

Abstract
Expansion in vitro prior to mesenchymal stem cells (MSCs) application is a necessary process. Functional and genomic stability has a crucial role in stem-cell-based therapies. However, the exact expression and co-expressed profiles of coding and non-coding RNAs in human bone marrow (BM)-MSCs in vitro aging are still lacking. In the present studies, the change of morphology, immunophenotype, and capacity of proliferation, differentiation, and immunoregulation of MSCs at passage (P) 4, P6, P8, P10, and P12 were investigated. RNA sequencing identified that 439 mRNAs, 65 long noncoding RNAs (lncRNAs), 59 microRNAs (miRNAs), and 229 circular RNAs (circRNAs) were differentially expressed (DE) in P12 compared with P4, with a similar trend in P6. Gene ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), and gene set enrichment analysis (GSEA) identified several significant biological processes and pathways, including binding, ossification, and Wnt and PPAR signaling pathways. Interaction and co-expression/localization analyses were performed for DE mRNAs and lncRNAs, and several key lncRNAs, circRNAs, and important pathways like autophagy and mitophagy were identified in the competing endogenous RNA (ceRNA) network. Some key RNAs found in the bioinformatics analysis were validated. Our studies indicate that replicative senescence of MSCs is a continuous process, including widespread alterations in biological characteristics and global gene expression patterns that need to be considered before therapeutic applications of MSCs.
Keywords: MSCs, passage, in vitro aging, RNA-seq, lncRNAs, circRNAs, ceRNA
Graphical abstract
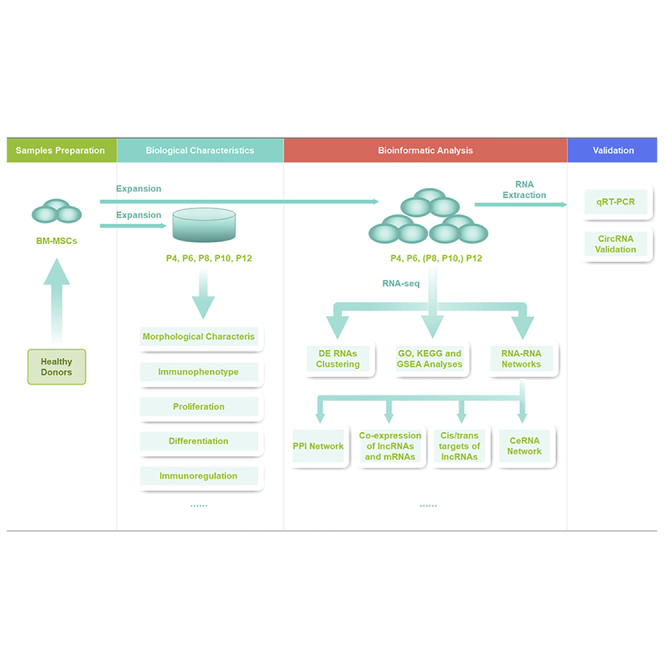
Probes into effects of long-term culture on the biological characteristics and ceRNA network-associated RNA profiles of human bone-marrow-derived mesenchymal stem cells (MSCs) demonstrated key molecules and networks, which might be applied potentially to MSC product manufacturing and quality assessment.
Introduction
Mesenchymal stem cells (MSCs) are an attractive candidate for clinical applications in cell therapy and regenerative medicine due to their self-renewal ability, multi-lineage differentiation potential, immunosuppressive properties, trophic effects, and paracrine function.1 Since the discovery of MSCs in bone marrow (BM) in 1966, various tissues, including umbilical cord (UC) blood, UC, synovial membrane, adipose tissues, dental pulp, and skeletal muscle, have been reported as sources of MSCs.2, 3, 4 Among the different sources, however, BM is still by far the best characterized source of MSCs. Moreover, due to significant ethical issues and safety considerations, MSCs are seen as more feasible and advantageous in clinical applications than embryonic stem cells.5 Thus, BM-MSCs have been broadly applied in the treatment of various diseases, including graft-versus-host disease (GVHD), Crohn’s disease (CD), diabetes mellitus (DM), multiple sclerosis (MS), myocardial infarction (MI), ankylosing spondylitis (AS), liver failure, and rejection after liver transplantation.6,7
Conventionally, the dose for MSC transplantation is 106 cells/kg body weight, and the total number of MSCs for one patient is approximately 108 per cell therapy in clinical trials.8 MSCs are a very rare population that accounts for only 0.01% to 0.001% of the cell numbers in BM.9 The low frequency and lack of high homogeneity of freshly extracted MSC populations necessitates extensive in vitro expansion following isolation prior to various clinical applications and essential manipulation. According to the “Hayflick limit,” MSCs have a limited replicative lifespan of approximately 50 population doublings in cell culture,10,11 and the functions of MSCs, like proliferative, differentiated, and immunoregulatory potential, are known to change with their in vitro expansion,12, 13, 14, 15 a process that may have a similar mechanism to that of aging in vivo.16 Therefore, it is important to consider the influence of in vitro aging on cellular characteristics when developing new MSC-based therapeutic strategies. Understanding the molecular processes controlling MSC proliferation, senescence, and commitment to specific differentiated cell lineages is not only essential for the development of therapeutic interventions but also crucial to determining the drivers and effectors of age-associated MSC dysfunction that can slow, or even reverse, age-related degenerative changes to enhance repair processes and maintain healthy function in aging tissues.
Long noncoding RNAs (lncRNAs) are a type of ncRNA greater than 200 nucleotides (nt) in length. Circular RNAs (circRNAs), a kind of ncRNA, are a large family of covalently closed ring-like RNA molecules. MicroRNAs (miRNAs) are short ncRNAs. It is known that miRNAs can induce gene silencing by binding to mRNAs, and lncRNAs/circRNAs can regulate gene expression by competitively binding to miRNAs.17,18 Recently, multiple studies have demonstrated that these ncRNAs are important epigenetic regulators and are widely involved in growth, immune system homeostasis, proliferation, and differentiation and development by controlling the fate of cells, including MSCs.19, 20, 21 Large intergenic nc RNAp21 (lincRNAp21) was reported to play a role in MSC senescence by interacting with the Wnt/β-catenin signaling pathway to attenuate oxidative stress.22 However, more lncRNAs and circRNAs involved in MSC aging need to be identified/characterized. Additionally, it is not known how senescence influences the overall expression of lncRNAs and circRNAs, and the epigenetic regulation of coding genes by lncRNAs and circRNAs in MSCs still needs to be analyzed.
With this in mind, we designed a study to analyze how the morphology, immunophenotype, proliferation, differentiation, and immunoregulatory capacity of MSCs are affected by in vitro expansion. In parallel, we analyzed how mRNA, miRNA, lncRNA, and circRNA expression profiles change upon culturing and in vitro propagation. We aimed to gain insight into the molecular mechanisms underlying MSC aging, which may have an impact on the quality control of MSC preparations, and identify strategies to rejuvenate senescent MSCs to broaden the range of therapeutic applications of MSCs.
Results
Changes in the morphological characteristics, population doublings, immunophenotype, differentiation, and immunoregulation of HBM-MSCs at different passages
A total of 20 healthy donors between the age of 20 and 30 years were selected for this study. MSCs from one donor were cultured separately as one sample. During primary culture, spindle-shaped, plastic-adherent, and fibroblast-like cells were observed around the adherent hematopoietic progenitor cells in all donor samples within the first day of culture. The cells grew, formed colonies, and appeared as a heterogeneous population of BM-MSCs at passage 0 (P0, Figure 1A), which were extended to P12 with subsequent subculture. P4 MSCs comprised a mostly pure population without hematopoietic progenitor cells. Proliferation and the number of cell population doublings (NCPD) decreased gradually in all samples over the course of long-term culture from P4 to P12 (Figures 1B and 1C). Replicative senescence led to the following previously observed typical morphological changes (Figures 1D and 1E): the cells became much larger with irregular and flat shapes, and the nuclei became more circumscribed by phase contrast microscopy. The cytoplasm became granular with many inclusions appearing as increased cell debris. The cell cycle analysis of MSCs from P4, P6, P8, P10, and P12 during long-term culture indicated that the percentages of cells in S and G2/M phases decreased gradually (Figure 1F; Table S1). Taken together, these results demonstrated the increased morphological heterogeneity and reduced proliferative capability of MSCs during in vitro aging.
Figure 1.

Morphologic and proliferative changes and immunophenotype upon senescence
(A and E) Representative images of the fibroblast-like morphology of MSCs at P0 (A) and P4, P6, P8, P10, and P12 (E) are presented. Scale bars, 100 μm. (B) CCK-8 assay of MSCs at P4, P6, P8, P10, and P12. (C) NCPD of MSCs from P4 to P12. (D) The continuous increase in cell size is reflected by the increasing forward-scatter signal in flow cytometry (FSC). (F) Representative cell cycle diagrams of MSCs during long-term culture at P4, P6, P8, P10, and P12. (G) Surface marker expression on MSCs at P4 and P12 analyzed using flow cytometry. Black lines represent isotype control. Data are presented as the means ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 compared with P4. n = 9 MSCs per passage.
Flow cytometric analysis indicated the presence of CD90, CD105, and CD73 expression and the absence of CD45, CD14, CD34, and human leukocyte antigen DR (HLA-DR) expression in MSCs at P4, P6, P8, P10, and P12, confirming their MSC immunophenotype (Figure 1G; Table 1). With increasing passages, the percentage of MSCs expressing CD45, CD14, CD34, and HLA-DR remained stable, while the percentage of MSCs expressing CD90, CD105, and CD73 decreased gradually, especially CD105, which was decreased from 94.05% (mean) in P4 to 47.38% in P12. These results show that the composition and expression levels of surface markers partly vary upon long-term expansion.
Table 1.
