

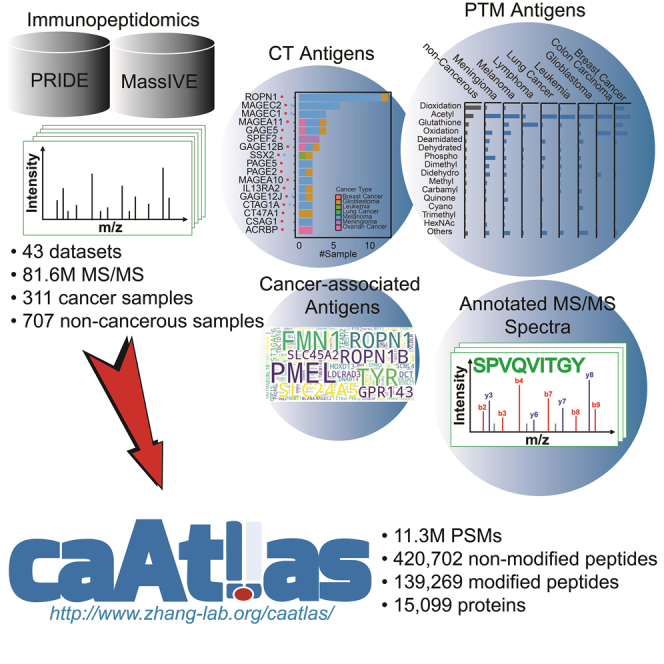
Summary
Comprehensive characterization of tumor antigens is essential for the design of cancer immunotherapies, and mass spectrometry (MS)-based immunopeptidomics enables high-throughput identification of major histocompatibility complex (MHC)-bound peptide antigens in vivo. Here we construct an immunopeptidome atlas of human cancer through an extensive collection of 43 published immunopeptidomic datasets and standardized analysis of 81.6 million MS/MS spectra using an open search engine. Our analysis greatly expands the current knowledge of MHC-bound antigens, including an unprecedented characterization of post-translationally modified antigens and their cancer-association. We also perform systematic analysis of cancer-testis antigens, cancer-associated antigens, and neoantigens. We make all these data together with annotated MS/MS spectra supporting identification of each antigen in an easily browsable web portal named cancer antigen atlas (caAtlas). caAtlas provides a central resource for the selection and prioritization of MHC-bound peptides for in vitro HLA binding assay and immunogenicity testing, which will pave the way to eventual development of cancer immunotherapies.
Subject areas: Immunology, Proteomics, Cancer
Graphical abstract
Highlights
-
•
Extensive collection of 43 immunopeptidomic datasets with 1018 samples
-
•
Standardized and rigorous identification of HLA-bound peptides, including PTM peptides
-
•
Comprehensive annotation of CT antigens and cancer-associated antigens
-
•
User-friendly data dissemination through the caAtlas web portal
Immunology; Proteomics; Cancer
Introduction
Immunotherapy that harnesses the human immune system to fight cancer has become an integral part of cancer treatment(Riley et al., 2019). At the core of immunotherapy is the T cell recognition of tumor antigens(Coulie et al., 2014). Thus, comprehensive characterization of tumor antigens is essential for the design of effective and safe cancer immunotherapy strategies.
Antigens are presented on the cell surface for T cell recognition by major histocompatibility complex (MHC) proteins, which are called human leukocyte antigen (HLA) proteins in humans. There are two classes of MHC proteins. MHC-class I (MHC-I) proteins, which are expressed in most nucleated cells, primarily present endogenously derived peptide antigens to CD8 T cells. MHC-class II (MHC-II) proteins are predominantly used by professional antigen-presenting cells to present exogenously-derived peptide antigens to CD4 T cells. However, many other cell types, including tumor cells, are also able to express MHC-II(Axelrod et al., 2019). There are three main HLA class I genes and six main class II genes in humans, and these genes are highly polymorphic, allowing each individual to have a unique set of HLA genes that can present a unique set of peptide antigens(Radwan et al., 2020).
Tumor antigens can be broadly classified into tumor-specific antigens and tumor-associated antigens(Haen et al., 2020; Ilyas and Yang, 2015). Neoantigens derived from oncoviruses and somatic mutations are exclusively presented on tumor cells but not on healthy cells, making them truly tumor-specific antigens. Next generation sequencing enables rapid and high throughput identification of somatic mutations altering the protein coding regions, and many computational tools have been developed to predict which mutation-derived peptides may be processed and presented by HLA proteins as neoantigens(Kreiter et al., 2015; Rizvi et al., 2015; Tran et al., 2015). This approach has predicted millions of putative mutation-derived neoantigens based on cancer genomic data(Wu et al., 2018), but the vast majority of them fail to show up in proteomic profiling of HLA-bound peptides(Bassani-Sternberg et al., 2016).
Cancer-testis (CT) antigens are derived from proteins with highly restricted expression in germline and trophoblastic cells under healthy conditions, but are abnormally expressed in cancer cells. CT antigens also have high tumor specificity because male germline cells and trophoblastic cells do not display HLA class I molecules on their surfaces (Haas Jr et al., 1988). The CT antigens database (http://www.cta.lncc.br/) provides a comprehensive collection of 276 putative CT antigen genes through manual curation based primarily on mRNA expression pattern of the genes, although protein expression data is also included in the database when available(Almeida et al., 2009). However, it is largely unknown whether and which peptides encoded by these putative CT antigen genes are presented by HLA proteins on cancer cells but not on healthy cells. It is also possible that the manual curation has missed some CT antigen genes.
Tumor associated antigens, including differentiation antigens and over-expressed antigens, have lower tumor specificity than neoantigens and CT antigens. Differentiation antigens are derived from lineage-specific proteins that are expressed on tumor cells and nonmalignant cells of the same cell lineage during at least some stage of differentiation. Overexpressed antigens are derived from proteins with minimal expression in healthy tissues but overexpressed in cancer cells. Targeting tumor-associated antigens requires the identification of a therapeutic window between tumor and normal tissues to mitigate the on-target off-tumor toxicity(Bonifant et al., 2016).
Mass spectrometry (MS)-based immunopeptidomics provide a high-throughput technique for direct identification of MHC-bound peptides in vivo(Purcell et al., 2019). This approach has been successfully applied to the analysis of cell lines(Abelin et al., 2017; Sarkizova et al., 2020), tumor and healthy tissues(Marcu et al., 2021; Schuster et al., 2017; Ternette et al., 2018), as well as plasma samples(Ritz et al., 2017; Shraibman et al., 2019). Importantly, more than a dozen mutation-derived neoantigens have been reported from immunopeptidomic analysis of cancer cell lines and tumor samples(Bassani-Sternberg et al., 2016; Bulik-Sullivan et al., 2019; Solleder et al., 2020). In addition to providing immunopeptidomic evidence for tumor antigens resulting from DNA sequence aberrations or dysregulated RNA expression, MS-based immunopeptidomics may also identify tumor antigens resulting from protein level alterations that cannot be predicted from DNA and RNA studies, such as post-translational modifications. When a post-translational modification is cancer-specific, it may give rise to tumor-associated post-translationally modified (PTM) antigens, a unique class of tumor-associated antigens(Haen et al., 2020). Phosphorylated antigens have been extensively characterized, and their potential as immunotherapeutic targets in cancer has been reported in multiple studies(Cobbold et al., 2013; Engelhard et al., 2020; Meyer et al., 2009; Mohammed et al., 2008; Petersen et al., 2009; Solleder et al., 2020; Zarling et al., 2006). Other types of PTM antigens have also been reported in isolated studies(Engelhard et al., 2006). However, the landscape of PTM antigens and their cancer specificity remains poorly understood.
By collecting and reanalyzing MS data from 16 published human immunopeptidomic studies, SysteMHC Atlas(Shao et al., 2018) provides a systematic, quality-controlled catalog of human peptide antigens, which distinguishes this resource from other antigen databases such as the Immune Epitope Database (IEDB) (Vita et al., 2019), TANTIGEN(Olsen et al., 2017), dePepNeo(Tan et al., 2020), and cancer antigen peptide database(Vigneron et al., 2013). However, although many studies included in SysteMHC Atlas were performed on cancer cell lines or tumor samples, the resource does not provide direct information on tumor antigens, limiting its utility in the cancer immunology community. Moreover, SysteMHC Atlas does not include any neoantigens and PTM antigens because mutations and PTMs were not considered in the analysis of MS data.
Here, we collected 43 published MS-based immunopeptidomic datasets with a total of 311 cancer samples covering nine cancer types and 707 non-cancerous samples. The non-cancerous samples, including 227 benign human tissue samples from 29 distinct tissue types analyzed in the recently published HLA Ligand Atlas study (Marcu et al., 2021), provide a comprehensive reference for defining tumor-associated antigens. Standardized application of a top-performing open search engine identified in this study to our dataset collection with rigorous quality control detected 59% more HLA class I peptides and 5.6 times more HLA class II peptides than those reported in SysteMHC Atlas. In particular, we identified 14,172 PTM antigens resulting from 146 distinct types of PTMs and investigated their molecular characteristics and cancer specificity. For CT antigens, our analysis provided direct evidence to support tumor specific-presentation of some previously annotated CT antigens, revealed non-tumor-specific presentation of other annotated CT antigens, and identified new CT antigen candidates. We also identified other tumor-associated antigens for different cancer types. Using a newly developed computational workflow, we systematically evaluated the possibility of detecting the 100,000 most frequently observed mutations in human cancer in our dataset collection. We provide all these data together with annotated MS/MS spectra supporting identification of each individual antigen in a publicly accessible, easily browsable web resource named cancer antigen atlas (caAtlas, http://www.zhang-lab.org/caatlas/).
