

Introduction
Medication-related osteonecrosis of the jaw (MRONJ) is a potentially debilitating condition seen in patients who have been treated with powerful antiresorptives (pARs) or angiogenesis inhibitors (AgIs). MRONJ is defined as exposed bone or bone that can be probed through an intra- or extraoral fistula in the maxillofacial region for more than eight weeks in patients who have been treated with pARs or AgIs and have no history of radiation therapy or metastatic disease in the jaws[1, 2].
pARs including nitrogen-containing bisphosphonates (N-BPs; e.g., zoledronic acid [ZOL], alendronate [ALN], etc.) and anti-RANKL antibodies (e.g., denosumab) are used to manage bone metastases in patients with cancer[3–6] or to prevent fragility fractures in patients with osteoporosis[7]. MRONJ associated with pARs is common in patients with cancer (1.8-5% incidence), but rare in patients with osteoporosis (0.01-0.03%)[1, 2, 8, 9]. Patients with MRONJ experience reduced oral health-related quality of life[10]. Clinical and preclinical data suggest that for MRONJ to occur, systemic risk factors (e.g., pARs or AgIs) and oral risk factors, such as tooth extraction and inflammatory dental disease (e.g., periodontitis, periapical infection) must co-occur[1, 2, 11–22]. MRONJ management can be challenging and the outcomes difficult to predict, often with problematic resolution[23–25].
Bone necrosis, the hallmark of MRONJ, is recognized histologically as an area of bone tissue with numerous contiguous empty osteocyte lacunae[26–28]. However, osteocytes die before this histologic pattern appears. Whereas the causes and mechanisms of osteocyte death have been studied in conditions like osteonecrosis of the femoral head (ONFH)[29], few studies of the causes and mechanisms of osteocyte death have been done in MRONJ. Improving the understanding of osteocyte death in MRONJ may be critical for preventing disease and developing treatment approaches. This review intends to provide insight into the biology of osteocytes, cell death in general, and osteocyte death in particular and discuss possible mechanisms for MRONJ-related osteocyte death.
Osteocyte biology
Osteocytes are the most numerous bone cell type in the adult skeleton. Osteocytes comprise 90-95% of the bone cells, while osteoclasts and osteoblasts make up the remaining 5-10%[30]. They terminally differentiate from osteoblasts, which themselves differentiate from mesenchymal precursors residing in the bone marrow and at bone surfaces[31]. As new bone is formed, osteoblasts synthesize osteoid at existing bone surfaces and undergo cellular transformations that involve changes in shape, size, and development of dendrite-like processes that extend into the mineralizing front of the osteoid to communicate with existing mature osteocytes[31–34]. As osteoblasts differentiate into osteocytes, they become encased in their surrounding recently-mineralized bone matrix[34, 35]. Their cell bodies reside within lacunae, and their dendritic processes, ranging from 40–100 per cell[35], run through 30-300nm diameter tunnels named canaliculi. The canaliculi traverse the mineralized bone matrix allowing intercellular communication among osteocytes. The physical structure of interconnected tunnels and lacunae is known as the osteocyte lacunar-canalicular network (LCN)[31, 35] (Figure 1). The connection among osteocyte cell processes within the LCN and osteoblasts at bone surfaces is attained via gap channel junctions[36–38]. Gap channel junctions are formed by connexins (Cx)[36–38], with Cx43 being the most abundant[39, 40]. Osteocytes and osteoblasts also express functional Cx43 hemichannels[41]. Hemichannels mediate communication, not only between adjacent cells but also with the extracellular matrix as it deforms. It has been proposed that gap junctions and hemichannels contribute to maintaining bone integrity and function by permitting the exchange of bone modulators and regulating signals elicited by mechanical stimulation through influencing bone modeling and/or remodeling[42–45]. Cx43 hemichannels may also play essential roles as transducers for the anti-apoptotic signals of bisphosphonates[46, 47].
Figure 1. Cartoon depicting the lacunar-canalicular network (LCN) and the functional syncytium.
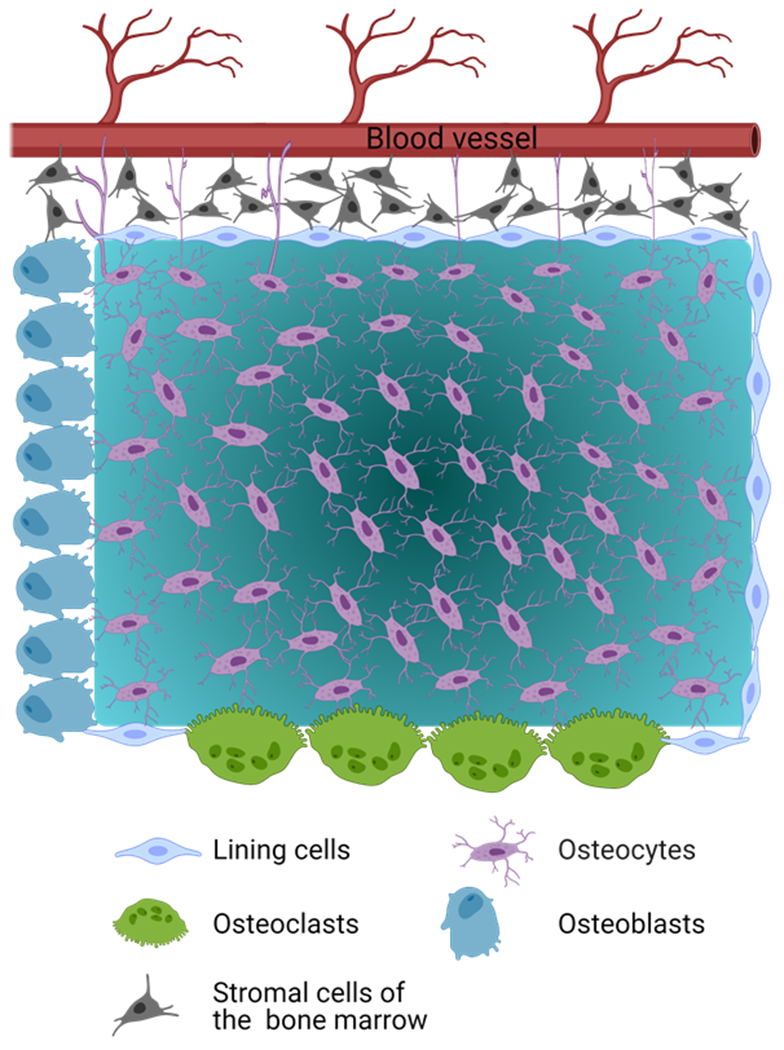
The LCN is the physical structure of interconnected tunnels and lacunae, where osteocytes reside. Osteocytes in the LCN form a functional syncytium with cells on the bone surfaces, including osteoblasts and lining cells, which in turn are in physical contact with stromal cells and hematopoietic stem cells in the marrow and endothelial cells of blood vessels. In addition, Osteocyte dendritic processes can extend beyond the cells in bone surfaces to directly interact with cells in the bone marrow and blood vessels[35], as depicted in the figure.
The osteocytes in the LCN form a functional syncytium with cells on the bone surfaces, including osteoblasts and lining cells, which in turn are in direct physical contact with endothelial cells of blood vessels, stromal cells, hematopoietic stem cells in the bone marrow, and nerves[48] (Figure 1). Notably, osteocyte dendritic processes can extend beyond the cells in bone surfaces to interact directly with blood vessels and cells in the bone marrow[35]. It has been proposed that this organization allows osteocytes to play a bidirectional role (receiver-transmitter system). Thus, osteocytes not only act as “receivers” of systemic signals (e.g., hormones, drugs) directly from blood vessels or local signals from the mineralized matrix as it is deformed, but also as “transmitters” of signals to the executor cells of bone modeling and/or remodeling (osteoclasts, osteoblasts, and lining cells). Hence, osteocytes are thought to operate as effective orchestrators of modeling and remodeling, integrating hormonal, pharmaceutical, and mechanical cues to regulate osteoblast and osteoclast function. It is proposed that the osteocyte’s centralized role in regulating responses to mechanical stimuli allows the skeleton to meet its mechanical and calcium and phosphorus homeostatic needs[35, 49, 50].
The control of bone remodeling requires the precise regulation of both osteoclast and osteoblast cell activity. Indeed, both osteocytes and osteoblasts control osteoclastogenesis and bone resorption by regulating RANK/RANKL/OPG signaling. Osteocytes and osteoblasts synthesize RANKL[51, 52]. Cells of the osteoclast lineage express RANK on their surfaces[53]. RANKL binding to its receptor RANK promotes differentiation of osteoclast precursors and the activation of mature osteoclasts that together increase bone resorption activity[54].
Interestingly, osteocytes and osteoblasts are also a source of osteoprotegerin (OPG), a soluble decoy receptor that binds to RANKL and prevents its binding to RANK, a process which in turn inhibits osteoclast-mediated bone resorption[55–57]. Thus, osteocytes regulate bone resorption, utilizing a unique molecular system based on the differential synthesis of OPG and RANKL[58].
Osteocytes also control osteoblast activity and bone formation by regulating the bone anabolic actions of the canonical Wnt/β-catenin signaling pathway[59]. This pathway is activated when Wnt proteins bind to receptor complexes that comprise frizzled proteins (receptors) and co-receptors low-density lipoprotein (LDL) receptor-related proteins (LRP) LRP 5 and/or 6[55, 60–62]. Though LRP5 and LRP6 are the more studied, other LDL receptor family members, including LRP4[61–63] and LRP8[64, 65], also function as co-receptors for Wnt ligands in the regulation of bone homeostasis. Mature osteocytes synthesize several modulators of Wnt signaling, including sclerostin and Dickkopf-related protein 1 (DKK-1)[63, 64]. Sclerostin and DKK-1 are potent, specific inhibitors of the Wnt/β-catenin pathway that bind to LRP4, LRP5, and LRP6[65] and prevent the binding of Wnt, playing a critical role in regulating bone formation[66]. At present, it is unclear whether LRP8 is inhibited by sclerostin or DKK1. By reducing the synthesis of these Wnt pathway inhibitors, osteocytes induce the upregulation and translocation of β-catenin to the nucleus and activate gene transcription signaling in osteoblasts to increase bone formation[66–68]. In contrast, increased expression of sclerostin and DKK1 by osteocytes, as occurs in skeletal unloading, suppresses bone formation[69, 70]. Osteocytes also mediate the effects of parathyroid hormone (PTH) on bone formation. These effects are in part attributable to the suppressive effect of PTH on sclerostin synthesis by osteocytes via transcriptional downregulation of the SOST gene that encodes sclerostin[71–73]. PTH also increases RANKL synthesis in osteocytes, indicating that PTH indirectly regulates bone resorption[74]. Furthermore, osteocytes are a source of diverse molecules that modulate bone remodeling. These molecules include mediators such as prostanoids, nitric oxide, nucleotides, and a broad spectrum of cytokines and growth factors such as insulin-like growth factor-1 (IGF-1), vascular endothelial cell growth factor (VEGF), and TGF-β[75–82]. Moreover, osteocytes are a significant source of fibroblast growth factor 23 (FGF-23), which decreases serum phosphorus levels by increasing renal phosphate excretion[83]. In addition, osteocytes are responsible for regulating the mineralization process as they become embedded in osteoid gradually mineralizing[84]. It has been proposed that the osteoid osteocyte is the cell primarily responsible for mineralization instead of osteoblasts on the bone surfaces. The SIBLING proteins DMP1 and MEPE and the protein PHEX are highly expressed in osteocytes[85–90] and are identified as essential molecules for bone mineralization[30, 91, 92]. DMP1 is expressed during the initial stages of mineralized matrix formation in bone and dentin[93]. It is expressed along and in the canaliculi of osteocytes in the bone matrix at gap regions between collagen type 1 fibrils[94, 95]. As a highly phosphorylated protein, DMP1 may be involved in the regulation of hydroxyapatite formation. The effects of DMP1 in the regulation of mineralization and osteocyte maturation appear to be predominantly due to its role in phosphate homeostasis regulation because the mineralization defects and the impairment in osteocyte maturation can be rescued by restoration of the normal phosphate homeostasis[96]. MEPE interacts with DMP1 and PHEX to affect FGF23 expression, regulating phosphate mineralization and bone turnover[97]. PHEX was initially described on the plasma membrane of osteoblasts and osteocytes[88], and loss-of-function mutations in this gene resulted in X Linked hypophosphatemic rickets[98].
Osteocytes of the craniofacial skeleton
Since MRONJ affects the jaws, it is pertinent to consider the possibility that the biology and regulation of osteocytes in the jaws might differ from that in non-jaw skeletal sites. The craniofacial skeleton differs in several ways from the better-studied postcranial skeleton, particularly concerning the embryologic origin, molecular regulation during skeletogenesis, and structural organization. The craniofacial skeleton has two embryonic origins[99]. The majority of the craniofacial bones, namely all facial bones and most cranial bones, including the maxilla and mandible, are derived from the cranial neural crest (NC). In contrast, the parietal and occipital bones of the calvarium are derived from the paraxial mesoderm[100–102]. Cranial NC cells originate from the anterior-dorsal aspect of the developing neural tube, contributing to most of the cartilage and bone of the cranial region[103, 104]. Most rostral cranial NC cells arise from the diencephalic and mesencephalic neural tube and form the skull’s frontonasal skeleton and membranous bones. The posterior cranial NC cells, coming from the posterior mesencephalon and hindbrain, occupy the pharyngeal arches and form the mandible, maxilla, middle ear bones, and hyoid bone.
Maxillae and mandibles develop from tissues of the first pharyngeal arch[104]. The mandible develops from the mandibular process and the maxilla within the maxillary process that expands from it. Though mandibular and maxillary primordia originate from similar NC cells, they develop into very different structural entities[104]. Once the positional identities of bone progenitor cells are defined by a unique combination of homeodomain transcription factors, the NC-derived mesenchymal stem cells differentiate into osteoblast lineage cells through the upregulation of BMPs and osteoinductive factors similar to those in the postcranial skeleton[103, 104].
Most craniofacial bones, such as the calvaria, some facial bones, and the mandible (except its condylar process), are formed through intramembranous ossification[105, 106]. On the other hand, the cranial base, the supporting platform for the development of the brain, is formed by endochondral ossification in the same manner as the appendicular skeleton and vertebrae[107].
It has been assumed that the biology of NC-derived osteoblasts is similar to that of mesoderm-derived osteoblasts. However, some studies have shown differences among osteoblasts from the two embryologic origins. Osteocytes derived from calvaria are commonly used for in vitro differentiation assays. As described above, these cells originate from NC or mesenchymal stem cells. Thus, their progenitors possess different bone-forming abilities[108]. Calvarial osteoblasts from the frontal bone, derived from NC stem cells, have better intrinsic osteogenic and tissue regeneration capacity than mesoderm-derived calvarial osteoblasts from the parietal bone[108, 109]. Furthermore, NC–derived frontal bone cells display a superior capacity to undergo osseous healing compared to the mesoderm-derived parietal bone cells due to greater activation of the canonical Wnt signaling pathway[109]. These studies suggest NC-derived osteoblasts and mesoderm-derived osteoblasts have different biologic features. However, it is unknown whether NC-derived osteocytes from the maxilla and mandible resemble NC-derived osteocytes from the frontal bones or mesoderm-derived bones[110]. Furthermore, it is unknown whether the regulatory mechanisms, biologic responses to cell survival signals, and cell death responses of NC-derived osteocytes are different from those in mesoderm-derived osteocytes.
Cell Death
Cell death is a terminal biologic event in which the affected cell ceases to carry out its functions. Dying cells are involved in a process that is reversible until the first irreversible event or “point-of-no-return” occurs[111]. In the absence of an accepted view of biochemical events considered as the point-of-no-return, the Nomenclature Committee on Cell Death (NCCD) recommended that a cell be considered dead when any of the following morphological criteria are met: a) permanent loss of the plasma membrane barrier; b) breakdown of a cell into discrete fragments, which are commonly referred to as apoptotic bodies; or c) engulfment of the cell by dedicated phagocytes or other cells with phagocytic activity[112]. Since this initial NCCD recommendation, additional cell death modalities have been described[111, 113, 114]. Subsequent reports from the NCCD have recommended limiting the definition of “dead” exclusively to cells that either exhibit irreversible plasma membrane permeabilization or have undergone complete fragmentation[115].
Although cell death can occur due to overwhelming damage, most cell death occurs actively through specific signaling pathways. Cell death has been operationally classified into two broad categories: “accidental” or “regulated”[115]. Accidental cell death (ACD) is caused by severe insults, including physical, chemical, and mechanical stimuli[115]. When exposed to extreme physicochemical or mechanical insults, cells die uncontrollably, losing their structural integrity and releasing damage-associated molecular patterns (DAMPs), endogenous molecules with immunomodulatory and sometimes, cytotoxic activity[116, 117]. Regulated cell death (RCD), in contrast, involves a genetically encoded molecular machinery[113, 118]. RCD occurs not only as a consequence of microenvironmental perturbations but also during post-embryonic development, immune responses, and inflammation[119]. While ACD is challenging to control, RCD can be modulated by inhibiting the transduction of death signals and enhancing the capacity of cells to mount adaptive responses to stress[115]. Indeed, the course of RCD can be modified by pharmacologic and/or genetic interventions that target its key components.
