

Abstract

The allene functionality has participated in one of the most exciting voyages in organic chemistry, from chemical curiosities to a recurring building block in modern organic chemistry. In the last decades, a special kind of allene, namely, allenol, has emerged. Allenols, formed by an allene moiety and a hydroxyl functional group with diverse connectivity, have become common building blocks for the synthesis of a wide range of structures and frequent motif in naturally occurring systems. The synergistic effect of the allene and hydroxyl functional groups enables allenols to be considered as a unique and sole functionality exhibiting a special reactivity. This Review summarizes the most significant contributions to the chemistry of allenols that appeared during the past decade, with emphasis on their synthesis, reactivity, and occurrence in natural products.
1. Introduction
Allenes are far considered the most useful and widely employed of the cumullenes. Since Van’t Hoff’s early predictions about structure and composition,1 chemists have produced a continuous stream of research in the allene field, facilitated by the perfect balance of reactivity and stability in the allene unit.2−13 Opposite to alkynes or alkenes, allenes show three reaction sites coupled to potential axial chirality. They can behave both as electrophiles and nucleophiles,14−17 and they can undergo cycloaddition reactions,18−21 thermal or radical rearrangements.22,23 Besides their synthetic utility, they are also recurring subjects in catalysis and theoretical studies.
In the last decades, a special kind of allene, namely, allenol, has emerged in both organic and physical organic chemistry, becoming a common building block for the synthesis of a wide range of structures..24−36 Allenols are formed by an allene and a hydroxyl functional group showing diverse connectivity. The synergistic effect of one functional group over the other when sharing the same skeleton makes the allenol unit a unique and sole functional group exhibiting a special reactivity. In one hand, allenols can be viewed as π-activated alcohols showing an extra reactivity toward eliminations, substitutions, or rearrangements. On the other hand, they can be viewed as allenes bearing extra electron pairs, which promote intramolecular cyclizations or provide an alternative metal-coordination site. This overview is focused in the most recent examples dealing with the enhanced chemical behavior of allenols, leaving aside the more particular situations where the allene and alcohol motifs react separately within the allenol molecules.
Herein we will discuss the most significant contributions to the allenol chemistry appeared during the past decade. Nevertheless, selected early works will be also mentioned to keep a critic and accurate review about the history of allenols. First of all, we will describe the most representative advances for the synthesis of allenols, specially focusing in the more challenging highly substituted structures and chiral allenes. Also, the different connectivity between the allene and hydroxyl moieties leading to α-, β-, γ-, or δ-allenols will be detailed. In a second chapter, synthetic utility of the allenol functional group will be discussed. This chapter will be organized according to the diverse reactivity of the allenol skeleton. Thus, in a first section, allenols as special π-activated alcohols will be considered, mainly showing hydroxyl units as leaving groups through elimination, substitution, aromatization, or rearrangement processes. A second section will describe the bidentate nature of the allenol, acting both as nucleophile and electrophile in annulation reactions. A third section is dedicated to all the examples where the OH group is not leaving or attacking the cumullene bonds. Instead, the alcohol unit is acting as a coordination site facilitating diverse transformations as additions, bond migrations, or isomerizations. A final section will contemplate the most recent achievements in chirality transfer using allenols through any kind of transformation. In addition, and to proof the extensive use of allenols in every field related to organic chemistry, a last chapter will be considered discussing the last contributions in natural product chemistry. Both allenols as key reaction intermediates and as motifs in the final structure will be presented.
2. Synthesis of Allenols
The extensive use of allenols in organic chemistry has also facilitated a considerable number of methodologies for their preparation. In addition to the most conventional routines, such as the allenation of terminal alkynes or the metal catalyzed addition of propargyl derivatives to aldehydes, different strategies have emerged during the past decade to provide more complex structures through more creative procedures. Among the diverse family of allenols, those bearing the hydroxyl unit at the α position, namely α-allenols, represent the widest number and focus the main part of investigations regarding both synthesis and applications. On the other hand, the synthesis of allenols exhibiting different connectivities such as β-, γ-, or δ-allenols have been frequently described following adapted methodologies from α-allenol synthesis. Thus, to get a concise discussion with a more homogeneous distribution, a classification according to the synthethic strategy is herein presented. Moreover, differently substituted allenols in both racemic and enantioenriched versions will be discussed.
2.1. Racemic Allenols
The most classical methodologies for the synthesis of racemic allenols start from alkynes, mainly including homologation of propargylic alcohols and addition of propargyl bromides to aldehydes (Scheme 1, reactions a and b). Also, activated allenes have been often used as starting materials in the aldol-type addition to carbonyl compounds (Scheme 1, reaction c).
Scheme 1. Classical Strategies for the Synthesis of Racemic Allenols.

Among the homologation procedures, allenation of terminal alkynes from propargylic alcohols is still one of the most common methodologies for the construction of the allenol skeleton. In the late 70s, Crabbé and co-workers reported the first synthesis of allenes from terminal alkynes, isopropylamine, paraformaldehyde, and CuBr as metal catalyst.37−39 The presence of a hydroxyl group in the alkyne unit seemed to activate the transformation providing α-allenols with excellent yields. Nevertheless, this transformation was limited to paraformaldehyde leading only to monosubstituted allenols as reaction products. In recent years, Ma research group has extensively investigated the scope of the allenation of terminal alkynes toward the synthesis of di- and trisubstituted allenes, by extending the methodology to diverse aldehydes and ketones.40 Thus, yields and scope were first improved by changing the original CuBr/propylamine pair for CuI/cyclohexylamine toward the optimized synthesis of terminal allenes 2. This methodology was successfully applied for the preparation of different allenols, such as β- or γ-allenols, normally showing low yields under original Crabbé’s reaction conditions (Scheme 2, reaction a).41−46 To extend the reaction to normal aldehydes, the same research group assumed that finding the proper metal salt/secondary amine combination should be the key for the direct allenylation. Fortunately, diverse matching combination such as ZnI2/morpholine or CuI/Bu2NH gave successful results from different starting materials (Scheme 2, reaction b); 1,3-disubstituted allenols 3 were therefore accessible.47,48 Further extension to trisubstituted allenols 4 was achieved by reaction with ketones using CdI2/pyrrolidine or the less toxic CuBr-ZnBr2/pyrrolidine reagent combination in a sequential addition procedure, or CuBr-ZnBr2–Ti(OEt)4/pyrrolidine in the one pot version (Scheme 2, reaction c).49−51
Scheme 2. Optimization of the Allenation of Terminal Alkynes for the Synthesis of Substituted Allenols.

An alternative approach for the homologation of terminal alkynes under copper catalysis employs differently substituted diazo compounds instead of aldehydes. First described by Wang and collaborators,52 this methodology has also been applied for the synthesis of substituted allenols by diverse research groups (Scheme 3).53−56 Alkyl and aryl-substituted allenes, allenoates, and TMS-disubstituted allenes are described in the recent literature following this strategy. In addition, both α- and β-allenols are accessed with no considerable decrease of yield.
Scheme 3. Substituted α- and β-Allenols from Terminal Alkynes and Diazo Compounds.

Allenylation of aldehydes with propargyl derivatives bearing an appropriate leaving group (normally bromides) represent another classical approach for the allenol synthesis. Many reports have appeared dealing with the regioselective control between propargylation and allenylation, including a wide variety of metal catalysts, such as Sn complexes,57−59 Zn,60−62 Bi, or Cd salts.63 Among all these well-known procedures, In-promoted allenylation in aqueous media has possibly provided the best results.64−67 In this field, Cr-catalyzed allenylation of aldehydes is probably the only contribution appeared during the past decade, allenols are prepared in a complete regioselective manner allowing both racemic and enantioselective synthesis, with the late being further discussed in the next section.85
In a similar approach, propargyl boronic esters have recently shown a good control in the regioselectivity toward the addition to aldehydes. Copper catalysis allowed the synthesis of both alkynol 9 and α-allenol structures 10 by switching the phosphine-based ligand (Scheme 4, reaction a).69,70 This study has been analyzed from both experimental and theoretical perspectives. Alternatively, the MOF (metal–organic framework)-supported mineral acid catalyst MIL-101 yielded similar α-allenols 10 as sole reaction products through a related transformation (Scheme 4, reaction b).71
Scheme 4. α-Allenol Synthesis through Propargylboronic Addition onto Carbonyl Compounds.

