

Summary
The mitochondrial unfolded protein response (UPRmt) is an organellar stress signaling pathway that functions to detect and restore disruption of mitochondrial proteostasis. The UPRmt is involved in a wide range of physiological and disease conditions, including aging, stem cell maintenance, innate immunity, neurodegeneration, and cancer. Here we report that the UPRmt is integral to zebrafish fin regeneration. Taking advantage of a novel zebrafish UPRmt reporter, we observed that UPRmt activation occurs in regenerating fin tissue shortly after injury. Through chemical and genetic approaches, we discovered that the Sirt1-UPRmt pathway, best known for its role in promoting lifespan extension, is crucial for fin regeneration. The metabolism of NAD+ is an important contributor to Sirt1 activity in this context. We propose that Sirt1 activation induces mitochondrial biogenesis in injured fin tissue, which leads to UPRmt activation and promotes tissue regeneration.
Subject areas: Molecular biology, Cell biology, Developmental biology
Graphical abstract
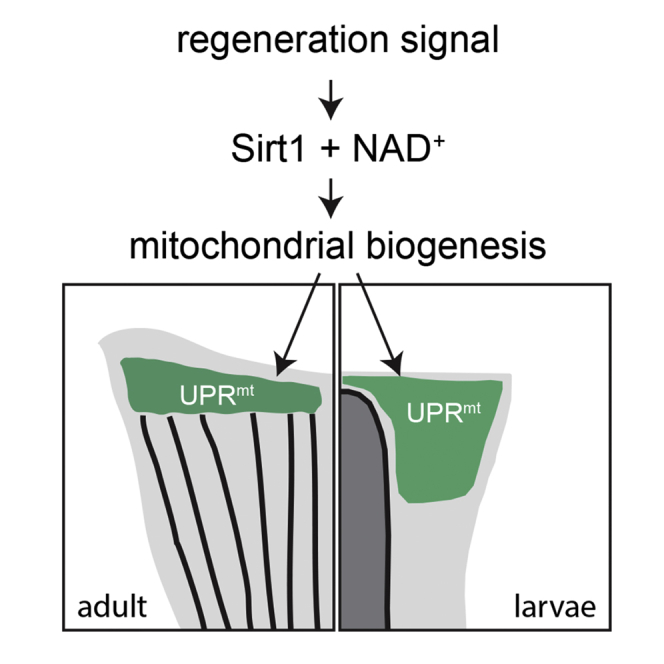
Highlights
-
•
The mitochondrial unfolded protein response is activated during fin regeneration
-
•
The pathway, requiring NAD+, is stimulated in both injured larval and adult fins
-
•
The sirtuin (Sirt1) pathway is also activated and required for regeneration
-
•
Sirtuin promotes mitochondrial biogenesis due to increased NAD+ in the blastema
Molecular biology; Cell biology; Developmental biology
Introduction
Mitochondria are dynamic organelles constantly being built or turned over according to the cellular energetic state or stress condition (Mishra and Chan, 2016). Because of this, the compartmental proteostasis is under considerable strain (Baker and Haynes, 2011). One pathway that has emerged as a key player in maintaining mitochondrial health is the mitochondrial unfolded protein response, or UPRmt. Triggered by an accumulation of unfolded proteins in the mitochondrial matrix, the UPRmt generates a transcriptional response that activates the expression of mitochondrial chaperone and protease genes to restore mitochondrial proteostasis (Jovaisaite and Auwerx, 2015; Shpilka and Haynes, 2018). Studies in C. elegans showed that the UPRmt plays an important role in aging, innate immunity and host-microbiome communication (Durieux et al., 2011; Han et al., 2017; Pellegrino et al., 2014). In mammals, the UPRmt is involved in a rapidly expanding list of conditions, including stem cell maintenance during aging, cancer metastasis, Alzheimer’s disease and cardiac injury (Kenny et al., 2019; Mohrin et al., 2015; Sorrentino et al., 2017; Wang et al., 2019; Zhang et al., 2016). These findings demonstrate that mitochondrial stress underlies diverse physiological and disease conditions. However, it is not well understood how the UPRmt is regulated to impact such diverse conditions.
Regenerating complex organs after injury is a remarkable feat that for some animal species is markedly robust, including the genetic model organism zebrafish (Chen and Poss, 2017). Unlike mammalian limbs, after fin injury zebrafish form a layer of progenitor cells underneath the wounded fin epithelium, referred to as a blastema. This layer of progenitor cells eventually forms the new tissue and restores the injured organ. During this relatively quick process (7–10 days), dramatic cell fate changes occur. To form the blastema, terminally differentiated bone cells in the spared fin undergo dedifferentiation and become the mesenchymal progenitor cells (Knopf et al., 2011; Sehring et al., 2016). These progenitor cells then proliferate and differentiate to restore the fin. Genetic studies have identified mutations in genes that impair fin regeneration. Although many of these genes belong to major developmental signaling pathways, there are also genes involved in organelle regulation, indicating that intricate cell biological remodeling takes place during blastema formation (Nechiporuk et al., 2003; Varga et al., 2014). A mutation in a mitochondrial protein chaperone gene, hspd1, was identified in a screen for genes required for the survival of blastema cells during fin regeneration (Makino et al., 2005). Since then, hspd1 has also been found essential for regeneration of the heart, retina and lateral line hair cells in zebrafish (Pei et al., 2016; Qin et al., 2009). hspd1 is a mitochondrial protein chaperone and, more importantly, the primary signaling output of the UPRmt, indicating that the UPRmt pathway is an integral part of the tissue regeneration signaling network. Here, we report that UPRmt activation during fin regeneration is regulated by Sirt1, one of the sirtuin protein deacetylase family members. The Sirt1-UPRmt pathway is evolutionarily conserved from worms to mammals, and best known for its effect in promoting lifespan extension (Mouchiroud et al., 2013; Zhang et al., 2016). Mechanistically, we propose that Sirt1 activates the UPRmt by triggering mitochondrial biogenesis, at least in part via enhancing mitochondrial translation. Our findings show that zebrafish rely on this innate mitochondrial protective pathway to regenerate injured organs.
Results
The UPRmt is part of the molecular network promoting fin regeneration in zebrafish
The expression of hspd1 in blastema cells suggests that the UPRmt, which encompasses transcriptional activation of a wide range of mito-protective genes, is part of the signaling network underlying tissue regeneration. To examine whether hspd1 expression is indeed part of the UPRmt response during regeneration, we examined by qRT-PCR the expression levels of a group of four well-characterized UPRmt signature genes in regenerating fins of adult zebrafish at 2 days post amputation (2 dpa, shortly after the blastema is formed). Besides hspd1, the group includes another mitochondrial protein chaperone hspa9, and two mitochondrial proteases, clpp and lonp1. Indeed all four genes are up-regulated, and in situ hybridization of lonp1 also confirms that the enhanced expression is in the blastema (Figure 1A). Zebrafish larvae have also been used as a model for studying fin regeneration. Interestingly, many genes and signaling pathways important for adult fin regeneration are also used during larval fin regeneration (Yoshinari et al., 2009). To examine whether it is also the case for the UPRmt, mRNA was collected from regenerating larval fins at 4 h post amputation (hpa) for qRT-PCR analysis. Again, all four UPRmt signature genes are up-regulated in the regenerating larval fin and lonp1 in situ hybridization assays corroborate with the qPT-PCR result, indicating that the enhanced expression occurs in the fin fold of injured larvae (Figure 1B). Based on these results, the UPRmt is activated during fin regeneration in larval fish. To get a better sense of which cell type the UPRmt takes place in the regenerating fin, a transgenic UPRmt fluorescent reporter line was generated using an hspd1 genomic DNA fragment (6 kb upstream of the translation start, Figure 1C-i) to direct expression of d2GFP (destabilized GFP). Confocal images of the transgenic adult fish show that after fin amputation fluorescent signal is concentrated in the blastema region, consistent with the reported hspd1 in situ hybridization data (Figures 1C-ii and 1C-iii). Interestingly, the confocal images of larval fins show that outside the strong background signal in the notochord, fluorescent signal appears to concentrate in the fin fold mesenchymal cells, based on their elongated shape and migratory nature upon injury (Figures 1C-iv to 1C-vi). Of note, the highest signal appears to be at the most distally located population of mesenchymal cells, which are thought to be equivalent to the blastema cells in injured adult fins (Mateus et al., 2012).