Results
Search engine performance comparison
High quality MS-based immunopeptidome characterization relies on sensitive and reliable peptide identification from immunopeptidomic data. Using previously published HLA class I data(Bassani-Sternberg et al., 2016) from two human melanoma samples (Mel15 and Mel16, Table S1), we compared the performance of four conventional search engines, including Comet(Eng et al., 2013), MaxQuant(Cox and Mann, 2008), MS-GF+(Kim and Pevzner, 2014), and X!Tandem(Craig and Beavis, 2004), and an open search engine open-pFind(Chi et al., 2018) that enables the identification of modified peptides. A fixed false discovery rate (FDR) of 1% was applied at both peptide spectrum match (PSM) and peptide levels. For open-pFind, FDRs were calculated for non-modified and modified identifications separately. We further compared these search results with those reported for the two samples in SysteMHC Atlas (Shao et al., 2018), in which multiple search engines were used for peptide identification. The comparison was limited to non-modified peptides with a length between 8 and 12 amino acids due to the peptide length restriction of SysteMHC Atlas in the analysis of these samples and the closed search strategy used by the four conventional search engines and SysteMHC Atlas. Open-pFind identified the largest numbers of unique non-modified peptides for both Mel15 (27,362) and Mel16 (14,461). These numbers were 24%–85% higher than those reported by the other search engines and SysteMHC Atlas for Mel15 and 57%–314% higher for Mel16 (Figures 1A and 1B;Table S1). Peptides uniquely identified by open-pFind constituted a significant portion of all peptides identified by the six methods, with 8% for Mel15 and 17% for Mel16. Moreover, open-pFind search results had a good coverage of the peptides identified by each of the other methods, ranging from 87%–96% for Mel15 and 79%–92% for Mel16 (Figures 1C and 1D;Table S1).
Figure 1.

Search engine comparison using HLA class I data from two melanoma cell lines (Left column: Mel15 and Right column: Mel16), see also Table S1
(A and B) The total number of non-modified peptides (length between 8 and 12) identified by each method, with different colors indicating the number of methods supporting a particular peptide identification.
(C and D) The percentage of non-modified peptides covered by open-pFind identifications for each method.
(E and F) HLA-binder percentages for the overlapping identifications of the six methods (blue bar) and for the total (black bar) and unique (green bar) identifications of each method, respectively.
(G and I) Venn diagrams comparing peptide sequences from modified (mod) and non-modified (non-mod) peptides.
(H and J) The percentages of peptides with single and multiple modification sites, with the singly modified sites further divided by localization probability (Loc. Pro.) computed by PTMiner.
To provide an independent evaluation of the quality of the peptide identification results, we used NetMHCpan v4.0 (Hoof et al., 2009) to predict the HLA binding affinity of the identified non-modified peptides and then classified them into HLA-binders and non-binders. Despite up to 1.9- and 4.1-fold differences in the total number of peptide identifications from the two samples respectively by the six methods, the percentages of predicted HLA-binders among all peptides identified by each method were similar, ranging from 78%–82% for Mel15 and 81%–84% for Mel16 (Figures 1E and 1F;Table S1). We further computed the HLA-binder percentages for both the overlap identifications of the six methods and the unique identifications of each identification method. As expected, the overlap identifications had the highest percentages of HLA-binders, with 84% for Mel15 and 83% for Mel16. For peptides uniquely identified by only one of the methods, the ones identified by open-pFind showed the highest percentages of HLA-binders (71% for Mel15 and 75% for Mel16).
Owing to the open search strategy, open-pFind also uniquely identified 1,924 modified peptides for Mel15 and 1,025 for Mel16, corresponding to 1,564 and 949 distinct peptide sequences, respectively (Figures 1G–1J;Table S1). Some of these sequences overlapped with the ones without modification, whereas 40% from Mel15 and 54% from Mel16 had unique sequences compared with non-modified peptides (Figures 1G and 1I). Almost all identified modified peptides, including 1,910 in Mel15 and 1,019 in Mel16, were singly modified (Figures 1H and 1I). Using PTMiner (An et al., 2019), we localized 1,654 (86%) of the modified sites from Mel15 and 937 (91%) from Mel16 with a posterior probability greater than 0.75 (STAR Methods).
Taken together, search engine choice has a significant impact on MS-based immunopeptidome characterization. Open-pFind showed relatively higher sensitivity and specificity in the identification of unmodified peptides compared with other tools, and it also had the unique advantage of identifying modified peptides. Therefore, open-pFind was selected for further analyses in this study.
A comprehensive immunopeptidome atlas of human cancer
To build a comprehensive immunopeptidome atlas of human cancer, we performed an extensive literature search and identified 43 human immunopeptidomic datasets (Figure 2A) with a total of 311 cancer samples covering 9 cancer types and 707 non-cancerous samples. Together, these datasets covered 109 HLA class I allele subtypes and 78 HLA class II allele subtypes (STAR Methods, Table S2). A total of 81.6 million MS/MS spectra from these studies, including 29.9 million from tumor samples and 51.7 million from non-cancerous samples, were downloaded from the Proteomics Identifications Database (PRIDE) and the Mass Spectrometry Interactive Virtual Environment (MassIVE) and searched using open-pFind. To ensure high quality of both non-modified and modified peptide identifications, 1% FDR was controlled at both PSM and peptide levels for modified and non-modified identifications separately across all datasets (STAR Methods, Figure S1A).
Figure 2.

Construction of a comprehensive immunopeptidome atlas of human cancer, see also Figure S1 and Table S2
(A) MS/MS spectra numbers of the immunopeptidomic datasets included in this study.
(B) The distributions of sample number, PSM number and non-modified and modified peptide number for the nine cancer types and non-cancerous samples, respectively.
(C) Length distributions of the identified HLA class I and HLA class II non-modified peptides.
(D) Length distributions of the identified HLA class I and HLA class II modified peptides.
(E) Venn diagram comparing unique HLA class I peptide sequences from this study and SysteMHC Atlas.
(F) Venn diagram comparing unique HLA class II peptide sequences from this study and SysteMHC Atlas.
(G) Cumulative number of all distinct HLA class I peptides as a function of dataset publication time. Each dataset is denoted as dataset ID: dataset public time (#peptides).
(H) Cumulative number of distinct peptides for each of the top nine HLA class I alleles as a function of dataset publication time.
Our analysis identified 11.3 million PSMs corresponding to 420,702 non-modified peptides and 139,269 modified peptides. These peptides had a total of 473,288 unique sequences, which could be attributed to a minimum of 15,099 proteins and a maximum of 20,502 proteins (STAR Methods, Figure 2B). The numbers of identified PSMs and peptides across different cancer types were not always proportional to the input MS/MS numbers (Figure 2B), which may be partially explained by resolution difference of the mass spectrometers used in different studies. In total, we identified 231,766 non-modified peptides and 55,710 modified peptides from the HLA class I peptidomes (252,974 unique sequences), and 229,099 non-modified peptides and 92,203 modified peptides from the HLA class II peptidomes (266,057 unique sequences), with 40,163 non-modified peptides and 8,644 modified peptides detected in both class I and class II samples. The Open-pFind score distribution of the modified peptide group was comparable to that of the non-modified peptide group (Figure S1B), suggesting high-quality of the modified peptide identifications. Both non-modified and modified peptide groups showed the characteristic length distributions for both class I and class II antigens (Figures 2C and 2D). Compared with SysteMHC Atlas (STAR Methods), a previously published database of MS-identified HLA peptides, our analysis identified 59% more HLA class I peptides sequences (Figure 2E) and 5.6 times more HLA class II peptides sequences (Figure 2F).
To assess the immunopeptidome size at the population level, we calculated the cumulative number of all identified distinct HLA class I peptide sequences as a function of dataset publication time to test for possible saturation. A similar analysis using combined data from all HLA class I alleles in SysteMHC Atlas showed that the saturation level had been reached. In contrast, our analysis showed continued increase of distinct peptide sequences (Figure 2G), likely due to recent addition of data from new cell and tissue types. In addition, when individual HLA class I alleles were considered, continued steep increase was found for the nine HLA class I allele subtypes with the largest numbers of identified peptides (Figure 2H). These results suggest that although our extensive dataset collection and the use of a more sensitive search engine have vastly increased the number of experimentally supported HLA peptides, our collection remains incomplete.
PTM antigens
The singly modified class I and class II peptides with modification sites confidently re-localized by PTMiner (STAR Methods, Figure S1A) were divided into 13 major modification classification categories according to the Unimod database (Figure 3A), including 146 distinct PTM types under the post-translational, O-linked glycosylation, N-linked glycosylation, and other glycosylation categories. The top 15 most frequently identified PTM types from class I and class II peptidomes respectively corresponded to a total of 19 PTM types (Figure 3B). The Open-pFind score distributions of peptides associated with these 19 PTM types were comparable to that of the non-modified peptides (Figure S1B). For each of the 19 PTM types, we further selected three representative annotated MS/MS spectra (57 in total) to manually evaluate the quality of the PSM results, and almost all the peaks with a high intensity could be explained by the identified peptide sequences (Figure S2). These results demonstrate high quality of the peptide identifications for individual PTM types.
Figure 3.

Classification and properties of modified antigens, see also Figures S1 and S2.
(A) Classification of modified class I and II antigens into 13 major modification classification categories annotated by Unimod. For each category, modified peptides were divided into the subset sharing the same sequence with non-modified peptides (Overlap) and another subset having unique sequences compared with non-modified peptides (Unique).
(B) Classification of class I and II PTM antigens into the 19 frequently identified PTM types.
(C) Length distributions of HLA class I and class II PTM antigens for the 16 PTM types with at least 20 distinct sequences for both HLA class I and class II.
(D) HLA-binder percentages for the non-modified peptides (black bar) and different categories of the modified peptides (blue bar), respectively.
(E) HLA-binder percentages for the non-modified peptides (black bar) and peptides with different types of PTMs (blue bar), respectively. For (D) and (E), the number of peptides for each modification category or PTM type is annotated on the top of each bar, and only PTMs with at least 20 distinct sequences were included in the analyses.
