

Abstract
Background & Aims
The inflammatory bowel diseases (IBDs), Crohn’s disease and ulcerative colitis, are caused in part by aberrant immune responses to resident intestinal bacteria. Certain dietary components, including carbohydrates, are associated with IBDs and alter intestinal bacterial composition. However, the effects of luminal carbohydrates on the composition and colitogenic potential of intestinal bacteria are incompletely understood. We hypothesize that carbohydrate metabolism by resident proinflammatory intestinal bacteria enhances their growth and worsens intestinal inflammation.
Methods
We colonized germ-free, wild-type, and colitis-susceptible interleukin-10 knockout mice (Il10-/-) with a consortium of resident intestinal bacterial strains and quantified colon inflammation using blinded histologic scoring and spontaneous secretion of IL12/23p40 by colon explants. We measured luminal bacterial composition using real-time 16S polymerase chain reaction, bacterial gene expression using RNA sequencing and real-time polymerase chain reaction, and luminal glucosamine levels using gas chromatography–mass spectrometry.
Results
We show that a consortium of 8 bacterial strains induces severe colitis in Il10-/- mice and up-regulates genes associated with carbohydrate metabolism during colitis. Specifically, Enterococcus faecalis strain OG1RF is proinflammatory and strongly up-regulates OG1RF_11616-11610, an operon that encodes genes of a previously undescribed phosphotransferase system that we show imports glucosamine. Experimental colitis is associated with increased levels of luminal glucosamine and OG1RF_11616 causes worse colitis, not by increasing E faecalis numbers, but rather by mechanisms that require the presence of complex microbiota.
Conclusions
Further studies of luminal carbohydrate levels and bacterial carbohydrate metabolism during intestinal inflammation will improve our understanding of the pathogenesis of IBDs and may lead to the development of novel therapies for these diseases.
Keywords: Inflammatory Bowel Diseases, Phosphotransferase System, Gene Expression
Abbreviations used in this paper: BHI, brain heart infusion; cDNA, complementary DNA; Ct, cycle threshold; IBD, inflammatory bowel disease; IL, interleukin; mRNA, messenger RNA; NCBI, National Center for Biotechnology Information; PCR, polymerase chain reaction; PTS, phosphotransferase system; RNA-seq, RNA sequencing; rRNA, ribosomal RNA; WT, wild-type; EIIA, enzyme IIA
Summary.
Dietary factors and resident intestinal bacteria are associated with inflammatory bowel diseases. We show that colonic glucosamine levels are increased during experimental colitis and that Enterococcus faecalis up-regulates glucosamine metabolism, which is associated with worse experimental colitis.
Inflammatory bowel diseases (IBDs), including Crohn’s disease and ulcerative colitis, are chronic, immune-mediated disorders of the gastrointestinal tract that afflict as many as 8 in 1000 people in Europe and North America and incur an average of US $30,000 per person annually in direct and indirect health care costs.1,2 Although the etiologies of IBDs are currently unclear, a prevailing hypothesis is that they are caused in part by aberrant immune responses to intestinal bacteria in genetically susceptible individuals.
Several studies have shown that IBDs are associated with altered composition of the normal intestinal bacterial community.3, 4, 5 In addition to shifts in general bacterial composition, IBDs also have been associated with alterations in the abundances of specific bacterial species. For example, Crohn’s disease is associated with increased numbers of mucosal and fecal Proteobacteria, particularly adherent-invasive Escherichia coli.6 Studies also have shown increased Enterococcus species7, 8, 9 and Ruminococcus species,10,11 but decreased Bacteroides species12,13 in feces from IBD patients vs controls. Despite well-documented associations between bacterial community composition and IBDs, it is still unknown whether these associations cause, or result from, IBDs. Moreover, other factors such as host genetic susceptibility and diet likely also contribute to the development of these diseases.
Dietary factors also are positively and negatively associated with the risk of developing IBDs. For example, consumption of fruit fiber correlates with a reduced risk of developing Crohn’s disease14 and decreased endoscopic activity of ulcerative colitis.15 High animal protein intake is associated with an increased risk of developing IBDs.16 Several cross-sectional studies have shown associations between increased sugar consumption and Crohn’s disease,17, 18, 19 although evidence for a similar connection in ulcerative colitis is lacking. Studies of diet on experimental murine colitis have shown a causal role of dietary components in intestinal inflammation, including certain fats, emulsifiers, and fiber.20, 21, 22, 23, 24 These findings suggest that dietary components may play a role in the pathogenesis of IBDs.
Diet may impact intestinal inflammation via its effects on the resident intestinal microbiota. For instance, interactions between diet and bacteria can produce characteristic changes in intestinal lumen metabolite profiles in IBDs. Untargeted metabolomic studies have shown increased levels of sphingolipids, acylcarnitines, certain primary bile acids, and polyunsaturated fatty acids, but decreased levels of short-chain fatty acids, triacylglycerols, tetrapyrroles, pantothenate, and nicotinate in feces from IBD patients compared with healthy controls.3,4 Although these studies have been informative, the metabolomic techniques used are unable to accurately quantify most carbohydrates.
Only a few studies have used more targeted methods to measure concentrations of specific carbohydrates in feces from IBD patients. For example, Le Gall et al25 detected increased levels of glucose in ulcerative colitis patients. Bacterial metabolism of luminal carbohydrates also may contribute to IBDs as shown by increased numbers of genes in carbohydrate metabolism pathways in fecal bacterial metagenomes from IBD patients vs controls.26 Given the epidemiologic evidence that connects sugar consumption with Crohn’s disease and the enrichment of fecal bacterial carbohydrate metabolism genes in patients with IBDs, further study of the roles of luminal carbohydrate levels and carbohydrate metabolism by bacteria in the pathogenesis of IBDs is warranted. Understanding these interactions in IBD patients could potentially lead to the development of new therapies or diet recommendations that improve disease activity.
In the current studies, we hypothesize that carbohydrate metabolism by resident proinflammatory intestinal bacteria enhances their growth and worsens intestinal inflammation. Because the complexity and variability of naturally occurring intestinal bacterial communities complicate mechanistic studies of this concept in human beings, we tested our hypothesis in a mouse model of chronic colitis in which we selectively colonized germ-free interleukin-10–deficient (Il10-/-) mice with a simplified consortium of 8 nonpathogenic, resident intestinal bacterial species. We report that experimental colitis is associated with increased levels of colonic luminal glucosamine and induces Enterococcus faecalis, a bacterial species known to cause experimental murine colitis,27,28 to increase expression of phosphotransferase genes that encode proteins that import glucosamine. We also found that the presence of an intact E faecalis glucosamine phosphotransferase pathway worsens experimental colitis and is associated with decreased Bacteroides species, but, contrary to our hypothesis, does not increase E faecalis numbers during colitis.
Results
Experimental Colitis Is Associated With Altered Luminal Bacteria Composition and Transcription in a Simplified Bacterial Community
We previously reported that chronic immune-mediated experimental colitis in Il10-/- mice induces differential expression of genes in luminal bacteria from mice monocolonized with resident intestinal bacterial strains.29,30 Other studies have reported that acute, chemically induced colitis induces changes in the luminal bacterial metatranscriptome in conventionally housed wild-type (WT) mice.31 However, little is known about how chronic, immune-mediated colitis affects the luminal bacterial metatranscriptome in mice colonized with a community of resident intestinal bacterial strains.
To determine this, we selectively colonized WT and Il10-/- mice for 10 weeks with a consortium of 8 bacterial species (Table 1) that represent the 4 major bacterial phyla in the mammalian intestine: Firmicutes, Bacteroidetes, Proteobacteria, and Actinobacteria. We selected these strains based on publicly available full-genome sequence at the time of the experiment and the association of several of the species with IBD or experimental colitis.10,27,28,32, 33, 34, 35, 36, 37 We inoculated germ-free mice by oral gavage of 200 μL of an equal-volume mixture of the anaerobically grown bacterial strains (see the Methods section). We previously reported that Il10-/- mice colonized with these strains have significantly greater histologic colitis and spontaneous secretion of the proinflammatory cytokine IL12/23p40 by colon explant cultures compared with WT mice.38 Specifically, average composite histologic inflammation scores were 8.9 (SEM, ±0.42) and 3.5 (SEM, ±0.42) in Il10-/- and WT mice, respectively (t test, P < .0005). Average IL12/23p40 secretion was 106 (SEM, ±6.7) and 2.41 (SEM, ±0.30) ng/mg tissue in Il10-/- and WT mice, respectively (t test, P < .0005). In the present studies, measurements of bacterial composition in cecal contents from these mice showed the presence of all species except Lactobacillus rhamnosus. Comparing the bacterial composition in the 2 groups of mice, we found significantly fewer Ruminococcus gnavus, Bacteroides thetaiotaomicron, E faecalis, and Faecalibacterium prausnitzii, slightly higher levels of Bifidobacterium longum, but unchanged levels of E coli and Bacteroides vulgatus in the cecal content of Il10-/- compared with WT mice (Figure 1A).
Table 1.
Bacterial Strains Used in the Current Studies
| Species | Strain | Source |
|---|---|---|
| Escherichia coli | NC101 | Mouse feces |
| Enterococcus faecalis | OG1RF | Human oral cavity |
| Bacteroides vulgatus | ATCC 8482 | Human feces |
| Bacteroides thetaiotaomicron | VPI-5482 | Human feces |
| Bifidobacterium longum | ATCC 15697 | Human feces |
| Lactobacillus rhamnosus | GG | Human feces |
| Ruminococcus gnavus | ATCC 29149 | Human feces |
| Faecalibacterium prausnitzii | A2-165 | Human feces |
Figure 1.
