

Summary
The release of excess glutamate following traumatic brain injury (TBI) results in glutamate excitotoxicity and metabolic energy failure. Endogenous mechanisms for reducing glutamate concentration in the brain parenchyma following TBI are poorly understood. Using multiple mass spectrometry approaches, we examined TBI-induced changes to glutamate metabolism. We present evidence that glutamate concentration can be reduced by glutamate oxidation via a “truncated” tricarboxylic acid cycle coupled to the urea cycle. This process reduces glutamate levels, generates carbon for energy metabolism, leads to citrulline accumulation, and produces nitric oxide. Several key metabolites are identified by metabolomics in support of this mechanism and the locations of these metabolites in the injured hemisphere are demonstrated by MALDI-MS imaging. The results of this study establish the advantages of multiple mass spectrometry approaches and provide insights into glutamate metabolism following TBI that could lead to improved treatment approaches.
Subject areas: Molecular physiology, Neuroscience, Metabolomics
Graphical abstract
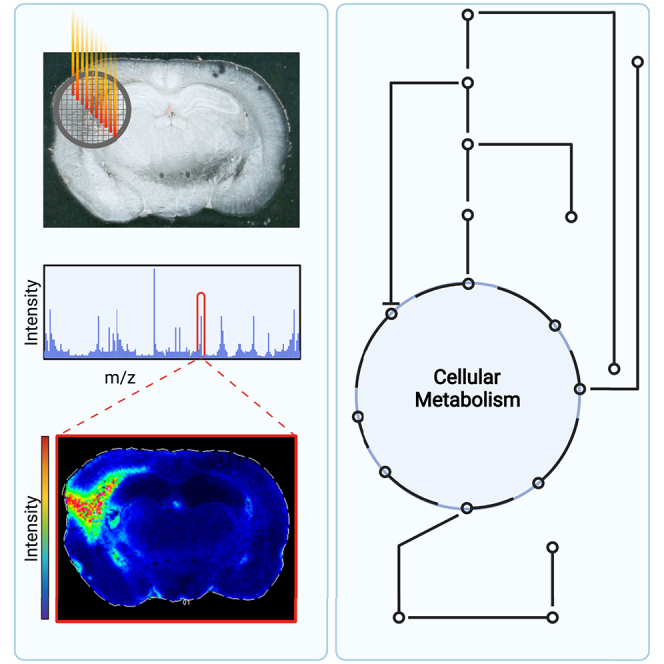
Highlights
-
•
Integration of mass spectrometry methods revealed unrecognized metabolic alterations
-
•
MALDI-MS imaging provides a region-specific view of metabolism changes after TBI
-
•
Glutamate metabolism is altered after TBI, suggesting future treatment avenues
-
•
3-phosphoserine and citrulline are new potential metabolic biomarkers for TBI
Molecular physiology; Neuroscience; Metabolomics
Introduction
Traumatic brain injury (TBI) is a growing public health issue worldwide (Maas et al., 2017). Complications from TBI can be immediately profound and result in lifelong disability. Prior studies have described a multitude of molecular events in TBI including oxidative and inflammation-mediated damage thought to result from glutamate excitotoxicity as well as altered cerebral blood flow (Mark et al., 2001; Yi and Hazell, 2006). Nevertheless, current management strategies are modestly effective, and treatment options are limited (Ginsberg, 2008; Ikonomidou and Turski, 2002). Further studies, which might reveal new treatment approaches, are therefore warranted.
Under normal physiologic conditions, glutamate concentrations are tightly regulated within the synaptic cleft by presynaptic and postsynaptic neurons as well as astrocytes (Danbolt, 2001; Levy et al., 1998; Zerangue and Kavanaugh, 1996). Astrocytes uptake most of the glutamate released into the synaptic cleft. Once glutamate is transported into a cell, it can be metabolized to glutamine, α-ketoglutarate (α-KG) or γ-aminobutyric acid (GABA), depending on the cell type. In astrocytes, glutamate is predominantly metabolized to glutamine and then released into the extracellular space to be taken in by neurons, where it is converted back to glutamate. This circuit is known as the glutamate-glutamine cycle. However, glutamate within astrocytes can also be oxidized to α-KG, part of the citric acid cycle (TCA cycle), which is used to generate high energy intermediates (NADH, FADH2) and GTP that drive the electron transport chain and oxidative phosphorylation. The conversion of glutamate to α-KG in astrocytes is believed to occur to balance the energy consumed by astrocytes for maintaining sodium and potassium pumps necessary for importing extracellular glutamate (Oz et al., 2012).
TBI results in stretching and tearing of fibers within the brain, resulting in an uncontrolled release of glutamate into the brain parenchyma and an increase in extracellular glutamate concentrations (Chamoun et al., 2010; Katayama et al., 1990; Mark et al., 2001; Palmer et al., 1993; Yi and Hazell, 2006). Released glutamate can bind to glutamate receptors, triggering Ca2+ influx, potentially inducing apoptosis in neurons in a process referred to as glutamate excitotoxicity (Chamoun et al., 2010; Katayama et al., 1990; Mark et al., 2001; Palmer et al., 1993; Yi and Hazell, 2006). The resulting ionic imbalances require activation of ATP-dependent ion pumps and increased glucose and oxygen demand to re-establish equilibrium. Increased demand on mitochondria can result in the release of reactive oxygen species, contributing to oxidative damage within the brain tissue (Giza and Hovda, 2014). Collectively, these metabolic events result in the death of neurons and other cells. Ultimately, the cascade of tissue damage may lead to diminished cognitive function.
Glutamate oxidation is a process by which glutamate is metabolized to α-KG and then cycled through the TCA cycle to ultimately generate more ATP (Deshmukh and Patel, 1980; McKenna, 2012; McKenna et al., 2002, 2016). McKenna et al. found that under normal homeostatic conditions, glutamate is primarily converted to glutamine rather than to α-KG (McKenna et al., 2002). However, as extracellular levels of glutamate increase, the balance can switch toward formation of α-KG (McKenna et al., 2002). Through glutamate oxidation, glutamate concentrations are reduced and provide a carbon source necessary for generating cellular energy to serve as an alternative energy source rather than glucose/pyruvate derived carbons.
Herein, we obtained brain tissues from rodents subjected to TBI using a fluid-percussion injury model to study perturbations in glutamate metabolism (Andersen et al., 2021; Wu et al., 2013). Tissues from one cohort of animals were used for the MALDI-imaging studies and metabolomics, whereas those from another cohort were used for proteomic studies. Changes in glutamate metabolism were measured at two time points, 24 h (24 h) and 2 weeks, following injury. We find that 24 h after TBI, excess glutamate is metabolized within a defined region of the injured hemisphere. Furthermore, we observed that TCA metabolites derived from glutamate enter the urea cycle resulting in the accumulation of citrulline, suggesting activation of nitric oxide synthases and production of nitric oxide. We propose glutamate oxidation under pathologic conditions may serve as an endogenous defense mechanism against glutamate excitotoxicity and/or energy depletion.
Results
Proteomics analysis reveals no consistent changes in glutamate metabolic enzymes in the damaged hemisphere
Prior studies (Deshmukh and Patel, 1980; McKenna, 2012; McKenna et al., 2002, 2016) have suggested that the neurotoxic effects of excess glutamate release following TBI could be attenuated by metabolizing glutamate to molecules that are not neurotransmitters. Glutamate can be enzymatically decarboxylated forming the inhibitory neurotransmitter GABA, aminated forming glutamine, or the glutamate amino group can be transferred to a series of metabolites forming other amino acids as shown in Figure 1.
Figure 1.

Levels of glutamate metabolic proteins are not uniformly increased after TBI
(A) Enzymes and metabolites involved in glutamate handling. Glutamate can be decarboxylated to form GABA, aminated to glutamine or transaminated to form alpha-ketoglutarate. Under homeostatic conditions in the brain, the conversion of glutamate to glutamine is the predominant reaction in astrocytes (bold arrow).
(B and C) The protein fold change ratios for TBI/Sham animals, derived from the cortex or hippocampus, from a parallel proteomics study at (B) 24 h and (C) 2 weeks for enzymes related to glutamate metabolism. Each data point represents a value from a different animal (4 animals in total). Red symbols represent statistically significant fold changes for that protein. Black symbols represent proteins that were confidently identified but did not have a statistically significant fold change. Gfap is shown to illustrate that the animals were injured and that the brain has elicited a consistent response to TBI among the majority of animals. Overall, no consistent change was observed for glutamate metabolizing enzymes at 24 h or 2 weeks in the cortex and hippocampus. In the cortex at 24 h, Glutamate decarboxylase 2 (GAD2) was increased in one animal. Glutamine synthase (Glns) was significantly increased in two of four injured animals, and glutamate dehydrogenase (Gdh) was decreased significantly in two. Aspartate aminotransferase 1 (Got1) was increased in one animal and decreased in another. The branched chain aminotransferase 1 (Bcat1) and 4-aminobutyrate aminotransferase (Abat1) were both reduced in animal 145. Glutamate decarboxylase 1 (GAD1), Aspartate aminotransferase 2 (Got2) and Phosphoserine aminotransferase (Psat1) were all unchanged. In the hippocampus at 24 h, GAD1 and GAD2 were both increased in an animal. Glns was increased in one animal and decreased in another. Got2 and Abat1 were each decreased in an animal. Gdh, Got2, Bcat1 and Psat1 were unchanged. At 2 weeks in the cortex, GAD2 and Glns were each increased in an animal and Gdh was decreased in one animal. At 2 weeks in the hippocampus, the only change observed was to Glns in one animal.
