

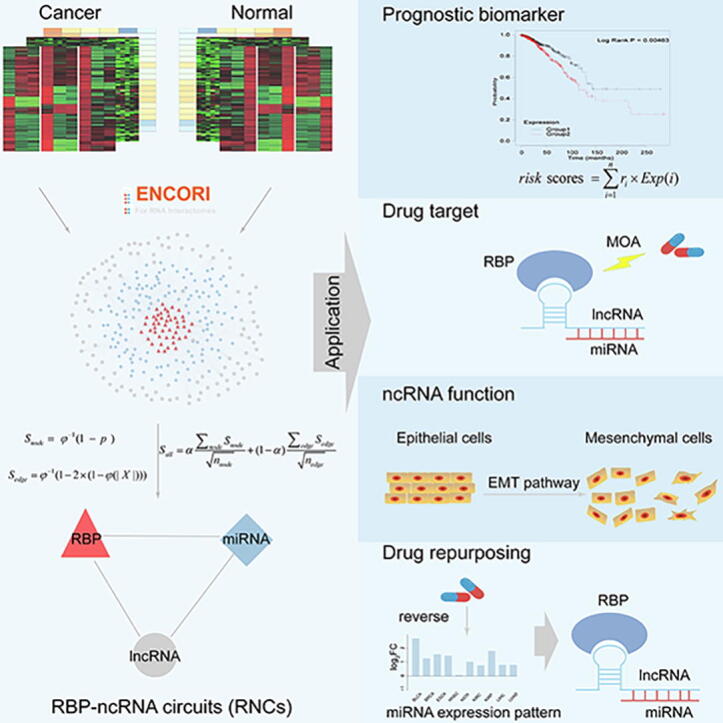
Graphical abstract
Keywords: RNA binding protein-ncRNA circuits, Gene regulation, RNA binding protein, MiRNA, LncRNA
Abstract
The interactions among non-coding RNA (ncRNA) and RNA binding protein (RBP) are increasingly recognized as one of basic mechanisms in gene regulation, and play a crucial role in cancer progressions. However, the current understanding of this regulation network, especially its dynamic spectrum according to the differentially expressed nodes (i.e. ncRNAs and RBP) is limited. Utilizing transcriptomics and interactomics resources, dysregulated RBP-ncRNA circuits (RNCs) are systematically dissected across 14 tumor types. We found these aberrant RNCs are robust and enriched with cancer-associated ncRNAs, RBPs and drug targets. Notably, the nodes in altered RNCs can jointly predict the clinical outcome while the individual node can’t, underscoring RNCs can serve as prognostic biomarkers. We identified 30 pan-cancer RNCs dysregulated at least in six tumor types. Pan-cancer RNC analysis can reveal novel mechanism of action (MOA) and repurpose for existing drugs. Importantly, our experiments elucidated the novel role of hsa-miR-224-5p, a member of the pan-cancer RNC hsa-miR-224-5p_MAGI2-AS3_MBNL2, in EMT program. Our analysis highlights the potential utilities of RNCs in elucidating ncRNA function in cancer, associating with clinical outcomes and discovering novel drug targets or MOA.
1. Introduction
Increasing evidence suggests the interactions between RNA-binding proteins (RBPs) and noncoding RNAs such as long noncoding RNAs (lncRNAs) and microRNA (miRNAs) play a fundamental role in gene post-transcriptional regulation [1], [2]. RBPs are a class of proteins that directly bind to RNA and control the synthesis, maturation, modification and degradation throughout the whole RNA life cycle [3]. The coupling of deep sequencing and mass spectrometry studies, such as RNA pull down assay and cross linking immunoprecipitation with high-throughput sequencing (HITS-CLIP), have greatly expanded the RNA-binding proteome [4]. It is estimated that there are ∼1500 RBPs in the human genome, most of which are ubiquitously expressed in many tissues [3]. Conservation analysis among different species indicated that RBPs are more evolutionary conserved compared to non-RBPs, especially those involved in various biological processes such as mRNA splicing and ribosome biogenesis [5].
When RBPs and a variety of ncRNAs are close enough physically, they can interact to form functional RBP-ncRNA circuits (RNC). For example, miRNAs bind with their RBP partners to form the RNA-induced silencing complex (RISC), thereby to repress the translation of mRNA and induces its degradation [6]. Jiang and his colleagues also found RNA-binding protein NONO-PSF heterodimer binds many expressed pri-miRNAs to affect miRNA biogenesis. At the same time, NONO-PSF also interacts with lncRNA, nuclear paraspeckle assembly transcript 1 (NEAT1), which harbors a 'pseudo pri-miRNA' structure to facilitate the miRNA processing [7]. Recently, dysregulated RNCs have been associated with several cancers [8], [9], [10], [11]. For example, the RBP ELAVL1/HuR was found to mediate the interaction between miR-124-3p and lncRNA NEAT1, which controls carcinogenesis in ovarian cancers [8]. This cooperative manner can also be identified in another lncRNA, metastasis-associated lung adenocarcinoma transcript 1(MALAT1). Several studies found MALAT interacts with multiple serine/arginine (SR) splicing factors and miRNAs such as miR-101, miR-217, thereby to control the alternative splicing of endogenous pre-mRNAs, and to inhibit proliferation and invasion in carcinoma [9], [10], [11].
Above experimental examples clearly demonstrated the regulatory role of RNCs in tumorgenesis. However, many questions have not been fully addressed. For example, can we identify the RNC computationally since the experimental approaches are usually time-consuming and labor-intensive? Is the RNC mediated regulation a generic mechanism across many cancers? More importantly, are the perturbations of RNC linked with prognostic and therapeutic significance? Answers to the above questions need to combine the individual RBP-miRNA, RBP-lncRNA and miRNA-lncRNA interactions into a regulation network; and to integrate other omics resources such transcriptomics and clinical data to delimit the dysregulated RNC from the basal regulation network. Several databases such as ENCORI and CLIPdb have archived the experimentally verified interactions among RBPs and ncRNAs [12], [13]. As the first step to investigate the RNC, we previously have also developed a computational pipeline, RBPvsMIR, to detect the competing miRNAs and RBP pairs that regulate the shared transcripts [14]. In this study, we utilized a systems biology approach to decipher RNCs associated with cancer progressions. First, an RBP-ncRNA regulation network was constructed by leveraging the available interactions from experimental data. Altered RNCs under different cancer conditions were then systematically characterized to understand how the perturbations impact these RNCs. Finally, we shown these dysregulated RBP-ncRNA circuits are associated with cancers mechanism and have valuable clinical utilities.
2. Results
Our pipeline utilized interactomics data among RBP and ncRNAs (miRNA and lncRNA) from the ENCORI database to construct RBP-miRNA-lncRNA regulation networks. Spurious interactions, which have <3 CLIP experimental evidence, were filtered out. Homogeneous primary cancer samples were selected from TCGA for transcriptomics analysis. A network motif finding algorithm, which simultaneously examined the dysregulation extent of both nodes and edges was used to infer the dynamic RNC changes under disease conditions. This pipeline is illustrated in Fig. 1 and is described in details in the Method section.
Fig. 1.

The identification of RNCs. Candidate miRNA-lncRNA, miRNA-RBP, and RBP–lncRNA pairs were obtained from the ENCORI database. The altered extent of nodes and edges were combined to identify dysregulated RNCs by using miRNA, lncRNA, and RBP expression profiles.