Kobayashi and co-workers have also reported an example of regiocontrol toward the allenylation versus propargylation of both aldehydes and ketones from boronic esters. In this case, Zn-propargyl or Zn-allenyl species are generated in situ from allenyl-boronic derivatives 11. The regioselectivity of the process was found to be temperature-dependent, yielding α-allenol species 12 as major reaction products at lower temperatures (Scheme 4, reaction c).72
Direct allene addition onto carbonyl compounds have also provided several examples of α-allenol synthesis through allene-aldol or Baylis-Hillman-type reactions, including both α- and γ-selective additions.73−75 In the recent literature, γ-addition has been reported from substituted allenoates 14 and 16 and diverse aldehydes through Morita-Baylis-Hillman additions using different catalysts (Scheme 5, reactions a and b).76,77 Direct addition of allenes to carbonyls frequently shows scope limitation as activated substrates are needed, normally allenoates. One rare example of this kind of transformation using a different activated allene employs tosyl derivatives 18. Treatment with nBuLi at low temperature yields the corresponding organolithium compound, which is reported to be trapped in the presence of different aldehydes generating α-allenols 19 (Scheme 5, reaction c).78 A wide variety of aldehydes or ketones and arylsulfones are tolerated, although the challenging trisubstitution pattern around the allene skeleton should be already present in the starting material.
Scheme 5. α-Allenol Synthesis through Activated Allene Addition to Aldehydes.

Besides the above-mentioned general strategies for the racemic allenol synthesis, the past decade has witnessed an increasing number of more specific transformation leading to more challenging α-allenol structures. Bäckvall research group has employed protected alkyndiols 20 and 22 through an iron-catalyzed cross-coupling reaction with Grignard reagents for the synthesis of di-, tri- and tetra-substituted allenols 21 and 23 (Scheme 6, reactions a and b).79 Acetate-protected hydroxyls act as the leaving group facilitating the cumullene generation, while TBS-protected OH remains unaltered in the final allenol skeleton. The two alcohol units could be placed in either opposite (20) or same (22) side of the alkyne moiety, providing a wider scope and versatility. Also, the mild reaction conditions allowed an extensive functional group compatibility in both the alkyndiol system and the Grignard reagent. Related work from Sherburn’s and Dou’s research groups have independently shown alkyndiol efficiency in the allenol synthesis (Scheme 6, reactions c and d). Pd-catalyzed Suzuki-Miyaura cross-coupling reaction from symmetrically substituted alkynes 24 allowed the synthesis of allenes 25. Nevertheless, only sterically hindered boronic acids were tolerated to avoid 2-fold addition processes (Scheme 6, reaction c).80 The use of rhodium catalysis in a similar transformation from diols 26 provided higher control toward the single addition reaction. Less hindered boronic acids were allowed, and unsymmetrically substituted alkyndiols 26 were also tolerated under Rh conditions (Scheme 6, reaction d).81
Scheme 6. Substituted α-Allenols from Alkyndiols.

Propargyl epoxides 28 have also been employed for the synthesis of substituted α-allenols through SN2′-type reactions in the presence of nucleophiles.82−87 While C-based nucleophiles such as Grignard reagents have been previously reported in classic methodologies, heteronucleophiles are much more scarcely described.88−91 Nevertheless, recent publications have started to focus in this transformation for the synthesis of allenes showing carbon-heteroatom bonds, not easily accessible through any other approach. Thus, B-,92 P-,93 and Sn-decorated allenols 29–31,94−96 have been synthesized using different transition metals as catalysts and mild reaction conditions (Scheme 7).
Scheme 7. Allenol Synthesis through Selective Ring Opening of Propargyl Epoxides.

Multicomponent reactions allow the synthesis of highly substituted and complex structures in one single step. This approach has been recently applied to the synthesis of β- and α-allenols from allenyl or propargyl boronic compounds, respectively. Petasis and co-workers have reported the synthesis of allenyl aminoalcohols 35, exhibiting a β-allenol motif, by a metal-free three-component process using allenyl boronic acids 32, amines 33, and hydroxyaldehydes 34.97 Regioselectivity (allenylation vs propargylation) was found to be dependent on the amine. Thus, secondary aliphatic amines selectively furnished the corresponding allenols (Scheme 8, reaction a), while primary and aromatic amines yielded the propargylation products. Alternatively, Thomson research group described a multicomponent reaction from alkynyl trifluoroborate salts 36, hydroxyaldehydes 34, and sulfonylhydrazines 37.98 The strategy was based in the in situ decomposition of the intermediate propargyl diazine 38 to yield the allenol compound 39 as sole reaction product through a so-called traceless Petasis-type process (Scheme 8, reaction b).
Scheme 8. Multicomponent Strategies for Allenol Synthesis.

More particular transformations to yield the allenol motif include the synthesis of exocyclic allenols through carbocyclization of both allenynes,99 or carbonyl enynes,100,101 the deoxygenation of pentadiyn diols,102 the aza-Cope-type rearrangement of propargyl indoles,103 one example of ethynylation and SN2′ reaction,104 or transformations from 1,3-enynes, such as alkylarylations,105 or hydromagnesiation.106
2.2. Enantioenriched Allenols
Asymmetric synthesis and chirality transfer processes have attracted much attention during the last years. Enantioenriched starting materials as chirality transfer agents represents one of the most common approaches. Thus, great interest has been shown in the design and synthesis of enantioenriched allenols, useful building blocks in asymmetric synthesis through diverse transformations as it will be later detailed. Because of the orthogonal distribution of cumullene molecular orbitals, allenes exhibit axial chirality when differently substituted. In addition, the presence of the extra alcohol unit in the allenol skeleton provides a potential stereogenic center. Synthesis of enantioenriched allenols may therefore contemplate axial chirality generation, central chiral generation, or both in the most complex cases. The principal methodologies for the synthesis of enantioenriched allenols that will be discussed in this section may be divided in three general groups; chirality transfer from enantioenriched starting materials, asymmetric synthesis using enantiopure catalysts, and dynamic resolution of racemic allenols.
2.2.1. Chirality Transfer and Chirality Induction from Enantioenriched Starting Materials
2.2.1.1. Allenols Showing Axial Chirality and Axial-Central Chirality
Allenation of terminal alkynes of terminal alkynols has also been investigated in the asymmetric version to yield optically pure allenols. Ma and co-workers have employed differently substituted prolinols 42 and 45 as chirality transfer agents providing practical yields and good to excellent enantioselectivities. TBS-protected alkynols 40 were first explored in combination with both (R)- and (S)-diphenylprolinol (42). Axial enantioselectivity is perfectly controlled from the absolute configuration of the amines 42 (Scheme 9, reactions a and b), while the stereochemistry of the hydroxyl carbon in the starting alkynol 40 is retained throughout the reaction when enantioenriched substrates were tested (Scheme 9, reaction c). The authors also point that the bulky TBS group in the alkynol 40 may have double role by avoiding the allene racemization and enhancing the enantioselectivity.107,108 In a later work, dimethylprolinol (45) was found to exhibit higher enantiodirection, allowing the use of unprotected alkynols 44 as starting materials and extending the scope to the obtention of β-, γ-, and δ-allenols (Scheme 9, reaction d).109
Scheme 9. Asymmetric Allenation of Terminal Alkynes Employing Chiral Amines.