Cell death can also be formally classified into three different forms based on morphologic features and mechanisms by which dead cells and their fragments are removed: a) type I cell death (apoptosis), b) type II cell death (autophagy), and c) type III cell death (necrosis)[111, 120]. Although these morphological classifications have limitations and caveats, they are extensively employed.
Type I cell death (apoptosis):
the term “apoptosis” was initially coined by Kerr et al.[121] to describe a RCD form with specific morphologic features that include cell shrinkage, retraction of cellular pseudopodia, reduction of cellular volume (pyknosis), chromatin condensation (karyopyknotic), nuclear fragmentation (karyorrhexis), little or no ultrastructural modifications of cytoplasmic organelles, and plasma membrane blebbing while the cell maintains its integrity until the final stages of the process[111]. Cells that die through apoptosis end by forming intact small vesicles, known as apoptotic bodies, which are phagocytosed by neighboring cells and degraded within lysosomes. There are several subtypes of apoptosis that are triggered through different biochemical routes, for instance, through “intrinsic” or “extrinsic” pathways[122, 123]. The subtypes of apoptosis are thoroughly reviewed elsewhere[124].
Type II cell death (autophagy):
the term “autophagy” derived from the Greek meaning “eating of self” was first coined by Duve[125]. It is an essential cellular mechanism for balancing energy sources at critical times during development or in response to nutrient stress or starvation[126]. Autophagy also plays a housekeeping role in removing misfolded or aggregated proteins, clearing damaged organelles, such as mitochondria, endoplasmic reticulum, peroxisomes, and eliminating intracellular pathogens[126]. Though autophagy is generally associated with survival mechanisms, its deregulation has also been linked to a non-apoptotic RCD mechanism. Morphologically, cell death by autophagy occurs in the absence of chromatin condensation and involves massive autophagic vacuolization of the cytoplasm[126, 127]. In contrast to apoptotic cells, whose clearance is ensured by phagocytosis and lysosomal degradation by neighboring phagocytic cells, cells that die by autophagy have little or no association with neighboring cells[128, 129]. Autophagy relies on the formation of autophagosomes and activation of the autophagic machinery. The fusion of autophagosomes with lysosomes generates autolysosomes, in which acidic lysosomal hydrolases degrade their luminal content.
Type III cell death (necrosis):
The term “necrosis,” from the ancient Greek νέκρωσις, nékrōsis, “death,” has been used for centuries to define drastic tissue changes visible to the naked eye[130]. Pathologists use this term to describe the presence of dead tissues, representing the sum of changes that occur in cells and tissues after they have died, regardless of the pre-lethal processes[131, 132]. Though the term has been typically used with this meaning, several investigators believe that necrosis is not a form of cell death but only refers to features that become apparent after cell death, which can occur by any cell death form, including apoptosis and autophagy[130, 132, 133]. However, with no consensus, the NCCD recommends that the term necrosis be used to mean a type of cell death that involves rupture of the plasma membrane without the hallmarks of apoptosis or autophagy[111–113, 115]. Different organelles and cellular processes are implicated in necrotic cell death than in apoptosis and autophagy. These primarily include a gain in cell volume (oncosis); swelling of the endoplasmic reticulum, mitochondria, and Golgi apparatus; increases in the cytosolic concentration of calcium (which results in mitochondrial overload and activation of non-caspase proteases); freed lysosomal hydrolases; degradation of nucleic acids, proteins, and lipids (e.g., calpains and cathepsins); and ultimately rupture of the plasma membrane and release of cellular content into the extracellular space causing inflammation[111, 134, 135]. Since oncosis precedes cell death and is accompanied by cellular swelling, organelle swelling, blebbing, and increased membrane permeability, it was proposed as the term to define the pre-lethal portion of the necrosis pathway[130, 136].
Necroptosis and the interrelationship with apoptosis and inflammation
For many years, necrotic cell death was considered only as a form of ACD. However, accumulating evidence showed that necrotic cell death could result from finely regulated sets of signal transduction pathways and catabolic mechanisms[135, 137–142]. The form of RCD displaying a necrotic cell death phenotype was named “necroptosis”[143]. A variety of triggers initiates necroptosis, including tumor necrosis factor-alpha (TNFα), other cytokines of the TNF superfamily, such as TNF-Related Apoptosis-Inducing Ligand (TRAIL/TNFSF10) and Fas Ligand (FasL/TNFSF6), interferons (IFNs), pathogen-associated molecular patterns (PAMPs), lipopolysaccharides (LPS), dsRNA, DNA damage, viral infections, anti-cancer drugs, etc. (Figure 2). These ligands bind to specific receptors, including FAS, TRAILR 1/2, and Toll-like receptors 2 and 4 (TLR2/4), respectively[144]. Most of the knowledge gained about necroptosis is based on TNFα-TNFR1 signaling[140].
Figure 2. TNFα/TNFR1 signaling activates necroptosis and triggers apoptosis and inflammation.
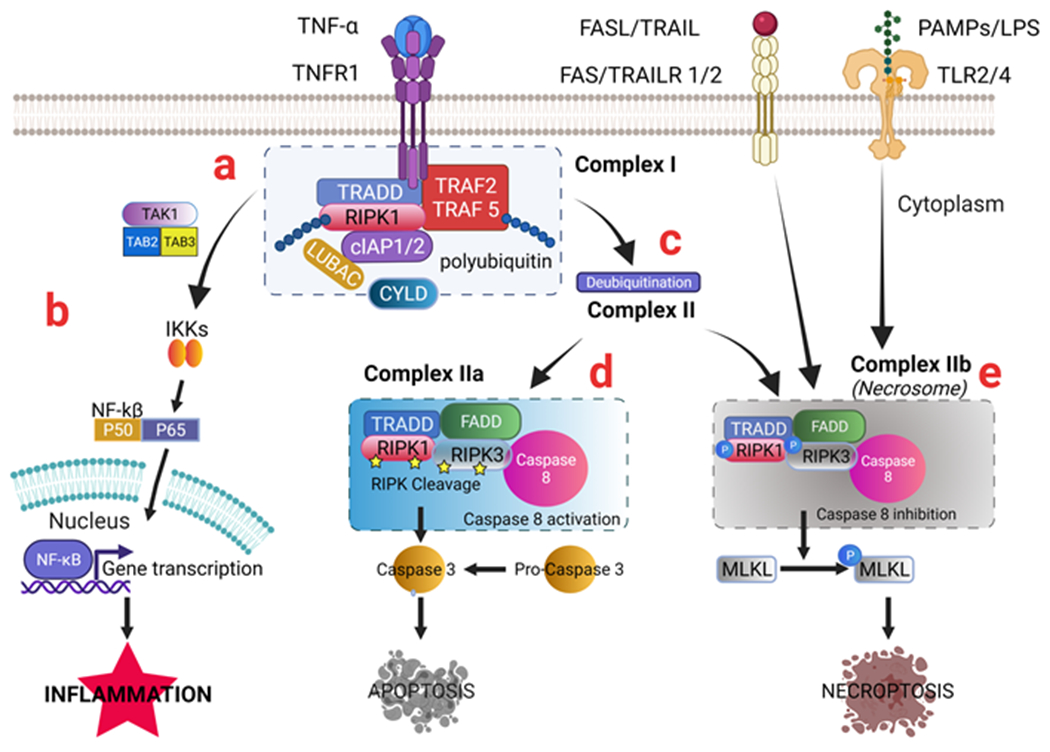
TNFα/TNFR1 molecular signaling is explained in more detail. Other signals such as FASL/FAS, TRAIL/TRAIL, PAMPS, or LPS/TLR2/4 can also trigger necroptosis. TNFα is released during inflammatory conditions. (a) TNFα binds to TNFR1, inducing recruitment of TRADD, RIPK1, TRAF 2, TRAF5, cIAP 1/2, and other molecules to form Complex I. (b) Upon polyubiquitinated RIPK1 in Complex I, the TNFα/TNFR1 signaling can activate IKKs, which triggers the NF-kB signaling pathway cascade that leads to gene expression of pro-inflammatory cytokines and inflammation. (c) deubiquitination and activation of RIPK1 by CYLD lead to the formation of Complex II. (d) TRADD and RIPK1 become modified and dissociate from TNFR1. The liberated death domain(DD) of TRADD (and/or RIPK1) binds to FADD, resulting in RIPK cleavage, caspase-8 recruitment (forming Complex IIa), activation of Caspase 8, which results in Caspase 3 activation and apoptosis. e) Inactivation of Caspase-8 in Complex II leads to the phosphorylation and activation of RIPK1, RIPK3, and subsequent phosphorylation and activation of MLKL during the necrosome assembly (Complex IIb), oligomerization of MLKL monomer leads to induction of necroptosis.
Abbreviations: TNF: tumor necrosis factor; TNFR1: TNF receptor 1; FAS: CD95/APO-1; FASL: FAS ligand; TRAIL: CD253 or TNFSF10; TRAILR1/2: TRAIL receptor 1/2; PAMPs: Pathogen-associated molecular patterns; LPS: lipopolysaccharide. TLR3/4, Toll-like receptors 3/4; TAK1: Transforming growth factor-β-activated kinase 1; TAB2: TGF-Beta Activated Kinase 1 (MAP3K7) binding protein 2; TAB3: TGF-Beta Activated Kinase 1 (MAP3K7) binding protein 3; IKKs: IKKα and IKKβ complex; NF-kB: nuclear factor kappa-light-chain-enhancer of activated B cells; P50:NF-κB1; P65: RelA; TRADD: TNFRSF1A-associated via death domain; RIPK: Receptor interacting serine/threonine kinase; TRAF: TNF receptor-associated factors; cIAP: Cellular inhibitor of apoptosis protein; CYLD: Deubiquitinase cylindromatosis; FADD: FAS-associated death domain; MLKL: mediator mixed-lineage kinase domain-like.
When TNFα binds to TNFR1, it causes trimerization of TNFR1 and the activation of death domains in the intracellular site via removal of SODD (Silencer of death domain). TNFR1 activation induces the formation of Complex I, which includes the receptor-interacting serine/threonine-protein kinase (RIPK1), adaptor proteins TNFR1-associated death domain (TRADD) and TNF receptor-associated factors 2 and 5 (TRAF2/TRAF5), and E3 ubiquitin ligases (cIAP1/cIAP2, and LUBAC complex)[145] (Figure 2). RIPK1 is regulated by multiple posttranslational modifications, being ubiquitination one of the most critical regulatory mechanisms. cIAP1/2 are recruited into Complex I, which with the help of TRAF2/5, mediate RIPK1 K63 ubiquitination. K63 ubiquitination of RIPK1 by cIAP1/2 facilitates the recruitment of the LUBAC complex, which performs further ubiquitination of RIPK1[146].
RIPK1 was the first protein demonstrated to be essential for TNFα-, Fas-, and TRAIL-induced necroptosis. The removal of ubiquitin chains from RIPK1 leads to its interaction with FADD, TRADD, RIPK3, and caspase-8, resulting in Complex II formation[138] (Figure 2). Under conditions of caspase-8 inactivation/depletion or cIAP deficiency, RIPK1 and RIPK3 are not cleaved and become phosphorylated. RIPK1 is activated via deubiquitination mediated by cylindromatosis (CYLD), which destabilizes Complex I and promotes activation of a cytosolic necrosome complex, also known as Complex IIb[147, 148] (Figure 2). This complex contains a hetero-oligomer of RIPK1 and RIPK3, which interact through their cognate RHIM domains[149–152]. Activated RIPK1 undergoes autophosphorylation at Ser14/15, Ser20, and Ser161/166[153]. Ser166 phosphorylation has emerged as a biomarker of RIPK1 activation[153–155]. RIPK1 autophosphorylation is then followed by RIPK3 autophosphorylation in the necrosomes on Ser227 (Thr231/Ser232 for mouse RIPK3)[140, 156]. Upon RIPK3 autophosphorylation, the mixed lineage kinase domain-like pseudokinase (MLKL) is phosphorylated at segment residues Thr357/Ser358[157, 158] (Ser 345 for mouse MLKL)[159, 160], causing pore-forming oligomers that puncture cell membranes, inducing cell death by necroptosis[138, 139, 158, 159, 161] (Figure 2). MKLK, which is downstream of kinases RIPK1 and RIPK3, is considered a more specific kinase for the necroptosis pathway[158, 159].
TNFα-TNFR1 signaling and RIPK1 are not exclusive to necroptosis since they are also involved in inflammation pathways, via kinase-dependent and independent functions, and in apoptosis[155, 162–166] (Figure 2). Indeed, polyubiquitinated RIPK1 in Complex I promotes the recruitment and activation of TAK1 kinase through the polyubiquitin binding adaptors TAB2/TAB3 and recruitment of the IKK complex, leading to NF-kB activation, gene expression of pro-inflammatory cytokines, and inflammation[167–169] (Figure 2). Furthermore, TRADD and RIPK1 can become modified and dissociate from TNFR1. The liberated death domain (DD) of TRADD (and/or RIPK1) binds to FADD, resulting in RIPK cleavage, caspase-8 recruitment (forming complex IIa), activation of caspase-8, which results in activation of caspase-3, and cell death by apoptosis[170–172] (Figure 2).
Unlike apoptosis[121], necroptosis can trigger or amplify inflammation[140, 173–175] and mediates a variety of inflammatory conditions, including periodontal, autoimmune, infectious cardiovascular, and pulmonary diseases[176–184], strongly suggesting that necroptosis plays a critical role in many other different disease processes[138–141, 161, 169, 176, 185].
Osteocyte Death
Osteocytes, as postmitotic cells, cannot replicate. However, they have developed adaptative mechanisms to ensure their survival under stressful conditions, such as immobilization, hypoxia, and disease[35]. However, when their survival capacity is overwhelmed, osteocytes can die. Osteocyte death has been associated with pathological conditions including osteoarthritis[186], inflammatory skeletal diseases[185, 187, 188], metastatic bone disease[189, 190], aging[191, 192], osteonecrosis of the femoral head[29], osteoradionecrosis[193], periodontitis[194–196], and MRONJ[2, 197].
All three forms of cell death (apoptosis, autophagy, and necrosis)[120] have been recognized in osteocytes. Osteocyte apoptosis was demonstrated under different conditions, including skeletal immobilization due to oxygen deprivation[198], osteonecrosis of the femoral head (ONFH)[29], estrogen withdrawal[199–201], and bone microcracks after bone fatigue[202–204]. It has also been associated with the natural process of aging, after menopause and bone unloading/weightlessness[205, 206]. Furthermore, increased osteocyte apoptosis plays an essential role in the decreased bone strength observed with glucocorticoid (GC) treatment[207]. Notably, treatment with N-BPs reduces osteocyte apoptosis in response to fatigue loading[208] and protects against GC-induced apoptosis by transiently increasing ERK phosphorylation[209]. A similar effect was observed with calcitonin and mechanical stimulation[210]. Mechanical stimulation also prevented osteocyte apoptosis[210], and treatment of OVX mice with a pan-caspase inhibitor inhibited OVX-induced osteocyte apoptosis and reduced bone resorption[201]. A study showed that young mice lacking FGFR1/FGFR2 or only FGFR1 are phenotypically normal. However, at age 6-12 weeks, mice developed a high bone mass phenotype and increased porosity preceded by a striking peak in osteocyte death, particularly by apoptosis[211]. The study identified a role for FGFR1 signaling in osteocytes and mature osteoblasts, which is required for osteocyte survival and the regulation of bone mass.
Osteocytes can also undergo autophagy[212–215]. GCs activate the autophagosomal pathway in osteocytes, increasing markers of autophagy[212]. This mechanism could be beneficial to repair damaged organelles or cell membranes. However, dexamethasone also reduced the number of metabolically normal osteocytes. This effect was augmented when autophagy was suppressed, suggesting that autophagy is an adaptative mechanism used by osteocytes to attenuate the impact of GCs[212]. The cell protective function of autophagy is likely to occur under short or moderate stress conditions. However, higher or more prolonged stress may result in an accumulation of autophagosomes and cell death[213]. This is not surprising since autophagy previously was suggested to act as a “double-edged sword” involved in both cell protection and cell death[216, 217]. Mechanical compression forces were also found to activate autophagy in osteocytic cells (MLO-Y4) in vitro and osteocytes in vivo, as demonstrated in an orthodontic tooth movement model[214]. Notably, suppression of osteocyte autophagy caused skeletal changes similar to those caused by aging, including decreased bone mass and strength[215].