2.1. Global characteristics of basal RBP-ncRNA regulation network
After eliminating redundancy, we obtained 116,451 regulations among 642 miRNAs, 3296 lncRNAs, and 1209 RBPs. Totally, 51,955 miRNA-lncRNA-RBP RNCs, consisting of 600 miRNAs, 601 lncRNAs, and 59 RBPs, were extracted and used in the following analysis (Table S1A). A global picture of the RBP-ncRNA network was shown in Fig. 2A. Examining the degree (i.e. the number of undirected edges for each node) distribution of the network in Cytoscape [15], we found RBP-ncRNA network had a typical scale-free structure, which is indicative of transcriptional regulatory networks (Fig. 2B, R square = 0.777) [16]. Next, comparing with known cancer-related RBPs from CGC and IntOGen databases [17], [18], we found the RBP-ncRNA network is significantly enriched with cancer-associated RBPs (Fisher exact Test: P-value = 5.88E-06). Similarly, the RBP-ncRNA network contains 108 lncRNAs and 511 miRNAs which have been linked with cancers (Fisher exact Test: of P-value of lncRNA = 7.26E-30, P-value of miRNA = 2.58E-87). The above results indicated the nodes in RBP-ncRNAs network tightly connected with cancer progression. Besides, we observed that all cancer-associated nodes, lncRNA nodes and miRNA nodes have significantly higher degrees than other non-cancer nodes (Fig. 2C, D, E, Wilcoxon rank-sum test). The average degree of cancer-related RBPs was higher than that of other RBP, although the difference was not significant (Fig. 2F). A higher degree indicated that these cancer-associated nodes were more likely to be the information transmission hubs in the network. Overall, the topological characteristics and enriched cancer nodes demonstrated that the constructed RBP-ncRNA regulation network is a plausible approximation and can provide insight into the cancer biology.
Fig. 2.

The global properties of basal RBP-ncRNA regulation network. (A) The overview of RBP-ncRNA regulation network; (B) The log-log plots show that the degree distributions of the RNCs network follow the power law. (C, D, E and F) Cancer-related nodes had a higher degree than other nodes.
2.2. Dysregulated RBP-ncRNA circuits can be recapitulated from other datasets
To investigate if the RNCs are biologically meaningful, we would like to see if the dysregulated RNC found from one dataset can be recapitulated from another validation dataset of the same cancer type. Due to the limited public available multiple omics datasets, we only focused on breast cancer. First, we randomly split the original TCGA breast data into Train data (4/5 of the original TCGA data) and Test data (the remaining 1/5 part). The dysregulated RNC were separately identified from both Train and Test data with the same method. As shown in Fig. S1A, 356 RNCs are overlapped between the two datasets, corresponding to 55% of Train data and 44.1% of the Test data. This result clearly indicated many of the dysregulated RNCs can be recapitulated (Fisher exact test: P-value = 2.2E-16). To further establish the validity of dysregulated RNCs, we also identified the RNCs from another independent breast cancer datasets downloaded from GEO. It should be noted that the new dataset is generated from oligos arrays, therefore there is inherent platform level noise when compared with RNA-seq based TCGA dataset. Despite this, there is still significant portion of RNCs overlapped between both datasets (Fig. S1B, Fisher exact test: P-value = 1.6E-16). Overall, the recurrent occurrence of RNCs from different datasets strongly suggested that the dysregulated RNC are related to the main theme in cancers.
2.3. Dysregulated RBP-ncRNA circuits in breast cancer and esophageal cancer
In cells, RBP dynamically modulated the signaling transduction cascade, thus we identified the aberrant RBP-ncRNAs by combining the differentially expressed nodes and edges between tumor and normal samples (See method for details). As a pilot study, we investigated the breast and esophageal cancer in details.
The 1116 dysregulated RNCs, which includes 47 RBPs, 109 lncRNAs and 138 miRNAs, were identified in esophageal cancer (Table S2). Similarly, the 787 aberrant RNCs including 37 RBP, 89 lncRNA and 161 miRNAs were obtained from the breast cancer (Table S3). There are 42 RNCs shared by both cancers, which respectively corresponds to 5.3% and 3.8% of the RNCs identified in breast and esophageal cancers. For example, the hsa-miR-18a-5p_SNHG15_EIF4A3 sub-network was identified from both cancers. Previously, hsa-miR-18a-5p was found over-expressed in different human cancers including breast and esophageal cancer [19], [20]. And SNHG15, a well-studied lncRNA, is identified as a key regulator in tumorigenesis and progression of various cancers [21], [22]. The RBP EIF4A3 is also found over-expressed in many cancers and can coordinate cell cycle and cell apoptosis [23], [24].
Most RNCs are cancer-specific and perturbated only in one cancer. For example, the RNC hsa-miR-101-3p_MALAT1_AUH was specifically altered in esophageal cancer. Concordantly, previous report found hsa-miR-101-3p represses the expression of the lncRNA MALAT1 to inhibit proliferation, migration, and invasion in esophageal squamous cell carcinoma cells [11]. Meanwhile, the RNC hsa-miR-148a-3p_HOTAIR_DDX54 was abnormal only in breast cancer. Consistently, Tao et al. found that miR-148a targets HOTAIR and is inhibited in breast cancer [25].
2.4. The nodes within the same dysregulated RBP-ncRNA circuits are functionally coherent
We examined the degree distribution of the breast specific and esophageal specific dysregulated RNCs (Fig. S2A and S2B). As shown in Fig. S2C and S2D, the power-law distribution was fitted well for the degrees in the two sub-networks (in breast cancer, R square = 0.862; in esophageal cancer, R square = 0.835). These results indicated that the two sub-networks approximated scale-free topology, which is the common feature of the most biological networks [26]. Then, we carried out Gene Ontology (GO) functional similarity analysis to investigate the nodes’ relationship (see Method section). The 243 and 330 RNCs, where every node has GO annotations information of the biological processes, were selected for breast and esophageal cancer, respectively. The results showed the functional similarities of nodes within the same RNCs are higher than that of nodes random generated (Wilcoxon rank-sum test, breast: P-value = 0.00441, esophageal: P-value = 0.00741, Table S4 listed the significant RNCs). As an example, the abnormal RNC hsa-miR-15a-5p_XIST_DDX3X in breast cancer has significantly closer function similarity (P-value = 0.0219). DDX3X is an ATP-dependent RNA helicase. Consistently, it has been reported that XIST colocalized with DDX3X and other chromatin binders to direct chromosome conformation [27]. The coherent functional spectrum among nodes within the same RNCs can help to efficiently establish orchestrated actions upon complex signaling. Thus, it is potentially to ascribe novel functions to RBP or ncRNA according to the other nodes within the same RNCs.
2.5. Dysregulated RBP-ncRNA circuits are associated with cancer pathways and enriched with drug targets
To further understand the functional significance of those aberrant RNCs in the two cancers, we performed enrichment analysis of KEGG pathway. A few interesting themes have emerged. We found that the RBPs in these RNCs were enriched in the pathways of spliceosome (breast cancer: P-value = 1.20E-13; esophageal cancer: P-value = 2.64E-09), RNA transport (breast cancer: P-value = 0.0732; esophageal cancer: P-value = 3.11E-04) and mRNA surveillance pathways (breast cancer: P-value = 7.61E-5; esophageal cancer: P-value = 7.34E-04). Our results are consistent with recent findings about the important role of abnormal RNA processing in cancers [28], [29]. For example, researches showed that spliceosome pathway is associated with the development of many types of cancer [30], [31]. Mis-spliced RNAs trigger an antiviral immune response in breast cancer [32], suggesting that targeted spliceosome therapy could serve as a research hotspot for cancer treatment [33]. Another enriched KEGG pathway is Herpes simplex infection (breast cancer: P-value = 0.0113; esophageal cancer: P-value = 0.0519). This is because several RBPs, which are general components of spliceosome, are interacted with and modulated by the viral protein during infection [34].
Besides, some of the RBPs were known therapeutic targets in cancers. For example, RNA binding fox-1 homolog 2 (RBFOX2) is found dysregulated in both breast and esophageal cancers. RBFOX2 is an essential regulator of alternative splicing, which leads to a higher degree of tissue invasiveness in multiple cancer models [35]. Choi et al. further demonstrated that resveratrol treatment inhibited specific RBFOX2 localization from nucleus to the cytoplasm, thereby to attenuate cancer progression and tumor growth [36]. Based on a recent summary of known therapeutic RBPs in cancers, we found the RBPs in those dysregulated RNCs are significantly enriched with these druggable RBPs (Fisher exact Test: breast P-value = 0.00027; esophageal P-value = 0.00976) [37]. Importantly, this result also implied that the other RBPs within dysregulated RNCs, though still unknown about their pharmaceutical usage, may potentially be developed as drug target for cancer treatments.
