

Abstract
Base editors are RNA-guided deaminases that enable site-specific nucleotide transitions. The targeting scope of these Cas-deaminase fusion proteins critically depends on the availability of a protospacer adjacent motif (PAM) at the target locus and is limited to a window within the CRISPR-Cas R-loop, where single-stranded DNA (ssDNA) is accessible to the deaminase. Here, we reason that the Cas9-HNH nuclease domain sterically constrains ssDNA accessibility and demonstrate that omission of this domain expands the editing window. By exchanging the HNH nuclease domain with a monomeric or heterodimeric adenosine deaminase, we furthermore engineer adenine base editor variants (HNHx-ABEs) with PAM-proximally shifted editing windows. This work expands the targeting scope of base editors and provides base editor variants that are substantially smaller. It moreover informs of potential future directions in Cas9 protein engineering, where the HNH domain could be replaced by other enzymes that act on ssDNA.
Keywords: CRISPR-Cas9, gene therapy, genome editing, base editing, HNHx-ABE
Graphical abstract
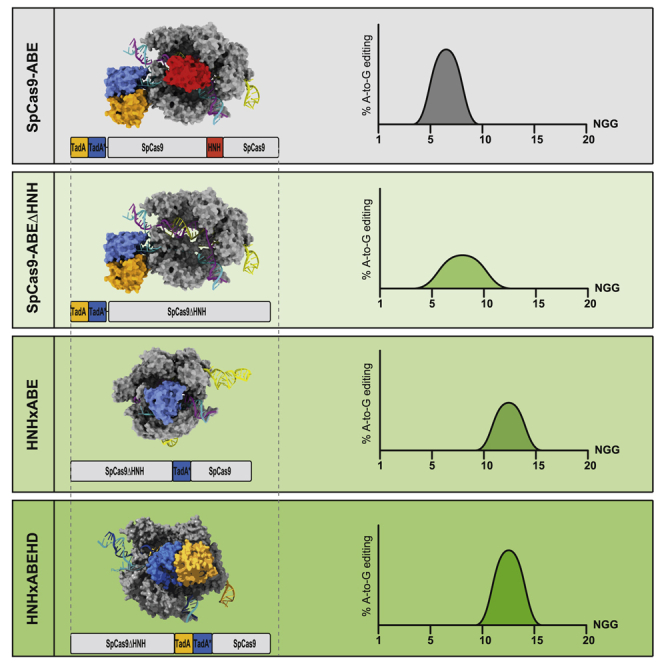
We reason that the Cas9-HNH nuclease domain sterically constrains ssDNA accessibility at PAM-proximal positions. Replacing the HNH domain of SpCas9 with adenosine deaminases results in size reduced base editors with PAM proximally shifted editing windows (HNHxABE). These base editors enable nucleotide transitions of previously difficult-to-target SNPs.
Introduction
CRISPR-Cas systems provide adaptive immunity to bacteria and archaea to defend them against foreign genetic elements. A small number of these systems have successfully been adopted for genome editing in mammalian cells, transforming biomedical research and therapeutics.1, 2, 3 A paradigmatic example is the Streptococcus pyogenes Cas9 (SpCas9) nuclease. Recruitment of SpCas9 to a desired genomic locus via a single guide RNA (sgRNA) allows facile and efficient genome editing by generating site-specific double-stranded DNA (dsDNA) breaks. Precise genome editing via such dsDNA breaks, however, relies on template DNA for homology-directed repair (HDR) that is inefficient in most mammalian cell types and leads to repair via alternative, error-prone end-joining pathways that generate frameshift mutations and gene knockouts.
Recent modifications to the highly modular SpCas9 protein resulted in sophisticated genome-editing technologies, such as base editors (BEs), which enable genome editing without dsDNA break formation. BEs allow precise conversion of A-to-G and C-to-T nucleobases or vice versa via nucleotide deamination independent of dsDNA breaks and HDR4, 5, 6 and thus have great potential for research and therapeutic applications.7, 8, 9 They are monodimers or heterodimers of ssDNA-specific deaminases fused N-terminally to catalytically impaired CRISPR class II effectors, such as Cas9 or Cas12a.4,5,10 Adenine BEs (ABEs) are based on laboratory-evolved E. coli tRNA adenine deaminase (TadA), and cytidine BEs (CBEs) have been generated from various deaminases, including APOBEC1, APOBEC3, AID, and CDA.11 Upon binding to a protospacer element, Cas9 complexed with the sgRNA forms an R-loop, making the non-target DNA strand (the target DNA strand is bound to the sgRNA) accessible to ssDNA-specific deaminases.