Osteonecrosis implies the death of bone cells. It can be caused by disease or trauma, such as a fracture, which negatively affects the blood supply to the bone. Osteonecrosis can also be idiopathic, but the pathological picture and resultant early clinical course are quite stereotypical[218]. The term osteonecrosis for certain skeletal conditions, such as aseptic, avascular, or ischemic necrosis, may be technically inaccurate, as it has not been demonstrated that the bone cells die by necrosis[29]. Komori[219] proposed that any form of osteocyte death, such as apoptosis or autophagy, ultimately results in secondary necrosis because dead osteocytes encased in the bone matrix cannot be immediately reached by phagocytic scavenger cells[220].
Necrosis ultimately leads to the rupture of the osteocyte cytoplasmic membrane, with most of the intracellular content being released into the extracellular environment[221]. Dying osteocytes release large amounts of DAMPs into the lacuna and adjacent canaliculi, including the histone deacetylase complex subunit SAP130, released and degraded cartilage matrix constituents, S100 family molecules, the high-mobility group box 1 (HMGB1) protein, purine metabolites, heat-shock proteins, and uric acid[185, 220]. DAMPS released into the canaliculi reach bone surfaces and vascular canals, initiating inflammatory responses by binding to various PRRs, such as the macrophage inducible C-type lectin receptor Mincle, TLR2/4, and RAGE on osteoclasts, macrophages, dendritic cells, monocytes, neutrophils[222–225].
Notably, necroptosis has also been identified as a RCD form in osteocytes under certain conditions[226–228]. Indeed, in addition to apoptosis, necroptosis was found in osteocytes under conditions of estrogen deficiency in OVX rats, suggesting the involvement of osteocyte necroptosis in the pathophysiology of postmenopausal osteoporosis[226]. Furthermore, necroptotic osteocytes and trabecular bone deterioration are related to the production of TNFα in OVX ratsl[227]. Besides apoptotic osteocytes, necroptotic osteocytes were also found in rats with GC-induced osteoporosis[228]. Notably, necrostatin-1 (Nec-1), a specific RIPK1 inhibitor that inhibits TNF-α induced necroptosis[143], ameliorated the skeletal effects of GCs[228]. The coexistence of apoptotic and necroptotic osteocytes is not surprising since it was previously suggested that apoptosis and necroptosis could co-occur[229].
Osteocyte death in MRONJ
Though bone necrosis is the hallmark of MRONJ, little attention has been paid to investigating the type of cell death afflicting osteocytes in MRONJ. Early studies[230] found focal areas of bone matrix necrosis in the mandible of dogs treated for three years with ALN. It has been shown that osteocyte death occurs as a physiologic end-stage of the skeleton’s life cycle[231], and that the prevalence of osteocyte death increases with skeletal aging[232, 233]. Investigators might assume from these findings that a systematic process for removing dead osteocytes, perhaps based on bone resorption, exists in the adult skeleton. Based on these ideas, the authors[230] suggested that jaw bone necrosis associated with ALN treatment resulted from dead osteocyte accumulation caused by the suppression of bone resorption by ALN.
In contrast, it was suggested that the necrotic alveolar bone would have been efficiently removed in the absence of an N-BP and a normal bone turnover rate, particularly in jawbones that appear to have higher basal bone turnover than the postcranial skeleton[234, 235]. These authors[230] also proposed an alternate theory in which ALN could have directly affected osteocyte viability, decreasing their lifespan and increasing the rate of bone necrosis. However, in contrast to this theory, preclinical in vivo studies have shown that clinical doses of N-BPs positively affect osteocyte viability, preventing osteocyte apoptosis induced by GCs in mice[209], or by fatigue cyclic loading in rats[208]. Supra-clinical doses of ALN have cytotoxic in vitro effects on fibroblasts[236] and endothelial cells[237]. Since N-BPs accumulate in the skeleton in a dose-dependent manner, these authors[230] also suggested that with prolonged exposure to ALN, the local bone accumulation of drug could reach levels that cause cytotoxic effects on osteocytes. However, a cytotoxic effect of N-BPs on osteocytes has never been proven.
Sustained activation of Nod-like receptor (NLR) family, pyrin domain-containing protein 3 (NLRP3) inflammasome contributes to persistent inflammation and impaired cutaneous wound healing in diabetic mice and humans[238]. One study[239] showed that macrophages at MRONJ-like lesions of diabetic mice harbor an up-regulated expression of NLRP3 inflammasome components and that ZOL augmented the persistent NLRP3 activation in diabetic macrophages, which may have contributed to the impaired oral socket wound healing and increased incidence and severity of MRONJ-like lesions in the diabetic mice. Though the study showed increased caspase-1 expression in cells within the MRONJ-like lesion, it did not specifically investigate cell death types and mechanisms in the osteocytes.
Herein, we propose a model that in the presence of systemic risk factors (e.g., pARs), inflammation associated with oral risk factors sustains molecular signaling pathways, largely TNFα/TNFR1 signaling, that enhance RCD-related osteocyte death, particularly necroptosis and apoptosis. These, in turn, promote the propagation of inflammatory signaling, accelerating soft and hard tissue necrosis to induce MRONJ (Figure 3).
Figure 3. A proposed model for MRONJ.
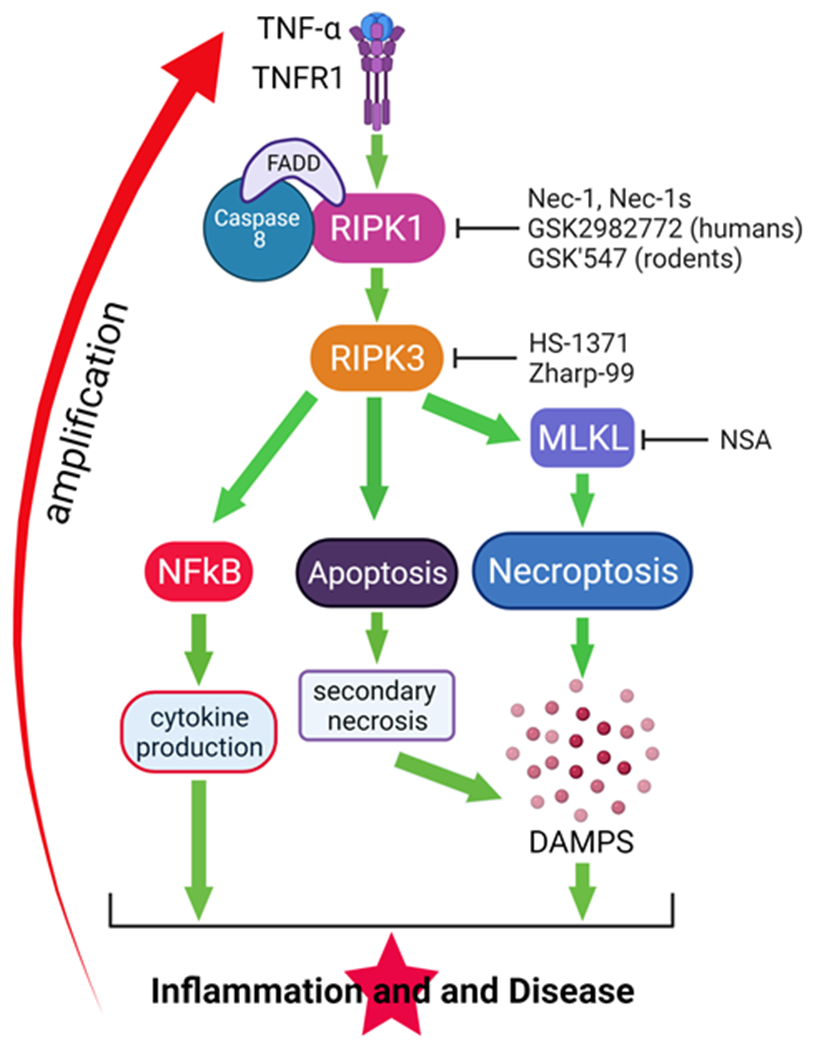
In the presence of systemic risk factors (e.g., pARs or AgIs), the inflammation associated with oral risk factors induces and sustains molecular signaling pathways, largely TNF-α -TNFR1, which enhance osteocyte death particularly by necroptosis but also apoptosis. TNF-α -TNFR1 signaling also promotes the activation of the NFkB cascade with the synthesis of pro-inflammatory cytokines. pARs prevent the resorption of bone, including necrotic bone. The accumulated necrotic osteocytes generate an accrued amount of DAMPs that further stimulate inflammation. Altogether these events amplify inflammation, oral soft and hard tissue destruction, and induction of MRONJ. Several inhibitors of RIPK1 (Nec-1, Nec-1s, GSL2982773, GSK’547); RIPK3 (HS-1371, Zharp-99); and MLKL (NSA) have been developed that, if apoptosis, necroptosis, or both are involved in MRONJ pathophysiology, will represent pharmacologic interventions to slow/stop MRONJ progression. Abbreviations: TNF: tumor necrosis factor; TNFR1: TNF receptor 1; NF-kB: nuclear factor kappa-light-chain-enhancer of activated B cells; RIPK: Receptor interacting serine/threonine kinase; FADD: FAS-associated death domain; MLKL: mediator mixed-lineage kinase domain-like; DAMPs: damage-associated molecular patterns; Nec-1: Necrostatin 1; NSA: necrosulfonamide.
Clinical and preclinical data indicate that for MRONJ to occur, systemic risk factors (e.g., pARs and AgIs) and oral risk factors, such as tooth extraction and inflammatory dental disease (e.g., periodontitis, periapical infection) must co-occur[1, 2, 11–22]. Oral risk factors associated with inflammation and/or infection induce local production of pro-inflammatory cytokines, such as TNF-α and IL-1, which stimulate inflammation and osteocyte death in alveolar bone[194–196]. Cell death associated with tissue infection and inflammation is linked to ACD[240]. However, as described earlier, strong evidence suggests that biomolecules that activate inflammation, like TNF-α and others, simultaneously activate cell death by RCD mechanisms[114, 143, 161, 174, 241–243], including necroptosis[138, 140, 141, 161, 169, 176, 244] and apoptosis[245, 246], and also stimulate inflammation[173] (Figure 3).
In cells that die by apoptosis, the apoptotic cell bodies are quickly taken up by neighboring cells and degraded within phagolysosomes. Therefore, and in contrast to necrosis, apoptosis might not induce an inflammatory reaction harmful to the host[247].
However, if cells at the terminal phases of apoptosis are not immediately engulfed by phagocytes, they can undergo secondary necrosis[130, 248]. Secondary necrosis has been suggested to occur in osteocytes dying by apoptosis and autophagy[219, 220]. In patients taking antiresorptives, the removal of necrotic bone is delayed or suppressed. Thus, it is possible that osteocytes dying by apoptosis or autophagy remain longer in the LCN of the persistent necrotic bone, increasing the probability of secondary necrosis[219, 220].
Since empty osteocyte lacunae are the distinctive histologic feature of MRONJ, the cellular remnants resulting from apoptosis, autophagy, or necrosis have to undergo a process of clearance from the LCN. Mobilization of apoptotic bodies, DAMPs, or cellular debris from the bone necrotic sites would be limited by the low permeability of the LCN to the movement of large solutes[249, 250]. Typical diameters of lacunae and canaliculi are ~10μm and 0.03-0.3 μm, respectively[251–254]. The annular fluid space surrounding the osteocyte cell processes inside canaliculi is much smaller, ~50–100 nm wide[249, 254, 255], and is filled with a gel-like matrix composed of proteoglycans and other matrix molecules[256]. Besides providing resistance to fluid flow[255, 256], the pericellular matrix of the LCN modulates solute transport behaving as a molecular sieve[250, 257]. Selective in vivo perfusion studies with various sized tracers demonstrate that the cut-off size of the LCN in adult bone lies between 7-12 nm[250, 254]. Interestingly, it is well accepted that convection due to mechanical loading augments solute diffusion in the bone as demonstrated in theory[258] and observed on histological sections[259, 260]. Apoptotic bodies range from 50 to 5000 nm in diameter[261]. Thus, even under mechanical loading conditions, they appear too large to circulate through the LCN[80]. As mentioned earlier, osteocytes dying by apoptosis, autophagy, or necrosis can undergo autolytic changes that result in the formation of DAMPs[219, 220, 262] (Figure 3). Andreev et al.[185] confirmed that high molecular weight DAMPs can circulate through the LCN. Thus, if DAMPs can circulate through the LCN, it is reasonable to suggest that dead osteocytes can be removed from the LCN in the form of DAMPs. Furthermore, DAMPs release into the canaliculi could reach bone surfaces and adjacent bone postcapillary venules, activating PRRs on osteoclasts, pericytes, and other types of phagocytic cells[222–225], enhancing innate immune responses and inflammation[263]. In addition, DAMPs could advance into the blood circulation, activate PRRs on immune cells in the circulation or lymphoid organs, and be cleared by macrophages in the red pulp of the spleen[264–267].
As seen in necrosis, necroptotic cells also manifest loss of membrane integrity and release of the cellular content, which function as DAMPs[140, 268] (Figure 3). Recently, Mincle was recognized as the PRR that more specifically senses another DAMP, SAP-130[269]. Dying osteocytes release SAP-130, and Mincle is highly expressed at skeletal sites of osteocyte death[185]. Mincle is specifically upregulated in osteoclasts in a RANK-RANKL-independent fashion, and its signaling appears to target bone resorption upon osteocyte death[185]. In patients taking N-BPs, though bone resorption is inhibited, the number of osteoclasts at bone surfaces does not decline[270]. When necrotic bone persists due to the inhibition of bone resorption by pARs, DAMPs, including SAP-130, would accumulate in the necrotic alveolar bone, suggesting that Mincle expression in N-BP-treated patients would be chronically elevated. Indeed, Mincle is highly expressed in necrotic bone areas of patients with MRONJ[185], and these authors suggested that SAP-130 and Mincle could be potential early markers for MRONJ. The DAMP molecule HMGB-1 activates TLR-2 and TLR-4, triggering an immune system response and inflammation in the extracellular milieu[271]. The pathophysiological significance of elevated expression of DAMPs and PRRs in the context of impaired bone resorption, as occurs in pAR-treated patients, has not been directly investigated.
After discovering necroptosis, several inhibitors of kinases involved in necroptosis and/or apoptosis signaling pathways, namely RIPK1, RIPK3 and MLKL, were developed[268, 272–274] (Figure 3). Necrostatins (Nec) are tryptophan-based compounds that inhibit RIPK1[153]. Nec-1 was first discovered during chemical screening for necroptosis antagonists[153]. However, Nec-1 has moderate potency, poor specificity, and poor pharmacokinetic properties[275, 276]. GSK2982772 is a highly selective inhibitor of RIPK1, being developed to treat chronic inflammatory diseases characterized by necroptosis and apoptosis[140]. GSK2982772 is currently being tested in clinical trials for psoriasis, rheumatoid arthritis, and ulcerative colitis. GSK’547[277] is a rodent-specific RIPK1 inhibitor that inhibits necroptosis, associated inflammation, and apoptosis[142, 153, 155, 272, 278]. RIPK3 provides the scaffold for RIPK1 that contributes to full caspase-8 activation independently of its kinase activity or intact RHIM domain to induce apoptosis[279] and NF-kB mediated inflammation[280]. The RIPK3 inhibitor HS-1371 suppresses TNF-induced necroptosis but does not inhibit TNF-induced apoptosis, indicating that HS-1371 specifically inhibits RIPK3-mediated necroptosis by suppressing RIPK3[281]. Another potent RIPK3 inhibitor (Zharp-99) was recently developed to ameliorate necroptosis-associated inflammatory injury[282]. Notably, necrosulfonamide (NSA) is an MLKL inhibitor that selectively inhibits necroptosis[159]. Thus, if necroptosis and/or apoptosis are indeed involved in MRONJ pathophysiology, as well as inflammation, inhibitors for these signaling pathways[142, 268, 272, 277, 283–286] represent pharmacologic interventions that could slow/stop the progression of MRONJ. They might be applied as monotherapy in early phases of MRONJ (stage 0) or as adjunctive therapy to existing effecting practices, such as the infection control measures used in stages 1-3, or the surgical interventions used to reduce the heavy burden of necrotic bone in more advanced cases.
Conclusions
MRONJ is a potentially debilitating condition that affects patients with cancer and patients with osteoporosis who have been treated with pARs or AgIs and have concurrent oral risk factors, including tooth extraction or inflammatory dental disease. Though several mechanisms have been proposed to explain the occurrence of MRONJ, the underlying pathophysiology has not been completely elucidated.