Quantitative profiles of surface marker expression of MSCs at P4, P6, P8, P10, and P12 (n = 9, means ± SD)
| Marker | P4 | P6 | P8 | P10 | P12 |
|---|---|---|---|---|---|
| CD105 | 94.05 ± 4.75 | 89.91 ± 11.13 | 68.27 ± 15.31 | 50.63 ± 16.25 | 47.38 ± 19.07 |
| CD90 | 98.19 ± 1.27 | 97.67 ± 1.95 | 95.67 ± 2.02 | 92.53 ± 4.27 | 92.78 ± 4.71 |
| CD73 | 97.48 ± 1.95 | 97.12 ± 2.95 | 93.17 ± 8.17 | 91.09 ± 3.17 | 89.76 ± 8.18 |
| CD14 | 0.32 ± 0.36 | 0.31 ± 0.31 | 0.07 ± 0.12 | 0.18 ± 0.25 | 0.10 ± 0.15 |
| CD34 | 0.01 ± 0.02 | 0.15 ± 0.22 | 0.04 ± 0.10 | 0.11 ± 0.16 | 0.05 ± 0.13 |
| CD45 | 0.70 ± 0.64 | 0.32 ± 0.39 | 0.11 ± 0.16 | 0.23 ± 0.31 | 0.89 ± 0.51 |
| HLA-DR | 3.38 ± 1.55 | 4.49 ± 7.29 | 4.54 ± 5.06 | 4.89 ± 4.59 | 3.62 ± 7.85 |
The potential for osteogenic and adipogenic differentiation was confirmed following standard protocols. Alizarin red S (ARS) and oil red O (ORO) staining showed that calcium deposition and fat formation were much more effective in MSCs at early passages than later passages, indicating that the osteogenic and adipogenic differentiation potential both decreased over the course of in vitro senescence (Figures 2A, 2B, 2D, and 2E). Furthermore, the degree of decrease in osteogenesis was much more dramatic than that in adipogenesis. Similarly, the percentage of senescence-associated β-galactosidase (SA-β-gal) positive cells and the staining intensity increased in the last passages of MSCs (Figures 2C and 2F), and the relative telomere length in MSCs decreased in P12 compared with P4 (Figure 2G). These results support the notion that long-term culture has an incremental impact on the differentiation potential of MSCs, which was accompanied by deepening of SA staining and shortening of telomere length of MSCs.
Figure 2.

In vitro differentiation and senescence detection of MSCs at P4, P6, P8, P10, and P12
(A–C) Adipogenic differentiation (ORO staining) (A); osteogenic differentiation (ARS staining) (B); SA-β-gal staining (C) of MSCs at P4, P6, P8, P10, and P12. Scale bars, 50 μm in (A) and (C), 100 μm in (B). (D) Quantification of ORO staining in (A). (E) Quantification of ARS staining in (B). (F) Percent of SA-β-gal staining in (C). (G) Relative telomere length detection of MSCs in P4, P6, and P12. Data are presented as the means ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 compared with P4. n = 9 MSCs per passage.
The potential of MSCs at different passages to inhibit CD4+ T cell and CD19+ B cell proliferation was analyzed by coculturing αCD3CD28-activated CD4+ T cells and LPS-stimulated CD19+ B cells with MSCs at P4, P6, P8, P10, and P12. The capacity of MSCs to suppress CD4+ T cell and B cell proliferation decreased during prolonged culture expansion. When cocultured with P4 and P6 MSCs, activated CD4+ T cells and B cells could be inhibited, while P8 and P10 MSCs almost completely lost the capacity to inhibit CD4+ T cell and CD19+ B cell proliferation. Moreover, P12 MSCs even enhanced the proliferation of CD4+ T cells and CD19+ B cells (Figures 3A–3D).
Figure 3.

Ability of MSCs at different passages to inhibit T cell and B cell proliferation
(A) Flow cytometric analysis of CD4+ T cell proliferation in the presence of MSCs at P4, P6, P8, P10, and P12. Nonactivated CD4+ T cells were defined as the blank group. The proliferation rate of CD4+ T cells was markedly elevated when the cells were stimulated by CD3/CD28. (B) Quantification of the CD4+ T cell proliferation rate in (A). (C) Flow cytometric analysis of CD19+ B cell proliferation in the presence of MSCs at P4, P6, P8, P10, and P12. Nonactivated CD19+ B cells were defined as the blank group. The proliferation rate of CD19+ B cells was markedly elevated upon stimulation with LPS. (D) Quantification of the CD19+ B cell proliferation rate in (C). Data are presented as the means ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. n = 9 MSCs per passage.
Differential gene-expression profiles of MSCs during in vitro aging
A total of nine samples consisted of three MSCs at P4, P6, and P12, respectively, were subjected to whole-transcriptome RNA sequencing (RNA-seq). The mRNAs, lncRNAs, miRNAs, and circRNAs that were differentially expressed (DE) in P12 compared with P4 (fold change |log2 [P12/P4]| ≥ 1, Q value [P12/P4] < 0.001), which have the same express trends in P6, and were abundantly expressed (mRNAs and circRNAs, FPKM ≥ 5; lncRNA and miRNA, FPKM ≥ 2) in MSCs from three donors were used for the subsequent analyses. A total of 792 RNAs were collected, among which there were 439 mRNAs (204 were upregulated and 235 were downregulated), 65 lncRNAs (48 were upregulated and 17 were downregulated), 229 circRNAs (202 were upregulated and 27 were downregulated), and 59 miRNAs (24 were upregulated and 35 were downregulated). The DE RNAs are depicted using a clustergram (Figure 4A–4D). The top 5 mRNAs, lncRNAs, miRNAs, and circRNAs that were upregulated or downregulated with the largest fold changes are shown in Tables 2, 3, 4, and 5, respectively.
Figure 4.

Heatmaps of DE RNAs and functional enrichment analysis of DE mRNAs
(A–D) Heatmaps of DE mRNAs (A), DE circRNAs (B), DE lncRNAs (C), and DE miRNAs (D) between the P4 and P12 groups and with a similar trend in P6 (n = 3 per passage). (E–G) DE mRNAs were clustered by GO analysis, and the top 20 terms of molecular function (E), biological process (F), and cellular component (G) are shown, respectively. (H) DE mRNAs were clustered by KEGG analysis, and the top 20 pathways are shown.
Table 2.
Characteristics of mRNAs with the largest fold changes (P12 versus P4)
| Gene ID | Gene symbol | Fold change (log2 [P12/P4]) | Regulation |
|---|---|---|---|
| 4741 | NEFM | 5.439747 | UP |
| 5055 | SERPINB2 | 5.239469 | UP |
| 3952 | LEP | 4.578009 | UP |
| 11075 | STMN2 | 4.034502 | UP |
| 730755 | KRTAP2-3 | 3.945871 | UP |
| 1043 | CD52 | −9.50693 | DOWN |
| 4318 | MMP9 | −8.25542 | DOWN |
| 165 | AEBP1 | −4.1185 | DOWN |
| 54 | ACP5 | −3.78729 | DOWN |
| 81035 | COLEC12 | −3.76893 | DOWN |
Table 3.
Characteristics of lncRNAs with the largest fold changes (P12 versus P4)
| Gene ID | Gene symbol | Fold change (log2 [P12/P4]) | Chromosome | Strand | Start | End | Regulation |
|---|---|---|---|---|---|---|---|
| 100750247 | HIF1A-AS2 | 4.959349 | NC_000014.9 | – | 61747039 | 61749089 | UP |
| 106480738 | OSTN-AS1 | 4.729266 | NC_000003.12 | – | 191212868 | 191235780 | UP |
| 109729169 | LINC02154 | 4.043334 | NC_000023.11 | + | 13246302 | 13329958 | UP |
| 152742 | LINC01085 | 2.826299 | NC_000004.12 | + | 14111968 | 14143592 | UP |
| 100506343 | INKA2-AS1 | 2.790145 | NC_000001.11 | + | 111738607 | 111753572 | UP |
| 107984611 | LOC107984611 | −5.35235 | NC_000013.11 | – | 42767224 | 42819890 | DOWN |
| 388011 | LINC01550 | −5.35235 | NC_000014.9 | + | 97925610 | 98542334 | DOWN |
| 102724678 | LINC01423 | −5.28412 | NC_000021.9 | + | 38323635 | 38346076 | DOWN |
| 105377043 | LOC105377043 | −5.18423 | NC_000003.12 | + | 40717410 | 40878958 | DOWN |
| 100192378 | ZFHX4-AS1 | −1.86988 | NC_000008.11 | + | 76491200 | 76683672 | DOWN |
Table 4.
Characteristics of miRNAs with the largest fold changes (P12 versus P4)
| Gene ID | Fold change (log2 [P12/P4]) | Chromosome | Strand | Start | End | Regulation |
|---|---|---|---|---|---|---|
| hsa-miR-302b-3p | 3.39128 | NC_000004.12 | – | 112648489 | 112648511 | UP |
| hsa-miR-302a-3p | 3.32416 | NC_000004.12 | – | 112648186 | 112648208 | UP |
| hsa-miR-302d-3p | 2.5962 | NC_000004.12 | – | 112648006 | 112648028 | UP |
| hsa-miR-483-5p | 2.03057 | NC_000011.10 | – | 2134181 | 2134202 | UP |
| hsa-miR-451a | 1.97568 | NC_000017.11 | – | 28861403 | 28861424 | UP |
| hsa-miR-122-5p | −1.47252 | NC_000018.10 | + | 58451088 | 58451109 | DOWN |
| hsa-miR-3613-5p | −1.50052 | NC_000013.11 | – | 49996465 | 49996486 | DOWN |
| hsa-miR-199b-5p | −2.84583 | NC_000009.12 | – | 128244783 | 128244805 | DOWN |
| hsa-miR-3529-3p | −6.02915 | NC_000015.10 | – | 88611856 | 88611879 | DOWN |
| hsa-miR-206 | −6.04024 | NC_000006.12 | + | 52144401 | 52144422 | DOWN |
Table 5.