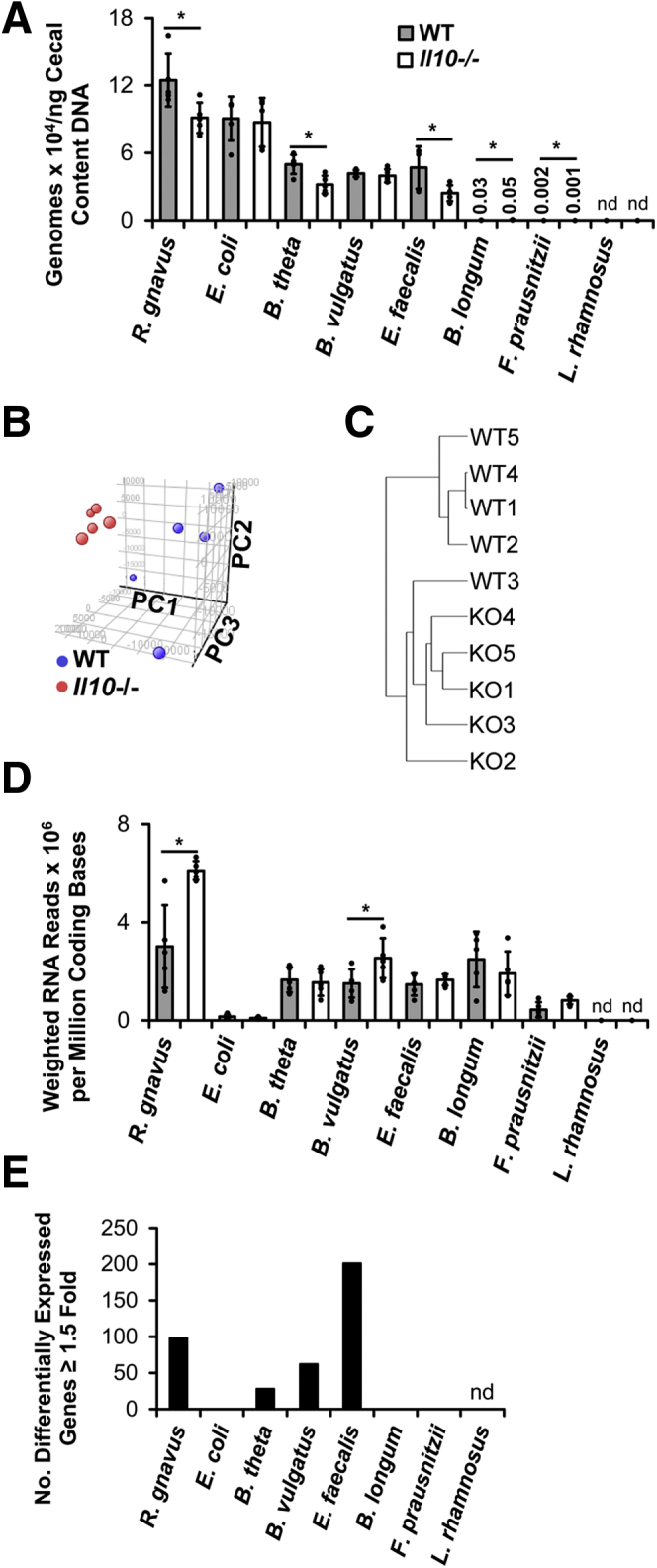
Experimental colitis is associated with altered luminal bacterial metatranscriptomes of a simplified consortium of resident intestinal bacterial strains. (A) Concentrations of bacterial species in cecal content from WT and Il10-/- mice colonized with the 8 indicated bacterial strains for 10 weeks. (B) Principle component analysis of bacterial metatranscriptomes in cecal content of the same mice. (C) Hierarchical clustering and Euclidean distances of bacterial metatranscriptomes from each mouse. (D) Transcriptional activity of bacterial strains in cecal content of the same mice. (E) Numbers of differentially expressed genes (≥1.5-fold) in bacterial strains in cecal content of the same mice (n = 5 mice/group, data presented as means ± SD, ∗P < .05 Il10-/- vs WT, Student t test). KO, Il10-/-; nd, not detected; PC, principle component.
Because half of the bacterial strains showed decreased relative proportions in cecal content from Il10-/- vs WT mice, we asked whether total bacterial density is lower in cecal content from Il10-/- mice. To answer this, we first measured total DNA concentrations in cecal contents from Il10-/- and WT mice (n = 6/group) selectively colonized for 8 weeks with the same bacterial strains as described earlier and detected an average of 1109 (SEM, ±133) and 384 (SEM, ±50) ng DNA/mg cecal contents in Il10-/- and WT mice, respectively (t test, P < .0005). To account for potential differences in the amount of mouse DNA in cecal contents from the 2 groups, we performed real-time polymerase chain reaction (PCR) on total DNA using universal bacterial 16S primers and found that cecal content DNA from Il10-/- mice contained 0.74-fold lower bacterial 16S copies than that from WT mice. Using this value to correct for differences in relative proportions of bacterial vs eukaryotic DNA, we estimate that cecal content from these Il10-/- mice contains approximately 820 ng bacterial DNA/mg cecal content compared with 384 ng bacterial DNA/mg cecal content in WT mice. Together these data suggest that total cecal content bacterial concentrations are actually higher in Il10-/- vs WT mice. The reasons why cecal bacterial DNA is increased in Il10-/- vs WT mice are unknown and will require further investigation.
Unsupervised analysis of the cecal bacterial metatranscriptomes has shown that those in the Il10-/- mice are significantly different from, and cluster more closely together than, those in the WT mice (Figure 1B). Hierarchical clustering by Euclidean distance confirms that metatranscriptomes from WT mice 1, 2, 4, and 5 cluster together and are distinct from the metatranscriptomes of the other mice (Figure 1C). These data suggest that the cecal environment in Il10-/- mice with colitis is distinct from, and more consistent than, that in WT mice. We then estimated overall transcriptional activity in each bacterial strain by dividing the number of mRNA reads for each strain by the number of coding bases in that strain’s genome and the fraction of that strain’s genomes in the sample. Transcriptional activity was lowest in E coli and F prausnitzii in both groups of mice and was significantly higher in R gnavus and B vulgatus in Il10-/- vs WT mice (Figure 1D).
To more fully characterize the transcriptional changes in bacteria from Il10-/- vs WT mice, we conducted a conserved domain analysis of the microbial RNA sequencing (RNA-seq) data sets. Briefly, we generated a custom database of National Center for Biotechnology Information (NCBI)-identified conserved protein domains for each bacterial gene in the consortium and then applied this database and the microbial RNA-seq data sets in gene set enrichment analysis to identify conserved protein domains that are enriched in cecal bacteria from Il10-/- vs WT mice. Several domains involved in protein translation, sugar metabolism, and other processes are enriched significantly in bacteria from Il10-/- vs WT mice (Table 2). Only 3 domains are enriched significantly in bacteria from WT compared with Il10-/- mice, 2 of which are important in DNA transposition (Table 3).
Table 2.
Top 15 Conserved Protein Domains Enriched in Cecal Bacteria From Il10-/- Mice With Colitis Vs Healthy WT Mice
| Conserved domain | Description | Normalized enrichment score | FDR q-value |
|---|---|---|---|
| CL00268 | Class II tRNA aminio-acyl synthetase-like catalytic core domain | 2.12 | 0.0010 |
| CL00015 | Nucleotidyl transferase superfamily | 2.05 | 0.0030 |
| CL02787 | Elongation factor Tu domain-like proteins | 2.05 | 0.0020 |
| CL12020 | Anticodon-binding domain of class Ia aminoacyl tRNA synthetases | 2.05 | 0.0018 |
| CL07225 | Domain of unknown function (DUF1735) | 1.93 | 0.0106 |
| CL03132 | Mur ligase family, catalytic domain | 1.92 | 0.0117 |
| CD06223 | Phosphoribosyl transferase domain | 1.90 | 0.0013 |
| CL00102 | Laminin G domain | 1.89 | 0.0128 |
| CL00266 | Histidyl, glycyl, threonyl, and prolyl anticodon binding domain | 1.89 | 0.0119 |
| CL00354 | Kyprides, Ouzounis, Woese motif | 1.89 | 0.0123 |
| CL00939 | Bacterial sugar transferase | 1.87 | 0.0149 |
| CL12026 | Gly radical superfamily | 1.86 | 0.0169 |
| CL00309 | Phosphoribosyl transferase domain | 1.85 | 0.0168 |
| CL15445 | PTS regulation domain | 1.84 | 0.0191 |
| CL09927 | Ribosomal protein S1-like RNA-binding domain | 1.83 | 0.0182 |
FDR, false-discovery rate; tRNA, transfer RNA.
Table 3.
Top 15 Conserved Protein Domains Enriched in Cecal Bacteria From Healthy WT Mice Vs Il10-/- Mice With Colitis
| Conserved domain | Description | Normalized enrichment score | FDR q-value |
|---|---|---|---|
| CL15789 | DDE superfamily endonuclease | -2.62 | 0.0000 |
| CL16926 | Transposase domain of unknown function (DUF772) | -2.54 | 0.0000 |
| CD02947 | Thioredoxin family | -1.92 | 0.0520 |
| CL02811 | E1-E2 ATPase superfamily | -1.92 | 0.0403 |
| CL04740 | FecR protein | -1.79 | 0.0991 |
| CL10482 | KefB superfamily | -1.70 | 0.1552 |
| CL06502 | Two-component regulator propeller | -1.62 | 0.2176 |
| CL00514 | Nitro FMN reductase superfamily | -1.61 | 0.2038 |
| CL08372 | Pyridine nucleotide-disulfide oxidoreductase, dimerization domain | -1.59 | 0.2151 |
| CL14891 | Putative helix-turn-helix domain of transposase IS66 | -1.55 | 0.2506 |
| CL00264 | Ferritin-like superfamily | -1.51 | 0.2884 |
| CL01155 | Porin superfamily | -1.50 | 0.2835 |
| CL14647 | Glycosyl hydrolase families GH43, 62, 32, 68, 117, 130 | -1.50 | 0.2709 |
| CD02966 | TlpA-like family | -1.44 | 0.3498 |
| CL00388 | Thioredoxin-like superfamily | -1.40 | 0.3992 |
ATPase, adenosine triphosphatase; FDR, false-discovery rate; FMN, flavin mononucleotide.
In supervised analyses, we identified bacterial genes that were up- or down-regulated significantly in bacteria from Il10-/- mice at least 1.5-fold and found that E faecalis had the highest number of differentially expressed genes whereas E coli, B longum, and F prausnitzii had no significantly differentially expressed genes (Figures 1E and 2). Of the 15 most highly down-regulated bacterial genes in Il10-/- mice, most were from B vulgatus, were down-regulated only modestly (ie, 3- to 5-fold), and encoded hypothetical proteins (Table 4, Supplementary Table 1). Of the 15 most highly up-regulated bacterial genes in Il10-/- mice, most were from R gnavus, were highly up-regulated (ie, 27- to 41-fold), and encoded proteins involved in DNA replication (Table 5, Supplementary Table 1). The only non–R gnavus gene in this set of 15 up-regulated genes was from E faecalis and encoded a putative protein predicted to belong to the phosphotransferase system (PTS) family, a family of molecules that import carbohydrates into bacteria.
Figure 2.