Bone necrosis represents the hallmark of MRONJ. However, we know very little about the precise timing and mechanisms involved in osteocyte death in the context of this disease. Osteocytes are postmitotic cells that have developed adaptative mechanisms to ensure their survival under stressful conditions. However, when their survival capacity is overwhelmed, osteocyte death occurs. All the three general forms of cell death (apoptosis, autophagy, and necrosis) have been recognized in osteocytes under different pathological conditions. Osteocyte death is, in a certain way, distinct from cell death in other cell types because osteocytes are isolated in the bone matrix, meaning that osteocytes that die by apoptosis or autophagy cannot be immediately phagocytized by scavenger cells. Thus, osteocytes that die by these cell death mechanisms persist for some time in the bone matrix, possibly ending in molecular and morphologic changes of secondary necrosis[219, 220]. Necrosis leads to the rupture of the osteocyte cytoplasmic membrane, with most of the intracellular content being released into the extracellular environment. Necrotic cell death can also occur as necroptosis, a form of RCD[143]. Unlike apoptosis[121], necroptosis triggers or amplifies inflammation[140, 173, 174] and mediates a variety of different inflammatory conditions[138–141, 161, 169, 176–185], suggesting that it might be involved in MRONJ pathophysiology.
A proposed hypothesis for MRONJ is that signaling pathways associated with oral risk factors, particularly TNFα/TNFR1 signaling, intensify the inflammatory response and the triggering of RCD mechanisms, including necroptosis, apoptosis, or both (Figure 3). In the presence of antiresorptives, DAMPS that accumulate in necrotic bone activate various PRRs (present on osteoclasts, phagocytic and antigen-presenting cells) that amplify the inflammatory response, inducing further osteocyte cell death and soft tissue necrosis. Several inhibitors of kinases involved in the necroptosis and/or apoptosis signaling pathways, namely RIPK1, MLKL, and RIPK3, have been developed[142, 268, 272, 277, 283–286]. Thus, if apoptosis, necroptosis, or both are involved in MRONJ pathophysiology, these inhibitors would represent pharmacologic interventions to slow/stop disease progression. In any case, improving our understanding of osteocyte death associated with MRONJ could be critical for developing more efficacious treatments.
Acknowledgments
This research was supported by NIH grant R01DE023783-01A from the National Institute of Dental and Craniofacial Research (NIDCR).
Abbreviations
- ACD
Accidental cell death
- AgIs
angiogenesis inhibitors
- ALN
alendronate
- BMPs
Bone morphogenetic proteins
- cIAP
Cellular inhibitor of apoptosis protein
- Cx
Connexins
- CYLD
Deubiquitinase cylindromatosis
- DAMPs
Damage-associated molecular patterns
- DD
Death domain
- DKK-1
Dickkopf-related protein 1
- DMP1
Dentin matrix protein-1
- ERK
Extracellular signal-regulated kinase
- FADD
FAS-associated death domain
- FasL/TNFSF6
Fas Ligand
- FGFR
Fibroblast growth factor receptor
- FGF23
fibroblast growth factor 23
- GC
Glucocorticoid
- HD
Homeodomain proteins
- HMGB1
High-mobility group box 1 protein
- IFNs
Interferons
- IGF-1
Insulin-like growth factor-1
- IKKs
IKKα and IKKβ complex
- LNC
Lacuna-canalicular (network)
- LRP
Low-density lipoprotein receptor-related protein
- LPS
lipopolysaccharide
- LUBAC
linear ubiquitin chain assembly complex
- MEPE
Matrix extracellular phosphoglycoprotein
- Mincle
Macrophage inducible C-type lectin receptor
- MLKL
Mixed lineage kinase domain-like pseudokinase
- MLO-Y4
Murine long bone osteocyte-like cell line
- MRONJ
Medication-related osteonecrosis of the jaw
- NF-kB
nuclear factor kappa-light-chain-enhancer of activated B cells
- ONFH
Osteonecrosis of the femoral head
- OVX
Ovariectomized
- N-BPs
nitrogen-containing bisphosphonates
- NCCD
Nomenclature Committee on Cell Death
- NC
neural crest
- Nec-1
Necrostatin-1
- NLR
Nod-like receptor
- NLRP3
NLR family, pyrin domain-containing protein 3
- NSA
Necrosulfonamide
- ONFH
Osteonecrosis of the femoral head
- OPG
Osteoprotegerin
- PAMPs
Pathogen-associated molecular patterns
- pARs
powerful antiresorptives
- PCD
Programmed cell death
- PGE2
Prostaglandin E2
- PHEX
Phosphate regulating endopeptidase homolog X-linked protein
- PRR
Pattern recognition receptors
- PTH
Parathyroid hormone
- P50
NF-κB1
- P65
RelA
- RAGE
Receptor for advanced glycation end-products
- RANK
receptor activator of nuclear factor kappa-Β
- RANKL
RANK ligand
- RCD
Regulated cell death
- RIPK
Receptor-interacting serine/threonine-protein kinase
- SAP130
Histone deacetylase complex subunit SAP130
- SIBLINGs
Small integrin-binding ligand N-linked glycoproteins
- SODD
Silencer of death domain
- S100
Soluble 100% protein
- TAB2
TGF-Beta Activated Kinase 1 (MAP3K7) binding protein 2
- TAB3
TGF-Beta Activated Kinase 1 (MAP3K7) binding protein 3
- TAK1
Transforming growth factor-β-activated kinase 1
- TGF-β
Transforming growth factor-beta
- TLR3/4
Toll-like receptors 3/4FAS: CD95/APO-1
- TNFα
Tumor necrosis factor-alpha
- TNFR1
TNF receptor 1
- TRADD
TNFRSF1A-associated via death domain
- TRAF
TNF receptor-associated factors
- TRAILR1/2
TRAIL receptor 1/2
- TRAIL/TNFSF10
TNF-Related Apoptosis-Inducing Ligand
- VEGF
vascular endothelial cell growth factor
- ZOL
zoledronic acid
Footnotes
Conflict of Interest
The authors have no conflicts of interest.
References
- [1].Khan AA, Morrison A, Hanley DA, Felsenberg D, McCauley LK, O’Ryan F, Reid IR, Ruggiero SL, Taguchi A, Tetradis S, Watts NB, Brandi ML, Peters E, Guise T, Eastell R, Cheung AM, Morin SN, Masri B, Cooper C, Morgan SL, Obermayer-Pietsch B, Langdahl BL, Al Dabagh R, Davison KS, Kendler DL, Sandor GK, Josse RG, Bhandari M, El Rabbany M, Pierroz DD, Sulimani R, Saunders DP, Brown JP, Compston J, J. International Task Force on Osteonecrosis of the, Diagnosis and management of osteonecrosis of the jaw: a systematic review and international consensus, J Bone Miner Res 30(1) (2015) 3–23. [DOI] [PubMed] [Google Scholar]
- [2].Ruggiero SL, Dodson TB, Fantasia J, Goodday R, Aghaloo T, Mehrotra B, O’Ryan F, American Association of Oral and Maxillofacial Surgeons position paper on medication-related osteonecrosis of the jaw--2014 update, J. Oral Maxillofac. Surg 72(10) (2014) 1938–1956. [DOI] [PubMed] [Google Scholar]
- [3].Van Poznak C, Somerfield MR, Barlow WE, Biermann JS, Bosserman LD, Clemons MJ, Dhesy-Thind SK, Dillmon MS, Eisen A, Frank ES, Jagsi R, Jimenez R, Theriault RL, Vandenberg TA, Yee GC, Moy B, Role of Bone-Modifying Agents in Metastatic Breast Cancer: An American Society of Clinical Oncology-Cancer Care Ontario Focused Guideline Update, J Clin Oncol 35(35) (2017) 3978–3986. [DOI] [PubMed] [Google Scholar]
- [4].Aapro M, Abrahamsson PA, Body JJ, Coleman RE, Colomer R, Costa L, Crino L, Dirix L, Gnant M, Gralow J, Hadji P, Hortobagyi GN, Jonat W, Lipton A, Monnier A, Paterson AH, Rizzoli R, Saad F, Thurlimann B, Guidance on the use of bisphosphonates in solid tumours: recommendations of an international expert panel, Ann Oncol 19(3) (2008) 420–32. [DOI] [PubMed] [Google Scholar]
- [5].Stopeck AT, Lipton A, Body JJ, Steger GG, Tonkin K, de Boer RH, Lichinitser M, Fujiwara Y, Yardley DA, Viniegra M, Fan M, Jiang Q, Dansey R, Jun S, Braun A, Denosumab compared with zoledronic acid for the treatment of bone metastases in patients with advanced breast cancer: a randomized, double-blind study, J. Clin. Oncol 28(35) (2010) 5132–5139. [DOI] [PubMed] [Google Scholar]
- [6].Van den Wyngaert T, Wouters K, Huizing MT, Vermorken JB, RANK ligand inhibition in bone metastatic cancer and risk of osteonecrosis of the jaw (ONJ): non bis in idem?, Support. Care Cancer 19(12) (2011) 2035–2040. [DOI] [PubMed] [Google Scholar]
- [7].Yu EW, Tsourdi E, Clarke BL, Bauer DC, Drake MT, Osteoporosis Management in the Era of COVID-19, J Bone Miner Res 35(6) (2020) 1009–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Rugani P, Walter C, Kirnbauer B, Acham S, Begus-Nahrman Y, Jakse N, Prevalence of Medication-Related Osteonecrosis of the Jaw in Patients with Breast Cancer, Prostate Cancer, and Multiple Myeloma, Dent J (Basel) 4(4) (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Coleman RE, Collinson M, Gregory W, Marshall H, Bell R, Dodwell D, Keane M, Gil M, Barrett-Lee P, Ritchie D, Bowman A, Liversedge V, De Boer RH, Passos-Coelho JL, O’Reilly S, Bertelli G, Joffe J, Brown JE, Wilson C, Tercero JC, Jean-Mairet J, Gomis R, Cameron D, Benefits and risks of adjuvant treatment with zoledronic acid in stage II/III breast cancer. 10 years follow-up of the AZURE randomized clinical trial (BIG 01/04), J Bone Oncol 13 (2018) 123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Miksad RA, Lai KC, Dodson TB, Woo SB, Treister NS, Akinyemi O, Bihrle M, Maytal G, August M, Gazelle GS, Swan JS, Quality of life implications of bisphosphonate-associated osteonecrosis of the jaw, Oncologist 16(1) (2011) 121–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wan JT, Sheeley DM, Somerman MJ, Lee JS, Mitigating osteonecrosis of the jaw (ONJ) through preventive dental care and understanding of risk factors, Bone Res 8 (2020) 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Aghaloo TL, Kang B, Sung EC, Shoff M, Ronconi M, Gotcher JE, Bezouglaia O, Dry SM, Tetradis S, Periodontal disease and bisphosphonates induce osteonecrosis of the jaws in the rat, J. Bone Miner. Res 26(8) (2011) 1871–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Aguirre JI, Akhter MP, Kimmel DB, Pingel JE, Williams A, Jorgensen M, Kesavalu L, Wronski TJ, Oncologic doses of zoledronic acid induce osteonecrosis of the jaw-like lesions in rice rats (Oryzomys palustris) with periodontitis, J. Bone Miner. Res 27(10) (2012) 2130–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].de Molon RS, Cheong S, Bezouglaia O, Dry SM, Pirih F, Cirelli JA, Aghaloo TL, Tetradis S, Spontaneous osteonecrosis of the jaws in the maxilla of mice on antiresorptive treatment: a novel ONJ mouse model, Bone 68 (2014) 11–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Soundia A, Hadaya D, Esfandi N, de Molon RS, Bezouglaia O, Dry SM, Pirih FQ, Aghaloo T, Tetradis S, Osteonecrosis of the jaws (ONJ) in mice after extraction of teeth with periradicular disease, Bone 90 (2016) 133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Messer JG, Jiron JM, Mendieta Calle JL, Castillo EJ, Israel R, Phillips EG, Yarrow JF, Van Poznak C, Kesavalu L, Kimmel DB, Aguirre JI, Zoledronate Treatment Duration Is Linked to Bisphosphonate-Related ONJ Prevalence in Rice Rats with Generalized Periodontitis, Oral Dis 25(4) (2019) 1116–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Messer JG, Mendieta Calle JL, Jiron JM, Castillo EJ, Van Poznak C, Bhattacharyya N, Kimmel DB, Aguirre JI, Zoledronic acid increases the prevalence of medication-related osteonecrosis of the jaw in a dose dependent manner in rice rats (Oryzomys palustris) with localized periodontitis, Bone 108 (2018) 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Katsarelis H, Shah NP, Dhariwal DK, Pazianas M, Infection and medication-related osteonecrosis of the jaw, J Dent Res 94(4) (2015) 534–9. [DOI] [PubMed] [Google Scholar]
- [19].Kang B, Cheong S, Chaichanasakul T, Bezouglaia O, Atti E, Dry SM, Pirih FQ, Aghaloo TL, Tetradis S, Periapical disease and bisphosphonates induce osteonecrosis of the jaws in mice, J. Bone Miner. Res 28(7) (2013) 1631–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Song M, Alshaikh A, Kim T, Kim S, Dang M, Mehrazarin S, Shin KH, Kang M, Park NH, Kim RH, Preexisting Periapical Inflammatory Condition Exacerbates Tooth Extraction-induced Bisphosphonate-related Osteonecrosis of the Jaw Lesions in Mice, J. Endod 42(11) (2016) 1641–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kikuiri T, Kim I, Yamaza T, Akiyama K, Zhang Q, Li Y, Chen C, Chen W, Wang S, Le AD, Shi S, Cell-based immunotherapy with mesenchymal stem cells cures bisphosphonate-related osteonecrosis of the jaw-like disease in mice, J Bone Miner. Res 25(7) (2010) 1668–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hasegawa T, Hayashida S, Kondo E, Takeda Y, Miyamoto H, Kawaoka Y, Ueda N, Iwata E, Nakahara H, Kobayashi M, Soutome S, Yamada SI, Tojyo I, Kojima Y, Umeda M, Fujita S, Kurita H, Shibuya Y, Kirita T, Komori T, Japanese M Study Group of Co-operative Dentistry with, Medication-related osteonecrosis of the jaw after tooth extraction in cancer patients: a multicenter retrospective study, Osteoporos Int 30(1) (2019) 231–239. [DOI] [PubMed] [Google Scholar]
- [23].Migliorati CA, Woo SB, Hewson I, Barasch A, Elting LS, Spijkervet FK, Brennan MT, A systematic review of bisphosphonate osteonecrosis (BON) in cancer, Support. Care Cancer 18(8) (2010) 1099–1106. [DOI] [PubMed] [Google Scholar]
- [24].Kuhl S, Walter C, Acham S, Pfeffer R, Lambrecht JT, Bisphosphonate-related osteonecrosis of the jaws - A review, Oral Oncol (2012). [DOI] [PubMed] [Google Scholar]
- [25].Ruggiero SL, Emerging Concepts in the Management and Treatment of Osteonecrosis of the Jaw, Oral and Maxillofacial Surgery Clinics of North America 25(1) (2013) 11–20. [DOI] [PubMed] [Google Scholar]
- [26].Franco-Pretto E, Pacheco M, Moreno A, Messa O, Gnecco J, Bisphosphonate-induced osteonecrosis of the jaws: clinical, imaging, and histopathology findings, Oral Surg Oral Med Oral Pathol Oral Radiol 118(4) (2014) 408–17. [DOI] [PubMed] [Google Scholar]
- [27].Zheng LZ, Wang JL, Kong L, Huang L, Tian L, Pang QQ, Wang XL, Qin L, Steroid-associated osteonecrosis animal model in rats, J Orthop Translat 13 (2018) 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yang L, Boyd K, Kaste SC, Kamdem Kamdem L, Rahija RJ, Relling MV, A mouse model for glucocorticoid-induced osteonecrosis: effect of a steroid holiday, J Orthop Res 27(2) (2009) 169–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Weinstein RS, Nicholas RW, Manolagas SC, Apoptosis of osteocytes in glucocorticoid-induced osteonecrosis of the hip, J Clin Endocrinol Metab 85(8) (2000) 2907–12. [DOI] [PubMed] [Google Scholar]
- [30].Bonewald LF, Osteocytes as dynamic multifunctional cells, Ann N Y Acad Sci 1116 (2007) 281–90. [DOI] [PubMed] [Google Scholar]
- [31].Bonewald LF, The amazing osteocyte, J Bone Miner Res 26(2) (2011) 229–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Palumbo C, Ferretti M, Marotti G, Osteocyte dendrogenesis in static and dynamic bone formation: an ultrastructural study, Anat. Rec. A Discov. Mol. Cell Evol. Biol 278(1) (2004) 474–480. [DOI] [PubMed] [Google Scholar]
- [33].Palumbo C, A three-dimensional ultrastructural study of osteoid-osteocytes in the tibia of chick embryos, Cell Tissue Res 246(1) (1986) 125–31. [DOI] [PubMed] [Google Scholar]