Characteristics of circRNAs with the largest fold changes (P12 versus P4)
| Gene ID | Fold change (log2 [P12/P4]) | Chromosome | Strand | Start | End | Gene symbol | Regulation |
|---|---|---|---|---|---|---|---|
| hsa_circ_0081567 | 2.473584 | NC_000007.14 | + | 101130421 | 101130654 | SERPINE1 | UP |
| hsa_circ_0081564 | 2.384603 | NC_000007.14 | + | 101128393 | 101128664 | SERPINE1 | UP |
| hsa_circ_0073686 | 2.371695 | NC_000005.10 | – | 122070494 | 122075541 | LOX | UP |
| hsa_circ_0081565 | 2.364576 | NC_000007.14 | + | 101128393 | 101132069 | SERPINE1 | UP |
| hsa_circ_0081574 | 2.358868 | NC_000007.14 | + | 101135494 | 101137084 | SERPINE1 | UP |
| hsa_circ_0135618 | −2.3529 | NC_000008.11 | + | 120147806 | 120168247 | COL14A1 | DOWN |
| hsa_circ_0116771 | −2.35738 | NC_000022.11 | + | 45541229 | 45542283 | FBLN1 | DOWN |
| hsa_circ_0063743 | −2.36496 | NC_000022.11 | + | 45525543 | 45528009 | FBLN1 | DOWN |
| hsa_circ_0063750 | −2.45822 | NC_000022.11 | + | 45541229 | 45543526 | FBLN1 | DOWN |
| hsa_circ_0063758 | −2.69773 | NC_000022.11 | + | 45562912 | 45563362 | FBLN1 | DOWN |
mRNA GO, KEGG, and GSEA analyses
We performed gene ontology (GO) analysis on the identified DE mRNAs. The top 20 GO terms related to biological processes, cellular components, and molecular functions are shown in Figures 4E–4G. The top 10 GO terms related to biological processes and DE mRNAs associated with these terms are shown in Table 6. Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis of the DE mRNAs determined that 32 pathways were significantly altered in MSCs during in vitro aging (p < 0.05). The top 20 enriched pathways are shown in Figure 4H. The top 10 pathways and DE mRNAs associated with these pathways are shown in Table 7. The top pathways included systemic lupus erythematosus, complement and coagulation cascades, Wnt signaling pathway, pathways in cancer, and Hedgehog signaling pathway, which are involved in immunity, signal transduction, proliferation, and differentiation. Gene set enrichment analysis (GSEA) identified 62 terms in MSCs of P12 compared with P4 (NOM p value < 0.05). Some important terms like DNA replication, Notch signaling pathway, T cell receptor signaling pathway, Wnt signaling pathway, etc. were illustrated in Figure S1. Specifically, we focused on the terms and DE mRNAs identified in the GO biological process analysis of DE mRNAs related to proliferation, differentiation, or the immune response and selected these DE mRNAs as the gene set for analysis. The top 10 terms with the largest significant differences are listed in Tables S2–S4.
Table 6.
Top 10 terms with the largest significant differences in the GO biological process analysis of DE mRNAs
| Term | Count | Rich ratio | p value | Candidate gene |
|---|---|---|---|---|
| Extracellular matrix organization | 25 | 0.090909 | 2.24E-11 | OLFML2B, NDNF, MMP11, OLFML2A, COL16A1, RECK, ADAM19, ITGA10, TNC, FBLN1, FGF2, MMP9, SERPINE1, COL5A3, EVI2A, TGFBI, THBS1, MFAP2, COMP, ERO1A, TNFRSF11B, COL14A1, KAZALD1, ABI3BP, COL4A1 |
| Ossification | 15 | 0.12605 | 2.79E-09 | TMEM119, LRRC17, IFITM1, MMP9, STC1, ACP5, MMP16, TWIST1, CLEC3B, PTN, COMP, RUNX2, KAZALD1, MGP, IGF2 |
| Cell adhesion | 38 | 0.048906 | 1.46E-08 | COL16A1, TINAGL1, S1PR1, RGMB, NRP2, PVR, ITGA10, TNC, EGFL7, JUP, THBS2, LOXL2, SCN1B, COL5A3, TGFBI, THBS1, MTSS1, PCDHGC3, CDH6, SPON2, BCAM, CXCL12, RND3, PERP, PCDHGB7, THBS3, COMP, CGREF1, PCDHGA10, COL14A1, CADM1, NRCAM, WISP2, SELENOP, MFAP4, GPNMB, EPHA2, PTPRU |
| Collagen fibril organization | 11 | 0.161765 | 2.64E-08 | MMP11, GREM1, PLOD2, LOXL2, COL5A3, EVI2A, LOX, COMP, COL14A1, LOXL4, P4HA1 |
| Nucleosome assembly | 15 | 0.088757 | 3.21E-07 | SMYD3, HIST1H4F, HIST1H1B, HIST1H4E, HIST1H2BI, HIST2H3D, HIST1H2BO, HIST1H4J, HIST1H4L, HIST1H4K, HIST1H4A, HIST1H3I, HIST1H3H, HIST1H3A, HIST1H2BN |
| Angiogenesis | 21 | 0.058496 | 1.73E-06 | LEP, NDNF, GREM1, ANGPTL4, S1PR1, NRP2, SLC38A1, EPGN, CXCL8, EPHB2, EGFL7, ADGRA2, SERPINE1, JAG1, TGFBI, FZD8, CAV1, CCBE1, NRCAM, EPHA2, COL4A1 |
| Cellular response to hypoxia | 13 | 0.089655 | 1.75E-06 | NDNF, MGARP, ANKRD1, STC1, TWIST1, FMN2, STC2, EDN1, PTN, NOP53, ZFP36L1, BNIP3, ERO1A |
| Skeletal system development | 14 | 0.075676 | 5.25E-06 | SH3PXD2B, FAM20C, NPR3, SOX4, PAPSS2, MMP9, ANKH, EDN1, AEBP1, COMP, RUNX2, TNFRSF11B, EPHA2, IGF2 |
| Negative regulation of blood coagulation | 5 | 0.294118 | 8.40E-06 | APOE, TFPI, SERPINE1, EDN1, SERPINE2 |
| Amino acid transport | 8 | 0.131148 | 1.09E-05 | SLC7A11, SLC38A1, SLC1A1, SLC7A14, SLC7A8, SLC38A5, PDPN, SLC43A1 |
Table 7.
Top 10 biological pathways with the largest significant differences in the KEGG pathway analysis of DE mRNAs
| Pathway | Count | Rich ratio | p value | Gene |
|---|---|---|---|---|
| Systemic lupus erythematosus | 21 | 0.094595 | 2.77E−10 | HIST1H4F, HIST1H4E, HIST1H2BI, HIST2H3D, HIST2H2AB, HIST1H2AH, HIST1H2BO, HIST1H4J, HIST1H4L, C1S, HIST1H4K, HIST1H4A, HIST2H2AC, HIST1H2AB, C1R, HIST1H3I, 105369230, HIST1H3H, HIST1H2AM, HIST1H3A, HIST1H2BN |
| Alcoholism | 20 | 0.096154 | 5.59E−10 | BDNF, HIST1H4F, HIST1H4E, HIST1H2BI, HIST2H3D, HIST2H2AB, HIST1H2AH, HIST1H2BO, HIST1H4J, HIST1H4L, HIST1H4K, SHC2, HIST1H4A, HIST2H2AC, HIST1H2AB, HIST1H3I, HIST1H3H, HIST1H2AM, HIST1H3A, HIST1H2BN |
| Malaria | 7 | 0.102941 | 1.67E−04 | CXCL8, THBS2, THBS1, GYPC, THBS3, COMP, SDC1 |
| Complement and coagulation cascades | 9 | 0.076271 | 2.07E−04 | CLU, TFPI, SERPINE1, C1S, C1R, CD55, CFI, SERPINB2, CFB |
| ECM-receptor interaction | 8 | 0.063492 | 0.001548 | ITGA10, TNC, THBS2, THBS1, THBS3, COMP, SDC1, COL4A1 |
| Wnt signaling pathway | 10 | 0.052632 | 0.001745 | GREM1, PLCB4, CCND1, FZD8, DKK1, DVL2, PORCN, SERPINF1, CCND2, SFRP4 |
| AGE-RAGE signaling pathway in diabetic complications | 8 | 0.061069 | 0.00198 | STAT5A, PLCB4, CXCL8, CCND1, SERPINE1, EDN1, STAT1, COL4A1 |
| Pathways in cancer | 24 | 0.032742 | 0.002047 | NOTCH3, DAPK2, STAT5A, SULF2, PLCB4, CXCL8, JUP, FGF2, MMP9, FGF5, CCND1, GLI3, JAG1, FZD8, GSTM4, STAT1, CXCL12, DVL2, SLC2A1, TXNRD1, ABI3BP, IGF2, COL4A1, CCND2 |
| Protein digestion and absorption | 7 | 0.066667 | 0.002287 | COL16A1, SLC1A1, COL5A3, SLC7A8, COL14A1, VWCE, COL4A1 |
| Bladder cancer | 5 | 0.089286 | 0.002755 | DAPK2, CXCL8, MMP9, CCND1, THBS1 |
PPI and co-expression network analyses
Altogether, 127 nodes (56 upregulated and 71 downregulated genes) and 311 interaction pairs were identified in the PPI network of the DE mRNAs (Figure 5A). The nodes with high topological scores were noted to possibly play important roles during in vitro aging of MSCs. In this study, the hub nodes matrix metallopeptidase 9 (MMP9), G1/S-specific Cyclin D1 (CCND1), FGF2, C-X-C motif chemokine ligand 8 (CXCL8, also known as interleukin-8 [IL-8]), serpin family E member 1 (SERPINE1), and LEP that interacted with many other DE mRNAs were regarded as the key genes of the network. In addition, a co-expression network was constructed for DE lncRNAs and DE mRNAs (Figure 5B). Finally, we obtained 128 co-expression pairs (including 32 mRNAs and 6 lncRNAs) according to a Pearson correlation coefficient ≥ 0.900 cut-off value. LINC01013 had the highest number of targets, including 13 DE RNAs. LINC00607 and LOC102723409 were also demonstrated to be important RNAs. The top 20 co-expression pairs are shown in Table 8. Next, we analyzed the possible target genes of the DE lncRNAs and identified a total of 784 target genes. A Venn diagram analysis demonstrated that 8 mRNAs were found at the intersection of the 439 DE mRNAs and the 784 DE lncRNA cis/trans-target genes (Figure 5C). These 8 mRNAs and the lncRNAs are shown in Figure 5D, which includes the LOC102723409-Rho GTPase activating protein 18 (ARHGAP18) trans-regulatory pair, a pair that was also identified in the co-expression analysis.
Figure 5.

Interaction and co-expression/co-localization network analyses
(A) Interactions between DE mRNAs. (B) Co-expression network of DE lncRNAs and DE mRNAs. (C) Venn diagram showing DE mRNAs and cis/trans-target genes of DE lncRNAs. Circles indicate coding genes, and rhombuses indicate lncRNAs. Red nodes indicate upregulated genes, and blue nodes indicate downregulated genes (P12/P4). The edges indicate the interaction between two nodes. Black arrows indicate the key lncRNAs.
Table 8.