As part of a longitudinal proteomics study of TBI induced by unilateral fluid percussion injury, proteins were extracted from the injured hemisphere of a series of four injured animals and from sham injured animals. Proteins were identified and quantified by LC-MS/MS and the fold-changes between injured and sham animals were determined for confidently identified proteins. Glial fibrillary acidic protein (Gfap), a non-enzymatic type II neurofilament expressed in the central nervous system, is considered a marker of brain injury. Gfap levels were observed to be increased in all the injured animals at both 24 h and two weeks, confirming the injury. Enzymes involved in the metabolism of glutamate shown in Figure 1 were also identified in animals at 24 h and two weeks following injury. Surprisingly, no consistent elevation in any of the glutamate-metabolizing enzymes was seen for a majority of the injured animals.
High-resolution liquid-chromatography-mass spectrometry (LC-MS/MS) identifies metabolites which increase in the injured hemisphere
To further probe markers of brain injury following TBI, the damaged and contralateral hemispheres were separated, and small molecule metabolites were extracted and analyzed. Small molecules were converted to their dansylated derivatives, a method particularly valuable for the analysis of amine-containing compounds including amino acids and analyzed by LC-MS/MS (Tables 1 and 2). Metabolites were identified based upon characteristic LC retention times and high-resolution mass and fragmentation patterns of the dansylated derivatives. The high mass accuracy of the dansylated derivatization is shown in Table S1.
Table 1.
Dansylated amino acid derivatives measured in TBI brains 24 h after TBI
| Metabolite | Average concentration injured hemisphere (pmole/mg tissue) | Average concentration contralateral hemisphere (pmole/mg tissue) | Difference in metabolite concentration | Fold change | p value |
|---|---|---|---|---|---|
| Aspartate | 1.11 E04 | 4.68 E03 | 6.42 E03 | 2.37 | 3.60E-12 |
| Ornithine | 7.48 E01 | 3.63 E01 | 3.85 E01 | 2.06 | 5.57E-05 |
| Glutamate | 1.19 E04 | 6.23 E03 | 5.72 E03 | 1.92 | 3.39E-12 |
| Citrulline | 3.07 E01 | 1.83 E01 | 1.24 E01 | 1.68 | 1.43E-09 |
| Glutamine | 3.02 E03 | 2.36 E03 | 6.61 E02 | 1.28 | 1.56E-06 |
| Serine | 1.47 E03 | 1.22 E03 | 2.55 E02 | 1.21 | 5.29E-09 |
| GABA | 1.22 E03 | 1.02 E03 | 2.03 E02 | 1.2 | 2.50E-02 |
| Isoleucine | 9.17 E01 | 7.77 E01 | 1.40 E01 | 1.18 | 2.43E-01 |
| Asparagine | 1.21 E02 | 1.08 E02 | 1.29 E01 | 1.12 | 1.40E-02 |
| Alanine | 2.30 E02 | 2.11 E02 | 1.90 E01 | 1.09 | 1.12E-01 |
| Valine | 1.04 E02 | 9.52 E01 | 8.57 E00 | 1.09 | 5.97E-01 |
| Leucine | 6.07 E01 | 6.13 E01 | -6.10 E-01 | 0.99 | 6.08E-01 |
| Arginine | 2.74 E01 | 3.19 E01 | -4.46 E00 | 0.86 | 1.63E-01 |
| Phosphoserine | 3.16 E00 | N/A | 3.16 E00 | - | - |
Table 2.
Dansylated Energy and Oxidative damage metabolites measured in TBI Brains 24 hours after TBI
| Metabolite | Average concentration injured hemisphere (pmole/mg tissue) | Average concentration contralateral hemisphere (pmole/mg tissue) | Difference in metabolite concentration | Fold change | p-value |
|---|---|---|---|---|---|
| GMP | 4.28E+02 | 1.13E+01 | 4.17E+02 | 3.81E+01 | 3.18E-09 |
| AMP | 3.09E+03 | 3.32E+02 | 2.76E+03 | 9.31E+00 | 3.73E-09 |
| cGMP | 5.80E-01 | 1.80E-01 | 4.00E-01 | 3.21E+00 | 1.70E-02 |
| Nitro-tyrosine | 2.14E+02 | 1.82E+02 | 3.28E+01 | 1.18E+00 | 1.90E-02 |
Glutamate levels were observed to be significantly elevated in the damaged hemisphere relative to the contralateral hemisphere. In addition, levels of the amino acids aspartate, ornithine, glutamine, citrulline, serine, asparagine, and GABA were significantly higher in the damaged hemisphere. Another product of glutamine transamination, phosphoserine (pSer) was observed only in the damaged hemisphere. The levels of isoleucine, alanine, valine, leucine, and arginine were not significantly different between hemispheres (Table 1). These measurements are consistent with increased flux through some, but not all the pathways shown in Figure 1.
The levels of some additional biomolecules were observed to change as well between injured and uninjured hemispheres (Table 2). Levels of the nucleotides GMP, AMP, and cGMP increased significantly in the damaged hemisphere. The level of 3-nitrotyrosine, a nonenzymatic product formed from tyrosine by reaction with nitric oxide, was also increased significantly.
Metabolites and their anatomic distribution can be revealed by using MALDI-MS imaging
To investigate TBI damage at the molecular level within the context of brain anatomy, brain tissues from injured and sham animals were sectioned and analyzed by MALDI-MS imaging. With this method, a series of MALDI mass spectra are obtained across the brain tissue. Regions of increased or diminished metabolites are indicated by constructing images at selected mass to charge ratios (m/z) and mapping its intensity.
Reduced levels of N-acetylaspartate demarcate the lesion site
We first examined the images for N-acetylaspartate (NAA). NAA levels are known to fall after TBI and reduced levels are considered to be a good marker of damage and metabolic energy status (Moffett et al., 2013; Croall et al., 2015). The mass to charge ratio (m/z) for NAA is 174. Images from brain sections corresponding to this ion are shown in Figure 2. A photographic image of this brain section is also shown in Figure 2, overlaid with labels demarcating anatomically defined brain regions. In the sham animal, ion intensities indicated by a color scale are symmetric in both hemispheres. However, in the injured animal 24 h following injury, a dark area of reduced NAA is observed in the damaged hemisphere. The region of greatest apparent damage is demarcated by the low NAA/black area encompassing a portion of the neocortex, the caudoputamen, corpus callosum and fimbria and will be referred to as the lesion site.
Figure 2.

N-acetylaspartate depletion occurs in the injured hemisphere of the rat brain 24 h and 2 weeks after TBI
MALDI-MS images for N-acetylaspartate (NAA, m/z (M-H) = 174.02) in (A) sham, (B) TBI at 24 h, and (C) TBI at 2 weeks.
(A) The green areas represent areas of elevated NAA in portions of the neocortex, thalamus, hypothalamus, and amygdala. The sham animal has a symmetric distribution between hemispheres.
(B) 24 h post injury, there is a reduction of NAA in the injured (left) and contralateral (right) neocortex. There is a dark region which represents the lesion site in the injured hemisphere and an area of little to no NAA. The thalamic and hypothalamic levels of NAA seem to be unaffected by TBI at the 24 h time point.
(C) 2 weeks post injury there is much more symmetry between the injured and contralateral hemisphere. However, there is still a small observed region in the neocortex and caudoputamen where NAA is reduced. (A–C) A representative scale bar of 5 mm is shown in white on the right hand-side mid-figure. Data are shown using a rainbow scale, normalized against total ion current.
(D and E) (D) The scanned image of the TBI brain and (E) overlaid with labels for general regions and the lesion site (LS) comprised of portions of the neocortex (CTX), caudoputamen (CP), corpus callosum (CC), and fimbria (Fi). (D and E) A representative scale bar of 5 mm is shown in white on the lower right-hand corner.
Although the lesion site is readily apparent at 24 h, the image obtained from an animal 2 weeks after injury has largely returned to normal. Diminished NAA at the lesion site and reduced NAA in the damaged hemisphere are observed in the images of the other injured animals in this series at 24 h, with recovery by two weeks as shown in Figures S1 and S2.
Enhanced conversion of glutamate to glutamine and pSer is observed at the lesion site
We next examined glutamate and glutamate metabolites identified by LC-MS/MS studies in the images of the injured animal at 24 h. Glutamate (m/z 146) is increased in the neocortex of the damaged hemisphere but appears to be decreased in the most damaged region defined by low NAA. The area of interest is indicated with a white dashed circle in Figure 3. Potential metabolites of glutamate include glutamine and α-ketoglutarate (α-KG) (m/z 145) which appear to be increased in the lesion site.
Figure 3.