- [34].Franz-Odendaal TA, Hall BK, Witten PE, Buried alive: how osteoblasts become osteocytes, Dev Dyn 235(1) (2006) 176–90. [DOI] [PubMed] [Google Scholar]
- [35].Dallas SL, Prideaux M, Bonewald LF, The osteocyte: an endocrine cell … and more, Endocr Rev 34(5) (2013) 658–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Knothe Tate ML, Adamson JR, Tami AE, Bauer TW, The osteocyte, Int J Biochem Cell Biol 36(1) (2004) 1–8. [DOI] [PubMed] [Google Scholar]
- [37].Jones SJ, Gray C, Sakamaki H, Arora M, Boyde A, Gourdie R, Green C, The incidence and size of gap junctions between the bone cells in rat calvaria, Anat Embryol (Berl) 187(4) (1993) 343–52. [DOI] [PubMed] [Google Scholar]
- [38].Doty SB, Morphological evidence of gap junctions between bone cells, Calcif Tissue Int 33(5) (1981) 509–12. [DOI] [PubMed] [Google Scholar]
- [39].Civitelli R, Cell-cell communication in the osteoblast/osteocyte lineage, Arch Biochem Biophys 473(2) (2008) 188–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Plotkin LI, Bellido T, Beyond gap junctions: Connexin43 and bone cell signaling, Bone 52(1) (2013) 157–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Romanello M, D’Andrea P, Dual mechanism of intercellular communication in HOBIT osteoblastic cells: a role for gap-junctional hemichannels, J Bone Miner Res 16(8) (2001) 1465–76. [DOI] [PubMed] [Google Scholar]
- [42].Batra N, Kar R, Jiang JX, Gap junctions and hemichannels in signal transmission, function and development of bone, Biochim Biophys Acta 1818(8) (2012) 1909–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Jiang JX, Siller-Jackson AJ, Burra S, Roles of gap junctions and hemichannels in bone cell functions and in signal transmission of mechanical stress, Front Biosci 12 (2007) 1450–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Cheng B, Zhao S, Luo J, Sprague E, Bonewald LF, Jiang JX, Expression of functional gap junctions and regulation by fluid flow in osteocyte-like MLO-Y4 cells, J Bone Miner Res 16(2) (2001) 249–59. [DOI] [PubMed] [Google Scholar]
- [45].Alford AI, Jacobs CR, Donahue HJ, Oscillating fluid flow regulates gap junction communication in osteocytic MLO-Y4 cells by an ERK1/2 MAP kinase-dependent mechanism, Bone 33(1) (2003) 64–70. [DOI] [PubMed] [Google Scholar]
- [46].Plotkin LI, Bellido T, Bisphosphonate-induced, hemichannel-mediated, anti-apoptosis through the Src/ERK pathway: a gap junction-independent action of connexin43, Cell Commun Adhes 8(4-6) (2001) 377–82. [DOI] [PubMed] [Google Scholar]
- [47].Plotkin LI, Manolagas SC, Bellido T, Transduction of cell survival signals by connexin43 hemichannels, J Biol Chem 277(10) (2002) 8648–57. [DOI] [PubMed] [Google Scholar]
- [48].Marotti G, The structure of bone tissues and the cellular control of their deposition, Ital J Anat Embryol 101(4) (1996) 25–79. [PubMed] [Google Scholar]
- [49].Bonewald LF, Johnson ML, Osteocytes, mechanosensing and Wnt signaling, Bone 42(4) (2008) 606–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Aarden EM, Burger EH, Nijweide PJ, Function of osteocytes in bone, J Cell Biochem 55(3) (1994) 287–299. [DOI] [PubMed] [Google Scholar]
- [51].Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-Hora M, Feng JQ, Bonewald LF, Kodama T, Wutz A, Wagner EF, Penninger JM, Takayanagi H, Evidence for osteocyte regulation of bone homeostasis through RANKL expression, Nat. Med 17(10) (2011) 1231–1234. [DOI] [PubMed] [Google Scholar]
- [52].Xiong J, Onal M, Jilka RL, Weinstein RS, Manolagas SC, O’brien CA, Matrix-embedded cells control osteoclast formation, Nat. Med 17(10) (2011) 1235–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Feng X, Teitelbaum SL, Osteoclasts: New Insights, Bone Res 1(1) (2013) 11–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Teitelbaum SL, Ross FP, Genetic regulation of osteoclast development and function, Nat Rev Genet 4(8) (2003) 638–49. [DOI] [PubMed] [Google Scholar]
- [55].Kramer I, Halleux C, Keller H, Pegurri M, Gooi JH, Weber PB, Feng JQ, Bonewald LF, Kneissel M, Osteocyte Wnt/beta-catenin signaling is required for normal bone homeostasis, Mol Cell Biol 30(12) (2010) 3071–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Ikeda T, Utsuyama M, Hirokawa K, Expression profiles of receptor activator of nuclear factor kappaB ligand, receptor activator of nuclear factor kappaB, and osteoprotegerin messenger RNA in aged and ovariectomized rat bones, J Bone Miner Res 16(8) (2001) 1416–25. [DOI] [PubMed] [Google Scholar]
- [57].Udagawa N, Takahashi N, Yasuda H, Mizuno A, Itoh K, Ueno Y, Shinki T, Gillespie MT, Martin TJ, Higashio K, Suda T, Osteoprotegerin produced by osteoblasts is an important regulator in osteoclast development and function, Endocrinology 141(9) (2000) 3478–84. [DOI] [PubMed] [Google Scholar]
- [58].Khosla S, Minireview: the OPG/RANKL/RANK system, Endocrinology 142(12) (2001) 5050–5. [DOI] [PubMed] [Google Scholar]
- [59].Tu X, Delgado-Calle J, Condon KW, Maycas M, Zhang H, Carlesso N, Taketo MM, Burr DB, Plotkin LI, Bellido T, Osteocytes mediate the anabolic actions of canonical Wnt/beta-catenin signaling in bone, Proc Natl Acad Sci U S A 112(5) (2015) E478–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Baron R, Rawadi G, Targeting the Wnt/beta-catenin pathway to regulate bone formation in the adult skeleton, Endocrinology 148(6) (2007) 2635–43. [DOI] [PubMed] [Google Scholar]
- [61].Tamai K, Semenov M, Kato Y, Spokony R, Liu C, Katsuyama Y, Hess F, Saint-Jeannet JP, He X, LDL-receptor-related proteins in Wnt signal transduction, Nature 407(6803) (2000) 530–5. [DOI] [PubMed] [Google Scholar]
- [62].Pinson KI, Brennan J, Monkley S, Avery BJ, Skarnes WC, An LDL-receptor-related protein mediates Wnt signalling in mice, Nature 407(6803) (2000) 535–8. [DOI] [PubMed] [Google Scholar]
- [63].Winkler DG, Sutherland MK, Geoghegan JC, Yu C, Hayes T, Skonier JE, Shpektor D, Jonas M, Kovacevich BR, Staehling-Hampton K, Appleby M, Brunkow ME, Latham JA, Osteocyte control of bone formation via sclerostin, a novel BMP antagonist, EMBO J 22(23) (2003) 6267–6276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Li J, Sarosi I, Cattley RC, Pretorius J, Asuncion F, Grisanti M, Morony S, Adamu S, Geng Z, Qiu W, Kostenuik P, Lacey DL, Simonet WS, Bolon B, Qian X, Shalhoub V, Ominsky MS, Zhu Ke H, Li X, Richards WG, Dkk1-mediated inhibition of Wnt signaling in bone results in osteopenia, Bone 39(4) (2006) 754–66. [DOI] [PubMed] [Google Scholar]
- [65].Choi HY, Dieckmann M, Herz J, Niemeier A, Lrp4, a novel receptor for Dickkopf 1 and sclerostin, is expressed by osteoblasts and regulates bone growth and turnover in vivo, PLoS One 4(11) (2009) e7930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Baron R, Kneissel M, WNT signaling in bone homeostasis and disease: from human mutations to treatments, Nat Med 19(2) (2013) 179–92. [DOI] [PubMed] [Google Scholar]
- [67].Tu X, Rhee Y, Condon KW, Bivi N, Allen MR, Dwyer D, Stolina M, Turner CH, Robling AG, Plotkin LI, Bellido T, Sost downregulation and local Wnt signaling are required for the osteogenic response to mechanical loading, Bone 50(1) (2012) 209–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Robling AG, Niziolek PJ, Baldridge LA, Condon KW, Allen MR, Alam I, Mantila SM, Gluhak-Heinrich J, Bellido TM, Harris SE, Turner CH, Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin, J Biol. Chem 283(9) (2008) 5866–5875. [DOI] [PubMed] [Google Scholar]
- [69].Lin C, Jiang X, Dai Z, Guo X, Weng T, Wang J, Li Y, Feng G, Gao X, He L, Sclerostin mediates bone response to mechanical unloading through antagonizing Wnt/beta-catenin signaling, J Bone Miner Res 24(10) (2009) 1651–61. [DOI] [PubMed] [Google Scholar]
- [70].Ke HZ, Richards WG, Li X, Ominsky MS, Sclerostin and Dickkopf-1 as therapeutic targets in bone diseases, Endocr. Rev 33(5) (2012) 747–783. [DOI] [PubMed] [Google Scholar]
- [71].Rhee Y, Allen MR, Condon K, Lezcano V, Ronda AC, Galli C, Olivos N, Passeri G, O’Brien CA, Bivi N, Plotkin LI, Bellido T, PTH receptor signaling in osteocytes governs periosteal bone formation and intracortical remodeling, J Bone Miner Res 26(5) (2011) 1035–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Keller H, Kneissel M, SOST is a target gene for PTH in bone, Bone 37(2) (2005) 148–58. [DOI] [PubMed] [Google Scholar]
- [73].Bellido T, Saini V, Pajevic PD, Effects of PTH on osteocyte function, Bone 54(2) (2013) 250–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Bohatyrewicz A, Burgdoerfer H, [Current principles of urological management of patients with injuries of the spine and spinal cord], Pol Tyg Lek 44(43-45) (1989) 937–40. [PubMed] [Google Scholar]
- [75].Yin J, Hao Z, Ma Y, Liao S, Li X, Fu J, Wu Y, Shen J, Zhang P, Li X, Wang H, Concomitant activation of the PI3K/Akt and ERK1/2 signalling is involved in cyclic compressive force-induced IL-6 secretion in MLO-Y4 cells, Cell Biol Int 38(5) (2014) 591–8. [DOI] [PubMed] [Google Scholar]
- [76].Kennedy OD, Laudier DM, Majeska RJ, Sun HB, Schaffler MB, Osteocyte apoptosis is required for production of osteoclastogenic signals following bone fatigue in vivo, Bone 64 (2014) 132–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Sheng MH, Lau KH, Baylink DJ, Role of Osteocyte-derived Insulin-Like Growth Factor I in Developmental Growth, Modeling, Remodeling, and Regeneration of the Bone, J Bone Metab 21(1) (2014) 41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Caballero-Alias AM, Loveridge N, Pitsillides A, Parker M, Kaptoge S, Lyon A, Reeve J, Osteocytic expression of constitutive NO synthase isoforms in the femoral neck cortex: a case-control study of intracapsular hip fracture, J Bone Miner Res 20(2) (2005) 268–73. [DOI] [PubMed] [Google Scholar]
- [79].Cheng B, Kato Y, Zhao S, Luo J, Sprague E, Bonewald LF, Jiang JX, PGE(2) is essential for gap junction-mediated intercellular communication between osteocyte-like MLO-Y4 cells in response to mechanical strain, Endocrinology 142(8) (2001) 3464–73. [DOI] [PubMed] [Google Scholar]
- [80].Schaffler MB, Cheung WY, Majeska R, Kennedy O, Osteocytes: master orchestrators of bone, Calcif Tissue Int 94(1) (2014) 5–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Heino TJ, Hentunen TA, Vaananen HK, Osteocytes inhibit osteoclastic bone resorption through transforming growth factor-beta: enhancement by estrogen, J Cell Biochem 85(1) (2002) 185–97. [DOI] [PubMed] [Google Scholar]
- [82].Lau KH, Baylink DJ, Zhou XD, Rodriguez D, Bonewald LF, Li Z, Ruffoni D, Muller R, Kesavan C, Sheng MH, Osteocyte-derived insulin-like growth factor I is essential for determining bone mechanosensitivity, Am J Physiol Endocrinol Metab 305(2) (2013) E271–81. [DOI] [PubMed] [Google Scholar]
- [83].Quarles LD, Skeletal secretion of FGF-23 regulates phosphate and vitamin D metabolism, Nat Rev Endocrinol 8(5) (2012) 276–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Barragan-Adjemian C, Nicolella D, Dusevich V, Dallas MR, Eick JD, Bonewald LF, Mechanism by which MLO-A5 late osteoblasts/early osteocytes mineralize in culture: similarities with mineralization of lamellar bone, Calcif Tissue Int 79(5) (2006) 340–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Liu S, Zhou J, Tang W, Jiang X, Rowe DW, Quarles LD, Pathogenic role of Fgf23 in Hyp mice, Am J Physiol Endocrinol Metab 291(1) (2006) E38–49. [DOI] [PubMed] [Google Scholar]
- [86].Nampei A, Hashimoto J, Hayashida K, Tsuboi H, Shi K, Tsuji I, Miyashita H, Yamada T, Matsukawa N, Matsumoto M, Morimoto S, Ogihara T, Ochi T, Yoshikawa H, Matrix extracellular phosphoglycoprotein (MEPE) is highly expressed in osteocytes in human bone, J Bone Miner Metab 22(3) (2004) 176–84. [DOI] [PubMed] [Google Scholar]
- [87].Thompson DL, Sabbagh Y, Tenenhouse HS, Roche PC, Drezner MK, Salisbury JL, Grande JP, Poeschla EM, Kumar R, Ontogeny of Phex/PHEX protein expression in mouse embryo and subcellular localization in osteoblasts, J Bone Miner Res 17(2) (2002) 311–20. [DOI] [PubMed] [Google Scholar]
- [88].Ruchon AF, Tenenhouse HS, Marcinkiewicz M, Siegfried G, Aubin JE, DesGroseillers L, Crine P, Boileau G, Developmental expression and tissue distribution of Phex protein: effect of the Hyp mutation and relationship to bone markers, J Bone Miner Res 15(8) (2000) 1440–50. [DOI] [PubMed] [Google Scholar]
- [89].Petersen DN, Tkalcevic GT, Mansolf AL, Rivera-Gonzalez R, Brown TA, Identification of osteoblast/osteocyte factor 45 (OF45), a bone-specific cDNA encoding an RGD-containing protein that is highly expressed in osteoblasts and osteocytes, J Biol Chem 275(46) (2000) 36172–80. [DOI] [PubMed] [Google Scholar]
- [90].Argiro L, Desbarats M, Glorieux FH, Ecarot B, Mepe, the gene encoding a tumor-secreted protein in oncogenic hypophosphatemic osteomalacia, is expressed in bone, Genomics 74(3) (2001) 342–51. [DOI] [PubMed] [Google Scholar]
- [91].Fisher LW, Fedarko NS, Six genes expressed in bones and teeth encode the current members of the SIBLING family of proteins, Connect Tissue Res 44Suppl 1 (2003) 33–40. [PubMed] [Google Scholar]
- [92].Schaffler MB, Kennedy OD, Osteocyte signaling in bone, Curr Osteoporos Rep 10(2) (2012) 118–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Feng JQ, Huang H, Lu Y, Ye L, Xie Y, Tsutsui TW, Kunieda T, Castranio T, Scott G, Bonewald LB, Mishina Y, The Dentin matrix protein 1 (Dmp1) is specifically expressed in mineralized, but not soft, tissues during development, J Dent Res 82(10) (2003) 776–80. [DOI] [PubMed] [Google Scholar]
- [94].He G, George A, Dentin matrix protein 1 immobilized on type I collagen fibrils facilitates apatite deposition in vitro, J Biol Chem 279(12) (2004) 11649–56. [DOI] [PubMed] [Google Scholar]
- [95].Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan B, Yu X, Rauch F, Davis SI, Zhang S, Rios H, Drezner MK, Quarles LD, Bonewald LF, White KE, Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism, Nat Genet 38(11) (2006) 1310–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Zhang R, Lu Y, Ye L, Yuan B, Yu S, Qin C, Xie Y, Gao T, Drezner MK, Bonewald LF, Feng JQ, Unique roles of phosphorus in endochondral bone formation and osteocyte maturation, J Bone Miner Res 26(5) (2011) 1047–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Zelenchuk LV, Hedge AM, Rowe PS, Age dependent regulation of bone-mass and renal function by the MEPE ASARM-motif, Bone 79 (2015) 131–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. The HYP Consortium, Nat Genet 11(2) (1995) 130–6. [DOI] [PubMed] [Google Scholar]
- [99].Helms JA, Schneider RA, Cranial skeletal biology, Nature 423(6937) (2003) 326–31. [DOI] [PubMed] [Google Scholar]
- [100].Jiang X, Iseki S, Maxson RE, Sucov HM, Morriss-Kay GM, Tissue origins and interactions in the mammalian skull vault, Dev Biol 241(1) (2002) 106–16. [DOI] [PubMed] [Google Scholar]