The numbers of class II modified antigens across different modification groups were not always proportional to those of class I modified antigens (Figures 3A and 3B). For example, the deamidated and didehydro groups showed relatively higher frequencies among class II modified antigens, whereas the dioxidation group showed an opposite trend. Class I and II PTM antigens also showed different amino acid preferences for some PTM types (Figure S3). For example, dioxidation preferentially occurred on the methionine residue for class I antigens but on the cysteine residue for class II antigens. Quinone modification preferentially occurred on the tyrosine and tryptophan for class I and II antigens, respectively.
Some modification groups, such as the artifact group, the dioxidation group, the oxidation group, the deamidated group, the dehydrated group, and the quinone group were dominated by peptides sharing the same sequence with the non-modified peptides; in contrast, other PTM groups, such as the acetyl group, the dimethyl group, and the carbamyl group were dominated by peptides with unique sequences compared with the non-modified peptides (Figures 3A and 3B). In addition, although modified antigens in general showed the characteristic length distributions for both class I and class II antigens (Figure 2D), several PTM groups showed distinct length distributions for class I antigens (Figure 3C). In particular, class I acetyl, dimethyl, methyl, and carbamyl antigens showed a clear enrichment in the region with longer peptide length compared with the characteristic length distribution. Together, these results suggest that some PTMs may interfere with MHC binding to peptides.
We used NetMHCpan v4.0 to predict HLA binding affinity of the peptide sequences of the modified class I antigens for different modification groups. The analysis was limited to the sequences not identified among the non-modified peptides (i.e., the unique portions in Figures 3A and 3B), and only modification groups with at least 20 distinct sequences with lengths between 8 and 12 amino acids were included in this analysis. The percentages of predicted HLA-binders in different modified antigen groups were lower than that in non-modified antigens in general (Figures 3D and 3E). Among the major categories, the artifact group, the chemical derivative group, and the multiple (e.g., both chemical and artifact) group showed relatively higher percentages of predicted HLA-binders (Figure 3D). Of note, modifications in these groups may occur after HLA-peptide binding, which may explain relatively higher percentages of HLA-binders predicted based on the non-modified sequences. Among different types of PTM antigens, some had relatively high percentages of predicted binders, but others, such as the acetyl antigens, showed very low percentages (Figure 3E). This may indicate varied impact of PTMs on MHC binding, which is not captured in the binding affinity prediction model trained using unmodified sequences.
For the dominant amino acid residues of each PTM (Figure S3), we performed positional frequency analysis for those with at least 20 distinct sequences to identify possible positional preference, using data from non-modified antigens as a reference (Figure 4). For phosphorylation, phosphorylated serine showed a clear preference for position 4 across different peptide lengths (Figure 4A), which is consistent with previous reports (Bassani-Sternberg et al., 2016; Gfeller and Bassani-Sternberg, 2018). Oxidation (Figure 4B), quinone modification (Figure 4C), dioxidation (Figure 4D), and didehydro modification (Figure 4E) of tyrosine and dioxidation of methionine (Figure 4H) showed a strong preference for the last position, whereas glutathione modification (Figure 4I) of cysteine showed a strong preference for the first position. N-terminal acetylation were significantly enriched for the alanine and serine residues but depleted for the methionine residue (Figure 4J). These data suggest possible selection of the modified peptide repertoire by the antigen processing and presentation machineries.
Figure 4.

Positional frequency analysis for different PTM sites, see also Figure S3
(A) Phospho [S].
(B) Oxidation [Y].
(C) Quinone [Y].
(D) Dioxidation[Y].
(E) Didehydro[Y].
(F) Methyl [E].
(G) Dehydrated [T].
(H) Dioxidation[M].
(I) Glutathione [C].
(J) Acetyl [Protein N-term]. The positional frequency distribution (%) of a PTM site (dark blue bars) was compared to the distribution of corresponding non-modified amino acid residues (light blue bars) using data from all HLA class I samples grouped by peptide length. p values were derived from the Fisher’s exact test and grouped into different categories as indicated in figure legend.
PTMs play important roles in cancer initiation and progression, and thus some PTM antigens may be cancer-associated. We compared the relative frequencies of different types of class I (Figure 5A) and class II (Figure 5B) PTM antigens in individual cancer types as well as non-cancerous samples. For class I antigens, acetylated antigens showed the highest frequency in almost all cancer types. An outlier was lymphoma, in which glutathione modification occupied 66% of all class I PTM antigens. In non-cancerous samples, the dominant modification was dioxidation, which occupied 41% of all class I PTM antigens. For the same cancer types, PTM type compositions were different between class I and class II antigens. For example, 32% of the class II PTM antigens in melanoma involved deamidation whereas the ratio was 5% for class I PTM antigens. Moreover, 28% of class II PTM antigens in non-cancerous samples were deamidated whereas the ratio was only 4% for class I PTM antigens.
Figure 5.

Distribution of different types of PTM antigens in different cancer types and the recurrently identified cancer-specific PTM antigens, see also Figure S4 and Table S3
(A and B) Distribution of different types of HLA class I (A) and II (B) PTM antigens in different cancer types and non-cancerous samples.
(C and D) Cancer-associated HLA class I (C) and II (D) PTM antigens recurrently identified in at least seven cancer samples. Each colored plus or minus symbol on the left of the bar plots indicates significantly higher or lower expression (multiple-test adjusted p < 0.01) in TCGA tumor samples compared with GTEx normal samples of the same tissue type (Note: Meningioma is not included in the TCGA datasets). p values were derived from Student’s t-test and then subjected to multiple-test adjustment.
To identify candidate cancer-associated PTM antigens, we performed qualitative differential detection analysis between cancerous and non-cancerous samples for HLA class I and class II PTM antigens, respectively. In total, 1,742 class I and 1,709 class II PTM antigens were detected in one or more cancer samples but were not detected in any non-cancerous samples except for testis (Figure S4;Table S3). Among them, 23 class I and 15 class II PTM antigens were identified in more than seven cancer samples (Figures 5C and 5D). The vast majority of these recurrently identified, cancer-associated PTM antigens were detected in leukemia and lymphoma samples, and the dominant PTM type was acetylation. Almost all source genes of these PTM antigens showed significantly higher expression in TCGA tumor samples of the same cancer types compared with corresponding GTEx normal tissues (multiple-test adjusted p < 0.01, Student’s t-test, Figures 5C and 5D), supporting their cancer relevance. Moreover, the source genes included some known cancer genes in the cancer gene census database, namely COL3A1, HLA-A, TOP1, BCL9L, and ASXL2 for HLA class I, and TOP1 for HLA class II PTM antigens.
Cancer-testis antigens
The CT antigens database (Almeida et al., 2009) lists 276 putative CT antigen genes. Recently, 512 proteins with distinct testicular protein expression patterns were identified by combining genome-wide transcriptomics analysis with immunohistochemistry (Pineau et al., 2019), and 178 of these also showed testis-specific expression in our analysis of the GTEx RNA-Seq data (STAR Methods). These testis-specific genes provide a complementary source of putative CT antigens. However, whether and which peptides encoded by these putative CT antigen genes or testis-specific genes are specifically presented by HLA on cancer cells is largely unknown.
The union of the CT antigen genes from the CT antigens database and the GTEx-supported testis-specific proteins included a total of 413 genes. We compared this list with the HLA class I antigen source genes identified in our analysis and found 92 with evidence of HLA class I bound peptides. Peptides from 61 genes, including 47 in the CT antigen database, were identified in non-cancerous samples, challenging their identities as bona fide CT antigen genes (Figure S5). Peptides from the remaining 31 genes were identified in cancer samples but not non-cancerous samples except for testis, and these genes are more likely to encode true CT antigens (Figure 6A;Table S4). Twenty-nine genes of this group are listed in the CT antigen database, including well established CT antigen genes such as ROPN1, MAGEC2, MAGEA11, MAGEC1, GAGE5, IL13RA2, etc. The other two, namely SPERT and DAZL are from the testis-specific proteins. These two genes have highly specific expression in testis among all normal human tissues analyzed by GTEx and may represent previously unrecognized CT antigens (Figures 6B and 6C). DAZL encodes an RNA-binding protein that has a central role in the early differentiation of primordial germ cells(Kee et al., 2009) and is a susceptibility gene for testicular cancer(Ruark et al., 2013). SPERT encodes a spermatid-associated protein and has been rarely studied.
Figure 6.

Putative CT antigen genes with evidence for tumor-specific presentation from the immunopeptidomic data, see also Figure S5 and Table S4
(A) The bar plot shows the number of tumor samples with MHC-bound peptides identified for each putative CT antigen gene. The color of the bars indicates different cancer types. The red and blue dots on the left indicate genes from the CT antigens database and the GTEx-supported testis-specific proteins, respectively.
(B and C) Gene expression pattern across the 54 normal tissues studied by GTEx for SPERT, and DAZL, respectively.
Among the 31 genes, SSX1, SSX2, and SSX4 are known cancer genes in the cancer gene census database. Eleven genes are amplified in more than 5% of the 9,958 tumors analyzed in the TCGA pan-cancer copy number study (Schaub et al., 2018) (STAR Methods, Table S4). For example, LY6K is amplified in 16% of the tumors. In addition, six genes are hypomethylated in more than 10% of the 9,664 tumors analyzed in the TCGA pan-cancer methylation study (Saghafinia et al., 2018) (STAR Methods, Table S4), with SPEF2, CCDC62 and ACRBP being hypomethylated in 83%, 60%, and 41% of the tumors, respectively. The two newly nominated CT antigens, DAZL and SPERT are amplified in 4%, and 2% of the tumors, respectively, and SPERT is hypomethylated in 3% of the samples. These data suggest potential oncogenic roles of these CT antigen genes in human cancer.
In summary, our analysis provided direct evidence to support cancer-specific presentation of 31 known and putative novel CT antigens and also revealed non-cancer-specific presentation of 47 previously annotated CT antigens.
Cancer-associated antigens
To identify cancer-associated antigens for each cancer type, we further performed a qualitative analysis as previously described(Schuster et al., 2017) to reveal antigen source genes that were detected in the cancer type but not non-cancerous samples except for testis.