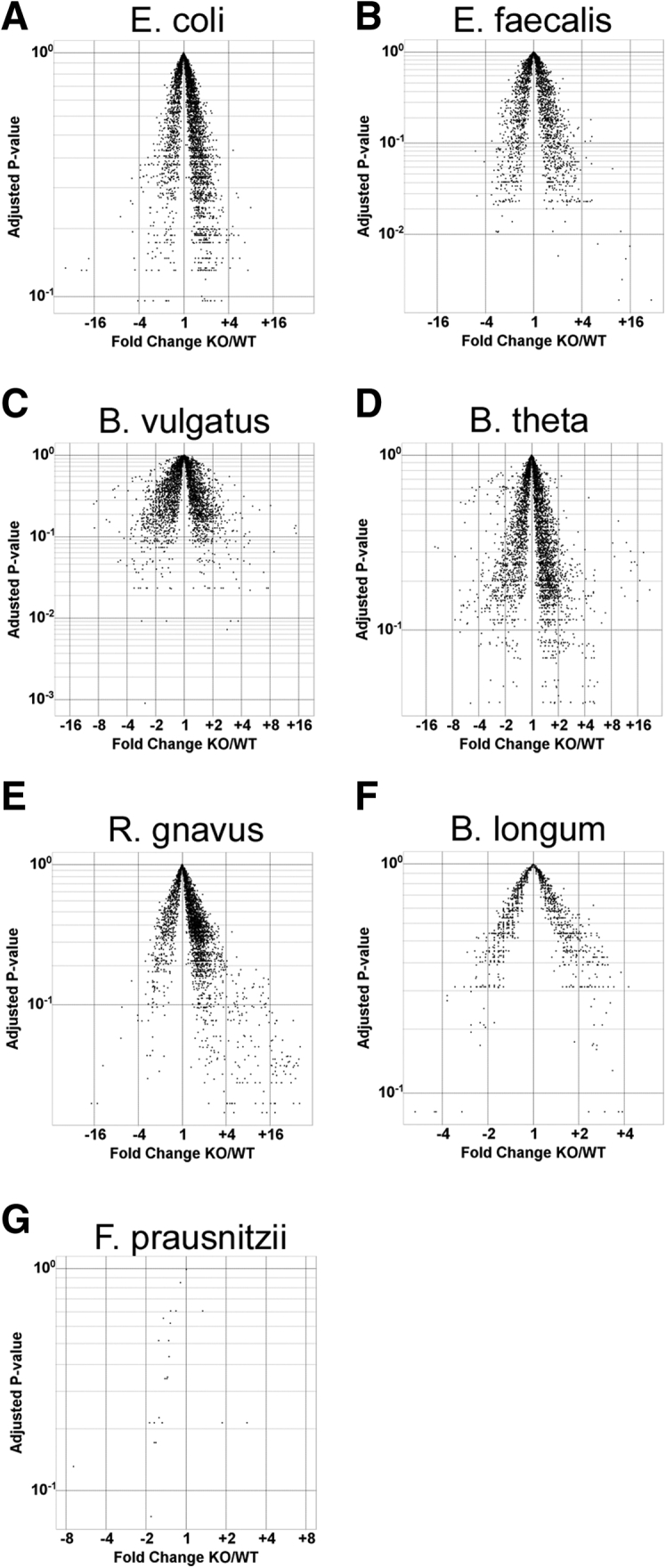
Resident intestinal bacteria differentially express genes during experimental colitis. Volcano plots of individual bacterial RNA-seq data sets (reads per kilobase per million) from the same cecal content samples as in Figure 1. X-axis represents fold-change (KO/WT) and Y-axis represents adjusted P values. KO, Il10-/-.
Table 4.
Top 15 Down-regulated Genes in Cecal Bacteria From Healthy WT Mice Vs Il10-/- Mice With Colitis
| Gene locus tag | Description | Fold change KO/WT |
|---|---|---|
| RUMGNA_00582 | Sigma-70 region 2 | 0.056 |
| RUMGNA_01937 | Hypothetical protein | 0.071 |
| RUMGNA_00581 | Hypothetical protein | 0.083 |
| OG1RF_11321 | Hypothetical protein | 0.196 |
| BT_0782 | Hypothetical protein | 0.200 |
| BVU_3435 | Hypothetical protein | 0.204 |
| BVU_1055 | Hypothetical protein | 0.213 |
| BVU_2182 | Hypothetical protein | 0.278 |
| BVU_3508 | Methylglyoxal synthase | 0.294 |
| OG1RF_12506 | Cell wall surface anchor family protein | 0.294 |
| BVU_3615 | Hypothetical protein | 0.294 |
| BVU_3614 | Hypothetical protein | 0.303 |
| OG1RF_10861 | Hypothetical protein | 0.303 |
| BVU_3100 | Hypothetical protein | 0.303 |
| BVU_2305 | Hypothetical protein | 0.313 |
KO, Il10-/-.
Table 5.
Top 15 Up-regulated Genes in Cecal Bacteria From Il10-/- Mice With Colitis Vs Healthy WT Mice
| Gene locus tag | Description | Fold change KO/WT |
|---|---|---|
| RUMGNA_01176 | Hypothetical protein | 41 |
| RUMGNA_02294 | Hypothetical protein | 40 |
| RUMGNA_02289 | Replication initiator protein A domain protein | 39 |
| RUMGNA_02295 | Hypothetical protein | 37 |
| RUMGNA_02290 | Hypothetical protein | 36 |
| RUMGNA_02293 | Hypothetical protein | 35 |
| RUMGNA_02573 | Coat F domain protein | 34 |
| RUMGNA_02287 | Replication initiator protein A domain protein | 34 |
| RUMGNA_02285 | Exonuclease | 31 |
| RUMGNA_02292 | CobQ/CobB/MinD/ParA nucleotide binding domain protein | 31 |
| RUMGNA_02288 | Hypothetical protein | 31 |
| RUMGNA_02291 | Hypothetical protein | 30 |
| OG1RF_11616 | PTS family mannose/fructose/sorbose porter component IIA | 29 |
| RUMGNA_02268 | Hypothetical protein | 28 |
| RUMGNA_02277 | Cna protein B-type domain protein | 27 |
KO, Il10-/-.
Together, these data show that chronic immune-mediated experimental colitis in mice colonized with a defined consortium of bacterial species is associated with significant shifts in luminal bacterial composition and function. In particular, colitis induces E faecalis to differentially express a large number of genes, including strong up-regulation of a putative PTS that may be involved in carbohydrate metabolism.
E faecalis Exacerbates Experimental Colitis
Because human IBDs and experimental colitis in Il10-/- mice are chronic and often progressive conditions, we next hypothesized that up-regulation of genes in colitogenic bacteria during colitis increases their fitness in the inflamed colon and exacerbates chronic inflammation in a positive feedback loop fashion. Because R gnavus is not easily genetically tractable, we chose to test this hypothesis by studying the effects of up-regulated E faecalis genes on colitis severity.
Others have shown previously that E faecalis OG1RF causes colitis in monocolonized Il10-/- mice and worsens colitis in E faecalis OG1RF–E coli NC101 dual-colonized Il10-/- mice.27,28,30 However, the colitogenic potential of E faecalis OG1RF in the more complex consortium of 8 bacterial species listed in Table 1 has not been explored. To examine this, we selectively colonized germ-free Il10-/- mice for 8 weeks with the bacterial species listed in Table 1, including or not including E faecalis OG1RF. We found that the inclusion of E faecalis OG1RF in the consortium caused increased histologic colon inflammation and spontaneous secretion of IL12/23p40 by colon explant cultures (Figure 3). Similar to previously reported findings in monocolonized Il10-/- mice, the presence of E faecalis OG1RF in this more complex bacterial consortium was associated with worse distal compared with proximal colitis (Figure 3).
Figure 3.
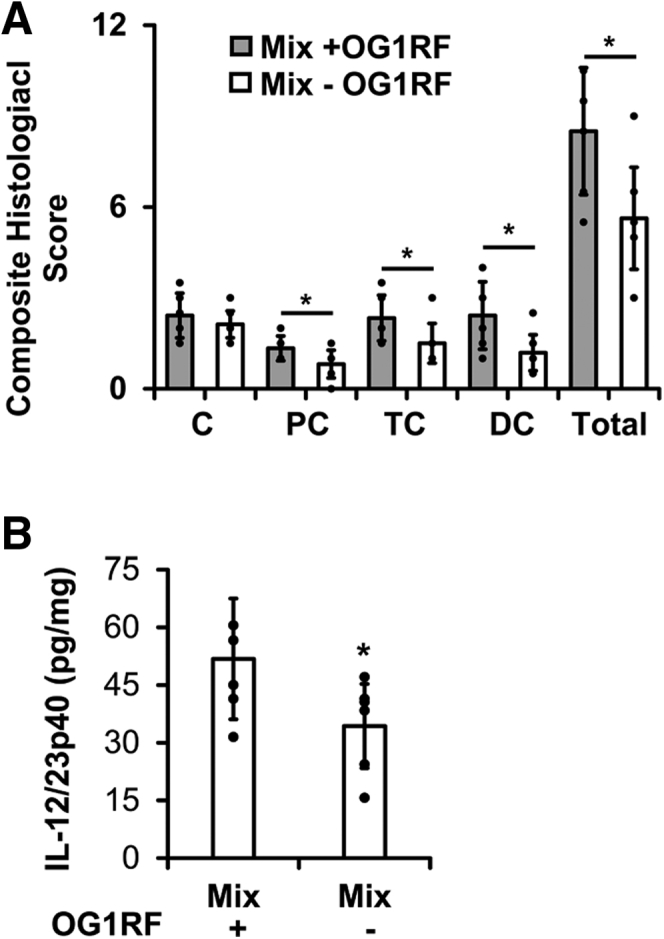
E faecalis worsens experimental colitis in mice colonized with a simplified consortium of resident intestinal bacterial strains. (A) Histologic colitis scores in cecum (C), proximal colon (PC), transverse colon (TC), and distal colon (DC) from Il10-/- mice colonized with the same resident intestinal bacterial strains as in Figure 1 with (Mix + OG1RF) or without (Mix - OG1RF) E faecalis. (B) IL12/23p40 secretion by colon explant cultures from the same mice (n = 6–8 mice/group, data presented as means ± SD, ∗P < .05 Mix + OG1RF vs Mix - OG1RF, Student t test).
E faecalis OG1RF Up-regulates the Putative PTS Operon OG1RF_11616-11610 During Experimental Colitis
Because E faecalis OG1RF in complex bacterial communities worsens experimental colitis and has the largest number of differentially expressed genes during colitis, we next sought to more fully characterize the most highly up-regulated E faecalis genes during colitis. The 7 most highly up-regulated E faecalis genes at 10 weeks after colonization all belonged to a putative PTS operon OG1RF_11616-11610 (Supplementary Table 1, Figure 4). A variety of gram-positive and gram-negative bacterial species use PTS to import and phosphorylate extracellular carbohydrates.39 Current NCBI annotation of the E faecalis OG1RF genome predicts 39 PTS operons, but the carbohydrate substrates for most of these, including OG1RF_11616-11610, have not been determined experimentally.
Figure 4.