2.6. Dysregulated RBP-ncRNA circuits are prognostic biomarkers
To test whether the expression of dysregulated RNCs correlated with patient survival, we used the Cox multivariate regression model for assessing their clinical usage in breast and esophageal cancer. 18.7% and 29.7% of dysregulated RNCs were found to correlate with survival in breast and esophageal cancer, respectively (Fig. 3A and 3B). For example, the RNC hsa-miR-429_LINC00667_SRSF1 was correlated with breast cancer patient survival (Fig. 3D, KM survival analysis, P-value = 0.000647). This agrees with previous studies showing that hsa-miR-429, LINC00667 and the RBP SRSF1 correlate with cancer patient survival [38], [39], [40]. Also, the RNC hsa-miR-125a-5p_NEAT1_FMR1 can stratify the two groups of patients with different clinical outcomes in esophageal cancer (Fig. 3E), which were consistent with previous researches [41], [42], [43]. In addition, the dysregulated RNCs could be used to identify the common RNCs shared by two or more tumors. As can be seen from Fig. 3C, there are 3 abnormal RNC shared by the breast and esophageal cancer. For example, hsa-miR-181b-5p_LINC00667_HNRNPC were significantly associated with the patient survival in breast and esophageal cancer with the P-value of 0.00417 and 0.0233, respectively (Fig. 3F).
Fig. 3.

The RNCs are potential prognostic biomarkers in breast and esophageal cancers. (A-C) The pie charts represent the number of dysregulated RNCs identified only in breast cancer, only in esophageal cancer and shared by both cancers. The number of RNC correlated with patient survival is also colored in light gray. (D-F) Kaplan-Meier survival analysis was performed on two groups of patients with different clinical outcomes. Representative KM plots are shown. The red lines represent the group with high risk, and the black lines represent the group with low risk. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
To test whether the whole RNC could better stratify patients than individual use of miRNAs node, lncRNA node or RBP node, Kaplan-Meier survival analysis was conducted and compared (see Method). In the majority of the RNCs (BRCA: 69%, ESCA: 65%), individual nodes were found unable to classify patients significantly (Table S5). For example, the three single nodes in the RNC hsa-miR-181b-5p_LINC00667_HNRNPC were not significantly correlated with patient survival in esophageal cancer, but the RNC can collectively predict the clinical outcome (Fig. 3F, P-value = 0.0233). These results suggested that RNC may serve as prognostic biomarkers even when the individual nodes are not associated with clinical features.
2.7. Pan-cancer RBP-ncRNA circuits reveal novel miRNA function
Having proved that the abnormal RBP-ncRNA network is functionally related to cancers and useful in clinical application, it would be natural to identify the common RNCs that are dysregulated in multiple tumor types. This investigation should shed light on the universal mechanism on tumorgenesis. Totally, 9421 RNCs significantly dysregulated in at least one tumor type were identified (Table S6). Thirty pan-cancer RNCs (pan-RNCs), which were dysregulated in at least six examined cancers, are found (Fig. 4A). The miRNA, RBP and lncRNA components, in the pan-RNCs are listed in Table 1. The fact that only a few RNCs are pan-RNCs suggested most RNC based post-transcriptional regulations are local and relevant to the specific tumor type. Of all the pan-RNCs, 93% (28/30) can be integrated into a big interlocking network (Fig. 4B), indicating that RBPs can be efficiently organized by their shared lncRNA or miRNA mediators.
Fig. 4.

Dysregulated pan-RNCs across cancers. (A). The number of dysregulated RNCs across tumor types. (B). The pan-cancer RNCs network. Red triangle, blue diamond, and gray ellipse nodes represent RBPs, miRNAs, and lncRNAs, respectively. (C). Western blot of vimentin and GAPDH after transfection with hsa-miR-224-5p mimics or inhibitor in TE1 cells. (D). hsa-miR-224-5p_MAGI2-AS3_MBNL2 was dysregulated in nine tumor types. Yellow asterisk indicated it is also a prognostic marker for this cancer type. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Table 1.
Summary of the pan-RNCs.
| MiRNA | LncRNA | RBP | #Number of tumor type |
|---|---|---|---|
| hsa-miR-224-5p | MAGI2-AS3 | MBNL2 | 9 |
| hsa-miR-31-5p | MAGI2-AS3 | MBNL2 | 8 |
| hsa-miR-18a-5p | SNHG1 | PTBP1 | 7 |
| hsa-miR-18a-5p | AC093010.3 | ELAVL1 | 7 |
| hsa-miR-18a-5p | AC093010.3 | MBNL2 | 7 |
| hsa-miR-182-5p | SNHG1 | MBNL2 | 7 |
| hsa-miR-224-5p | MAGI2-AS3 | HNRNPC | 7 |
| hsa-miR-224-5p | AC093010.3 | MBNL2 | 7 |
| hsa-miR-195-5p | SNHG1 | HNRNPA2B1 | 7 |
| hsa-miR-374b-5p | MAGI2-AS3 | MBNL2 | 7 |
| hsa-miR-19a-3p | SNHG20 | CBX7 | 6 |
| hsa-miR-30a-5p | AC239868.2 | MOV10 | 6 |
| hsa-miR-31-5p | MAGI2-AS3 | RBFOX2 | 6 |
| hsa-miR-139-5p | AC093010.3 | MBNL2 | 6 |
| hsa-miR-1-3p | MIR4435-2HG | PTBP1 | 6 |
| hsa-miR-1-3p | LINC00641 | HNRNPA2B1 | 6 |
| hsa-miR-1-3p | LINC00641 | IGF2BP3 | 6 |
| hsa-miR-133a-3p | FGD5-AS1 | PTBP1 | 6 |
| hsa-miR-145-5p | SNHG1 | DDX54 | 6 |
| hsa-miR-195-5p | FGD5-AS1 | MBNL2 | 6 |
| hsa-miR-195-5p | SNHG1 | UPF1 | 6 |
| hsa-miR-195-5p | SNHG1 | PTBP1 | 6 |
| hsa-miR-195-5p | SNHG1 | DDX54 | 6 |
| hsa-miR-195-5p | AC024075.2 | HNRNPA2B1 | 6 |
| hsa-miR-374a-5p | MAGI2-AS3 | MBNL2 | 6 |
| hsa-miR-381-3p | AC093010.3 | ELAVL1 | 6 |
| hsa-miR-381-3p | AC093010.3 | HNRNPA2B1 | 6 |
| hsa-miR-140-3p | SNHG1 | PTBP1 | 6 |
| hsa-miR-450b-5p | LINC00641 | MBNL2 | 6 |
| hsa-miR-374b-5p | MAGI2-AS3 | IGF2BP2 | 6 |
We further compared the common properties of 30 pan-RNCs with the other thousands of dysregulated RNCs, in terms of network topology and clinical application. We found the degree is significantly high for pan-RNCs in each cancer type (Table S7). This comparison indicated that nodes in pan-RNCs tended to be the hubs in the basal RBP-ncRNA network, implying their important functional roles. In addition, twenty-nine of the pan-RNCs can significantly separate the patients in at least one cancer type. As a comparison, only about half of the other aberrant RNC (4818/9391) can predict the clinical outcome well. This result suggested that pan-RNCs are also versatile prognostic biomarkers in multiple cancers (Fisher exact test: breast P-value = 6.192E-08).