Top 10 co-expression pairs of DE mRNAs and lncRNAs
| Source ID | Gene symbol | Target ID | Gene symbol | Correlation coefficient |
|---|---|---|---|---|
| 7057 | THBS1 | 100507254 | LINC01013 | 0.972 |
| 1901 | S1PR1 | 100507254 | LINC01013 | 0.963 |
| 9411 | ARHGAP29 | 100507254 | LINC01013 | 0.952 |
| 100507254 | LINC01013 | 51232 | CRIM1 | 0.949 |
| 114902 | C1QTNF5 | 100507254 | LINC01013 | 0.947 |
| 102723761 | LINC02773 | 7123 | CLEC3B | 0.946 |
| 729178 | STXBP5-AS1 | 158471 | PRUNE2 | 0.943 |
| 11217 | AKAP2 | 100507254 | LINC01013 | 0.941 |
| 79674 | VEPH1 | 100507254 | LINC01013 | 0.939 |
| 79642 | ARSJ | 646324 | LINC00607 | 0.939 |
| 25825 | BACE2 | 100507254 | LINC01013 | 0.938 |
| 5166 | PDK4 | 102723761 | LINC02773 | 0.938 |
| 5552 | SRGN | 646324 | LINC00607 | 0.935 |
| 5176 | SERPINF1 | 102723761 | LINC02773 | 0.935 |
| 102723409 | LOC102723409 | 93663 | ARHGAP18 | 0.933 |
| 100507254 | LINC01013 | 25925 | ZNF521 | 0.933 |
| 7436 | VLDLR | 102723761 | LINC02773 | 0.933 |
| 9411 | ARHGAP29 | 646324 | LINC00607 | 0.930 |
| 5552 | SRGN | 102723409 | LOC102723409 | 0.928 |
| 102723409 | LOC102723409 | 9411 | ARHGAP29 | 0.924 |
ceRNA networks
lncRNAs and circRNAs act as competing endogenous RNAs (ceRNAs) to exert their biological function; thus, we constructed ceRNA networks with lncRNAs and circRNAs as decoys, miRNAs as centers, and mRNAs as targets from all of the DE mRNAs, lncRNAs, and miRNAs to identify possible regulatory lncRNA/circRNA-miRNA-mRNA axes. A total of 21 miRNAs, 53 lncRNAs, and 130 mRNAs involving 204 regulatory relationships (Figures 6A–6C) and 16 miRNAs, 59 circRNAs, and 98 mRNAs involving 330 regulatory relationships (Figures 7A–7C) were obtained. We divided the ceRNA regulatory networks into two groups based on their gene expression patterns: down-up-down (Figures 6B and 7B) and up-down-up (Figures 6C and 7C). MIR99AHG, SMILR, LOC105747689, LINC01013, STXBP5-AS1, and LINC01503 were enriched in the ceRNA network. Next, we performed KEGG pathway analysis on these genes to identify pathway clusters. The related RNAs and the relationships between these pathways are shown in Figure 7D and include the autophagy and mitophagy signaling pathways, extracellular matrix (ECM)-receptor interactions, and PI3K-Akt signaling pathway. LOC102723409 and ITGA6-AS1 are the key lncRNAs involved in this network. INKA2-AS1, MIR99AHG, STXBP5-AS1, SMILR, LINC01503, and LOC105747689 are important lncRNAs involved in autophagy and the mitophagy pathways. Some circRNAs like circ_0005773 and circ_0058476 were identified in the circRNAs ceRNA network. The characteristics, like position, length, and parental genes, of the circRNAs involved in the prediction analysis were listed in Table S5.
Figure 6.

ceRNA network of lncRNA-miRNA-mRNA interactions
(A) Venn diagram showing the intersection of DE mRNAs and target mRNAs of DE miRNAs and the intersection of DE lncRNAs and target lncRNAs of DE miRNAs. (B and C) ceRNA regulatory network of lncRNA-miRNA-mRNA interactions. (D) Genes involved in the ceRNA network were clustered using KEGG pathway analysis. Squares indicate miRNAs, circles indicate coding genes, and rhombuses indicate lncRNAs. Red indicates upregulation, and blue indicates downregulation (P12/P4). Black arrows indicate the key lncRNAs.
Figure 7.

ceRNA network of circRNA-miRNA-mRNA interactions
(A) Venn diagram showing the intersection of DE mRNAs and target mRNAs of DE miRNAs and the intersection of DE circRNAs and target circRNAs of DE miRNAs. (B and C) ceRNA regulatory network of circRNA-miRNA-mRNA interactions. Squares indicate miRNAs, circles indicate coding genes, and triangles indicate circRNAs. Red indicates upregulation, and blue indicates downregulation (P12/P4). Black arrows indicate the key circRNAs.
Validation of the key RNAs expression
To verify the RNA-seq data and major findings from computational analysis, we performed quantitative real-time PCR for some key RNAs selected from the RNA networks. The expression levels of those RNAs were measured in 10 human BM-MSCs in a different passage. We designed specific divergent primers spanning the back-splice junction sites of circRNAs (Table S6). The qPCR results showed that the expression of miR-210-5p, miR-431-5p, ITGA6-AS1, INKA2-AS1, STXBP5-AS1, LOC102723409, LOC105747689, LINC01013, circ_0005773, circ_0058476, circ_0081571, circ_0081562, COMP, STC1, ADAMTS1, INKA2 CDH6, STC2, and TINAGL1 were upregulated and that the expression of miR-1908-5p, miR-16-5p, miR-122-5p, miR-193a-3p, miR-199b-5p, miR-339-5p, MIR99AHG, LINC01503, circ_0078715, circ_0078711, and SPON2 were downregulated (Figures 8A–8D). Other qPCR results were illustrated in Figure S2. To verify the back-splice junction sequences of circRNAs, we sent circ_0005773 quantitative real-time PCR amplified products to Sanger sequencing, and the sequencing result confirmed the back-splice junction sites (Figure 8E). We confirmed that circ_0005773 was resistant to RNase R, whereas its linear SERPINE2 mRNA level was significantly decreased after RNase R treatment (Figure 8F). Bioinformatic analyses indicated that circ_0005773 could potentially bind to miR-17-5p. qPCR results showed the upregulated expression of circ_0005773, downregulated expression of miR-17-5p, and upregulated expression of mRNA LIF, the possible targets of miR-17-5p, in MSCs of P12 compared with P4, consistent with the results of deep sequencing (Figures 8G–8I). The circ_0005773/miR17-5p/LIF axis deserves further attention. All qPCR results were consistent with the RNA-seq results, confirming the reliability of the sequencing results, while more work still need to be done to validate the RNA network relationship.
Figure 8.

Validation of the key RNAs
(A–D) Comparison of log2FC in eight DE miRNAs (A), eight DE mRNAs (B), eight lncRNAs (C), and six circRNAs (D) between RNA-seq and quantitative real-time PCR. n = 3 MSCs per passage in RNA-seq. n = 10 per passage in quantitative real-time PCR. (E) Schematic illustration showed the circularization of SERPINE2 exons 3–4, which resulted in the formation of circ_0005773 (black arrow). Sanger sequencing confirmed the back-splice junction sites of circ_0005773. Red arrow represents head-to-tail circ_0005773 splicing sites. (F) The expression of circ_0005773 and SERPINE2 mRNA in MSCs treated with or without RNase R was detected by quantitative real-time PCR. The relative levels of circ_0005773 and SERPINE2 mRNA were normalized to the values measured for mock treatment. n = 10 MSCs. (G–I) qPCR validation of the expression levels of circ_0005773 (G), miR-17-5p (H), and LIF (I). Data are presented as the means ± SEM. ∗p < 0.05, ∗∗p < 0.01. FC, fold change. n = 10 MSCs per passage.
Discussion
The present study identified a series of MSC aging-related mRNAs, miRNAs, lncRNAs, and circRNAs that are potential mediators of the changes in biological characteristics, such as decreased proliferation, differentiation, and immunosuppression abilities after long-term culture. Using a series of bioinformatics analyses, LOC1027234409, ITGAS6-AS1, MIR99AHG, LINC01013, etc., and some circRNAs were identified based on our prediction. By constructing a ceRNA network with lncRNAs/circRNAs, miRNAs, and mRNAs, we identified the links between these RNA species. Further functional analyses suggested a potential mechanism through which these ceRNAs mediated MSC aging. This is the first systematic analysis of lncRNAs and circRNAs and their functional networks in human MSCs at early and late passages, which emphasize the important role of lncRNAs and circRNAs in the replicative senescence of BM-MSCs, although more research is necessary to confirm our findings.
Many studies have reported that MSCs undergo the typical Hayflick phenomenon of cellular senescence, characterized by decreased proliferation and changes in cell morphology, and exhibit an aberrant phenotype of irregular fattened geometry and enlarged size during long-term culture.13 Consistent with this, we found that senescence obviously occurred after P6–P8, which is compatible with expansion rates of 106-fold to 108-fold (ranging from 16 to 24 population doublings). The decreased CD105 was detected, while CD90 and CD73 were relatively stable after prolonged passaging, consistent with previous reports.15,23 CD105, which is the same as Stro-1, has been considered to be more applicable to be used as phenotypic markers of MSCs multipotency.24 Typical MSC markers such as CD90 and CD73 appear not enough or specific in indicating maintenance of “stemness,” and that multiparametric immunophenotyping, including CD105 or other stem cell markers, should be supplemented. In addition, decreased adipogenic differentiation abilities of MSCs during in vitro aging were observed in our studies, in accordance with the previous reports.10,13 However, the propensity for osteogenic differentiation of MSCs during in vitro aging were still discordant. Wagner et al.10 suggested that osteogenic differentiation increases at higher passages. Yu et al.25 reported that the osteogenic differentiation potential increased up to P10 and decreased thereafter. Yang et al.13 suggested that osteogenic differentiation abilities of MSCs during in vitro aging were decreased as a generation, consistent with our studies. These conflicting results of the differentiation ability of MSCs during long-term culture demonstrated that further study is needed. Consistent with our results, De Witte et al.14 reported that long-term expansion induced aging of human UC MSCs (hUC-MSCs), which exhibited reduced immunosuppressive properties from P4 to P12.
Moreover, recent studies found that changes in transcriptome and epigenetic regulation in hUC-MSCs occurred during long-term expansion.26, 27, 28 mRNA expression profiles reflect the biological behaviors and functions of cells.14 These changes in mRNA expression profiles are closely related to the functional changes during in vitro aging.29 We identified 768 DE mRNAs between P12 and P4, with a consistent trend in P6. Among these, DE mRNAs, NEFM, SERPINB2, CD52, and MMP9 were the most significantly changed. In addition, GO analysis demonstrated enrichment of specific biological processes, such as ossification and cell differentiation. KEGG and GSEA identified some environmental information processing signaling pathways, including ECM-receptor interaction, Wnt signaling pathway, Hedgehog signaling pathway, PPAR signaling pathway, and Jak-STAT signaling pathway. Part of these findings and analysis were confirmed in Figure S2. These signal transduction pathways play important roles in SA functional changes.30,31 Notably, the SLE and AGE-RAGE signaling pathways in diabetic complications and insulin resistance were also among the pathways identified as having the most significant difference by KEGG analysis. These results are consistent with those of a previous study and emphasize the importance of MSC senescence in the pathogenesis and clinical treatment of autoimmune and metabolic diseases.