Because Cas9 and Cas12 proteins require protospacer adjacent motif (PAM) sites for DNA binding, the targeting scope of BEs is restricted to nucleobases that lie within a defined proximity to these motifs. To address this limitation, deaminases have been fused to Cas variants with less restrictive PAM requirements.12, 13, 14, 15, 16 However, because no PAMless Cas9 and Cas12 variants are yet available, several pathogenic T>C and G>A single-nucleotide polymorphisms (SNPs) remain untargetable. Pursuing a different approach, recent work led to the expansion of the BE targeting scope by generating variants with slightly extended editing windows: Huang et al.17 fused deaminases to circularly permuted Cas9 proteins, and Wang et al.18 incorporated the cytidine deaminase rAPOBEC1 into the PAM-interacting (PI) domain of SpCas9. Broadening the editing window, however, can induce additional bystander mutations, and this approach is therefore applicable only at certain loci.
Here, we report novel ABE variants, where the HNH nuclease domain of SpCas9 is replaced by monomeric or heterodimeric (HD) TadA. These variants are significantly reduced in size and have a PAM-proximally shifted editing window, enabling correction of several pathogenic SNPs that were not well targetable by classical ABEs.
Results
The HNH domain of SpCas9 restrains ssDNA accessibility at PAM-proximal positions
In our first attempt to shift the editing window of BEs, we applied the strategy of Wang et al.18 to ABEs and incorporated the codon-optimized TadA7.10 deaminase19 into the PI domain of SpCas9. Similar to CBEs,18 also for ABEs this approach did not lead to a shift but rather to a PAM-proximal extension of the editing window (ABEmax7.10 PI; Figures 1A and 1B). However, no editing was observed at nucleotides beyond position 12 (counting from PAM-distal end of the protospacer), suggesting that ssDNA accessibility in this region is blocked. Structural data suggest that the HNH nuclease domain (amino acids 775–908) in SpCas9 is likely responsible for preventing the deaminase from accessing the ssDNA substrate at positions 13–15 (Figure 1C). Importantly, the HNH domain is not required for Cas9 to bind its target DNA and form an R-loop, and nickase activity is not essential for base editing (Figure 1E).20 We therefore reasoned that omission of the HNH domain might improve accessibility and editing at PAM-proximal positions. To test this hypothesis, we engineered ABEmax7.10 ΔHNH, where a heterodimer of wild-type (WT) and evolved TadA7.10 is linked to the N terminus of an SpCas9 variant that lacks the HNH domain (Figure 1D). Although the highest editing rates remained at positions that are also efficiently targeted with ABEmax7.10, ABEmax7.10 ΔHNH allowed editing at positions 12 and 14 (Figure 1E). Our data therefore suggest that the HNH domain of SpCas9 indeed constrains access of TadA to PAM-proximal ssDNA nucleobases.
Figure 1.

Approaches to increase substrate accessibility for base editing at PAM-proximal bases
(A) Schematic domain organization of ABEmax7.10 and ABEmax7.10 PI1-3. ABEmax7.10 PI1-3 comprise an engineered SpCas9 (D10A) construct, where the TadA deaminase is integrated within the PI domain. ABEmax7.10 PI1, PI2, and PI3 use different linker lengths flanking the TadA deaminase. (B) Editing efficiencies of ABEmax7.10 PI constructs after transfection into HEK293T cells are quantified by high-throughput sequencing at three different endogenous loci. Values represent mean of three independent biological replicates performed on separate days ± SD. (C) Structural data (PDB: 6VPC) of adenine base editors with (left) and without (right) an HNH domain (indicated in red). TadA deaminases are indicated in orange and blue and target the ssDNA substrate (turquoise). (D) Schematic domain organization of ABEmax7.10 ΔHNH, where the HNH domain is replaced by a glycine-serine linker (Gly-Gly-Ser-Gly-Gly-Ser). (E) High-throughput sequencing data compare editing efficiencies of ABEmax7.10, dABEmax7.10, and ABEmax7.10 ΔHNH in endogenous loci in HEK293T cells. ABEmax7.10 retains nickase activity, nicking the target strand. Elimination of the nickase activity in ABEmax7.10 by introducing a H840A mutation in the HNH domain leads to dABEmax7.10. Complete removal of the HNH domain (that also abolishes nickase activity) in ABEmax7.10 results in ABEmax7.10 ΔHNH. Values represent mean of three independent biological replicates performed on separate days ± SD.