Using melanoma as an example, the analysis identified 470 melanoma-associated antigen source genes (Figure 7A), including 201 that were identified in at least two melanoma samples (Table S5). These recurrently identified, melanoma-associated antigen source genes were highly enriched among the genes that were over-expressed in the 472 TCGA skin cutaneous melanoma (SKCN) samples compared to 1,809 normal skin tissue samples from GTEx (p < 0.01, Kolmogorov–Smirnov test, Figure 7B, STAR Methods). Individually, 32 of these genes were identified in more than five melanoma samples and showed significantly higher expression in the TCGA melanoma samples compared with GTEx normal skin tissues (multiple-test adjusted p < 0.01, ttest, Figure 7C). This list included well-known melanoma-associated CT antigens such as ROPN1 and MAGEC2, as well as melanoma-associated differentiation or overexpressed antigens such as PMEL(Zhang et al., 2021), TYR(Gudbjartsson et al., 2008; Weinstein et al., 2014), DCT(Pak et al., 2004), SLC45A2(Fernandez et al., 2008), and MLANA (MART1)(Leisegang et al., 2016; Pitcovski et al., 2017). It also included many putative novel melanoma-associated antigens, such as ROPN1B (Rhophilin associated tail protein 1B), which was detected in 13 melanoma samples. Although not included in the CT antigen database and the list of testis-specific proteins, ROPN1B shows restricted mRNA expression in testis among all normal tissues (Figure S6A) and is selectively expressed in melanoma among different cancer types (Figure S6B). Thus, ROPN1B may be a new melanoma-associated CT antigen.
Figure 7.

Tumor associated antigen analysis for melanoma, see also Figures S6, S7, and Table S5
(A) Differential immunopeptidomic analysis of antigen source genes from melanoma HLA class I samples (n = 28) and comparative profiling to non-cancerous HLA class I samples (n = 373). Profiling is based on the detection frequency for individual genes. The numbers of genes that are unique to melanoma samples, unique to non-cancerous samples, and shared between melanoma and non-cancerous samples are indicated above the graph, together with the respective area under the curve in percentage of total area (i.e., the sum of sample frequency for each gene divided by that for all genes). Genes are sorted according to the sample numbers within each of the three groups.
(B) All genes quantified in both TCGA and GTEx studies were sorted by signed −log10(pValue) in the top panel. The locations of the 201 recurrently identified, melanoma-associated antigen source genes in the sorted list are annotated by the black bars in the bottom panel. p values were derived from Student’s t-test.
(C) Bar plot showing the number of tumor samples for each of the 32 melanoma-associated antigen source genes identified in more than five melanoma samples and with significantly higher expression in the TCGA melanoma samples compared to GTEx normal skin tissues. The red and blue dots on the left indicate genes from the CT antigens database and the GTEx-supported testis-specific proteins, respectively.
Analyses of other cancer types identified 168, 23, 56, 20, 48, 6, and 1 recurrently identified cancer-associated antigen source genes for meningioma, lymphoma, glioblastoma, leukemia, ovarian cancer, breast cancer, and lung cancer, respectively (Figure S7;Table S5). The top recurrently identified genes included PNMA2 (10 samples) and CHSY3 (9 samples) for meningioma, FCRL3 (12 samples) and FCRL2 (11 samples) for lymphoma, MUC17 (11 samples) and HEPACAM (7 samples) for glioblastoma, SH2D4B (4 samples) and GUSBP1 (3 samples) for leukemia, PTPRU and LRP11 (3 samples for each) for ovarian cancer, RET and KLK8 (2 samples for each) for breast cancer, and PTPN21 (2 samples) for lung cancer. Some of these genes have previously reported roles in the corresponding cancer types.
To systematically evaluate cancer relevance of the recurrently identified cancer associated antigen genes, we first overlapped them with those in the cancer gene census and revealed some known cancer genes, including MN1, JAZF1, LRP1B, OMD, SFRP4 and PRDM16 for meningioma, HOXD13, PAX3, SFRP4, and JAZF1 for melanoma, DDIT3, and SOX21 for glioblastoma, and RET for both leukemia and breast cancer. Using TCGA copy number and methylation data, we further identified many amplified or hypomethylated genes in the corresponding cancer types (Table S5). For example, the known melanoma-associated CT antigen MAGEC2 and melanoma-associated differentiation antigen MLANA are amplified in 4% and 5% of the 436 TCGA melanoma samples, and METTL23 and TNRC6C are amplified in 18% of the TCGA melanoma samples, respectively. FCRLB is amplified in 23% of the TCGA lymphoma samples. ANO8 and EDN1, which were detected in 2 ovarian samples in our dataset, are amplified in 32% of the 404 TCGA ovarian tumors and hypomethylated in 100% of 10 TCGA ovarian tumors with methylation data, respectively.
In summary, our data provided direct immunopeptidomic evidence to support known and putative novel tumor-associated antigen genes.
Neoantigens
The peptides identified above do not include any neoantigens because mutations were not considered in the MS data analysis. To identify neoantigens from the cancer immunopeptidomes, we developed the NeoQuery workflow, which comprises three quality control steps (Figure S8A, STAR Methods). The first step uses a modified version of PepQuery(Wen et al., 2019) to identify preliminary immunopeptidomic evidence for a list of genomic mutations. Stringent filtering criteria were implemented, including 1% FDR estimated by approximating the null distribution with the PSM scores of matches to non-cancerous samples and competitive filtering based on unrestricted modification searching results. The second step incorporates variant peptides identified in the first step in sample-specific databases to perform customized database searching. The third step further filters the search result using MHC binding affinity predicted by NetMHCpan.
To identify immunopeptidomics evidence for the highly recurrently observed mutations in human cancer studies, we performed a systematic analysis for the top 100,000 somatic mutations with the highest mutation frequencies in the International Cancer Genome Consortium (ICGC, sorted by #donors affected) database (STAR Methods). Among these mutations, the 7,439 unique missense mutations that are not included in the dbSNP database(Sherry et al., 2001) were queried against all MS/MS spectra in our HLA class I data using NeoQuery. As positive controls, we also included in our analysis an additional 11 somatic mutations for which neoantigens have been previously reported in one of our HLA class I dataset by searching immunopeptidomic data from each sample in the melanoma dataset against sample-specific protein databases derived from matched whole exome sequencing (WES) data(Bassani-Sternberg et al., 2016).
For the positive controls, our analysis identified immunopeptidomics evidence for five out of the 11 mutations (Figure S8B) without using WES-derived sample-specific protein databases. Remarkably, although these mutations were searched against all 40 HLA class I datasets including a total of 579 samples, the matches were only identified in the same melanoma samples as reported in the original study, and the identified mutant peptide sequences were the same as the ones reported in the original study, suggesting high specificity of NeoQuery. Our analysis missed six previously reported neoantigens; however, those identifications were made using a 5% FDR threshold in the original study and they were filtered out in our FDR control step with the 1% threshold. We manually checked the identifications derived from these six somatic mutations and found that these sequences had better matchings to spectra from non-cancerous samples than those from the previously reported melanoma samples, further indicating potential false discoveries. These results demonstrate the sensitivity and specificity of NeoQuery in validating mutation-derived neoantigen candidates using our large collection of HLA class I data, without using sample-specific protein databases derived from matched WES data.
Despite encouraging results from the positive controls, our analysis of the 7,439 highly recurrently observed missense mutations identified no immunopeptidomics evidence. Because of the demonstrated sensitivity of our workflow on positive controls, this result may suggest possible selection against the most frequent somatic mutations in antigen processing and presentation.
Data dissemination through caAtlas
We built a web portal caAtlas (http://www.zhang-lab.org/caatlas/) to facilitate the retrieval of antigens together with annotated MS/MS spectra through a user-friendly interface. All antigens are searchable by gene symbol, protein name, UniProt ID, peptide sequence, or HLA allele. Moreover, PTM antigens, CT antigens, and cancer type-specific antigens are browsable in three separate tables, in which the antigen source genes can be sorted by sample number or associated cancer type. For each antigen source gene, caAtlas lists all the MHC-bound peptides and visualizes the position of these peptides in the protein sequence, with protein domains annotated based on the Pfam database (El-Gebali et al., 2019). In addition, caAtlas also provides links to human protein atlas (HPA), which will allow users to quickly check mRNA and protein expression of the antigen source gene across normal tissues using data from GTEx and HPA. For each peptide, caAtlas presents all identification information, such as cancer type, spectrum ID, dataset ID, HLA allele, predicted binding affinity, and the annotated MS/MS spectra generated by PDV(Li et al., 2019). This allows users to manually check and download the mass spectrum matching results for all antigens identified in this study, which facilitates the prioritization of the most promising peptides for further experimental and translational cancer research.
Discussion
We have constructed an immunopeptidome atlas of human cancer through an extensive collection of published immunopeptidomic datasets, standardized processing of the datasets using a top-performing search engine identified in this study, and comprehensive analyses of PTM antigens, CT antigens, cancer-associated antigens, and neoantigens. This new resource is available to the cancer and immunology research communities through caAtlas, a user-friendly web portal.
One unique contribution of our study is the systematic identification and characterization of PTM antigens. Applying an open-search strategy to 43 immunopeptidomic datasets without specific PTM enrichment, our analysis identified 146 types of PTM antigens. PTMs did not seem to alter peptide length distribution of HLA class II peptides, but some PTMs showed a strong impact on the length distribution of the HLA class I modified peptides, shifting the distribution toward longer peptide length. Positional frequency analysis of different PTM sites on HLA class I antigens revealed strong positional preference of the PTM sites, suggesting a role of antigen processing and presentation pathways in shaping the PTM antigen repertoire. Comparative analysis between data from cancer and non-cancerous samples identified cancer-associated PTM antigens, which were dominated by N-terminal acetylation. N-terminal acetylation is believed to occur on most human proteins and has been implicated in cancer (Kalvik and Arnesen, 2013). A recent study showed that N-terminal acetyltransferases are frequently upregulated in tumors, and most of them are among the most essential genes in cancer cells (Koufaris and Kirmizis, 2020). Our analysis not only provides a general resource for PTM antigen research but also identifies cancer-associated PTM antigens for future investigation.