In the interactions between proteins encoded by DE, mRNAs, MMP9, CXCL8, SERPINE1, and LEP were identified as the key genes that interact with many other DE mRNAs in this network. MMP9 is involved in the breakdown of extracellular matrix in normal physiological processes and integrates multiple immunoregulatory pathways that determine the high suppressive activity of hMSCs.32 CXCL8 was demonstrated to play an important role in neutrophil activation and migration and could also recruit T leukomonocytes, monocytes, etc.33 CXCL8 mediates CD4+ T cell migration regulated by MSCs through autophagy.34 SERPINE1 has been reported to be involved in cellular and replicative senescence35 and in the regulation of odontoblast differentiation of periodontal ligament stem cells.36 LEP can maintain mitochondrial integrity and prolong the survival of hMSCs.37 The MMP family members and some kinds of chemokines as the SA secretory phenotype (SASP) components have been reported to be consistently upregulated in senescent cells,38 consistent with our sequencing results. The expression of these molecules was partly validated in Figure S2. Therefore, these proteins may be used as indicators for the characterization of MSCs after long-term culturing. However, whether these proteins can be used as markers in clinical detection remains to be further studied.
mRNA expression profiles are under the control of a series of epigenetic regulators, of which ncRNAs, such as lncRNAs, miRNAs, and circRNAs, are important components. Numerous studies have confirmed that lncRNAs and miRNAs regulate the aging of MSCs,21,22 and the function of circRNAs in MSC fate decisions has emerged.19,39 However, the circRNAs involved in the aging of MSCs remain unknown, and a comprehensive analysis of lncRNAs involved in MSC aging is also lacking. In this study, we first determined the ncRNA expression profiles involved in MSC long-term culture and found that 65 lncRNAs, 59 miRNAs, and 229 circRNAs were DE in MSCs at P12 compared with P4. These DE lncRNAs/circRNAs may contribute to the differential expression of mRNAs and thus may be involved in aging-related changes in MSCs. What’s more, the interacting ncRNA cascades can serve as a novel and potential therapeutic target for MSCs application and molecular therapy. The expression of some key ncRNAs involved in the predicted analysis were validated in Figure 8.
lncRNAs can function in cis, at the site of transcription, or in trans and as such participate in multiple networks regulating gene expression and function.40 We identified three pairs of genes that interact through cis-regulation and five pairs that interact through trans-regulation, which may be involved in the regulation of MSC aging. In addition, the ARHGAP18-LOC102723409 pair was identified in the co-expression and target analysis, and the ARHGAP29-LINC01013 pair was identified in the co-expression analysis. The ARHGAP genes encode a family of at least 32 members of the RhoGAP protein family with variable functions,41 including maintenance and control of the actin cytoskeleton, angiogenesis, cell proliferation, differentiation, and survival.42 However, their functions in MSCs, especially in the aging process, have not yet been determined, and in our study, the increased levels of ARHGAP29 and ARHGAP18 during expansion and the relationship with lncRNAs need more attention in the future.
One of the most critical roles of lncRNAs and circRNAs is their ability to act as ceRNAs for miRNAs and relieve mRNAs from miRNA targeting.43 Successful applications of molecular network analysis have been reported in multiple diseases and drug development.44 The cross-talk between ceRNAs through shared miRNAs represents a novel layer of gene regulation that plays an important role in the aging of MSCs.45 However, the specific lncRNAs/circRNAs involved in the immunoregulatory, proliferative, and differentiation capacities of MSCs during aging require further research. Therefore, a core ceRNA network was constructed between DE mRNAs, lncRNAs/circRNAs, and miRNAs based on DE miRNAs targeting interactions, which allowed us to identify several key lncRNAs and circRNAs. The synergically expressed mRNAs and lncRNAs/circRNAs but divergently expressed miRNAs represented the core ceRNAs.44 Therefore, we analyzed the up-down-up and down-up-down relationships of DE lncRNAs/circRNAs, miRNAs, and mRNAs for ceRNA network construction. Combining the ceRNA and KEGG analyses of these genes, we identified several key lncRNAs, including LINC01013, LOC102723409, iTGA6-AS1, INKA2-AS1, MIR99AHG, LINC01503, SMILR, STXBP5-AS1, and LOC105747689, which were not explored in BM-MSCs in previous studies.
Among them, LINC01013, INKA2-AS1, ITGA6-AS1, LOC105747689, and STXBP5-AS1 all showed intimate interactions with miR-339-5p and miR-1908-5p, both of which have been reported to regulate the osteogenic differentiation of BM-MSCs.46,47 Besides, LINC01013 had the highest number of co-expression targets and the top correlation coefficient. It has been reported to negatively regulate the chondrogenic differentiation potential of stem cells.48 INKA2-AS1 was also predicted to cis-target INKA2, which is a novel, direct downstream target of p53 that potentially decreases cell growth by inhibiting the PAK4/β-catenin pathway49 and is involved in mechanisms regulating the actin cytoskeleton and actin-driven processes.50 ITGA6-AS1/PDK1 is another cis-target gene pair. ITGA6-AS1, also named AC078883.3, is enriched in ion binding, transport, cell-cell signaling, and plasma membrane parts and is mainly related to the calcium signaling pathway and the mitogen-activated protein kinase signaling pathway.51 LOC105747689, also named AK130181, which is highly expressed in CD4+ T lymphocytes, suppresses nuclear factor-κB (NF-κB) activity.52 STXBP5-AS1 was reported to inhibit cell proliferation53 and also coexpressed with PRUNE2 in our co-expression analysis. To our knowledge, the role of these lncRNAs in MSC aging has not yet been reported. Future research could focus on these lncRNAs to identify their role in MSC functional changes during aging.
Our trans-regulation results indicated the MIR99AHG and its target gene ZNF608, and the ceRNAs network reminded that the miR-134-5p may be a regulator involved in the MIR99AHG-ZNF608 pair. MIR99AHG has been reported to have a role in cell proliferation and differentiation.54 Moreover, MIR99AHG-ZNF608 was found to be involved in the autophagy and mitophagy signaling pathways in the KEGG analysis. LOC102723409 connected with and regulated several important pathways based on the KEGG analysis, which may be mediated through sponging miR-16-5p, miR-122-5p, and miR-17-5p based on the ceRNA prediction or through ARHGAP18 etc. co-expression mRNAs. LINC01503 could promote proliferation by acting as a ceRNA,55 and miR-210, a pro-survival and anti-apoptotic factor,56 miR-432-5p, and miR-1228-5p may deserve further attention to explore the role and mechanism of LINC01503 in MSC aging.
In addition to lncRNAs, circRNAs can also act as ceRNAs to sponge miRNAs and regulate pathological and physiological processes, including aging-related processes.57,58 circRNAs are a new class of noncoding single-stranded RNAs that differ from linear lncRNAs in that they form covalently closed loop structures without free 3′ poly(A) tails or 5′ caps. Due to the stability of circRNAs, their potential adsorption of miRNAs is stronger than that of lncRNAs and mRNAs.59 Our RNA-seq analysis showed that MSC aging was accompanied by the upregulation of many circRNAs (Figure 4B). Whether these circRNAs function as miRNA sponges to counteract miRNA-mediated repression of mRNAs during MSC aging deserves our attention. circRNAs, generated from genes of THBS1, THBS2, SERPINE1, SERPINE2, LOXL2, LDHA, CCND1, etc. were predicted in the ceRNAs network. Some of circRNAs’ function generated from these genes have been investigated. circSERPINE2, which is formed by the circularization of exons 2–4 of Serpine2 gene, alleviated IL-1β-induced chondrocytes apoptosis and extracellular matrix degradation.60,61 circTHBS1 and circ-CCND1 (hsa_circ_0023303) promote cell proliferation.62, 63, 64 Circ_0005773 was existed and highly expressed in MSCs. The expression of the predicted circ_0005773/miR-17-5p/LIF axis was validated (Figure 8). However, the fuction of many other circRNAs has not been demonstrated yet, not even in the MSC aging.
In conclusion, MSCs expansion is accompanied with a series of biological changes and transcriptome drift, which deserve more concern, especially in MSCs manufacturing protocol. The key RNAs reported here may allow us not only to interrogate for biomarkers and to evaluate relative levels of desired genes to refine parameters for maximum MSCs expansion without compromising therapeutic efficacy but also to identify strategies to rejuvenate senescent MSCs to broaden the range of therapeutic applications of MSCs.
Materials and methods
Human BM-MSC isolation and passage
This study was approved by the Ethics Committee of Eighth Affiliated Hospital, Sun Yat-sen University (Shenzhen, People’s Republic of China). All donors in our study were informed of the possible risks and the study objectives and signed informed consent forms. 20 healthy donors (10 males, 10 females) between the ages of 20 and 30 years who had no history of any significant illness were selected for the study. BM was extracted from the posterior superior iliac spine under sterile conditions, and MSCs were isolated and purified according to a previously reported method.7 MSCs were always passaged at the same density of approximately 70% confluence and cultured with Dulbecco’s modified Eagle’s medium (DMEM; GIBCO) containing 10% fetal bovine serum (FBS; GIBCO) and supplemented with growth factors (FGF) at 37°C in a 5% CO2 atmosphere.
Flow cytometry
MSCs were detached and incubated for 30 min at room temperature with the following specific antibodies: PE mouse anti-human CD90 (immunoglobulin G [IgG], k, clone: 5E10), FITC mouse anti-human CD73 (IgG1, k; clone: MAR4), PE rat anti-human CD14 (IgG2b, k; clone: G44-26), PE mouse anti-human CD105 (IgG1, k; clone: 266), APC rat anti-human CD45 (IgG2b, k; clone: 30-F11), PE mouse anti-human CD34 (IgG1, k; clone: 563) and PE mouse anti-human HLA-DR (IgG2a, k; clone: G46-6; all from BD Biosciences, San Jose, CA, http://www.bdbiosciences.com). Finally, the cells were assayed using a BD Influx Cell Sorter (BD Biosciences). For the cell cycle analysis, MSCs were trypsinized and fixed with 80% cold alcohol at 4°C for 12 h. After centrifugation, MSCs were resuspended in 50 μL of RNase and 450 μL of propidium iodide (PI; Sigma-Aldrich) staining solution, and the phases of the cell cycle in each sample after 30 min of incubation were analyzed by flow cytometry.