- [101].Jiang Y, Jahagirdar BN, Reinhardt RL, Schwartz RE, Keene CD, Ortiz-Gonzalez XR, Reyes M, Lenvik T, Lund T, Blackstad M, Du J, Aldrich S, Lisberg A, Low WC, Largaespada DA, Verfaillie CM, Pluripotency of mesenchymal stem cells derived from adult marrow, Nature 418(6893) (2002) 41–49. [DOI] [PubMed] [Google Scholar]
- [102].Kuratani S, Cephalic neural crest cells and the evolution of craniofacial structures in vertebrates: morphological and embryological significance of the premandibular-mandibular boundary, Zoology (Jena) 108(1) (2005) 13–25. [DOI] [PubMed] [Google Scholar]
- [103].Franceschi RTG, C., ; Wilson CG, Osteoblasts of Craniofacial bone, in: McCauley LKS, M.J. (Ed.), Mineralized tissues in oral and craniofacial science. Biological principles and clinical correlates, Wiley-Blackwell, Ames, Iowa, USA., 2012. [Google Scholar]
- [104].Nanci AM, P., Embryology of craniofacial bones, in: McCauley LKS, M.J. (Ed.), Mineralized tissues in oral and craniofacial science. Biological principles and clinical correlates, Wiley-Blackwell, Ames, Iowa, USA., 2012. [Google Scholar]
- [105].Wei X, Thomas N, Hatch NE, Hu M, Liu F, Postnatal Craniofacial Skeletal Development of Female C57BL/6NCrl Mice, Front Physiol 8 (2017) 697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Goldberg M, Embryology and Development: Mandible, Maxillary, Deciduous and Permanent Teeth, JSM Dentistry 7(1) (2019) 1116. [Google Scholar]
- [107].Rengasamy Venugopalan S, Van Otterloo E, The Skull’s Girder: A Brief Review of the Cranial Base, J Dev Biol 9(1) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Doro D, Liu A, Grigoriadis AE, Liu KJ, The Osteogenic Potential of the Neural Crest Lineage May Contribute to Craniosynostosis, Mol Syndromol 10(1-2) (2019) 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Quarto N, Wan DC, Kwan MD, Panetta NJ, Li S, Longaker MT, Origin matters: differences in embryonic tissue origin and Wnt signaling determine the osteogenic potential and healing capacity of frontal and parietal calvarial bones, J Bone Miner Res 25(7) (2010) 1680–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Bonewald L, Cell biology of craniofacial bone: osteocytes, in: McCauley LKS, M.J. (Ed.), Mineralized tissues in oral and craniofacial science. Biological principles and clinical correlates, Wiley-Blackwell, Ames, Iowa, USA., 2012. [Google Scholar]
- [111].Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS, Golstein P, Green DR, Hengartner M, Knight RA, Kumar S, Lipton SA, Malorni W, Nunez G, Peter ME, Tschopp J, Yuan J, Piacentini M, Zhivotovsky B, Melino G, D. Nomenclature Committee on Cell, Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009, Cell Death Differ 16(1) (2009) 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Kroemer G, El-Deiry WS, Golstein P, Peter ME, Vaux D, Vandenabeele P, Zhivotovsky B, Blagosklonny MV, Malorni W, Knight RA, Piacentini M, Nagata S, Melino G, Nomenclature D Committee on Cell, Classification of cell death: recommendations of the Nomenclature Committee on Cell Death, Cell Death Differ 12Suppl 2 (2005) 1463–7. [DOI] [PubMed] [Google Scholar]
- [113].Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, Gottlieb E, Green DR, Hengartner MO, Kepp O, Knight RA, Kumar S, Lipton SA, Lu X, Madeo F, Malorni W, Mehlen P, Nunez G, Peter ME, Piacentini M, Rubinsztein DC, Shi Y, Simon HU, Vandenabeele P, White E, Yuan J, Zhivotovsky B, Melino G, Kroemer G, Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012, Cell Death Differ 19(1) (2012) 107–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW, Annicchiarico-Petruzzelli M, Antonov AV, Arama E, Baehrecke EH, Barlev NA, Bazan NG, Bernassola F, Bertrand MJM, Bianchi K, Blagosklonny MV, Blomgren K, Borner C, Boya P, Brenner C, Campanella M, Candi E, Carmona-Gutierrez D, Cecconi F, Chan FK, Chandel NS, Cheng EH, Chipuk JE, Cidlowski JA, Ciechanover A, Cohen GM, Conrad M, Cubillos-Ruiz JR, Czabotar PE, D’Angiolella V, Dawson TM, Dawson VL, De Laurenzi V, De Maria R, Debatin KM, DeBerardinis RJ, Deshmukh M, Di Daniele N, Di Virgilio F, Dixit VM, Dixon SJ, Duckett CS, Dynlacht BD, El-Deiry WS, Elrod JW, Fimia GM, Fulda S, Garcia-Saez AJ, Garg AD, Garrido C, Gavathiotis E, Golstein P, Gottlieb E, Green DR, Greene LA, Gronemeyer H, Gross A, Hajnoczky G, Hardwick JM, Harris IS, Hengartner MO, Hetz C, Ichijo H, Jaattela M, Joseph B, Jost PJ, Juin PP, Kaiser WJ, Karin M, Kaufmann T, Kepp O, Kimchi A, Kitsis RN, Klionsky DJ, Knight RA, Kumar S, Lee SW, Lemasters JJ, Levine B, Linkermann A, Lipton SA, Lockshin RA, Lopez-Otin C, Lowe SW, Luedde T, Lugli E, MacFarlane M, Madeo F, Malewicz M, Malorni W, Manic G, Marine JC, Martin SJ, Martinou JC, Medema JP, Mehlen P, Meier P, Melino S, Miao EA, Molkentin JD, Moll UM, Munoz-Pinedo C, Nagata S, Nunez G, Oberst A, Oren M, Overholtzer M, Pagano M, Panaretakis T, Pasparakis M, Penninger JM, Pereira DM, Pervaiz S, Peter ME, Piacentini M, Pinton P, Prehn JHM, Puthalakath H, Rabinovich GA, Rehm M, Rizzuto R, Rodrigues CMP, Rubinsztein DC, Rudel T, Ryan KM, Sayan E, Scorrano L, Shao F, Shi Y, Silke J, Simon HU, Sistigu A, Stockwell BR, Strasser A, Szabadkai G, Tait SWG, Tang D, Tavernarakis N, Thorburn A, Tsujimoto Y, Turk B, Vanden Berghe T, Vandenabeele P, Vander Heiden MG, Villunger A, Virgin HW, Vousden KH, Vucic D, Wagner EF, Walczak H, Wallach D, Wang Y, Wells JA, Wood W, Yuan J, Zakeri Z, Zhivotovsky B, Zitvogel L, Melino G, Kroemer G, Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018, Cell Death Differ 25(3) (2018) 486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Galluzzi L, Bravo-San Pedro JM, Vitale I, Aaronson SA, Abrams JM, Adam D, Alnemri ES, Altucci L, Andrews D, Annicchiarico-Petruzzelli M, Baehrecke EH, Bazan NG, Bertrand MJ, Bianchi K, Blagosklonny MV, Blomgren K, Borner C, Bredesen DE, Brenner C, Campanella M, Candi E, Cecconi F, Chan FK, Chandel NS, Cheng EH, Chipuk JE, Cidlowski JA, Ciechanover A, Dawson TM, Dawson VL, De Laurenzi V, De Maria R, Debatin KM, Di Daniele N, Dixit VM, Dynlacht BD, El-Deiry WS, Fimia GM, Flavell RA, Fulda S, Garrido C, Gougeon ML, Green DR, Gronemeyer H, Hajnoczky G, Hardwick JM, Hengartner MO, Ichijo H, Joseph B, Jost PJ, Kaufmann T, Kepp O, Klionsky DJ, Knight RA, Kumar S, Lemasters JJ, Levine B, Linkermann A, Lipton SA, Lockshin RA, Lopez-Otin C, Lugli E, Madeo F, Malorni W, Marine JC, Martin SJ, Martinou JC, Medema JP, Meier P, Melino S, Mizushima N, Moll U, Munoz-Pinedo C, Nunez G, Oberst A, Panaretakis T, Penninger JM, Peter ME, Piacentini M, Pinton P, Prehn JH, Puthalakath H, Rabinovich GA, Ravichandran KS, Rizzuto R, Rodrigues CM, Rubinsztein DC, Rudel T, Shi Y, Simon HU, Stockwell BR, Szabadkai G, Tait SW, Tang HL, Tavernarakis N, Tsujimoto Y, Vanden Berghe T, Vandenabeele P, Villunger A, Wagner EF, Walczak H, White E, Wood WG, Yuan J, Zakeri Z, Zhivotovsky B, Melino G, Kroemer G, Essential versus accessory aspects of cell death: recommendations of the NCCD 2015, Cell Death Differ 22(1) (2015) 58–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Zelenay S, Reis e Sousa C, Adaptive immunity after cell death, Trends Immunol 34(7) (2013) 329–35. [DOI] [PubMed] [Google Scholar]
- [117].Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ, Circulating mitochondrial DAMPs cause inflammatory responses to injury, Nature 464(7285) (2010) 104–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Galluzzi L, Kepp O, Krautwald S, Kroemer G, Linkermann A, Molecular mechanisms of regulated necrosis, Semin Cell Dev Biol 35 (2014) 24–32. [DOI] [PubMed] [Google Scholar]
- [119].Fuchs Y, Steller H, Programmed cell death in animal development and disease, Cell 147(4) (2011) 742–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Galluzzi L, Maiuri MC, Vitale I, Zischka H, Castedo M, Zitvogel L, Kroemer G, Cell death modalities: classification and pathophysiological implications, Cell Death Differ 14(7) (2007) 1237–43. [DOI] [PubMed] [Google Scholar]
- [121].Kerr JF, Wyllie AH, Currie AR, Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics, Br J Cancer 26(4) (1972) 239–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Kroemer G, Galluzzi L, Brenner C, Mitochondrial membrane permeabilization in cell death, Physiol Rev 87(1) (2007) 99–163. [DOI] [PubMed] [Google Scholar]
- [123].Danial NN, Korsmeyer SJ, Cell death: critical control points, Cell 116(2) (2004) 205–19. [DOI] [PubMed] [Google Scholar]
- [124].Elmore S, Apoptosis: a review of programmed cell death, Toxicol Pathol 35(4) (2007) 495–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Deter RL, De Duve C, Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes, J Cell Biol 33(2) (1967) 437–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Glick D, Barth S, Macleod KF, Autophagy: cellular and molecular mechanisms, J Pathol 221(1) (2010) 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, Choi AM, Chu CT, Codogno P, Colombo MI, Cuervo AM, Debnath J, Deretic V, Dikic I, Eskelinen EL, Fimia GM, Fulda S, Gewirtz DA, Green DR, Hansen M, Harper JW, Jaattela M, Johansen T, Juhasz G, Kimmelman AC, Kraft C, Ktistakis NT, Kumar S, Levine B, Lopez-Otin C, Madeo F, Martens S, Martinez J, Melendez A, Mizushima N, Munz C, Murphy LO, Penninger JM, Piacentini M, Reggiori F, Rubinsztein DC, Ryan KM, Santambrogio L, Scorrano L, Simon AK, Simon HU, Simonsen A, Tavernarakis N, Tooze SA, Yoshimori T, Yuan J, Yue Z, Zhong Q, Kroemer G, Molecular definitions of autophagy and related processes, EMBO J 36(13) (2017) 1811–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Clarke PG, Developmental cell death: morphological diversity and multiple mechanisms, Anat Embryol (Berl) 181(3) (1990) 195–213. [DOI] [PubMed] [Google Scholar]
- [129].Baehrecke EH, Autophagy: dual roles in life and death?, Nat Rev Mol Cell Biol 6(6) (2005) 505–10. [DOI] [PubMed] [Google Scholar]
- [130].Majno G, Joris I, Apoptosis, oncosis, and necrosis. An overview of cell death, Am J Pathol 146(1) (1995) 3–15. [PMC free article] [PubMed] [Google Scholar]
- [131].Schwartz SM, Bennett MR, Death by any other name, Am J Pathol 147(2) (1995) 229–34. [PMC free article] [PubMed] [Google Scholar]
- [132].Fink SL, Cookson BT, Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells, Infect Immun 73(4) (2005) 1907–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Levin S, Apoptosis, necrosis, or oncosis: what is your diagnosis? A report from the Cell Death Nomenclature Committee of the Society of Toxicologic Pathologists, Toxicol Sci 41(2) (1998) 155–6. [DOI] [PubMed] [Google Scholar]
- [134].Nicotera P, Bernassola F, Melino G, Nitric oxide (NO), a signaling molecule with a killer soul, Cell Death Differ 6(10) (1999) 931–3. [DOI] [PubMed] [Google Scholar]
- [135].Golstein P, Kroemer G, Cell death by necrosis: towards a molecular definition, Trends Biochem Sci 32(1) (2007) 37–43. [DOI] [PubMed] [Google Scholar]
- [136].Trump BF, Berezesky IK, Chang SH, Phelps PC, The pathways of cell death: oncosis, apoptosis, and necrosis, Toxicol Pathol 25(1) (1997) 82–8. [DOI] [PubMed] [Google Scholar]
- [137].Festjens N, Vanden Berghe T, Vandenabeele P, Necrosis, a well-orchestrated form of cell demise: signalling cascades, important mediators and concomitant immune response, Biochim Biophys Acta 1757(9-10) (2006) 1371–87. [DOI] [PubMed] [Google Scholar]
- [138].Dhuriya YK, Sharma D, Necroptosis: a regulated inflammatory mode of cell death, J Neuroinflammation 15(1) (2018) 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Christofferson DE, Yuan J, Necroptosis as an alternative form of programmed cell death, Curr Opin Cell Biol 22(2) (2010) 263–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Pasparakis M, Vandenabeele P, Necroptosis and its role in inflammation, Nature 517(7534) (2015) 311–20. [DOI] [PubMed] [Google Scholar]
- [141].Choi ME, Price DR, Ryter SW, Choi AMK, Necroptosis: a crucial pathogenic mediator of human disease, JCI Insight 4(15) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Degterev A, Ofengeim D, Yuan J, Targeting RIPK1 for the treatment of human diseases, Proc Natl Acad Sci U S A 116(20) (2019) 9714–9722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J, Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury, Nat Chem Biol 1(2) (2005) 112–9. [DOI] [PubMed] [Google Scholar]
- [144].Grootjans S, Vanden Berghe T, Vandenabeele P, Initiation and execution mechanisms of necroptosis: an overview, Cell Death Differ 24(7) (2017) 1184–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Ofengeim D, Yuan J, Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death, Nat Rev Mol Cell Biol 14(11) (2013) 727–36. [DOI] [PubMed] [Google Scholar]
- [146].Dondelinger Y, Darding M, Bertrand MJ, Walczak H, Poly-ubiquitination in TNFR1-mediated necroptosis, Cell Mol Life Sci 73(11-12) (2016) 2165–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Pop C, Oberst A, Drag M, Van Raam BJ, Riedl SJ, Green DR, Salvesen GS, FLIP(L) induces caspase 8 activity in the absence of interdomain caspase 8 cleavage and alters substrate specificity, Biochem J 433(3) (2011) 447–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].O’Donnell MA, Perez-Jimenez E, Oberst A, Ng A, Massoumi R, Xavier R, Green DR, Ting AT, Caspase 8 inhibits programmed necrosis by processing CYLD, Nat Cell Biol 13(12) (2011) 1437–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, Damko E, Moquin D, Walz T, McDermott A, Chan FK, Wu H, The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis, Cell 150(2) (2012) 339–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Weber K, Roelandt R, Bruggeman I, Estornes Y, Vandenabeele P, Nuclear RIPK3 and MLKL contribute to cytosolic necrosome formation and necroptosis, Communications Biology 1(1) (2018) 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X, Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha, Cell 137(6) (2009) 1100–11. [DOI] [PubMed] [Google Scholar]
- [152].Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK, Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation, Cell 137(6) (2009) 1112–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, Hedrick SM, Gerber SA, Lugovskoy A, Yuan J, Identification of RIP1 kinase as a specific cellular target of necrostatins, Nat Chem Biol 4(5) (2008) 313–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Laurien L, Nagata M, Schunke H, Delanghe T, Wiederstein JL, Kumari S, Schwarzer R, Corona T, Kruger M, Bertrand MJM, Kondylis V, Pasparakis M, Autophosphorylation at serine 166 regulates RIP kinase 1-mediated cell death and inflammation, Nat Commun 11(1) (2020) 1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Berger SB, Kasparcova V, Hoffman S, Swift B, Dare L, Schaeffer M, Capriotti C, Cook M, Finger J, Hughes-Earle A, Harris PA, Kaiser WJ, Mocarski ES, Bertin J, Gough PJ, Cutting Edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice, J Immunol 192(12) (2014) 5476–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Ting AT, Tools in the Art of Studying Necroptosis, Methods Mol Biol 1857 (2018) 1–9. [DOI] [PubMed] [Google Scholar]