Our extensive dataset collection also allowed us to thoroughly annotate CT antigens and cancer-associated antigens using immunopeptidomic data. Our analysis provided direct evidence to support tumor-specific presentation of some known and novel CT antigens and tumor associated antigens. We also revealed presentation of some previously annotated CT antigens in non-testis benign tissues. Some non-tumor samples used in immunopeptidomics research are Epstein-Barr virus-transformed B cell lines and the process of transformation may drive unusual protein expression. However, we also identified presentation of some previously annotated CT antigens in other non-cancerous samples such as primary fibroblast cells and normal tissue samples. This information is critical to the prioritization of antigens for vaccine development, because the on-target off-tumor normal cell toxicity is a major concern for vaccines targeting CT antigens and tumor associated antigens.
Next-generation sequencing of human tumors combined with advanced computational tools for MHC binding prediction has enabled high-throughput in silico neoantigen discoveries. However, only a few dozen of these computationally predicted MHC-bound neoantigens have been experimentally validated to date. Using our extensive dataset collection and the NeoQuery workflow, we systematically analyzed 100,000 most frequently reported mutations in the ICGC database, but no immunopeptidomics evidence was found. There are a few possible explanations for this result. First, these mutations may not exist in the samples we studied. However, one of our datasets (Bassani-Sternberg et al., 2016), in which WES-based somatic mutation data are publicly available, included 59 of these frequent mutations. Therefore, we would expect a much larger number of the frequent mutations in our complete data collection. Second, the detection rate is limited by both the immunopeptidomic technology and the computational algorithm used for neoantigen identification. However, because we were able to successfully identify neoantigens derived from rare, patient-specific mutations using the same workflow, this may not completely explain the extremely low detection rate. Another possible explanation is that the recurrent driver oncogenic mutations are selected against in MHC presentation, as suggested in a previous study(Marty et al., 2017). If this is true, we would expect that the vast majority of neoantigens would be patient-specific, limiting their utility as prefabricated vaccines. In contrast, cancer-specific PTM antigens, CT antigens, and cancer-type specific antigens reported in this study may allow more convenient establishment of vaccines based on “off-the-shelf” peptides.
Limitations of study
There are several limitations of this work. First, although caAtlas provides the most comprehensive coverage of experimentally supported HLA peptides to date, our collection remains incomplete based on the saturation analysis (Figures 2F and 2G). Moreover, there is an apparent bias for certain cancer types, such as glioblastoma, in the published immunopeptidomics studies. Immunopeptidomics is an emerging research field, and we expect that the number of immunopeptidomic datasets will grow substantially in the near future. We will continue collecting new datasets and adding new analysis results to caAtlas. Second, bioinformatic analysis of immunopeptidomics data remains a key challenge as illustrated by the moderate overlap among peptide identification results from different search engines (Figure 1A). This may be improved by using more advanced analysis methods, such as the recently published DeepRescore method that leverages deep learning and semi-supervised classification to improve peptide identification in immunopeptidomic data analysis (Li et al., 2020). Third, using a computational prediction model trained based on unmodified HLA binding sequences, many modified peptides identified in this study, especially acetyl peptides, were not predicted to bind HLAs (Figure 3E). Although our modified peptide identifications, including acetyl peptide identifications, are of high quality (Figures S1B andS2), in vitro HLA-peptide binding assay is needed to fully validate the binding affinity of these modified peptides. After that, new predictors for modified HLA peptides should be developed. Finally, immunogenicity of the peptides in caAtlas remains unknown. Nevertheless, we believe caAtlas may serve as a powerful resource for the selection and prioritization of peptides for in vitro HLA-peptide binding assay and immunogenicity testing, which will pave the way to eventual development of cancer immunotherapies.
STAR★Methods
Key resources table
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Bing Zhang (bing.zhang@bcm.edu).
Materials availability
This study did not generate new reagents
Methoddetails
Data collection and curation
Human immunopeptidomic datasets were collected from peer-reviewed immunopeptidomics publications and public data repositories. We first searched the PubMed database using the search terms ‘MHC’, ‘HLA’, and ‘immunopeptidomics’, and then manually reviewed all returned publications to identify the ones reporting immunopeptidomics studies on human cell lines or tissue samples. Then, we further selected the publications in which the dataset is publicly available. To ensure high quality of the MS data, we only considered studies published after January 2015. Meanwhile, we searched the MS data repositories Proteomics Identifications Database (PRIDE) using the keywords, ‘MHC’ and ‘HLA’ and set the organism option as ‘homo sapiens’. Similarly, we only considered datasets submitted after 2015 and further selected immunopeptidomic datasets by reading corresponding publications.
After collecting the human immunopeptidomic datasets, we further curated them by: (1) associating each dataset with cell line and tissue samples; (2) classifying all the samples into cancer and non-cancerous samples; (3) classifying all the samples into HLA class I and HLA class II samples; and (4) assigning HLA alleles to each sample based on information available in the publications or through direct communication with the authors.
Search engine performance comparison
Two HLA class I human melanoma samples (Mel15 and Mel16, Table S1) from previously published immunopeptidomics dataset(Bassani-Sternberg et al., 2016) generated by the Q Exactive HF mass spectrometers (Thermotems) were used to compare the performance of search engines. The MS/MS data in the RAW format were converted to MGF files using MSconvert (Adusumilli and Mallick, 2017) (ProteoWizard, version 3.0.19014). The MS/MS data were searched using five search engines (Comet v201801, MaxQuant v1.6.5, MS-GF+ v2019.02.28, Open-pFind v3.1.5, and X!Tandem v2017.2) against the UniProt database supplemented with 245 frequently observed contaminants (such as human keratins, bovine serum proteins and proteases) and the reversed decoy sequences of the same size. The following parameters were used for all search engines: fixed modification: carbamidomethyl [C]; variable modification: oxidation [M]; enzyme specificity: no cleavage specificity; precursor mass matching tolerance: 10 ppm; fragment mass matching tolerance: 0.02Da. ‘Open search’ mode was used for open-pFind. Peptide length was set to 8-25 amino acids. Both peptide spectrum match (PSM) and peptide levels False discovery rate (FDR) was computed by the PGA package(Wen et al., 2016) for Comet, MaxQuant, MS-GF+ and X!Tandem identifications respectively, and FDR of 1% was required for both groups. Modified (except Oxidation[M] and Carbamidomethyl [C]) and non-modified (include Oxidation[M] and Carbamidomethyl[C]) identifications of open-pFind were filtered using FDR calculated by the PGA package separately and FDR was controlled at 1% at both PSM and peptide levels. All PSM and peptide identifications passing the FDR threshold were combined.
Peptide identification for all immunopeptidomic datasets
To identify peptides from all immunopeptidomic datasets, we used Open-pFind v3.1.5(Chi et al., 2018), which showed the best performance in our evaluation. The MS/MS data in raw format were converted to MGF files using MSconvert (ProteoWizard, version 3.0.19014). The MS/MS data in wiff format were converted to MGF files using MSDataConverter(v1.3) implemented in Sciex. The UniProt database supplemented with 245 frequently observed contaminants such as human keratins, bovine serum proteins, and proteases was used as the reference database. The enzyme specificity was set as no cleavage specificity. Oxidation [M] was set as the variable modification. No fixed modifications were set for search. Peptide length was set to 7-50 amino acids. Among the 43 immunopeptidomic datasets, 41 are high-precision datasets and the remaining two (PRIDE ID: PXD007635 and PXD008984) are low-precision. For the high-precision datasets, the precursor mass matching tolerance was set as 20 ppm and the fragment mass matching tolerance was set as 0.05 Da. ‘Open search’ mode of open-pFind was used for these high-precision datasets. For the low-precision datasets, the precursor mass matching tolerance was set as 5 ppm and the fragment mass matching tolerance was set as 0.5 Da. ‘Close search’ mode of open-pFind was used for the low-precision datasets.
Quality control of the peptide identifications
We used rigorous quality control strategies to ensure high quality of both non-modified and modified peptide identifications in the whole caAtlas database. As shown in Figure S1A, our quality control workflow includes four major steps:
-
(1)
FDR control for individual datasets. Open-pFind identifications, including both modified and non-modified identifications, were controlled at 1% peptide level FDR for each dataset separately.
-
(2)
Joint FDR control across all datasets for modified and non-modified peptides, respectively. Peptides identified from all datasets were combined and then separated into a modified peptide group and a non-modified peptide group. Oxidation[M] was included in the non-modified group instead of the modified group because it was considered as a variable modification in database searching. For each group, a joint FDR control across all datasets was performed using the PGA package(Wen et al., 2016), and an FDR threshold of 1% was applied at both PSM and peptide levels.
-
(3)
Modification site localization confidence filtering. For modified peptides passing the joint FDR control, we further used PTMiner v1.0 to assess the confidence of modified site localization. Due to the software restriction, the analysis was not performed for a small number of peptides with more than one modification, which were discarded from downstream analyses. Open-pFind identifications were transferred into the PTMiner format (∗.txt). For the PTMiner analysis, all identified modifications were set as target modifications and the target sites included all sites listed in the Unimod database for each identified modification except for Oxidation [M]. Modification sites with a localized posterior probability >75% were regarded as confident modification sites, and the others were discarded.
-
(4)
PSM visualization. Annotated spectra for all peptide identification results were generated by PDV v1.0 (Li et al., 2019) for manual examination.
HLA binding affinity prediction
HLA binding affinity was used to evaluate the quality of non-modified peptides identified. HLA binding affinity was also computed for sequences of the modified peptides. NetMHCpan v4.0 was used for HLA binding affinity prediction, and only peptides with length between 8 and 12 were used for prediction. Binding affinity of 500nM (standard settings in NetMHCpan v4.0) was used as the threshold to distinguish HLA-binders from non-binders.
