 4 (5), 484–488. [DOI] [PubMed] [Google Scholar]
- 17.Lo YI, Relationships between the hydrophilic-lipophilic balance values of pharmaceutical excipients and their multidrug resistance modulating effect in Caco-2 cells and rat intestines. J Control Release 2003, 90 (1), 37–48. [DOI] [PubMed] [Google Scholar]
- 18.Jain A; Jain A; Garg NK; Tyagi RK; Singh B; Katare OP; Webster TJ; Soni V, Surface engineered polymeric nanocarriers mediate the delivery of transferrin-methotrexate conjugates for an improved understanding of brain cancer. Acta Biomater 2015, 24, 140–51. [DOI] [PubMed] [Google Scholar]
- 19.Xu QG; Ensign LM; Boylan NJ; Schon A; Gong XQ; Yang JC; Lamb NW; Cai ST; Yu T; Freire E; Hanes J, Impact of Surface Polyethylene Glycol (PEG) Density on Biodegradable Nanoparticle Transport in Mucus ex Vivo and Distribution in Vivo. Acs Nano 2015, 9 (9), 9217–9227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang M; Lai SK; Yu T; Wang YY; Happe C; Zhong W; Zhang M; Anonuevo A; Fridley C; Hung A; Fu J; Hanes J, Nanoparticle penetration of human cervicovaginal mucus: the effect of polyvinyl alcohol. J Control Release 2014, 192, 202–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu Q; Boylan NJ; Cai S; Miao B; Patel H; Hanes J, Scalable method to produce biodegradable nanoparticles that rapidly penetrate human mucus. J Control Release 2013, 170 (2), 279–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Curtis C; Toghani D; Wong B; Nance E, Colloidal stability as a determinant of nanoparticle behavior in the brain. Colloids Surf B Biointerfaces 2018, 170, 673–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Altuntas E; Schubert US, “Polymeromics”: Mass spectrometry based strategies in polymer science toward complete sequencing approaches: a review. Anal Chim Acta 2014, 808, 56–69. [DOI] [PubMed] [Google Scholar]
- 24.Nance E; Timbie K; Miller GW; Song J; Louttit C; Klibanov AL; Shih TY; Swaminathan G; Tamargo RJ; Woodworth GF; Hanes J; Price RJ, Non-invasive delivery of stealth, brain-penetrating nanoparticles across the blood-brain barrier using MRI-guided focused ultrasound. J Control Release 2014, 189, 123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hering I; Eilebrecht E; Parnham MJ; Gunday-Tureli N; Tureli AE; Weiler M; Schafers C; Fenske M; Wacker MG, Evaluation of potential environmental toxicity of polymeric nanomaterials and surfactants. Environ Toxicol Pharmacol 2020, 76, 103353. [DOI] [PubMed] [Google Scholar]
- 26.Banks WA; Freed EO; Wolf KM; Robinson SM; Franko M; Kumar VB, Transport of human immunodeficiency virus type 1 pseudoviruses across the blood-brain barrier: role of envelope proteins and adsorptive endocytosis. J Virol 2001, 75 (10), 4681–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cox A; Andreozzi P; Dal Magro R; Fiordaliso F; Corbelli A; Talamini L; Chinello C; Raimondo F; Magni F; Tringali M; Krol S; Jacob Silva P; Stellacci F; Masserini M; Re F, Evolution of Nanoparticle Protein Corona across the Blood-Brain Barrier. Acs Nano 2018, 12 (7), 7292–7300. [DOI] [PubMed] [Google Scholar]
- 28.Chen YC; Hsieh WY; Lee WF; Zeng DT, Effects of surface modification of PLGA-PEG-PLGA nanoparticles on loperamide delivery efficiency across the blood-brain barrier. J Biomater Appl 2013, 27 (7), 909–22. [DOI] [PubMed] [Google Scholar]
- 29.Mittal G; Carswell H; Brett R; Currie S; Kumar MN, Development and evaluation of polymer nanoparticles for oral delivery of estradiol to rat brain in a model of Alzheimer’s pathology. J Control Release 2011, 150 (2), 220–8. [DOI] [PubMed] [Google Scholar]
- 30.Chaturvedi M; Molino Y; Sreedhar B; Khrestchatisky M; Kaczmarek L, Tissue inhibitor of matrix metalloproteinases-1 loaded poly(lactic-co-glycolic acid) nanoparticles for delivery across the blood-brain barrier. Int J Nanomedicine 2014, 9, 575–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gelperina S; Maksimenko O; Khalansky A; Vanchugova L; Shipulo E; Abbasova K; Berdiev R; Wohlfart S; Chepurnova N; Kreuter J, Drug delivery to the brain using surfactant-coated poly(lactide-co-glycolide) nanoparticles: influence of the formulation parameters. Eur J Pharm Biopharm 2010, 74 (2), 157–63. [DOI] [PubMed] [Google Scholar]