Figure 1.

The UPRmt is activated in adult and larval injured fins
(A and B) Representative in situ hybridization images (left panels) show that lonp1 (a UPRmt gene) is up-regulated during fin regeneration in adult (A, partial fin view, posterior toward top) and larval (B, partial body view, posterior toward the right) fish at 2 dpa or 4 hpa, respectively. qRT-PCR assays (right panels) show that the expression of all four UPRmt signature genes are enhanced in adult and larval injured fin tissue.
(C-i) Diagram indicates the hspd1 genomic fragment (2.6 kb upstream of the transcription start (+1) to 3 kb downstream until the translation start) employed for driving the expression of the destabilized GFP. (C-ii and C-iii) Adult fin of the UPRmt reporter fish, Tg(hspd1:d2gfp), exhibits induced fluorescence at the injury zone during regeneration. Higher magnification image (iii; white rectangle in ii) indicates that the fluorescent signal marks the blastema of the injured fin. (C-iv to C-vi) A time series of images shows the dynamics of the UPRmt reporter activation in larval fin upon injury. Note fluorescent migrating mesenchymal-like cells.
(D) Representative images of hspd1 in situ hybridization assays show that hspd1 expression is diminished in SU5402 treated injured larvae at 4 hpa. (Error bars indicate standard deviation. Student’s t test was performed to determine statistical significance. ∗p < 0.05). Scale bars are as indicated.
Therefore, these expression patterns strongly support the idea that in both adult and larval zebrafish hspd1 (and thus UPRmt) activation is part of the tissue regeneration signaling network. Moreover, given the conserved reporter activities under the control of a relatively short genomic fragment (6 kb), the transcriptional regulatory mechanisms targeting hspd1 (leading to UPRmt) are likely shared between adult and larval fins. Recently, different aspects of mitochondrial regulation have been shown to be involved in wound healing, for example, mtROS production and mitochondrial dynamics (Fu et al., 2020; Xu and Chisholm, 2014). Hspd1 has even been shown to act extracellularly as a chemoattractant for immune cells during wound healing and regeneration in zebrafish (Pei et al., 2016). To validate that the UPRmt is genuinely involved in regeneration rather than wound healing, a larval regenerating wound was converted to a non-regenerating wound by treating injured larvae with an FGF receptor inhibitor, SU5402, given that FGF signaling is essential for regeneration but not wound healing (Owlarn et al., 2017). In situ hybridization results showed that hspd1 expression is strongly diminished in the SU5402 treated larvae (Figure 1D), indicating that UPRmt activation in larval fin is specifically associated with regeneration rather than generic wound healing. Together, these results show that the UPRmt is part of the signaling network underlying fin regeneration in both adult and larval zebrafish.
Transcript profiling reveals that mitochondrial biogenesis is part of the cellular process of fin regeneration
To identify possible mechanisms of UPRmt activation during fin regeneration, we examined by RNA sequencing the transcriptome of the larval fin fold during regeneration. RNA samples were collected at 4 hpa when the UPRmt activity is at its peak according to the in situ hybridization data. Many genes known to be impacted during larval fin regeneration are shown to be up-regulated in our analysis, validating that the assay is sensitive enough to study gene expression changes during regeneration (Figure 2A, top half). A number of genes associated with the UPRmt are also up-regulated according to this analysis (Figure 2A, bottom half). To explore the potential transcriptional mechanism we focused on known transcriptional factors associated with the UPRmt response in mammalian cells, i.e., CHOP (ddit3 in zebrafish) (Zhao et al., 2002), ATF4 (atf4a and atf4b in zebrafish) (Quiros et al., 2017), ATF5 (atf5a and atf5b in zebrafish) (Fiorese et al., 2016), but found none of them are up-regulated in the regenerating fin. Subsequent qRT-PCR assays corroborated this finding (Figure S1A). FOXO3a has been reported to activate antioxidant defense by inducing expression of a mitochondrial superoxide dismutase, sod2, in response to mitochondrial stress (Mouchiroud et al., 2013; Papa and Germain, 2014). However, neither RNA-seq profiling nor qRT-PCR analysis found sod2 up-regulated (Figure S1A).
Figure 2.

Chemical inhibition of Sirt1 impairs UPRmt activation in injured larval fins
(A) Heatmap of relative gene expression levels derived from the transcriptional profiling data demonstrates that previously studied regeneration genes (top half) as well as the UPRmt genes (bottom half) are all up-regulated in the injured fins during regeneration.
(B) GSEA indicates that mitochondrial translation and transcription are among the cellular processes that are most enhanced during fin regeneration.
(C) qRT-PCR assays show that the expression of two mitochondrial transcriptional machinery components, polrmt and tf2bm, are enhanced in zebrafish adult fins during regeneration.
(D) The schematic depicts the mechanism of Sirt1 inhibition by various small molecule inhibitors used in subsequent experiments.
(E) Representative images of hspd1 in situ hybridization.
(F) qRT-PCR assays show that Sirt1 inhibition via Sirtinol treatment or Nampt inhibition via FK866 treatment suppresses the UPRmt gene expression in the injured larval fin. (Error bars indicate standard deviation. Student’s t test was performed to determine statistical significance. ∗p < 0.05.)
Because known mito-stress genes were not changed, we identified those that are up-regulated to a similar extent as the UPRmt signature genes (at least 1.5-fold increase in injured tissue, Figure S1B) and performed GO enrichment analysis. The top 15 enriched processes were identified (Figure S1C). Among these, the nuclear genome encoded genes involved in mitochondrial translation function were highlighted. GSEA analysis further revealed that mitochondrial translation (GO: 0032543) and mitochondrial transcription (GO: 0006390) gene sets are enriched in the injured samples (Figure 2B). The transcriptional signature strongly suggests that mitochondrial biogenesis is taking place during regeneration and is perhaps is linked with UPRmt activation, which would be consistent with a recent finding showing that the nuclear genome encoded mitochondrial ribosomal function related genes are up-regulated under UPRmt hyperactivation in C. elegans (Shpilka et al., 2021). To probe whether genes associated with mitochondrial biogenesis are also part of the transcriptional program in adult fin regeneration, two central factors of the mitochondrial transcriptional machinery were examined by qRT-PCR in adult injured fins at 2 dpa. Indeed, polrmt and tfb2m are both significantly up-regulated during regeneration (Figure 2C). Based on these results we conclude that mitochondrial biogenesis is a key cellular process occurring during regeneration, and it is likely associated with activation of the UPRmt.