Comparison with SysteMHC Atlas
We downloaded all the SysteMHC Atlas (Shao et al., 2018) peptide identifications in the csv format from the website (https://systemhcatlas.org/stats). Only data from human samples were included in our comparison(Marcu et al., 2021).
Differential expression analysis of cancer and non-cancerous samples
We downloaded relevant RNASeq data from TCGA and GTEx to evaluate the melanoma-specific antigen source genes and cancer-specific PTM antigen source genes. For TCGA, we included 1,101 available breast invasive carcinoma (BRCA) samples, 478 available colon adenocarcinoma (COAD) samples, 174 available glioblastoma multiforme (GBM) samples, 152 available acute myeloid leukemia (LAML) samples, 534 available lung adenocarcinoma (LUAD) samples, 48 available lymphoid neoplasm diffuse large B-cell lymphoma (DLBC) samples, 472 available skin cutaneous melanoma (SKCM) samples, and 374 available ovarian serous cystadenocarcinoma (OV) samples. For GTEx, we included 459 available breast tissue samples, 779 available colon tissue samples, 2,642 available brain tissue samples, 929 available blood tissue samples, 578 available lung tissue samples, 174 lymphocyte samples, 1,809 available skin tissue samples, and 180 available ovary tissue samples. To make expression data comparable between TCGA and GTEx, we reprocessed TCGA data using the same protocol as used in the GTEx data analysis. Specifically, gene-level expression was quantified using RNA-SeQC v2.3.5 (DeLuca et al., 2012) based on the annotation of Gencode 26. Because RNA-SeQC does not support full annotation gtfs, we used the python script downloaded from the RNA-SeQC github website (https://github.com/getzlab/rnaseqc/issues/34) to collapse the annotation for each gene. To compare gene expression between normal and tumor samples, a p-value was calculated for each gene based on the Student’s t-test. The Benjamini & Hochberg (BH) correction was then used for FDR calculation to account for multiple testing. Genes with an FDR≤1% were regarded as differentially expressed between tumor and normal samples.
Identification of testis-specific proteins
We downloaded a list of 512 testicular proteins identified using genome-wide transcriptomic analysis and immunohistochemistry in a recent publication (Pineau et al., 2019). Next, we downloaded RNA-Seq data from GTEx for all 54 normal tissues and manually checked the RNA-Seq expression levels of the 512 testicular proteins across all normal tissues. Proteins with obvious mRNA expression in non-testis tissues were removed, resulting in a list of 180 testis-specific proteins.
Copy number amplification data analysis
The TCGA pan-cancer copy number alteration (CNA) data were downloaded from the output of GISTIC2 from a TCGA PanCanAtlas study(Schaub et al., 2018). The CNA data from all the cancer types were used to study the CNA of CT antigen genes. CNA data from the matched cancer type were used to study the CNA of cancer-associated antigen genes although data was not available for meningioma. Following previous publications, we used a threshold of 0.3 to determine amplified or deleted genes. The copy number amplification ratio for one gene was calculated as the number of patients with the gene amplification divided by the number of all patients analyzed.
Methylation data analysis
The TCGA pan-cancer methylation data (450K only) were downloaded from a TCGA PanCanAtlas study (Saghafinia et al., 2018). The methylation data from all the cancer types were used to study the methylation of CT antigen genes. Methylation data from the matched cancer type were used to study the methylation of cancer-associated antigen genes although data was not available for meningioma. Data from all CpG sites of one gene were averaged to get gene-level methylation quantification for all genes. Following previous publications, we used a threshold of 0.2 to determine hypo- or hyper-methylated genes. The hypomethylation ratio for one gene was calculated as the number of patients with hypomethylation divided by the number of all patients analyzed.
NeoQuery for neoantigen identification
The NeoQuery pipeline includes six major steps, which are illustrated in Figure S8A and described in detail below.
-
(1)
To check for overlap with the dbSNP database. Each single nucleotide variant (SNV) identified based on chromosome location and reference and alternative alleles was checked against the GRCh37 dbSNP database. SNVs included in the dbSNP database were not pursued further.
-
(2)
To retrieve protein sequences for an SNV. For each SNV passing the dbSNP filtering, corresponding gene name, transcript names, and single amino acid variant (SAAV) information were used to get the protein names, full-length wild-type protein sequences from the Ensembl database release 75, and corresponding mutant protein sequences.
-
(2)
To get candidate neoantigen sequences. For each SAAV site in a protein sequence, a sliding window was applied to get all possible peptide sequences with a length of 8-12 amino acids that contains the SAAV site.
-
(4)
PSM scoring and SAAV site filtering. Each candidate neoantigen sequence derived from one SAAV site was compared against all spectra from all HLA class I studies, including spectra from both cancer and non-cancerous samples. PSM scoring was based on the Hyperscore used in X!Tandem(Craig and Beavis, 2004) as described in PepQuery v1.2(Wen et al., 2019). In order to estimate FDR, a new approach was used based on the fact that all neoantigens identified in the non-cancerous samples are incorrect identifications. Assuming that the distribution of the incorrect neoantigen identifications in the cancer samples is the same as that of the identifications in non-cancerous samples for each SAAV site, we used the score distribution of the identifications from the non-cancerous samples to estimate the score distribution of the incorrect neoantigen identifications. The interactive semi-parametric method was used to estimate the distribution parameters, which is the same as the method used in PetideProphet(Choi et al., 2008; Ma et al., 2012) and transfer PEP(Yi et al., 2020). Only those PSMs derived from one SAAV site with an FDR <1% were retained. Furthermore, any PSMs involving a spectrum with a better match to a reference peptide sequence or a modified reference peptide sequence were disqualified. Because the reference database searching was performed using open-pFind that enables unrestricted modification searching, this filtering removes potential false positives caused by incorrect interpretation of spectra resulted from reference peptides with modifications.
-
(5)
Patient-specific database construction and neoantigen identification. Based on the qualified SAAV sites identified from different tumor samples, patient-specific database was built using Customprodbj v1.0 (https://github.com/bzhanglab/customprodbj)(Wen et al., 2020). The Ensembl release 75 protein database was used as the reference database. Open-pFind was used as the search engine to identify neoantigens. The parameters used were the same as search engine performance comparison: fixed modification: carbamidomethyl [C]; variable modification: oxidation [M]; enzyme specificity: no cleavage specificity; precursor mass matching tolerance: 10 ppm; fragment mass matching tolerance: 0.02Da. ‘Open search’ mode was used for open-pFind. Peptide length was set to 8-25 amino acids. Modified (except Oxidation[M] and Carbamidomethyl [C]) and non-modified (included Oxidation[M] and Carbamidomethyl[C]) identifications of open-pFind were filtered using FDR calculated by the PGA package separately and FDR was controlled at 1% at both PSM and peptide levels. All PSM and peptide identifications passing the FDR threshold were combined. The identified peptides were used as input for NetMHCpan v4.0 analysis and only those with a predicted MHC binding affinity of <500nM are reported as neoantigen identifications.
Identifying neoantigen candidates using NeoQuery
We sorted all mutations in the ICGC data portal by #donors affected and selected the top 100,000 mutations. We downloaded all these mutations in json format provided by the ICGC API ENDPOINTS (https://docs.icgc.org/portal/api-endpoints/#!/mutations/countDonors_0). Only the 7,464 unique missense mutations that are not included in the dbSNP database were retained for the NeoQuery analysis as described above. As positive controls, we also included in our analysis an additional 11 somatic mutations for which neoantigens have been previously reported in one of our HLA class I dataset by searching immunopeptidomic data from each sample in the dataset against sample-specific protein databases derived from matched WES data(Bassani-Sternberg et al., 2016).
Quantification and statistical analysis
For search engine performance comparison and peptide identification for all immunopeptidomics datasets, 1% peptide level and PSM level FDR was calculated for modified and non-modified identifications, separately. For differential expression analysis of cancer and non-cancerous samples, p values were calculated using Kolmogorov-Smirnov test for enrichment analysis and Student’s t-test for differential expression analysis. Bonferroni & Hochberg (BH) correction was used for multiple test correction. For positional frequency analysis for different PTM sites, p values were derived from Fisher’s exact test. For the FDR estimation of neoantigen identifications, the interactive semi-parametric method was used to estimate the distribution parameters.
Acknowledgments
This study was supported by the Cancer Prevention & Research Institutes of Texas (CPRIT) award RR160027, grants U24 CA210954 and R01 CA245903 from the National Cancer Institute (NCI), and funding from the McNair Medical Institute at The Robert and Janice McNair Foundation. B.Z. is a CPRIT Scholar in Cancer Research and a McNair Scholar.
Authorcontributions
B.Z. conceived the study. X.Y. performed all analyses with help from B.W., K.L., Y.D., S.S., and B.Z. Y.L. implemented the web portal. X.Y. and B.Z. wrote the manuscript. All authors read and approved the final manuscript.
Declarationofinterests
The authors declare no competing interests.
Published: October 22, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103107.
Supplemental information
Data and code availability
-
•
All data are publicly available through the caAtlas portal (http://www.zhang-lab.org/caatlas/).
-
•
This paper does not report original code.