Replacing the HNH domain with TadA7.10 shifts the ABE editing window PAM proximally
Simply expanding the editing window of BEs has the disadvantage that it can generate additional undesired non-target nucleotide (bystander) edits within the ssDNA of the R-loop. This led us to investigate whether directly replacing the HNH domain with the adenine deaminase (HNHx-ABE) leads to a shift rather than a broadening of the editing window (Figures 2A and 2B). Before incorporating a deaminase domain in place of the HNH domain in SpCas9, we first exchanged the HNH domain for a superfolder GFP (sfGFP). Transfection of these constructs into HEK293T cells demonstrated localization of GFP to the nucleus (Figure 2C), indicating that the HNH domain can be exchanged with another protein domain without substantial misfolding and degradation of Cas9. In a next step, we engineered HNHx-ABEmax7.10 by incorporating the deaminase domain from ABEmax7.10 with 20 different peptide linker combinations into SpCas9 lacking the HNH domain. Using high-throughput sequencing (HTS), editing efficiencies of constructs with different linker combinations were compared 2 days after transfection into HEK293T cells (Figure 2D). In the most promising construct, a GGS-linker was used to join SpCas9 S793 to the TadA7.10 N terminus, and a SGG-linker was used to join the TadA7.10 C terminus to SpCas9 R919. This construct (HNHx-ABEmax7.10) exhibited a distinct shift in the editing window PAM-proximally, with up to 13.7% editing at positions >12 at 5 days after transfection (Figure S1A) and 32.5% editing at 7 days after transfection (Figure 3A). A similar shift in the editing window was obtained when HNHx-ABEmax7.10 was transfected into the K562 human erythroleukemic cell line (Figure 3A). Notably, when we exchanged the HNH domain with the cytosine deaminases FERNY, a laboratory-evolved APOBEC variant,21 we observed a similar shift in the editing window, albeit with very low editing rates (Figure S2A). Although these could be increased by exchanging FERNY with the cytidine deaminase PmCDA1, HNHx-PmCDA1 constructs induced broad deamination across the entire protospacer region, leading to significant bystander edits (Figure S2B). Likewise, when we adapted the HNHx-ABE architecture to the smaller Cas9 ortholog from Staphylococcus aureus (SaCas9-KKH22) using various linker combinations, we also did not obtain substantial deamination in the protospacer (Figure S3).
Figure 2.

HNH domain substitution with sfGFP and ssDNA-specific deaminase domains
(A) Schematic domain organization of Cas9 variants, where the HNH domain is replaced by a sfGFP or a ssDNA-specific deaminase domain (TadA∗). (B) Structural data of hypothetical Cas9 (PDB: 5F9R) constructs, where the HNH domain is omitted or replaced by sfGFP (PDB: 2B3P) or a TadA deaminase (PDB: 6VPC). Nucleotides (14–17) indicated in red are outside of a typical editing window of ABEmax7.10. (C) Fluorescence microscopy of HEK293T expressing Cas9, where the HNH domain is replaced with sfGFP, with (left panel) and without (right panel) nuclear localization signals. Blue: Hoechst; green: GFP. Scale bar: 20 μm. (D) Heatmap depicting different flanking amino acids of Cas9 and linkers to incorporate the TadA deaminase in place of the HNH domain. The TadA deaminase reading frame is as listed in the supplemental information. Editing efficiencies were quantified by high-throughput sequencing indicated as % A-to-G conversion 2 days after transfection. The x axis defines base conversions at adenine base positions within the protospacer.
Figure 3.

Targeting of endogenous loci
(A and B) Editing efficiencies of different adenine bases within the protospacer region comparing ABEmax7.10 and HNHx-ABEmax7.10 (A) or ABE8e and HNHx-ABE8e (B) in endogenous loci in HEK293T (left) and K562 (right) cells. Numbering starts with PAM-distal nucleotides. Values represent mean of three independent biological replicates performed on separate days ± SD. (C) Editing of a disease-causing c.3188G>A mutation in the FANCA gene comparing different ABEmax7.10, ABE8e, and HNHx-ABE8e constructs. Green indicates the target base, orange a synonymous mutation, and red a non-synonymous mutation. Values represent mean of three independent biological replicates performed on separate days ± SD.
Replacing HNH with TadA8e increases the efficiency and broadens the editing window of HNHx-ABE
Phage-assisted non-continuous and continuous evolution has enabled the development of TadA variants with increased deaminase activity.23 To assess whether the use of these variants would improve editing rates of HNHx-ABEs, we next exchanged the TadA7.10 deaminase domain with hyperactive TadA8e, resulting in HNHx-ABE8e. As expected, we observed higher editing rates with HNHx-ABE8e compared with HNHx-ABEmax7.10, up to 19.8% at 5 days after transfection (Figure S1B) and up to 40.4% at 7 days after transfection (Figure 3B). Higher editing efficiencies, however, came at the cost of lower specificity, because the editing window was significantly broadened (Figures 3B and S1B). Although this is a potential limitation due to the increase in bystander editing, targeting the pathogenic c.3188G>A mutation in FANCA provides an example for a disease-causing locus where HNHx-ABE8e has a benefit over classical ABEs with N-terminally fused TadA. Here, the only available canonical NGG PAM site positions the disease-causing mutation outside of the editing window of classical ABEs, and HNHx-ABE8e led to considerably higher on-target editing compared with ABE8e or ABEmax7.10 (Figure 3C). Notably, similar on-target editing rates were obtained when using ABE8e-SpRY,14 where an evolved Cas9 binds a downstream NCC PAM site and positions the disease-causing mutation within reach of classical ABEs (Figure 3C), but compared with HNHx-ABE8e this variant resulted in more non-synonymous bystander mutations.