Inhibiting Sirt1 blocks the activation of UPRmt in the regenerating fin
A well-known mitochondrial biogenesis regulator is Sirt1, which promotes mitochondrial biogenesis by modifying PGC1α post-translationally (Chang and Guarente, 2014). Sirt1 activity is also important for UPRmt activation in the context of lifespan regulation in C. elegans and mice. Because we discovered that the UPRmt signaling and mitochondrial biogenesis are both prominent in regenerating fins, we hypothesized that Sirt1 plays a key role in regulating mitochondria during regeneration. To test whether Sirt1 activity is required for the UPRmt during regeneration, zebrafish larvae, upon receiving fin amputation, were treated with a Sirt1/Sirt2 inhibitor, Sirtinol (Figure 2D), from 1 to 4 hpa. Evaluation of hspd1 expression levels by in situ hybridization show that regeneration-induced hspd1 expression is blocked in the Sirtinol treated larvae (Figure 2E). However, another mammalian Sirt1 inhibitor, EX527, showed no effect, perhaps due to species differences. Sirtuins use NAD+ to catalyze protein deacetylation, which produces de-acetylated protein and nicotinamide (NAM). A system of enzymes then reverts NAM back to NAD+ in a pathway called the NAD+ salvage pathway (Figure 2D). To verify that the phenotype of the Sirtinol treated larvae is indeed due to sirtuin inhibition, as well as to probe the question whether intracellular NAD+ level is critical for regeneration, we attempted to inhibit sirtuin activity indirectly via blocking the NAD+ salvage pathway. We employed FK866, which inhibits the activity of Nampt, an enzyme in the salvage pathway. Indeed, hspd1 in situ hybridization assays showed that FK866 effectively blocks the regeneration-induced hspd1 expression (Figure 2E). To show that the UPRmt is blocked by these small molecule treatments, we performed qRT-PCR for all four UPRmt signature genes. The result showed that in general the UPRmt genes are less activated in the Sirtinol and FK866 treated fins, albeit to varying degrees (Figure 2F). Together, the data support the idea that Sirt1 is a regulator of the UPRmt during fin regeneration.
Sirt1 inhibition causes fin regeneration defects
To test the effect of Sirtinol on fin regeneration, amputated larvae were incubated in Sirtinol for the duration of full regeneration (up to 3 dpa). However, extended incubation (more than 6 h) in Sirtinol results in non-specific toxicity and larval death. Washing out Sirtinol at 4 hpa produced a moderate but significant regeneration growth defect at 3 dpa (Figure 3A). We attributed the mild phenotype to inadequate inhibition of Sirt1 by the short Sirtinol treatment. NAM, which is one of the end products of sirtuin enzymatic reaction, works at a high concentration as a non-competitive inhibitor for sirtuins (Bitterman et al., 2002). Because NAM is more tolerable to fish larvae than Sirtinol, we incubated amputated larvae in a high concentration of NAM (20 mM) for the duration of regeneration. At day 3, fin regeneration growth was moderately but significantly blocked (Figure 3B). To maximize Sirt1 inhibition we then sought to inhibit Sirt1 orthogonally with Sirtinol in conjunction with a low concentration of NAM (10 mM). During regeneration, we incubated Sirtinol-treated larvae in low NAM after washing out Sirtinol from 4 hpa until 3 dpa. Remarkably, the combination of Sirtinol (1–4 hpa) and 10 mM NAM (5–72 hpa) treatment (but not NAM alone) produced a very strong inhibitory effect on regeneration without causing larval death (Figure 3A). This synergistic interaction between Sirtinol and NAM further illustrates the critical role of the NAD+ salvage pathway to remove NAM and maintain Sirt1 activity during fin regeneration. A similar regeneration defect was also seen in the larvae treated with FK866 and NAM (Figure 3A).
Figure 3.

Sirt1 inhibition causes a fin regeneration defect in larval fish
(A) Representative images of larval fins show the outcomes of the regenerative fin growth in the control and the Sirtinol + NAM treatment conditions at 3 dpa. (The white dashed line in the left panel indicates the resecting line.) The quantification in the graph on the right shows that Sirt1 inhibition by Sirtinol alone or combination of Sirtinol or FK866 with NAM significantly impairs regenerative fin growth in larval fish.
(B) The quantification shows that Sirt1 inhibition by a high concentration of NAM significantly impairs regenerative fin growth in larval fish.
(C) The schematic shows the alignment of genomic DNA sequence of the wild type and a sirt1 mutation containing a 28 bp insertion in exon 1 generated by CRISPR/Cas9, which results in a premature stop codon (indicated by an asterisk). The official allele name for this mutation is sirt1wcm18/wcm18. The agarose gel image shows that PCR of genomic DNA generates a larger band from a homozygous sirt1 mutant fish compared to wildtype (WT).
(D) Representative images (left) and the quantification result (right) show that sirt1 mutant fish fail to regenerate fins when incubated in NAM. Each dot represents fin regeneration measurement from one fish. Error bars indicate standard deviation. Student’s t test was performed to determine statistical significance. ∗p < 0.05.)
The function of Sirt1 is regeneration specific
Many regeneration regulators also play important roles in embryonic development. To determine whether Sirt1 promotes regeneration by regulating developmental growth or processes specific for regeneration, we sought to examine the effect of Sirt1 inhibition on embryonic fin development. Because Sirtinol causes non-specific toxicity to fish larvae over a long incubation, a genetic approach was carried out instead by targeted mutation of the sirt1 gene. To this end, we employed CRISPR/Cas9 technology to generate sirt1 mutant fish, and obtained a sirt1 mutation, with a 28 bp insertion in exon 1, which results in a premature translation termination and presumably a null allele (Figure 3B). However, despite the presumably strong lesion, the sirt1 mutant fish upon fin amputation show no regeneration defect in larvae (Figure S2A). We considered that the lack of phenotype might be because of a rescue from other sirtuin genes, via a genetic compensation mechanism seen in other zebrafish mutants harboring a premature termination codon leading to RNA nonsense mediated decay (El-Brolosy et al., 2019). Because, like in mammals, there are 7 sirtuin genes in the zebrafish genome (sirt1-7), genetic compensation might reasonably account for the lack of regeneration phenotype, but we hypothesized that a stronger phenotype might manifest under a more stressful condition. Indeed, when the amputated sirt1 mutant larvae were incubated in a low concentration of NAM, they exhibit a strong fin regeneration phenotype (Figure 3D) as seen in the Sirtinol treatment experiment (Figure 3A). To ensure the specificity of the mutant phenotype, a second independent sirt1 mutant allele was identified, which carries a 5 bp deletion and also results in a premature stop codon (Figure S2B). The fish larvae harboring this second mutation also showed no innate phenotype in fin regeneration, but exhibited a severe fin regeneration defect when treated with low NAM (Figure S2C). This was phenocopied when the sirt1 mutant larvae were also treated with Sirtinol, and this did not make the fin regeneration phenotype any worse (Figure S2D). Taken together, we conclude that the fin regeneration defect can be confidently attributed to sirt1 loss of function. Interestingly, the sirt1 mutant fish do not exhibit any defects in fin development in 3 day-old larvae even when incubated in NAM (data not shown). This result supports the idea that Sirt1 regulates a specific process in fin regeneration as opposed to developmental fin growth.