- [157].Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, Wang X, Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase, Cell 148(1–2) (2012) 213–27. [DOI] [PubMed] [Google Scholar]
- [158].Yoon S, Kovalenko A, Bogdanov K, Wallach D, MLKL, the Protein that Mediates Necroptosis, Also Regulates Endosomal Trafficking and Extracellular Vesicle Generation, Immunity 47(1) (2017) 51–65 e7. [DOI] [PubMed] [Google Scholar]
- [159].Wu J, Huang Z, Ren J, Zhang Z, He P, Li Y, Ma J, Chen W, Zhang Y, Zhou X, Yang Z, Wu SQ, Chen L, Han J, Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis, Cell Res 23(8) (2013) 994–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Rodriguez DA, Weinlich R, Brown S, Guy C, Fitzgerald P, Dillon CP, Oberst A, Quarato G, Low J, Cripps JG, Chen T, Green DR, Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis, Cell Death Differ 23(1) (2016) 76–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Wallach D, Kang TB, Dillon CP, Green DR, Programmed necrosis in inflammation: Toward identification of the effector molecules, Science 352(6281) (2016) aaf2154. [DOI] [PubMed] [Google Scholar]
- [162].Lukens JR, Vogel P, Johnson GR, Kelliher MA, Iwakura Y, Lamkanfi M, Kanneganti TD, RIP1-driven autoinflammation targets IL-1alpha independently of inflammasomes and RIP3, Nature 498(7453) (2013) 224–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Chan FK, Luz NF, Moriwaki K, Programmed necrosis in the cross talk of cell death and inflammation, Annu Rev Immunol 33 (2015) 79–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Silke J, Rickard JA, Gerlic M, The diverse role of RIP kinases in necroptosis and inflammation, Nat Immunol 16(7) (2015) 689–97. [DOI] [PubMed] [Google Scholar]
- [165].Newton K, RIPK1 and RIPK3: critical regulators of inflammation and cell death, Trends Cell Biol 25(6) (2015) 347–53. [DOI] [PubMed] [Google Scholar]
- [166].Webster JD, Kwon YC, Park S, Zhang H, Corr N, Ljumanovic N, Adedeji AO, Varfolomeev E, Goncharov T, Preston J, Santagostino SF, Patel S, Xu M, Maher J, McKenzie BS, Vucic D, RIP1 kinase activity is critical for skin inflammation but not for viral propagation, J Leukoc Biol 107(6) (2020) 941–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Kanayama A, Seth RB, Sun L, Ea CK, Hong M, Shaito A, Chiu YH, Deng L, Chen ZJ, TAB2 and TAB3 activate the NF-kappaB pathway through binding to polyubiquitin chains, Mol Cell 15(4) (2004) 535–48. [DOI] [PubMed] [Google Scholar]
- [168].Zhao H, Jaffer T, Eguchi S, Wang Z, Linkermann A, Ma D, Role of necroptosis in the pathogenesis of solid organ injury, Cell Death Dis 6 (2015) e1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Baidya R, Crawford DHG, Gautheron J, Wang H, Bridle KR, Necroptosis in Hepatosteatotic Ischaemia-Reperfusion Injury, Int J Mol Sci 21(16) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Micheau O, Tschopp J, Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes, Cell 114(2) (2003) 181–90. [DOI] [PubMed] [Google Scholar]
- [171].Declercq W, Vanden Berghe T, Vandenabeele P, RIP kinases at the crossroads of cell death and survival, Cell 138(2) (2009) 229–32. [DOI] [PubMed] [Google Scholar]
- [172].Tummers B, Green DR, Caspase-8: regulating life and death, Immunol Rev 277(1) (2017) 76–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G, Molecular mechanisms of necroptosis: an ordered cellular explosion, Nat Rev Mol Cell Biol 11(10) (2010) 700–14. [DOI] [PubMed] [Google Scholar]
- [174].Kearney CJ, Martin SJ, An Inflammatory Perspective on Necroptosis, Mol Cell 65(6) (2017) 965–973. [DOI] [PubMed] [Google Scholar]
- [175].Davidovich P, Kearney CJ, Martin SJ, Inflammatory outcomes of apoptosis, necrosis and necroptosis, Biol Chem 395(10) (2014) 1163–71. [DOI] [PubMed] [Google Scholar]
- [176].Khoury MK, Gupta K, Franco SR, Liu B, Necroptosis in the Pathophysiology of Disease, Am J Pathol 190(2) (2020) 272–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Li J, Ke X, Yan F, Lei L, Li H, Necroptosis in the periodontal homeostasis: Signals emanating from dying cells, Oral Dis 24(6) (2018) 900–907. [DOI] [PubMed] [Google Scholar]
- [178].Shi J, Li J, Su W, Zhao S, Li H, Lei L, Loss of periodontal ligament fibroblasts by RIPK3-MLKL-mediated necroptosis in the progress of chronic periodontitis, Sci Rep 9(1) (2019) 2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Ke X, Lei L, Li H, Li H, Yan F, Manipulation of necroptosis by Porphyromonas gingivalis in periodontitis development, Mol Immunol 77 (2016) 8–13. [DOI] [PubMed] [Google Scholar]
- [180].Polykratis A, Martens A, Eren RO, Shirasaki Y, Yamagishi M, Yamaguchi Y, Uemura S, Miura M, Holzmann B, Kollias G, Armaka M, van Loo G, Pasparakis M, A20 prevents inflammasome-dependent arthritis by inhibiting macrophage necroptosis through its ZnF7 ubiquitin-binding domain, Nat Cell Biol 21(6) (2019) 731–742. [DOI] [PubMed] [Google Scholar]
- [181].Jhun J, Lee SH, Kim SY, Ryu J, Kwon JY, Na HS, Jung K, Moon SJ, Cho ML, Min JK, RIPK1 inhibition attenuates experimental autoimmune arthritis via suppression of osteoclastogenesis, J Transl Med 17(1) (2019) 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Ou L, Sun T, Cheng Y, Huang L, Zhan X, Zhang P, Yang J, Zhang Y, Zhou Z, MicroRNA-214 contributes to regulation of necroptosis via targeting ATF4 in diabetes-associated periodontitis, J Cell Biochem 120(9) (2019) 14791–14803. [DOI] [PubMed] [Google Scholar]
- [183].Gupta K, Phan N, Wang Q, Liu B, Necroptosis in cardiovascular disease - a new therapeutic target, J Mol Cell Cardiol 118 (2018) 26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Mizumura K, Maruoka S, Gon Y, Choi AM, Hashimoto S, The role of necroptosis in pulmonary diseases, Respir Investig 54(6) (2016) 407–412. [DOI] [PubMed] [Google Scholar]
- [185].Andreev D, Liu M, Weidner D, Kachler K, Faas M, Gruneboom A, Schlotzer-Schrehardt U, Munoz LE, Steffen U, Grotsch B, Killy B, Kronke G, Luebke AM, Niemeier A, Wehrhan F, Lang R, Schett G, Bozec A, Osteocyte necrosis triggers osteoclast-mediated bone loss through macrophage-inducible C-type lectin, J Clin Invest 130(9) (2020) 4811–4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Prasadam I, Farnaghi S, Feng JQ, Gu W, Perry S, Crawford R, Xiao Y, Impact of extracellular matrix derived from osteoarthritis subchondral bone osteoblasts on osteocytes: role of integrinbeta1 and focal adhesion kinase signaling cues, Arthritis Res Ther 15(5) (2013) R150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Metzger CE, Narayanan SA, The Role of Osteocytes in Inflammatory Bone Loss, Front Endocrinol (Lausanne) 10 (2019) 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Claes L, Recknagel S, Ignatius A, Fracture healing under healthy and inflammatory conditions, Nat Rev Rheumatol 8(3) (2012) 133–43. [DOI] [PubMed] [Google Scholar]
- [189].Ross MH, Esser AK, Fox GC, Schmieder AH, Yang X, Hu G, Pan D, Su X, Xu Y, Novack DV, Walsh T, Colditz GA, Lukaszewicz GH, Cordell E, Novack J, Fitzpatrick JAJ, Waning DL, Mohammad KS, Guise TA, Lanza GM, Weilbaecher KN, Bone-Induced Expression of Integrin beta3 Enables Targeted Nanotherapy of Breast Cancer Metastases, Cancer Res 77(22) (2017) 6299–6312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Wang W, Sarazin BA, Kornilowicz G, Lynch ME, Mechanically-Loaded Breast Cancer Cells Modify Osteocyte Mechanosensitivity by Secreting Factors That Increase Osteocyte Dendrite Formation and Downstream Resorption, Front Endocrinol (Lausanne) 9 (2018) 352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Hemmatian H, Bakker AD, Klein-Nulend J, van Lenthe GH, Aging, Osteocytes, and Mechanotransduction, Curr Osteoporos Rep 15(5) (2017) 401–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Holguin N, Brodt MD, Silva MJ, Activation of Wnt Signaling by Mechanical Loading Is Impaired in the Bone of Old Mice, J Bone Miner Res 31(12) (2016) 2215–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Marx RE, Osteoradionecrosis: a new concept of its pathophysiology, J Oral Maxillofac Surg 41(5) (1983) 283–8. [DOI] [PubMed] [Google Scholar]
- [194].Fu YW, He HB, Apoptosis of periodontium cells in streptozototocin- and ligature-induced experimental diabetic periodontitis in rats, Acta Odontol Scand 71(5) (2013) 1206–15. [DOI] [PubMed] [Google Scholar]
- [195].Jun HK, Jung YJ, Choi BK, Treponema denticola, Porphyromonas gingivalis, and Tannerella forsythia induce cell death and release of endogenous danger signals, Arch Oral Biol 73 (2017) 72–78. [DOI] [PubMed] [Google Scholar]
- [196].Huang X, Xie M, Xie Y, Mei F, Lu X, Li X, Chen L, The roles of osteocytes in alveolar bone destruction in periodontitis, Journal of Translational Medicine 18(1) (2020) 479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Khan AA, Morrison A, Kendler DL, Rizzoli R, Hanley DA, Felsenberg D, McCauley LK, O’Ryan F, Reid IR, Ruggiero SL, Taguchi A, Tetradis S, Watts NB, Brandi ML, Peters E, Guise T, Eastell R, Cheung AM, Morin SN, Masri B, Cooper C, Morgan SL, Obermayer-Pietsch B, Langdahl BL, Dabagh RA, Davison KS, Sandor GK, Josse RG, Bhandari M, El Rabbany M, Pierroz DD, Sulimani R, Saunders DP, Brown JP, Compston J, International J Task Force on Osteonecrosis of the, Case-Based Review of Osteonecrosis of the Jaw (ONJ) and Application of the International Recommendations for Management From the International Task Force on ONJ, J Clin Densitom 20(1) (2017) 8–24. [DOI] [PubMed] [Google Scholar]
- [198].Dodd JS, Raleigh JA, Gross TS, Osteocyte hypoxia: a novel mechanotransduction pathway, Am J Physiol 277(3) (1999) C598–602. [DOI] [PubMed] [Google Scholar]
- [199].Tomkinson A, Reeve J, Shaw RW, Noble BS, The death of osteocytes via apoptosis accompanies estrogen withdrawal in human bone, J Clin. Endocrinol. Metab 82(9) (1997) 3128–3135. [DOI] [PubMed] [Google Scholar]
- [200].Tomkinson A, Gevers EF, Wit JM, Reeve J, Noble BS, The role of estrogen in the control of rat osteocyte apoptosis, J Bone Miner. Res 13(8) (1998) 1243–1250. [DOI] [PubMed] [Google Scholar]
- [201].Emerton KB, Hu B, Woo AA, Sinofsky A, Hernandez C, Majeska RJ, Jepsen KJ, Schaffler MB, Osteocyte apoptosis and control of bone resorption following ovariectomy in mice, Bone 46(3) (2010) 577–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Verborgt O, Tatton NA, Majeska RJ, Schaffler MB, Spatial distribution of Bax and Bcl-2 in osteocytes after bone fatigue: complementary roles in bone remodeling regulation?, J Bone Miner Res 17(5) (2002) 907–14. [DOI] [PubMed] [Google Scholar]
- [203].Noble BS, Reeve J, Osteocyte function, osteocyte death and bone fracture resistance, Mol. Cell Endocrinol 159(1-2) (2000) 7–13. [DOI] [PubMed] [Google Scholar]
- [204].Cardoso L, Herman BC, Verborgt O, Laudier D, Majeska RJ, Schaffler MB, Osteocyte apoptosis controls activation of intracortical resorption in response to bone fatigue, J Bone Miner Res 24(4) (2009) 597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Aguirre JI, Plotkin LI, Stewart SA, Weinstein RS, Parfitt AM, Manolagas SC, Bellido T, Osteocyte apoptosis is induced by weightlessness in mice and precedes osteoclast recruitment and bone loss, J Bone Miner. Res 21(4) (2006) 605–615. [DOI] [PubMed] [Google Scholar]
- [206].Cabahug-Zuckerman P, Frikha-Benayed D, Majeska RJ, Tuthill A, Yakar S, Judex S, Schaffler MB, Osteocyte Apoptosis Caused by Hindlimb Unloading is Required to Trigger Osteocyte RANKL Production and Subsequent Resorption of Cortical and Trabecular Bone in Mice Femurs, J Bone Miner Res 31(7) (2016) 1356–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].O’Brien CA, Jia D, Plotkin LI, Bellido T, Powers CC, Stewart SA, Manolagas SC, Weinstein RS, Glucocorticoids act directly on osteoblasts and osteocytes to induce their apoptosis and reduce bone formation and strength, Endocrinology 145(4) (2004) 1835–41. [DOI] [PubMed] [Google Scholar]
- [208].Follet H, Li J, Phipps RJ, Hui S, Condon K, Burr DB, Risedronate and alendronate suppress osteocyte apoptosis following cyclic fatigue loading, Bone 40(4) (2007) 1172–7. [DOI] [PubMed] [Google Scholar]
- [209].Plotkin LI, Weinstein RS, Parfitt AM, Roberson PK, Manolagas SC, Bellido T, Prevention of osteocyte and osteoblast apoptosis by bisphosphonates and calcitonin, J Clin Invest 104(10) (1999) 1363–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Plotkin LI, Mathov I, Aguirre JI, Parfitt AM, Manolagas SC, Bellido T, Mechanical stimulation prevents osteocyte apoptosis: requirement of integrins, Src kinases, and ERKs, Am J Physiol Cell Physiol 289(3) (2005) C633–C643. [DOI] [PubMed] [Google Scholar]
- [211].McKenzie J, Smith C, Karuppaiah K, Langberg J, Silva MJ, Ornitz DM, Osteocyte Death and Bone Overgrowth in Mice Lacking Fibroblast Growth Factor Receptors 1 and 2 in Mature Osteoblasts and Osteocytes, J Bone Miner Res 34(9) (2019) 1660–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Xia X, Kar R, Gluhak-Heinrich J, Yao W, Lane NE, Bonewald LF, Biswas SK, Lo WK, Jiang JX, Glucocorticoid-induced autophagy in osteocytes, J Bone Miner Res 25(11) (2010) 2479–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Yao W, Dai W, Jiang JX, Lane NE, Glucocorticoids and osteocyte autophagy, Bone 54(2) (2013) 279–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Li W, Zhao J, Sun W, Wang H, Pan Y, Wang L, Zhang WB, Osteocytes promote osteoclastogenesis via autophagy-mediated RANKL secretion under mechanical compressive force, Arch Biochem Biophys 694 (2020) 108594. [DOI] [PubMed] [Google Scholar]
- [215].Onal M, Piemontese M, Xiong J, Wang Y, Han L, Ye S, Komatsu M, Selig M, Weinstein RS, Zhao H, Jilka RL, Almeida M, Manolagas SC, O’Brien CA, Suppression of autophagy in osteocytes mimics skeletal aging, J Biol Chem 288(24) (2013) 17432–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Tanida I, Ueno T, Kominami E, LC3 conjugation system in mammalian autophagy, Int J Biochem Cell Biol 36(12) (2004) 2503–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Tsujimoto Y, Shimizu S, Another way to die: autophagic programmed cell death, Cell Death Differ 12Suppl 2 (2005) 1528–34. [DOI] [PubMed] [Google Scholar]
- [218].Mankin HJ, Nontraumatic necrosis of bone (osteonecrosis), N Engl J Med 326(22) (1992) 1473–9. [DOI] [PubMed] [Google Scholar]
- [219].Komori T, Cell Death in Chondrocytes, Osteoblasts, and Osteocytes, Int J Mol Sci 17(12) (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Komori T, Functions of the osteocyte network in the regulation of bone mass, Cell Tissue Res 352(2) (2013) 191–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Zong WX, Thompson CB, Necrotic death as a cell fate, Genes Dev 20(1) (2006) 1–15. [DOI] [PubMed] [Google Scholar]