E faecalis up-regulates expression of a putative PTS operon, OG1RF_11616-11610, during experimental colitis. (A) Organization of open reading frames in the putative PTS operon, OG1RF_11616-11610, in E faecalis. NCBI annotations are indicated below, and fold change (Il10-/- vs WT mice from Figure 1) is indicated above, each open reading frame. (B) E faecalis OG1RF_116616 mRNA abundance in cecal content from WT and Il10-/- mice colonized with the 8 bacterial strains as in Figure 1 for 5–10 weeks (n = 4–6 mice/group, data presented as means ± SD, ∗P < .05 Il10-/- vs WT, Student t test).
Il10-/- mice colonized with the earlier-mentioned bacterial consortium for 10 weeks developed severe colitis, and E faecalis OG1RF in these mice significantly up-regulated OG1RF_11616-11610. However, the effect of mild colitis on OG1RF_11616-11610 expression is unknown. To determine this, we measured OG1RF_11616 mRNA in cecal contents from WT and Il10-/- mice at 5, 8, and 10 weeks after colonization using quantitative PCR. We previously reported composite histologic inflammation scores and spontaneous secretion of IL12/23p40 by colon explant cultures in these mice.38 The average composite histologic inflammation scores at 5, 8, and 10 weeks were 6.75 (SEM, ±0.43), 9.17 (SEM, ±0.44), and 8.90 (SEM, ±0.43), respectively, in Il10-/- mice, and 2.75 (SEM, ±0.48), 2.17 (SEM, ±0.21), and 3.5 (SEM, ±0.35) in WT mice. We detected increased OG1RF_11616 mRNA in Il10-/- mice at all 3 time points after colonization, but the difference was only statistically significant at 10 weeks (Figure 4). Therefore, we conclude that E faecalis increases OG1RF_11616-11610 transcription primarily during more severe colitis in Il10-/- mice.
E faecalis OG1RF_11616-11610 Encodes PTS Proteins That Import Glucosamine
The function of OG1RF_11616-11610 is poorly understood. NCBI annotations indicate that this operon may encode subunits of a PTS transporter belonging to the fructose/mannose/sorbose family. To experimentally test whether proteins in this operon transport glucose, fructose, mannose, or sorbose, we measured the growth of E faecalis OG1RF or E faecalis OG1RF lacking OG1RF_11616 (OG1RFΔ11616) in minimal media containing these carbohydrates as the primary carbon source. We chose to delete only OG1RF_11616, the predicted Enzyme IIA (EIIA) subunit of the operon, because the EIIA subunit is essential to the optimal function of PTS in general.40 Although there were statistically significant differences in growth between the 2 strains at some time points in glucose-, fructose-, and mannose-containing media, the absolute differences were relatively small and unlikely to have pathophysiologic consequences (Figure 5).
Figure 5.

E faecalis OG1RF_11616 encodes PTS-glucosamine. (A–F) Growth curves of E faecalis OG1RF or OG1RFΔ11616 in minimal media containing the indicated carbohydrate as the primary carbon source. (n = 3 cultures per strain, data are presented as means ± SD although error bars are not visible because of their small height, ∗P < .05). (G) OG1RF_11616 mRNA abundance in E faecalis growing in minimal media containing the indicated carbohydrate as the primary carbon source (n = 3 cultures per group per time point, data presented as means ± SD, ∗P < .05 glucosamine vs others, Student t test).
We then performed a protein Basic Local Alignment Search Tool (BLAST) search using each open reading frame in the operon to query the nonredundant microbial database in NCBI and found that OG1RF_11612 and OG1RF_11611 nucleotide sequences were similar to genes from other E faecalis strains that are annotated as glucosamine-fructose-6-phosphate aminotransferase genes. Therefore, we repeated the growth assays in minimal media containing glucosamine as the primary carbon source. Growth of OG1RFΔ11616 was slightly impaired in glucosamine-containing media in aerobic conditions and moderately impaired in anaerobic conditions (Figure 5). These data suggest that OG1RF_11616-11610 encode PTS proteins that import glucosamine, although other glucosamine utilization pathways also likely are present in E faecalis.
If OG1RF_11616-11610 encode a PTS that imports glucosamine, we hypothesized that E faecalis would up-regulate genes in this operon when glucosamine is the primary carbon source. To test this hypothesis, we grew E faecalis in minimal media containing various carbohydrate sources including glucosamine and measured OG1RF_11616 mRNA abundance. We detected increased levels of OG1RF_11616 mRNA at 2 and 3 hours after inoculation only in E faecalis grown in glucosamine-containing media (Figure 5). Together, these results suggest that OG1RF_11616-11610 encodes a glucosamine-specific PTS (PTS-glucosamine).
Luminal Glucosamine Levels Are Increased During Experimental Colitis
We next hypothesized that E faecalis increases PTS-glucosamine expression in Il10-/- mice owing to increased concentrations of luminal glucosamine during colitis. To test this, we used gas chromatography–mass spectrometry to quantify glucosamine levels in cecal content from WT and Il10-/- mice colonized with either the 8 strains of bacteria used previously (Table 1) or specific pathogen-free microbiota. In both cases, we detected increased concentrations of glucosamine in cecal contents in Il10-/- mice with colitis compared with healthy WT mice (Figure 6). Whether the luminal glucosamine arises from dietary or host sources and the mechanisms by which it increases during colitis are currently unclear.
Figure 6.
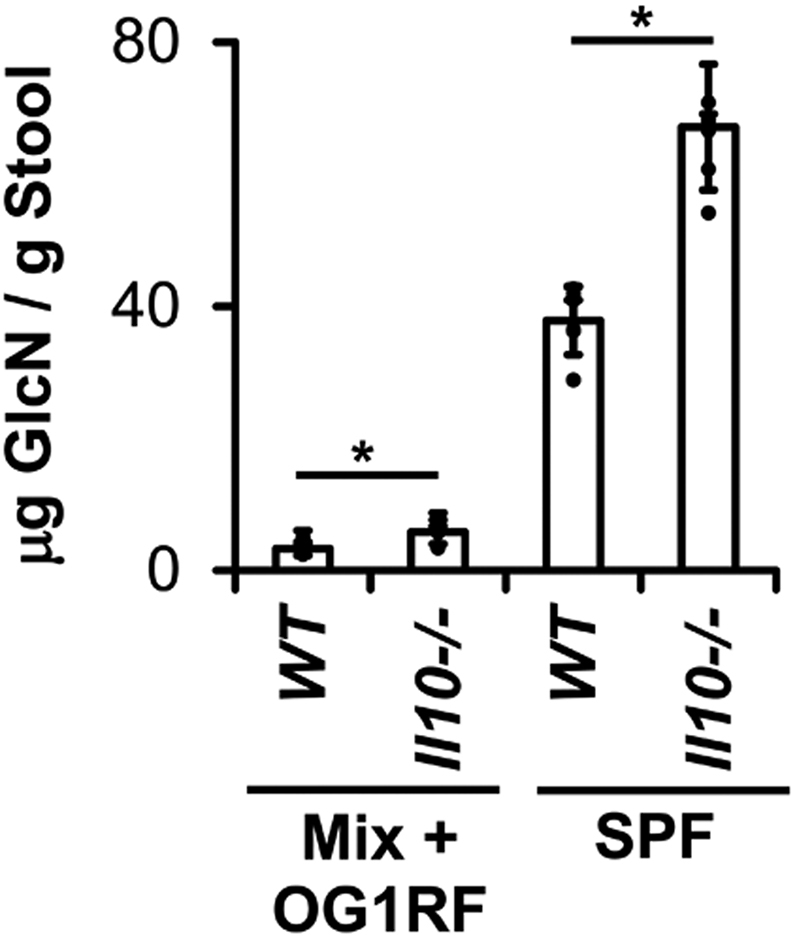
Cecal glucosamine is increased during experimental colitis. Glucosamine (GlcN) concentrations in WT and Il10-/- mice colonized with 8 bacterial strains as in Figure 1 (Mix + OG1RF) for 10 weeks or specific pathogen-free (SPF) microbiota for 4 weeks (n = 4–6 mice/group, data presented as means ± SD, ∗P < .05 Il10-/- vs WT, Student t test).
E faecalis PTS-Glucosamine Is Associated With Worse Experimental Colitis and Decreased Abundance of B vulgatus and B thetaiotaomicron
Having established that E faecalis OG1RF worsens experimental colitis and that glucosamine concentrations and OG1RF PTS-glucosamine expression are increased in cecal contents during experimental colitis, we next hypothesized that OG1RF PTS-glucosamine enhances E faecalis growth and colitis severity.
To test this, we colonized germ-free WT and Il10-/- mice with the 8-member bacterial consortium containing either E faecalis OG1RF or OG1RFΔ11616. After 8 weeks, we detected numerically fewer E faecalis in cecal contents of Il10-/- mice colonized with the consortium including OG1RFΔ11616 vs OG1RF, but this difference was not statistically significant (P = .08) (Figure 7). On the other hand, concentrations of B thetaiotaomicron and B vulgatus were both increased significantly in Il10-/- mice colonized with the consortium containing OG1RFΔ11616 vs OG1RF (Figure 7). Histologic colon inflammation and spontaneous IL12/23p40 secretion by colon explant cultures both were decreased in Il10-/- mice colonized with the consortium containing OG1RFΔ11616 vs OG1RF at both 4 and 8 weeks after colonization (Figure 7). No signs of inflammation were present in WT mice from either group. These data indicate that OG1RF PTS-glucosamine exacerbates experimental colitis with only subtle effects on E faecalis growth, and decreases Bacteroides species concentrations in Il10-/- mice.
Figure 7.