High levels of ABE expression can lead to substantial sgRNA-independent off-target deamination on the transcriptome.24, 25, 26, 27, 28 Considering that in the HNHx-ABE architecture TadA is not terminally fused but instead integrated into Cas9, and thus potentially constrained by the HNHx-ABE architecture, we reasoned that off-target editing on the transcriptome might be reduced. However, RNA sequencing (RNA-seq) did not show significant differences in adenine deamination between HNHx-ABE8e- and ABE8e-treated cells and between HNHx-ABEmax7.10- and ABEmax7.10-treated cells (Figure S4). Thus, also with HNHx-ABE variants, excessive overexpression should be avoided in applications where off-target deamination is of concern.
Replacing the HNH domain with HD TadA increases HNHx-ABE efficiency
Recent studies demonstrate that in ABE variants with monomeric TadA, dimerization occurs with a second TadA domain contributed in trans from another ABE molecule, suggesting that TadA dimerization is essential for ABE activity.29 Because in trans TadA dimerization might be impeded by steric hinderance in HNHx-ABE constructs (Figure S5), we hypothesized that replacement of the HNH domain by HD TadA could improve editing rates. We therefore incorporated WT TadA linked to TadA7.10 or TadA8e into Cas9 lacking the HNH domain, resulting in HNHx-ABEmax7.10HD and HNHx-ABE8eHD (Figure 4A). Importantly, at the three analyzed loci, both variants still led to a PAM-proximal shift in adenine deamination (Figures 4B and 4C), but with an elevation in editing rates compared with monomeric TadA HNHx-ABE variants (Figures 3A, 3B, 4B, and 4C). To further refine editing characteristics of the different HNHx-ABE constructs generated in our study, we next transfected a pool of HEK293T cells carrying a stably integrated self-targeting library, where each cell contains one of the 100 target loci and also expresses the corresponding sgRNA (Figure S6). HTS of the target loci 10 days post-transfection confirmed the PAM-proximal shift of the editing window in all HNHx-ABE constructs, with substantially higher editing when the HNH domain was replaced with TadA heterodimers compared with monomers (mean editing rates with HNHx-ABE8eHD and HNHx-ABEmax7.10HD were 33.1% and 20.5% compared with 14.8% and 4.9% with HNHx-ABE8e and HNHx-ABEmax7.10; Figure 4D; Figures S7 and S8). Taken together and exemplified at two different pathogenic loci (c.3612G>A in CFTR and c.14911C>T USH2A) where disease-causing mutations were repaired with higher efficiency and precision (Figure 4E), the here-generated HNHx-ABE variants are a valuable extension to the ABE toolkit.
Figure 4.

HNH domain substitution with heterodimeric ssDNA-specific deaminase domains
(A) Schematic domain organization of the HNHx-ABEHD variant, where the HNH domain is replaced by a heterodimeric ssDNA-specific deaminase domain (TadA-TadA∗). (B and C) Editing efficiencies of different adenine bases within the protospacer region comparing ABEmax7.10 and HNHx-ABEmax7.10HD (B) or ABE8e and HNHx-ABE8eHD (C) in endogenous loci in HEK293T (left) and K562 (right) cells. Numbering starts with PAM-distal nucleotides. Values represent mean of three independent biological replicates performed on separate days ± SD. (D) High-throughput sequencing data comparing editing efficiencies of ABEmax7.10, HNHx-ABEmax7.10, and HNHx-ABEmax7.10HD (left) and ABE8e, HNHx-ABE8e, and HNHx-ABE8eHD (right) on a library of 100 self-targeting loci in HEK293T cells. Solid line represents mean of two independent biological replicates performed on separate days, with dotted lines indicating ± SD. (E) Editing of the disease-causing c.3612G>A mutation in CFTR (top) and the c.14911C>T mutation in the USH2A (bottom) using the different ABE constructs. Green indicates the target base, orange a synonymous mutation, and red a non-synonymous mutation. Values represent mean of two independent biological replicates performed on separate days ± SD.