MSC propagation
Two methods were conducted to detect the proliferative capability of MSCs during in vitro aging. Cells collected at the end of each passage were counted using a trypan blue exclusion method. The NCPD13 was then calculated based on the following equation: NCPD = 3.33 × log(Nt/Ni). Nt and Ni are the cell numbers at a specific time point t (day 14) and at initial seeding (day 0), respectively. In addition, MSCs at P4, P6, P8, P10, and P12 were seeded in 96-well microplates and subjected to different treatments for the indicated times. A cell counting kit-8 (CCK-8) assay (Dojindo Molecular Technologies, Rockville, MD, http://www.dojindo.com) was used to assess the cell proliferation ability at days 0, 1, 2, 3, 4, and 5 according to the manufacturer’s protocol.
MSC adipogenic and osteogenic differentiation
MSCs were seeded at a concentration of 0.5 × 104 cells per well in 12-well plates, and osteogenic differentiation was induced by the addition of DMEM supplemented with 10% FBS, 100 IU/mL penicillin, 100 IU/mL streptomycin, 0.1 mM dexamethasone, 10 mM β-glycerol phosphate, and 50 mM ascorbic acid (Sigma-Aldrich, St. Louis, MO, https://www.sigmaaldrich.com). The medium was replaced every 3 days for 21 days. Differentiated cells were stained with ARS to detect the de novo formation of bone matrix. For adipogenic induction, MSCs were induced in adipogenic medium consisting of high-glucose DMEM supplemented with 10% FBS, 1 mM dexamethasone (Sigma-Aldrich), 10 mg/mL insulin (Sigma-Aldrich), 0.5 mM 3-isobutyl-1-methylxanthine (Sigma-Aldrich), and 0.2 mM indomethacin (Sigma-Aldrich). The cells were stained with ORO on day 21.
SA-β-gal staining and relative telomere length detection
The expression of pH-dependent SA-β-gal activity was analyzed in MSCs at different passages using the SA-β-gal staining kit (Cell Signaling Technology, #9860) according to previous report.23 We used quantitative PCR proposed by Cawthon in 2002 to measure the telomere length.65 Genomic DNAs of MSCs of P4, P6 and P12 were extracted. The ratio of telomere (T) and single copy gene (S), named T/S ration = [2Ct(Telomeres)/2Ct(S)]−1 = 2−ΔCt, was determined using the Telomere length detection kit (Ji Pu, #GP1501). Since the T/S ratio is proportional to the telomere length, the relative length of the telomere can be obtained by the T/S ratio.
CD4+ T cell and CD19+ B cell proliferation assay
PBMCs were collected and purified from the peripheral blood of 6 healthy donors (3 males, 3 females) using Ficoll-Hypaque gradient centrifugation (GE Healthcare Life Sciences, Marlborough, MA, http://www.gelifesciences.com). CD4+ T cells and CD19+ B cells were purified with CD4 and CD19 immunomagnetic bead antibodies, respectively, incubated with 5 mM carboxyfluorescein succinimidyl ester (CFSE, Thermo Fisher Scientific, Waltham, MA, USA, http://www.thermofisher.com) for 15 min, washed with PBS containing 10% FBS, and then added to the coculture system with 2 mL medium at a ratio of 1:5 or 1:1 (MSCs [0.5 × 105 cells]:CD4+ T [2.5 × 105 cells] or CD19+ B cells [0.5 × 105 cells]). The cells were cocultured in RPMI-1640 medium containing purified anti-CD3 (0.2 mg/mL, BD Biosciences) and anti-CD28 (1 mg/mL, BD Biosciences) antibodies to stimulate the T cells and LPS (2.5 μg/mL, Sigma-Aldrich, L2630) to stimulate the B cells to proliferate for 5 days. CD4+ T cell and CD19+ B cell proliferation was measured by fluorescence-activated cell sorting.
Library construction and high-throughput sequencing
A total of nine samples consisted of three MSCs at P4, P6, and P12, respectively, were separately treated with TRIzol (TAKARA), and then two libraries were constructed: lncRNA library and miRNA library. In the lncRNA-seq, after removing rRNA using Ribo-Zero Magnetic Kit (Epicenter), the retrieved RNA was fragmented by adding First Strand Master Mix (Invitrogen) using random primers reverse transcription followed by a second-strand cDNA synthesis. Then the cDNA fragments were subjected end-repair and then were 3′ adenylated following by adaptor ligation and enrichment with several rounds of PCR amplification. The library products were evaluated using the Agilent 2100 bioanalyzer (Thermo Fisher Scientific, USA) and then sequenced on Illumina Hiseq X-ten platform with paired-end 100 bp at BGI. Tech (BGI-Shenzhen, China). The sequencing data were filtered with SOAPnuke (v1.5.2) in FASTQ format. The total number of clean reads is about 120M for one sample. The clean reads were mapped to GCF_000001405.38_GRCh38.p12 using HISAT2. Particularly, we analyzed circRNAs, classical genes, and lncRNAs, separately. Database of circbase (http://circrna.org/) was used to identify circRNAs by BLAST. The expression level of gene was calculated by RSEM (v.1.2.12). The following all analyses including heatmap, GO, KEGG, and GSEA et al. of annotated DE gene were conducted with the online bioinformatic platform Dr. Tom (https://biosys.bgi.com/) provided by BGI.
In the miRNA sequencing, total RNA was purified by electrophoretic separation on a 15% urea denaturing polyacrylamide gel electrophoresis (PAGE) gel and small RNA regions corresponding to the 18–30 nt bands in the marker lane (14–33 single-stranded RNA [ssRNA] Ladder Marker, TAKARA) were excised and recovered. The purified RNAs were then ligated with 3′ RNA adaptor and followed by 5′ adaptor ligation. The adaptor-ligated small RNAs were subsequently transcribed into cDNA and several rounds of PCR amplification were performed. The PCR products were selected by PAGE with target fragments 100–120 bp and purified by QIAquick Gel Extraction Kit (QIAGEN, CA). After being evaluated using the Agilent 2100 bioanalyzer (Thermo Fisher Scientific, USA), the purified library products were then sequenced with a 50 bp single-end run on a BGISEQ 500 platform at BGI. Tech (BGI-Shenzhen, China). After sequencing, the raw reads were filtered by FASTQC to get clean reads. The total number of clean reads is about 20M for one sample. After filtering, the clean reads were mapped to the reference genome (GCF_000001405.38_GRCh38.p12) and other small RNA (sRNA) database including miRbase, small interfering RNA (siRNA), Piwi-interactiing RNA (piRNA), and small nucleolar RNA (snoRNA) with Bowtie2. The miRNA expression was calculated by RPM (reads per million) values. Differential expression analysis was performed using the DEGseq according to the criteria of |log2 (fold change)| ≥ 1 and Q-value < 0.001. TargetScan, RNAhybrid, miRanda were used to predict targets gene of selected miRNA on the online tool Dr. Tom software (BGI-Shenzhen, China).
Protein-protein interaction network analysis
Dr. Tom (http://biosys.bgi.com) was used to evaluate the protein-protein interaction (PPI) network of the DE coding genes. The PPI score was set as 0.9 (highest confidence). All the nodes in the PPI network were DE mRNAs. The network was visualized using Cytoscape version 3.6.1 (http://www.cytoscape.org/). The topological property of degree centrality was used to analyze the node score in the network.
Co-expression network analysis
The Pearson’s correlation coefficient of each DE lncRNA and mRNA was calculated, and the correlation test was performed on Dr. Tom (http://biosys.bgi.com) to identify lncRNA-mRNA pairs. The mRNA-lncRNA pairs with Pearson correlation > 0.900 and p value < 0.01 were collected for co-expression network construction.
Analysis of lncRNAs targeting mRNAs
To identify cis-regulatory interactions, we investigated lncRNAs that act on neighboring target genes. We searched for coding genes 20 k upstream and downstream of each lncRNA. The binding energy was used to identify trans-regulatory targets of lncRNAs. Binding energy ≤ −100 was as the criterion of trans-regulatory interactions.
ceRNA network analysis
Along with the identified DE mRNAs, lncRNAs, and miRNAs, we constructed a ceRNA network. First, we used starBase (http://starbase.sysu.edu.cn/)66 and Dr. Tom (http://biosys.bgi.com) to predict the DE miRNA-targeted lncRNAs with scores > 0.75, which were then intersected with the DE lncRNAs. Next, we used TargetScan (http://www.targetscan.org/) and Dr. Tom to identify miRNA-targeted mRNAs, which were then intersected with the DE mRNAs. Finally, the interaction network was constructed and visualized using Cytoscape software based on the screening of lncRNA-miRNA-mRNA pairs.
Quantitative real-time PCR
Total RNA was isolated using TRIzol reagent (Invitrogen, Scotland) and RNA Rapid Extraction Kit (Yishan, China) according to the manufacturer’s instructions. RNA was quantified with a NanoDrop ND-1000 Spectrophotometer (NanoDrop Technologies, USA). The Prime ScriptRT reagent kit (Takara, Japan) was used to obtain cDNA. For miRNA assays, the reverse primer specific to miRNAs was provided in the Mir-X miRNA First-Strand Synthesis Kit (TaKaRa, USA), and cDNA was synthesized according to the manufacturer’s instructions. For circRNAs, total RNA was digested by RNase R (GENESEED, China) or not according to the manufacturer’s instructions. The circRNA real-time PCR reagent kit (GENESEED, China) was used to obtain cDNA. The cycling parameters were as follows: 10 min at 25°C followed by 30 min at 42°C and 5 min at 85°C. Specific divergent primers spanning the back-splice junction sites of circRNAs were designed. qPCR was performed on an ABI PRISM 7700HT Sequence Detection System Instrument (Applied Biosystems, Germany) using SYBR Premix Ex Taq (TaKaRa). The cycling parameters were as follows: 30 s at 95°C followed by 40 cycles of 5 s at 95°C and 20 s at 60°C. Relative changes in RNA levels were assessed using the 2–ΔΔCt method and normalized to the levels of GAPDH or U6. The sequences of primers used in the qPCR assay are listed in Table S1.
Statistical analysis
Statistical data are presented as the means ± standard error of the mean. Student’s t test was used for comparisons between two groups, or one-way analysis of variance in conjunction with a Bonferroni post-test was used for multiple comparisons using GraphPad Prism 5 software. Spearman correlation was used to determine the relationship between lncRNAs and their target genes. p values < 0.05 were considered significant.
Data availability statement
The sequencing data analyzed in this study have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO: GSE178514 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE178514).