By integrating these RNCs, we constructed a pan-RNC network, which consisted of 16 miRNAs, 9 lncRNAs and 12 RBPs. In the pan-cancer network (Fig. 4B), the RBP muscleblind-like splicing regulator 2 (MBNL2), acted as a central hub with 14 partners, indicating that it was involved in many pan-RNCs. This gene is originally identified as a member of the muscleblind protein family which is involved in myotonic dystrophy [44], [45]. It encodes a C3H-type zinc finger protein that modulates alternative splicing of pre-mRNAs. However, increasing evidence suggested its pivotal role in the invasive properties of cancer cells. The expression of MBNL2 in breast and lung carcinoma tumor tissues was significantly lower compared to normal tissues. MBNL class of splicing factor is also a driver of EMT in breast cancer [46]. Furthermore, MBNL2-mediated anti-metastasis regulation was significantly correlated with the EMT in breast and lung carcinoma tumor tissues [47]. But there is no report on how it interacts with ncRNAs in tumors to date.
Also, the lncRNA MAGI2-AS3 was with high connections with RBPs and miRNAs in the pan-cancer network (Degree = 8, Fig. 4B). Recent reports indicated it is an EMT-associated lncRNA in breast cancer, bladder cancer and gastric cancers [48], [49], [50]. In the pan-RNC network, MAGI2-AS3_MBNL2 interaction is totally involved in 4 RNCs (Table 1). Among their miRNA partners (hsa-miR-224-5p, hsa-miR-31-5p and hsa-miR-374a/b-5p), both hsa-miR-31-5p and hsa-miR-374a/b-5p have been found to regulate the malignant transformation. For example, miR-31 overexpression reduced glioma cell invasion in mesenchymal stem cells [51]. Sun et al. also found miR-374 promotes the proliferation and migration of rat bone marrow-derived mesenchymal stem cells [52]. Thus, the MAGI2-AS3_MBNL2 associated pan-RNCs might underlie cancer development by participating in the EMT pathway. Compared with hsa-miR-31-5p and hsa-miR-374a/b-5p, little is known about miR-224 except that it is a predictive biomarker in hepatocellular carcinoma [53]. Considering its interaction with the MAGI2-AS3_MBNL2 axis, it is reasonable to predict the role of miR-224 in EMT. To verify this hypothesis, we either over-expressed or inhibited hsa-miR-224-5p in an esophageal squamous cell line TE1. As shown in Fig. 4C, ectopic expression of hsa-miR-224-5p could up-regulate vimentin protein, which is a mesenchymal cells marker; on the contrary, expression of vimentin was significantly decreased after depletion of miRNA by inhibitor. These results demonstrated that hsa-miR-224-5p facilitates primary cancer cells to gain mesenchymal properties and turns into migratory and invasive cancer cells. Furthermore, the pan-RNC hsa-miR-224-5p_MAGI2-AS3_MBNL2 was differentially altered in 9 out of 14 surveyed tumor types. Survival analysis indicated that it is a prognostic marker in UCEC, LUSC and BRCA with Log Rank P-value 0.0284, 0.0392, and 0.0293, respectively (Fig. 4D). All above analysis pinpointed that this pan-RNC plays an important role in EMT program across multiple cancers.
2.8. Pan-cancer RBP-ncRNA circuits predict potential drug MOA and repurposing
In order to study the role of pan-RNC in revealing drug mechanism, we analyze the anticancer drugs or small molecules that targeted the RBPs or affected miRNA expression in the pan-RNCs networks. First, we found that compared with the basal regulatory network (18.6%), the pan-RNCs (50%) had the higher proportion of therapeutic target RBPs (Fisher exact Test: P-value = 0.0304), which indicated that pan-cancer RNCs preferred to be druggable [37] (Table 2). Second, RNC analysis suggested a novel drug MOA. As shown in Fig. 5, previous studies indicated a cumarin-derived small molecule named CMLD-2, inhibits ELAVL1 (HuR) and exhibits anticancer activity in cancer cells [54], [55]. But the MOA of CMLD-2 under physiological condition is currently not completely understood. In this study, we found that ELAVL1 interacts with ncRNAs to form two RNCs, hsa-miR-381-3p_AC093010.3_ELAVL1 and RNC hsa-miR-18a-5p_AC093010.3_ELAVL1. Although ELAVL1 was upregulated in almost all 14 cancer tissues, the perturbation pattern of these two RNCs varied in different cancers (Fig. 5). For example, RNC hsa-miR-381-3p_AC093010.3_ELAVL1 but not RNC hsa-miR-18a-5p_AC093010.3_ELAVL1, is dysregulated in PRAD. These results indicated CMLD-2 may play an anti-cancer effect via regulation of the RNC hsa-miR-381-3p_AC093010.3_ELAVL1 in prostate cancer. Next, we obtained the drugs that can affect miRNA expression from the SM2miR database, which archives the experimentally confirmed influences of small molecule on miRNA expression [56]. We considered drugs coded with code L01 (Antineoplastic agents) according to the Anatomical Therapeutic Chemical classification, as anticancer agents. As shown in Fig. 5, some molecules such as Fluorouracil, Azacitidine, Trastumab, Vorinostat and Cisplatin, are known anticancer drugs. In addition, we found that the expression of hsa-miR-18a-5p and hsa-miR-381-3p could be reversed by many small molecule drugs, suggesting that they might play the anticancer role via regulating the pan-RNCs related mechanism. This inspired us to further repurpose the other small molecules as potential anticancer drugs. For example, Ginsenoside Rh2 reverse the expression of hsa-miR-18a-5p, which is up-regulated in almost all cancer types (Fig. 5) [57]. Ginsenoside Rh2 is one of the major bioactive ginsenosides in Panax ginseng. It has long been used to promote longevity and keep heath in traditional Chinese medicine [58]. Modern pharmaceutical studies have found Ginsenosides can improve brain function and enhance cardiovascular health. The investigation has confirmed its role in anti-proliferation, anti-invasion, anti-metastasis properties in cancers [58]. Also, the small molecule Bicalutamide upregulated the expression of hsa-miR-381a-3p, which was down-regulated in nearly all cancer types (Fig. 5). Bicalutamide was an oral non-steroidal antagonist of androgen receptor. It has been clinically used in androgen-deprivation therapy for the treatment of advanced prostate cancers. Recent study indicated that Bicalutamide also inhibits the proliferation and invasion in triple negative breast cancer, thus it is a promising drug repurposed for breast cancer treatment [59].
Table 2.
The potential druggable RBPs in pan-RNCs.
| RNA regulation mechanism | DRUGa | Therapeutic strategya | PMID | Expression in cancersb | |
|---|---|---|---|---|---|
| MBNL2 | Alternative splicing | Neobractatin | Small molecule inducer | 31320607 | Down-regulated in cancers |
| ELAVL1 | mRNA translation, mRNA stability | Dehydromutactin, Androstanolone, MS-444, CMLD-2 | Small molecule inhibitor | 17632515; 11356683; 27677075; 25750985 | Upregulated in cancers |
| RBFOX2 | Alternative splicing | resveratrol | Small molecule inhibitor | 23435423, 31028247; 23149937; 19448617 | Down-regulated in most cancers |
| IGF2BP2 | mRNA localization, mRNA stability | Gene knockdown | 30513526; 29510198; 30220054 | Upregulated in most cancers | |
| IGF2BP3 | mRNA localization, mRNA stability | Gene knockdown | 26158423; 26974154; 29847788 | Upregulated in most cancers | |
| HNRNPA2B1 | Alternative splicing | Copperc, Dihydroartemisininc | Gene knockdown | 21586613; 14633690 | Upregulated in most cancers |
The drug targeted to the RBPs and its therapeutic strategy was extracted from the biomedical literatures.
The expression pattern of the RBPs across cancer types in this study.
The drug was targeted to the RBPs based on the DRUGBANK database (https://go.drugbank.com/).
Fig. 5.