-
•
All additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Abelin J.G., Keskin D.B., Sarkizova S., Hartigan C.R., Zhang W., Sidney J., Stevens J., Lane W., Zhang G.L., Eisenhaure T.M. Mass spectrometry profiling of HLA-associated peptidomes in mono-allelic cells enables more accurate epitope prediction. Immunity. 2017;46:315–326. doi: 10.1016/j.immuni.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adusumilli R., Mallick P. Proteomics. Springer; 2017. Data conversion with ProteoWizard msConvert; pp. 339–368. [DOI] [PubMed] [Google Scholar]
- Almeida L.G., Sakabe N.J., Deoliveira A.R., Silva M.C.C., Mundstein A.S., Cohen T., Chen Y.-T., Chua R., Gurung S., Gnjatic S. CTdatabase: a knowledge-base of high-throughput and curated data on cancer-testis antigens. Nucleic Acids Res. 2009;37:D816–D819. doi: 10.1093/nar/gkn673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An Z., Zhai L., Ying W., Qian X., Gong F., Tan M., Fu Y. PTMiner: localization and quality control of protein modifications detected in an open search and its application to comprehensive post-translational modification characterization in human proteome. Mol. Cell Proteom. 2019;18:391–405. doi: 10.1074/mcp.RA118.000812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelrod M.L., Cook R.S., Johnson D.B., Balko J.M. Biological consequences of MHC-II expression by tumor cells in cancer. Clin. Cancer Res. 2019;25:2392–2402. doi: 10.1158/1078-0432.CCR-18-3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassani-Sternberg M., Bräunlein E., Klar R., Engleitner T., Sinitcyn P., Audehm S., Straub M., Weber J., Slotta-Huspenina J., Specht K. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat.Commun. 2016;7:1–16. doi: 10.1038/ncomms13404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifant C.L., Jackson H.J., Brentjens R.J., Curran K.J. Toxicity and management in CAR T-cell therapy. Mol. Therapy-Oncolytics. 2016;3:16011. doi: 10.1038/mto.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulik-Sullivan B., Busby J., Palmer C.D., Davis M.J., Murphy T., Clark A., Busby M., Duke F., Yang A., Young L. Deep learning using tumor HLA peptide mass spectrometry datasets improves neoantigen identification. Nat.Biotechnol. 2019;37:55–63. doi: 10.1038/nbt.4313. [DOI] [PubMed] [Google Scholar]
- Chi H., Liu C., Yang H., Zeng W.-F., Wu L., Zhou W.-J., Wang R.-M., Niu X.-N., Ding Y.-H., Zhang Y. Comprehensive identification of peptides in tandem mass spectra using an efficient open search engine. Nat.Biotechnol. 2018;36:1059–1061. doi: 10.1038/nbt.4236. [DOI] [PubMed] [Google Scholar]
- Choi H., Ghosh D., Nesvizhskii A.I. Statistical validation of peptide identifications in large-scale proteomics using the target-decoy database search strategy and flexible mixture modeling. J.Proteome Res. 2008;7:286–292. doi: 10.1021/pr7006818. [DOI] [PubMed] [Google Scholar]
- Cobbold M., De La Peña H., Norris A., Polefrone J.M., Qian J., English A.M., Cummings K.L., Penny S., Turner J.E., Cottine J. MHC class I–associated phosphopeptides are the targets of memory-like immunity in leukemia. Sci.Transl.Med. 2013;5:203ra125. doi: 10.1126/scitranslmed.3006061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulie P.G., Van den Eynde B.J., van der Bruggen P., Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat. Rev. Cancer. 2014;14:135–146. doi: 10.1038/nrc3670. [DOI] [PubMed] [Google Scholar]
- Cox J., Mann M. MaxQuant enables high peptide identification rates, individualized ppb-range mass accuracies and proteome-wide protein quantification. Nat.Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Craig R., Beavis R.C. TANDEM: matching proteins with tandem mass spectra. Bioinformatics. 2004;20:1466–1467. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- DeLuca D.S., Levin J.Z., Sivachenko A., Fennell T., Nazaire M.-D., Williams C., Reich M., Winckler W., Getz G. RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics. 2012;28:1530–1532. doi: 10.1093/bioinformatics/bts196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Gebali S., Mistry J., Bateman A., Eddy S.R., Luciani A., Potter S.C., Qureshi M., Richardson L.J., Salazar G.A., Smart A. The Pfam protein families database in 2019. Nucleic Acids Res. 2019;47:D427–D432. doi: 10.1093/nar/gky995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng J.K., Jahan T.A., Hoopmann M.R. Comet: an open-source MS/MS sequence database search tool. Proteomics. 2013;13:22–24. doi: 10.1002/pmic.201200439. [DOI] [PubMed] [Google Scholar]
- Engelhard V.H., Altrich-Vanlith M., Ostankovitch M., Zarling A.L. Post-translational modifications of naturally processed MHC-binding epitopes. Curr.Opin.Immunol. 2006;18:92–97. doi: 10.1016/j.coi.2005.11.015. [DOI] [PubMed] [Google Scholar]
- Engelhard V.H., Obeng R.C., Cummings K.L., Petroni G.R., Ambakhutwala A.L., Chianese-Bullock K.A., Smith K.T., Lulu A., Varhegyi N., Smolkin M.E. MHC-restricted phosphopeptide antigens: preclinical validation and first-in-humans clinical trial in participants with high-risk melanoma. J. Immunother. Cancer. 2020;8:e000262. doi: 10.1136/jitc-2019-000262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez L., Milne R., Pita G., Aviles J., Lazaro P., Benitez J., Ribas G. SLC45A2: a novel malignant melanoma-associated gene. Hum.Mutat. 2008;29:1161–1167. doi: 10.1002/humu.20804. [DOI] [PubMed] [Google Scholar]
- Gfeller D., Bassani-Sternberg M. Predicting antigen presentation—what could we learn from a million peptides? Front.Immunol. 2018;9:1716. doi: 10.3389/fimmu.2018.01716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudbjartsson D.F., Sulem P., Stacey S.N., Goldstein A.M., Rafnar T., Sigurgeirsson B., Benediktsdottir K.R., Thorisdottir K., Ragnarsson R., Sveinsdottir S.G. ASIP and TYR pigmentation variants associate with cutaneous melanoma and basal cell carcinoma. Nat. Genet. 2008;40:886–891. doi: 10.1038/ng.161. [DOI] [PubMed] [Google Scholar]
- Haas G., Jr., D'Cruz O., De Bault L. Distribution of human leukocyte antigen-ABC and-D/DR antigens in the unfixed human testis. Am. J. Reprod. Immunol. Microbiol. 1988;18:47–51. doi: 10.1111/j.1600-0897.1988.tb00234.x. [DOI] [PubMed] [Google Scholar]
- Haen S.P., Löffler M.W., Rammensee H.-G., Brossart P. Towards new horizons: characterization, classification and implications of the tumour antigenic repertoire. Nat. Rev. Clin. Oncol. 2020;17:595–610. doi: 10.1038/s41571-020-0387-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoof I., Peters B., Sidney J., Pedersen L.E., Sette A., Lund O., Buus S., Nielsen M. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics. 2009;61:1. doi: 10.1007/s00251-008-0341-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard T., Barker D., Birney E., Cameron G., Chen Y., Clark L., Cox T., Cuff J., Curwen V., Down T. The Ensembl genome database project. Nucleic Acids Res. 2002;30:38–41. doi: 10.1093/nar/30.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilyas S., Yang J.C. Landscape of tumor antigens in T cell immunotherapy. J. Immunol. 2015;195:5117–5122. doi: 10.4049/jimmunol.1501657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalvik T.V., Arnesen T. Protein N-terminal acetyltransferases in cancer. Oncogene. 2013;32:269–276. doi: 10.1038/onc.2012.82. [DOI] [PubMed] [Google Scholar]
- Kee K., Angeles V.T., Flores M., Nguyen H.N., Pera R.A.R. Human DAZL, DAZ and BOULE genes modulate primordial germ-cell and haploid gamete formation. Nature. 2009;462:222–225. doi: 10.1038/nature08562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S., Pevzner P.A. MS-GF+ makes progress towards a universal database search tool for proteomics. Nat.Commun. 2014;5:5277. doi: 10.1038/ncomms6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koufaris C., Kirmizis A. N-terminal acetyltransferases are cancer-essential genes prevalently upregulated in tumours. Cancers (Basel) 2020;12:2631. doi: 10.3390/cancers12092631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreiter S., Vormehr M., Van de Roemer N., Diken M., Löwer M., Diekmann J., Boegel S., Schrörs B., Vascotto F., Castle J.C. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature. 2015;520:692–696. doi: 10.1038/nature14426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leisegang M., Kammertoens T., Uckert W., Blankenstein T. Targeting human melanoma neoantigens by T cell receptor gene therapy. J.Clin.Invest. 2016;126:854–858. doi: 10.1172/JCI83465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K., Jain A., Malovannaya A., Wen B., Zhang B. DeepRescore: leveraging deep learning to improve peptide identification in immunopeptidomics. Proteomics. 2020;20:e1900334. doi: 10.1002/pmic.201900334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K., Vaudel M., Zhang B., Ren Y., Wen B. PDV: an integrative proteomics data viewer. Bioinformatics. 2019;35:1249–1251. doi: 10.1093/bioinformatics/bty770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma K., Vitek O., Nesvizhskii A.I. A statistical model-building perspective to identification of MS/MS spectra with PeptideProphet. BMC Bioinform. 2012;13:S1. doi: 10.1186/1471-2105-13-S16-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcu A., Bichmann L., Kuchenbecker L., Kowalewski D.J., Freudenmann L.K., Backert L., Mühlenbruch L., Szolek A., Lübke M., Wagner P. HLA Ligand Atlas: a benign reference of HLA-presented peptides to improve T-cell-based cancer immunotherapy. J.Immunother.Cancer. 2021;9:e002071. doi: 10.1136/jitc-2020-002071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty R., Kaabinejadian S., Rossell D., Slifker M.J., van de Haar J., Engin H.B., de Prisco N., Ideker T., Hildebrand W.H., Font-Burgada J. MHC-I genotype restricts the oncogenic mutational landscape. Cell. 2017;171:1272–1283.e1215. doi: 10.1016/j.cell.2017.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer V.S., Drews O., Gunder M., Hennenlotter J., Rammensee H.G., Stevanovic S. Identification of natural MHC class II presented phosphopeptides and tumor-derived MHC class I phospholigands. J. Proteome Res. 2009;8:3666–3674. doi: 10.1021/pr800937k. [DOI] [PubMed] [Google Scholar]