Levels of glutamate metabolites increase in the injured hemisphere 24 h after TBI
(A–C) The coronal tissue section displays ions that correspond to (A) glutamate, and related metabolites, (B) glutamine and (C) pSer, 24 h after TBI. Higher levels of glutamate were observed in the injured cortex and thalamus in the TBI brain. However, within the ipsilateral hemisphere, glutamate was noticeably reduced from parts of the cortex, fimbria, caudate putamen, and corpus callosum (referred to as the lesion site). The region corresponding to decreased glutamate in the cortex overlaps with the regions where glutamine and 3-phosphoserine (pSer) accumulate in the cortex. (A–C) A representative scale bar of 5 mm is shown in white on the lower left-hand corner.
(D and E) (D) pSer is a novel metabolite within the context of TBI and (E) was validated by high-resolution mass spectrometry.
(F) pSer was only measured in the ipsilateral hemisphere and was significantly increased compared to the contralateral hemisphere. Glns, glutamine synthetase; Psat1, phosphoserine aminotransferase 1. ∗∗∗∗p ≤ 0.001.
In MALDI-MS images obtained with a low-resolution instrument, metabolites with similar mass such as glutamine and α-KG, which differ in mass by less than 0.05 mass units, cannot be distinguished. We therefore obtained images from another injured animal in the series at 24 h with a higher mass resolution instrument. Both the low-resolution and high-resolution images of this animal are shown in Figure S3. The higher-intensity metabolite at the lesion site, identified as either glutamine or α-ketoglutarate in the low-resolution image, is consistent with glutamine in the high-resolution image. Indeed, α-KG appears to be diminished at the lesion site.
In the high mass-resolution LC-MS/MS study described previously, pSer, a metabolite in serine biosynthesis, was significantly increased in the damaged hemisphere but was unobservable in the contralateral hemisphere. Consistent with this finding, we observe enhanced pSer (m/z 184) in the MALDI-MS images in the damaged, but not contralateral hemisphere in a region colocalizing with the lesion site (Figure 3). By two weeks, glutamine and pSer levels at the lesion site have decreased substantially but are still apparent (Figure S4).
Biological replicates reveal similar patterns of increased accumulation of pSer in the injured hemisphere 24 h after injury, but not in the sham animals (Figure S5). The levels of pSer decline substantially by two weeks following injury (Figure S6). These data confirm the reproducibility of the MALDI-imaging method and its utility in establishing the anatomic location of selected metabolites.
Changes in the levels of additional amino acids and their metabolites are observed by MALDI-MS imaging at the lesion site
Metabolites with altered intensity in and around the lesion site include aspartate, leucine/isoleucine, valine, pyruvate, and oxalate or lactate. As shown in Figure 4, the levels of aspartate and the branched chain amino acids leucine/isoleucine and valine as well as pyruvate are diminished in the lesion site whereas levels of oxalate or lactate are increased.
Figure 4.

Additional altered metabolites 24 h after TBI
Within the lesion site three additional metabolites that participate in transamination reactions with glutamate were observed to be decreased in the lesion site 24 h after injury (A) aspartate (Asp), (B) leucine (Leu), isoleucine (Ile), (C) valine (Val).
(D and E)(D) Pyruvate and (E) lactate were elevated in the injured hemisphere relative to the contralateral hemisphere, especially in the neocortex extending toward the amygdala. Lactate was most increased in the lesion site, elevated in portions of the ipsilateral thalamus, and bilaterally in the hypothalamus relative to the rest of the brain. A representative scale bar of 5 mm for the figure is shown in white on the lower left-hand corner.
The ions attributed to the metabolites lactate and oxalate have similar mass which differ by less than 0.04 mass units, and therefore cannot be distinguished using a low-resolution instrument. We therefore obtained images at higher mass resolution as shown in Figure S7. Oxalate and lactate can be distinguished with higher mass resolution, and both appear to be present at the lesion site. However, the increased intensity observed in the low-resolution image at the lesion site (Figure 4) is more consistent with the anatomic distribution of lactate (Figure S7). In Table S2, we present a list of metabolites identified with the high-resolution instrument.
An unanticipated increased level of citrulline is observed at the lesion site
Unlike several of the metabolites described above, citrulline is not a direct metabolite of glutamate. Citrulline was, however, identified as its dansylated derivative by high resolution LC-MS/MS with significantly increased levels in the damaged hemisphere. The MALDI-MS image of m/z 176 corresponding to citrulline is shown in Figure 6 and reveals increased intensity at the lesion site. Increased citrulline at the lesion site was also observed in the two additional injured animals in this series (Figure S8). In images obtained from three injured animals at two weeks, citrulline levels have decreased at the lesion site but are still apparent in some animals (Figure S9).
Figure 6.

two-way ANOVA quantification of metabolites from MALDI-MS imaging data
Quantification of metabolites identified by MALDI-MS imaging in the lesion site compared to (A) the contralateral hemisphere of the TBI brain (TBI-con) or (B) the sham animal. (A/B) metabolites increased relative to TBI-con or sham include 3-phosphoserine, citrulline, and lactate. Metabolites decreased in the lesion site relative to TBI-con include NAA, leucine, isoleucine, and valine. Metabolites decreased in the lesion site relative to sham include aspartate, NAA, leucine, isoleucine, and valine. Glutamine, asparagine, pyruvate, glutamate, GABA and fumarate remained unchanged in the lesion site relative to TBI-con and sham. Error bars represent the standard deviation of n = 3 biological replicates. ∗q-value < 0.1, ∗∗< 0.01.
Quantification of changes in metabolite levels in the damaged region
Although MALDI-MS imaging provides an unprecedented view of metabolic changes resulting from TBI with anatomic resolution, quantification of the magnitude of the changes is not provided directly by the MALDI images. To be more quantitative, we developed an approach common to imaging modalities. In this method, the lesion region was demarcated using ImageJ. The mean signal intensity within this demarcated region was then compared to a similar region of the contralateral, undamaged hemisphere of the same animal as well as a corresponding region of the sham animal. A 2-way analysis of variance (ANOVA) was then performed, and multiple comparisons were controlled using a False-discovery rate-two state method of Benjamin, Kreiger and Yekuti. This process was repeated for each of the metabolites observed in the higher resolution MALDI-MS imaging studies (Table S2).
The results of the image quantification are tabulated in Table S3 and shown graphically in Figure 6. Levels of pSer, citrulline, and lactate are significantly increased in the lesion site, relative to the contralateral hemisphere and sham animal. Pyruvate is increased in the lesion site relative to the contralateral hemisphere. Levels of NAA, leucine/isoleucine, fumarate and valine are significantly diminished at the lesion site relative to both the contralateral hemisphere and sham animal. The level of aspartate is significantly reduced when comparing the lesion site with a sham animal. The levels of glutamine are significantly increased only in the cortical region of the lesion site, relative to the contralateral hemisphere and sham animal (Figure S11). The levels of several other metabolites did not show significant changes.
Discussion
Further studies are required to provide new directions for the treatment of traumatic brain injury
Traumatic brain injury is a growing and significant health challenge. More patients are surviving initial trauma; however, those experiencing traumatic brain injury often suffer declining neurological function and accelerated neurodegeneration. Prior studies using multiple methods have suggested that stretch-induced release of glutamate stored in neurons binds to glutamate receptors, inducing glutamate excitotoxicity and potential neuronal apoptosis. Glutamate excitotoxicity is coupled to a metabolic energy crisis in the injured brain resulting in reduced neurological function and the death of neurons and supporting cells (Chamoun et al., 2010; Giza and Hovda, 2014; Katayama et al., 1990; Palmer et al., 1993; Yi and Hazell, 2006).
Although studies have revealed aspects of initial TBI molecular pathology, mechanisms leading to progressive neurological decline are yet unknown. Potential mechanisms for reducing levels of endogenous excess glutamate are poorly understood as are mechanisms for reducing glutamate-induced injury. More fundamental studies are needed to explore the underlying molecular basis for TBI that will lead to more effective treatment strategies.
In this study we used multiple mass spectrometry approaches to investigate an experimental rodent traumatic brain injury model
The first approach used here was a standard proteomics approach to investigate changes in protein expression in injured versus sham animals at 24 h. We observed increases in the levels of glial fibrillary acidic protein (gfap), a non-enzymatic type II neurofilament expressed in numerous cell types in the central nervous system, in all injured animals, consistent with prior studies. Surprisingly, we saw no consistent changes in proteins involved in glutamate metabolism (Figure 1B) at 24 h post-injury.
We next used a high-mass resolution LC-MS/MS method (Tang et al., 2019) to identify dansylated derivatives of amine-containing brain metabolites. We observed increases in aspartate, ornithine, glutamate, citrulline, glutamate, serine, GABA, isoleucine, and asparagine in the injured hemisphere. We also observed increases in GMP, AMP, cGMP and 3-nitrotyrosine (Tables 1 and 2).
Increased glutamate levels are consistent with previous studies (Chamoun et al., 2010; Giza and Hovda, 2014; Katayama et al., 1990; Palmer et al., 1993; Yi and Hazell, 2006). However, this result is somewhat confusing. The release of glutamate from neurons into the brain parenchyma could induce glutamate neurotoxicity. However, the redistribution of glutamate into the brain parenchyma caused by neuron stretching would not be expected to significantly increase total glutamate in the injured brain, as measured by LC-MS/MS. Hawkins et al. has reported that the concentration of glutamate in the extracellular space of the normal brain brain is 0.5–2 μM whereas in the plasma, the concentration is 50–100 μM (Hawkins, 2009). The observation of a net increase in glutamate levels in the damaged hemisphere suggests that compromise of the blood-brain barrier may contribute to excess brain glutamate following TBI.