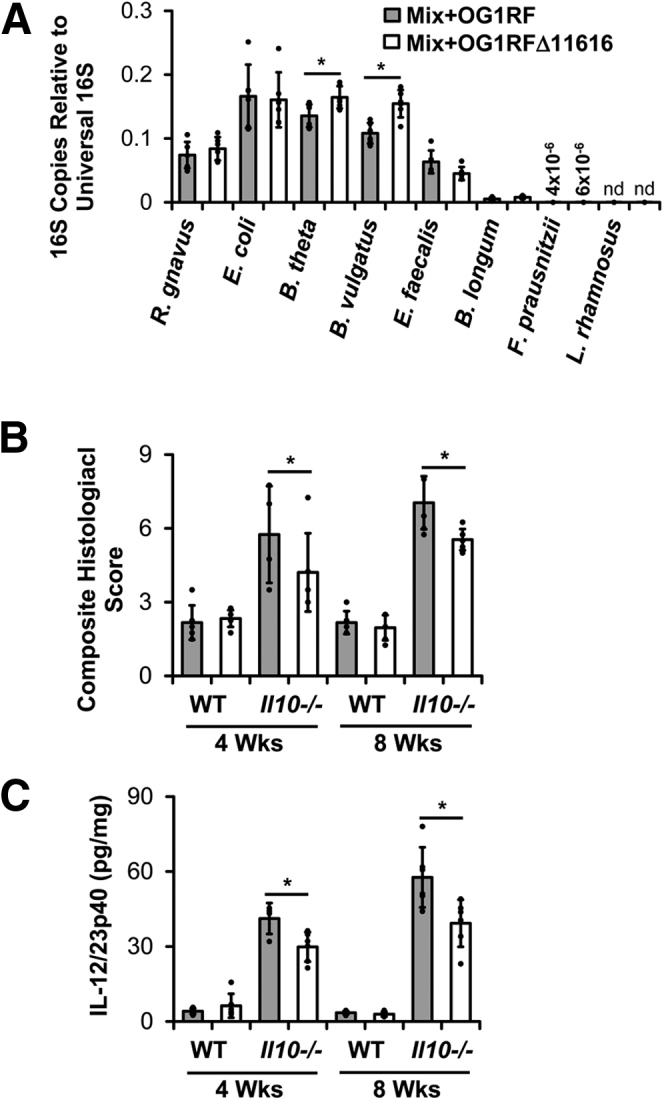
E faecalis OG1RF_11616 causes worse colitis and decreased abundance of Bacteroides species. (A) Relative abundance of bacterial strains in cecal contents from Il10-/- mice colonized with the bacterial strains as in Figure 1 containing either parental E faecalis OG1RF or OG1RF_11616-deficient E faecalis OG1RF (OG1RFΔ11616) for 8 weeks. (B) Composite histologic colitis scores in WT and Il10-/- mice colonized with the bacterial strains as in panel A for 4 and 8 weeks. (C) IL12/23p40 secretion by colon explant cultures from the same mice as in panel B (n = 4–6 mice/group, data presented as means ± SD, ∗P < .05 Mix+OG1RF vs Mix+OG1RFΔ11616, Student t test). nd, not detected.
Proinflammatory Effects of E faecalis PTS-Glucosamine Require the Presence of Complex Microbiota
Because OG1RF PTS-glucosamine does not increase luminal E faecalis concentrations significantly, we hypothesized that OG1RF PTS-glucosamine worsens experimental colitis by altering the intrinsic virulence of E faecalis. To test this, we monocolonized germ-free Il10-/- mice with E faecalis OG1RF or OG1RFΔ11616 for 20 weeks. We colonized the mice for this longer period of time because others previously have shown that E faecalis OG1RF induces relatively slow onset colitis in monocolonized IL10-/- mice.27 We detected no differences in cecal content bacterial concentrations, composite histologic scores, or spontaneous secretion of IL12/23p40 by colon explant cultures between the 2 groups of mice at 10 and 20 weeks after colonization (Figure 8). These data indicate that OG1RF PTS-glucosamine does not affect the colitogenic potential or luminal fitness of E faecalis OG1RF in monocolonized Il10-/- mice.
Figure 8.

E faecalis OG1RF_11616-associated worsening of colitis requires complex microbiota. (A–C) Composite histologic colitis scores, spontaneous IL12/23p40 secretion by colon explant cultures, and E faecalis concentrations in cecal contents from Il10-/- mice monocolonized with E faecalis OG1RF or E faecalis OG1RFΔ11616 for 10 and 20 weeks (n = 5–6 mice/group, data presented as means ± SD, no differences between the 2 groups were statistically significant). (D–F) Composite histologic colitis scores, spontaneous IL2/23p40 secretion by colon explant cultures, and E faecalis concentrations in cecal contents from Il10-/- mice colonized with 5 bacterial strains (R gnavus, E coli, L rhamnosus, F prausnitzii, and B longum) and either E faecalis OG1RF or E faecalis OG1RFΔ11616 for 10 weeks (n = 7–9 mice/group, data presented as means ± SD, no differences between 2 groups were statistically significant). (G–I) Composite histologic colitis scores, spontaneous IL12/23p40 secretion by colon explant cultures, and E faecalis concentrations in cecal contents from Il10-/- mice colonized with B thetaiotaomicron (Bt), B vulgatus (Bv), and either E faecalis OG1RF or E faecalis OG1RFΔ11616 for 10 weeks (n = 6–7 mice/group, data presented as means ± SD, no differences between 2 groups were statistically significant). CFU, colony-forming unit.
We next wanted to determine whether OG1RF PTS-glucosamine enhances colitis indirectly via its effects on other species in the bacterial community. Because luminal concentrations of B thetaiotaomicron and B vulgatus are decreased in the presence of OG1RF PTS-glucosamine, we hypothesized that OG1RF PTS-glucosamine exerts proinflammatory effects via these 2 bacterial species. To test this, we colonized germ-free Il10-/- mice for 10 weeks with E faecalis OG1RF or OG1RFΔ11616 along with B thetaiotaomicron and B vulgatus. Under these conditions, we detected no statistically significant differences in the cecal content of E faecalis concentrations, composite histologic scores, or spontaneous secretion of IL12/23p40 by colon explant cultures between the 2 groups of mice (Figure 8). We also colonized germ-free Il10-/- mice for 10 weeks with the same consortia of bacteria as in Figure 7, but omitted B thetaiotaomicron and B vulgatus and detected no statistically significant differences in the cecal content of E faecalis concentrations, composite histologic scores, or spontaneous secretion of IL12/23p40 by colon explant cultures between the 2 groups of mice (Figure 8). Based on these data, we conclude that the increased colitis severity associated with OG1RF PTS-glucosamine requires the presence of complex microbiota and that the presence of the non–Bacteroides species or Bacteroides species alone is insufficient. The mechanisms by which E faecalis OG1RF PTS-glucosamine and the other microbial species interact with one another to affect intestinal immune responses remain to be determined.
Discussion
In the current studies, we show that E faecalis worsens experimental colitis in mice colonized with a simplified consortium of bacteria and that colitis causes significant transcriptional changes in luminal bacteria, including up-regulation of an E faecalis PTS that imports glucosamine. Furthermore, we show that luminal glucosamine levels are higher during experimental colitis and that E faecalis PTS-glucosamine exacerbates colitis severity in a microbiota-dependent fashion.
IBDs are associated with altered composition of resident intestinal bacteria including increased Ruminococci, Enterococci, and E coli, and decreased F prausnitzii and Bacteroides.6, 7, 8, 9, 10, 11, 12, 13 However, in our studies, we show that experimental colitis is associated with decreased numbers of R gnavus, E faecalis, B thetaiotaomicron, F prausnitzii, and B longum, and no change in E coli. These differences could be owing to several factors including host species, cecal vs fecal specimens, complexity of the bacterial community, diet, and mechanisms of inflammation. It is particularly notable that cecal E faecalis numbers are lower during colitis even though we and others have shown that E faecalis causes colitis in this model. This finding indicates that the quantity of a proinflammatory bacterial species may not be as important as its mere presence when determining colitogenic potential. Alternatively, it is possible that the ability of a species to cause colitis is independent of its growth response to intestinal inflammation.
In our studies, bacterial transcription activity was highest in R gnavus in both the uninflamed and inflamed colon. Moreover, transcription activity significantly increased in R gnavus during colitis, but was unchanged in most other bacterial species in our model. These findings are consistent with those of Schirmer et al,41 who showed that transcriptional activity of fecal R gnavus strongly correlates with human IBDs. Given the increased transcription activity and abundance of R gnavus in human IBDs,10,42,43 further studies of the mechanisms by which R gnavus may worsen intestinal inflammation are warranted. Indeed, others recently have shown that R gnavus secretes a complex glucorhamnan polysaccharide with proinflammatory properties.37 Because, to our knowledge, the R gnavus genome currently is genetically intractable, for now it is difficult to determine how specific R gnavus genes might impact its growth and virulence during colitis.
In the current studies of experimental colitis, we detected the highest number of differentially expressed genes in E faecalis. Lengfelder et al. also have examined gene expression profiles of E faecalis during experimental colitis in Il10-/- mice.44 Sixty-two of the differentially expressed E faecalis genes in our studies also were differentially expressed in E faecalis monocolonized WT vs Il10-/- mice with colitis, 37 of which were regulated in the same direction.44 On the other hand, 50 of the differentially expressed E faecalis genes in our studies were differentially expressed in Il10-/- mice colonized with a similar consortium of bacterial species, 47 of which were regulated in the same direction.44 The differences in colitis-associated E faecalis gene regulation in the different studies may be owing to differences in housing facilities (chow), sample source (colon vs cecal), duration of colonization (10 vs 16 wk), and the presence of different bacterial species. However, it is notable that the most highly up-regulated E faecalis gene in our studies, OG1RF_11616, also was up-regulated during experimental colitis in mice colonized with a similar consortium of bacteria.44
We show that the operon including OG1RF_11616 encodes proteins of a glucosamine PTS in E faecalis. Bacterial PTS primarily function to import carbohydrates for metabolic purposes. However, we and others have shown that some PTS also regulate bacterial virulence.38,45,46 We previously have shown that E faecalis up-regulate PTS-gluconate during experimental colitis and that PTS-gluconate enhances survival of intramacrophage E faecalis.38 Bacterial PTS genes and genes of other carbohydrate metabolic pathways also are enriched in fecal metagenomic DNA from Crohn’s disease patients vs controls.26 Further studies of bacterial carbohydrate metabolism, particularly PTS, in experimental colitis, bacterial virulence, and IBDs are warranted.
We hypothesized that increased luminal concentration of glucosamine during colitis caused E faecalis to up-regulate PTS-glucosamine, proliferate, and worsen colitis. However, we found that although the presence of PTS-glucosamine in E faecalis exacerbated experimental colitis, it did not facilitate E faecalis growth in the inflamed or noninflamed colon. The ability of PTS-glucosamine to worsen colitis in this model depended on the presence of complex microbiota through unknown mechanisms. Because the concentrations of B thetaiotaomicron and B vulgatus were significantly higher in the absence of E faecalis PTS-glucosamine, and because others have shown that B thetaiotaomicron attenuates colitis in Il10-/- mice,33 we had hypothesized that B thetaiotaomicron or B vulgatus occupied a new niche in the intestine to suppress inflammation in the absence of E faecalis PTS-glucosamine. However, our data did not support this hypothesis. Instead, the combination of the 2 Bacteroides species with the other bacterial species in the consortium was necessary for the E faecalis PTS-glucosamine–associated worsening of colitis.
Lastly, the connection between glucosamine and IBDs/experimental colitis is underexplored. Because epidemiologic studies have indicate that 4% of IBD patients consume glucosamine-containing nutritional supplements,47 we believe that this is an important area of investigation. Two studies have shown that dietary glucosamine supplementation attenuates acute chemically induced murine colitis, but whether dietary glucosamine affects IBD severity is unclear.48,49 Moreover, published metabolomic studies to date in human IBD patients have not quantified luminal glucosamine concentrations or bacterial glucosamine metabolism. Therefore, whether luminal glucosamine or bacterial glucosamine metabolism impact the course of human IBDs remains to be determined. Answering these questions could not only enhance our understanding of the pathogenesis of IBDs, but may lead to the development of novel therapies targeting glucosamine–bacteria interactions.