Yu’s research group has envisioned an aldol allenoate addition to aldehydes 47 promoted by chiral bromoboranes 48 in the presence of tertiary amines. Both regiochemical and stereochemical outcomes of the reaction are explained through a Curtin-Hammet-type transition state 49, selectively favoring γ-addition products 50 and providing excellent chiral and central enantioselectivities (Scheme 10). The methodology was also applied for the kinetic resolution of racemic aldehydes, and further generation of the butenolide core of the natural product (+)-xilogiblactone A.110−113
Scheme 10. Aldol Allenoate Addition to Aldehydes Promoted by Chiral Bromoboranes.

Enantioenriched oxiranes have also been employed for the synthesis of di- and trisubstituted allenols with good diastereoselectivites.114−116 Two related approaches based in metal catalysis and organoboron reagents have been described. Propargyl epoxides 51 undergo a ring-opening through a syn-hydride borane addition using MeOH as proton shuttle, followed by selective syn-elimination catalyzed by copper salts. The proper phosphine-base combination seemed to be crucial for the axial selectivity toward allenols 52 (Scheme 11, reaction a).117 On the other hand, enynyl oxiranes 53 have been reported to react through a formal SN2′ mechanism in the presence of aryl boronic esters and palladium catalysts to provide enantioenriched allenols 54 (Scheme 11, reaction b).118
Scheme 11. Chiral Oxiranes as α- and γ-Allenol Precursors.

Another example of a central-to-axial chirality transfer uses optically pure ethynyl β-lactams 55 and different aldehydes for the asymmetric synthesis of structurally complex allene diols 56. The process includes initial in situ generation of the propargyl indium reagent, and further addition onto aldehydes. Although complete selectivity in the generation of the new α-hydroxy chiral center was not achieved, a reasonable dr = 11:89 could be attained (Scheme 12).119
Scheme 12. Central-to-Axial Chirality Transfer from Ethynyl β-Lactams.

Ma and colleagues have accomplished the synthesis of enantioenriched β- and γ-allenols 58 taking advantage of the reduction of optically pure allenoic acids 57 with LiAlH4 (Scheme 13, reaction a).120−122 The preparation of enantioenriched α-allenol 60 from allenoic acid 59 required an esterification followed by reduction with DIBAL-H (Scheme 13, reaction b).123 The above processes occur with efficient chirality transfer, which shows the high synthetic potential of this methodology in asymmetry synthesis. The DIBAL-H-promoted reduction of racemic α-allenoates into α-allenols can be conveniently achieved,124 while the LiAlH4-assisted reduction of enantioenriched α-allenoates 61 (Scheme 13, reaction c125 and the double 1,2-addition of allyl magnesium chloride to axially chiral α-allenoate 63 (Scheme 13, reaction d)126 generated with retained chirality the corresponding optically active α-allenols 62 and 64, respectively.
Scheme 13. Chiral Allenoic Acids and Allenoates as α-, β-, and γ-Allenol Precursors.

2.2.1.2. Allenols Showing Central Chirality
[2,3]-Wittig rearrangement of propargylic ethers provides the α-allenol skeleton in one single synthetic step. A first approach to the asymmetric version of the Wittig rearrangement was applied to the synthesis of a pharmacologically attractive α-hydroxy γ-amino acid 66 bearing an allene unit, despite in poor yield (Scheme 14, reaction a).127 More recently, a Wittig-based methodology was also employed for the synthesis of a family of substituted asymmetric α-allenols 68. In this case, a remote chiral sulfoxide in the starting propargylic ether 67 was responsible of the stereochemistry, behaving as a chiral inductor rather than a chirality transfer agent (Scheme 14, reaction b).128 In both cases, good diastereoselectivities were achieved, although no axial chirality is described, and the lack of a wider scope in the methodology leaves much work yet to be explored in this field.
Scheme 14. Asymmetric Wittig Rearrangement in α-Allenol Synthesis.

Other examples dealing only with central chirality in the newly formed hydroxyl carbon use the asymmetric version of the allenyl boronate addition onto aldehydes. Zn catalysis provides complete regioselectivity toward the allenylation versus propargylation processes, while chiral α-amino aldehydes 69 are responsible of the stereoselectivity observed as chiral inductors. Diastereoselectivity can be tuned by simply modifying the aldehyde substitution. NHBoc-substituted aldehydes led to syn amino allenols 71 though a Cram-chelation model 71′, while NBnBoc-substituted aldehydes yielded anti isomers 72 through a Felkin-Ahn addition model 72′ (Scheme 15).129
Scheme 15. Asymmetric Allenyl Boronate Addition to Aldehydes.

More specific transformations include the asymmetric propargylboration of aldehydes using 10-trimethylsilyl-9-borabicyclo[3.3.2]decanes,130 or Barluenga’s multicomponent reaction of chromium carbenes 73, oxazolidine-2-ones lithium enolates 74, and Grignard reagents 75 to yield highly substituted cyclohexenones 76 bearing allenolic units.131 Central-to-central chirality transfer is reported, using optically pure oxazolidines as chiral inductors, and yielding allenols 76 showing up to 99% ee (Scheme 16).
Scheme 16. Multicomponent Asymmetric Reaction for the Synthesis of Allenyl Cyclohexenones.

2.2.2. Kinetic Resolution of Racemic Allenols
Because of the more effective and economic chirality transfer approaches based in enantiopure catalysts, the past decade has experienced a decay in the number of contributions dealing with kinetic resolution strategies. Nevertheless, pioneer research groups in this field such as Bäckvall’s,132−135 have continued their interest in kinetic resolution strategies proposing new alternatives and more efficient procedures.
Dynamic kinetic resolution (DKR) by means of thermal or chemical induced isomerization has been extensively used to overcome the limited yield disadvantage inherent to KR. Axially chiral trisubstituted α-allenols 78 were obtained through esterification of 77 in the presence of lipase from porcine pancreas and vinyl butyrate. DKR was achieved by using palladium catalysis, inducing the allene 77 isomerization through the corresponding π-allyl palladium complex. The reported hybrid chemo-enzymatic methodology led to improved yields up to 83%, and good enantioselectivities (Scheme 17).136
Scheme 17. DKR of α-Allenols Using Palladium Catalysis.

One different conceptual approach to get access to chiral allenols with high yields employs prochiral starting materials in desymmetrization processes. Thus, allene diols 79 react selectively with vinyl butyrate in the presence of Lipase from porcine pancreas as sole catalyst to yield optically pure monoesters 80. Yields were good to excellent for allenes bearing aromatic susbtituents, and expectedly lower for aliphatic systems, though practical high enantioselectivities up to 99% were reported (Scheme 18).137
Scheme 18. Desymmetrization of Allendiols.

The group of Ma reported in 2002 the use of Novozym-435 as a convenient biocatalyst for the kinetic resolution of racemic α-allenols, giving rise to enantioenriched (S)-(−)-α-allenols and (R)-(+)-α-allenyl acetates in an efficient way.138 Hong and co-workers have contributed to the asymmetric synthesis of allenols with central chirality by developing different KR methodologies. In this regard, both enzymatic and chemical alternatives have been studied. Acetylation of α-allenols 81 in the presence of the appropriate lipase allowed the preparation of optically pure compounds (R)-81 and (S)-82 with yields up to 50% and enantiomeric excesses above 99%. After an enzyme screening, lipase AK (Pseudomonas fluorescens) was identified as the best candidate to achieve this transformation. The methodology was expanded to many differently substituted terminal allenols, including alkyl-, alkenyl-, and aryl-decorated structures (Scheme 19).139
Scheme 19. Enzymatic KR of Centrally Chiral α-Allenols.

