

Abstract
Isocitrate dehydrogenase (IDH) mutations increase (R)-2-hydroxyglutarate (R-2HG) production; however, functional mechanisms of R-2HG in regulating cholangiocarcinoma (CCA) development remain to be further investigated. We first applied the CRISPR-Cas9 gene-editing system to create IDH1R132H-mutated CCA cells. Interestingly, our data showed that R-2HG could function through downregulating estrogen receptor alpha (ERα) and Yes-associated protein 1 (YAP1) pathways to decrease CCA growth. Detailed mechanistic studies revealed that R-2HG could target and degrade the fat mass and obesity-associated protein (FTO), the first identified mRNA demethylase. This reduced FTO can increase the N6-methyladenosine (m6A) to methylate the mRNA of ERα, and consequently decrease protein translation of the ERα. Further mechanistic studies revealed that ERα could transcriptionally suppress miR-16-5p expression, which could then increase YAP1 expression due to the reduced miR-16-5p binding to the 3′ UTR of YAP1. Furthermore, data from the pre-clinical animal model with implantation of IDH1R132H QBC939 cells demonstrated that R-2HG generated by the IDH1 mutation could downregulate ERα and YAP1 to suppress CCA tumor growth. Taken together, our new findings suggested that IDH1 mutation-induced R-2HG could suppress CCA growth via regulating the FTO/m6A-methylated ERα/miR16-5p/YAP1 signaling pathway. Upregulating R-2HG or downregulating the ERα signal by short hairpin RNA ERα (shERα) or antiestrogen could be effective strategies to inhibit CCA.
Keywords: (R)-2-hydroxyglutarate, cholangiocarcinoma, ERα, miR16-5p, YAP1
Graphical abstract
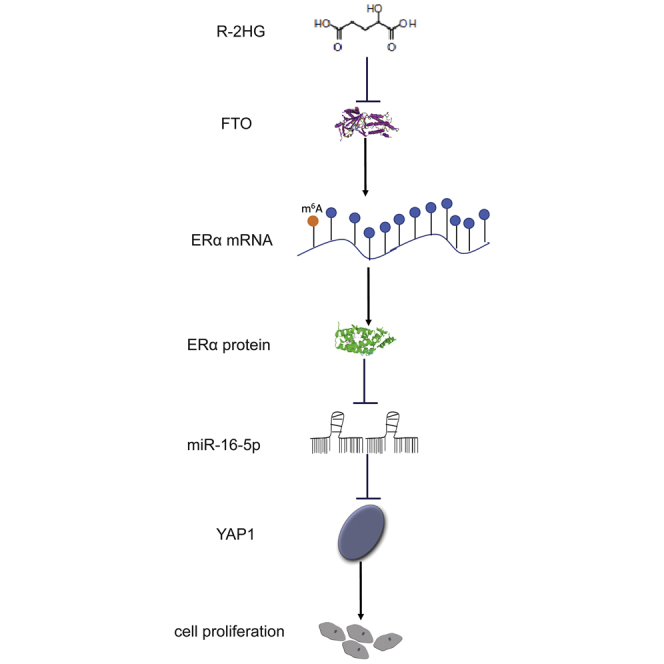
Our study demonstrates that (R)-2-hydroxyglutarate (R-2HG), produced by IDH1/2 mutations, could suppress tumor growth in cholangiocarcinoma through regulating the FTO/m6A-methylated ERα/miR16-5p/YAP1 pathway.
Introduction
Cholangiocarcinoma (CCA) arises from malignant growth of the epithelial lining of bile ducts and represents the second most common primary liver malignancy.1 Based on anatomical criteria, CCA can be subtyped as intrahepatic CCA (ICC) and extrahepatic CCA (ECC).2 Painless jaundice is the main reason for the first doctor visits for CCA patients. The poor prognosis and low median survival time (approximately 1 year) are its main characteristics.3, 4, 5 Complete surgical resection with histological negative resection margins and liver transplantation are still considered as the mainstay of curative treatment.1,2,6 According to recently reported data, even though the mortality of ECC has leveled off, the incidence and mortality of ICC are still increasing worldwide.7,8 Due to improved understanding of molecular mechanisms and pathogenesis, a growing body of evidence has shown specific gene derangements that could contribute to the development of carcinogenesis. Also, several genetic mutation profiles in CCA have been identified.9,10 Among these gene derangements, the isocitrate dehydrogenase 1/2 (IDH1/2) mutations were identified in glioma, acute myeloid leukemia (AML), chondrosarcomas, and CCA, suggesting an important role of the IDH1/2 mutations in the occurrence and prognosis of CCA.11, 12, 13, 14 IDH1/2 play important roles in the tricarboxylic acid (TCA) cycle. Subsequent findings showed that IDH1/2 could increase (R)-2-hydroxyglutarate (R-2HG), which leads to inhibiting the classic alpha-ketoglutarate (α-KG)-dependent dioxygenases involved in epigenetic regulation, extracellular matrix maturation, and cell signaling. Functional studies also uncovered that R-2HG promoted carcinogenesis and may be regarded as an oncometabolite.15, 16, 17, 18 Interestingly, after surgical resection, CCA patients with IDH1/2 mutations tend to have better overall survival than do those without IDH1/2 mutations.19,20 Thus, R-2HG appears to exhibit complex effects and unclear functions during the initiation and progression of CCA.
Estrogen is a well-known biological sex steroid, which can elevate the progression of various types of cancers through binding to estrogen receptors (ERs), such as in breast, endometrial, and ovarian carcinomas.21, 22, 23 Females are significantly more susceptible than males to suffer from several biliary tract diseases.24,25 Further research also revealed that estrogen promoted CCA initiation and progression, acting through ER alpha (ERα).26 However, the detailed mechanisms of ERα in CCA progression remain largely unclear. These emerging studies set a course for further exploring the functional mechanisms and signaling pathway of ERα in CCA.
Yes-associated protein 1 (YAP1), a co-transcriptional regulator, plays an important role in the Hippo pathway that can activate the transcription of genes involved in cell proliferation and suppress apoptotic gene expression.27,28 Phosphorylation of YAP1 functionally sequesters YAP1 in the cytoplasm and limits its activity as a transcriptional co-activator. Dephosphorylated or overexpressed YAP1 can be transported into the nuclei and bound to transcription factors, subsequently enhancing transcription.29 In CCA, many genes related to cell proliferation are regulated by YAP1, such as cyclin D1, Bcl-xL, and BIRC5.30,31 Evaluation of YAP1 expression levels and subcellular localization in human CCA specimens revealed that most YAP1 expression was localized in the nuclei.27 Clinical studies have shown that high expressions of YAP1 significantly decreased overall survival in CCA patients.32 Those studies suggest that YAP1 plays a key role in the growth of the CCA tumor mass. However, the functional connections between YAP1, ERα, and R-2HG are an underinvestigated area.
In the present study, we focused on the biological effects of the oncometabolite R-2HG. The new findings showed that R-2HG could function as a tumor suppressor to downregulate ERα and YAP1 signals to inhibit CCA through the FTO/m6A/ERα/miR16-5p/YAP1 axis.
Results
R-2HG suppresses cell proliferation in CCA
To study the R-2HG function in CCA cell growth, and since R-2HG can accumulate to millimolar levels in tumors,15 we first exposed two CCA cell lines to 300 μM R-2HG for 48 h. The results from the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assays revealed that treating with R-2HG led to suppressing CCA cell proliferation in both CCA QBC939 and HuCCT-1 cells (Figures 1A and 1C). Similar results were also obtained when we replaced the MTT assays with colony formation assays (Figures 1B and 1D), suggesting that treatment with R-2HG can suppress CCA cell proliferation.
Figure 1.

Oncometabolite R-2HG inhibits CCA cell proliferation in QBC939 and HuCCT1 cell lines
(A and C) MTT assays were conducted to detect cell growth in QBC939 cells (A) and HuCCT1 cells (C). (B and D) Colony formation was used as another strategy to confirm the R-2HG effect on cell growth in QBC939 cells (B) and HuCCT1 cells (D). (E) R-2HG from overexpression of IDH1 mutation impacted QBC939 cell growth. (F) A colony formation assay confirmed the reduced growth of QBC939 cells with the IDH1 mutation. (G) Schematic representation of the CRISPR-Cas9 gene-editing technique in mutating the IDH1 gene. (H) Sequencing result of the IDH1 gene before (upper) and after (lower) the CRISP-Cas9 gene editing. (I) An MTT assay was applied to detect the cell growth difference between wild-type IDH1 (IDH1WT) and IDH1R132H QBC939 cells. (J) Colony formation was conducted to confirm cell growth between IDH1WT and IDH1R132H QBC939 cells. For (B), (D), (F), and (J), quantitation is at the right and presented as mean ± SD (n = 3). ∗p < 0.05, ∗∗p < 0.01; NS, not significant.