Acknowledgments
The authors thank American Journal Experts for providing English language editing of the manuscript. This study was supported by the National Natural Science Foundation of China (31700884, 81672097), the Key-Area Research and Development Program of Guangdong Province (2019B020236001), and the Shenzhen Key Laboratory of Ankylosing Spondylitis (ZDSYS20190902092851024).
Author contributions
S.W., Z.W., and H. Su designed the study, performed the experiments, and analyzed the data; Y.W. and H. Shen revised the manuscript; M.M., X.W., and F.C. collected the data; G.Y., W.Y., S.C., R.M., W.D., P.F., and C.Z. contributed the reagents/materials/analysis tools; and S.W., Z.W., and H. Shen wrote the manuscript. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.08.013.
Contributor Information
Huiyong Shen, Email: shenhuiy@mail.sysu.edu.cn.
Yanfeng Wu, Email: wuyf@mail.sysu.edu.cn.
Supplemental information
References
- 1.Song N., Scholtemeijer M., Shah K. Mesenchymal Stem Cell Immunomodulation: Mechanisms and Therapeutic Potential. Trends Pharmacol. Sci. 2020;41:653–664. doi: 10.1016/j.tips.2020.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Musina R.A., Bekchanova E.S., Sukhikh G.T. Comparison of mesenchymal stem cells obtained from different human tissues. Bull. Exp. Biol. Med. 2005;139:504–509. doi: 10.1007/s10517-005-0331-1. [DOI] [PubMed] [Google Scholar]
- 3.Paniushin O.V., Domaratskaia E.I., Starostin V.I. [Mesenchymal stem cells: sources, phenotype, and differentiation potential] Izv. Akad. Nauk Ser. Biol. 2006;1:6–25. [PubMed] [Google Scholar]
- 4.Ceccarelli S., Pontecorvi P., Anastasiadou E., Napoli C., Marchese C. Immunomodulatory Effect of Adipose-Derived Stem Cells: The Cutting Edge of Clinical Application. Front. Cell Dev. Biol. 2020;8:236. doi: 10.3389/fcell.2020.00236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Y., Zhang Z., Chi Y., Zhang Q., Xu F., Yang Z., Meng L., Yang S., Yan S., Mao A. Long-term cultured mesenchymal stem cells frequently develop genomic mutations but do not undergo malignant transformation. Cell Death Dis. 2013;4:e950. doi: 10.1038/cddis.2013.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saeedi P., Halabian R., Imani Fooladi A.A. A revealing review of mesenchymal stem cells therapy, clinical perspectives and Modification strategies. Stem Cell Investig. 2019;6:34. doi: 10.21037/sci.2019.08.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang P., Li Y., Huang L., Yang J., Yang R., Deng W., Liang B., Dai L., Meng Q., Gao L. Effects and safety of allogenic mesenchymal stem cell intravenous infusion in active ankylosing spondylitis patients who failed NSAIDs: a 20-week clinical trial. Cell Transplant. 2014;23:1293–1303. doi: 10.3727/096368913X667727. [DOI] [PubMed] [Google Scholar]
- 8.Galipeau J., Sensébé L. Mesenchymal Stromal Cells: Clinical Challenges and Therapeutic Opportunities. Cell Stem Cell. 2018;22:824–833. doi: 10.1016/j.stem.2018.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi Y., Wang Y., Li Q., Liu K., Hou J., Shao C., Wang Y. Immunoregulatory mechanisms of mesenchymal stem and stromal cells in inflammatory diseases. Nat. Rev. Nephrol. 2018;14:493–507. doi: 10.1038/s41581-018-0023-5. [DOI] [PubMed] [Google Scholar]
- 10.Wagner W., Horn P., Castoldi M., Diehlmann A., Bork S., Saffrich R., Benes V., Blake J., Pfister S., Eckstein V., Ho A.D. Replicative senescence of mesenchymal stem cells: a continuous and organized process. PLoS ONE. 2008;3:e2213. doi: 10.1371/journal.pone.0002213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baxter M.A., Wynn R.F., Jowitt S.N., Wraith J.E., Fairbairn L.J., Bellantuono I. Study of telomere length reveals rapid aging of human marrow stromal cells following in vitro expansion. Stem Cells. 2004;22:675–682. doi: 10.1634/stemcells.22-5-675. [DOI] [PubMed] [Google Scholar]
- 12.Fu X., Xu B., Jiang J., Du X., Yu X., Yan Y., Li S., Inglis B.M., Ma H., Wang H. Effects of cryopreservation and long-term culture on biological characteristics and proteomic profiles of human umbilical cord-derived mesenchymal stem cells. Clin. Proteomics. 2020;17:15. doi: 10.1186/s12014-020-09279-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang Y.K., Ogando C.R., Wang See C., Chang T.Y., Barabino G.A. Changes in phenotype and differentiation potential of human mesenchymal stem cells aging in vitro. Stem Cell Res. Ther. 2018;9:131. doi: 10.1186/s13287-018-0876-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Witte S.F.H., Lambert E.E., Merino A., Strini T., Douben H.J.C.W., O’Flynn L., Elliman S.J., de Klein A.J.E.M.M., Newsome P.N., Baan C.C., Hoogduijn M.J. Aging of bone marrow- and umbilical cord-derived mesenchymal stromal cells during expansion. Cytotherapy. 2017;19:798–807. doi: 10.1016/j.jcyt.2017.03.071. [DOI] [PubMed] [Google Scholar]
- 15.Ruparel N.B., de Almeida J.F.A., Henry M.A., Diogenes A. Characterization of a stem cell of apical papilla cell line: effect of passage on cellular phenotype. J. Endod. 2013;39:357–363. doi: 10.1016/j.joen.2012.10.027. [DOI] [PubMed] [Google Scholar]
- 16.Wagner W., Bork S., Horn P., Krunic D., Walenda T., Diehlmann A., Benes V., Blake J., Huber F.X., Eckstein V. Aging and replicative senescence have related effects on human stem and progenitor cells. PLoS ONE. 2009;4:e5846. doi: 10.1371/journal.pone.0005846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anastasiadou E., Faggioni A., Trivedi P., Slack F.J. The Nefarious Nexus of Noncoding RNAs in Cancer. Int. J. Mol. Sci. 2018;19:2072. doi: 10.3390/ijms19072072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anastasiadou E., Jacob L.S., Slack F.J. Non-coding RNA networks in cancer. Nat. Rev. Cancer. 2018;18:5–18. doi: 10.1038/nrc.2017.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhuang Z., Jia L., Li W., Zheng Y. The emerging roles of circular RNAs in regulating the fate of stem cells. Mol. Cell. Biochem. 2021;476:231–246. doi: 10.1007/s11010-020-03900-w. [DOI] [PubMed] [Google Scholar]
- 20.Xie Z.Y., Wang P., Wu Y.F., Shen H.Y. Long non-coding RNA: The functional regulator of mesenchymal stem cells. World J. Stem Cells. 2019;11:167–179. doi: 10.4252/wjsc.v11.i3.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Puvvula P.K. LncRNAs Regulatory Networks in Cellular Senescence. Int. J. Mol. Sci. 2019;20:2615. doi: 10.3390/ijms20112615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xia W., Zhuang L., Deng X., Hou M. Long noncoding RNA-p21 modulates cellular senescence via the Wnt/β-catenin signaling pathway in mesenchymal stem cells. Mol. Med. Rep. 2017;16:7039–7047. doi: 10.3892/mmr.2017.7430. [DOI] [PubMed] [Google Scholar]
- 23.Bakopoulou A., Apatzidou D., Aggelidou E., Gousopoulou E., Leyhausen G., Volk J., Kritis A., Koidis P., Geurtsen W. Isolation and prolonged expansion of oral mesenchymal stem cells under clinical-grade, GMP-compliant conditions differentially affects “stemness” properties. Stem Cell Res. Ther. 2017;8:247. doi: 10.1186/s13287-017-0705-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang D., Kilian K.A. The effect of mesenchymal stem cell shape on the maintenance of multipotency. Biomaterials. 2013;34:3962–3969. doi: 10.1016/j.biomaterials.2013.02.029. [DOI] [PubMed] [Google Scholar]
- 25.Yu Y., Park Y.S., Kim H.S., Kim H.Y., Jin Y.M., Jung S.C., Ryu K.H., Jo I. Characterization of long-term in vitro culture-related alterations of human tonsil-derived mesenchymal stem cells: role for CCN1 in replicative senescence-associated increase in osteogenic differentiation. J. Anat. 2014;225:510–518. doi: 10.1111/joa.12229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Witte S.F.H., Peters F.S., Merino A., Korevaar S.S., Van Meurs J.B.J., O’Flynn L., Elliman S.J., Newsome P.N., Boer K., Baan C.C., Hoogduijn M.J. Epigenetic changes in umbilical cord mesenchymal stromal cells upon stimulation and culture expansion. Cytotherapy. 2018;20:919–929. doi: 10.1016/j.jcyt.2018.05.005. [DOI] [PubMed] [Google Scholar]
- 27.Wiese D.M., Ruttan C.C., Wood C.A., Ford B.N., Braid L.R. Accumulating Transcriptome Drift Precedes Cell Aging in Human Umbilical Cord-Derived Mesenchymal Stromal Cells Serially Cultured to Replicative Senescence. Stem Cells Transl. Med. 2019;8:945–958. doi: 10.1002/sctm.18-0246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peffers M.J., Collins J., Fang Y., Goljanek-Whysall K., Rushton M., Loughlin J., Proctor C., Clegg P.D. Age-related changes in mesenchymal stem cells identified using a multi-omics approach. Eur. Cell. Mater. 2016;31:136–159. doi: 10.22203/ecm.v031a10. [DOI] [PubMed] [Google Scholar]