Taking advantage of the transition metals ability to catalyze allenol cycloisomerizations, which will be further discussed in the next chapter, a chemical KR of allenols was envisioned. The chiral silver phosphate 84 allowed the selective oxycyclization of the (S)-enantiomers from the racemic mixture of allenols 83. Thus, enantioenriched dihydrofurans 85 were obtained, while unreacted (R)-allenols 83 were recovered. Both species were easily separable after column chromatography, providing aryl-substituted terminal allenols with yields up to 50% and up to 99% ee (Scheme 20).140,141
Scheme 20. Chemical Kinetic Resolution of Aryl-Substituted Terminal Allenols.

2.2.3. Asymmetric Synthesis Using Enantiopure Catalysts
The use of enantiopure catalysts represents a strategy of increasing interest in asymmetric synthesis. The possibility to avoid the preparation of optically pure starting materials in large scale, along with the catalyst recycling facilitates more economic synthetic routes. Regarding the synthesis of enantioenriched allenols, both enantiopure ligands in metal catalysis and enantiopure organocatalysts are described.
2.2.3.1. Enantiopure Ligands in Metal Catalysis: Axial and Axial-Central Chirality
Ma and co-workers have studied the asymmetric allenation of terminal alkynes using enantiopure ligands as chirality transfer agents in the synthesis of enantioenriched α-allenols. Readily available propargylic alcohols 86 were first submitted to Cu catalysis using pyrrolidine (88) as amine and (R,Ra)-PINAP as ligand. To achieve practical reaction conversions, cocatalysts ZnBr2 or CdI2 were needed, describing a one pot/two steps and a one pot/one step procedures respectively (Scheme 21, reaction a).142,143 Further investigations on this transformation revealed that increasing the amine ring size (90) led to high enantioselectivities under CuI as sole catalytic species in a more efficient and economic manner (Scheme 21, reaction b). Also, reversal enantioselectivity was easily achieved by using enantiomer (R,Sa)-PINAP as chiral ligand.144 Good axial enantioselectivities were reported through a versatile and practical methodology.
Scheme 21. Asymmetric Allenation of Terminal Alkynes Using Chiral Ligands.

Aldol-type additions of both propargylic and allenyl substrates onto carbonyls also find a stereoselective variant based on the use of chiral ligands in transition metal catalysis for the synthesis of α-allenols. Alkynylogous aldol reaction from propargylic carboxylates 91 catalyzed by copper salts and (R)-DTBM-SEGPHOS (94) as chiral ligand was found to be very effective for the synthesis of 2,3-allenols 93 from aromatic aldehydes (Scheme 22, reaction a). On the other hand, aliphatic aldehydes showed better stereoselectivities in the presence of (R,R)-Ph-BPE (95) as chiral ligand, which provided opposite central enantioselectivity. In both cases, high diastereo- and enantioselectivities were obtained (Scheme 22, reaction a).145,146 Au (III) salts have been reported to promote the aldol-type addition of allenic esters 96 onto isatin 97. In this case, a chiral N,N-dioxide 99 was used as chirality transfer agent, providing tri- and tetra-substituted allenols 98 in good to excellent yields and good enantioselectivities (Scheme 22, reaction b).147 In a different approach, condensation of boronic acids with α-hydroxycarbonyls 100 formed 1,3-dioxaboroles 102, which can be used as electrophiles in asymmetric allenylation reactions for the synthesis of β-allenols. Thus, racemic allenes 101 were transformed into enantioenriched allenols 103 using palladium catalysis and enantiopure phosphine ligands (Scheme 22, reaction c).148
Scheme 22. Asymmetric Aldol-type Synthesis of Substituted α- and β-Allenols.

1,3-Enynes have also been employed as common starting materials for the asymmetric synthesis of allenols exhibiting diverse allene-hydroxyl connectivity. Challenging tri- and tetra-substituted allenols 107 showing axial chirality are accessible through a cooperative Cu–Pd arylboration of enynes 105. Treatment of boron intermediates 106 with NaBO3 eventually yielded the expected α-allenols 107. The appropriate use of both metal catalyst and the noncommercial chiral sulfoxide 112 as ligand is reported to be responsible of the high enantioselectivity observed, avoiding racemization of allenyl copper intermediates (Scheme 23, reaction a).149 In a different contribution, related 2-trifluoromethyl enynes 108 decorated with a hydroxyl group, provided the allenol skeleton through a similar Cu-catalyzed 1,4-protoborylation or 1,4-protosylilation. In this case, new designed chiral bisoxazoline ligands 113 showed the best results yielding up to 97% ee (Scheme 23, reaction b).150 Alternatively, copper hydride semireduction of enynes 110 provided axially chiral disubstituted allenols 111. Mild reaction conditions and the use of water as proton source allowed a wide functional group compatibility. Commercially available 1,2-bis((2S,5S)-2,5-diphenyl-phospholano)ethane [(S,S)-Ph-BPE] (114) showed enantiomeric excesses above 99% (Scheme 23, reaction c).151
Scheme 23. Axially Chiral Allenols from Enynes.

2.2.3.2. Enantiopure Ligands in Metal Catalysis: Central Chirality
Besides the above-mentioned coupling reaction with enynes, organoboron reagents have shown great versatility toward allenol preparation, both starting from diverse substrates and through different transition metal-catalyzed reaction mechanisms. Cross-coupling reaction between propargylic carbonates 115 and boronate complexes 116 under Pd catalysis afforded β-boryl allenes 117 as reaction intermediates. Again, boron functionalization was effectively used as hydroxyl precursors by treatment with NaBO3. (S,S)-MandyPhos ligand (119) was employed to induce central chirality, providing β-allenols 118 with good yields and enantioselectivities up to 98% (Scheme 24, reaction a).152 A related approach reacted vinyl arenes 121 and bis(pinacolato)diboron B2(pin)2 with propargylic phosphates 120 under Cu catalysis and (R,S)-Josiphos (124) as chirality transfer agent. Following a similar strategy, the hydroxyl group was obtained after treatment of the corresponding β-boryl allenes 122 with NaBO3 (Scheme 24, reaction b).153
Scheme 24. Cross-Coupling Reactions of Borane Complexes and Propargylic Compounds.

More particular transformations based on enantiopure ligands include reaction between diazoesters 125 and propargylic compounds 126 through a tandem ylide formation/[2,3]-sigmatropic rearrangement (Scheme 25, reaction a),154 [2,3]-Wittig rearrangement of propargylic isatins 128 (Scheme 25, reaction b),155 Wacker-type oxyallenylation of cyclic alkenes 131 (Scheme 25, reaction c),156 or a Cr-catalyzed addition of propargyl bromides 136 onto aldehydes (Scheme 25, reaction d).157
Scheme 25. Allenol Asymmetric Synthesis from Propargylic Derivatives through Diverse Procedures.

2.2.3.3. Enantiopure Organocatalysts
Enantiopure organocatalysts have been scarcely reported for the synthesis of allenols. Nevertheless, during the last years, different research groups have started to apply this methodology to the asymmetric addition of propargyl and allenyl compounds to carbonyls. This transformation unravelling intriguing catalytic strategies allows the synthesis of both axially and centrally chiral allenols with good yields. Hoveyda’s research group has described a general methodology for the asymmetric nucleophilic addition to carbonyls controlled by fluorine–ammonium electrostatic interactions. The organocatalyzed procedure was applied to the addition of allenyl boronic complexes 140 to trifluoromethyl ketones 139, yielding α-allenols 142. An enantiopure organocatalyst 141 showing the appropriate electronic features delivered allenols 142 in excellent yields and enantioselectivities above 99% (Scheme 26, reaction a).158 Chen and co-workers have also studied a related organoboron addition onto carbonyls for the asymmetric approach to the allenol skeleton. In this case, propargylic boronates 144 were added to aldehydes in the presence of a chiral phosphoric acid 145, providing α-allenols 146 with good to excellent yields and showing central chirality with up to 99% ee (Scheme 26, reaction b).159
Scheme 26. Organocatalyzed Addition of Allenyl and Propargyl Boronic Complexes to Carbonyls.