To further verify the results with endogenous versus exogenous R-2HG, we applied the lentivirus system to create overexpression of wild-type and mutant (MUT) IDH1R132H in CCA QBC939 cell lines. The results from MTT and colony formation assays showed similar outcomes as those for treatment with exogenous R-2HG (Figures 1E and 1F). Although the effect of overexpressing mutant IDH1 confirmed the exogenous R-2HG result, there was little change of growth rate in the IDH1 wild-type overexpressed cells. We then conducted the CRISPR-Cas9 genome-editing technique to create a heterozygous IDH1R132H mutation (IDH1R132H) in CCA QBC939 cell lines (Figure 1G). The sequencing data show the cells with wild-type (upper) versus cells with a heterozygous IDH1 point mutation (lower) (Figure 1H). We used MTT and colony formation assays to study the endogenous R-2HG function in CCA QBC939 cells. As expected, the endogenously elevated R-2HG, generated by IDH1R132H, could suppress CCA QBC939 cell proliferation and colony formation significantly (Figures 1I and 1J).
Taken together, the results from Figures 1A–1J suggest that increased R-2HG via an ectopic supply or due to the IDH1 mutation could effectively inhibit CCA cell growth.
Mechanism dissection of how R-2HG can suppress CCA cell growth via decreasing ERα expression
Next, we focused on dissecting the molecular mechanism of how R-2HG can suppress CCA cell growth. Since either the IDH1 or IDH2 mutation could result in elevated R-2HG, we used the online tool GEO2R to conduct expression difference analysis of the GEO: GSE107102 microarray dataset, which contained IDH1/2 mutated human samples from the Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) and found 738 differential genes (Figure 2A). Then, we applied DAVID 6.8 to perform pathway enrichment analysis and obtained the top 20 enrichment pathways (Figure 2B). Subsequently, we chose the top four proliferation-related pathways as a priority for our study and eventually obtained three genes, ERα, HIF-1α, and GJA, with consistent expression changes (Figure 2C). We then further characterized the potential clinical linkage of those genes via analyzing the human CCA samples survey from The Cancer Genome Atlas (TCGA) data (https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga), including 43 CCAs with six samples having IDH1/2 mutation (Table S1). Results revealed that CCA patients with better overall survival (more than 1 year) have a lower ERα, but they had no significant changes in HIF-1α and GJA1 expressions in CCA tissues (Figure 2D). We hypothesized that ERα expression may be differential between CCAs with wild-type versus mutant IDH1/2. Although the numbers of IDH1/2 mutation cases are limited, the results indeed suggested higher ERα expressions in CCAs with wild-type IDH1/2 than with IDH1/2 mutant (Figure 2E). We then focused on studying ERα since early reports indicated a higher ERα expression in CCA patients (versus non-malignant patients) and that higher ERα expression promotes CCA cell growth.26,33,34 We then applied the western blot assay to detect the protein expression between mock (DMSO)-treated versus R-2HG-treated CCA cells and results demonstrated that treatment with R-2HG could decrease the expression of ERα in two CCA cell lines, QBC939 and HuCCT1 (Figure 2F).
Figure 2.

ERα is involved in R-2HG suppression of CCA cell growth via decreasing ERα expression
(A) We extracted 738 differential genes from a microarray dataset (GEO: GSE107102). Red represents high overexpression, and green represents lower expression of ERα. (B) Top 20 enrichment pathways from the 738 differential genes. (C) ERα, HIF-1α, and GJA1 genes were obtained through overlapping the top four pathways of size related to proliferation. (D) According to overall survival (12 months [12MTH]), TCGA data was divided into two groups; then, we checked the differential expression of three genes (ERα, GJA1, and HIF-1α) between the two groups. (E) ERα expression comparing patients with IDH1/2WT versus mutant IDH1/2 (IDH1/2MUT). (F) Western blot was conducted to examine ERα expression between DMSO and R-2HG treatment in QBC939 and HuCCT1 cell lines. Quantitation in the lower panel. (G and H) Cell proliferation results of HuCCT1 cells transfected with/without (w/wo) oeERα (overexpressing ERα) and subsequently treated with DMSO or R-2HG are shown by an MTT assay (G) and sphere formation with quantitation at the right (H). (I) Western blot was used to detect AR, ERα, and TR4 expression in QBC939 cells treated with R-2HG. (J) QBC939 cells transfected w/wo shERα (knocked down ERα) and subsequently treated with DMSO or R-2HG, after which an MTT assay was performed to examine cell growth. (K) QBC939 cells transfected w/wo shERα and subsequently electro-transfected w/wo Cas9-IDH1-Puro+template, after which an MTT assay was performed to examine cell growth. Data are presented as mean ± SD (n = 3). ∗p < 0.05, ∗∗p < 0.01; NS, not significant.
We further applied the immunoblot to examine the ERα expression in CCA QBC939 and HuCCT-1 cell lines, and the results revealed lower ERα expression in HuCCT1 cells and higher expression in QBC939 cells (Figure S1A). We then overexpressed ERα with ERα-cDNA (oeERα) in HuCCT-1 cells or knocked down ERα with ERα-short hairpin RNA (shERα) in QBC939 cells via the lentiviral system. Results showed that oeERα in HuCCT-1 cells could significantly increase CCA cell proliferation (Figure S1B), and knocking down ERα in QBC939 cells could significantly decrease the CCA cell growth (Figure S1C).
Similar results were also obtained when we induced ERα activity with estradiol (E2) treatment. Compared to mock treatment, 10 nM E2 treatment could increase HuCCT-1 cell proliferation (Figure S1D), and treating with methyl-piperidino-pyrazole (MPP, 1 μM), a specific ERα antagonist, could suppress the growth of the QBC939 cells (Figure S1E).
Importantly, results from the MTT assay using the interruption approach also revealed that overexpressing ERα led to the partial rescue/reverse of R-2HG-suppressed cell growth in HuCCT1 cells (Figure 2G). This interruption effect was further confirmed using a sphere formation assay (Figure 2H). To demonstrate that ERα is the specific R-2HG-targeting downstream gene, we first treated CCA cells with R-2HG for 48 h, and western blot data showed that among three different nuclear receptors, ERα, AR, and TR4, only ERα is selectively reduced (Figure 2I). Next, we treated ERα knocked-down QBC939 cells with R-2HG. As expected, after knocking down ERα, the sensitivity of QBC939 cells toward R-2HG treatment was diminished significantly (Figure 2J). We also knocked down ERα in IDH1R132H QBC939 cells. Data from MTT assays showed that ERα knockdown can eliminate the IDH1R132H effect (Figure 2K), which is similar to the shERα-QBC939 cells treated with R-2HG (Figure 2J).
Taken together, the results from Figures 2A–2K and Figures S1A–S1E suggest that R-2HG can suppress CCA cell growth via specifically decreasing ERα expression.
R-2HG/ERα can suppress CCA cell growth via altering the downstream gene YAP1 expression
Next, to study the downstream signs of how R-2HG/ERα can suppress CCA cell growth, we examined those genes that are modulated in IDH1/2 mutated tumors as well as regulated by ERα. We tested which IDH1/2 mutant-modulated genes can also be regulated by ERα, and found that knocking down ERα led to decreased YAP1 protein expression in CCA QBC939 cells (Figure 3A). Consistently, overexpression of ERα led to an increase of YAP1 expression in CCA HuCCT1 cells (Figure 3B). Results from western blot assays also confirmed that R-2HG treatment could result in decreasing YAP1 expression (Figure S2A) in HuCCT1 cells, as well as in IDH1R132H QBC939 cells (Figure S2B).
Figure 3.

R-2HG-reduced ERα can suppress CCA cell growth via altering downstream gene YAP1 expression
(A and B) A western blot assay was performed to detect the four potential downstream genes that might be regulated by ERα in QBC939 cells w/wo ERα knockdown (A), and in HuCCT1 cells w/wo ERα overexpression (B). (C–E) QBC939 cells were transfected w/wo shERα and subsequently transfected w/wo oeYAP1, after which an MTT assay (C) and colony formation (D) were performed to examine cell growth. A western blot assay (E) was used to reveal ERα and YAP1 expressions. (F–H) HuCCT1 cells were transfected w/wo oeERα and subsequently transfected w/wo shYAP1, after which an MTT assay (F) and colony formation (G) were performed to examine cell proliferation, and ERα and YAP1 expressions were detected by a western blot assay (H). (I–K) The results of an MTT assay (I) and colony formation (J) demonstrate the efficiency of cell growth in QBC939 cells w/wo IDH1R132H and transfected w/wo oeYAP1, and ERα and YAP1 expressions were detected by a western blot assay (K) . For (D), (G), and (J), quantitations are at the right and presented as mean ± SD (n = 3). ∗p < 0.05, ∗∗p < 0.01; NS, not significant.