The ELAVL1-related RNCs were affected by CMLD-2. These bar plots represent the expression pattern of ELAVL1 (top left), hsa-miR-18a-5p and hsa-miR-381-3p (bottom) across 14 tumor types. The molecules colored in red are anticancer drugs. The table at top right summarizes the dysregulation of the RNCs in corresponding cancer; 1 and 0 denote dysregulation and no dysregulation, respectively. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3. Discussion
RNA-binding proteins, representing 7.5% of all protein-coding genes, play a key regulatory role in the post-transcriptional events [60]. A thorough analysis indicated only 2% of RBP families are tissue-specific [3]. This observation raised the question of how do RBPs precisely establish and maintain the distinct programs in highly differentiated cells? In recent years, hundreds of RBPs are characterized via either RNA-centric approaches (i.e. detecting proteins bound to an RNA of interest), or protein-centric methods (i.e. examining RNAs bound to a protein of interest). Analyses of these experimental data indicate that RBPs usually interact with multiple ncRNAs such as miRNAs and lncRNAs, to work in concert. Conversely, one single ncRNA such as miRNA, can target the mRNAs of multiple RBPs. Therefore, the combinatorial diversity among RBP-ncRNA interactions contribute, in part, to the diversity of cellular programs [2], [5], [61].
Particularly relevant in cancer, ncRNAs-RBP interactions have been identified as key determinants. For example, we recently found the binding of TRA2A with a ∼8Kbp long lncRNA MALAT1, triggers carcinogenesis in esophageal cancer cells [62]. RBPs usually regulate lncRNA stability, transportation, and localization [63]. RBPs also play a role as “RNA scaffold” to recruit lncRNA to participate in gene transcription and post-transcriptional translation [64]. In addition, Pol II, RNase III enzymes and RBPs in splicing machinery are actively involved in the miRNA processing. Disruption of RNA binding proteins impacts multiple steps of the RNA life cycle in cancers [28], [65]. On the other hand, numerous miRNAs bind with complementary sites in the 3′-untranslated region and guided the degradation of transcripts by interacting with RBPs in RNA-induced silencing complexes [6]. In addition to this canonical mechanism, miRNAs post-transcriptionally regulate the target mRNA/lncRNA expression mostly through shared miRNA response elements (MREs), which form the basis for lncRNAs and mRNAs crosstalk [66]. Investigating the RBP-ncRNA circuits formed by complex ncRNAs-RBP interactions would provide a deeper understanding of the robustness of cancer system to perturbations, thus to design better therapeutic interventions.
In this study, we first constructed the human RNC network based on experimentally identified RBP-ncRNA interactions. We only considered the interactions among RBP, miRNA and lncRNA, whereas it is far more complicated in the real RBP-ncRNA network. It may contain other types of nodes (such as circular RNAs), more nodes engaged in the altered RNC (i.e. the number of nodes in RNC larger than 3), and sometimes the edges are with directions. Nevertheless, we have shown the resulted networks demonstrate typical scale free property of transcriptional networks, and the function of nodes relates to cancer biology. Combining with the differentially expressed analyses of lncRNA, miRNA, and RBP in each tumor type, we comprehensively dissected dysregulated RBP-ncRNA circuits across 14 tumor types. Survival analyses suggested that RNCs can serve as prognostic biomarkers. Furthermore, the RBPs and ncRNAs within aberrant RNCs are also promising drug targets. In addition, we identified 30 pan-RNCs that are dysregulated in multiple cancers. They are usually with high links with other nodes, which underlies their involvement in cancer development. Compared with the other abnormal RNCs in individual cancer, these ‘conserved’ RNCs are good prognostic biomarkers in many cancers suggesting they have wide clinical applications. We showed that the nodes within the same dysregulated RNC often share similar functionality, which can be used to understand the molecular mechanism and predict novel functions. Specifically, we demonstrated with experiments that hsa-miR-224-5p affects EMT program. Analyzing the wiring pattern of RNC also inspires novel MOA of CMLD-2, an ELAVL1 inhibitor, and repurpose Ginsenoside Rh2 and Bicalutamide with new indications.
4. Conclusions
Our analysis indicated RNC mediated post-transcriptional regulation is a generic organizing principle across many tumor types, highlighting the potential of RBP-ncRNA circuits as a new dimension to elucidate the pathogenesis of cancer, and to develop anticancer drugs.
5. Materials and methods
5.1. Data collection and pre-processing
The expression data of miRNA, lncRNAs and mRNA and the corresponding clinical data of samples, were collected from the TCGA project [67]. Cancer samples used in our expression analysis are labelled with barcode (01) in TCGA database, which indicated they are homogeneous and all from primary cancer. The sample sizes across 14 cancer types ranged from 89 to 1176 (Table S1B). For miRNA expression, we calculated the read counts of each mature miRNA from the isoform quantification files, in which mature miRNA IDs are updated based on miRBase release 22. For gene expression, genes in these samples were annotated by the Ensembl 90 annotation of the human genome, and the lncRNA IDs were extracted based on the gene biotype in the annotation information. Furthermore, the lncRNAs, miRNAs, or mRNAs that expressed (expression value >0) in at least half of the samples were retained for further analysis in each type of cancer.
The mRNA/lncRNA and miRNA expression profiling of breast tumors were also downloaded from the Gene Expression Omnibus (GEO, GSE28884). It included 127 breast carcinomas samples and 11 normal samples. We obtained mRNA/lncRNA expression data based on the re-annotation of probes on the NKI-CMF 35 k oligo array to the human genome (GRCH38). Limma package was used for expression normalization and to obtain the significance of expression changes [68].
5.2. Construction of the basal miRNA-lncRNA-RBP network
The mutual interactions among miRNAs, mRNAs (RBPs) and lncRNAs were extracted from ENCORI with a high stringency, where the number of the supported CLIP experimental evidence is 3 or greater (miRNA-target relationships are from Ago CLIP-seq data) [12]. During the construction of regulatory network, the lncRNA and gene names were mapped to Ensembl IDs, the miRNA names were mapped to miRBase accession numbers of mature miRNAs. The RBPs, miRNAs, and lncRNAs that did not map to these IDs were discarded. As a result, 642 miRNAs, 3296 lncRNAs, and 1209 RBPs constitute 116,451 interactions. In this study, we only consider RBP-ncRNA sub-networks containing one mRNA, one miRNA and one lncRNA. Totally there are 51,955 RBP-miRNA-lncRNA circuits, which consist of 600 miRNAs, 601 lncRNAs and 59 RBPs. The network was visualized with the Network Analyzer plug-in of Cytoscape [15].
5.3. Identification of the dysregulated RNCs
A sub-network finding algorithm was used to identify abnormal RNCs [69]. This method outperforms other methods in that it considers the dysregulation extent of both nodes and edges. Briefly, each node score was calculated through the formula (1), which was based on the significance of differential expression between the corresponding cancer samples and normal samples.
| (1) |
where P-value indicated the significance of expression change determined by edgeR package [70]. The φ−1 represents the inverse normal cumulative distribution function. Secondly, each edge score was calculated according to the change of gene co-expression in cancer and normal samples using the following equations separately.
| (2) |
| (3) |
| (4) |
where rtumor and rnormal are the Spearman correlation coefficient of gene expression based on normalized read counts between cancer and normal samples, respectively. The correlations were then computed with Fisher transformation (2). The edge was scored according to the difference between correlations in tumor samples and normal samples through Eqs. (3), (4).
Subsequently, the score of a candidate RNC is calculated by combining the node scores and the edge scores as follows:
| (5) |
where nnode and nedge denote the number of nodes and edges in the network. The parameter α∈(0,1) is used to control the weight of node score and edge score. Here, we set α = 0.5 to the equal contribution of nodes and edges score.
Permutation analysis was performed to estimate the significance of each sub-network score. Firstly, three molecules (one mRNA, one miRNA and one lncRNA) were randomly selected to construct a background RNC. This process was repeated 100,000 times. Next, the score for each random RNC was calculated according to the equations above and generated the null distribution of random RNCs.
The P-value for an observed RNC was defined as the proportion of random RNC scores (random) larger than the observed RNC score (S): P-value = (Nrandom > S)/Np, where Nrandom > S is the number of random RNC that have larger scores than the observed network, and Np is the number of permutations. In this study, only RNCs with a nominal P-value < 0.05 were considered as dysregulated in cancer.