- 29.Bellayr I.H., Catalano J.G., Lababidi S., Yang A.X., Lo Surdo J.L., Bauer S.R., Puri R.K. Gene markers of cellular aging in human multipotent stromal cells in culture. Stem Cell Res. Ther. 2014;5:59. doi: 10.1186/scrt448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cai J., Miao X., Li Y., Smith C., Tsang K., Cheng L., Wang Q.F. Whole-genome sequencing identifies genetic variances in culture-expanded human mesenchymal stem cells. Stem Cell Reports. 2014;3:227–233. doi: 10.1016/j.stemcr.2014.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Neri S., Borzì R.M. Molecular Mechanisms Contributing to Mesenchymal Stromal Cell Aging. Biomolecules. 2020;10:340. doi: 10.3390/biom10020340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lavini-Ramos C., Silva H.M., Soares-Schanoski A., Monteiro S.M., Ferreira L.R.P., Pacanaro A.P., Gomes S., Batista J., Faé K., Kalil J., Coelho V. MMP9 integrates multiple immunoregulatory pathways that discriminate high suppressive activity of human mesenchymal stem cells. Sci. Rep. 2017;7:874. doi: 10.1038/s41598-017-00923-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krupa A., Fol M., Dziadek B.R., Kepka E., Wojciechowska D., Brzostek A., Torzewska A., Dziadek J., Baughman R.P., Griffith D., Kurdowska A.K. Binding of CXCL8/IL-8 to Mycobacterium tuberculosis Modulates the Innate Immune Response. Mediators Inflamm. 2015;2015:124762. doi: 10.1155/2015/124762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cen S., Wang P., Xie Z., Yang R., Li J., Liu Z., Wang S., Wu X., Liu W., Li M. Autophagy enhances mesenchymal stem cell-mediated CD4+ T cell migration and differentiation through CXCL8 and TGF-β1. Stem Cell Res. Ther. 2019;10:265. doi: 10.1186/s13287-019-1380-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kortlever R.M., Higgins P.J., Bernards R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat. Cell Biol. 2006;8:877–884. doi: 10.1038/ncb1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jin H., Choung H.W., Lim K.T., Jin B., Jin C., Chung J.H., Choung P.H. Recombinant Human Plasminogen Activator Inhibitor-1 Promotes Cementogenic Differentiation of Human Periodontal Ligament Stem Cells. Tissue Eng. Part A. 2015;21:2817–2828. doi: 10.1089/ten.tea.2014.0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang F., Wu R., Jiang Z., Chen J., Nan J., Su S., Zhang N., Wang C., Zhao J., Ni C. Leptin increases mitochondrial OPA1 via GSK3-mediated OMA1 ubiquitination to enhance therapeutic effects of mesenchymal stem cell transplantation. Cell Death Dis. 2018;9:556. doi: 10.1038/s41419-018-0579-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coppé J.P., Desprez P.Y., Krtolica A., Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Della Bella E., Menzel U., Basoli V., Tourbier C., Alini M., Stoddart M.J. Differential Regulation of circRNA, miRNA, and piRNA during Early Osteogenic and Chondrogenic Differentiation of Human Mesenchymal Stromal Cells. Cells. 2020;9:398. doi: 10.3390/cells9020398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Batista P.J., Chang H.Y. Long noncoding RNAs: cellular address codes in development and disease. Cell. 2013;152:1298–1307. doi: 10.1016/j.cell.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.He Y., Luo J., Chen Y., Zhou X., Yu S., Jin L., Xiao X., Jia S., Liu Q. ARHGAP18 is a novel gene under positive natural selection that influences HbF levels in β-thalassaemia. Mol. Genet. Genomics. 2018;293:207–216. doi: 10.1007/s00438-017-1377-2. [DOI] [PubMed] [Google Scholar]
- 42.Peck J., Douglas G., 4th, Wu C.H., Burbelo P.D. Human RhoGAP domain-containing proteins: structure, function and evolutionary relationships. FEBS Lett. 2002;528:27–34. doi: 10.1016/s0014-5793(02)03331-8. [DOI] [PubMed] [Google Scholar]
- 43.Thomson D.W., Dinger M.E. Endogenous microRNA sponges: evidence and controversy. Nat. Rev. Genet. 2016;17:272–283. doi: 10.1038/nrg.2016.20. [DOI] [PubMed] [Google Scholar]
- 44.Silverman E.K., Schmidt H.H.H.W., Anastasiadou E., Altucci L., Angelini M., Badimon L., Balligand J.L., Benincasa G., Capasso G., Conte F. Molecular networks in Network Medicine: Development and applications. Wiley Interdiscip. Rev. Syst. Biol. Med. 2020;12:e1489. doi: 10.1002/wsbm.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li M., Xie Z., Cai Z., Su F., Zheng G., Li J., Wang S., Cen S., Liu W., Tang S. lncRNA-mRNA expression profiles and functional networks of mesenchymal stromal cells involved in monocyte regulation. Stem Cell Res. Ther. 2019;10:207. doi: 10.1186/s13287-019-1306-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou J., Nie H., Liu P., Wang Z., Yao B., Yang L. Down-regulation of miR-339 promotes differentiation of BMSCs and alleviates osteoporosis by targeting DLX5. Eur. Rev. Med. Pharmacol. Sci. 2019;23:29–36. doi: 10.26355/eurrev_201901_16744. [DOI] [PubMed] [Google Scholar]
- 47.Chen Y.S., Kang X.R., Zhou Z.H., Yang J., Xin Q., Ying C.T., Zhang Y.P., Tao J. MiR-1908/EXO1 and MiR-203a/FOS, regulated by scd1, are associated with fracture risk and bone health in postmenopausal diabetic women. Aging (Albany NY) 2020;12:9549–9584. doi: 10.18632/aging.103227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang H., Cao Y., Zhang J., Liang Y., Su X., Zhang C., Liu H., Han X., Ge L., Fan Z. DLX5 and HOXC8 enhance the chondrogenic differentiation potential of stem cells from apical papilla via LINC01013. Stem Cell Res. Ther. 2020;11:271. doi: 10.1186/s13287-020-01791-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu Y.Y., Tanikawa C., Ueda K., Matsuda K. INKA2, a novel p53 target that interacts with the serine/threonine kinase PAK4. Int. J. Oncol. 2019;54:1907–1920. doi: 10.3892/ijo.2019.4786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Akiyama H., Iwasaki Y., Yamada S., Kamiguchi H., Sakakibara S.I. Control of cell migration by the novel protein phosphatase-2A interacting protein inka2. Cell Tissue Res. 2020;380:527–537. doi: 10.1007/s00441-020-03169-x. [DOI] [PubMed] [Google Scholar]
- 51.Lei B., Yu L., Jung T.A., Deng Y., Xiang W., Liu Y., Qi S. Prospective Series of Nine Long Noncoding RNAs Associated with Survival of Patients with Glioblastoma. J. Neurol. Surg. A Cent. Eur. Neurosurg. 2018;79:471–478. doi: 10.1055/s-0038-1655549. [DOI] [PubMed] [Google Scholar]
- 52.Li H., Chi X., Li R., Ouyang J., Chen Y. A Novel lncRNA, AK130181, Contributes to HIV-1 Latency by Regulating Viral Promoter-Driven Gene Expression in Primary CD4+ T Cells. Mol. Ther. Nucleic Acids. 2020;20:754–763. doi: 10.1016/j.omtn.2020.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang J., Xie N., Huang H., Yao J., Hu W. Long noncoding RNA STXBP5-AS1 inhibits cell proliferation, migration, and invasion via preventing the PI3K/AKT against STXBP5 expression in non-small-cell lung carcinoma. J. Cell. Biochem. 2018;2018:18. doi: 10.1002/jcb.28023. [DOI] [PubMed] [Google Scholar]
- 54.Meng Q., Wang X., Xue T., Zhao Q., Wang W., Zhao K. Long noncoding RNA MIR99AHG promotes gastric cancer progression by inducing EMT and inhibiting apoptosis via miR577/FOXP1 axis. Cancer Cell Int. 2020;20:414. doi: 10.1186/s12935-020-01510-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shen Q., Sun Y., Xu S. LINC01503/miR-342-3p facilitates malignancy in non-small-cell lung cancer cells via regulating LASP1. Respir. Res. 2020;21:235. doi: 10.1186/s12931-020-01464-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bavelloni A., Ramazzotti G., Poli A., Piazzi M., Focaccia E., Blalock W., Faenza I. MiRNA-210: A Current Overview. Anticancer Res. 2017;37:6511–6521. doi: 10.21873/anticanres.12107. [DOI] [PubMed] [Google Scholar]
- 57.Ren S., Lin P., Wang J., Yu H., Lv T., Sun L., Du G. Circular RNAs: Promising Molecular Biomarkers of Human Aging-Related Diseases via Functioning as an miRNA Sponge. Mol. Ther. Methods Clin. Dev. 2020;18:215–229. doi: 10.1016/j.omtm.2020.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hansen T.B., Jensen T.I., Clausen B.H., Bramsen J.B., Finsen B., Damgaard C.K., Kjems J. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495:384–388. doi: 10.1038/nature11993. [DOI] [PubMed] [Google Scholar]
- 59.Memczak S., Jens M., Elefsinioti A., Torti F., Krueger J., Rybak A., Maier L., Mackowiak S.D., Gregersen L.H., Munschauer M. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–338. doi: 10.1038/nature11928. [DOI] [PubMed] [Google Scholar]
- 60.Shen S., Wu Y., Chen J., Xie Z., Huang K., Wang G., Yang Y., Ni W., Chen Z., Shi P. CircSERPINE2 protects against osteoarthritis by targeting miR-1271 and ETS-related gene. Ann. Rheum. Dis. 2019;78:826–836. doi: 10.1136/annrheumdis-2018-214786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang Q., Qiao X., Xia W. CircSERPINE2 weakens IL-1β-caused apoptosis and extracellular matrix degradation of chondrocytes by regulating miR-495/TGFBR2 axis. Biosci. Rep. 2020;40 doi: 10.1042/BSR20201601. BSR20201601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ma Y., Shan Z., Liu Y., Shao H., Xin Y., He K., Jiang S., Wang Y. CircTHBS1 targeting miR-211/CCND2 pathway to promote cell proliferation and migration potential in primary cystitis glandularis cells. Biosci Rep. 2020;41 doi: 10.1042/BSR20201164. BSR20201164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen H., Wu G., Chen G., Wang W., Ruan F. Circular RNA THBS1 promotes proliferation and apoptosis of non-small cell lung cancer cells by sponging miR-129-5p and regulating SOX4 expression. J. BUON. 2020;25:1721–1727. [PubMed] [Google Scholar]
- 64.Zang Y., Li J., Wan B., Tai Y. circRNA circ-CCND1 promotes the proliferation of laryngeal squamous cell carcinoma through elevating CCND1 expression via interacting with HuR and miR-646. J. Cell. Mol. Med. 2020;24:2423–2433. doi: 10.1111/jcmm.14925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cawthon R.M. Telomere measurement by quantitative PCR. Nucleic Acids Res. 2002;30:e47. doi: 10.1093/nar/30.10.e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li J.H., Liu S., Zhou H., Qu L.H., Yang J.H. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014;42:D92–D97. doi: 10.1093/nar/gkt1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing data analyzed in this study have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO: GSE178514 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE178514).