Following with the extensive use of unsaturated organoboron compounds, and continuing with the applications of Petasis-type reactions, Thomson and co-workers have developed a chiral organocatalyzed version of the multicomponent reaction between propargyl boronates 147, protected aldehyde 148, and sulfonyl hydrazine 149. Enantiopure biphenol 150 gave access to enantioenriched allenols 151 displaying axial chirality, with moderate to good yields and up to 99% ee (Scheme 27).160
Scheme 27. Organocatalyzed Asymmetric Traceless-Petasis for the Synthesis of Allenols.

Organocatalyzed alkynylogous Mukaiyama aldol reaction also constitutes a feasible methodology for the asymmetric allenol preparation. List and collaborators have recently reported an enyne addition onto aldehydes catalyzed by a newly designed chiral disulfonimide 154. Challenging tetrasubstituted allenols 155 were prepared, exhibiting both axial and central chirality. The scope of the transformation includes aromatic aldehydes 152 and differently substituted alkyl enynes 153. Mild reaction conditions are reported, leading to moderate or excellent yields, diasteroselectivities up to 27:1, and enantiomeric excesses up to 98.5% (Scheme 28).161
Scheme 28. Organocatalyzed Alkynylogous Mukaiyama Aldol Synthesis of Allenols.

3. Synthetic Utility
3.1. Allenols as π-Activated Alcohols
Hydroxyl units are traditionally considered bad leaving groups in organic chemistry, unless previous OH-activation has been made. π-Activated alcohols are a special class of hydroxylic compounds in which the positive charge at the α-carbon is stabilized by the presence of conjugated π-orbitals. Taking advantage of this particular reactivity, π-activated α-allenols have been reported to undergo a wide number of transformations where the C–O bond cleaves at one certain point of the reaction mechanism. In this context, reactivity of allenols may be divided in two main groups: (i) those where the OH leaves the molecule at the first steps of the mechanism, leading normally to the diene, enone or enyne skeletons, and (ii) those where the OH loss occurs at the final stages of the process, which are normally found in tandem reactions for the synthesis of aromatic rings and alkaloids. Thus, either when a 1,3-migration reaction takes place or an external nucleophile attacks the central allenic carbon promoting the extrusion of the previously activated alcohol in a SN2′-type reaction, diene/enone skeletons may be formed (Scheme 29, path a). On the other hand, if a base abstracts a terminal allenic proton, rearrangement and activated alcohol elimination can take place to yield the enyne motif (Scheme 29, path b). In addition, when the allenic carbocation resulting from C–O bond dissociation is stable enough, an allene transfer process may happen by nucleophilic trapping, retaining the allene moiety (Scheme 29, path c). Finally, C–OH cleavage can take place through a further dehydratation step after carbo- or heterocyclization processes, leading normally to aromatic or heteroaromatic compounds (Scheme 29, path d).
Scheme 29. General Reaction Mechanisms for C–OH Cleavage in α-Allenols.

3.1.1. OH as a Leaving Group in the First Stage of the Reaction
3.1.1.1. Synthesis of Dienes and Enones
Dienes are easily generated from allenols and protected allenols trough 1,3-rearrangement processes. Several methodologies including acid or base promoted isomerizations and metal promoted reactions have been recently reported.162−185
Starting from allenols 156 and the appropriate sulfonyl chloride, the Alcaide and Almendros research group described a novel [3,3]-sigmatropic rearrangement of nonisolable α-allenic methanesulfonates and arylsulfonates 157. The formal OH migration to yield dienes 159 is proposed to proceed through a six membered ring transition state 158 (Scheme 30).186,187 DFT calculations supported an aromatic transition state in accordance to a pericyclic reaction mechanism, in view of a negative nucleus independent chemical shift obtained at the ring critical point of the electron density (NICS = −6.5 ppm). Also, a low calculated activation barrier of only 17.7 kcal/mol shows coincidence with the mild experimental reaction conditions needed for the transformation.
Scheme 30. Methyl and Arylsulfonyl Chloride Promoted Synthesis of Dienes and Its Application toward the Synthesis of Polycyclic Structures.

The methodology was extended to a wide number of substituted allenols 156 and applied to the preparation of different fused polycyclic structures. In one hand, a tandem [3,3]-sigmatropic rearrangement/Diels–Alder reaction provided optically pure tricyclic β-lactams such as 160 (Scheme 30, reaction a).186 On the other hand, related [3,3]-sigmatropic transposition of arylsulfonyl allenes produced dienes 159b or 159c, which are employed to the synthesis of enantiopure polycyclic sultones such as 162b or 162c trough a two-step sequence (Scheme 30, reactions b and c).187
Wang’s research group has presented an alternative procedure for the [3,3]-sigmatropic allenol rearrangement using sulfonic acids 164 instead of sulfonyl chlorides. The methodology was applied to di- and trisubstituted allenols 163 (Scheme 31, reaction a).188 Related work from Lee and collaborators provided E-dienes 167 with good yields and good to excellent stereoselectivities using trimethylsilyl triflate or trimethylsilyl chlorides 166. In this case, triflate- and chlorine-decorated dienes 167 were respectively prepared, expanding the scope of diene functionalization. DFT calculations also pointed to a similar six membered aromatic transition state, based on hydrogen bonding. Nevertheless, the change of 1,3-migration reagent from sulfur- to TMS-derivatives seemed to induce a slight loss of stereoselectivity, leading to E/Z mixtures in rates depending on the allenol substitution (Scheme 31, reaction b).189
Scheme 31. Scope of Metal-Free [3,3]-Sigmatropic Rearrangement of α-Allenols.

Different 1,3-migration strategies involving a previous reaction of the OH unit in α-allenols with coupling or protecting reagents have appeared. Thus, reaction of allenols 163 with TsNCO yielded the corresponding allenic N-tosylcarbamates 168. Taking advantage of the thermal instability of these N-tosylcarbamates, a decarboxylative aza-Michael addition/elimination sequence generating dienes 169 has been induced by heating at 125 °C in the presence of a basic catalyst (Scheme 32, reaction a). The 4-ethoxycarbonyl substitution on the allene moiety seems to be crucial for the transformation, although both alkyl and aryl substituents at the carbinolic core are well tolerated, and excellent steroselectivities achieved.190
Scheme 32. Metal-Free 1,3-Migration Strategies for the Synthesis of Dienes from α-Allenols.

Protection of the hydroxyl group as acetate led to acetoxyallenes 170, which were described to undergo an Ireland-Claisen-type rearrangements in the presence of a base. A [3,3]-sigmatropic reaction mechanism was therefore proposed, proceeding via a six membered chair-type transition state 172 (Scheme 32, reaction b). It was also described the use of N,N-dimethylacetamide dimethylacetal as protective reagent instead of acetic acid, promoting an Eschenmoser-Claisen rearrangement leading to similar results.191
Reaction of the alcohol unit in allenols 174 under Mitsunobu conditions using N-isopropylidine-N′-2-nitrobenzenesulfonyl hydrazine (175) as nucleophile, led to allenyl diazenes 176. Those substrates were envisioned as precursors of 1,3-dienes through a reductive transposition, via a retro-ene-type transition state 177. The above methodology generated unsubstituted dienes 178 in moderate to good yields. Opposite to previously mentioned [3,3]-sigmatropic rearrangements, the reaction course proceeded with a notable lack of stereoselectivity, yielding cis/trans dienes in rates from 3:1 to 1:1 (Scheme 32, reaction c).192
Preprepared HCl or HBr solutions in ether or ethyl acetate are reported to promote the isomerization of (1-hydroxybuta-2,3-dien-2-yl)diphenylphosphine oxides 179 into chlorinated or brominated phosphinoyl 1,3-butadienes 180. The methodology is applied to primary alcohols, and no stereocontrol is stated at the C3–C4 double bond. Added value to this acid-promoted metal-free methodology is given by further applicability of halogenated dienes 180 on epoxidation or Suzuki-type reactions, providing tetrasubstituted epoxides 181 and aryl-substituted dienes 182, respectively (Scheme 33).193,194
Scheme 33. Acid Promoted Synthesis of Halogenated Phosphinoyl Dienes and Synthetic Applications.