Discussion
In this work, we demonstrate that replacing the HNH domain of SpCas9 with a deaminase domain shifts the editing window of BEs PAM proximally (from positions 4–8 to 7–14 for TadA7.10). This expands the targeting scope of currently available BEs and enables targeting of additional disease-causing mutations. Replacement of the HNH domain with TadA furthermore reduces the size of ABEs from 5.4 kb to below 4.4 kb, allowing the construction of single adeno-associated virus (AAV) vectors with minimal promoters for in vivo delivery. Although SaCas9 fused to a TadA monomer, in principle, also fits on a single AAV, applicability of such variants is limited by their broader editing window and the requirement of NNGRRT PAM sites.11 Interestingly, editing rates of HNHx-ABEs were lower compared with classical ABEs with N-terminally fused TadA when the HNH domain was replaced by monomeric Tad. In part, this could be due to the absence of the HNH domain, because ABEmax7.10 lacking the HNH domain (ABEmax7.10 ΔHNH) also exhibited a reduction in editing rates as compared with classical ABEmax7.10. However, because replacing the HNH domain with HD TadA increased editing rates to levels comparable with N-terminally fused TadA ABEs, and because TadA dimerization is likely essential for enzymatic activity,29 we speculate that in trans TadA dimerization from another ABE molecule was inefficient in monomeric HNHx-ABE variants, potentially due to inaccessibility of the dimerization interface.
In the future, directed protein evolution or rational protein engineering strategies may refine HNHx-ABE constructs and further increase editing specificity or efficiencies. In addition, our approach of replacing the HNH domain with proteins up to 370 amino acids could be extended to other effector enzymes that act on ssDNA, thereby further expanding the Cas9-based genome editing toolbox.
Materials and methods
General methods and cloning
PCR was performed using Q5 High-Fidelity DNA Polymerase (New England Biolabs). All BE constructs were assembled using NEBuilder HiFi DNA Assembly (New England Biolabs). pCMV_ABEmax, ABE8e, pBT280, and SaKKH-ABEmax were a gift from David Liu (Addgene plasmid no. 112095, 138489, 122610, and 119815). pcDNA3.1_pCMV-nCas-PmCDA1-ugi pH1-gRNA(HPRT) was a gift from Akihiko Kondo (Addgene plasmid no. 79620). Plasmids expressing sgRNAs were cloned using T4 DNA Ligase (New England Biolabs). Lenti_gRNA-Puro was a gift from Hyongbum Kim (Addgene plasmid no. 84752). LentiGuide-Puro was a gift from Feng Zhang (Addgene plasmid no. 52963). sgRNA sequences used are listed in Table S1.
Cell culture and HTS
All cell lines were cultured at 37°C and 5% CO2 in tissue culture incubators. HEK293T cells (ATCC CRL-3216) were cultured in Dulbecco’s modified Eagle’s medium GlutaMAX and K562 cells in RPMI-1640 medium (Thermo Fisher Scientific), both supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% penicillin-streptomycin (Thermo Fisher Scientific). Cells were maintained at confluency below 90% and seeded on 96-well cell culture plates (Greiner). At 12–16 h after seeding, at approximately 70% confluency, cells were transfected using 0.5 μL Lipofectamine 2000 (Thermo Fisher Scientific) and 400 ng BE plasmid DNA and 100 ng sgRNA plasmids. Cells were incubated for 5 days, unless specified otherwise. Genomic DNA was isolated by adding 10 μL lysis buffer (10 mM Tris-HCl at pH 8.0, 2% Triton X and 1 mM EDTA, and 25 μg/mL Proteinase K) to 30 μL cell suspension. The lysate was incubated at 60°C for 60 min, followed by a 95°C incubation for 10 min. The lysate was diluted with double-distilled H2O (ddH2O) to a final volume of 100 μL. 2 μL of the diluted lysate was used for subsequent PCRs of 10 μL using NEBNext High-Fidelity 2× PCR Master Mix. The PCR product was purified using Agencourt AMPure XP beads (Beckman Coulter) and amplified with primers containing sequencing adapters. The products were gel purified and quantified using the Qubit 3.0 fluorometer with the dsDNA HS assay kit (Thermo Fisher Scientific). Samples were sequenced on an Illumina MiSeq.
HTS data analysis
Sequencing reads were demultiplexed using MiSeq Reporter (Illumina) and analyzed using a MATLAB script as previously described.5 Values are shown as n = 3 independent biological replicates over different days with mean ± SD.