- [222].Liu-Bryan R, Terkeltaub R, Emerging regulators of the inflammatory process in osteoarthritis, Nat Rev Rheumatol 11(1) (2015) 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Takeuchi O, Akira S, Pattern recognition receptors and inflammation, Cell 140(6) (2010) 805–20. [DOI] [PubMed] [Google Scholar]
- [224].Ibrahim ZA, Armour CL, Phipps S, Sukkar MB, RAGE and TLRs: relatives, friends or neighbours?, Mol Immunol 56(4) (2013) 739–44. [DOI] [PubMed] [Google Scholar]
- [225].Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, Abraham E, Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein, J Biol Chem 279(9) (2004) 7370–7. [DOI] [PubMed] [Google Scholar]
- [226].Cui H, Zhu Y, Jiang D, The RIP1-RIP3 Complex Mediates Osteocyte Necroptosis after Ovariectomy in Rats, PLoS One 11(3) (2016) e0150805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Cui H, Zhu Y, Yang Q, Zhao W, Zhang S, Zhou A, Jiang D, Necrostatin-1 treatment inhibits osteocyte necroptosis and trabecular deterioration in ovariectomized rats, Sci Rep 6 (2016) 33803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Feng M, Zhang R, Gong F, Yang P, Fan L, Ni J, Bi W, Zhang Y, Wang C, Wang K, Protective effects of necrostatin-1 on glucocorticoid-induced osteoporosis in rats, J Steroid Biochem Mol Biol 144Pt B (2014) 455–62. [DOI] [PubMed] [Google Scholar]
- [229].Galluzzi L, Joza N, Tasdemir E, Maiuri MC, Hengartner M, Abrams JM, Tavernarakis N, Penninger J, Madeo F, Kroemer G, No death without life: vital functions of apoptotic effectors, Cell Death Differ 15(7) (2008) 1113–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Allen MR, Burr DB, Mandible matrix necrosis in beagle dogs after 3 years of daily oral bisphosphonate treatment, J Oral Maxillofac. Surg 66(5) (2008) 987–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Enlow DH, Functions of the Haversian system, Am J Anat 110 (1962) 269–305. [DOI] [PubMed] [Google Scholar]
- [232].Frost HM, In vivo osteocyte death, J Bone Joint Surg Am 42-A (1960) 138–43. [PubMed] [Google Scholar]
- [233].Enlow DH, Osteocyte necrosis in normal bone, J Dent Res 45(1) (1966) 213. [DOI] [PubMed] [Google Scholar]
- [234].Huja SS, Fernandez SA, Hill KJ, Li Y, Remodeling dynamics in the alveolar process in skeletally mature dogs, Anat. Rec. A Discov. Mol. Cell Evol. Biol 288(12) (2006) 1243–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Garetto LP, Chen J, Parr JA, Roberts WE, Remodeling dynamics of bone supporting rigidly fixed titanium implants: a histomorphometric comparison in four species including humans, Implant Dent 4(4) (1995) 235–43. [DOI] [PubMed] [Google Scholar]
- [236].Correia Vde F, Caldeira CL, Marques MM, Cytotoxicity evaluation of sodium alendronate on cultured human periodontal ligament fibroblasts, Dent Traumatol 22(6) (2006) 312–7. [DOI] [PubMed] [Google Scholar]
- [237].Moreira MS, Katayama E, Bombana AC, Marques MM, Cytotoxicity analysis of alendronate on cultured endothelial cells and subcutaneous tissue. a pilot study, Dent Traumatol 21(6) (2005) 329–35. [DOI] [PubMed] [Google Scholar]
- [238].Mirza RE, Fang MM, Weinheimer-Haus EM, Ennis WJ, Koh TJ, Sustained inflammasome activity in macrophages impairs wound healing in type 2 diabetic humans and mice, Diabetes 63(3) (2014) 1103–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [239].Zhang Q, Yu W, Lee S, Xu Q, Naji A, Le AD, Bisphosphonate Induces Osteonecrosis of the Jaw in Diabetic Mice via NLRP3/Caspase-1-Dependent IL-1beta Mechanism, J Bone Miner Res 30(12) (2015) 2300–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Graves DT, Cochran D, The contribution of interleukin-1 and tumor necrosis factor to periodontal tissue destruction, J Periodontol 74(3) (2003) 391–401. [DOI] [PubMed] [Google Scholar]
- [241].Schwarzer R, Laurien L, Pasparakis M, New insights into the regulation of apoptosis, necroptosis, and pyroptosis by receptor interacting protein kinase 1 and caspase-8, Curr Opin Cell Biol 63 (2020) 186–193. [DOI] [PubMed] [Google Scholar]
- [242].Sun R, Zhang Y, Lv Q, Liu B, Jin M, Zhang W, He Q, Deng M, Liu X, Li G, Li Y, Zhou G, Xie P, Xie X, Hu J, Duan Z, Toll-like receptor 3 (TLR3) induces apoptosis via death receptors and mitochondria by up-regulating the transactivating p63 isoform alpha (TAP63alpha), J Biol Chem 286(18) (2011) 15918–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [243].Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G, The molecular machinery of regulated cell death, Cell Res 29(5) (2019) 347–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [244].Christofferson DE, Li Y, Hitomi J, Zhou W, Upperman C, Zhu H, Gerber SA, Gygi S, Yuan J, A novel role for RIP1 kinase in mediating TNFalpha production, Cell Death Dis 3 (2012) e320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [245].Rock KL, Kono H, The inflammatory response to cell death, Annu Rev Pathol 3 (2008) 99–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [246].Ru JY, Wang YF, Osteocyte apoptosis: the roles and key molecular mechanisms in resorption-related bone diseases, Cell Death Dis 11(10) (2020) 846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [247].Haanen C, Vermes I, Apoptosis and inflammation, Mediators Inflamm 4(1) (1995) 5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Silva MT, do Vale A, dos Santos NM, Secondary necrosis in multicellular animals: an outcome of apoptosis with pathogenic implications, Apoptosis 13(4) (2008) 463–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Wang L, Wang Y, Han Y, Henderson SC, Majeska RJ, Weinbaum S, Schaffler MB, In situ measurement of solute transport in the bone lacunar-canalicular system, Proc Natl Acad Sci U S A 102(33) (2005) 11911–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [250].Wang L, Ciani C, Doty SB, Fritton SP, Delineating bone’s interstitial fluid pathway in vivo, Bone 34(3) (2004) 499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [251].Reilly GC, Knapp HF, Stemmer A, Niederer P, Knothe Tate ML, Investigation of the morphology of the lacunocanalicular system of cortical bone using atomic force microscopy, Ann Biomed Eng 29(12) (2001) 1074–81. [DOI] [PubMed] [Google Scholar]
- [252].McCreadie BR, Hollister SJ, Schaffler MB, Goldstein SA, Osteocyte lacuna size and shape in women with and without osteoporotic fracture, J Biomech 37(4) (2004) 563–72. [DOI] [PubMed] [Google Scholar]
- [253].Cooper RR, Milgram JW, Robinson RA, Morphology of the osteon. An electron microscopic study, J Bone Joint Surg Am 48(7) (1966) 1239–71. [PubMed] [Google Scholar]
- [254].Wang L, Solute Transport in the Bone Lacunar-Canalicular System (LCS), Curr Osteoporos Rep 16(1) (2018) 32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [255].You LD, Weinbaum S, Cowin SC, Schaffler MB, Ultrastructure of the osteocyte process and its pericellular matrix, Anat Rec A Discov Mol Cell Evol Biol 278(2) (2004) 505–13. [DOI] [PubMed] [Google Scholar]
- [256].Weinbaum S, Cowin SC, Zeng Y, A model for the excitation of osteocytes by mechanical loading-induced bone fluid shear stresses, J Biomech 27(3) (1994) 339–60. [DOI] [PubMed] [Google Scholar]
- [257].Knothe Tate ML, Niederer P, Knothe U, In vivo tracer transport through the lacunocanalicular system of rat bone in an environment devoid of mechanical loading, Bone 22(2) (1998) 107–17. [DOI] [PubMed] [Google Scholar]
- [258].Piekarski K, Munro M, Transport mechanism operating between blood supply and osteocytes in long bones, Nature 269(5623) (1977) 80–2. [DOI] [PubMed] [Google Scholar]
- [259].Ciani C, Sharma D, Doty SB, Fritton SP, Ovariectomy enhances mechanical load-induced solute transport around osteocytes in rat cancellous bone, Bone 59 (2014) 229–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [260].Tami AE, Schaffler MB, Knothe Tate ML, Probing the tissue to subcellular level structure underlying bone’s molecular sieving function, Biorheology 40(6) (2003) 577–90. [PubMed] [Google Scholar]
- [261].Crescitelli R, Lasser C, Szabo TG, Kittel A, Eldh M, Dianzani I, Buzas EI, Lotvall J, Distinct RNA profiles in subpopulations of extracellular vesicles: apoptotic bodies, microvesicles and exosomes, J Extracell Vesicles 2 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [262].Kono H, Rock KL, How dying cells alert the immune system to danger, Nat Rev Immunol 8(4) (2008) 279–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [263].Sun S, Sursal T, Adibnia Y, Zhao C, Zheng Y, Li H, Otterbein LE, Hauser CJ, Itagaki K, Mitochondrial DAMPs increase endothelial permeability through neutrophil dependent and independent pathways, PLoS One 8(3) (2013) e59989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [264].Rock KL, Latz E, Ontiveros F, Kono H, The sterile inflammatory response, Annu Rev Immunol 28 (2010) 321–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [265].Medzhitov R, Origin and physiological roles of inflammation, Nature 454(7203) (2008) 428–35. [DOI] [PubMed] [Google Scholar]
- [266].Komai K, Shichita T, Ito M, Kanamori M, Chikuma S, Yoshimura A, Role of scavenger receptors as damage-associated molecular pattern receptors in Toll-like receptor activation, Int Immunol 29(2) (2017) 59–70. [DOI] [PubMed] [Google Scholar]
- [267].Borges da Silva H, Fonseca R, Pereira RM, Cassado Ados A, Alvarez JM, D’Imperio Lima MR, Splenic Macrophage Subsets and Their Function during Blood-Borne Infections, Front Immunol 6 (2015) 480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [268].Weisel K, Scott NE, Tompson DJ, Votta BJ, Madhavan S, Povey K, Wolstenholme A, Simeoni M, Rudo T, Richards-Peterson L, Sahota T, Wang JG, Lich J, Finger J, Verticelli A, Reilly M, Gough PJ, Harris PA, Bertin J, Wang ML, Randomized clinical study of safety, pharmacokinetics, and pharmacodynamics of RIPK1 inhibitor GSK2982772 in healthy volunteers, Pharmacol Res Perspect 5(6) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [269].Yamasaki S, Ishikawa E, Sakuma M, Hara H, Ogata K, Saito T, Mincle is an ITAM-coupled activating receptor that senses damaged cells, Nat Immunol 9(10) (2008) 1179–88. [DOI] [PubMed] [Google Scholar]
- [270].Weinstein RS, Roberson PK, Manolagas SC, Giant osteoclast formation and long-term oral bisphosphonate therapy, N Engl J Med 360(1) (2009) 53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [271].Lotze MT, Tracey KJ, High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal, Nat Rev Immunol 5(4) (2005) 331–42. [DOI] [PubMed] [Google Scholar]
- [272].Harris PA, Berger SB, Jeong JU, Nagilla R, Bandyopadhyay D, Campobasso N, Capriotti CA, Cox JA, Dare L, Dong X, Eidam PM, Finger JN, Hoffman SJ, Kang J, Kasparcova V, King BW, Lehr R, Lan Y, Leister LK, Lich JD, MacDonald TT, Miller NA, Ouellette MT, Pao CS, Rahman A, Reilly MA, Rendina AR, Rivera EJ, Schaeffer MC, Sehon CA, Singhaus RR, Sun HH, Swift BA, Totoritis RD, Vossenkamper A, Ward P, Wisnoski DD, Zhang D, Marquis RW, Gough PJ, Bertin J, Discovery of a First-in-Class Receptor Interacting Protein 1 (RIP1) Kinase Specific Clinical Candidate (GSK2982772) for the Treatment of Inflammatory Diseases, J Med Chem 60(4) (2017) 1247–1261. [DOI] [PubMed] [Google Scholar]
- [273].Bai Y, Lam HC, Lei X, Dissecting Programmed Cell Death with Small Molecules, Acc Chem Res 53(5) (2020) 1034–1045. [DOI] [PubMed] [Google Scholar]
- [274].Liao DS, L.; Liu W; He S; Wang X; Lei X, Necrosulfonamide inhibits necroptosis by selectively targeting the mixed lineage kinase domain-like protein, Med. Chem. Commun 5 (2014) 333–337. [Google Scholar]
- [275].Degterev A, Maki JL, Yuan J, Activity and specificity of necrostatin-1, small-molecule inhibitor of RIP1 kinase, Cell Death Differ 20(2) (2013) 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [276].Teng X, Degterev A, Jagtap P, Xing X, Choi S, Denu R, Yuan J, Cuny GD, Structure-activity relationship study of novel necroptosis inhibitors, Bioorg Med Chem Lett 15(22) (2005) 5039–44. [DOI] [PubMed] [Google Scholar]
- [277].Wang W, Marinis JM, Beal AM, Savadkar S, Wu Y, Khan M, Taunk PS, Wu N, Su W, Wu J, Ahsan A, Kurz E, Chen T, Yaboh I, Li F, Gutierrez J, Diskin B, Hundeyin M, Reilly M, Lich JD, Harris PA, Mahajan MK, Thorpe JH, Nassau P, Mosley JE, Leinwand J, Kochen Rossi JA, Mishra A, Aykut B, Glacken M, Ochi A, Verma N, Kim JI, Vasudevaraja V, Adeegbe D, Almonte C, Bagdatlioglu E, Cohen DJ, Wong KK, Bertin J, Miller G, RIP1 Kinase Drives Macrophage-Mediated Adaptive Immune Tolerance in Pancreatic Cancer, Cancer Cell 34(5) (2018) 757–774 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [278].Humphries F, Yang S, Wang B, Moynagh PN, RIP kinases: key decision makers in cell death and innate immunity, Cell Death Differ 22(2) (2015) 225–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [279].Dondelinger Y, Aguileta MA, Goossens V, Dubuisson C, Grootjans S, Dejardin E, Vandenabeele P, Bertrand MJM, RIPK3 contributes to TNFR1-mediated RIPK1 kinase-dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition, Cell Death & Differentiation 20(10) (2013) 1381–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [280].Moriwaki K, Chan FK, The Inflammatory Signal Adaptor RIPK3: Functions Beyond Necroptosis, Int Rev Cell Mol Biol 328 (2017) 253–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [281].Park H-H, Park S-Y, Mah S, Park J-H, Hong S-S, Hong S, Kim Y-S, HS-1371, a novel kinase inhibitor of RIP3-mediated necroptosis, Experimental & Molecular Medicine 50(9) (2018) 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [282].Xia K, Zhu F, Yang C, Wu S, Lin Y, Ma H, Yu X, Zhao C, Ji Y, Ge W, Wang J, Du Y, Zhang W, Yang T, Zhang X, He S, Discovery of a Potent RIPK3 Inhibitor for the Amelioration of Necroptosis-Associated Inflammatory Injury, Front Cell Dev Biol 8 (2020) 606119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [283].Jiao J, Wang Y, Ren P, Sun S, Wu M, Necrosulfonamide Ameliorates Neurological Impairment in Spinal Cord Injury by Improving Antioxidative Capacity, Front Pharmacol 10 (2019) 1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [284].Zhang QX, Guo D, Wang FC, Ding WY, Necrosulfonamide (NSA) protects intervertebral disc degeneration via necroptosis and apoptosis inhibition, Eur Rev Med Pharmacol Sci 24(5) (2020) 2683–2691. [DOI] [PubMed] [Google Scholar]
- [285].Motawi TMK, Abdel-Nasser ZM, Shahin NN, Ameliorative Effect of Necrosulfonamide in a Rat Model of Alzheimer’s Disease: Targeting Mixed Lineage Kinase Domain-like Protein-Mediated Necroptosis, ACS Chem Neurosci 11(20) (2020) 3386–3397. [DOI] [PubMed] [Google Scholar]
- [286].Dong WZ, M.; Zhu Y; Chen Y; Zhao X; Li R; Zhang L; Ye Z; Liang X, Protective effect of NSA on intestinal epithelial cells in a necroptosis model, Oncotarget 8(49) (2017) 86726–86735. [DOI] [PMC free article] [PubMed] [Google Scholar]