5.4. Survival analysis
For each aberrant RNCs, a univariate Cox regression analysis was used to evaluate the association between survival and expression of each node in corresponding cancer. The summary risk scores of the RNC, were calculated based on the combination of the expressions and the values of the regression coefficients from the univariate Cox regression analysis as follows.
| (6) |
where ri is the Cox regression coefficient of node i in the network (n = 3), n is the number of nodes, and Exp(i) is the normalized expression value of node i. The median risk score first was used as a cut-off to classify patients into high and low-risk groups. Then, a Kaplan-Meier survival analysis was used to assess the clinical significance between the two groups.
5.5. Cancer-related mRNA, lncRNA and miRNA
For evaluation, cancer-related lncRNAs were derived from LncRNADisease v2.0 and lnc2cancer v3.0 [71], [72], whereas cancer-related miRNAs were obtained from HMDD v2.0 [73]. Besides, we also collected the cancer-related gene from the Cancer Gene Census (CGC) and IntOGen databases, which archive manually annotated causal genes in cancer [17], [18].
5.6. Functional similarity analysis
We used Gene Ontology similarity in the biological processes to measure the functional relationship between nodes in the network. Firstly, GO terms of the gene, miRNA, and lncRNA were downloaded from the NCBI gene (gene2go), miRWalk v2.0, and the RISE database, respectively [74], [75]. Next, the similarity score was calculated by the sum of similarities among the miRNA, lncRNA, and gene nodes in each RNC with the method of mgoSim as follows [76].
| (7) |
Sn denote the set of nodes in the network (n = 3). Lastly, the permutation was used to estimate the statistical significance of each sub-network score. Nodes with expression values and the GO annotations, were selected randomly to construct a random sub-network population, and the random go-similarity score was calculated based on the same method. This process was repeated 10,000 times. The percentage in the background network GO similarity scores that is greater than the observed go-similarity score was reported as nominal P-value.
5.7. Transfection and Western blot
Esophageal squamous cell line TE1 was cultured in as previous described [14]. hsa-miR-224-5p mimics and inhibitor oligonucleotides were designed and synthesized by GenePharma (Suzhou, China). The transfections were performed using Lipofectamine 3000 (Invitrogen, USA) according to the manufacturer’s recommendations. After 48 h, cells were placed on ice for 30 min in a RIPA lysis buffer and PMSF (Solarbio, Beijing, China). After protein extraction, 30 μg protein per sample was loaded to 10% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride membranes (Immobilon P, Millipore, Billerica, USA). After blocking with 5% non-fat milk, the membranes were incubated with primary antibody at 4 °C overnight, followed by incubation with secondary antibodies at room temperature for 1 h. The antibodies including GAPDH, E-cadherin and Vimentin were purchased from Cell Signaling Technology (USA).
CRediT authorship contribution statement
Leiming Jiang: Methodology, Investigation. Qiuyang Chen: Investigation, Writing – original draft. Mingrong Bei: Investigation. Mengting Shao: Investigation. Jianzhen Xu: Conceptualization, Methodology, Writing – original draft, Writing - review & editing, Supervision, Funding acquisition.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
We thank members of Computational Systems Biology Laboratory, SUMC, for helpful discussion. This work has been supported in part by the National Natural Science Foundation of China (No. 81673037).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2021.09.019.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Supplementary figure 1.

RNCs detected from different breast datasets. (A) Train: RNCs from 4/5 of the original TCGA data; Test: RNCs from the remaining 1/5 samples. (B) GEO: RNCs from oligos arrays datasets; TCGA: RNCs from RNA-seq datasets. Numbers represent the identified RNCs in each scenario.
Supplementary figure 2.

The RNCs in breast and esophageal cancer. (A, B) The breast and esophageal specific RNCs network. Red triangle, blue diamond, and gray ellipse nodes represent RBPs, miRNAs, and lncRNAs, respectively. (C, D) The log-log plots show that the degree distributions follow the power law.
Summary of the RBP-ncRNA regulation network and the samples used in this study.
The significantly dysregulated RNCs in esophageal cancer.
The significantly dysregulated RNCs in breast cancer.
RNCs with similar nodes function in breast and esophageal cancers.
Survival analysis of RNCs in breast cancer and esophageal cancer.
Dysregulated RNCs across 14 tumor types and their association with clinical outcomes.
Comparison of node degree between the pan-RNC and the other dysregulated RNC in each cancer.
References
- 1.Mukherjee N., Corcoran D.L., Nusbaum J.D., Reid D.W., Georgiev S. Integrative regulatory mapping indicates that the rna-binding protein hur couples pre-mrna processing and mrna stability. Mol Cell. 2011;43:327–339. doi: 10.1016/j.molcel.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Licatalosi D.D., Darnell R.B. Rna processing and its regulation: global insights into biological networks. Nat Rev Genet. 2010;11:75–87. doi: 10.1038/nrg2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gerstberger S., Hafner M., Tuschl T. A census of human rna-binding proteins. Nat Rev Genet. 2014;15:829–845. doi: 10.1038/nrg3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ramanathan M, Porter DF, Khavari PA. Methods to study rna-protein interactions. 2019, 16, 225–234. [DOI] [PMC free article] [PubMed]
- 5.Neelamraju Y., Hashemikhabir S., Janga S.C. The human rbpome: from genes and proteins to human disease. J Proteomics. 2015;127:61–70. doi: 10.1016/j.jprot.2015.04.031. [DOI] [PubMed] [Google Scholar]
- 6.Gregory R.I., Chendrimada T.P., Cooch N., Shiekhattar R. Human risc couples microrna biogenesis and posttranscriptional gene silencing. Cell. 2005;123:631–640. doi: 10.1016/j.cell.2005.10.022. [DOI] [PubMed] [Google Scholar]
- 7.Jiang L., Shao C., Wu Q.J., Chen G., Zhou J. Neat1 scaffolds rna-binding proteins and the microprocessor to globally enhance pri-mirna processing. Nat Struct Mol Biol. 2017;24:816–824. doi: 10.1038/nsmb.3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chai Y., Liu J., Zhang Z., Liu L. Hur-regulated lncrna neat1 stability in tumorigenesis and progression of ovarian cancer. Cancer Med. 2016;5:1588–1598. doi: 10.1002/cam4.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu J., Peng W.X., Mo Y.Y., Luo D. Malat1-mediated tumorigenesis. Front Biosci (Landmark Ed) 2017;22:66–80. doi: 10.2741/4472. [DOI] [PubMed] [Google Scholar]
- 10.Tripathi V., Ellis J.D., Shen Z., Song D.Y., Pan Q. The nuclear-retained noncoding rna malat1 regulates alternative splicing by modulating sr splicing factor phosphorylation. Mol Cell. 2010;39:925–938. doi: 10.1016/j.molcel.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang X., Li M., Wang Z., Han S., Tang X. Silencing of long noncoding rna malat1 by mir-101 and mir-217 inhibits proliferation, migration, and invasion of esophageal squamous cell carcinoma cells. J Biol Chem. 2015;290:3925–3935. doi: 10.1074/jbc.M114.596866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li J.H., Liu S., Zhou H., Qu L.H., Yang J.H. Starbase v2.0: Decoding mirna-cerna, mirna-ncrna and protein-rna interaction networks from large-scale clip-seq data. Nucleic Acids Res. 2014;42:D92–97. doi: 10.1093/nar/gkt1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang Y.C., Di C., Hu B., Zhou M., Liu Y. Clipdb: A clip-seq database for protein-rna interactions. BMC Genomics. 2015;16:51. doi: 10.1186/s12864-015-1273-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao X., Chen D., Cai Y., Zhang F., Rbpvsmir X.J. A computational pipeline to identify competing mirnas and rna-binding protein pairs regulating the shared transcripts. Genes (Basel) 2018;9 doi: 10.3390/genes9090426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Assenov Y., Ramírez F., Schelhorn S.E., Lengauer T., Albrecht M. Computing topological parameters of biological networks. Bioinformatics. 2008;24:282–284. doi: 10.1093/bioinformatics/btm554. [DOI] [PubMed] [Google Scholar]