- 32.Kulkarni SA; Feng SS, Effects of surface modification on delivery efficiency of biodegradable nanoparticles across the blood-brain barrier. Nanomedicine (Lond) 2011, 6 (2), 377–94. [DOI] [PubMed] [Google Scholar]
- 33.Tahara K; Miyazaki Y; Kawashima Y; Kreuter J; Yamamoto H, Brain targeting with surface-modified poly(D,L-lactic-co-glycolic acid) nanoparticles delivered via carotid artery administration. Eur J Pharm Biopharm 2011, 77 (1), 84–8. [DOI] [PubMed] [Google Scholar]
- 34.Helmbrecht H; Joseph A; McKenna M; Zhang M; Nance E, Governing Transport Principles for Nanotherapeutic Application in the Brain. Curr Opin Chem Eng 2020, 30 (12), 112–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nance E; Kambhampati SP; Smith ES; Zhang Z; Zhang F; Singh S; Johnston MV; Kannan RM; Blue ME; Kannan S, Dendrimer-mediated delivery of N-acetyl cysteine to microglia in a mouse model of Rett syndrome. J Neuroinflammation 2017, 14 (1), 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Canton I; Battaglia G, Endocytosis at the nanoscale. Chem Soc Rev 2012, 41 (7), 2718–39. [DOI] [PubMed] [Google Scholar]
- 37.Hutter E; Boridy S; Labrecque S; Lalancette-Hebert M; Kriz J; Winnik FM; Maysinger D, Microglial response to gold nanoparticles. Acs Nano 2010, 4 (5), 2595–606. [DOI] [PubMed] [Google Scholar]
- 38.Zensi A; Begley D; Pontikis C; Legros C; Mihoreanu L; Wagner S; Buchel C; von Briesen H; Kreuter J, Albumin nanoparticles targeted with Apo E enter the CNS by transcytosis and are delivered to neurones. J Control Release 2009, 137 (1), 78–86. [DOI] [PubMed] [Google Scholar]
- 39.Gref R; Luck M; Quellec P; Marchand M; Dellacherie E; Harnisch S; Blunk T; Muller RH, ‘Stealth’ corona-core nanoparticles surface modified by polyethylene glycol (PEG): influences of the corona (PEG chain length and surface density) and of the core composition on phagocytic uptake and plasma protein adsorption. Colloids Surf B Biointerfaces 2000, 18 (3–4), 301–313. [DOI] [PubMed] [Google Scholar]
- 40.Hrabetova S; Masri D; Tao L; Xiao F; Nicholson C, Calcium diffusion enhanced after cleavage of negatively charged components of brain extracellular matrix by chondroitinase ABC. J Physiol 2009, 587 (Pt 16), 4029–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wagner T; Kroll A; Haramagatti CR; Lipinski HG; Wiemann M, Classification and Segmentation of Nanoparticle Diffusion Trajectories in Cellular Micro Environments. PLoS One 2017, 12 (1), e0170165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Illum L; Jacobsen LO; Muller RH; Mak E; Davis SS, Surface Characteristics and the Interaction of Colloidal Particles with Mouse Peritoneal-Macrophages. Biomaterials 1987, 8 (2), 113–117. [DOI] [PubMed] [Google Scholar]
- 43.Liao R; Wood TR; Nance E, Superoxide dismutase reduces monosodium glutamate-induced injury in an organotypic whole hemisphere brain slice model of excitotoxicity. J Biol Eng 2020, 14, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saunders NR; Liddelow SA; Dziegielewska KM, Barrier mechanisms in the developing brain. Front Pharmacol 2012, 3, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grontoft O, Intracranial haemorrhage and blood-brain barrier problems in the new-born; a pathologico-anatomical and experimental investigation. Acta Pathol Microbiol Scand Suppl 1954, 100, 8–109. [PubMed] [Google Scholar]
- 46.Ek CJ; Dziegielewska KM; Stolp H; Saunders NR, Functional effectiveness of the blood-brain barrier to small water-soluble molecules in developing and adult opossum (Monodelphis domestica). J Comp Neurol 2006, 496 (1), 13–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Millen JW; Hess A, The blood-brain barrier: an experimental study with vital dyes. Brain 1958, 81 (2), 248–57. [DOI] [PubMed] [Google Scholar]
- 48.Nance E; Porambo M; Zhang F; Mishra MK; Buelow M; Getzenberg R; Johnston M; Kannan RM; Fatemi A; Kannan S, Systemic dendrimer-drug treatment of ischemia-induced neonatal white matter injury. J Control Release 2015, 214, 112–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Curtis C; Rokem A; Nance E, diff_classifier: Parallelization of multi-particle tracking video analyses. J Open Source Softw 2019, 4 (36). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data is available upon request.