Sirt1 inhibition blocks the UPRmt and causes a fin regeneration defect in adult fish
The conserved UPRmt activation in both adult and larval injured fins predicts that Sirt1 is also required for adult fin regeneration. To investigate if that is the case, we blocked Sirt1 activity by treating amputated adult fish with a high concentration of NAM (20 mM, see STAR Methods) added to the water. At 5 dpa the amount of growth is significantly reduced in the treated fish (Figure 4A). According to hspd1 in situ hybridization assays, the regeneration defect in the NAM treated fish is likely a result of UPRmt suppression, as hspd1 expression levels are much reduced in fin blastema at 2 dpa, before the regeneration growth defect is apparent (Figure 4B). To further demonstrate that Sirt1 regulates fin regeneration via UPRmt activation, we performed a genetic interaction experiment. Taking advantage of the fact that the nbl (no blastema) mutant allele of hspd1 is temperature-sensitive (Makino et al., 2005), homozygous nbl fish upon fin amputation were incubated at a semi-permissive temperature (30°C) and a lower dose of NAM (10 mM). The result showed that each single condition did not cause any fin regeneration defect. However, in the context of the mutation, low level NAM treatment exerts a strong inhibitory effect on regeneration growth (Figure 4C). Therefore, Sirt1 regulates the UPRmt to promote fin regeneration in adult zebrafish.
Figure 4.

Sirt1 inhibition impairs adult fin regeneration via reduction of the UPRmt
(A) Representative images (left) and quantification (right) show that inhibiting Sirt1 by adding NAM to fish water impairs fin regeneration in adult zebrafish.
(B) Representative in situ hybridization images show that NAM treatment reduces hspd1 expression levels in the blastema of an injured adult fin.
(C) The representative images (left) and quantification (right) show that incubation of injured hspd1 mutant fish at a semi-permissive temperature in conjunction with a low concentration of NAM results in significant inhibition of fin regeneration.
(D) Quantification of regeneration growth shows that sirt1 mutant larval fish exhibit a fin regeneration defect when treated with doxycycline. Each dot represents a fin regeneration measurement from one fish. Error bars indicate standard deviation. Student’s t test was performed to determine statistical significance. ∗p < 0.05.).
Inhibiting mitochondrial translation exacerbates the fin regeneration phenotype of the sirt1 mutant
The RNA sequencing data indicates that mitochondrial translation is enhanced during fin regeneration; particularly many of the mitochondrial ribosomes subunits are up-regulated. To investigate whether mitochondrial translation plays a role in fin regeneration, we blocked mitochondrial translation in injured larvae. Doxycycline, an antibiotic, perturbs mitochondrial ribosome and inhibits mitochondrial translation. Wild type larvae treated with doxycycline within the sublethal dose range did not exhibit any obvious regeneration defect. Strikingly, when the same treatment was performed on the sirt1 mutant larvae, a severe fin regeneration defect was observed (Figure 4D). The synergistic interaction between sirt1 mutation and doxycycline treatment indicates that enhanced mitochondrial translation is one of the downstream events of Sirt1 activation during regeneration. Collectively, our data suggest a model in which the UPRmt is a conserved pathway promoting fin regeneration in both adult and larval zebrafish. The UPRmt is likely activated to cope with the enhanced mitochondrial translation and the resulting biogenesis in blastema or fin fold mesenchymal cells, which is induced by Sirt1 under the condition of increased intracellular NAD+.
Discussion
The UPRmt is involved in a broad range of physiological and disease conditions, which demonstrates the versatility of this ancient pathway. Here we show that the UPRmt, via the regulation of Sirt1, is important for fin regeneration in zebrafish. Abrogating the function of Sirt1 via chemical inhibitors or genetic mutations represses UPRmt activation, and consequently, tissue regeneration. The Sirt1-UPRmt pathway is evolutionarily conserved in its role in regulating lifespan from worms to mammals. In the context of lifespan extension Sirt1 promotes mitochondrial biogenesis along with activating the UPRmt to preserve metabolic activity and mitochondrial integrity against the aging process (Mouchiroud et al., 2013). Analogous molecular processes occur during tissue regeneration, as we show that mitochondrial biogenesis, indicated by increased expression of the nuclear genome encoded mitochondrial translation and transcription genes, takes place simultaneously with UPRmt activation. Mitochondrial biogenesis associated with UPRmt activation has also been observed during embryonic development and stem cell differentiation (Mohrin et al., 2015; Shpilka et al., 2021). Although in some aspects tissue regeneration is regarded as a reiteration of embryonic developmental growth, we believe that the Sirt1-UPRmt pathway is uniquely required for regeneration based on the regeneration specific phenotype in the sirt1 mutant fish. In keeping with this idea, a mutation in zebrafish polg, a mitochondrial DNA polymerase, exhibits defects in fin regeneration but not in embryonic development (Rahn et al., 2015). Therefore, regenerative growth imposes special challenges to mitochondrial homeostasis possibly in the form of a surge of mitochondrial biogenesis, and it would be interesting to determine whether other mitochondrial biogenesis regulators are also required for tissue regeneration.
Besides inducing expression of mitochondrial protein chaperones and proteases, Sirt1 also activates an antioxidant defense response via the FOXO3a/SOD2 pathway (Mouchiroud et al., 2013). In the context of lifespan extension, the antioxidant defense likely functions to protect cells from ROS produced from the aged mitochondria. A similar connection between the UPRmt and antioxidant defense is also discovered in mitochondrial stressed cancer cells, which intriguingly involves a different sirtuin, Sirt3 (Papa and Germain, 2014). The antioxidant defense is now thought to constitute a separate axis of the URPmt signaling because of its prevalence and apparent independence to the canonical UPRmt pathway that promotes mitochondrial proteostasis (Munch, 2018). Although we did not detect activation of SOD2 at the onset of regeneration, it is possible that SOD2 activation does not take place simultaneously with the canonical UPRmt but at a later stage of regeneration, as the case in lifespan extension (Mouchiroud et al., 2013). Moreover, the ability of Sirt3 to mediate SOD2 activation in stressed cancer cells suggests that Sirt3 might compensate for loss of Sirt1 and rescue fin regeneration.
The multiple signaling modalities of the UPRmt signify that the response of a cell to mitochondrial stress could vary according to factors, such as the nature and severity of the stress and metabolic activity. For instance, when cultured cells are treated with exogenous mitochondrial stressors, repression of mitochondrial translation is one of the main responses, which takes place in parallel with the canonical UPRmt axis (Munch and Harper, 2016; Quiros et al., 2017). However, in Sirt1-mediated lifespan extension, an increase in the mitochondrial encoded OXPHOS protein is thought to be the trigger of the canonical UPRmt axis. A recent study in worms demonstrated that elevated mitochondrial protein synthesis is associated with UPRmt hyperactivation (Shpilka et al., 2021). The seeming discrepancy highlights the fact that our view of mitochondrial stress response based on treating cultured cells with exogenous stressors might be incomplete. Due to the signature of increased mitochondrial translation, our study suggests that the Sirt1-UPRmt pathway in fin regeneration, along with its role in life span extension, and the embryonically activated UPRmt, represent a distinct mode of UPRmt activation when cells are under mitochondrial network expansion. Further studies of the UPRmt in physiological conditions, such as zebrafish fin regeneration, might provide valuable new information regarding how cells respond to mitochondrial stress in vivo.