The observed increased levels of other amino acids, and in particular aspartate, might suggest metabolic pathways for reducing excess glutamate in the injured brain (Divakaruni et al., 2017; Hertz and Rothman, 2017; McKenna et al., 2002, 2016; Skytt et al., 2012). Increases in GMP and AMP are consistent with consumption of ATP and overall metabolic energy crisis. Increased cGMP and 3-nitrotyrosine, a chemical reaction product from nitric oxide (NO) and tyrosine residues, suggest increased NO generation as previously described (Kozlov et al., 2017; Russwurm and Koesling, 2004).
Both proteomics and LC-MS/MS methods provide an overall impression of changes in protein or metabolite levels in injured tissues. However, they cannot provide information on metabolite distribution within the injured hemisphere or identify localized areas where injury might be more severe. A previous study using magnetic resonance spectroscopy was able to localize more severe alterations in metabolite levels following TBI (Harris et al., 2012). Within the brain regions captured by the cortical voxel, NAA, glutamate, aspartate, and GABA were found to be reduced, whereas lactate and glutamine were increased. In the hippocampal voxel, there was a less pronounced decrease in NAA and increase in glutamine and lactate (Harris et al., 2012). The discordant findings in metabolite levels between more global approaches (proteomics, LC-MS/MS) and more region-selective approaches led us to examine MALDI-MS imaging as a modality to advance our understanding of TBI.
MALDI-MS imaging reveals the lesion site and changes in metabolite levels within the lesion site
We obtained a series of MALDI-MS images of injured brain sections which revealed a lesion site. The first metabolite we investigated was N-acetylaspartate (NAA) as shown in Figure 2. Reduced NAA has been used previously as a marker of neurologic injury and an indicator of a reduced energy status (Baslow, 2002; Croall et al., 2015; Moffett et al., 2013; Di Pietro et al., 2014). At 24 h post-injury, a region of reduced NAA is observed in the injured (left) and contralateral (right) neocortex, relative to sham. In the injured hemisphere, a profound dark area (reduced NAA) is observed which is identified here as the lesion site. The reduced NAA is consistent with increased AMP and GMP observed in the injured hemisphere and an impending metabolic energy crisis.
We observed an uneven distribution of other metabolites in and around the lesion site (Figure 3). Levels of glutamate were increased in the cortex adjacent to the lesion site but decreased within the lesion. The glutamate metabolite, pSer, showed high intensity within the lesion site, consistent with the observation of pSer only in the damaged hemisphere by LC-MS/MS. The enhanced pSer was seen in all three injured animals at 24 h and was substantially reduced at 2 weeks. To our knowledge, pSer has not previously been discussed as a marker of TBI damage.
Other metabolites of glutamate include glutamine and α-KG. The low mass resolution MALDI-MS image (Figure 3) revealed increased intensity of an ion that could be either metabolite due to the similarity in their molecular weight. A subsequent higher resolution MALDI-MS image (Figure S3) revealed that glutamine was increased whereas α-KG was decreased within the lesion. The increased glutamine in the cortex portion of the lesion site (Figures 3 and S11) is consistent with the increased glutamine in the damaged hemisphere measured by LC-MS/MS. The apparent decrease in α-KG, a TCA intermediate, is consistent with reduced NAA in the lesion site and increased AMP in the damaged hemisphere.
Glutamate can serve as an amine donor in the synthesis of several amino acids, as shown in Figure 1, and therefore transamination reactions could serve as a general mechanism to reduce excess glutamate levels in neurons and astrocytes (Divakaruni et al., 2017; Hertz and Rothman, 2017; McKenna, 2012; McKenna et al., 2016; Skytt et al., 2012). In the LC-MS/MS study, aspartate was the most increased amino acid in the damaged hemisphere. Surprisingly, the MALDI-MS imaging data (Figure 4) reveals aspartate is reduced within the lesion site relative to sham. Other potential products of glutamate transamination include the branched chain amino acids (BCAA), leucine, isoleucine, and valine. The levels of all of the BCAA are decreased within the lesion site. Therefore, some but not all the glutamate transamination pathways shown in Figure 1 are operational within the lesion site.
Metabolites important in energy generation such as pyruvate and lactate can also be examined by MALDI-MS imaging. Glucose can be metabolized via glycolysis to yield pyruvate which enters the TCA cycle. Previous studies have indicated that the transfer of carbon atoms from pyruvate to oxaloacetate, with formation of citrate, is impaired following TBI (Kilbaugh et al., 2015). If the pyruvate carbon atoms cannot be transferred to citrate, pyruvate can be reduced to lactate to allow glycolysis to proceed even when oxygen is abundant. Our MALDI-MS imaging result (Figure 4) shows decreased pyruvate within the lesion site, but increased pyruvate in the cortex portion of the lesion site. In the low resolution MALDI-MS images, lactate and oxalate, oxidation products of vitamin C, cannot be distinguished due to similar molecular weights. In the higher resolution image (Figure S7), an increase in oxalate can be observed throughout the cortex of the damaged hemisphere. However, the metabolite with increased intensity localized to the lesion site is lactate, consistent with diminished flux of pyruvate into the TCA cycle.
Unanticipated metabolites such as citrulline can be observed by MALDI-MS imaging. In the LC-MS/MS study, citrulline was observed to be significantly increased in the damaged hemisphere. We therefore examined the MALD-MS images corresponding to citrulline (Figure 5) and observed significant signal intensity within the lesion site. Enhanced citrulline was observed within the lesion site for all damaged animals examined in this study at 24 h (Figure S8). Levels diminished in the injured animals at 2 weeks, although it was still evident in some animals.
Figure 5.

Citrulline accumulates in the lesion site 24 h after injury
(A) MALDI-MS image for citrulline. A representative scale bar of 5 mm is shown in white on the lower left-hand corner.
(B) Citrulline was significantly elevated in the injured hemisphere relative to the contralateral hemisphere by LC-MS/MS.
(C) LC-MS Chromatogram of danysl-derivitized citrulline.
(D) MS/MS fragmentation pattern of the dansyl derivatized citrulline shows characteristic fragments of the citrulline molecule providing further verification. ∗∗∗∗p ≤ 0.001.
Metabolite levels in MALDI-images of damaged brains can be quantified
Ion intensity levels from the mass spectra obtained across a brain section can be reconstructed to create images as shown above. Signal intensity reflected by a pseudo color scale is useful for identifying regions of increased or decreased metabolite levels within a given image. Furthermore, we developed an approach common to imaging modalities shown graphically in Figure 6. The metabolites pSer, citrulline and lactate are significantly increased in the damaged hemisphere, relative to the contralateral hemisphere and sham animal. The metabolites NAA and the BCAA leucine, isoleucine and valine are significantly decreased in the lesion site relative to the contralateral hemisphere and sham animals. Significantly decreased aspartate within the lesion was observed only when comparing the damaged hemisphere with the corresponding sham animal.
Significant changes can be observed in metabolite levels when comparing the lesion site with the rest of the hemisphere; however, they can also vary significantly within the lesion site. The MALDI-MS image shown in Figure 3 indicates increased glutamine in or near the lesion site. In contrast, the quantitative method applied above indicated increased glutamine within the lesion site, but the increase did not achieve statistical significance when compared with the contralateral hemisphere or sham animal. We therefore narrowed the region of interest to the cortex of the lesion area. As shown in Figure S11, glutamine levels are significantly increased in the cortex of the lesion area when compared to the corresponding contralateral or sham areas.
Measurements of metabolites by multiple mass spectrometry methods can be integrated to identify altered metabolic pathways
Previous studies have presented changes in the levels of various metabolites within the damaged hemispheres of experimental TBI animals (Baker et al., 2018; Chitturi et al., 2018; Harris et al., 2012; McGuire et al., 2019). In this study, we observed changes in the levels of a sufficient number of metabolites to suggest how normal metabolic pathways have been disturbed and result in alternative metabolic pathways not previously described within the context of TBI. Our results could begin to unravel the metabolic changes induced by TBI.
In the normal brain, glucose is metabolized to pyruvate which then enters the TCA cycle. Intermediates of the TCA cycle then drive oxidative phosphorylation and ATP generation when perfusion is adequate. Continuous ATP generation is needed to drive ATP-dependent ion pumps required to maintain membrane potentials. Extracellular glutamate levels are controlled by the glutamate-glutamine shuttle where extracellular glutamate is taken up by astrocytes, converted to glutamine and then returned to neurons (Bak et al., 2006; Bartnik-Olson et al., 2010; Danbolt, 2001).
TBI results in the stretching and tearing of brain tissues and the release of glutamate into the extracellular space, and likely influx through the damaged blood-brain barrier (Chodobski et al., 2011; Dorsett et al., 2017; Hawkins, 2009; Thompson et al., 2005). Within the lesion site, NAA levels fall and AMP levels increase as ATP is depleted (Croall et al., 2015; Moffett et al., 2013; Di Pietro et al., 2014). Proteins involved in shuttling pyruvate into the TCA cycle are damaged after TBI, resulting in a metabolic energy crisis (Lazzarino et al., 2019; Opii et al., 2007; Xing et al., 2009). Diminished NAA levels demarcate the lesion site as observed by MALDI-MS imaging.