A conceptually different approach for the synthesis of dienes was based on a SN2′ reaction in 1-acetoxy-2-allenoates 183. DABCO addition onto the central allenic carbon induced the generation of 1,3-dienes 184 facilitated by extrusion of the AcO group. Tong and co-workers envisioned intermediate 1,3-diene-2-amonium species 184 as adequate 1,3-bis(electrophiles) for the reaction with 1,3-bis(nucleophiles) in a formal (3 + 3) ring closing reaction. Indoline-2-thiones 185 were selected as ideal candidates, showing 1,3-bisnucleophilic nature in the presence of a weak base. Thus, reaction of 1-acetoxy-2-allenoates 183 and indolines 185 in the presence of DABCO provided dihydrothiocarbazoles 186 through a SN2′–SN2′ reaction sequence (Scheme 34).195,196
Scheme 34. Dihydrothiocarbazole Synthesis by Reaction of Indoline-2-thiones and In Situ Generated Dienes.

Metal species are also reported to promote isomerization of allenols to dienes in both equimolecular and catalytic manner. The cobalt-catalyzed regioselective C8 dienylation of quinoline N-oxides with allenylcarbinol carbonates has been reported by Volla and co-workers, while the use of unprotected allenylcarbinols as the dienylating agents resulted in diminished yields.197 Alcaide and Almendros research group has described the use of FeBr3 or FeCl3 for the halogenation/rearrangement of 2-indolinone-tethered allenols 187 yielding 2-halo-1,3-dienes 188 (Scheme 35, reaction a). The transformation tolerated different substitution on the aromatic ring and exhibited complete Z-selectivity in every case. The high stereoselectivity observed could be explained considering a pseudopericyclic transition state 189, rather than a stepwise reaction mechanism. Thus, reaction course could be initiated by coordination of the OH group to the metal salt acting as a Lewis acid. Then, a six-membered chair-type transition state 189 facilitates the cleavage of the hydroxyl with concomitant halogen delivery. Extra coordination of the metal ion with the C=O group displayed in axial position could also support the high stereoselectivity found for this transformation. Further Suzuki-Miyaura coupling reaction from 2-halo-1,3-dienes 188 and aryl boronic acids 190 provided the corresponding aryl-substituted dienes 191, showing the synthetic applicability of the methodology.198
Scheme 35. Iron-Promoted Halogenation/Rearrangement of Allenols and Synthetic Applications.

Following a similar idea, Lin and co-workers developed a FeBr3-mediated bromination/rearrangement reaction to yield related 2,5-dibromo-4-aryl-1,3-pentadienes 193. Further one-pot N-alkylation/Diels-Alder reaction with tosyl-amines 194 provided products 195 bearing the hexahydro-1-H-isoindole skeleton with good yields and high diastereoselectivities (Scheme 35, reaction b).199
The ability of α-allenols to easily isomerize to dienes through 1,3-migration reactions was also illustrated during the attempts to oxidize allenyl vinyl alcohols to the corresponding allenyl vinyl ketones. Harmata and collaborators envisioned the synthesis of ketones 198 as starting materials for Nazarov cycloadditions using PCC as mild oxidant. Surprisingly, Cr-mediated 1,3-migration took place leading to unexpected α′-hydroxydienones 197 (Scheme 36). According to the authors, a mechanistic explanation for this result could start from formation of the chromate ester 199, followed by 1,3-transposition through the habitual six-membered chair-type transition state 200 to give the chromium enolate 201. Spontaneous (2,3)-sigmatropic rearrangement could produce the new chromate ester 202, which could yield the observed dienyl ketones 197 after hydrolysis. The methodology was extended to a wide number of structures with different substituents, exhibiting moderate to excellent yields and excellent stereoselectivity.78
Scheme 36. Chromium-Mediated Rearrangement of Allenyl Vinyl Alcohols.

Metal species in catalytic amounts have also been reported to promote allenol transformations into dienes showing several advantages. As previously mentioned, phosphinoyl allenols were described to react in the presence of acid solutions yielding phosphinoyl dienes despite of low efficiency and lack of stereoselectivity (179 to yield 180 in Scheme 33). Nevertheless, related 4-phosphoryl-2,3-allenols 203 have been recently found to provide the corresponding 1-phosphoryl 1,3-butadienes 205 in excellent diastereoselectivities using palladium catalysis through a Suzuki-Miyaura cross-coupling reaction with aryl boronic acids 204 (Scheme 37). A plausible reaction mechanism could start with coordination of the metal species to the terminal C–C double bond, and simultaneous activation of the OH groups with the boronic acid in complex 206. C–O bond cleavage would then take place generating the corresponding π-allyl palladium complex 207. The high diasteroselectivity resulting from this transformation could be explained by the extra coordination of the metal unit with the P(O) group, leading to the stabilized vinyl palladium complex 208. Transmetalation and reductive elimination in species 209 would provide observed phosphinoyl dienes 205 and regenerate Pd(0) to the catalytic cycle (Scheme 37).200
Scheme 37. Palladium-Catalyzed Synthesis of 1-Phosphoryl 1,3-butadienes.

Palladium and platinum catalysis has also been employed in the transformation of simple allenols and boronic acids. Exploring the addition of arylboronic acids onto a wide variety of allenes, one example of 1,3-diene synthesis is reported when allenol 210 is submitted to Pd or Pt conditions in the presence of boronic acid 211. Nevertheless, chemoselectivity is not complete, and addition of boronic acids without dehydratation is observed (Scheme 38, reaction a).201
Scheme 38. Metal-Catalyzed Synthesis of Dienes in the Presence of Boronic Acids.

A rhodium-catalyzed alternative for this transformation was described using 4-arylbuta-2,3-dien-1-ols (214) as starting material and different aryl boronic acids 215. In this case, carbometalation across the phenyl-substituted allenic bond, followed by δ-elimination of Rh(I)–OH was proposed as mechanistic rationale. Again, metal catalysis provided higher selectivity toward the Z-dienes 216 compared with the metal-free analogous transformation. Nevertheless, the lack of an extra coordination site such as the phosphoryl group in allenols 203 resulted in a slight decrease in the Z/E ratio, observing mixtures of diastereomers (from 89:11 to 95:5) in dienes 216 (Scheme 38, reaction b).202
The palladium-catalyzed preparation of (1Z)-1,2-dihalo-3-vinyl-1,3-dienes 220 has been accomplished in a stereoselective manner through the coupling between allenol esters, namely 2,3-butadienyl acetates 218, and haloalkynes 219 in the presence of lithium bromide (Scheme 39, top). Particularly interesting is the finding that haloalkynes show increase reactivity in comparison with allenes or acetylenes under the halopalladation reaction conditions. A plausible reaction path is depicted in Scheme 39 (bottom). The initial formation of alkenyl-palladium intermediates I should occur by trans-addition of the halide toward haloalkynes 219. Next, the carbopalladation reaction with allenol acetates 218 should form allyl-palladium intermediates II. β-Heteroatom elimination releases trienes 220 with concomitant regeneration of the catalytic species.203
Scheme 39. Palladium-Catalyzed Synthesis of Trienes from Allenyl Acetates and Mechanistic Rationale.