Regulation of sirtuin activity is closely tied to cellular metabolism, because NAD+, a key metabolite participating in many metabolic pathways, is the obligatory cofactor of the protein deacetylation reaction (Canto and Auwerx, 2012). In addition, the expression of nampt, an enzyme in the NAD+ salvage pathway, is influenced by changes in the organismal energy homeostasis, for example in diabetes (Yoshino et al., 2011), to exert influence on Sirt1 activity. Given that Sirt1 and Nampt inhibition both result in fin regeneration defects, our data suggest that activation of the Sirt1-UPRmt axis is likely tied to certain forms of metabolic remodeling during fin regeneration. Interestingly, a number of recent studies have suggested that tissue regeneration depends on a global overhaul of energy usage. Neuroendocrine systems, such as leptin and melanocortin, which normally regulate organismal energy homeostasis and feeding behavior, are involved in tissue regeneration (Kang et al., 2016; Zhang et al., 2018). In zebrafish, a leptin gene, lepb, is highly up-regulated in fin blastema cells. In the Xenopus tadpole, melanocortin signaling is necessary for limb regeneration through modulating mitochondrial energy output in injured limb tissue. However, the connections between remodeling of cellular metabolism and regeneration growth remain largely unknown. Future studies focused on identifying metabolic pathways that contribute to the NAD+ increase and Sirt1 activation could provide critical insights to this emerging area of regeneration biology.
The evolutionarily conserved role of the Sirt1-UPRmt axis in lifespan regulation draws an interesting parallel between aging and tissue regeneration. Certainly, the cell dedifferentiation process that drives the formation of the blastema has been compared to iPS cell formation, a process that rejuvenates cells (Studer et al., 2015). Because aging doesn’t significantly impede fin regeneration capacity in zebrafish (Itou et al., 2012), it is plausible that this rejuvenation process resets the “age” of blastema cells. A recent study showing that telomerase and elongation of telomeres are important to heart regeneration in zebrafish further supports this idea (Bednarek et al., 2015). Interestingly, these two subcellular structures have long been shown interacting during the course of aging, where telomere shortening leads to impairment of mitochondrial biogenesis and, consequently, cell senescence (Sun et al., 2016). Therefore, it would be very interesting to probe the relationship between telomere regulation and mitochondrial biogenesis during regeneration and determine how the two processes might be coordinated. The findings may shed light on the intriguing relationship between aging and tissue regeneration. Furthermore, strategies to elevate intracellular NAD+ have been under intense research effort because of the effect on mitochondrial health and lifespan extension (Gariani et al., 2016; Rajman et al., 2018). The zebrafish fin regeneration model potentially provides a useful platform for identifying new strategies to raise NAD+ levels in vivo to prevent aging or mitigate other disease conditions where the Sirt1-UPRmt axis plays a prominent role.
Limitations of study
For technical reasons due to lack of reagents or sensitivity, we tried but were unable to compare MTOC/SDHA ratios in control and regenerating fins, or to evaluate if there is nuclear localization of acetylated Foxo3 during the process. We sought to create a ubl-5 loss of function mutant, as Ubl-5 has been shown to be transported to the nucleus in the UPRmt activated cells. Unfortunately, zebrafish ubl-5 (pin1) has not been annotated or even mapped to the zebrafish genome, so it is not yet feasible to perform CRISPR-Cas9 mutagenesis. We instead took a cell biological approach and created a transgenic fish expressing Ubl-5-GFP in larval fin fold mesenchymal cells. However, we did not detect an increase of nuclear localization of Ubl-5 upon injury (not shown). This result suggests that Ubl-5 is not involved in UPRmt activation during fin regeneration, but a genetic experiment is still required to definitively rule out ubl-5. Finally, rescuing regeneration by activating UPRmt downstream of the SIRT-NAM axis would support the model. Unfortunately, there is currently no chemical tool that can ectopically activate the UPRmt besides mitochondrial toxins. Treating larval zebrafish with a mitochondrial toxin, such as the Complex I inhibitor rotenone, resulted in a strong general toxic effect, which prohibited regeneration experiments (not shown).
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| One Shot TOP10 Chemically Competent E. coli | Invitrogen | C404006 |
| Chemicals, Peptides and Recombinant Proteins | ||
| Tricaine (MS-222) | Syndel | Tricaine-S |
| SU5402 | Selleckchem | S7667 |
| Nicotinamide (NAM) | Sigma | N0636 |
| Sirtinol | Sigma | 566321 |
| FK-866 | Cayman Chemical | 13287 |
| Doxycycline | Sigma | D9891 |
| Cas9 protein | PNA Bio | CP01-200 |
| T7 nuclease | New England Biolabs | M0302L |
| Pfusion Taq polymerase | New England Biolabs | M0535L |
| Critical Commercial Assays | ||
| mMESSAGE mMACHINE | Invitrogen | AM1340 |
| MEGAshortscript Kit | ThermoFisher SCIENTIFIC | AM1354 |
| RNeasy Mini Kit | QIAGEN | 74104 |
| Superscript VILO cDNA Synthesis Kit | Invitrogen | 11754050 |
| LightCycler 480 SYBR Green master mix | Roche | 04-887-352-001 |
| TruSeq RNA Library Prep Kit v2 | Illumina | RS-122-2001 |
| Gateway LR Clonase II Enzyme mix | ThermoFisher SCIENTIFIC | 11791020 |
| Trizol Reagent | ThermoFisher SCIENTIFIC | 15596018 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO:GSE182113 |
| Experimental Models: Organisms/Strains | ||
| Zebrafish hspd1 mutant strain (nbl) | ZIRC | ZL737 |
| Zebrafish strain sirt1wcm18/wcm18 | This paper | sirt1wcm18/wcm18 |
| Zebrafish strain sirt1wcm19/wcm19 | This paper | sirt1wcm19/wcm19 |
| Zebrafish strain Tg(hspd1:d2-gfp)wcm20 | This paper | Tg(hspd1:d2-gfp)wcm20 |
| Oligonucleotides | ||
| PCR primer sequences | IDT | See Table S1 |
| sirt1 mutant genotyping primers | IDT | See Table S1 |
| hspd1 promoter cloning primers | IDT | See Table S1 |
| Mouse ODC D2 fragment cloning primers | IDT | See Table S1 |
| sirt1 gRNA for CRISPR-Cas9 mutagenesis | IDT | See Table S1 |
| Recombinant DNA | ||
| pCR4Blunt-TOPO | ThermoFisher SCIENTIFIC | K287520 |
| pDestTol2pA2 | Addgene | http://tol2kit.genetics.utah.edu |
| p5E-MCS | Addgene | http://tol2kit.genetics.utah.edu |
| pCS2FA-transposase | Addgene | http://tol2kit.genetics.utah.edu |
| pME-EGFP | Addgene | http://tol2kit.genetics.utah.edu |
| Software and Algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| R | Cran | https://www.R-project.org/ |
| Star V2.3 | rna-star | http://code.google.com/p/rna-star/ |
| DESeq2 | Bioconductor | https://doi.org/10.18129/B9.bioc.DESeq2 |
| Prism7 | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
| GSEA | UC-SanDiego, Broad Institute | https://www.gsea-msigdb.org/gsea/ |
Resource availability
Lead contact
Further information and requests for reagents should be directed to and will be fulfilled by the Lead Contact, Todd Evans (tre2003@ med.cornell.edu).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Experimental model and subject details
Zebrafish were used with approval by the Weill Cornell Medicine College (WCMC) and National Health Research Institutes (NHRI) institutional animal care and use committees. We used 0–6 days post fertilization (dpf) larval zebrafish and 6- to 12-month-old adult zebrafish for this study, bred on an AB/TUB background. Zebrafish were raised under standard conditions at 28C and morphologically staged. No statistical methods were used to predetermine sample size, and animal selection was not randomized or blinded. Mutant fish lines utilized in this study include sirt1wcm18/wcm18 and sirt1wcm19/wcm19. The transgenic reporter line Tg(hspd1:d2-gfpwcm20) was used for imaging UPRmt activation in larval and adult injured fins during regeneration. Embryos selected for experiments were typically less than 6 dpf, a stage at which sex cannot be determined and is unlikely to influence the biological processes under study. For experiments using adult fish, both male and female animals were included in the analysis, and results were not affected by sex.