In a compensatory response that would reduce the levels of excitotoxic glutamate, the glutamate amino group can be transferred to oxaloacetate, generating aspartate and α-KG (McKenna et al., 2016; Skytt et al., 2012; Yudkoff et al., 1994). Significantly increased aspartate is seen in the damaged hemisphere by LC-MS/MS, but decreased levels are seen within the lesion site by MALDI-MS imaging perhaps due to further metabolism. In the damaged brain, the carbon backbone of glutamate can replace carbon from pyruvate to fuel a truncated TCA cycle (Figure 7). Pyruvate can be converted to lactate in the lesion site to allow continued glycolysis. Enhanced conversion of glutamate to glutamine and pSer is also observed within the lesion site by MALDI-MS imaging (Figures 3 and S11).
Figure 7.

Glutamate oxidation via the TCA cycle coupled to the urea cycle after TBI
This schematic illustrates how glutamate can be used as an alternative fuel source when pyruvate carbons are prevented from entering the TCA cycle (bold arrows). The blue shaded metabolites represent the metabolites participating in glutamate metabolism. Bolded metabolites represent molecules measured in this study. Pyruvate and lactate metabolites are shaded in purple to illustrate that glycolysis and the TCA cycle are decoupled from one another, following acute TBI. Select enzymes discussed are italicized. TBI is known to damage and impair pyruvate dehydrogenase and coenzyme A (CoASH). This pathway bypasses the need for the first several steps of the TCA cycle that couple glucose metabolism to the TCA cycle and are blocked in the context of TBI. The TCA cycle continues on to generate high energy intermediates (red and dark purple) that facilitate oxidative phosphorylation. This pathway ultimately produces citrulline, nitric oxide (NO) by nitric oxide synthases and urea via the arginine to ornithine branch. This figure was created with BioRender.com.
What might be the fate of the aspartate generated from glutamate? Aspartate can enter the urea cycle generating ornithine and citrulline (Figure 7), both of which are increased in the damaged hemisphere as measured by LC-MS/MS. The conversion of aspartate derived arginine to citrulline generates nitric oxide, consistent with increased 3-nitrotyrosine in the damaged hemisphere and increased citrulline within the lesion site.
Conclusions
This study demonstrates that multiple mass spectrometry approaches can provide complementary information on the metabolic disruptions resulting from TBI. LC-MS/MS demonstrated increased levels of several metabolites globally in the damaged hemisphere, which can occur in the absence of significant changes in metabolic protein levels. MALDI-MS imaging was able to define the lesion site with decreased NAA levels which coincide with increased levels of lactate, pSer and citrulline. Collectively, these results could be integrated to reveal alternative metabolic pathways that would function to reduce excess glutamate while providing an alternative energy source. MALDI-MS imaging studies at two weeks demonstrate that these metabolic perturbations can resolve over time. Increased understanding of these pathways might lead to approaches to monitor changes, enhance recovery rates and reduce long-term damage in people who have had a TBI.
Limitations of the study
A general limitation of our study includes the small number of animals used. Three animals were used in each the naive, sham, and injured groups to ensure experimental reproducibility, but more animals would increase the statistical validity of the experimental results.
Multiple mass spectrometry methods were used to provide a more in-depth view of metabolic changes; however, each method has advantages and limitations. The choice of a specific mass spectrometry method limits the range of molecules that can be examined by that method. Proteomic studies were performed on tissues obtained from the entire damaged or contralateral hemisphere. While using tissue from an entire cortex increases the number of confidently identified proteins, anatomic resolution within the cortex is lost. Similarly, analysis of small molecules extracted from an entire cortex using the LC/MS-MS method described here results in loss of anatomic resolution. Although derivatization for LC/MS-MS improves chromatographic resolution and analyte identification, the selection of a given derivatization method limits the range of molecules that can be detected. The MALDI-MS method here provides unprecedented anatomic resolution. However, initial studies with a one mass unit-resolution instrument resulted in ambiguous identification of some metabolites. Subsequent MALDI-MS studies with a higher resolution instrument resolved most metabolite identifications. Even with the high mass resolution instrument, metabolites with identical molecular formulas can not be resolved, and analysis is limited to small molecules.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| 9-Amino Acridine | MilliporeSigma | Cat.# 8,183,620,010 |
| 2,5-Dihydroxybenzoid acid | MilliporeSigma | Cat.# 149,357 |
| Dansyl chloride | MilliporeSigma | Cat.#D2625 |
| Critical commercial assays | ||
| TMT6plex Isobaric label reagent set | Thermo Fisher | Cat.# 90,064 |
| Deposited data | ||
| Open data commons for TBI – animal dataset DOI | http://odc-tbi.org | 10.34945/F50P40 |
| Open Data Commons for TBI – proteomics TMT dataset DOI | http://odc-tbi.org | 10.34945/F5W011 |
| Experimental models: organisms/strains | ||
| Adult male, outbred, Sprague–Dawley rats | Charles Rivers Laboratories | https://www.criver.com/products-services/find-model/sas-sprague-dawley-rat?region=3611 |
| Software and algorithms | ||
| FlexImaging 3.0.54 | Bruker | N/A |
| SciLS Lab 9.02.12704 | Bruker | https://scils.de/ |
| PRISM 9.2.0 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ 1.53e | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Proteome Discoverer 2.2 | Thermo Fisher | N/A |
| Other | ||
| QExactive Mass Spectrometer | Thermo Fisher | N/A |
| Ultraflextreme MALDI-Tof/Tof | Bruker | N/A |
| Lateral fluid percussion trauma device | Virginia Commonwealth University, VA | N/A |
| CM1950 cryostat | Leica Microsystems GmbH | N/A |
| ITO coated glass slides | Hudson Surface Technology, Inc. | Cat.#PL-IC-000010-P100 |
Resource availability
Lead contact
Further information and requests for resources and regents should be directed to and will be fulfilled by the lead contact, James Sowers (jlsowers@utmb.edu)
Materials availability
This study did not generate new unique reagents
Experimental model and subject details
Animals
This study was conducted in a facility approved by the American Association for the Accreditation of Laboratory Animal Care (AAALAC). All experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (8th edition, National Research Council) and approved by the Institutional Animal Care and Use Committee of the University of Texas Medical Branch (UTMB). Adult 3-month-old male, outbred, Sprague–Dawley rats (Charles Rivers Laboratories, Inc., Portland, ME), 250–400 g, were group housed (two rats of similar injury status per cage) and had access to food and water ad libitum in a vivarium with these constant conditions: light cycle (6:00–18:00) temperature (21°C–23°C), and humidity (40%–50%). Unless noted, all animals were provided with enrichment materials, such as a cardboard tube, in their home cage.
Model: fluid percussion injury
Animals were anesthetized with 4% isoflurane in an anesthetic chamber, intubated, and mechanically ventilated with 1.5–2.0% isoflurane in O2: room air (70:30) using a volume ventilator (EDCO Scientific, Chapel Hill, NC). Rats were prepared for parasagittal fluid-percussion injury (FPI) as previously described (Andersen et al., 2021; Gao et al., 2016; Wang et al., 2011; Wu et al., 2013). Briefly, animals were placed in a stereotaxic head frame and the scalp was sagittally incised. A 4.0 mm diameter hole was trephined into the skull 2.0 mm to the right of the sagittal suture and midway between lambda and bregma. A modified 20-gauge Luerlok syringe hub (Becton-Dickinson, Franklin Lakes, NJ) was then placed over the exposed dura, bonded in place with cyanoacrylic adhesive and covered with dental acrylic. Animals with punctured dura were excluded from the study. Isoflurane was temporarily discontinued and rats were connected to the fluid percussion trauma device (Custom Design and Fabrication, Virginia Commonwealth University, VA) using a long tube (high-pressure tubing length 41cm, volume 2mL, Baxter #2C5643) connected at one end to the FPI device and the other end fit securely into the Luerlok syringe hub of the rat still in the stereotaxic head holder. They were subjected to FPI [266–320 mV oscilloscope (Tektronix TDS 1002 (60MHz, 2-channel digital real time) with Trauma Inducer Pressure Transducer Amplifier] readings, 1.81–2.17 atm range calculated, consistently held at 15.5 cm pendulum height, and pressure pulse length set at 25 ms) immediately after the return of a withdrawal reflex to paw pinch.
Prior to FPI induction, the device and connected tubing were filled with sterile water and checked that they were free of air bubbles. The device was prepared for the injury by delivering approximately 3 test pulses (confirmed by a smooth waveform on the oscilloscope) while the Luerlok at the end of the tubing was in the closed position. After TBI or sham injury, rats were disconnected from the fluid percussion device and righting reflex was assessed until a normal righting reflex was observed three times (and the time at third righting was recorded). Rats were then placed on 2% isoflurane while wound sites were infused with bupivicaine and skin was closed with wound clips. The animals received approximately 100mg/kg acetaminophen suppository before emerging from anesthesia. Isoflurane was discontinued and the rats were extubated and allowed to recover in a warm, humidified incubator.