Target library screen
One hundred self-targeting constructs were designed and ordered as gene blocks via TWIST Bioscience (Table S2). Subsequently, the gene blocks were pooled together in equimolar ratio, and PCR amplification was performed. Amplified pool was Gibson assembled into a BsmBI-digested Lenti_gRNA-Puro backbone. Assembly was electroporated with a BioRad GenePulser II Electroporation System into ElectroMAX Stbl4 (Thermo Fisher Scientific). Grown colonies (>100,000 colonies for 1,000× coverage) were harvested by scraping colonies from LB Agar plates and performing Miniprep (GeneJET) for plasmid extraction. Lentivirus was produced in HEK293T by lipofection of the plasmid library together with pMD2.G and psPAX2 in a 1:3:8.8 (weight) ratio. pMD2.G and psPAX2 were a gift from Didier Trono (Addgene plasmid no. 12259 and 12260). After 3 days, supernatant-containing lentivirus library was harvested and filtered (0.45 μm) and used for subsequent transduction of HEK293T at low MOI (<0.3). HEK293T cells containing library sequences were transfected (n = 2 independent biological replicates on separate days) with ABEmax7.10, ABE8e, HNHx-ABEmax7.10, HNHx-ABE8e, HNHx-ABEmax7.10HD, or HNHx-ABE8eHD on a Tol2-backbone together with pCMV-Tol2. p2T-CMV-ABEmax-BlastR was a gift from David Liu (Addgene plasmid no. 152989), and pCMV-Tol2 was a gift from Stephen Ekker (Addgene plasmid no. 31823). After 1 day, medium was changed to selection medium containing Blasticidin S (7.5 μg/mL; InvivoGen), and selection was continued for 9 days. During the whole experiment, live cell number was always kept above 100,000 to maintain a 1,000× coverage. Genomic DNA was extracted as described above and continued with PCR and NGS on a MiSeq (Illumina) system. Sequencing data were analyzed by using an in-house Python script available on GitHub (https://github.com/mathinic/HNHx_ABE_Library). All sequences with less than 50 reads per editor construct were omitted.
Stable integration of FANCA (c.3188G>A) locus into HEK293T
The sequence of the FANCA (c.3188G>A; chr16:89,749,762–89,749,797) mutation was cloned between LTR sequences of a lentiviral plasmid (containing Hygromycin B resistance gene). Lentivirus was produced as stated above, and the filtrate was used to transduce HEK293T with a low MOI (<0.3), which was subsequently selected with Hygromycin B.
Microscopy
HEK293T cells were transfected with 50 ng GFP-expressing plasmids in a 96-well plate, counterstained with Hoechst 33342, and imaged using a Zeiss Apotome. Imaging conditions and intensity scales were matched for all images. Images were analyzed using Fiji ImageJ software (v.1.51n).
Linker determination and testing
Structural data from SpCas9 (PDB: 5F9R) or SaCas9 (PDB: 5CZZ) were used to estimate linker lengths flanking deaminases in PyMol version 2.3.4. Different constructs with combinations of N- and C-terminal linkers were tested, and editing efficiencies and activity windows were determined by HTS.
Molecular graphics and analyses were done with UCSF ChimeraX, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from National Institutes of Health grant R01-GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases.
RNA-seq experiments and data analysis
RNA library preparation was performed using the TruSeq Stranded Total RNA kit (Illumina) with a ribosomal RNA (rRNA) deletion step. RNA-seq libraries were sequenced on an Illumina Novaseq machine with a sequencing depth of 140–200 Mio reads per sample.
Quality control, pre-processing, and alignment of RNA-seq reads
Quality of Illumina PE RNA-seq reads was evaluated by FastQC v.0.11.7 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Possible contaminations (genomic DNA, rRNA, Mycoplasma, etc.) were screened for using FastqScreen v.0.11.1 (https://www.bioinformatics.babraham.ac.uk/projects/fastq_screen/) against a customized database that consists of SILVA rRNA (https://www.arb-silva.de/), UniVec (https://www.ncbi.nlm.nih.gov/tools/vecscreen/univec/), refseq mRNA sequences, and selected genome sequences (human, mouse, Arabidopsis, bacteria, virus, phix, lambda, and mycoplasma) (https://www.ncbi.nlm.nih.gov/refseq/). Illumina PE reads were pre-processed using Trimmomatic version 0.36 to trim off sequencing adaptors and low-quality ends (average quality lower than 20 within a 4-nt window). Flexbar version 3.0.3 was used to remove the first 6 bases of each read, which showed priming bias introduced during library preparation. PE RNA-seq reads were generated with different read length (2X51 and 2X151). After adaptor and quality trimming, if the read length was longer than 50 nt, only the first 50 nt were kept for downstream STAR mapping and variant calling. Quality-controlled reads (average quality 20 and above, read length 20 and above) were aligned to the reference genomes (human reference genome: GRCh38.p10, Ensembl release 91) using STAR version 2.7.0e with 2-passes mode. PCR duplicates were marked using Picard version 2.9.0. Read alignments were comprehensively evaluated in terms of different aspects of RNA-seq experiments, such as sequence quality, gDNA and rRNA contamination, GC/PCR/sequence bias, sequencing depth, strand specificity, coverage uniformity, and read distribution over the genome annotation, using R scripts in ezRun (https://github.com/uzh/ezRun/) developed at the Functional Genomics Center Zurich.