Methods details
Fish strains, transgenesis and mutagenesis
All zebrafish strains were maintained at 28.5°C in WCMC (USA) and NHRI (Taiwan) zebrafish facilities, and all procedures carried out as approved by the WCMC and NHRI IACUCs. The hspd1 mutant fish (nbl) was obtained from ZIRC. The transgenic zebrafish, Tg(hspd1:d2gfpwcm20), was generated via the Tol2 transposon method (Clark et al., 2011). The official allele name for this strain is wcm20. To generate this line, the ∼6 kb hspd1 genomic sequence spanning from 2666 bp upstream of the transcription start to 2971 bp downstream until the translation start (including the first noncoding exon) was cloned from wildtype genomic DNA. The d2GFP cassette was created by fusing DNA encoding amino acids 422-461 of mouse ornithine decarboxylase, cloned from mouse cardiomyocyte cDNA, to the C-terminus of GFP. After cloning these elements into the destination vector pDestTol2pA2, 25 pg DNA was co-injected with 25 pg RNA encoding Tol2 recombinase (in 2 nl volume) into 1-2 cell embryos that were subsequently grown to adulthood. Potential F0 founders were outcrossed and tested for transmission of the transgene. Several independent F1 lines were isolated and one line maintained as Tg(hspd1:d2gfpwcm20). Primers used for generating the construct are given in Table S1. The two sirt1 mutant lines were created via the CRISPR/Cas9 procedure, with a previously validated sgRNA (Kim et al., 2019), which is listed in Table S1. For this purpose, the guide RNA (gRNA) sequence was ordered from IDT as a double stranded oligomer cassette with a T7 promoter at the 5’ end. The gRNA was generated with the MEGAshortscript kit (Thermo Fisher) and purified through ammonium acetate precipitation as per the manufacturer’s instructions. The mRNA was quantified with a Nanodrop and analyzed by electrophoresis through a 1% agarose gel with ethidium bromide in order to verify that there was no degradation. 250 ng/μl gRNA was combined with 500 ng/μl recombinant Cas9 protein (PNA BIO) and 0.2M KCl, and incubated at room temperature for 15 minutes to allow RNA-protein complexes to form. 1 nl of the gRNA-Cas9 complex solution was injected into embryos at the 1-2 cell stage.
Putative F0 founder fish were raised to adulthood and outcrossed to wildtype fish (AB/Tub hybrids). Several samples of genomic DNA from each clutch (typically 2-3 samples from 5-10 embryos) were analyzed for mutant alleles by T7 endonuclease assays following PCR (Pfusion Taq, NEB). When mutant alleles were detected, the remaining embryos were raised to adulthood, and F1 heterozygous fish were identified by genotyping similarly from fin-clips. PCR products were also cloned into a TOPO vector (ThermoFischer) and sequenced to define each mutant allele. The two mutant lines, which resulted from a single guide RNA but independent Cas9 cutting events, were isolated from two independent founder fish. Homozygous mutant fish were identified by PCR (primers shown in Table S1) of genomic DNA and PCR products sequenced to confirm the lesion. The mutant allele with a 28 bp insertion (Figure 3) is designated sirt1wcm18/wcm18. The mutant allele with a 5 bp deletion (Figure S2) is designated sirt1wcm19/wcm19.
Larval fish amputation, small molecule treatment and regeneration assay
Wild-type and sirt1 mutant larvae at 3 dpf were anesthetized in 0.2 mg/ml tricaine for 20 min before amputation with a resection line past the rostral tip of the notochord to remove tail fin fold tissue. Amputated larvae were recovered in E3 for 40 min to 1 hour before incubation in Sirtinol (20μM, Sigma), FK-866 (100μM, Cayman) or SU5402 (5μM, Selleckchem) for 4 hours for the in situ hybridization and the qRT-PCR experiments. For the regeneration growth assay, the treated larvae were then transferred to NAM (10 mM, Sigma) and incubated for 3 days before evaluated for regeneration. 3 dpf amputated sirt1 mutant larvae were recovered in E3 for 2 hours before incubation in 10 mM NAM or 60 μg/mL doxycycline (Sigma) for 3 days. Regeneration length was measured as the distance from the tip of the notochord to the point where the body A-P axis intersects with the edge of the newly formed larval fin fold. All data were derived from 3 biologically independent experiments. For the sirt1 mutant fin development analyses, 1 dpf mutant embryos were incubated in 10 mM NAM for 48 hours until larval fin fold is formed.
Adult fish amputation, NAM treatment and regeneration assay
Wild-type adult fish aged from 6 to 9 months were subjected to fin amputation following anesthesia with tricaine (0.2mg/ml) in which tail fins were cut with a razor blade along a line that roughly intersects at the middle of and is perpendicular to the shortest fin array. Amputated fish were then recovered in system water at a density of 1 per 100 ml. For NAM treatment, fish were transferred to 50 mM NAM at 3 hpa for 6 hours and then switched to 20 mM NAM (high NAM) for the duration of regeneration until 5 dpa, during which time fish were fed once a day and water containing NAM was changed twice a day. For hspd1 mutant fish, a similar procedure was followed except fish were incubated with 10 mM NAM (low NAM) at a semi-permissive temperature of 30°C for 2 days. The regeneration outcome of adult fish was measured as average growth per fin array, which sums the distance measurement of each fin array from the tip of new tissue to the border of the old tissue, based on pigmentation, and divided by the number of fin arrays. All data were derived from 3 biologically independent experiments.
RNA sequencing, qRT-PCR analysis and in situ hybridization
RNA samples were extracted from the regenerating fin along with a small amount of the adjacent tissue using Trizol. In larval experiments, fin fold tissue was collected from the resected line towards anterior for approximately 100 μm, where the intersection of ventral notochord pigmentation and the caudal vasculature serves as a landmark. (The collected fin fold tissue includes the terminal tip of the notochord.) Approximately 100 fin folds were collected for preparing each RNA sample. For transcript profiling, whole embryo RNA samples were used to prepare libraries with the TruSeq RNA Library Prep Kit v2 (Illumina) and submitted to WCMC Genomics Resources Core Facility for sequencing. Paired end reads were aligned using Star v2.3 to the zebrafish genome (GRCz10) using the Ensembl transcriptome. Analysis of differential gene expression was performed using DESeq2. Differentially expressed genes were defined by log2 fold change greater than 2 or less than −2 and an adjusted p-value<0.05. GSEA was performed on RNAseq data using normalized counts. Signatures represented in the results are the top 10, FDR < 25%, nominal p-value<5%. RNA-seq data is available in the GEO data repository (GSE182113). Heatmaps of relative gene expression were generated based on z-score calculated with the normalized gene counts generated with the DESeq2 package in R. For whole mount in situ hybridization, zebrafish embryos at the desired stages were fixed in 4% paraformaldehyde (PFA) before hybridization to antisense RNA probes, according to standard protocols (Jowett, 1999). Whole embryo RNA was isolated with the RNeasy Mini kit (Qiagen). For quantitative RT-PCR (qPCR) at least 5 larvae were used for each biological replicate. RNA was reverse transcribed with the Superscript Vilo Synthesis System (Invitrogen). The qPCR analysis was performed on a LightCycler 480 II (Roche) using LightCycler 480 Sybr Green master mix (Roche). Relative gene expression levels were determined using eif1a (eef1a1l1) as a reference gene (McCurley and Callard, 2008). Primers for generating the in situ hybridization probes and for the qRT-PCR assays are listed in Table S1. All data were derived from 3 biologically independent experiments.