To verify that YAP1 can mediate CCA cell growth, we manipulated YAP1 expression in CCA cell lines and found that overexpressing YAP1 can increase cell proliferation in CCA QBC939 cells (Figure S2C), and knocking down YAP1 (using shYAP1#1 and shYAP1#2) can both decrease cell proliferation in HuCCT1 cells (Figure S2D). Importantly, the results of MTT (Figure 3C) and sphere formation assays (Figure 3D), from an interruption approach, also revealed that overexpressing YAP1 can reverse the downregulation of ERα-mediated growth reduction in CCA QBC939 cells. Protein expressions of ERα and YAP1 for the above interruption assay were validated and are shown in Figure 3E. Consistently, using these assays, we demonstrate that knocking down YAP1 can block the ERα overexpression-increased cell growth in HuCCT1 cells (Figures 3F–3H). These rescue experiments were also conducted in IDH1R132H QBC939 cells, and, as expected, altering YAP1 expression can partially block the function of the endogenously IDH1R132H-increased R-2HG (Figures 3I–3K).
Taken together, the results from Figures 3A–3K and Figures S2A–S2D suggest that R-2HG treatments can lead to suppressing cell growth via altering the ERα/YAP1 axis in CCA cells.
R-2HG/ERα can decrease YAP1 expression via altering miR16-5p expression
To dissect the mechanism of how R-2HG/ERα can alter YAP1 expression in CCA cells, we examined the mRNA expression of YAP1, and results from quantitative real-time PCR assay showed that downregulation of ERα in QBC939 cells could decrease YAP1 expression at the protein level (see Figure 3A), but not at the mRNA level (Figure 4A). Overexpression of ERα in HuCCT1 cells could increase YAP1 expression at the protein level (see Figure 3B), but not at the mRNA level (Figure 4A). Additionally, these results were also observed in R-2HG-treated cell lines (Figure 4B). We then focused on the non-coding RNAs (ncRNAs), as recent studies indicated that microRNAs (miRNAs, miRs) could function as crucial regulators to bind to the 3′ UTR of the targeted gene’s mRNAs and affect their protein expressions to alter cancer progression.35,36
Figure 4.

R-2HG/ERα can decrease YAP1 expression via altering miR16-5P expression
(A) Real-time PCR showed YAP1 mRNA expression of shERα in QBC939 cells and oeERα in HuCCT1 cells. (B) Real-time PCR detected YAP1 expression in QBC939 and HuCCT1 cells treated w/wo R-2HG. (C) RIP assay to detect the YAP1 mRNA levels in the Argonaute 2 (Ago2) complex in QBC939 cells transfected with pLKO or shERα (left) and in HuCCT1 cells transfected with pWPI or oeERα (right). (D) Quantitative real-time PCR was used to screen seven potential miRNAs that might be able to regulate YAP1 in QBC939 cells transfected with ERα-shRNA or pLKO (left) and HuCCT1 cells transfected with oeERα or pWPI (right). (E) Western blot was used to examine YAP1 expression from the four groups of QBC939 cells as indicated (with shERα+miR16-5p inhibitor, pLKO+miR16-5p inhibitor, shERα+Ctrl, or pLKO+Ctrl) (left), and another four groups in HuCCT1 cells as indicated (oemiR16-5p+pWPI, oemiR16-5p+oeERα, pLKO+pWPI, pLKO+oeERα) (right). (F) MTT (left) and colony formation (middle) assays were performed in QBC939 cells transfected w/wo shERα and w/wo miR16-5p inhibitor as indicated (left) and in four groups of HuCCT1 cells as indicated (middle). (G) MTT (left) and colony formation (middle) assays were performed in 4 groups of HuCCT1 cells as indicated. For (E, F, and G), quantitations are at the right and presented as mean ± SD (n = 3). ∗p < 0.05, ∗∗p < 0.01; NS, not significant.
To examine whether the binding capacity of miRNAs to 3′ UTR of YAP1 mRNA changes and is involved in the ERα-regulated YAP1 expression, we performed an Argonaute 2 (Ago2)-RNA immunoprecipitation (RIP) assay on both cell lines (Figures S3A and S3B). The results revealed that knocking down ERα in QBC939 cells could increase the YAP1-3′ UTR level, and adding ERα-cDNA could decrease the YAP1-3′ UTR level in the Ago2 complex (Figure 4C). Using the bioinformatics prediction databases tool (TargetScan), we found seven candidate miRNAs (miR15b-5p, miR6838-5p, miR15a-5p, miR16-5p, miR424-5p, miR195-5p, and miR497-5p) that might be able to bind to the 3′ UTR of YAP1 and regulate YAP1 expression. Importantly, results from quantitative real-time PCR analysis demonstrated that miR16-5p could be regulated by ERα, showing that overexpressing ERα decreased miR16-5p expression in HuCCT-1 cells, and knocking down ERα increased the expression of miR16-5p in QBC939 cells (Figure 4D).
We then applied the functional assay to examine the consequence of altered miR16-5p expression. The results revealed that altering miR16-5p with inhibitor treatment (Figure S3C, left) resulted in overexpression of YAP1 protein and increasing cell proliferation in QBC939 cells (Figure S3D, left), and overexpressing miR16-5p (Figure S3C, right) resulted in the reduction of YAP1 protein and suppression of cell growth in HuCCT1 cells (Figure S3D, right). To further verify that ERα can function through miR16-5p to modulate YAP1 expression and cell proliferation, we conducted a reverse assay in CCA cell lines. Western blotting analysis indicated that suppressing miR16-5p could rescue YAP1 protein expression in ERα knocked down (shERα) QBC939 cells (Figure 4E, left) and overexpressing miR16-5p also can block YAP1 protein expression in ERα overexpressed HuCCT1 cells (Figure 4E, right). In Figure 4F, the results of MTT (left) and sphere formation assays (middle) also show that targeting miR16-5p could rescue cell growth suppressed by knocking down ERα in QBC939 cells and enhancing miR16-5p level could reverse/block ERα-induced cell proliferation in HuCCT1 cells (Figure 4G).
In summary, the results from Figures 4A–4G and Figures S3A–S3D suggest that ERα may increase cell proliferation via altering miR16-5p/YAP1 signaling in CCA cells.
R-2HG/ERα can regulate miR16-5p expression via transcriptional regulation
To uncover the molecular mechanism of how R-2HG/ERα can decrease miR16-5p expression at the transcriptional level, we first applied the Ensembl website with JASPAR database approaches to search for the potential estrogen response elements (EREs) on the 3-kb region of miR16-1 and miR16-2 promoters, which are the precursor (pre-)miRNAs of mature miR16-5p. We found only two putative EREs located within the miR16-1 promoter region (Figure 5A). We then applied the chromatin immunoprecipitation (ChIP) in vivo binding assay to verify their capacity of binding to ERα, and the results revealed that ERα could bind to ERE1/2 (Figure 5B).
Figure 5.

ERα may transcriptionally regulate miR16-5p, and miR16-5p may directly target the 3′ UTR of YAP1-mRNA to suppress its protein expression
(A) Two putative EREs predicted by JASPAR from the miR16-1 promoter. (B) ChIP assay results of ERE1/2 of the miR16-1 promoter in QBC939 cells. (C) Wild-type and mutant pGL3-miR16-1 promoter reporter constructs. (D) Luciferase activity after transfection of wild-type or mutant EREs on miR16-1 promoter reporter construct in HuCCT1 cells (left) transfected with ERα-cDNA (oeERα) or pWPI and QBC939 cells (right) transfected with ERα-shRNA (shERα) or vector pLKO. (E) Sequence alignment of the YAP1 3′ UTR with wild-type versus mutant potential miR16-5p targeting sites. (F) Luciferase reporter activity after transfection of wild-type or mutant YAP1 3′ UTR reporter construct in HuCCT1 cells w/wo oemiR16-5p (left) and QBC939 cells treated w/wo miR16-5p inhibitor (right). For (D) and (F), quantitations are presented as mean ± SD. ∗p < 0.05; NS, not significant.
We also constructed the miR16-1 gene promoter luciferase reporter by inserting a 3.0-kb 5′ promoter region of miR16-1 containing ERE1/2 into the pGL3 luciferase backbone as well as a version with the mutated ERE1/2 (Figure 5C). The results revealed that oeERα significantly decreased luciferase activity in HuCCT-1 cells transfected with wild-type miR16-1 promoter, but not in the cells with the mutant miR16-1 promoter (Figure 5D, left). Knocking down ERα dramatically increased luciferase activity in QBC939 cells transfected with the wild-type miR16-1 promoter, but not in cells with the mutant miR16-1 promoter (Figure 5D, right).
Taken together, the results from Figures 5A–5D suggest that R-2HG/ERα can suppress miR16-5p expression via transcriptional regulation via binding to ERE1/2 on the miR-16-1 promoter region.