RNA sequence variant calling and filtering
Variant calling from RNA-seq reads was performed according to GATK Best Practices https://gatkforums.broadinstitute.org/gatk/discussion/3891/calling-variants-in-rnaseq. In detail, GATK (version 4.1.2.0) tool SplitNCigarReads was applied to post-processed read alignments. Afterward, variants were called using HaplotypeCaller (GATK version 4.1.2.0) on PCR-deduplicated, post-processed aligned reads. Variant loci in BE overexpression experiments were filtered to exclude sites without high-confidence reference genotype calls in the control experiment. For a given SNV, the read coverage in the control experiment should be >90th percentile of the read coverage across all SNVs in the corresponding overexpression experiment. Only loci having at least 99% of reads containing the reference allele in the control experiment were kept. Only sites with more than 10 reads mapping in the overexpression experiment were kept. The cut-off for RNA A-to-I conversion frequency was set to 0.1.
Data availability
All data will be made available upon reasonable request. Amino acid sequences of BE constructs are shown in Data S1. Plasmids encoding HNHx-ABEmax7.10HD and HNHx-ABE8eHD are deposited on Addgene: 176477and 176478, respectively.
Accession numbers
HTS data from all experiments are deposited online under accession number GEO: GSE161293.
Acknowledgments
We would like to thank Weihong Qi of the Functional Genomics Center Zurich for the analysis of RNA-seq experiments. This work was supported by a Swiss National Science Foundation grant (180257) and a PHRT grant (528).
Author contributions
L.V. and L.S. designed the study, performed experiments, analyzed data, and wrote the manuscript. N.M., T.R., and K.M. performed experiments and analyzed data. G.S. designed and supervised the research and wrote the manuscript. All authors approved the final version.
Declaration of interests
L.V. and G.S. have filed a patent application based on these constructs.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.08.025.
Supplemental information
References
- 1.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zetsche B., Gootenberg J.S., Abudayyeh O.O., Slaymaker I.M., Makarova K.S., Essletzbichler P., Volz S.E., Joung J., van der Oost J., Regev A. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015;163:759–771. doi: 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ran F.A., Cong L., Yan W.X., Scott D.A., Gootenberg J.S., Kriz A.J., Zetsche B., Shalem O., Wu X., Makarova K.S. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gaudelli N.M.M., Komor A.C.C., Rees H.A.A., Packer M.S.S., Badran A.H.H., Bryson D.I.I., Liu D.R.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature. 2017;551:464–471. doi: 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee S., Ding N., Sun Y., Yuan T., Li J., Yuan Q., Liu L., Yang J., Wang Q., Kolomeisky A.B. Single C-to-T substitution using engineered APOBEC3G-nCas9 base editors with minimum genome- and transcriptome-wide off-target effects. Sci. Adv. 2020;6:eaba1773. doi: 10.1126/sciadv.aba1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yeh W.H., Chiang H., Rees H.A., Edge A.S.B., Liu D.R. In vivo base editing of post-mitotic sensory cells. Nat. Commun. 2018;9:2184. doi: 10.1038/s41467-018-04580-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Villiger L., Grisch-Chan H.M., Lindsay H., Ringnalda F., Pogliano C.B., Allegri G., Fingerhut R., Häberle J., Matos J., Robinson M.D. Treatment of a metabolic liver disease by in vivo genome base editing in adult mice. Nat. Med. 2018;24:1519–1525. doi: 10.1038/s41591-018-0209-1. [DOI] [PubMed] [Google Scholar]
- 9.Ryu S.M., Koo T., Kim K., Lim K., Baek G., Kim S.T., Kim H.S., Kim D.E., Lee H., Chung E., Kim J.S. Adenine base editing in mouse embryos and an adult mouse model of Duchenne muscular dystrophy. Nat. Biotechnol. 2018;36:536–539. doi: 10.1038/nbt.4148. [DOI] [PubMed] [Google Scholar]
- 10.Li X., Wang Y., Liu Y., Yang B., Wang X., Wei J., Lu Z., Zhang Y., Wu J., Huang X. Base editing with a Cpf1-cytidine deaminase fusion. Nat. Biotechnol. 2018;36:324–327. doi: 10.1038/nbt.4102. [DOI] [PubMed] [Google Scholar]
- 11.Anzalone A.V., Koblan L.W., Liu D.R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020;38:824–844. doi: 10.1038/s41587-020-0561-9. [DOI] [PubMed] [Google Scholar]