Quantification and statistical analysis
Statistical analysis was performed using Graphpad Prism 7 unless otherwise indicated. Data comparing two samples were analyzed using Student's t test. The significance is indicated as ∗p < 0.05.
Acknowledgments
We thank the Zebrafish International Resource Center for providing zebrafish strains, the Genomics core facility at Weil Cornell Medical College for RNA sequencing and the Taiwan Zebrafish Core facility at the National Health Research Institutes for assistance in zebrafish shipment and husbandry. This study was supported by the Department of Surgery, WCM (T.E.). T.E. is supported by an Outstanding Investigator Award from the NHLBI (R35 HL135778). J.S. is supported by a training grant from the New York State Department of Health (NYSTEM C32558GG). Y.-F.L. was supported by a grant from the Ministry of Science and Technology in Taiwan (109-2311-B-007-009-MY).
Author contributions
Y-F.L. and T.E. designed research, analyzed results and wrote the paper. Y-F.L. performed most of the experiments. J.S. also performed experiments.
Declaration of interests
The authors declare no competing interests.
Published: October 22, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103118.
Contributor Information
Yi-Fan Lin, Email: yflin@life.nthu.edu.tw.
Todd Evans, Email: tre2003@med.cornell.edu.
Supplemental information
Data and code availability
-
•
Data. RNA-sequencing data has been deposited at GEO and are available as of the date of publication, under accession number GSE182113, which is also indicated in the key resources table.
-
•
Code. This paper does not report original code.
-
•
All other items. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Baker B.M., Haynes C.M. Mitochondrial protein quality control during biogenesis and aging. Trends Biochem. Sci. 2011;36:254–261. doi: 10.1016/j.tibs.2011.01.004. [DOI] [PubMed] [Google Scholar]
- Bednarek D., Gonzalez-Rosa J.M., Guzman-Martinez G., Gutierrez-Gutierrez O., Aguado T., Sanchez-Ferrer C., Marques I.J., Galardi-Castilla M., de Diego I., Gomez M.J. Telomerase is essential for zebrafish heart regeneration. Cell Rep. 2015;12:1691–1703. doi: 10.1016/j.celrep.2015.07.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitterman K.J., Anderson R.M., Cohen H.Y., Latorre-Esteves M., Sinclair D.A. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J. Biol. Chem. 2002;277:45099–45107. doi: 10.1074/jbc.M205670200. [DOI] [PubMed] [Google Scholar]
- Canto C., Auwerx J. Targeting sirtuin 1 to improve metabolism: all you need is NAD(+)? Pharmacol. Rev. 2012;64:166–187. doi: 10.1124/pr.110.003905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H.C., Guarente L. SIRT1 and other sirtuins in metabolism. Trends Endocrinol. Metab. 2014;25:138–145. doi: 10.1016/j.tem.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.H., Poss K.D. Regeneration genetics. Annu. Rev. Genet. 2017;51:63–82. doi: 10.1146/annurev-genet-120116-024554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark K.J., Urban M.D., Skuster K.J., Ekker S.C. Transgenic zebrafish using transposable elements. Methods Cell Biol. 2011;104:137–149. doi: 10.1016/B978-0-12-374814-0.00008-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durieux J., Wolff S., Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011;144:79–91. doi: 10.1016/j.cell.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Brolosy M.A., Kontarakis Z., Rossi A., Kuenne C., Gunther S., Fukuda N., Kikhi K., Boezio G.L.M., Takacs C.M., Lai S.L. Genetic compensation triggered by mutant mRNA degradation. Nature. 2019;568:193–197. doi: 10.1038/s41586-019-1064-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorese C.J., Schulz A.M., Lin Y.F., Rosin N., Pellegrino M.W., Haynes C.M. The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Curr. Biol. 2016;26:2037–2043. doi: 10.1016/j.cub.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H., Zhou H., Yu X., Xu J., Zhou J., Meng X., Zhao J., Zhou Y., Chisholm A.D., Xu S. Wounding triggers MIRO-1 dependent mitochondrial fragmentation that accelerates epidermal wound closure through oxidative signaling. Nat. Commun. 2020;11:1050. doi: 10.1038/s41467-020-14885-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gariani K., Menzies K.J., Ryu D., Wegner C.J., Wang X., Ropelle E.R., Moullan N., Zhang H., Perino A., Lemos V. Eliciting the mitochondrial unfolded protein response by nicotinamide adenine dinucleotide repletion reverses fatty liver disease in mice. Hepatology. 2016;63:1190–1204. doi: 10.1002/hep.28245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han B., Sivaramakrishnan P., Lin C.J., Neve I.A.A., He J., Tay L.W.R., Sowa J.N., Sizovs A., Du G., Wang J. Microbial genetic composition tunes host longevity. Cell. 2017;169:1249–1262.e1213. doi: 10.1016/j.cell.2017.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itou J., Kawakami H., Burgoyne T., Kawakami Y. Life-long preservation of the regenerative capacity in the fin and heart in zebrafish. Biol. Open. 2012;1:739–746. doi: 10.1242/bio.20121057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovaisaite V., Auwerx J. The mitochondrial unfolded protein response-synchronizing genomes. Curr. Opin. Cell Biol. 2015;33:74–81. doi: 10.1016/j.ceb.2014.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jowett T. Analysis of protein and gene expression. Methods Cell Biol. 1999;59:63–85. doi: 10.1016/s0091-679x(08)61821-x. [DOI] [PubMed] [Google Scholar]
- Kang J., Hu J., Karra R., Dickson A.L., Tornini V.A., Nachtrab G., Gemberling M., Goldman J.A., Black B.L., Poss K.D. Modulation of tissue repair by regeneration enhancer elements. Nature. 2016;532:201–206. doi: 10.1038/nature17644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny T.C., Craig A.J., Villanueva A., Germain D. Mitohormesis primes tumor invasion and metastasis. Cell Rep. 2019;27:2292–2303.e2296. doi: 10.1016/j.celrep.2019.04.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D.H., Jung I.H., Kim D.H., Park S.W. Knockout of longevity gene Sirt1 in zebrafish leads to oxidative injury, chronic inflammation, and reduced life span. PloS One. 2019;14:e0220581. doi: 10.1371/journal.pone.0220581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knopf F., Hammond C., Chekuru A., Kurth T., Hans S., Weber C.W., Mahatma G., Fisher S., Brand M., Schulte-Merker S. Bone regenerates via dedifferentiation of osteoblasts in the zebrafish fin. Dev. Cell. 2011;20:713–724. doi: 10.1016/j.devcel.2011.04.014. [DOI] [PubMed] [Google Scholar]