R-2HG/ERα/miR16-5p axis can decrease YAP1 expression via the binding of miR16-5p to the 3′ UTR of YAP1
To further dissect the molecular mechanism of how the R-2HG/ERα/miR16-5p axis can decrease YAP1 expression, we applied a luciferase assay to determine whether miR-16-5p might directly target the YAP1 mRNA 3′ UTR to suppress its expression. We identified some potential binding sites located on the 3′ UTR of YAP1 (http://www.targetscan.org) and then constructed the reporter plasmids with the psiCHECK-2 vector carrying the wild-type and mutant miRNA target sites (Figure 5E). As expected, the luciferase assay results revealed that overexpressing miR16-5p suppressed luciferase activity in HuCCT1 cells transfected with wild-type YAP1 3′ UTR but not in the mutant YAP1 3′ UTR (Figure 5F, left), and decreasing miR-16-5p, with inhibitor treatment, could increase luciferase activity in QBC939 cells transfected with the wild-type YAP1 3′ UTR, but not within the mutant YAP1 3′ UTR (Figure 5F, right).
Taken together, the results from Figures 5E and 5F suggest that R-2HG/ERα/miR16-5p axis can decrease YAP1 expression via direct targeting of the 3′ UTR of YAP1-mRNA.
R-2HG can suppress ERα expression via inducing high RNA-m6A methylation
To further dissect the molecular mechanism of how R-2HG can regulate ERα expression, we used quantitative real-time PCR to detect ERα mRNA expression comparing DMSO- and R-2HG-treated cells, and the results indicated that the ERα mRNA expression had no dramatic differences (Figure 6A). Even no significant difference was shown between CRISPR-Cas9 genome-edited wild-type IDH1 (IDH1WT) and IDH1R132H QBC939 cells (Figure 6B). There was little difference of the ERα mRNA stability between the DMSO- and R-2HG-treated HuCCT1 cells (data not shown). We then conducted a RIP assay using anti-Ago2 antibody and insulin-like growth factor 2 mRNA binding protein 3 antibody (anti-IGF2BP3). Many studies reported that Ago2 protein is involved in the post-transcriptional regulation through RNA-induced silencing complex (RISC), and IGF2BP3 can enhance this procedure.37, 38, 39, 40 Unexpectedly, the results opposed the hypothesis that R-2HG regulated ERα translation through miRNA-induced post-transcription regulation (Figure 6C). Also as expected, in the IDH1R132H QBC939 cells, the result from the RIP assay was consistent with that of R-2HG-treated HuCCT1 cells (Figure S4A). We then switched to study the ERα protein stability, using cycloheximide (CHX) treatment for 0, 1, 2, 4, 6, and 8 h, and the result of the western blot assay demonstrated that ERα protein has slightly more stability in the R-2HG treated group than that in the DMSO-treated group (Figure 6D), and a similar tendency was found in the IDH1WT QBC939 cells compared to the IDH1R132H QBC939 cells (Figure S4B). We then studied whether the ERα expression may be regulated at the translational level. To test this possibility, we examined the binding of 18S ribosomal RNA (rRNA), a translation marker,41 to ERα mRNA using an ERα mRNA-biotin pull-down assay (Figure S4C). The quantitative real-time PCR results from the pull-down assay demonstrated that after R-2HG treatment, the 18S rRNA level decreased significantly in the ERα mRNA-biotin complex in QBC939 cells (Figure 6E).
Figure 6.

R-2HG can suppress ERα expression via inducing high RNA-m6A methylation
(A) Quantitative real-time PCR was conducted to detect ERα mRNA expression in HuCCT1 cells and QBC939 cells treated with R-2HG. (B) Quantitative real-time PCR was conducted to detect ERα mRNA expression in QBC939 cells with/without (w/wo) IDH1R132H. (C) The ERα mRNA level was detected in the Ago2 complex and IGF2BP3 complex using a RIP assay in HuCCT1 cells treated with DMSO or R-2HG. (D) ERα protein stability test using cycloheximide (CHX) treatment in HuCCT1 cells treated w/wo R-2HG (left) and quantitation at the right. (E) Quantitative real-time PCR showed 18S rRNA levels in the ERα mRNA-biotin pull-down complex in QBC939 cells treated w/wo R-2HG. (F) The ERα mRNA level was detected in the anti-m6A complex using a RIP assay in QBC939 cells treated w/wo R-2HG. (G) FTO expression in QBC939 (upper) and HuCCT1 (lower) cells after R-2HG treatment. (H) QBC939 cells transfected w/wo shFTO and subsequently treated w/wo R-2HG, after which an MTT assay was performed to examine cell growth. (I) The methylation of ERα mRNA could change its protein expression. A western blot was used to examine ERα expression from the four groups as indicated in QBC939 cells (upper) with quantitation in the lower panel. (J) We knocked down FTO in QBC939 cells and then divided cells into three groups and transfected them with pWPI, oeERαWT, or mutant oeERα (oeERαMT). A western blot was used to detect ERα expression. (K) MTT assay was conducted to examine cell growth among the three groups as indicated in QBC939 cells. Quantitations are presented as mean ± SD. ∗p < 0.05, ∗∗p < 0.01; NS, not significant.
To further study how R-2HG can impact ERα protein expression at the translational level, we focused on altering the global cell RNA-m6A methylation to influence protein translation.15,42 We first conducted a RIP assay using anti-m6A antibody (Figure S4D), and the results showed higher ERα mRNA expression in the m6A antibody complex in QBC939 cells treated with R-2HG (Figure 6F). This result also was reproduced in the IDH1R132H QBC939 cells (Figure S4E). We then examined the FTO expression in R-2HG-treated CCA cells, as a recent study indicated that R-2HG might induce a high m6A level via degrading FTO, which is the first mRNA demethylase studied.43,44 We checked the FTO expression in the R-2HG-treated CCA cells, and the results of western blot (Figure 6G) showed that FTO expression decreased in the R-2HG-treated QBC939 (upper) and HuCCT1 (lower) cells. After manipulating FTO expression with overexpression of FTO or FTO-shRNA, MTT results revealed that cell proliferation was significantly enhanced or suppressed, respectively (Figures S4F and S4G). Furthermore, knockdown of FTO in R-2HG-sensitive QBC939 cells significantly abolished their sensitivity to R-2HG (Figure 6H), and western blot results demonstrated that after knocking down FTO in QBC939 cells, ERα expression showed no difference between DMSO and R-2HG treatment (Figure 6I). To further determine the mechanisms of the regulatory effects of the m6A-ERα mRNA, the topology of m6A RNA methylomes revealed that m6A is especially enriched around the stop codons of mRNAs with a consensus sequence of RRACH (R = G or A; H = A, C, or U).45, 46, 47 The ERα mRNA-m6A methylated sites are located at amino acids 563–565 in the ligand-binding domain of ERα. To translate the same amino acids, we mutated and swapped these 3 aa codons ACG-GAC-CAA with ACU-GAU-CAG and defined this as the codon swapping mutant, ERα563–565 CDSmt (Figure S4H). In QBC939 cells with FTO knockdown, m6A methylation of ERα mRNA could decrease the ERα protein translation in cells containing wild-type ERα, but not in cells with ERα563–565 CDSmt (Figure 6J). Subsequently, the functional assay also showed that this mutant ERα could rescue/reverse the FTO knockdown-mediated cell growth reduction (Figure 6K).
Taken together, Figures 6A–6K and Figures S4A–S4H results suggest that R-2HG can degrade FTO and then increase ERα mRNA m6A methylation, which may then result in suppressing ERα protein expression via translational regulation.
Increasing R-2HG or inhibition of ERα/YAP1 signals can suppress tumor growth in the preclinical mouse CCA model
To confirm the above in vitro cell line data in the in vivo mouse CCA model, we randomly divided mice into four groups for subcutaneous injections of QBC939 cells with IDH1WT+pLKO (group 1), IDH1WT+shERα (group 2), IDH1R132H+pLKO (group 3), and IDH1R132H+shERα (group 4). We measured the length and width of tumors to monitor tumor growth every week. After 8 weeks, mice injected with cells with the elevated R-2HG due to IDH1R132H had a suppressed tumor growth rate; however, after knocking down ERα, the effect of R-2HG-suppressed tumor growth of QBC939-IDH1R132H was abolished as compared with the QBC939-IDH1WT group (Figure 7A). We then sacrificed the mice and measured the size and weight of each tumor (Figure 7B). The results showed that the tumor weights of the IDH1WT+pLKO group were higher than those of the IDH1WT pLKO group, but no difference was detected between the IDH1WT+shERα and IDH1R132H +shERα groups (Figure 7C).
Figure 7.