When each rat was fully recovered, it was returned to its home cage with food and water ad libitum. All animals were housed in pairs with its pre-injury cage mate that had similar injury status (two TBI rats were housed together rather than a TBI rat and a naive rat, due to previous observations that the naive rat displays dominant behaviors to the TBI rat that may negatively impact behavior studies). All animals were monitored for signs of infection, severe neurological injury or discomfort. Signs of discomfort or pain in rodents include persistent dormouse position and unwillingness to move, refusal to eat or drink, vocalizations when handled, posturing, aggressiveness, polyphagia of bedding. Rats exhibiting these symptoms are humanely euthanized immediately (4% isoflurane in an anesthetic chamber followed by decapitation) to prevent pain and distress.
Method details
Proteomics procedures
For this study, two brain regions (cortex and hippocampus) ipsilateral to the injury were collected from rats 24 h and 2 weeks after injury. There were three groups of animals at each time point (naive, sham and TBI) each with 4 animals per group. At the 24 h time point, the cortices and hippocampus came from different animals. At the 2-week time point, the cortices and hippocampi came from the same animals. Collected brain tissues were homogenized separately and labeled with tandem-mass-tags (TMT) from 126–131. Labeled peptide samples were separated via reversed-phase liquid chromatography and analyzed on a QExactive mass spectrometer. Proteins were identified with the Proteome Discoverer (PD) 2.2 platform (version 2.2.0.388, Thermo Fisher Scientific) using the Sequest HT search engine and the uniprot-proteome_UP000002494_RAT Uniprot Proteome and ISOFORMS protein sequence database (September 2018 release).
Experimental groups for MALDI-MS imaging
Animals for the MALDI-MS imaging study were randomly divided into experimental subgroups (TBI, Sham, naive; n = 3 per group) at 24 h and 2 weeks after injury. The 24 h post-injury animals were assigned four-digit identifiers: 1365, 1366 and 1474 for the naive group, 1135, 1136 and 1471 for the sham group, and 1355, 1324 and 1472 for the TBI group. The 2 week time point animals were 1475, 1476 and 1477 for the naive group, 1469, 1470 and 1473 for the sham group, and 1468, 1467 and 1507 for the TBI group.
Metabolite extraction and Primary amine dansyl chloride derivatization
Adjacent 10 μm thick coronal frozen sections, approximately at −3.3 and −4.8 bregma, were collected such that 4.4 mg of injured brain tissue and 5.0 mg of the matched contralateral brain tissue were obtained. These were then separately mechanically homogenized with 400μL of metabolite extraction solvent (methanol/isoprolanol/water: 3/3/2 in volume) with a 1 mL Dounce homogenizer. Homogenates were then sonicated with a micro-tip solicitor (Qsonica Sonicators, newtown, CT) at amplitude set to 30 for 10 pulses. Next the sample was vortexed at 24°C for 2hr and centrifuged at 10,000 RPM for 10 min. The supernatant was transferred to a new tube and dried using a SpeedVac. Metabolites were brought up in 60% acetonitrile in ddH2O, briefly vortexed, and then centrifuged at 10,000 RPM for 10 min. The supernatant was filtered through a 3000 MW cut-off membrane (Amicon Ultra – 0.5 mL Centrifugal Filters, Ref# UFC500324, Merk Millipore, Ireland). The filtrate was dried again and reconstituted in water (100 μL). An aliquot of each sample (7 μL) was mixed with of an internal standard mixture (5 μL) containing three stable isotope amino acids, glycine, serine, and methionine all at 0.2mM. These were purchased from Cambridge Isotopes 13C2, 2H2, 15N-Glycine Ca#: CDNLM-6799-0.25; 13C3, 2H3, 15N-Serine, Ca#: CDNLM-6813-0.25; 2H3-Methionine, Ca#: DLM-431-1.
To this mixture, 50 μL of 0.5M Na2CO3/NaHCO3 and 1.25 mg of dansyl chloride in acetone (200 ul) were added, as previously described (Guo and Li, 2009). The mixture was vortexed at 55°C for 1hr. This was centrifuged and the supernatant was dried with SpeedVac and re-dissolved in 20 μL of 88% formic acid and 180 μL 1% formic acid. 20 μL of this solution was diluted to 100 μL with 1% formic acid. 2 μL was injected for each LC-MS/MS run. The dansylated products were separated by a 25 cm (length) x 75 μm (ID) capillary column packed with a mixture of Waters XSelect HSS T3 (100Å, 5 μm particle size) and Waters YMC ODS-AQ S-5, 120 Å) on the Thermo's Easy-nLC 1000. The flow-rate was 400 nL/Min and a gradient from 5% acetonitrile to 98% acetonitrile over one hour with three steps (5–25%, 25–45%, and 45–98%). Mass spectrometry was run on QExactive using top 15 data-dependent tandem mass scan (ddMS2) and positive electrospray ionization.
Tissue sectioning for MALDI-MS imaging and LC-MS validation
Serial coronal rat brain sections were obtained for MALDI-imaging. Rat brains were fixed atop a drop of distilled water on the cutting stage in upright orientation for coronal sectioning. All tissues were sectioned at 10 μm thickness using a Leica CM1950 cryostat (Leica Microsystems GmbH, Wetzlar, Germany) at −20°C and thaw mounted onto indium tin oxide (ITO) coated glass slides, (ITO glass, Type II, 0.7mm) (Hudson Surface Technology, Inc., West New York, NJ; cat. # PL-IC-000010-P100 (formerly PSI 1207000)). Each tissue slice for MALDI imaging was followed by a slice collected for morphological/immunohistochemical staining and mounted on a regular Superfrost Plus microscope slide (Fisher Scientific, Pittsburgh, PA; cat.# 12-550-15). From each brain, 20 consecutive slices were collected starting at approximately the bregma −3.3 mm coordinate. Each slide was stored in a 50 mL plastic conical tube at −80°C. Before matrix application, the slides were transferred to −20°C for 20 min and then placed into a vacuum desiccator for approximately 25 min before matrix application.
Matrix application and MALDI-MS imaging
2,5-Dihydroxybenzoic acid (DHB) (MilliporeSigma, St. Louis. MO; cat.# 149,357) or 9-aminoacridine (9-AA) (MilliporeSigma, St. Louis. MO; cat. # 8,183,620,010) were applied by sublimation under vacuum using a procedure as previously described (Wildburger, 2017).
Briefly, sublimation of DHB was performed for 1 min at 120°C, and sublimation of 9-AA was performed for 2.5 min at 130°C under 10–15 mTorr. After that, the slide was stored at −80°C. Prior to MALDI-MS imaging, the slides were transferred to −20°C for 20 min, and then quickly transferred into a vacuum desiccator and brought to room temperature under vacuum for approximately 10 min.
An Ultraflextreme MALDI-TOF/TOF MS (Bruker Daltonics, Billerica, MA) equipped with a smartbeam Nd: YAG 355 nm laser was utilized for MALDI analysis. The laser was fired at 1000 Hz, and the analyzer was operated in reflector mode. The positive-ion mass spectra in the reflector mode were collected with a pulsed ion extraction time of 120 ns, an accelerating voltage of 25.00 kV, an extraction voltage of 22.4 kV, a lens voltage of 7.85 kV, and a reflector voltage of 26.4 kV. The negative-ion mass spectra in the reflector mode were collected with a pulsed ion extraction time of 140 ns, an accelerating voltage of 20.00 kV, an extraction voltage of 17.4 kV, a lens voltage of 5.85 kV, and a reflector voltage of 21.15 kV. The laser spot size was set at medium focus (∼50 μm laser spot diameter), and laser power was optimized at the start of each run and then fixed for the whole experiment. The mass spectra data were acquired over a mass range of m/z 70–700 Da. Mass calibration was performed with an external standard prior to data acquisition.
Calibration in the negative ion mode, 70–700 Da mass range, was performed using 1 mM xanthine and 1 mM Leu-enkephaline with 9-aminoacridine matrix. Calibration in positive ion mode, 70–700 Da mass range, was performed using an aqueous solution of 1 mM γ-aminobutyric acid, 1 mM caffeine and 1 mM MRFA peptide with α-cyano-4-hydroxycinnamic acid matrix.
For imaging mass spectrometry analysis, imaging spatial resolution was set to 100 μm. Each spectrum was a sum of 500 laser shots. Regions of interest were manually defined in the flexImaging 3.0.54 software using a scan of the slide with the tissue sections acquired at 1200 dpi using an Epson 3170 photo scanner. MALDI mass spectra were processed with the total ion current (TIC) normalization, and the signal intensity of each imaging data was represented as the normalized intensity.
High-resolution mass spectrometry imaging
Mass spectrometry imaging was performed using a Bruker timsTOF fleX MALDI qTOF mass spectrometer (Bruker Daltonics, Bremen, Germany) operated in either negative ion mode or positive ion mode with TIMS off. Data were collected from m/z 50 to m/z 1000, with tune parameters optimized for sensitivity of analytes < m/z 300 as follow: Funnel 1 RF of 50 Vpp; Funnel 2 RF of 100 Vpp; Multipole RF of 150 Vpp; Collision RF of 600 Vpp; Transfer Time of 35 μs; and Pre Pulse Storage of 2 μs. Images were collected with 100 μm spatial resolution with a beam scan of 96 μm. A total of 1200 laser shots were summed per pixel with the laser operating at 10 kHz. Data were visualized using SCiLS Lab 2021b (Version 9.01.12514).