- Makino S., Whitehead G.G., Lien C.L., Kim S., Jhawar P., Kono A., Kawata Y., Keating M.T. Heat-shock protein 60 is required for blastema formation and maintenance during regeneration. Proc. Natl. Acad. Sci. U. S. A. 2005;102:14599–14604. doi: 10.1073/pnas.0507408102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateus R., Pereira T., Sousa S., de Lima J.E., Pascoal S., Saude L., Jacinto A. In vivo cell and tissue dynamics underlying zebrafish fin fold regeneration. PloS One. 2012;7:e51766. doi: 10.1371/journal.pone.0051766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCurley A.T., Callard G.V. Characterization of housekeeping genes in zebrafish: male-female differences and effects of tissue type, developmental stage and chemical treatment. BMC Mol. Biol. 2008;9:102. doi: 10.1186/1471-2199-9-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra P., Chan D.C. Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 2016;212:379–387. doi: 10.1083/jcb.201511036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohrin M., Shin J., Liu Y., Brown K., Luo H., Xi Y., Haynes C.M., Chen D. Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science. 2015;347:1374–1377. doi: 10.1126/science.aaa2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouchiroud L., Houtkooper R.H., Moullan N., Katsyuba E., Ryu D., Canto C., Mottis A., Jo Y.S., Viswanathan M., Schoonjans K. The NAD(+)/Sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell. 2013;154:430–441. doi: 10.1016/j.cell.2013.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munch C. The different axes of the mammalian mitochondrial unfolded protein response. BMC Biol. 2018;16:81. doi: 10.1186/s12915-018-0548-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munch C., Harper J.W. Mitochondrial unfolded protein response controls matrix pre-RNA processing and translation. Nature. 2016;534:710–713. doi: 10.1038/nature18302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechiporuk A., Poss K.D., Johnson S.L., Keating M.T. Positional cloning of a temperature-sensitive mutant emmental reveals a role for sly1 during cell proliferation in zebrafish fin regeneration. Dev. Biol. 2003;258:291–306. doi: 10.1016/s0012-1606(03)00129-5. [DOI] [PubMed] [Google Scholar]
- Owlarn S., Klenner F., Schmidt D., Rabert F., Tomasso A., Reuter H., Mulaw M.A., Moritz S., Gentile L., Weidinger G. Generic wound signals initiate regeneration in missing-tissue contexts. Nat. Commun. 2017;8:2282. doi: 10.1038/s41467-017-02338-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papa L., Germain D. SirT3 regulates the mitochondrial unfolded protein response. Mol. Cell Biol. 2014;34:699–710. doi: 10.1128/MCB.01337-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei W., Tanaka K., Huang S.C., Xu L., Liu B., Sinclair J., Idol J., Varshney G.K., Huang H., Lin S. Extracellular HSP60 triggers tissue regeneration and wound healing by regulating inflammation and cell proliferation. NPJ Regen. Med. 2016;1:1–11. doi: 10.1038/npjregenmed.2016.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrino M.W., Nargund A.M., Kirienko N.V., Gillis R., Fiorese C.J., Haynes C.M. Mitochondrial UPR-regulated innate immunity provides resistance to pathogen infection. Nature. 2014;516:414–417. doi: 10.1038/nature13818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Z., Barthel L.K., Raymond P.A. Genetic evidence for shared mechanisms of epimorphic regeneration in zebrafish. Proc. Natl. Acad. Sci. U. S. A. 2009;106:9310–9315. doi: 10.1073/pnas.0811186106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiros P.M., Prado M.A., Zamboni N., D'Amico D., Williams R.W., Finley D., Gygi S.P., Auwerx J. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 2017;216:2027–2045. doi: 10.1083/jcb.201702058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahn J.J., Bestman J.E., Stackley K.D., Chan S.S. Zebrafish lacking functional DNA polymerase gamma survive to juvenile stage, despite rapid and sustained mitochondrial DNA depletion, altered energetics and growth. Nucleic Acids Res. 2015;43:10338–10352. doi: 10.1093/nar/gkv1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajman L., Chwalek K., Sinclair D.A. Therapeutic potential of NAD-boosting molecules: the in vivo evidence. Cell Metab. 2018;27:529–547. doi: 10.1016/j.cmet.2018.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehring I.M., Jahn C., Weidinger G. Zebrafish fin and heart: what's special about regeneration? Curr. Opin. Genet. Dev. 2016;40:48–56. doi: 10.1016/j.gde.2016.05.011. [DOI] [PubMed] [Google Scholar]
- Shpilka T., Du Y., Yang Q., Melber A., Uma Naresh N., Lavelle J., Kim S., Liu P., Weidberg H., Li R. UPR(mt) scales mitochondrial network expansion with protein synthesis via mitochondrial import in Caenorhabditis elegans. Nat. Commun. 2021;12:479. doi: 10.1038/s41467-020-20784-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shpilka T., Haynes C.M. The mitochondrial UPR: mechanisms, physiological functions and implications in ageing. Nat. Rev. Mol. Cell Biol. 2018;19:109–120. doi: 10.1038/nrm.2017.110. [DOI] [PubMed] [Google Scholar]
- Sorrentino V., Romani M., Mouchiroud L., Beck J.S., Zhang H., D'Amico D., Moullan N., Potenza F., Schmid A.W., Rietsch S. Enhancing mitochondrial proteostasis reduces amyloid-beta proteotoxicity. Nature. 2017;552:187–193. doi: 10.1038/nature25143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studer L., Vera E., Cornacchia D. Programming and reprogramming cellular age in the era of induced pluripotency. Cell Stem Cell. 2015;16:591–600. doi: 10.1016/j.stem.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun N., Youle R.J., Finkel T. The mitochondrial basis of aging. Mol. Cel. 2016;61:654–666. doi: 10.1016/j.molcel.2016.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga M., Sass M., Papp D., Takacs-Vellai K., Kobolak J., Dinnyes A., Klionsky D.J., Vellai T. Autophagy is required for zebrafish caudal fin regeneration. Cell Death Differ. 2014;21:547–556. doi: 10.1038/cdd.2013.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.T., Lim Y., McCall M.N., Huang K.T., Haynes C.M., Nehrke K., Brookes P.S. Cardioprotection by the mitochondrial unfolded protein response requires ATF5. Am. J. Physiol. Heart Circ. Physiol. 2019;317:H472–H478. doi: 10.1152/ajpheart.00244.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S., Chisholm A.D. C. elegans epidermal wounding induces a mitochondrial ROS burst that promotes wound repair. Dev. Cell. 2014;31:48–60. doi: 10.1016/j.devcel.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshinari N., Ishida T., Kudo A., Kawakami A. Gene expression and functional analysis of zebrafish larval fin fold regeneration. Dev. Biol. 2009;325:71–81. doi: 10.1016/j.ydbio.2008.09.028. [DOI] [PubMed] [Google Scholar]
- Yoshino J., Mills K.F., Yoon M.J., Imai S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14:528–536. doi: 10.1016/j.cmet.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Ryu D., Wu Y., Gariani K., Wang X., Luan P., D'Amico D., Ropelle E.R., Lutolf M.P., Aebersold R. NAD(+) repletion improves mitochondrial and stem cell function and enhances life span in mice. Science. 2016;352:1436–1443. doi: 10.1126/science.aaf2693. [DOI] [PubMed] [Google Scholar]
- Zhang M., Chen Y., Xu H., Yang L., Yuan F., Li L., Xu Y., Chen Y., Zhang C., Lin G. Melanocortin receptor 4 signaling regulates vertebrate limb regeneration. Dev. Cell. 2018;46:397–409.e395. doi: 10.1016/j.devcel.2018.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q., Wang J., Levichkin I.V., Stasinopoulos S., Ryan M.T., Hoogenraad N.J. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002;21:4411–4419. doi: 10.1093/emboj/cdf445. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Data. RNA-sequencing data has been deposited at GEO and are available as of the date of publication, under accession number GSE182113, which is also indicated in the key resources table.
-
•
Code. This paper does not report original code.
-
•
All other items. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.