- 16.Sumazin P., Yang X., Chiu H.S., Chung W.J., Iyer A. An extensive microrna-mediated network of rna-rna interactions regulates established oncogenic pathways in glioblastoma. Cell. 2011;147:370–381. doi: 10.1016/j.cell.2011.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martinez-Jimenez F., Muinos F., Sentis I., Deu-Pons J., Reyes-Salazar I. A compendium of mutational cancer driver genes. Nat Rev Cancer. 2020;20:555–572. doi: 10.1038/s41568-020-0290-x. [DOI] [PubMed] [Google Scholar]
- 18.Sondka Z., Bamford S., Cole C.G., Ward S.A., Dunham I. The cosmic cancer gene census: describing genetic dysfunction across all human cancers. Nat Rev Cancer. 2018;18:696–705. doi: 10.1038/s41568-018-0060-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu H., Zhang Q., Lou Q., Zhang X., Cui Y. Differential analysis of lncrna, mirna and mrna expression profiles and the prognostic value of lncrna in esophageal cancer. Pathol Oncol Res. 2020;26:1029–1039. doi: 10.1007/s12253-019-00655-8. [DOI] [PubMed] [Google Scholar]
- 20.Shen K., Cao Z., Zhu R., You L., Zhang T. The dual functional role of microrna-18a (mir-18a) in cancer development. Clin Transl Med. 2019;8:32. doi: 10.1186/s40169-019-0250-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zimta A.A., Tigu A.B., Braicu C., Stefan C., Ionescu C. An emerging class of long non-coding rna with oncogenic role arises from the snorna host genes. Front Oncol. 2020;10:389. doi: 10.3389/fonc.2020.00389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shuai Y, Ma Z, Lu J, Feng J. Lncrna snhg15: A new budding star in human cancers. 2020, 53, e12716. [DOI] [PMC free article] [PubMed]
- 23.Lin Y., Zhang J., Cai J., Liang R., Chen G. Systematic analysis of gene expression alteration and co-expression network of eukaryotic initiation factor 4a–3 in cancer. J Cancer. 2018;9:4568–4577. doi: 10.7150/jca.27655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lu G, Chen L, Wu S, Feng Y, Lin T. Comprehensive analysis of tumor-infiltrating immune cells and relevant therapeutic strategy in esophageal cancer. 2020, 2020, 8974793. [DOI] [PMC free article] [PubMed]
- 25.Tao S., He H., Chen Q. Estradiol induces hotair levels via gper-mediated mir-148a inhibition in breast cancer. J Transl Med. 2015;13:131. doi: 10.1186/s12967-015-0489-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barabási A.L., Oltvai Z.N. Network biology: understanding the cell's functional organization. Nat Rev Genet. 2004;5:101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- 27.Minajigi A., Froberg J., Wei C., Sunwoo H., Kesner B. Chromosomes. A comprehensive xist interactome reveals cohesin repulsion and an rna-directed chromosome conformation. Science. 2015;349 doi: 10.1126/science.aab2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Obeng E.A., Stewart C., Abdel-Wahab O. Altered rna processing in cancer pathogenesis and therapy. Cancer Discov. 2019;9:1493–1510. doi: 10.1158/2159-8290.CD-19-0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolin S.L., Maquat L.E. Cellular rna surveillance in health and disease. Science. 2019;366:822–827. doi: 10.1126/science.aax2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wahl M.C., Will C.L., Luhrmann R. The spliceosome: design principles of a dynamic rnp machine. Cell. 2009;136:701–718. doi: 10.1016/j.cell.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 31.Wang E., Aifantis I. Rna splicing and cancer. Trends Cancer. 2020;6:631–644. doi: 10.1016/j.trecan.2020.04.011. [DOI] [PubMed] [Google Scholar]
- 32.Bowling E.A., Wang J.H., Gong F., Wu W., Neill N.J. Spliceosome-targeted therapies trigger an antiviral immune response in triple-negative breast cancer. Cell. 2021;184(384–403) doi: 10.1016/j.cell.2020.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bonnal S., Vigevani L., Valcarcel J. The spliceosome as a target of novel antitumour drugs. Nat Rev Drug Discov. 2012;11:847–859. doi: 10.1038/nrd3823. [DOI] [PubMed] [Google Scholar]
- 34.Sandri-Goldin R.M. The many roles of the regulatory protein icp27 during herpes simplex virus infection. Front Biosci. 2008;13:5241–5256. doi: 10.2741/3078. [DOI] [PubMed] [Google Scholar]
- 35.Braeutigam C., Rago L., Rolke A., Waldmeier L., Christofori G. The rna-binding protein rbfox2: an essential regulator of emt-driven alternative splicing and a mediator of cellular invasion. Oncogene. 2014;33:1082–1092. doi: 10.1038/onc.2013.50. [DOI] [PubMed] [Google Scholar]
- 36.Choi S., Sa M., Cho N., Kim K.K., Park S.H. Rbfox2 dissociation from stress granules suppresses cancer progression. Exp Mol Med. 2019;51:1–12. doi: 10.1038/s12276-019-0246-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mohibi S., Chen X., Zhang J. Cancer the'rbp'eutics-rna-binding proteins as therapeutic targets for cancer. Pharmacol Ther. 2019;203 doi: 10.1016/j.pharmthera.2019.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dai W, He J. Mir-148b-3p, mir-190b, and mir-429 regulate cell progression and act as potential biomarkers for breast cancer. 2019, 22, 219–236. [DOI] [PMC free article] [PubMed]
- 39.Jiang L., Huang J., Higgs B.W., Hu Z., Xiao Z. Genomic landscape survey identifies srsf1 as a key oncodriver in small cell lung cancer. PLoS Genet. 2016;12 doi: 10.1371/journal.pgen.1005895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pang B., Wang Q., Ning S., Wu J., Zhang X. Landscape of tumor suppressor long noncoding rnas in breast cancer. J Exp Clin Cancer Res. 2019;38:79. doi: 10.1186/s13046-019-1096-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao Y., Ma K., Yang S., Zhang X., Wang F. Microrna-125a-5p enhances the sensitivity of esophageal squamous cell carcinoma cells to cisplatin by suppressing the activation of the stat3 signaling pathway. Int J Oncol. 2018;53:644–658. doi: 10.3892/ijo.2018.4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen X., Kong J., Ma Z., Gao S., Feng X. Up regulation of the long non-coding rna neat1 promotes esophageal squamous cell carcinoma cell progression and correlates with poor prognosis. Am J Cancer Res. 2015;5:2808–2815. [PMC free article] [PubMed] [Google Scholar]
- 43.Peters C.J., Rees J.R., Hardwick R.H., Hardwick J.S., Vowler S.L. A 4-gene signature predicts survival of patients with resected adenocarcinoma of the esophagus, junction, and gastric cardia. Gastroenterology. 2010;139:1995–2004.e1915. doi: 10.1053/j.gastro.2010.05.080. [DOI] [PubMed] [Google Scholar]