In vivo mouse tumor model showing that the R-2HG-downregulated ERα signals could modulate tumor growth
(A) The QBC939 cells transfected with IDH1WT+pLKO, IDH1WT+shERα, IDH1R132H+pLKO, or IDH1R132H+shERα were subcutaneously implanted into female nude mice, and tumor sizes were measured weekly. After 8 weeks, the mice were sacrificed and the tumor growth rates of different groups were compared. (B) Gross comparison of CCA tumor size in the four groups of mice. (C) Quantification of tumor weights. (D) Representative images of the immunohistochemistry staining of YAP1 in each group. Brown signals indicate the positive YAP1 staining. (E) Two samples from each tumor group were randomly chosen to detect YAP1 and ERα using a western blot. (F) Schematic model of modulating the FTO/m6A methylated ERα/miR16-5p/YAP1 signaling by the oncometabolite R-2HG to suppress CCA growth. Quantitations are presented as mean ± SD. ∗p < 0.05, ∗∗p < 0.01; NS, not significant.
Our immunohistochemistry (IHC) staining demonstrated that YAP1 was decreased after IDH1 was mutated or ERα was knocked down (Figure 7D). Importantly, results from assaying two tumor samples from each group also revealed that endogenously elevated R-2HG, due to the R132H mutation, could decrease the ERα and YAP1 expressions, which is consistent with our in vitro studies showing that R-2HG can function via altering the ERα to suppress YAP1 expression (Figure 7E).
Taken together, these results of the animal experiments in Figures 7A–7E were consistent with the in vitro data and revealed that endogenous R-2HG suppresses CCA tumor growth; however, after knocking down ERα, the effect of suppressing tumor growth induced by R-2HG was abolished. R-2HG could suppress CCA proliferation by modulation of ERα/miR16-5p/YAP1 signaling.
Discussion
An increase of R-2HG is the primary mechanism of mutant IDHs contributing to tumorigenesis. During tumorigenesis, R-2HG degrades TET2 and inhibits its catalytic activity, which suppresses cell differentiation and promotes malignant transformation through enhancing DNA methylation.48 However, recent studies revealed that IDH1 mutation decreased tumor progression in glioma and suppressed AML cell proliferation. Mechanism dissection displayed that R-2HG functioned through m6A RNA modification regulated by FTO.43 R-2HG inhibited HNF4α activity through histone methylation to interrupt hepatocyte differentiation and promote CCA genesis.13 However, our study first found that R-2HG also played an anti-tumor role during CCA progression through ERα/miR16-5p/YAP1 signaling (Figure 7F). Our data confirmed the previously published paper, demonstrating that R-2HG functions through FTO to induce high levels of m6A modifications. High levels of m6A can recruit m6A “readers” to bind to the RNA and decide the fate of m6A-modified RNA. Among these readers, YTHDF1, YTHDF3, YTHDC1, and YTHDC2 could enhance RNA stability, and, in contrast, YTHDF2 could degrade RNA.47 However, in our study, R-2HG did not dramatically decrease ERα mRNA levels (Figures 6A and 6B). Thus, the decreasing translation of ERα mRNA could not be ascribed to the m6A reader YTHDF2. Several studies reported that m6A modification could decrease translation through blocking m6A-methylated mRNAs combining with ribosomes.49,50 Our data on 18S RNA detected from ERα-biotin pull-down demonstrated that m6A reduced the binding capacity of mRNA and ribosomes. Moreover, after mutating the potential m6A modification sites of ERα mRNA, this effect induced by m6A was blocked.
It is well known that ERα plays crucial regulatory roles facilitating the tumorigenesis process by regulation of CCA.26,34,51 One published study revealed higher ERα expression in CCA compared with normal biliary duct tissues and found that the change of ERα expression can serve as a prediction marker for the prognosis of CCA patients.52 Our study demonstrates that R-2HG could functionally connect between the IDH1/2 mutations and change of ERα expression. Then, ERα could regulate the YAP1 level through controlling miR16-5p expression. The miR16 family has been investigated in several human cancers and was verified to be involved in tumor suppressive and antimetastatic properties.53,54 In our study, ERα could decrease the pre-miRNA of miR16-5p (miR16-1) expression at a transcriptional level via binding to ERE1/2 located in the 5′ promoter region of its precursor, and miR16-5p could directly bind to the 3′ UTR of YAP1 mRNA to suppress its protein expression. Our findings support the notion that miR16-5p could suppress CCA cell growth and R-2HG in CCA, with the IDH1/2 mutants mainly using the ERα/miR16-5p/YAP1 signaling pathway to regulate cell proliferation.
During tumorigenesis, YAP1 functions as a coactivator combined with transcriptionally enhanced associated domain transcription factors to promote tumor progression.55 Connected to this role, it could regulate cellular proliferation, invasion, chemoresistance, and angiogenesis.28,31,56 In addition, YAP1 plays vital roles in the immune system by regulating PD-L1 expression in human thoracic cancer.57 Several clinical studies showed that YAP1 overexpression was correlated with poor prognosis in CCA.27,32 In human astrocytes, IDH1 mutation could suppress cell proliferation through decreasing YAP1 expression; however, the mechanism of how IDH1 mutation affected YAP1 expression was unclear.58 Our data uncovered that decreased YAP1 expression in IDH1 mutated cells was ascribed to R-2HG, which is the main product of the mutated IDH enzyme.
Conclusions
This study provides novel insights into the molecular mechanisms of anti-tumor activity of R-2HG generated from IDH1/2 mutations, the cross-talk between R-2HG- and ERα-regulated signals, and may help in the development of novel therapeutic strategies to treat CCA with IDH mutations.
Materials and methods
Microarray data source
Gene expression profile data (GEO: GSE107102) was downloaded from Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo). Four CCA IDH1/2 mutation samples and 20 wild-type samples were included. The array data were acquired from Illumina HumanHT-12 WG-DASL v4.0 R2 (GPL14951; transcript [gene] version).
Analysis of DEGs
GEO2R (https://www.ncbi.nlm.nih.gov/geo/geo2r/) was applied to compare two groups of samples in a GEO series to identify the differentially expressed genes (DEGs) under the same experimental conditions. |log2 fold change (FC)| ≥ 0.5 and p < 0.01 were chosen as cutoff criteria and defined a statistically significant difference. A volcano plot map of DEGs was generated using ggplot2.
GO enrichment analysis
Gene Ontology (GO) enrichment analysis was performed by DAVID 6.8.59 DAVID contains a series of functional annotation programs to explore abundant biological messages of genes, and four proliferation-related terms were chosen for overlapping. The Venn diagram was conducted using an online tool (https://bioinfogp.cnb.csic.es/tools/venny/index.html).
Clinical sample description
We used the public TCGA (https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga) database as our primary source of samples. To analyze the data generated by TCGA, we directly downloaded all of the CCA patients’ clinical information. In total, 43 tumors with clinical data were profiled for survival analysis. We also performed class discovery and class prediction analyses by comparing gene expressions.
Cell culture and reagents
QBC939 cells were donated by Dr. Xiangcheng Li’s lab,60 HuCCT1 cells were obtained from the Japanese Collection of Research Bioresources Cell Bank, and HEK293T cell lines were purchased from the American Type Culture Collection. All three cell lines were maintained in Dulbecco’s modified Eagle’s medium (Invitrogen, Grand Island, NY, USA) with 1% penicillin and streptomycin antibiotics and 10% fetal bovine serum (FBS). All cells were incubated in a humidified 5% CO2 environment at 37°C. All cells were shown to be mycoplasma negative as detected by PCR. QBC939 and HuCCT1 cells were treated with 300 μM R-2HG (#16366, Cayman Chemical, Ann Arbor, MI, USA) for the indicated time points.
CRISPR-Cas9-mediated IDH1 editing
The pSpCas9(BB)-2A-Puro (PX459) v2.0 was a gift from Dr. Feng Zhang61 (Addgene plasmid #62988; http://addgene.org/62988). The guide RNA (gRNA), 5′-GCAUGACGACCUAUGATGAUAGG-3′, was chosen through using an online tool developed by Dr. Zhang’s lab (https://zlab.bio/guide-design-resources). The gRNA was cloned into a Cas9 vector using Quick Ligase (New England Biolabs, Ipswich, MA, USA). We designed the 84-bp DNA template with a single base mutation (CGT-CAT) synthesized by Integrated DNA Technologies (IDT). Plasmids and templates were electro-transfected into QBC939 cells. After 3 days, the transfected cells were then subjected to puromycin selection for 3 days and viable cells were serially diluted to generate single-cell clones. The genomic mutation was screened by Genewiz sequencing.