- Mohammed F., Cobbold M., Zarling A.L., Salim M., Barrett-Wilt G.A., Shabanowitz J., Hunt D.F., Engelhard V.H., Willcox B.E. Phosphorylation-dependent interaction between antigenic peptides and MHC class I: a molecular basis for the presentation of transformed self. Nat. Immunol. 2008;9:1236–1243. doi: 10.1038/ni.1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen L.R., Tongchusak S., Lin H., Reinherz E.L., Brusic V., Zhang G.L. TANTIGEN: a comprehensive database of tumor T cell antigens. Cancer Immunol. Immunother. 2017;66:731–735. doi: 10.1007/s00262-017-1978-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak B.J., Lee J., Thai B.L., Fuchs S.Y., Shaked Y., Ronai Z.e., Kerbel R.S., Ben-David Y. Radiation resistance of human melanoma analysed by retroviral insertional mutagenesis reveals a possible role for dopachrome tautomerase. Oncogene. 2004;23:30–38. doi: 10.1038/sj.onc.1207007. [DOI] [PubMed] [Google Scholar]
- Petersen J., Wurzbacher S.J., Williamson N.A., Ramarathinam S.H., Reid H.H., Nair A.K., Zhao A.Y., Nastovska R., Rudge G., Rossjohn J. Phosphorylated self-peptides alter human leukocyte antigen class I-restricted antigen presentation and generate tumor-specific epitopes. Proc. Natl. Acad. Sci. 2009;106:2776–2781. doi: 10.1073/pnas.0812901106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineau C., Hikmet F., Zhang C., Oksvold P., Chen S., Fagerberg L., Uhlén M., Lindskog C. Cell type-specific expression of testis elevated genes based on transcriptomics and antibody-based proteomics. J. Proteome Res. 2019;18:4215–4230. doi: 10.1021/acs.jproteome.9b00351. [DOI] [PubMed] [Google Scholar]
- Pitcovski J., Shahar E., Aizenshtein E., Gorodetsky R. Melanoma antigens and related immunological markers. Crit.Rev.Oncol./Hematol. 2017;115:36–49. doi: 10.1016/j.critrevonc.2017.05.001. [DOI] [PubMed] [Google Scholar]
- Purcell A.W., Ramarathinam S.H., Ternette N. Mass spectrometry–based identification of MHC-bound peptides for immunopeptidomics. Nat.Protoc. 2019;14:1687. doi: 10.1038/s41596-019-0133-y. [DOI] [PubMed] [Google Scholar]
- Radwan J., Babik W., Kaufman J., Lenz T.L., Winternitz J. Advances in the evolutionary understanding of MHC polymorphism. Trends Genet. 2020;36:298–311. doi: 10.1016/j.tig.2020.01.008. [DOI] [PubMed] [Google Scholar]
- Riley R.S., June C.H., Langer R., Mitchell M.J. Delivery technologies for cancer immunotherapy. Nat.Rev. Drug Discov. 2019;18:175–196. doi: 10.1038/s41573-018-0006-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz D., Gloger A., Neri D., Fugmann T. Purification of soluble HLA class I complexes from human serum or plasma deliver high quality immuno peptidomes required for biomarker discovery. Proteomics. 2017;17:1600364. doi: 10.1002/pmic.201600364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizvi N.A., Hellmann M.D., Snyder A., Kvistborg P., Makarov V., Havel J.J., Lee W., Yuan J., Wong P., Ho T.S. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruark E., Seal S., McDonald H., Zhang F., Elliot A., Lau K., Perdeaux E., Rapley E., Eeles R., Peto J. Identification of nine new susceptibility loci for testicular cancer, including variants near DAZL and PRDM14. Nat. Genet. 2013;45:686–689. doi: 10.1038/ng.2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saghafinia S., Mina M., Riggi N., Hanahan D., Ciriello G. Pan-cancer landscape of aberrant DNA methylation across human tumors. CellRep. 2018;25:1066–1080. doi: 10.1016/j.celrep.2018.09.082. e1068. [DOI] [PubMed] [Google Scholar]
- Sarkizova S., Klaeger S., Le P.M., Li L.W., Oliveira G., Keshishian H., Hartigan C.R., Zhang W., Braun D.A., Ligon K.L. A large peptidome dataset improves HLA class I epitope prediction across most of the human population. Nat. Biotechnol. 2020;38:199–209. doi: 10.1038/s41587-019-0322-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaub F.X., Dhankani V., Berger A.C., Trivedi M., Richardson A.B., Shaw R., Zhao W., Zhang X., Ventura A., Liu Y. Pan-cancer alterations of the MYC oncogene and its proximal network across the cancer genome atlas. CellSyst. 2018;6:282–300.e282. doi: 10.1016/j.cels.2018.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster H., Peper J.K., Bösmüller H.-C., Röhle K., Backert L., Bilich T., Ney B., Löffler M.W., Kowalewski D.J., Trautwein N. The immunopeptidomic landscape of ovarian carcinomas. Proc. Natl. Acad. Sci. 2017;114:E9942–E9951. doi: 10.1073/pnas.1707658114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao W., Pedrioli P.G., Wolski W., Scurtescu C., Schmid E., Vizcaíno J.A., Courcelles M., Schuster H., Kowalewski D., Marino F. The SysteMHC atlas project. Nucleic Acids Res. 2018;46:D1237–D1247. doi: 10.1093/nar/gkx664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry S.T., Ward M.-H., Kholodov M., Baker J., Phan L., Smigielski E.M., Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shraibman B., Barnea E., Kadosh D.M., Haimovich Y., Slobodin G., Rosner I., López-Larrea C., Hilf N., Kuttruff S., Song C. Identification of tumor antigens among the HLA peptidomes of glioblastoma tumors and plasma. Mol. Cell Proteomics. 2019;18:1255–1268. doi: 10.1074/mcp.RA119.001524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solleder M., Guillaume P., Racle J., Michaux J., Pak H.-S., Müller M., Coukos G., Bassani-Sternberg M., Gfeller D. Mass spectrometry based immunopeptidomics leads to robust predictions of phosphorylated HLA class I ligands. Mol. Cell Proteom. 2020;19:390–404. doi: 10.1074/mcp.TIR119.001641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan X., Li D., Huang P., Jian X., Wan H., Wang G., Li Y., Ouyang J., Lin Y., Xie L. dbPepNeo: amanually curated database for human tumor neoantigen peptides. Database. 2020;2020:baaa004. doi: 10.1093/database/baaa004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ternette N., Olde Nordkamp M.J., Müller J., Anderson A.P., Nicastri A., Hill A.V., Kessler B.M., Li D. Immunopeptidomicprofiling of HLA-A2-positive triple negative breast cancer identifies potential immunotherapy target antigens. Proteomics. 2018;18:1700465. doi: 10.1002/pmic.201700465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran E., Ahmadzadeh M., Lu Y.-C., Gros A., Turcotte S., Robbins P.F., Gartner J.J., Zheng Z., Li Y.F., Ray S. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science. 2015;350:1387–1390. doi: 10.1126/science.aad1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigneron N., Stroobant V., Van den Eynde B.J., van der Bruggen P. Database of T cell-defined human tumor antigens: the 2013 update. Cancer Immun. Arch. 2013;13:15. [PMC free article] [PubMed] [Google Scholar]
- Vita R., Mahajan S., Overton J.A., Dhanda S.K., Martini S., Cantrell J.R., Wheeler D.K., Sette A., Peters B. The immune epitope database (IEDB): 2018 update. Nucleic Acids Res. 2019;47:D339–D343. doi: 10.1093/nar/gky1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein D., Leininger J., Hamby C., Safai B. Diagnostic and prognostic biomarkers in melanoma. J.Clin.Aesthet.Dermatol. 2014;7:13. [PMC free article] [PubMed] [Google Scholar]
- Wen B., Li K., Zhang Y., Zhang B. Cancer neoantigen prioritization through sensitive and reliable proteogenomics analysis. Nat.Commun. 2020;11:1–14. doi: 10.1038/s41467-020-15456-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen B., Wang X., Zhang B. PepQuery enables fast, accurate, and convenient proteomic validation of novel genomic alterations. Genome Res. 2019;29:485–493. doi: 10.1101/gr.235028.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen B., Xu S., Zhou R., Zhang B., Wang X., Liu X., Xu X., Liu S. PGA: an R/Bioconductor package for identification of novel peptides using a customized database derived from RNA-Seq. BMC Bioinform. 2016;17:244. doi: 10.1186/s12859-016-1133-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J., Zhao W., Zhou B., Su Z., Gu X., Zhou Z., Chen S. TSNAdb: a database for tumor-specific neoantigens from immunogenomics data analysis. Genom.Proteom.Bioinform. 2018;16:276–282. doi: 10.1016/j.gpb.2018.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi X., Gong F., Fu Y. Transfer posterior error probability estimation for peptide identification. BMC Bioinform. 2020;21:1–17. doi: 10.1186/s12859-020-3485-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarling A.L., Polefrone J.M., Evans A.M., Mikesh L.M., Shabanowitz J., Lewis S.T., Engelhard V.H., Hunt D.F. Identification of class I MHC-associated phosphopeptides as targets for cancer immunotherapy. Proc. Natl. Acad. Sci. U. S. A. 2006;103:14889–14894. doi: 10.1073/pnas.0604045103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Bajari R., Andric D., Gerthoffert F., Lepsa A., Nahal-Bose H., Stein L.D., Ferretti V. The international cancer genome consortium data portal. Nat.Biotechnol. 2019;37:367–369. doi: 10.1038/s41587-019-0055-9. [DOI] [PubMed] [Google Scholar]
- Zhang S., Chen K., Liu H., Jing C., Zhang X., Qu C., Yu S. PMEL as a prognostic biomarker and negatively associated with immune infiltration in skin cutaneous melanoma (SKCM) J. Immunother. 2021;44:214–223. doi: 10.1097/CJI.0000000000000374. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data are publicly available through the caAtlas portal (http://www.zhang-lab.org/caatlas/).
-
•
This paper does not report original code.
-
•
All additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.