- 12.Nishimasu H., Shi X., Ishiguro S., Gao L., Hirano S., Okazaki S., Noda T., Abudayyeh O.O., Gootenberg J.S., Mori H. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science. 2018;361:1259–1262. doi: 10.1126/science.aas9129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu J.H., Miller S.M., Geurts M.H., Tang W., Chen L., Sun N., Zeina C.M., Gao X., Rees H.A., Lin Z., Liu D.R. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature. 2018;556:57–63. doi: 10.1038/nature26155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kleinstiver B.P., Prew M.S., Tsai S.Q., Topkar V.V., Nguyen N.T., Zheng Z., Gonzales A.P.W.W., Li Z., Peterson R.T., Yeh J.-R.R.J. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature. 2015;523:481–485. doi: 10.1038/nature14592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller S.M., Wang T., Randolph P.B., Arbab M., Shen M.W., Huang T.P., Matuszek Z., Newby G.A., Rees H.A., Liu D.R. Continuous evolution of SpCas9 variants compatible with non-G PAMs. Nat. Biotechnol. 2020;38:471–481. doi: 10.1038/s41587-020-0412-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walton R.T., Christie K.A., Whittaker M.N., Kleinstiver B.P. Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science. 2020;368:290–296. doi: 10.1126/science.aba8853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang T.P., Zhao K.T., Miller S.M., Gaudelli N.M., Oakes B.L., Fellmann C., Savage D.F., Liu D.R. Circularly permuted and PAM-modified Cas9 variants broaden the targeting scope of base editors. Nat. Biotechnol. 2019;37:626–631. doi: 10.1038/s41587-019-0134-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y., Zhou L., Liu N., Yao S. BE-PIGS: a base-editing tool with deaminases inlaid into Cas9 PI domain significantly expanded the editing scope. Signal Transduct. Target. Ther. 2019;4:36. doi: 10.1038/s41392-019-0072-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koblan L.W., Doman J.L., Wilson C., Levy J.M., Tay T., Newby G.A., Maianti J.P., Raguram A., Liu D.R. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat. Biotechnol. 2018;36:843–846. doi: 10.1038/nbt.4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sternberg S.H., LaFrance B., Kaplan M., Doudna J.A. Conformational control of DNA target cleavage by CRISPR-Cas9. Nature. 2015;527:110–113. doi: 10.1038/nature15544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thuronyi B.W., Koblan L.W., Levy J.M., Yeh W.H., Zheng C., Newby G.A., Wilson C., Bhaumik M., Shubina-Oleinik O., Holt J.R., Liu D.R. Continuous evolution of base editors with expanded target compatibility and improved activity. Nat. Biotechnol. 2019;37:1070–1079. doi: 10.1038/s41587-019-0193-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kleinstiver B.P., Prew M.S., Tsai S.Q., Nguyen N.T., Topkar V.V., Zheng Z., Joung J.K. Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat. Biotechnol. 2015;33:1293–1298. doi: 10.1038/nbt.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Richter M.F., Zhao K.T., Eton E., Lapinaite A., Newby G.A., Thuronyi B.W., Wilson C., Koblan L.W., Zeng J., Bauer D.E. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat. Biotechnol. 2020;38:883–891. doi: 10.1038/s41587-020-0453-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rees H.A., Wilson C., Doman J.L., Liu D.R. Analysis and minimization of cellular RNA editing by DNA adenine base editors. Sci. Adv. 2019;5:eaax5717. doi: 10.1126/sciadv.aax5717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grünewald J., Zhou R., Garcia S.P., Iyer S., Lareau C.A., Aryee M.J., Joung J.K. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature. 2019;569:433–437. doi: 10.1038/s41586-019-1161-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou C., Sun Y., Yan R., Liu Y., Zuo E., Gu C., Han L., Wei Y., Hu X., Zeng R. Off-target RNA mutation induced by DNA base editing and its elimination by mutagenesis. Nature. 2019;571:275–278. doi: 10.1038/s41586-019-1314-0. [DOI] [PubMed] [Google Scholar]
- 27.Villiger L., Rothgangl T., Witzigmann D., Oka R., Lin P.J.C., Qi W., Janjuha S., Berk C., Ringnalda F., Beattie M.B. In vivo cytidine base editing of hepatocytes without detectable off-target mutations in RNA and DNA. Nat. Biomed. Eng. 2021;5:179–189. doi: 10.1038/s41551-020-00671-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rothgangl T., Dennis M.K., Lin P.J.C., Oka R., Witzigmann D., Villiger L., Qi W., Hruzova M., Kissling L., Lenggenhager D. In vivo adenine base editing of PCSK9 in macaques reduces LDL cholesterol levels. Nat. Biotechnol. 2021;39:949–957. doi: 10.1038/s41587-021-00933-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lapinaite A., Knott G.J., Palumbo C.M., Lin-Shiao E., Richter M.F., Zhao K.T., Beal P.A., Liu D.R., Doudna J.A. DNA capture by a CRISPR-Cas9-guided adenine base editor. Science. 2020;369:566–571. doi: 10.1126/science.abb1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data will be made available upon reasonable request. Amino acid sequences of BE constructs are shown in Data S1. Plasmids encoding HNHx-ABEmax7.10HD and HNHx-ABE8eHD are deposited on Addgene: 176477and 176478, respectively.