Methods
Animals and Bacteria
Germ-free WT and Il10-/- mice on the 129S6/SvEv background were obtained from the National Gnotobiotic Rodent Resource Center (University of North Carolina–Chapel Hill). All mice were housed in sterilized flexible film isolators and provided with sterilized food and drinking water ad libitum. Adult germ-free mice (age, 8–16 wk) were colonized by oral gavage with 200 μL stationary-phase, anaerobically grown cultures of the indicated bacterial strains. In cases of multistrain colonization experiments, mice were gavaged with 200 μL equivolume mixture of stationary-phase bacterial strains. Animal protocols were performed in accordance with the American Association for Laboratory Animal Care standards and were approved by the University of North Carolina Institutional Animal Care and Use Committee.
We used 8 IBD-relevant bacterial strains as described previously (Table 1).38 OG1RF_11616 was deleted from the E faecalis genome to create E faecalis OG1RFΔ11616 by allelic replacement using the temperature-sensitive shuttle vector pJRS233 as previously described.50 Briefly, a 1-kb region of genomic DNA exactly upstream of the OG1RF_11616 open reading frame was PCR-amplified using the primer pair PTSIIA-1.A-EcoR1 5'-tatagaattcTCAAGTAAGCCTTTAATGGTG-3' and PTSIIA-1.B-BamHI 5'-gcgcggatccAGAAGAAGAATTTTAGGAGG-3', and a 1-kb region of genomic DNA exactly downstream from the OG1RF_11616 open reading frame was PCR-amplified using the primer pair PTSIIA-2.A-BamHI 5'-tataggatccTCACTTACTTCTTTACATAGC-3' and PTSIIA-2.B-NcoI 5'-gcgcccatggTGGAACGTCAAGAAACGAGG-3'. These 2 PCR products were sequentially cloned into pJRS233 (a kind gift from Michael Caparon, Washington University St. Louis, St. Louis, MO) using standard cloning techniques to create the targeting vector pJH185. Electrocompetent E faecalis OG1RF were transformed with pJH185 and transformants were plated onto brain heart infusion (BHI) agar containing 25 μg/mL erythromycin and incubated at 30°C for 48 hours. A single clone then was serially cultured in BHI + erythromycin at 42°C × 96 hours to force plasmid integration onto the chromosome followed by BHI at 30°C × 96 hours to select for bacteria lacking OG1RF_11616. In-frame deletion of OG1RF_11616 was confirmed by PCR amplification and Sanger sequencing of the entire operon.
Growth Curves
M9-based E faecalis minimal media was prepared to contain the following: 1× M9 salts, 2 mmol/L MgSO4, 0.1 mmol/L CaCl2, 2× MEM nonessential amino acids (Gibco, Gaithersburg, MD), 1× MEM amino acids (Gibco), and 2× MEM vitamin solution (Gibco). Indicated carbohydrates were purchased from Sigma-Aldrich (St. Louis, MO) and added to a final concentration of 1% (wt/vol). Three milliliters minimal media was inoculated with 30 μL overnight culture of the indicated E faecalis strain grown in BHI broth at 37°C. At the indicated time points, 100 μL of each culture was transferred to a 96-well plate, and absorbance at 600 nm was measured using a BioTek Synergy HTX plate reader (Winooski, VT).
Real-Time PCR
For in vitro gene expression experiments, E faecalis OG1RF was grown overnight at 37°C in BHI, diluted 1:100 in M9-based minimal media containing 0.02% glucose (wt/vol) to a final volume of 2 mL, incubated for 2 hours at 37°C, and then the indicated carbohydrate was added to a final concentration of 0.2% (wt/vol). At the indicated time points after adding the carbohydrate, bacteria were rapidly pelleted, pellets were resuspended in 0.5 mL Bacterial RNAProtect (Qiagen, Germantown, MD), and then frozen at -80°C for later use. Pellets were thawed, centrifuged at 14,000 × g for 2 minutes, and supernatant was discarded. Bacterial RNA was isolated using a combination of phenol–chloroform extraction, mechanical bead beating, ethanol precipitation, and silica gel column purification as described previously, and complementary DNA (cDNA) was synthesized as described previously.29 Relative transcript abundance of OG1RF_11616 was determined by SYBR Green (Bio-Rad, Hercules, CA) real-time PCR using primers Efa16SF 5’-CGCGGTGCATTAGCTAGTTG-3', Efa16SR 5’-TCACCCTCTCAGGTCGGCTAT-3', Ef11616F 5'-TGCATTGCTTGTTTCCCCAC-3', and Ef11616R 5'-AGCCAAGACGGTCAAAGACA-3', and results were expressed relative to OG1RF 16S ribosomal RNA (rRNA) transcripts using the delta cycle threshold (Ct) method as described previously.29
To determine the abundance of each bacterial species in cecal contents, we isolated bacterial DNA from cecal contents and quantified 16S rRNA gene abundance for each bacterial species using SYBR Green real-time PCR as described previously.38,51,52 Relative abundances are presented relative to universal bacterial 16S rDNA using the delta Ct method. Absolute abundances were calculated from standard curves (Ct vs genome copy number) that were generated by SYBR Green real-time PCR as described previously, but using purified genomic DNA from each bacterial strain as a PCR template.
Quantifying Colitis Severity
Histologic inflammation was quantified by blinded histologic scoring of formalin-fixed, H&E-stained sections of the cecum, proximal colon, midcolon, and distal colon as described previously.29 Composite histologic inflammation scores represent the sum of 4 individually scored colon pieces (cecum, proximal colon, transverse colon, and distal colon). Each piece was scored on a scale of 0 to 4, based on crypt hyperplasia, lamina propria mononuclear cell infiltration, goblet cell dropout, and transmural inflammation. Mouse IL12/23p40 was measured in supernatants of colon explant cultures using an enzyme-linked immunosorbent assay as described previously.29
Microbial RNA Isolation and RNA Sequencing Analysis
Total bacterial RNA was isolated from the cecal content of WT and Il10-/- mice using a combination of bead beating and phenol:chloroform extraction as described previously.53 To deplete total microbial community RNA of 16S, 23S, 5S rRNA, and transfer RNA species before synthesis of cDNA with random hexanucleotide primers, each fecal RNA preparation was subjected to column-based size selection and hybridization to custom biotinylated oligonucleotides directed at conserved regions of bacterial rRNA genes present in human gut communities, followed by streptavidin-bead based capture of the hybridized RNA sequences.54 Briefly, RNA samples were treated with MICROBExpress (Thermo Fischer Scientific, Waltham, MA) rRNA capture beads according to the manufacturer’s instructions, followed by a second round of bacterial rRNA depletion using biotinylated custom oligonucleotides that have been reported previously.54 Barcoded Illumina (San Diego, CA) sequencing libraries were prepared from double-stranded cDNA generated from each of the mRNA-enriched samples, and the 10-sample pool was sequenced on 1 lane of an Illumina HiSeq2000 sequencer 2 × 50. After barcode removal, each of the RNA-seq data sets was composed of 38-nucleotide-long reads (mean, 3.57 ± 0.8 × 107/mRNA reads per sample). Transcript abundances were normalized separately for each of the 10 species in each sample to reads per kilobase per million.
Principal component analysis of the luminal bacterial metatranscriptomes and differential expression analysis of individual bacterial genes were performed using GeneSpring GX version 11.0 (Agilent Technologies, Santa Clara, CA). Statistically significant differences in bacterial gene expression grouped by mouse genotype were identified using the Welch t test, with a P value cut-off of .05, and multiple testing correction using the Benjamini and Hochberg false-discovery rate. Hierarchical clustering was performed on log-transformed, normalized, centered data using HierarchicalClustering, version 6 (cloud.genepattern.org) to determine Euclidean distance with pairwise average linkage. Dendrograms were viewed on HierarchicalClusteringViewer version 11.3 (cloud.genepattern.org). Volcano plots were generated from reads per kilobase per million data for each bacterial species using Multiplot Studio version 1.5.62 (cloud.genepattern.org) applying the Benjamini-Hochberg false-discovery rate to calculate P values.
For conserved protein domain analysis, RNA-seq reads aligned to the genomes of the 8 bacterial strains were categorized by conserved protein domain based on the NCBI conserved protein domain database queried on August 15, 2012.
We then performed gene set enrichment analysis (Broad Institute, available from:http://software.broadinstitute.org/gsea/index.jsp) to determine conserved domains that were differentially enriched in a given mouse genotype.55 Transcription activity of luminal bacteria was determined by dividing the number of mRNA reads for each strain by the number of coding bases in that strain’s genome and the fraction of that strain’s genomes in the sample.
Glucosamine Measurement in Colon Contents
One hundred microliters of stable isotope labeled internal standard mixture (1 μmol/L), 600 μL mixture of isopropanol:acetonitrile:water (3:3:2), and 30-mg glass beads were added to approximately 20 mg colon content in a 1.5-mL microcentrifuge tube. Samples were processed in a TissueLyser (Qiagen) at 50 Hz for 10 minutes, sonicated for 10 minutes in a 25°C water bath, and centrifuged for 10 minutes at 15,000 rpm. The supernatant was saved and the pellet was extracted a second time as described previously. The 2 supernatants were combined and dried in a SpeedVac (Thermo Fisher Scientific, Waltham, MA) vacuum concentrator. The dried extract then was oximated with 100 μL methoxyamine hydrochloride (30 mg/mL) at 70°C for 2 hours. After oximation, 100 μL N,O-bis(trimethylsilyl)trifluoroacetamide with 1% chlorotrimethylsilane was added and the mixture was incubated at 70°C for 2 hours to form trimethylsilyl derivatives. The mixture was centrifuged at 25°C and 10,000 rpm for 5 minutes and supernatant was used for instrument injection. An Agilent gas chromatograph (7820A; Agilent Technologies) and mass spectrometer (5977B MSD; Agilent Technologies) were used for sample analysis. Splitless injection was used (1 μL), along with a DB-5 (Agilent Technologies, Santa Clara, CA) column (30 m × 0.25 mm inner diameter × 0.25-μm film thickness). The gas chromatography column oven temperature was programmed to increase at a rate of 8°C/min from an initial temperature of 80°C to 300°C, and then held for 5 minutes. The temperatures of the inlet heater, transfer line, and ion source were held at 280°C, 300°C, and 230°C, respectively. The amount of sugar was determined using the calibration curve generated with authentic analytical and internal standards.
Statistical Analysis
P values were calculated using a 2-tailed Student t test when 2 experimental groups were compared. All data are presented as the means ± SD.