A different mechanistic pathway was proposed to explain the results observed from the reaction of different α-allenols 221 in the presence of catalytic amounts of iron triflate or iron trichloride. Opposite to the above-mentioned halogenation/rearrangement reaction of allenols promoted by iron halides in equimolecular manner (compounds 191 and 193 in Scheme 35), catalytic addition of similar metal species yielded the enone skeleton 222 through a Meyer-Schuster-type rearrangement (Scheme 40). The methodology showed best results for aryl- and heteroaryl-substituted allenols, and Fe(OTf)3 as metal catalyst, avoiding halogenated byproducts as observed in the presence of FeCl3, or decomposition products when acid catalysis was employed. The high E-selectivity observed in final enones 222 was found to be independent of the geometry in the starting materials, pointing to a stepwise reaction mechanism. Thus, first coordination of the metal species to the hydroxyl group in intermediate 223 could led to carbocation 224 by C–O bond cleavage. Addition of one molecule of water could then generate the metalated complex 225, evolving to the experimentally observed enones 222 through sequential demetalation/isomerization processes (Scheme 40). A related mechanistic pathway has been recently proposed by Gao and Xu et al., for the acid-catalyzed synthesis of ketophosphine oxides, although in both cases an alternative mechanistic pathway describing the inverse addition/elimination sequence may not be discarded.204,205
Scheme 40. Synthesis of Enones by Iron-Catalyzed Meyer-Schuster Rearrangement and Mechanistic Rationale.

Trost’s research groups have extensively investigated the metal-catalyzed Meyer-Schuster rearrangement of allenols 227 from a different perspective. Instead of delivering the corresponding enones from demetalation/isomerization of species 228, intermediates 228 were envisioned as coupling reagents with both electrophiles and nucleophiles, providing a wide and diverse family of functionalized ketones. Thus, reaction of vanadium enolate 228 with vinyl epoxides 229 as masked aldehydes in the presence of a Lewis acid, provided aldol products 230 (Scheme 41, right).206 Vanadium enolates in the presence of diazocompounds 231 as electrophiles provided ketones 232 from a direct sigmatropic amination reaction (Scheme 41, bottom).207 Noteworthy, reaction of allenes with diazocompounds are well-known to yield vinyl diazines through Alder-ene mechanisms.208,209 On the other hand, reaction of vanadium enolates 228 with different nucleophile halogen sources such as NCS or NFSI provided the corresponding α-haloketones 233 with moderate to excellent yields (Scheme 41, left).210
Scheme 41. Vanadium-Catalyzed Transformations of α-Allenols.

The allenol-enone metal-catalyzed isomerization has been recently proposed as an intermediate step toward the synthesis of spirocyclic scaffolds in compounds 237. First, treatment of sulfonyl allenols 234 with a Pd catalyst yielded the corresponding enones 235, through a 1,3-migration rearrangement via the corresponding metal-enolate 238. Enones 235 evolved in the presence of a base and p-quinone methides 236 to the observed adducts 237 through a cascade Michael-type addition/ring closing reaction (Scheme 42).211
Scheme 42. One-Pot Synthesis of Spirocyclic Compounds from Allenols Involving Palladium-Mediated Enone Formation.

Bis(trifyl)enones 242 have been recently prepared from α-allenols 239 unravelling a reversal regioselectivity for the reaction of allenes with electrophiles. Almendros et al. have reported the reaction of allenols 239 with the in situ generated strong electrophile Tf2C=CH2 (241).212 The reaction proceeds under mild reaction conditions in the absence of catalysts or additives (Scheme 43, reaction a). Interestingly, complete selectivity was found in the reaction of substrates 239a or 239b bearing an alkene or an extra allene unit (Scheme 43, reaction b and c, respectively), illustrating the divergent chemical behavior of allenes versus allenols. Thus, addition of 2-(2-fluoropyridinium-1-yl)-1,1-bis(triflyl)ethan-1-ide (240) to the reaction media spontaneously generates bis(trifyl)ethene (241), which undergoes a selective electrophilic attack toward the terminal allenic carbon in 239, opposite to the commonly reported central carbon atom electrophilic attack. The proposed reaction mechanism is followed by addition of water to the central positively charged carbon in 243 and by a 1,5-proton shift in the resulting species 244 to generate intermediate 245. Dehydratation would finally yield the experimentally observed enones 242 (Scheme 43, bottom). Computed calculations for the reaction profile supported the mechanistic hypothesis, stating a rare electrophile-mediated transformation of allenols into enones instead of the usual nucleophile-based procedures. Also, the unexpected regioselectivity leaves a door open for a further exploration of the yet intriguing reactivity of the allenol moiety.
Scheme 43. Allenol-enone Transformation by Electrophilic Attack of Bis(triflyl)ethene.

The ability of allenols to isomerize into dienes has been applied to more particular transformations in the context of the intermolecular addition of phenols. Ga(OTf)3 catalyzes a cascade process from oxindole-based allenols 187 and phenols 247 providing dihydrobenzofuran compounds 248 with moderate to good yields and practical diasetereoselectivities (Scheme 44). A mechanistic proposal to explain this result could start by a double coordination of the Lewis acid catalyst with both the OH unit from the phenol and the inner double bond from the allenol in complex 249. Then nucleophilic attack from the ortho position of the phenol to the central allenic carbon could generate intermediate 250. Loss of water would then lead to the diene skeleton 251, which may undergo intramolecular oxycyclization to build the dihydrobenzofuran system in oxonium ion 252. TfOH extrusion and demetalation would then provide the observed oxindole-functionalized dihydrobenzofurans 248 (Scheme 44).213
Scheme 44. Gallium-Catalyzed Synthesis of Dihydrobenzafurans by Phenol Addition to α-Allenols.

3.1.1.2. Synthesis of Enynes
During the attempts to oxidize the alcohol group in different allenols employing the Swern protocol, Ma and co-workers developed a straightforward methodology for the transformation of α-allenols 254 into conjugated enynes 255 and chlorinated dienes 256. It was found that the presence of triethylamine as a base notably favored the enyne 255 synthesis, while the presence of DMSO promoted the halogenation/isomerization process toward the diene 256 generation (Scheme 45, reaction a).214 Nevertheless, complete selectivity was only achieved in certain cases. Besides, this divergent protocol was restricted to a particular type of 2,3-allenols, namely, allenols having a 2-ethoxycarbonyl substituent. This example clearly illustrates the divergent behavior of allenols under subtle modifications on the experimental reaction conditions.
Scheme 45. Synthesis of Conjugated Enynes by Metal-Free OH-Activation/Elimination.

Alcaide and Almendros research group has presented an alternative procedure for the same transformation avoiding competitive halogenation/isomerization processes. Related OH-activation/elimination strategy was described, employing different reagents and bases. Thus, reaction of differently substituted α-allenols 259 with acetyl chloride and NaOH in aqueous media yielded conjugated E-1,3-enynes 260 in good yields and complete regio- and stereoselectivity. In addition, the methodology was compatible with a wide range of functional groups, and extended to hindered tertiary alcohols, which are frequently unreactive in acetylation processes (Scheme 45, reaction b).215−217
Sawama and collaborators have developed a phosphate-mediated synthesis of related conjugated E-enynes 263 from allenols 262. Trimethyl phosphate was used as activator of the hydroxyl group, while NaH acted as the basic reagent favoring the enyne synthesis by H2 release. Terminal substitution at the allene moiety 262 was tolerated, providing the synthesis of challenging inner alkynes and allowing therefore the extension of the methodology, despite of a slight decrease in the E/Z selectivity (Scheme 45, reaction c).218 Lee has described an efficient protocol for the direct and stereoselective conversion of allenyl acetates into (E)-α-ethynyl-α,β-unsaturated esters 255 using DABCO in catalytic amounts (10 mol %).219
Metal catalysis has been scarcely reported for the allenol-enyne transformation. One rare contribution described the use of Cu(OTf)2 acting both as hydroxyl activator and proton catcher. Thus, α-allenols 265 reacted in the presence of catalytic amounts of copper triflate providing enynes 266 in good to excellent yields and complete E-selectivity (Scheme 46, reaction a). The methodology was reported to be useful starting from secondary aryl-substituted allenols and was also extended to the synthesis of dienynes and enedyines. The reaction is proposed to require an initial C–O bond cleavage promoted by the Lewis acidic nature of the metal salt, yielding allenic carbocation species 269 and metal complex Cu(OTf)(OH) 268. Loss of water would then generate observed enynes 266 and regenerate the metal catalyst Cu(OTf)2 (Scheme 46, bottom). Further Z-E isomerization experiments on Z-enyne 266a′ under similar reaction conditions showed that formation of the observed E-enynes 266 should be thermodynamically controlled (Scheme 46, reaction b). Zwitterionic allenyl copper species Cu-270 and Cu-270′ are proposed as intermediates in the observed alkene isomerization.220
Scheme 46. Copper-Catalyzed Synthesis of Enynes from α-Allenols.