Lentivirus packaging and cell transfection
The shRNA or overexpression cDNA plasmids were co-transfected with packaging plasmid psPAX2 and envelope plasmid pMD2.G into HEK293T cells following the calcium phosphate cell transfection protocol. After a 48-h incubation, the virus supernatants were harvested for immediate use and/or frozen at −80°C for later use. The oligonucleotide sequences for the construction of the two shRNAs against ERα follow. The first forward primer was 5′-CCG GCT ACA GGC CAA ATT CAG ATA ATT GGA TCC GTT ATC TGA ATT TGG CCT GTA GTT TTT G-3′, and the first reverse primer was 5′-AAT TCA AAA ACT ACA GGC CAA ATT CAG ATA ACG GAT CCA ATT ATC TGA ATT TGG CCT GTA G-3′. The second pair of primers were forward, 5′-CCG GGC AGG ATT GTT GTG GCT ACT ATT GGA TCC GTA GTA GCC ACA ACA ATC CTG CTT TTT G-3′, and reverse, 5′-AAT TCA AAA AGC AGG ATT GTT GTG GCT ACT ACG GAT CCA ATA GTA GCC ACA ACA ATC CTG C-3′. The oligonucleotide sequences for the construction of the two shRNAs against FTO were as follows. The first pairs of primers were forward, 5′-CCG GTC ACC AAG GAG ACT GCT ATT TGG ATC CAA ATA GCA GTC TCC TTG GTG ATT TTT G-3′, and reverse, 5′-AAT TCA AAA ATC ACC AAG GAG ACT GCT ATT TGG ATC CAA ATA GCA GTC TCC TTG GTG A-3′. The second pair of primers were forward, 5′-CCG GCG GTT CAC AAC CTC GGT TTA GGG ATC CCT AAA CCG AGG TTG TGA ACC GTT TTT G-3′, and reverse, 5′-AAT TCA AAA ACG GTT CAC AAC CTC GGT TTA GGG ATC CCT AAA CCG AGG TTG TGA ACC G-3′. The oligonucleotide sequences for the construction of the two shRNAs against YAP1 were as follows. The first pair of primers were forward, 5′-CCG GCC CAG TTA AAT GTT CAC CAA TGG ATC CAT TGG TGA ACA TTT AAC TGG GTT TTT G-3′, and reverse, 5′-AAT TCA AAA ACC CAG TTA AAT GTT CAC CAA TGG ATC CAT TGG TGA ACA TTT AAC TGG G-3′. The second pair of primers were forward, 5′-CCG GGC CAC CAA GCT AGA TAA AGA AGG ATC CTT CTT TAT CTA GCT TGG TGG CTT TTT G-3′, and reverse, 5′-AAT TCA AAA AGC CAC CAA GCT AGA TAA AGA AGG ATC CTT CTT TAT CTA GCT TGG TGG C-3′. The miR-16-5p antisense inhibitor, 5′-CGC CAA UAU UUA CGU GCU GCU A-3′, was ordered from IDT (Newark, NJ, USA).
RNA extraction and quantitative real-time PCR analysis
Total RNAs were extracted using TRIzol reagent (Invitrogen, Grand Island, NY, USA). One microgram of total RNA was subjected to reverse transcription using SuperScript III transcriptase (Invitrogen). Real-time PCR was conducted using a Bio-Rad CFX96 system with SYBR Green to determine the mRNA expression level of a gene of interest. Expression levels were normalized to GAPDH level using the 2−ΔΔCt method.
Western blot
Cells were washed twice with cold PBS and lysed in radioimmunoprecipitation assay (RIPA) buffer, and proteins (30–50 μg) were separated on 6%–10% SDS-PAGE gel and then transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA, USA). After blocking with 5% BSA, the PVDF membranes were incubated with primary antibodies and then horseradish peroxidase (HRP)-conjugated secondary antibodies. Subsequently, The enhanced chemiluminescence (ECL) system (Thermo Fisher Scientific, Rochester, NY, USA) was used for visualization. The primary antibodies used for western blot were ERα (#ab75635, Abcam, Cambridge, MA, USA), GAPDH (#sc-166574, Santa Cruz, Paso Robles, CA, USA), YAP1 (#sc-376830, Santa Cruz, Paso Robles, CA, USA), FTO (#ab124892, Abcam, Cambridge, MA, USA), Ago2 (#2897, Cell Signaling Technology, Boston, MA, USA), and immunoglobulin (Ig)G (#sc-2027, Santa Cruz, Paso Robles, CA, USA).
RIP
QBC939 and HuCCT1 cells after different treatments were fixed by 4% paraformaldehyde and were lysed in ice-cold lysis buffer supplemented with RNase inhibitor. After centrifugation, 1 mL of the supernatants was pre-cleared by protein A/G beads for 1 h. After that, samples were incubated with Ago2, IGF2BP3, or m6A antibody overnight at 4°C. The RNA/antibody complexes were washed four times by RIPA buffer supplemented with RNase inhibitor and protease inhibitor cocktail. The RNA was extracted using TRIzol (Invitrogen) according to the manufacturer’s protocol and subjected to quantitative real-time PCR analysis.
ERα mRNA-18S rRNA pull-down assay
The cells were collected in cell lysis buffer after receiving the designated treatments for 48 h. The cell lysate mixture was rotated overnight at 4°C after adding 1.5 μL of RNase inhibitor and 500 pM biotin-labeled anti-sense oligonucleotides against ERα mRNA (5′-TTC CCT GGT TCC TGT CCA AGA GCA-3′). The lysate mixture was rotated for 1 h at 4°C after adding 10 μL of streptavidin agarose beads. The mixture was centrifuged at 3,000 rpm for 2 min, and then the beads were washed with cell lysis buffer five times. Total RNAs were extracted by TRIzol (Invitrogen) according to the manufacturer’s protocol and reverse transcribed. Quantitative real-time PCR was used to test the levels of ERα to ensure that each group was loaded equally for the following step. 18S rRNA was detected using quantitative real-time PCR analysis from the complex.
ChIP assay
The cross-linked protein-DNA complexes were fixed by 1% formaldehyde and quenched using 125 mM glycine. Cells were harvested in lysis buffer and subjected to sonication. Normal IgG and protein A-agarose were used sequentially to preclear the cell lysates. We then added 2.0 μg of anti-ERα antibody to the cell lysates and incubated them overnight at 4°C. IgG was used in the reaction for a negative control. Specific primer sets were designed to amplify a target sequence within the miR-16-1 promoter, and agarose gel electrophoresis was used to identify the PCR products.
Luciferase reporter assay
A 3-kb length of the miR-16-1 promoter with wild-type or mutant ERα-responsive elements was cloned into the pGL3-basic luciferase reporter vector (Promega, Madison, WI, USA). Then, 500-bp-fragment nucleotides of the YAP1 3′ UTR with wild-type or mutant miRNA-binding sites were cloned into the psiCHECK-2 vector (Promega) downstream of the Renilla luciferase open reading frame (ORF). HuCCT1 and QBC939 cells were plated in 24-well plates and the plasmids were transfected with Lipofectamine 3000 transfection reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Luciferase activity was measured 36 h after transfection by a Dual-Luciferase assay (Promega) according to the manufacturer’s manual.
Preclinical mouse tumor study
Thirty-two 6-week-old female nude mice were purchased from the National Cancer Institute (NCI) and divided into four groups for injection of QBC939 cells transfected/engineered with (1) IDH1WT+pLKO, (2) IDH1WT+shERα, (3) IDH1R132H+pLKO, and (4) IDH1R132H+shERα. The prepared stable QBC939 cells (mixed with Matrigel, 1:1) were subcutaneously injected at 1 × 106 into the mouse flanks. The growth rates of CCA tumors were monitored by measuring the length and width of the tumors once a week. The mice were sacrificed after 8 weeks. Tumors were removed for studies. Animal experiments were approved by institutional animal care at the Univeristy of Rochester Medical Center.
IHC
IHC was performed on the xenografted tumors. Formalin-fixed and paraffin-embedded tissue slides (5 μm) were deparaffinized and rehydrated. Antigen retrieval was performed by incubating the tissue slides in 0.01 M citric acid buffer at 100°C for 10 min. After blocking with 3% H2O2 and 5% FBS, the slides were incubated with a monoclonal antibody against YAP1 (1:100, #sc-376830, Santa Cruz, Paso Robles, CA, USA) at 4°C overnight. The slides were then reacted with polymer HRP reagent. The peroxidase activity was visualized with diaminobenzidine tetrahydrochloride solution. The sections were counterstained with hematoxylin. Dark brown cytoplasmic staining of at least 1% of tumor cells was defined as positive, and no staining or less than 1% of cells stained was defined as negative.
Statistical analysis
Statistical analysis was performed using SPSS software (version 23.0; SPSS, Chicago, IL, USA) or GraphPad Prism 6 (GraphPad, La Jolla, CA, USA). The Mann-Whitney U test was applied to compare the two independent groups when the dependent variable was continuous. A Kruskal-Wallis test with a Dunn-Bonferroni post hoc test was applied to test the multiple group’s continuous data. All experiments were conducted with three technical replicates and performed at least three times. The data values were presented as the mean ± SD. Differences in mean values between two groups were analyzed by a two-tailed Student’s t test, and the mean values of more than two groups were compared with one-way ANOVA. p < 0.05 was considered to indicate a statistically significant difference.