- 44.Charizanis K., Lee K.Y., Batra R., Goodwin M., Zhang C. Muscleblind-like 2-mediated alternative splicing in the developing brain and dysregulation in myotonic dystrophy. Neuron. 2012;75:437–450. doi: 10.1016/j.neuron.2012.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee K.Y., Li M., Manchanda M., Batra R., Charizanis K. Compound loss of muscleblind-like function in myotonic dystrophy. EMBO Mol Med. 2013;5:1887–1900. doi: 10.1002/emmm.201303275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shapiro I.M., Cheng A.W., Flytzanis N.C., Balsamo M., Condeelis J.S. An emt-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genet. 2011;7 doi: 10.1371/journal.pgen.1002218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang J., Zheng Z., Wu M., Zhang L., Wang J. The natural compound neobractatin inhibits tumor metastasis by upregulating the rna-binding-protein mbnl2. Cell Death Dis. 2019;10:554. doi: 10.1038/s41419-019-1789-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang Y., Yang H., Xu M., Zhang H., Sun M. Long non-coding rna (lncrna) magi2-as3 inhibits breast cancer cell growth by targeting the fas/fasl signalling pathway. Hum Cell. 2018;31:232–241. doi: 10.1007/s13577-018-0206-1. [DOI] [PubMed] [Google Scholar]
- 49.Wang F., Zu Y., Zhu S., Yang Y., Huang W. Long noncoding rna magi2-as3 regulates ccdc19 expression by sponging mir-15b-5p and suppresses bladder cancer progression. Biochem Biophys Res Commun. 2018;507:231–235. doi: 10.1016/j.bbrc.2018.11.013. [DOI] [PubMed] [Google Scholar]
- 50.Li D., Wang J., Zhang M., Hu X., She J. Lncrna magi2-as3 is regulated by brd4 and promotes gastric cancer progression via maintaining zeb1 overexpression by sponging mir-141/200a. Mol Ther Nucleic Acids. 2020;19:109–123. doi: 10.1016/j.omtn.2019.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kurogi R., Nakamizo A., Suzuki S.O., Mizoguchi M., Yoshimoto K. Inhibition of glioblastoma cell invasion by hsa-mir-145-5p and hsa-mir-31-5p co-overexpression in human mesenchymal stem cells. J Neurosurg. 2018;130:44–55. doi: 10.3171/2017.8.JNS1788. [DOI] [PubMed] [Google Scholar]
- 52.Sun Z., Chen J., Zhang J., Ji R., Xu W. The role and mechanism of mir-374 regulating the malignant transformation of mesenchymal stem cells. Am J Transl Res. 2018;10:3224–3232. [PMC free article] [PubMed] [Google Scholar]
- 53.Amr K.S., Elmawgoud Atia H.A., Elazeem Elbnhawy R.A., Ezzat W.M. Early diagnostic evaluation of mir-122 and mir-224 as biomarkers for hepatocellular carcinoma. Genes Dis. 2017;4:215–221. doi: 10.1016/j.gendis.2017.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Allegri L., Baldan F., Roy S., Aube J., Russo D. The hur cmld-2 inhibitor exhibits antitumor effects via mad2 downregulation in thyroid cancer cells. Sci Rep. 2019;9:7374. doi: 10.1038/s41598-019-43894-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Muralidharan R., Mehta M., Ahmed R., Roy S., Xu L. Hur-targeted small molecule inhibitor exhibits cytotoxicity towards human lung cancer cells. Sci Rep. 2017;7:9694. doi: 10.1038/s41598-017-07787-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu X., Wang S., Meng F., Wang J., Zhang Y. Sm2mir: A database of the experimentally validated small molecules' effects on microrna expression. Bioinformatics. 2013;29:409–411. doi: 10.1093/bioinformatics/bts698. [DOI] [PubMed] [Google Scholar]
- 57.Wu N., Wu G.C., Hu R., Li M., Feng H. Ginsenoside rh2 inhibits glioma cell proliferation by targeting microrna-128. Acta Pharmacol Sin. 2011;32:345–353. doi: 10.1038/aps.2010.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li X., Chu S., Lin M., Gao Y., Liu Y. Anticancer property of ginsenoside rh2 from ginseng. Eur J Med Chem. 2020;203 doi: 10.1016/j.ejmech.2020.112627. [DOI] [PubMed] [Google Scholar]
- 59.Kong Y., Qu F., Yuan X., Yan X., Yu W. Effect of bicalutamide on the proliferation and invasion of human triple negative breast cancer mda-mb-231 cells. Medicine (Baltimore) 2020;99 doi: 10.1097/MD.0000000000019822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pereira B., Billaud M., Almeida R. Rna-binding proteins in cancer: Old players and new actors. Trends. Cancer. 2017;3:506–528. doi: 10.1016/j.trecan.2017.05.003. [DOI] [PubMed] [Google Scholar]
- 61.Li Y., Zhuang L., Wang Y., Hu Y., Wu Y. Connect the dots: a systems level approach for analyzing the mirna-mediated cell death network. Autophagy. 2013;9:436–439. doi: 10.4161/auto.23096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhao X., Chen Q., Cai Y., Chen D., Bei M. Tra2a binds with lncrna malat1 to promote esophageal cancer progression by regulating ezh2/beta-catenin pathway. J Cancer. 2021;12:4883–4890. doi: 10.7150/jca.55661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jonas K., Calin G.A., Pichler M. Rna-binding proteins as important regulators of long non-coding rnas in cancer. Int J Mol Sci. 2020;21 doi: 10.3390/ijms21082969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xie W., Zhu H., Zhao M., Wang L., Li S. Crucial roles of different rna-binding hnrnp proteins in stem cells. Int J Biol Sci. 2021;17:807–817. doi: 10.7150/ijbs.55120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li L., Xu J., Yang D., Tan X., Wang H. Computational approaches for microrna studies: a review. Mamm Genome. 2010;21:1–12. doi: 10.1007/s00335-009-9241-2. [DOI] [PubMed] [Google Scholar]
- 66.Karreth F.A., Pandolfi P.P. Cerna cross-talk in cancer: when ce-bling rivalries go awry. Cancer Discov. 2013;3:1113–1121. doi: 10.1158/2159-8290.CD-13-0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hutter C., Zenklusen J.C. The cancer genome atlas: creating lasting value beyond its data. Cell. 2018;173:283–285. doi: 10.1016/j.cell.2018.03.042. [DOI] [PubMed] [Google Scholar]
- 68.Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W. Limma powers differential expression analyses for rna-sequencing and microarray studies. Nucleic Acids Res. 2015;43 doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jiang W., Mitra R., Lin C.C., Wang Q., Cheng F. Systematic dissection of dysregulated transcription factor-mirna feed-forward loops across tumor types. Brief Bioinform. 2016;17:996–1008. doi: 10.1093/bib/bbv107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Robinson M.D., McCarthy D.J., Edger S.G.K. A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bao Z., Yang Z., Huang Z., Zhou Y., Cui Q. Lncrnadisease 2.0: An updated database of long non-coding rna-associated diseases. Nucleic Acids Res. 2019;47:D1034–d1037. doi: 10.1093/nar/gky905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gao Y., Wang P., Wang Y., Ma X., Zhi H. Lnc2cancer v2.0: updated database of experimentally supported long non-coding rnas in human cancers. Nucleic Acids Res. 2019;47:D1028–d1033. doi: 10.1093/nar/gky1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li Y., Qiu C., Tu J., Geng B., Yang J. Hmdd v2.0: a database for experimentally supported human microrna and disease associations. Nucleic Acids Res. 2014;42:D1070–1074. doi: 10.1093/nar/gkt1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gong J., Shao D., Xu K., Lu Z., Lu Z.J. Rise: a database of rna interactome from sequencing experiments. Nucleic Acids Res. 2018;46:D194–d201. doi: 10.1093/nar/gkx864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dweep H., Gretz N. Mirwalk2.0: a comprehensive atlas of microrna-target interactions. Nat Methods. 2015;12:697. doi: 10.1038/nmeth.3485. [DOI] [PubMed] [Google Scholar]
- 76.Yu G., Li F., Qin Y., Bo X., Wu Y. Gosemsim: an r package for measuring semantic similarity among go terms and gene products. Bioinformatics. 2010;26:976–978. doi: 10.1093/bioinformatics/btq064. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Summary of the RBP-ncRNA regulation network and the samples used in this study.
The significantly dysregulated RNCs in esophageal cancer.
The significantly dysregulated RNCs in breast cancer.
RNCs with similar nodes function in breast and esophageal cancers.
Survival analysis of RNCs in breast cancer and esophageal cancer.
Dysregulated RNCs across 14 tumor types and their association with clinical outcomes.
Comparison of node degree between the pan-RNC and the other dysregulated RNC in each cancer.