All authors had access to the study data and reviewed and approved the final manuscript
Acknowledgments
The authors acknowledge the Gnotobiotic, Histology, and Immunotechnology Cores at the University of North Carolina Center for Gastrointestinal Biology and Disease. The authors also thank Gary Port and Dr Jeffrey I. Gordon at Washington University at St. Louis for technical advice during the construction of the E faecalis deletion mutant and material support for the microbial RNA-seq, respectively.
CRediT Authorship Contributions
Ting-Jia Fan (Conceptualization: Supporting; Data curation: Lead; Formal analysis: Lead; Investigation: Lead; Methodology: Lead; Writing – review & editing: Supporting)
Laura Goeser (Data curation: Supporting; Investigation: Supporting; Methodology: Supporting)
Kun Lu (Data curation: Supporting; Formal analysis: Equal; Investigation: Equal; Methodology: Equal)
Jeremiah Faith (Data curation: Equal; Formal analysis: Equal; Methodology: Equal; Writing – review & editing: Supporting)
Jonathan James Hansen (Conceptualization: Lead; Data curation: Lead; Formal analysis: Lead; Funding acquisition: Lead; Investigation: Lead; Methodology: Lead; Project administration: Lead; Resources: Lead; Validation: Lead; Writing – original draft: Lead; Writing – review & editing: Equal)
Footnotes
Conflicts of interest The authors disclose no conflicts.
Funding The Gnotobiotic, Histology, and Immunotechnology Cores at the University of North CarolinaCenter for Gastrointestinal Biology and Disease were supported by National Institutes of Health grants P30DK34987 and P40OD 010995, and the Crohn’s and Colitis Foundation.
Supplementary Material
References
- 1.Ng S.C., Shi H.Y., Hamidi N., Underwood F.E., Tang W., Benchimol E.I., Panaccione R., Ghosh S., Wu J.C.Y., Chan F.K.L., Sung J.J.Y., Kaplan G.G. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. 2018;390:2769–2778. doi: 10.1016/S0140-6736(17)32448-0. [DOI] [PubMed] [Google Scholar]
- 2.Manceur A.M., Ding Z., Muser E., Obando C., Voelker J., Pilon D., Kinkead F., Lafeuille M.H., Lefebvre P. Burden of Crohn's disease in the United States: long-term healthcare and work-loss related costs. J Med Econ. 2020;23:1092–1101. doi: 10.1080/13696998.2020.1789649. [DOI] [PubMed] [Google Scholar]
- 3.Lloyd-Price J., Arze C., Ananthakrishnan A.N., Schirmer M., Avila-Pacheco J., Poon T.W., Andrews E., Ajami N.J., Bonham K.S., Brislawn C.J., Casero D., Courtney H., Gonzalez A., Graeber T.G., Hall A.B., Lake K., Landers C.J., Mallick H., Plichta D.R., Prasad M., Rahnavard G., Sauk J., Shungin D., Vazquez-Baeza Y., White R.A., 3rd, Braun J., Denson L.A., Jansson J.K., Knight R., Kugathasan S., McGovern D.P.B., Petrosino J.F., Stappenbeck T.S., Winter H.S., Clish C.B., Franzosa E.A., Vlamakis H., Xavier R.J., Huttenhower C. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature. 2019;569:655–662. doi: 10.1038/s41586-019-1237-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Franzosa E.A., Sirota-Madi A., Avila-Pacheco J., Fornelos N., Haiser H.J., Reinker S., Vatanen T., Hall A.B., Mallick H., McIver L.J., Sauk J.S., Wilson R.G., Stevens B.W., Scott J.M., Pierce K., Deik A.A., Bullock K., Imhann F., Porter J.A., Zhernakova A., Fu J., Weersma R.K., Wijmenga C., Clish C.B., Vlamakis H., Huttenhower C., Xavier R.J. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat Microbiol. 2019;4:293–305. doi: 10.1038/s41564-018-0306-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pascal V., Pozuelo M., Borruel N., Casellas F., Campos D., Santiago A., Martinez X., Varela E., Sarrabayrouse G., Machiels K., Vermeire S., Sokol H., Guarner F., Manichanh C. A microbial signature for Crohn's disease. Gut. 2017;66:813–822. doi: 10.1136/gutjnl-2016-313235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hansen J., Gulati A., Sartor R.B. The role of mucosal immunity and host genetics in defining intestinal commensal bacteria. Curr Opin Gastroenterol. 2010;26:564–571. doi: 10.1097/MOG.0b013e32833f1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fite A., Macfarlane S., Furrie E., Bahrami B., Cummings J.H., Steinke D.T., Macfarlane G.T. Longitudinal analyses of gut mucosal microbiotas in ulcerative colitis in relation to patient age and disease severity and duration. J Clin Microbiol. 2013;51:849–856. doi: 10.1128/JCM.02574-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nemoto H., Kataoka K., Ishikawa H., Ikata K., Arimochi H., Iwasaki T., Ohnishi Y., Kuwahara T., Yasutomo K. Reduced diversity and imbalance of fecal microbiota in patients with ulcerative colitis. Dig Dis Sci. 2012;57:2955–2964. doi: 10.1007/s10620-012-2236-y. [DOI] [PubMed] [Google Scholar]
- 9.Kang S., Denman S.E., Morrison M., Yu Z., Dore J., Leclerc M., McSweeney C.S. Dysbiosis of fecal microbiota in Crohn's disease patients as revealed by a custom phylogenetic microarray. Inflamm Bowel Dis. 2010;16:2034–2042. doi: 10.1002/ibd.21319. [DOI] [PubMed] [Google Scholar]
- 10.Hall A.B., Yassour M., Sauk J., Garner A., Jiang X., Arthur T., Lagoudas G.K., Vatanen T., Fornelos N., Wilson R., Bertha M., Cohen M., Garber J., Khalili H., Gevers D., Ananthakrishnan A.N., Kugathasan S., Lander E.S., Blainey P., Vlamakis H., Xavier R.J., Huttenhower C. A novel Ruminococcus gnavus clade enriched in inflammatory bowel disease patients. Genome Med. 2017;9:103. doi: 10.1186/s13073-017-0490-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nagayama M., Yano T., Atarashi K., Tanoue T., Sekiya M., Kobayashi Y., Sakamoto H., Miura K., Sunada K., Kawaguchi T., Morita S., Sugita K., Narushima S., Barnich N., Isayama J., Kiridooshi Y., Shiota A., Suda W., Hattori M., Yamamoto H., Honda K. TH1 cell-inducing Escherichia coli strain identified from the small intestinal mucosa of patients with Crohn's disease. Gut Microbes. 2020;12 doi: 10.1080/19490976.2020.1788898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gevers D., Kugathasan S., Denson L.A., Vazquez-Baeza Y., Van Treuren W., Ren B., Schwager E., Knights D., Song S.J., Yassour M., Morgan X.C., Kostic A.D., Luo C., Gonzalez A., McDonald D., Haberman Y., Walters T., Baker S., Rosh J., Stephens M., Heyman M., Markowitz J., Baldassano R., Griffiths A., Sylvester F., Mack D., Kim S., Crandall W., Hyams J., Huttenhower C., Knight R., Xavier R.J. The treatment-naive microbiome in new-onset Crohn's disease. Cell Host Microbe. 2014;15:382–392. doi: 10.1016/j.chom.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frank D.N., St Amand A.L., Feldman R.A., Boedeker E.C., Harpaz N., Pace N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A. 2007;104:13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thornton J.R., Emmett P.M., Heaton K.W. Diet and Crohn's disease: characteristics of the pre-illness diet. Br Med J. 1979;2:762–764. doi: 10.1136/bmj.2.6193.762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Magee E.A., Edmond L.M., Tasker S.M., Kong S.C., Curno R., Cummings J.H. Associations between diet and disease activity in ulcerative colitis patients using a novel method of data analysis. Nutr J. 2005;4:7. doi: 10.1186/1475-2891-4-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jantchou P., Morois S., Clavel-Chapelon F., Boutron-Ruault M.C., Carbonnel F. Animal protein intake and risk of inflammatory bowel disease: the E3N prospective study. Am J Gastroenterol. 2010;105:2195–2201. doi: 10.1038/ajg.2010.192. [DOI] [PubMed] [Google Scholar]
- 17.Katschinski B., Logan R.F., Edmond M., Langman M.J. Smoking and sugar intake are separate but interactive risk factors in Crohn's disease. Gut. 1988;29:1202–1206. doi: 10.1136/gut.29.9.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tragnone A., Valpiani D., Miglio F., Elmi G., Bazzocchi G., Pipitone E., Lanfranchi G.A. Dietary habits as risk factors for inflammatory bowel disease. Eur J Gastroenterol Hepatol. 1995;7:47–51. [PubMed] [Google Scholar]
- 19.Mayberry J.F., Rhodes J., Newcombe R.G. Increased sugar consumption in Crohn's disease. Digestion. 1980;20:323–326. doi: 10.1159/000198454. [DOI] [PubMed] [Google Scholar]
- 20.Llewellyn S.R., Britton G.J., Contijoch E.J., Vennaro O.H., Mortha A., Colombel J.F., Grinspan A., Clemente J.C., Merad M., Faith J.J. Interactions between diet and the intestinal microbiota alter intestinal permeability and colitis severity in mice. Gastroenterology. 2018;154:1037–1046 e2. doi: 10.1053/j.gastro.2017.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen L., He Z., Iuga A.C., Martins Filho S.N., Faith J.J., Clemente J.C., Deshpande M., Jayaprakash A., Colombel J.F., Lafaille J.J., Sachidanandam R., Furtado G.C., Lira S.A. Diet modifies colonic microbiota and CD4(+) T-cell repertoire to induce flares of colitis in mice with myeloid-cell expression of interleukin 23. Gastroenterology. 2018;155:1177–1191 e16. doi: 10.1053/j.gastro.2018.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chassaing B., Koren O., Goodrich J.K., Poole A.C., Srinivasan S., Ley R.E., Gewirtz A.T. Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature. 2015;519:92–96. doi: 10.1038/nature14232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Devkota S., Wang Y., Musch M.W., Leone V., Fehlner-Peach H., Nadimpalli A., Antonopoulos D.A., Jabri B., Chang E.B. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/- mice. Nature. 2012;487:104–108. doi: 10.1038/nature11225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh V., Yeoh B.S., Walker R.E., Xiao X., Saha P., Golonka R.M., Cai J., Bretin A.C.A., Cheng X., Liu Q., Flythe M.D., Chassaing B., Shearer G.C., Patterson A.D., Gewirtz A.T., Vijay-Kumar M. Microbiota fermentation-NLRP3 axis shapes the impact of dietary fibres on intestinal inflammation. Gut. 2019;68:1801–1812. doi: 10.1136/gutjnl-2018-316250. [DOI] [PubMed] [Google Scholar]