Acknowledgments
This work was partially supported by the University of Rochester George Whipple Endowment, the National Natural Science Foundation of China (no. 81672469), and the endowment from the Division of General Surgery of Changzhou No. 2 People’s Hospital. We thank Karen Wolf for editing the manuscript.
Author contributions
S.Y., X.Q., and Y.G. designed and conceived the experiments. Y.G., X.O., and Y.S. performed the experiments. Y.G., S.Y., X.O., L.Z., Y.X., X.Q., and C.C. contributed to the acquisition, analysis, and interpretation of data. Y.G., S.Y., Y.S., and C.C. wrote the paper. All authors read and approved the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2021.06.017.
Contributor Information
Xihu Qin, Email: czeyxh@njmu.edu.cn.
Shuyuan Yeh, Email: shuyuan_yeh@urmc.rochester.edu.
Supplemental information
References
- 1.Razumilava N., Gores G.J. Cholangiocarcinoma. Lancet. 2014;383:2168–2179. doi: 10.1016/S0140-6736(13)61903-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khan S.A., Thomas H.C., Davidson B.R., Taylor-Robinson S.D. Cholangiocarcinoma. Lancet. 2005;366:1303–1314. doi: 10.1016/S0140-6736(05)67530-7. [DOI] [PubMed] [Google Scholar]
- 3.Watanapa P. Cholangiocarcinoma in patients with opisthorchiasis. Br. J. Surg. 1996;83:1062–1064. doi: 10.1002/bjs.1800830809. [DOI] [PubMed] [Google Scholar]
- 4.Dreyer C., Le Tourneau C., Faivre S., Qian Z., Degos F., Vuillerme M.P., Paradis V., Hammel P., Ruszniewski P., Cortes A. [Cholangiocarcinoma: epidemiology and global management] Rev. Med. Interne. 2008;29:642–651. doi: 10.1016/j.revmed.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 5.El-Diwany R., Pawlik T.M., Ejaz A. Intrahepatic cholangiocarcinoma. Surg. Oncol. Clin. N. Am. 2019;28:587–599. doi: 10.1016/j.soc.2019.06.002. [DOI] [PubMed] [Google Scholar]
- 6.Rizvi S., Gores G.J. Pathogenesis, diagnosis, and management of cholangiocarcinoma. Gastroenterology. 2013;145:1215–1229. doi: 10.1053/j.gastro.2013.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bertuccio P., Malvezzi M., Carioli G., Hashim D., Boffetta P., El-Serag H.B., La Vecchia C., Negri E. Global trends in mortality from intrahepatic and extrahepatic cholangiocarcinoma. J. Hepatol. 2019;71:104–114. doi: 10.1016/j.jhep.2019.03.013. [DOI] [PubMed] [Google Scholar]
- 8.Khan S.A., Tavolari S., Brandi G. Cholangiocarcinoma: Epidemiology and risk factors. Liver Int. 2019;39(Suppl 1):19–31. doi: 10.1111/liv.14095. [DOI] [PubMed] [Google Scholar]
- 9.Vogelstein B., Papadopoulos N., Velculescu V.E., Zhou S., Diaz L.A., Jr., Kinzler K.W. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zabron A., Edwards R.J., Khan S.A. The challenge of cholangiocarcinoma: Dissecting the molecular mechanisms of an insidious cancer. Dis. Model. Mech. 2013;6:281–292. doi: 10.1242/dmm.010561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dang L., Su S.M. Isocitrate dehydrogenase mutation and (R)-2-hydroxyglutarate: From basic discovery to therapeutics development. Annu. Rev. Biochem. 2017;86:305–331. doi: 10.1146/annurev-biochem-061516-044732. [DOI] [PubMed] [Google Scholar]
- 12.Peraldo-Neia C., Ostano P., Cavalloni G., Pignochino Y., Sangiolo D., De Cecco L., Marchesi E., Ribero D., Scarpa A., De Rose A.M. Transcriptomic analysis and mutational status of IDH1 in paired primary-recurrent intrahepatic cholangiocarcinoma. BMC Genomics. 2018;19:440. doi: 10.1186/s12864-018-4829-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saha S.K., Parachoniak C.A., Ghanta K.S., Fitamant J., Ross K.N., Najem M.S., Gurumurthy S., Akbay E.A., Sia D., Cornella H. Mutant IDH inhibits HNF-4α to block hepatocyte differentiation and promote biliary cancer. Nature. 2014;513:110–114. doi: 10.1038/nature13441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saha S.K., Parachoniak C.A., Bardeesy N. IDH mutations in liver cell plasticity and biliary cancer. Cell Cycle. 2014;13:3176–3182. doi: 10.4161/15384101.2014.965054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Losman J.A., Kaelin W.G., Jr. What a difference a hydroxyl makes: Mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013;27:836–852. doi: 10.1101/gad.217406.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dang L., White D.W., Gross S., Bennett B.D., Bittinger M.A., Driggers E.M., Fantin V.R., Jang H.G., Jin S., Keenan M.C. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Figueroa M.E., Abdel-Wahab O., Lu C., Ward P.S., Patel J., Shih A., Li Y., Bhagwat N., Vasanthakumar A., Fernandez H.F. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu W., Yang H., Liu Y., Yang Y., Wang P., Kim S.H., Ito S., Yang C., Wang P., Xiao M.T. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kipp B.R., Voss J.S., Kerr S.E., Barr Fritcher E.G., Graham R.P., Zhang L., Highsmith W.E., Zhang J., Roberts L.R., Gores G.J., Halling K.C. Isocitrate dehydrogenase 1 and 2 mutations in cholangiocarcinoma. Hum. Pathol. 2012;43:1552–1558. doi: 10.1016/j.humpath.2011.12.007. [DOI] [PubMed] [Google Scholar]
- 20.Wang P., Dong Q., Zhang C., Kuan P.F., Liu Y., Jeck W.R., Andersen J.B., Jiang W., Savich G.L., Tan T.X. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene. 2013;32:3091–3100. doi: 10.1038/onc.2012.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yager J.D., Davidson N.E. Estrogen carcinogenesis in breast cancer. N. Engl. J. Med. 2006;354:270–282. doi: 10.1056/NEJMra050776. [DOI] [PubMed] [Google Scholar]
- 22.Richardson G.S. Endometrial cancer as an estrogen-progesterone target. N. Engl. J. Med. 1972;286:645–647. doi: 10.1056/NEJM197203232861207. [DOI] [PubMed] [Google Scholar]
- 23.Brown S.B., Hankinson S.E. Endogenous estrogens and the risk of breast, endometrial, and ovarian cancers. Steroids. 2015;99(Pt A):8–10. doi: 10.1016/j.steroids.2014.12.013. [DOI] [PubMed] [Google Scholar]
- 24.Alvaro D., Mancino M.G., Onori P., Franchitto A., Alpini G., Francis H., Glaser S., Gaudio E. Estrogens and the pathophysiology of the biliary tree. World J. Gastroenterol. 2006;12:3537–3545. doi: 10.3748/wjg.v12.i22.3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gershwin M.E., Selmi C., Worman H.J., Gold E.B., Watnik M., Utts J., Lindor K.D., Kaplan M.M., Vierling J.M., USA PBC Epidemiology Group Risk factors and comorbidities in primary biliary cirrhosis: A controlled interview-based study of 1032 patients. Hepatology. 2005;42:1194–1202. doi: 10.1002/hep.20907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Isse K., Specht S.M., Lunz J.G., 3rd, Kang L.I., Mizuguchi Y., Demetris A.J. Estrogen stimulates female biliary epithelial cell interleukin-6 expression in mice and humans. Hepatology. 2010;51:869–880. doi: 10.1002/hep.23386. [DOI] [PubMed] [Google Scholar]
- 27.Wu H., Liu Y., Jiang X.W., Li W.F., Guo G., Gong J.P., Ding X. Clinicopathological and prognostic significance of Yes-associated protein expression in hepatocellular carcinoma and hepatic cholangiocarcinoma. Tumour Biol. 2016;37:13499–13508. doi: 10.1007/s13277-016-5211-y. [DOI] [PubMed] [Google Scholar]
- 28.Zhao B., Li L., Lei Q., Guan K.L. The Hippo-YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev. 2010;24:862–874. doi: 10.1101/gad.1909210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sugihara T., Werneburg N.W., Hernandez M.C., Yang L., Kabashima A., Hirsova P., Yohanathan L., Sosa C., Truty M.J., Vasmatzis G. YAP tyrosine phosphorylation and nuclear localization in cholangiocarcinoma cells are regulated by LCK and independent of LATS activity. Mol. Cancer Res. 2018;16:1556–1567. doi: 10.1158/1541-7786.MCR-18-0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rosenbluh J., Nijhawan D., Cox A.G., Li X., Neal J.T., Schafer E.J., Zack T.I., Wang X., Tsherniak A., Schinzel A.C. β-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell. 2012;151:1457–1473. doi: 10.1016/j.cell.2012.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cao X., Pfaff S.L., Gage F.H. YAP regulates neural progenitor cell number via the TEA domain transcription factor. Genes Dev. 2008;22:3320–3334. doi: 10.1101/gad.1726608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sugimachi K., Nishio M., Aishima S., Kuroda Y., Iguchi T., Komatsu H., Hirata H., Sakimura S., Eguchi H., Bekki Y. Altered expression of Hippo signaling pathway molecules in intrahepatic cholangiocarcinoma. Oncology. 2017;93:67–74. doi: 10.1159/000463390. [DOI] [PubMed] [Google Scholar]