Quantification and statistical analysis
Proteomics statistical analysis
Perseus (Cox and Mann, 2012; Tyanova et al., 2016) version 1.6.0.7 was used to process the data files exported from the PD2.2 software. Each dataset was comprised of approximately 19,000 proteins. The datasets were filtered first by removing proteins that had a false discovery rate (FDR) value of >0.05 meaning that only proteins that were identified with high (FDR ≤0.01) and medium (0.01 < FDR ≤0.05) confidence were kept. The second filtering step removed proteins that were not identified at least once by each TMT 6 plex label (TMT 126–131). Fold change values and adjusted p values were calculated in Perseus. Statistics were calculated using a modified two-sided t test with multiple test corrections (a permutation-based method that controls the FDR), using the default settings (FDR ≤0.05, S0 = 0.1). For the remaining list of approximately 2,000 proteins. The complete dataset from the entire longitudinal study can be found at Open Data Commons for TBI (ODC-TBI), http://odc-tbi.org.
High-resolution dansyl chloride derivatization mass spectrometry statistical analysis
NanoLC-MS Metabolomics statistical analysis was done by manually integrating MS1 peak areas of metabolites validated by either tandem mass spectra, and or reference standard. Peaks were quantified in Xcalibur (Version 2.4) and exported to a spreadsheet software where peak areas were normalized to the isotope labeled serine internal standard peak area as well as the mass of each tissue sample. Fold changes and a two-sided t test were done in Excel using technical replicates (n = 6).
Quantification of metabolites in MALDI-MS images
A region around the lesion site defined by reduced NAA was demarcated. Gray scale images within the demarcated area at a selected m/z were then generated with the Fleximaging software. ImageJ software was used to quantify the mean gray scale within the demarcated area. A symmetric region was then demarcated in the undamaged contralateral hemisphere as well as in the left hemisphere of the sham animal (Figure S10). This procedure was repeated for glutamine and aspartate specifically for just the cortex region of the injury site (Figure S11). Mean metabolite intensities were tabulated in the PRISM software which performed a two-way ANOVA analysis (Table S3). Multiple comparisons were controlled using a false discovery rate in PRISM using the two-stage Benjamini, Kreiger and Yekutieli procedure.
Acknowledgments
We thank Dr. Lawrence Sowers for conceptual discussions as well as suggestions on editing of the manuscript. We thank Dr. Erin Seeley and the mass spectrometry imaging facility at the University of Texas at Austin. This work was supported by the Moody Project for Translational Traumatic Brain Injury Research, resources from the UTMB Department of Pharmacology and Toxicology and funding from the UTMB MD-PhD combined degree program (J.L.S., M.L.S.).
Author contributions
Conceptualization of Methodology: All authors; Formal analysis and validation, data curation and visualization: J.L.S., M.L.S., A.S.S., K.Z; Resources: B.E.H., D.S.D., D.S.P., K.Z.; Writing – original draft: J.L.S.; Writing – review and editing: All authors; Supervision, administration, and funding acquisition: D.S.D, D.S.P., K.Z. All authors approved the final version of the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: October 22, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103108.
Supplemental information
Data and code availability
-
•
The proteomic data have been deposited at Open Data Commons for TBI (ODC-TBI). The DOI number for the animal dataset information is, 10.34945/F50P40 and the proteomics TMT dataset is, 10.34945/F5W011. The metabolite data and MALDI-MS imaging data are included within the manuscript
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Andersen C.R., Wolf J., Jennings K., Prough D.S., Hawkins B.E. Accelerated failure time survival model to analyze morris water maze latency data. J. Neurotrauma. 2021;38:435–445. doi: 10.1089/neu.2020.7089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bak L.K., Schousboe A., Waagepetersen H.S. The glutamate/GABA-glutamine cycle: aspects of transport, neurotransmitter homeostasis and ammonia transfer. J. Neurochem. 2006;98:641–653. doi: 10.1111/j.1471-4159.2006.03913.x. [DOI] [PubMed] [Google Scholar]
- Baker E.W., Henderson W.M., Kinder H.A., Hutcheson J.M., Platt S.R., West F.D. Scaled traumatic brain injury results in unique metabolomic signatures between gray matter, white matter, and serum in a piglet model. PLoS One. 2018;13:1–21. doi: 10.1371/journal.pone.0206481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartnik-Olson B.L., Oyoyo U., Hovda D.A., Sutton R.L. Astrocyte oxidative metabolism and metabolite trafficking after fluid percussion brain injury in adult rats. J. Neurotrauma. 2010;27:2191–2202. doi: 10.1089/neu.2010.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baslow M.H. Evidence supporting a role for N-acetyl-L-aspartate as a molecular water pump in myelinated neurons in the central nervous system: an analytical review. Neurochem. Int. 2002;40:295–300. doi: 10.1016/s0197-0186(01)00095-x. [DOI] [PubMed] [Google Scholar]
- Chamoun R., Suki D., Gopinath S.P., Goodman J.C., Robertson C. Role of extracellular glutamate measured by cerebral microdialysis in severe traumatic brain injury: clinical article. J. Neurosurg. 2010;113:564–570. doi: 10.3171/2009.12.JNS09689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitturi J., Li Y., Santhakumar V., Kannurpatti S.S. Early behavioral and metabolomic change after mild to moderate traumatic brain injury in the developing brain. Neurochem. Int. 2018;120:75–86. doi: 10.1016/j.neuint.2018.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chodobski A., Zink B.J., Szmydynger-Chodobska J. Blood-brain barrier pathophysiology in traumatic brain injury. Transl. Stroke Res. 2011;2:492–516. doi: 10.1007/s12975-011-0125-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J., Mann M. 1D and 2D annotation enrichment: a statistical method integrating quantitative proteomics with complementary high-throughput data. BMC Bioinformatics. 2012;13:S12. doi: 10.1186/1471-2105-13-S16-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croall I., Smith F.E., Blamire A.M. Magnetic resonance spectroscopy for traumatic brain injury. Top. Magn. Reson. Imaging. 2015;24:267–274. doi: 10.1097/RMR.0000000000000063. [DOI] [PubMed] [Google Scholar]
- Danbolt N.C. Glutamate uptake. Prog. Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Deshmukh D., Patel M. Age-dependent CHanges in glutamate oxidation by non-synaptic and synaptic mitochondria from rat brain. Mech. Ageing Dev. 1980;13:75–81. doi: 10.1016/0047-6374(80)90131-1. [DOI] [PubMed] [Google Scholar]
- Divakaruni A.S., Wallace M., Buren C., Martyniuk K., Andreyev A.Y., Li E., Fields J.A., Cordes T., Reynolds I.J., Bloodgood B.L. Inhibition of the mitochondrial pyruvate carrier protects from excitotoxic neuronal death. J. Cell Biol. 2017;216:1091–1105. doi: 10.1083/jcb.201612067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsett C.R., McGuire J.L., Depasquale E.A.K., Gardner A.E., Floyd C.L., McCullumsmith R.E. Glutamate neurotransmission in rodent models of traumatic brain injury. J. Neurotrauma. 2017;34:263–272. doi: 10.1089/neu.2015.4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J., Grill R.J., Dunn T.J., Bedi S., Labastida J.A., Hetz R.A., Xue H., Thonhoff J.R., Dewitt D.S., Donald S. Human neural stem cell transplantation-mediated alteration of microglial/macrophage phenotypes after traumatic brain injury. Cell Transpl. 2016;25:1863–1877. doi: 10.3727/096368916X691150. [DOI] [PubMed] [Google Scholar]
- Ginsberg M.D. Neuroprotection for ischemic stroke: past, present and future. Neuropharmacology. 2008;55:363–389. doi: 10.1016/j.neuropharm.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giza C.C., Hovda D.A. The new neurometabolic cascade of concussion. Neurosurgery. 2014;75:S24–S33. doi: 10.1227/NEU.0000000000000505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo K., Li L. Differential 12C-/13C-isotope dansylation labeling and fast liquid chromatography/mass spectrometry for absolute and relative quantification of the metabolome. Anal. Chem. 2009;81:3919–3932. doi: 10.1021/ac900166a. [DOI] [PubMed] [Google Scholar]