- 25.Le Gall G., Noor S.O., Ridgway K., Scovell L., Jamieson C., Johnson I.T., Colquhoun I.J., Kemsley E.K., Narbad A. Metabolomics of fecal extracts detects altered metabolic activity of gut microbiota in ulcerative colitis and irritable bowel syndrome. J Proteome Res. 2011;10:4208–4218. doi: 10.1021/pr2003598. [DOI] [PubMed] [Google Scholar]
- 26.Nayfach S., Bradley P.H., Wyman S.K., Laurent T.J., Williams A., Eisen J.A., Pollard K.S., Sharpton T.J. Automated and accurate estimation of gene family abundance from shotgun metagenomes. PLoS Comput Biol. 2015;11 doi: 10.1371/journal.pcbi.1004573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim S.C., Tonkonogy S.L., Albright C.A., Tsang J., Balish E.J., Braun J., Huycke M.M., Sartor R.B. Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology. 2005;128:891–906. doi: 10.1053/j.gastro.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 28.Kim S.C., Tonkonogy S.L., Karrasch T., Jobin C., Sartor R.B. Dual-association of gnotobiotic IL-10-/- mice with 2 nonpathogenic commensal bacteria induces aggressive pancolitis. Inflamm Bowel Dis. 2007;13:1457–1466. doi: 10.1002/ibd.20246. [DOI] [PubMed] [Google Scholar]
- 29.Patwa L.G., Fan T.J., Tchaptchet S., Liu Y., Lussier Y.A., Sartor R.B., Hansen J.J. Chronic intestinal inflammation induces stress-response genes in commensal Escherichia coli. Gastroenterology. 2011;141:1842–1851 e1–10. doi: 10.1053/j.gastro.2011.06.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ocvirk S., Sava I.G., Lengfelder I., Lagkouvardos I., Steck N., Roh J.H., Tchaptchet S., Bao Y., Hansen J.J., Huebner J., Carroll I.M., Murray B.E., Sartor R.B., Haller D. Surface-associated lipoproteins link Enterococcus faecalis virulence to colitogenic activity in IL-10-deficient mice independent of their expression levels. PLoS Pathog. 2015;11 doi: 10.1371/journal.ppat.1004911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berry D., Schwab C., Milinovich G., Reichert J., Ben Mahfoudh K., Decker T., Engel M., Hai B., Hainzl E., Heider S., Kenner L., Muller M., Rauch I., Strobl B., Wagner M., Schleper C., Urich T., Loy A. Phylotype-level 16S rRNA analysis reveals new bacterial indicators of health state in acute murine colitis. ISME J. 2012;6:2091–2106. doi: 10.1038/ismej.2012.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bloom S.M., Bijanki V.N., Nava G.M., Sun L., Malvin N.P., Donermeyer D.L., Dunne W.M., Jr., Allen P.M., Stappenbeck T.S. Commensal Bacteroides species induce colitis in host-genotype-specific fashion in a mouse model of inflammatory bowel disease. Cell Host Microbe. 2011;9:390–403. doi: 10.1016/j.chom.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Delday M., Mulder I., Logan E.T., Grant G. Bacteroides thetaiotaomicron ameliorates colon inflammation in preclinical models of Crohn's disease. Inflamm Bowel Dis. 2019;25:85–96. doi: 10.1093/ibd/izy281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Edwards L.A., Lucas M., Edwards E.A., Torrente F., Heuschkel R.B., Klein N.J., Murch S.H., Bajaj-Elliott M., Phillips A.D. Aberrant response to commensal Bacteroides thetaiotaomicron in Crohn's disease: an ex vivo human organ culture study. Inflamm Bowel Dis. 2011;17:1201–1208. doi: 10.1002/ibd.21501. [DOI] [PubMed] [Google Scholar]
- 35.Hansen J.J., Huang Y., Peterson D.A., Goeser L., Fan T.J., Chang E.B., Sartor R.B. The colitis-associated transcriptional profile of commensal Bacteroides thetaiotaomicron enhances adaptive immune responses to a bacterial antigen. PLoS One. 2012;7 doi: 10.1371/journal.pone.0042645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sokol H., Pigneur B., Watterlot L., Lakhdari O., Bermudez-Humaran L.G., Gratadoux J.J., Blugeon S., Bridonneau C., Furet J.P., Corthier G., Grangette C., Vasquez N., Pochart P., Trugnan G., Thomas G., Blottiere H.M., Dore J., Marteau P., Seksik P., Langella P. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A. 2008;105:16731–16736. doi: 10.1073/pnas.0804812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Henke M.T., Kenny D.J., Cassilly C.D., Vlamakis H., Xavier R.J., Clardy J. Ruminococcus gnavus, a member of the human gut microbiome associated with Crohn's disease, produces an inflammatory polysaccharide. Proc Natl Acad Sci U S A. 2019;116:12672–12677. doi: 10.1073/pnas.1904099116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fan T.J., Goeser L., Naziripour A., Redinbo M.R., Hansen J.J. Enterococcus faecalis gluconate phosphotransferase system accelerates experimental colitis and bacterial killing by macrophages. Infect Immun. 2019;87 doi: 10.1128/IAI.00080-19. e00080-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deutscher J., Ake F.M., Derkaoui M., Zebre A.C., Cao T.N., Bouraoui H., Kentache T., Mokhtari A., Milohanic E., Joyet P. The bacterial phosphoenolpyruvate:carbohydrate phosphotransferase system: regulation by protein phosphorylation and phosphorylation-dependent protein-protein interactions. Microbiol Mol Biol Rev. 2014;78:231–256. doi: 10.1128/MMBR.00001-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Robillard G.T., Broos J. Structure/function studies on the bacterial carbohydrate transporters, enzymes II, of the phosphoenolpyruvate-dependent phosphotransferase system. Biochim Biophys Acta. 1999;1422:73–104. doi: 10.1016/s0304-4157(99)00002-7. [DOI] [PubMed] [Google Scholar]
- 41.Schirmer M., Franzosa E.A., Lloyd-Price J., McIver L.J., Schwager R., Poon T.W., Ananthakrishnan A.N., Andrews E., Barron G., Lake K., Prasad M., Sauk J., Stevens B., Wilson R.G., Braun J., Denson L.A., Kugathasan S., McGovern D.P.B., Vlamakis H., Xavier R.J., Huttenhower C. Dynamics of metatranscription in the inflammatory bowel disease gut microbiome. Nat Microbiol. 2018;3:337–346. doi: 10.1038/s41564-017-0089-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Willing B.P., Dicksved J., Halfvarson J., Andersson A.F., Lucio M., Zheng Z., Jarnerot G., Tysk C., Jansson J.K., Engstrand L. A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology. 2010;139:1844–1854 e1. doi: 10.1053/j.gastro.2010.08.049. [DOI] [PubMed] [Google Scholar]
- 43.Joossens M., Huys G., Cnockaert M., De Preter V., Verbeke K., Rutgeerts P., Vandamme P., Vermeire S. Dysbiosis of the faecal microbiota in patients with Crohn's disease and their unaffected relatives. Gut. 2011;60:631–637. doi: 10.1136/gut.2010.223263. [DOI] [PubMed] [Google Scholar]
- 44.Lengfelder I., Sava I.G., Hansen J.J., Kleigrewe K., Herzog J., Neuhaus K., Hofmann T., Sartor R.B., Haller D. Complex bacterial consortia reprogram the colitogenic activity of Enterococcus faecalis in a gnotobiotic mouse model of chronic, immune-mediated colitis. Front Immunol. 2019;10:1420. doi: 10.3389/fimmu.2019.01420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hondorp E.R., Hou S.C., Hause L.L., Gera K., Lee C.E., McIver K.S. PTS phosphorylation of Mga modulates regulon expression and virulence in the group A streptococcus. Mol Microbiol. 2013;88:1176–1193. doi: 10.1111/mmi.12250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Peng Z., Ehrmann M.A., Waldhuber A., Niemeyer C., Miethke T., Frick J.S., Xiong T., Vogel R.F. Phosphotransferase systems in Enterococcus faecalis OG1RF enhance anti-stress capacity in vitro and in vivo. Res Microbiol. 2017;168:558–566. doi: 10.1016/j.resmic.2017.03.003. [DOI] [PubMed] [Google Scholar]
- 47.Rawsthorne P., Clara I., Graff L.A., Bernstein K.I., Carr R., Walker J.R., Ediger J., Rogala L., Miller N., Bernstein C.N. The Manitoba Inflammatory Bowel Disease Cohort Study: a prospective longitudinal evaluation of the use of complementary and alternative medicine services and products. Gut. 2012;61:521–527. doi: 10.1136/gutjnl-2011-300219. [DOI] [PubMed] [Google Scholar]
- 48.Bak Y.K., Lampe J.W., Sung M.K. Effects of dietary supplementation of glucosamine sulfate on intestinal inflammation in a mouse model of experimental colitis. J Gastroenterol Hepatol. 2014;29:957–963. doi: 10.1111/jgh.12485. [DOI] [PubMed] [Google Scholar]
- 49.Yomogida S., Kojima Y., Tsutsumi-Ishii Y., Hua J., Sakamoto K., Nagaoka I. Glucosamine, a naturally occurring amino monosaccharide, suppresses dextran sulfate sodium-induced colitis in rats. Int J Mol Med. 2008;22:317–323. [PubMed] [Google Scholar]
- 50.Perez-Casal J., Price J.A., Maguin E., Scott J.R. An M protein with a single C repeat prevents phagocytosis of Streptococcus pyogenes: use of a temperature-sensitive shuttle vector to deliver homologous sequences to the chromosome of S. pyogenes. Mol Microbiol. 1993;8:809–819. doi: 10.1111/j.1365-2958.1993.tb01628.x. [DOI] [PubMed] [Google Scholar]
- 51.Fan T.J., Tchaptchet S.Y., Arsene D., Mishima Y., Liu B., Sartor R.B., Carroll I.M., Miao E.A., Fodor A.A., Hansen J.J. Environmental factors modify the severity of acute DSS colitis in caspase-11-deficient mice. Inflamm Bowel Dis. 2018;24:2394–2403. doi: 10.1093/ibd/izy244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tchaptchet S., Fan T.J., Goeser L., Schoenborn A., Gulati A.S., Sartor R.B., Hansen J.J. Inflammation-induced acid tolerance genes gadAB in luminal commensal Escherichia coli attenuate experimental colitis. Infect Immun. 2013;81:3662–3671. doi: 10.1128/IAI.00355-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Faith J.J., McNulty N.P., Rey F.E., Gordon J.I. Predicting a human gut microbiota's response to diet in gnotobiotic mice. Science. 2011;333:101–104. doi: 10.1126/science.1206025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rey F.E., Faith J.J., Bain J., Muehlbauer M.J., Stevens R.D., Newgard C.B., Gordon J.I. Dissecting the in vivo metabolic potential of two human gut acetogens. J Biol Chem. 2010;285:22082–22090. doi: 10.1074/jbc.M110.117713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.