- 33.Mancino A., Mancino M.G., Glaser S.S., Alpini G., Bolognese A., Izzo L., Francis H., Onori P., Franchitto A., Ginanni-Corradini S. Estrogens stimulate the proliferation of human cholangiocarcinoma by inducing the expression and secretion of vascular endothelial growth factor. Dig. Liver Dis. 2009;41:156–163. doi: 10.1016/j.dld.2008.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hunsawong T., Singsuksawat E., In-chon N., Chawengrattanachot W., Thuwajit C., Sripa B., Paupairoj A., Chau-in S., Thuwajit P. Estrogen is increased in male cholangiocarcinoma patients’ serum and stimulates invasion in cholangiocarcinoma cell lines in vitro. J. Cancer Res. Clin. Oncol. 2012;138:1311–1320. doi: 10.1007/s00432-012-1207-1. [DOI] [PubMed] [Google Scholar]
- 35.Iorio M.V., Croce C.M. MicroRNA involvement in human cancer. Carcinogenesis. 2012;33:1126–1133. doi: 10.1093/carcin/bgs140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin S., Gregory R.I. MicroRNA biogenesis pathways in cancer. Nat. Rev. Cancer. 2015;15:321–333. doi: 10.1038/nrc3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ennajdaoui H., Howard J.M., Sterne-Weiler T., Jahanbani F., Coyne D.J., Uren P.J., Dargyte M., Katzman S., Draper J.M., Wallace A. IGF2BP3 modulates the interaction of invasion-associated transcripts with RISC. Cell Rep. 2016;15:1876–1883. doi: 10.1016/j.celrep.2016.04.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hutvagner G., Simard M.J. Argonaute proteins: Key players in RNA silencing. Nat. Rev. Mol. Cell Biol. 2008;9:22–32. doi: 10.1038/nrm2321. [DOI] [PubMed] [Google Scholar]
- 39.Filipowicz W., Jaskiewicz L., Kolb F.A., Pillai R.S. Post-transcriptional gene silencing by siRNAs and miRNAs. Curr. Opin. Struct. Biol. 2005;15:331–341. doi: 10.1016/j.sbi.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 40.Miyoshi K., Tsukumo H., Nagami T., Siomi H., Siomi M.C. Slicer function of Drosophila Argonautes and its involvement in RISC formation. Genes Dev. 2005;19:2837–2848. doi: 10.1101/gad.1370605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Both G.W., Moyer S.A., Banerjee A.K. Translation and identification of the mRNA species synthesized in vitro by the virion-associated RNA polymerase of vesicular stomatitis virus. Proc. Natl. Acad. Sci. USA. 1975;72:274–278. doi: 10.1073/pnas.72.1.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao B.S., Roundtree I.A., He C. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 2017;18:31–42. doi: 10.1038/nrm.2016.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Su R., Dong L., Li C., Nachtergaele S., Wunderlich M., Qing Y., Deng X., Wang Y., Weng X., Hu C. R-2HG exhibits anti-tumor activity by targeting FTO/m6A/MYC/CEBPA signaling. Cell. 2018;172:90–105.e23. doi: 10.1016/j.cell.2017.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao X., Yang Y., Sun B.F., Shi Y., Yang X., Xiao W., Hao Y.J., Ping X.L., Chen Y.S., Wang W.J. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 2014;24:1403–1419. doi: 10.1038/cr.2014.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dominissini D., Moshitch-Moshkovitz S., Schwartz S., Salmon-Divon M., Ungar L., Osenberg S., Cesarkas K., Jacob-Hirsch J., Amariglio N., Kupiec M. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–206. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- 46.Meyer K.D., Saletore Y., Zumbo P., Elemento O., Mason C.E., Jaffrey S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149:1635–1646. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang X., Lu Z., Gomez A., Hon G.C., Yue Y., Han D., Fu Y., Parisien M., Dai Q., Jia G. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117–120. doi: 10.1038/nature12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Losman J.A., Looper R.E., Koivunen P., Lee S., Schneider R.K., McMahon C., Cowley G.S., Root D.E., Ebert B.L., Kaelin W.G., Jr. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science. 2013;339:1621–1625. doi: 10.1126/science.1231677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Qi S.T., Ma J.Y., Wang Z.B., Guo L., Hou Y., Sun Q.Y. N6-methyladenosine sequencing highlights the involvement of mRNA methylation in oocyte meiotic maturation and embryo development by regulating translation in Xenopus laevis. J. Biol. Chem. 2016;291:23020–23026. doi: 10.1074/jbc.M116.748889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Slobodin B., Han R., Calderone V., Vrielink J.A.F.O., Loayza-Puch F., Elkon R., Agami R. Transcription impacts the efficiency of mRNA translation via co-transcriptional N6-adenosine methylation. Cell. 2017;169:326–337.e12. doi: 10.1016/j.cell.2017.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sritana N., Suriyo T., Kanitwithayanun J., Songvasin B.H., Thiantanawat A., Satayavivad J. Glyphosate induces growth of estrogen receptor alpha positive cholangiocarcinoma cells via non-genomic estrogen receptor/ERK1/2 signaling pathway. Food Chem. Toxicol. 2018;118:595–607. doi: 10.1016/j.fct.2018.06.014. [DOI] [PubMed] [Google Scholar]
- 52.Alvaro D., Barbaro B., Franchitto A., Onori P., Glaser S.S., Alpini G., Francis H., Marucci L., Sterpetti P., Ginanni-Corradini S. Estrogens and insulin-like growth factor 1 modulate neoplastic cell growth in human cholangiocarcinoma. Am. J. Pathol. 2006;169:877–888. doi: 10.2353/ajpath.2006.050464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maximov V.V., Akkawi R., Khawaled S., Salah Z., Jaber L., Barhoum A., Or O., Galasso M., Kurek K.C., Yavin E., Aqeilan R.I. miR-16-1-3p and miR-16-2-3p possess strong tumor suppressive and antimetastatic properties in osteosarcoma. Int. J. Cancer. 2019;145:3052–3063. doi: 10.1002/ijc.32368. [DOI] [PubMed] [Google Scholar]
- 54.Liu G.P., Wang W.W., Lu W.Y., Shang A.Q. The mechanism of miR-16-5p protection on LPS-induced A549 cell injury by targeting CXCR3. Artif. Cells Nanomed. Biotechnol. 2019;47:1200–1206. doi: 10.1080/21691401.2019.1593998. [DOI] [PubMed] [Google Scholar]
- 55.Marti P., Stein C., Blumer T., Abraham Y., Dill M.T., Pikiolek M., Orsini V., Jurisic G., Megel P., Makowska Z. YAP promotes proliferation, chemoresistance, and angiogenesis in human cholangiocarcinoma through TEAD transcription factors. Hepatology. 2015;62:1497–1510. doi: 10.1002/hep.27992. [DOI] [PubMed] [Google Scholar]
- 56.Sugihara T., Isomoto H., Gores G., Smoot R. YAP and the Hippo pathway in cholangiocarcinoma. J. Gastroenterol. 2019;54:485–491. doi: 10.1007/s00535-019-01563-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hsu P.C., Jablons D.M., Yang C.T., You L. Epidermal growth factor receptor (EGFR) pathway, Yes-associated protein (YAP) and the regulation of programmed death-ligand 1 (PD-L1) in non-small cell lung cancer (NSCLC) Int. J. Mol. Sci. 2019;20:3821. doi: 10.3390/ijms20153821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wei S., Wang J., Oyinlade O., Ma D., Wang S., Kratz L., Lal B., Xu Q., Liu S., Shah S.R. Heterozygous IDH1R132H/WT created by “single base editing” inhibits human astroglial cell growth by downregulating YAP. Oncogene. 2018;37:5160–5174. doi: 10.1038/s41388-018-0334-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 60.Wang Y., Jiang W., Li C., Xiong X., Guo H., Tian Q., Li X. Autophagy suppression accelerates apoptosis induced by norcantharidin in cholangiocarcinoma. Pathol. Oncol. Res. 2020;26:1697–1707. doi: 10.1007/s12253-019-00719-9. [DOI] [PubMed] [Google Scholar]
- 61.Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.