- Harris J.L., Yeh H.W., Choi I.Y., Lee P., Berman N.E., Swerdlow R.H., Craciunas S.C., Brooks W.M. Altered neurochemical profile after traumatic brain injury: 1 H-MRS biomarkers of pathological mechanisms. J. Cereb. Blood Flow Metab. 2012;32:2122–2134. doi: 10.1038/jcbfm.2012.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins R.A. The blood-brain barrier and glutamate. Am. J. Clin. Nutr. 2009;90:867S–874S. doi: 10.3945/ajcn.2009.27462BB. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz L., Rothman D.L. Glutamine-glutamate cycle flux is similar in cultured astrocytes and brain and both glutamate production and oxidation are mainly catalyzed by aspartate aminotransferase. Biology (Basel) 2017;6:1–21. doi: 10.3390/biology6010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomidou C., Turski L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol. 2002;1:383–386. doi: 10.1016/s1474-4422(02)00164-3. [DOI] [PubMed] [Google Scholar]
- Katayama Y., Becker D.P., Tamura T., Hovda D.A. Massive increases in extracellular potassium and the indiscriminate release of glutamate following concussive brain injury. J. Neurosurg. 1990;73:889–900. doi: 10.3171/jns.1990.73.6.0889. [DOI] [PubMed] [Google Scholar]
- Kilbaugh T.J., Karlsson M., Byro M., Bebee A., Ralston J., Sullivan S., Duhaime A.C., Hansson M.J., Elmér E., Margulies S.S. Mitochondrial bioenergetic alterations after focal traumatic brain injury in the immature brain. Exp. Neurol. 2015;271:136–144. doi: 10.1016/j.expneurol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlov A.V., Bahrami S., Redl H., Szabo C. Alterations in nitric oxide homeostasis during traumatic brain injury. Biochim. Biophys. Acta. 2017;1863:2627–2632. doi: 10.1016/j.bbadis.2016.12.020. [DOI] [PubMed] [Google Scholar]
- Lazzarino G., Amorini A.M., Signoretti S., Musumeci G., Lazzarino G., Caruso G., Pastore F.S., Di Pietro V., Tavazzi B., Belli A. Pyruvate dehydrogenase and tricarboxylic acid cycle enzymes are sensitive targets of traumatic brain injury induced metabolic derangement. Int. J. Mol. Sci. 2019;20:5774. doi: 10.3390/ijms20225774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy L.M., Warr O., Attwell D. Stoichiometry of the glial glutamate transporter GLT-1 expressed inducibly in a Chinese hamster ovary cell line selected for low endogenous Na+-dependent glutamate uptake. J. Neurosci. 1998;18:9620–9628. doi: 10.1523/JNEUROSCI.18-23-09620.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas A.I.R., Menon D.K., Adelson P.D., Andelic N., Bell M.J., Belli A., Bragge P., Brazinova A., Büki A., Chesnut R.M. Traumatic brain injury: integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 2017;16:987–1048. doi: 10.1016/S1474-4422(17)30371-X. [DOI] [PubMed] [Google Scholar]
- Mark L.P., Prost R.W., Ulmer J.L., Smith M.M., Daniels D.L., Strottmann J.M., Brown W.D., Hacein-Bey L. Pictorial review of glutamate excitotoxicity: fundamental concepts for neuroimaging. Am. J. Neuroradiol. 2001;22:1813–1824. [PMC free article] [PubMed] [Google Scholar]
- McGuire J.L., DePasquale E.A.K., Watanabe M., Anwar F., Ngwenya L.B., Atluri G., Romick-Rosendale L.E., McCullumsmith R.E., Evanson N.K. Chronic dysregulation of cortical and subcortical metabolism after experimental traumatic brain injury. Mol. Neurobiol. 2019;56:2908–2921. doi: 10.1007/s12035-018-1276-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna M.C. Substrate competition studies demonstrate oxidative metabolism of glucose, glutamate, glutamine, lactate and 3-hydroxybutyrate in cortical astrocytes from rat brain. Neurochem. Res. 2012;37:2613–2626. doi: 10.1007/s11064-012-0901-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna M.C., Sonnewald U., Huang X., Stevenson J., Zielke H.R. Exogenous glutamate concentration regulates the metabolic fate of glutamate in astrocytes. J. Neurochem. 2002;66:386–393. doi: 10.1046/j.1471-4159.1996.66010386.x. [DOI] [PubMed] [Google Scholar]
- McKenna M.C., Stridh M.H., McNair L.F., Sonnewald U., Waagepetersen H.S., Schousboe A. Glutamate oxidation in astrocytes: roles of glutamate dehydrogenase and aminotransferases. J. Neurosci. Res. 2016;94:1561–1571. doi: 10.1002/jnr.23908. [DOI] [PubMed] [Google Scholar]
- Moffett J.R., Arun P., Ariyannur P.S., Namboodiri A.M.A. N-Acetylaspartate reductions in brain injury: impact on post-injury neuroenergetics, lipid synthesis, and protein acetylation. Front. Neuroenergetics. 2013;5:1–19. doi: 10.3389/fnene.2013.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opii W.O., Nukala V.N., Sultana R., Pandya J.D., Day K.M., Merchant M.L., Klein J.B., Sullivan P.G., Butterfield D.A. Proteomic identification of oxidized mitochondrial proteins following experimental traumatic brain injury. J. Neurotrauma. 2007;24:772–789. doi: 10.1089/neu.2006.0229. [DOI] [PubMed] [Google Scholar]
- Oz G., Okar D., Henry P. In: Neural Metabolism in Vivo. Choi I., Gruetter R., editors. Springer; 2012. Glutamate-glutamine cycle and anaplerosis; pp. 921–946. [Google Scholar]
- Palmer A.M., Marion D.W., Botscheller M.L., Swedlow P.E., Styren S.D., DeKosky S.T. Traumatic brain injury-induced excitotoxicity assessed in a controlled cortical impact model. J. Neurochem. 1993;61:2015–2024. doi: 10.1111/j.1471-4159.1993.tb07437.x. [DOI] [PubMed] [Google Scholar]
- Di Pietro V., Amorini A.M., Tavazzi B., Vagnozzi R., Logan A., Lazzarino G., Signoretti S., Lazzarino G., Belli A. The molecular mechanisms affecting N-acetylaspartate homeostasis following experimental graded traumatic brain injury. Mol. Med. 2014;20:147–157. doi: 10.2119/molmed.2013.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russwurm M., Koesling D. NO activation of guanylyl cyclase. EMBO J. 2004;23:4443–4450. doi: 10.1038/sj.emboj.7600422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C., Rasband W., Eliceiri K. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skytt D.M., Klawonn A.M., Stridh M.H., Pajecka K., Patruss Y., Quintana-Cabrera R., Bolaños J.P., Schousboe A., Waagepetersen H.S. SiRNA knock down of glutamate dehydrogenase in astrocytes affects glutamate metabolism leading to extensive accumulation of the neuroactive amino acids glutamate and aspartate. Neurochem. Int. 2012;61:490–497. doi: 10.1016/j.neuint.2012.04.014. [DOI] [PubMed] [Google Scholar]
- Tang W., Chen J., Zhou J., Ge J., Zhang Y., Li P., Li B. Quantitative MALDI imaging of spatial distributions and dynamic changes of tetrandrine in multiple organs of rats. Theranostics. 2019;9:932–944. doi: 10.7150/thno.30408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson H.J., Lifshitz J., Marklund N., Grady M.S., Graham D.I., Hovda D.A., McIntosh T.K. Lateral fluid percussion brain injury: a 15-year review and evaluation. J. Neurotrauma. 2005;22:42–75. doi: 10.1089/neu.2005.22.42. [DOI] [PubMed] [Google Scholar]
- Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M.Y., Geiger T., Mann M., Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods. 2016;13:731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- Wang E., Gao J., Yang Q., Parsley M.O., Dunn T.J., Zhang L., DeWitt D.S., Denner L., Prough D.S., Wu P. Molecular mechanisms underlying effects of neural stem cells against traumatic axonal injury. J. Neurotrauma. 2011;29:295–312. doi: 10.1089/neu.2011.2043. [DOI] [PubMed] [Google Scholar]
- Wildburger N.C. Springer US; 2017. Lipidomics, Neuromethods, Chapter 4: MALDI-imaging Mass Spectrometry of Brain Lipids. [Google Scholar]
- Wu P., Zhao Y., Haidacher S.J., Wang E., Parsley M.O., Gao J., Sadygov R.G., Starkey J.M., Luxon B.A., Spratt H. Detection of structural and metabolic changes in traumatically injured hippocampus by quantitative differential proteomics. J. Neurotrauma. 2013;30:775–788. doi: 10.1089/neu.2012.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing G., Ren M., Watson W.A., O’Neil J.T., Verma A. Traumatic brain injury-induced expression and phosphorylation of pyruvate dehydrogenase: a mechanism of dysregulated glucose metabolism. Neurosci. Lett. 2009;454:38–42. doi: 10.1016/j.neulet.2009.01.047. [DOI] [PubMed] [Google Scholar]
- Yi J.H., Hazell A.S. Excitotoxic mechanisms and the role of astrocytic glutamate transporters in traumatic brain injury. Neurochem. Int. 2006;48:394–403. doi: 10.1016/j.neuint.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Yudkoff M., Nelson D., Daikhin Y., Erecinska M. Tricarboxylic acid cycle in rat brain synaptosomes. Fluxes and interactions with aspartate aminotransferase and malate/aspartate shuttle. J. Biol. Chem. 1994;269:27414–27420. [PubMed] [Google Scholar]
- Zerangue N., Kavanaugh M.P. Flux coupling in a neuronal glutamate transporter. Nature. 1996;383:634–637. doi: 10.1038/383634a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The proteomic data have been deposited at Open Data Commons for TBI (ODC-TBI). The DOI number for the animal dataset information is, 10.34945/F50P40 and the proteomics TMT dataset is, 10.34945/F5W011. The metabolite data and MALDI-MS imaging data are included within the manuscript
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.