Enynes have been also proposed as reaction intermediates in the transformation of allenols into the 2H-pyran-2-one skeleton, or into substituted benzene rings. 3-Hydroxy-4,5-dienoates 271 can be converted into differently substituted pyranones 272 under protic acid catalysis. Protonation of the hydroxyl group followed by elimination of water is proposed to generate 1,4-enynes 273 as reaction intermediates. Addition of water to the terminal propargylic carbon atom on 273 and further intramolecular transesterification could explain the obtained 2H-pyran-2-ones 272 (Scheme 47, reaction a).221
Scheme 47. Enynes as Reaction Intermediates in the Synthesis of 2H-Pyran-2-ones and Substituted Benzenes from α-Allenols.

Iodine is well-known to efficiently promote the dehydratation of tertiary alcohols. When propargylic allenols 274 were treated with I2 in refluxing acetonitrile, iodobenzaldehydes or iodoarylketones 275 were synthesized, depending on the alkynyl substitution in 274. In this case, allenyl enynes 276 are described as plausible intermediates for this transformation. Further iodine activation of the triple bond followed by allene–alkyne cyclization would furnish the aromatic core in 275, while the observed carbonyl functional groups may proceed from oxidation of the iodine substituent by atmospheric oxygen (Scheme 47, reaction b).222
In a related approach, allenols 274 reacted with thiophenols 277 under transition metal catalysis generating 1,3,5-trisubstituted benzene rings 278. Ring closing and aromatization reaction is triggered by the nucleophilic addition of thiophenols 277 to the central allenic carbon in intermediates 279. InI3 exhibited a dual behavior as σ- and π-acid because it was used as metal source, activating both the hydroxyl group in 279 and the alkyne functional group in enyne intermediate 280 (Scheme 47, reaction c).223
3.1.1.3. Allene Transfer Reactions
The cleavage of the C–O bond in the allenol skeleton after the appropriate hydroxyl activation may result in the generation of an allenic carbocation. When this kind of carbocation is trapped by a nucleophile without isomerization or rearrangement, the overall process results in a formal allene transfer reaction. Because of the high reactivity of the allene functional group, transfer reactions where the allene moiety remains unaltered are still rare. Nevertheless, recent reports have appeared dealing with this transformation and its synthetic applications.
Ma and collaborators have used diverse α-arylallenols 281 as precursors of stabilized allenic carbocations 284 under acid catalysis. Thus, treatment of 281 with p-toluenesulfonic acid in the presence of indoles 281 as nucleophiles, yielded 3-allenyl indoles 283 with moderate to excellent yields through an allene transfer process. 3-Allenyl indoles 283 were employed as precursors for the synthesis of a family of heteroaromatic compounds 285 showing the carbazole scaffold.224,225 The synthetic strategy included a carbocyclization process catalyzed by gold, followed by oxidation of the resulting dihydrocarbazoles with DDQ to yield the fully aromatic structure in compounds 285 (Scheme 48).
Scheme 48. Synthesis of 3-Allenyl Indoles through Acid-Mediated Allene Transfer and Synthetic Applications.

Tsukamoto’s research group has developed a metal-catalyzed variant of the allene transfer reaction, employing primary alcohols 286 and diverse pronucleophiles (287, 290). The reaction is reported to possible proceed through a π-allyl palladium complex intermediate 289, generated by the oxidative addition of Pd(0) species to allenols 286. Nucleophilic addition toward the unsubstituted carbon on the π-allyl complex would furnish the new allene structure 288 (Scheme 49, reaction a). When the reaction takes place with ketones 290 bearing electron-withdrawing groups as pronucleophiles allenones 291 are obtained, which in situ undergo a palladium-catalyzed oxycyclization providing vinyl dihydrofurans 292 (Scheme 49, reaction b).226
Scheme 49. Palladium-Catalyzed Allene Transfer and Application toward the Synthesis of Vinyl Dihydrofurans.

Oshima and collaborators have reported a different strategy to achieve the allene transfer process. Copper carbene complexes 295 have shown great activity promoting a challenging C(sp3)-C(sp3) bond cleavage (Scheme 50, top, path b) in allenol structures 293, instead of the more frequent C–OH dissociation (Scheme 50, top, path a). Coordination of the metal species with both the OH group and the cumullene is reported to generate metal intermediate 297, which may evolve through PhCOMe elimination providing copper propargyl complex 298. This strategy induced an umpolung on the normal electronic charges in allene transfer processes, allowing the reaction of allenols with electrophiles such as imines 294. As a result, allenyl amines 296 were synthesized and in situ submitted for aza-cyclization reaction yielding the pyrrole scaffold in compounds 299 with good to excellent yields (Scheme 50).227,228
Scheme 50. Copper-Catalyzed Allene Transfer for the Synthesis of Allenamines and In Situ Aza-Cyclization Reactions.

3.1.2. OH as a Leaving Group in the Last Stages of the Reaction
Although less frequent, the C–OH bond dissociation can happen at the last stages of the reaction pathway, opposite to what has been so far reported in diene, enone, enyne or allene transfer procedures. Late OH release takes normally place in the form of dehydration leading to aromatic or conjugated systems, and it usually constitutes the driving force of the transformation.
This methodology has been extensively used for the synthesis of different structures exhibiting the carbazole motif, a natural occurring alkaloid showing a wide range of biological and pharmacological activities. Both platinum and gold catalysis have been found to catalyze the carbocyclization/dehydration of indole-tethered allenols to yield the carbazole skeleton in a highly efficient manner.229−231 Ma and collaborators have invested much effort in developing synthetic routes to carbazole-based natural products through this approach, later discussed in the natural products section.232,233
The Alcaide and Almendros research group has focused its research in this field on the mechanistic insights of this transformation under gold and palladium catalysis. Indole-tethered allenols 300 may exhibit three possible reaction sites, leading therefore to the corresponding carbo-, oxy-, or aza-cyclization products 301, 302, and 303, respectively. Despite the ability of gold salts to promote oxy-cyclization reactions, complete selectivity toward the carbo-cyclization process (compounds 301) was found in the presence of AuCl as metal catalyst (Scheme 51, top). The transformation succeeded for both methyl- and sterically hindered phenyl-substituted allenols 300, leading to the carbazole core 301 with good yields. The reaction mechanism was proposed to start with coordination of the metal to the terminal allenic bond, followed by a 6-endo carboauration process generating the zwitterionic vinyl gold specie 305. Loss of HCl would then lead to the neutral complex 302. A final dehydration and protodemetalation step should furnish the experimentally observed carbazoles 301 and return AuCl to the catalytic cycle (Scheme 51, bottom).234
Scheme 51. Au-Catalyzed Synthesis of Carbazoles from Indole-Tethered Allenols.

Taking advantage of the more π-coordinating nature of palladium ions, a tandem reaction including a similar carbo-cyclization process followed by cross-coupling reactions of allenols 300 with allyl bromides 307 was envisioned (Scheme 52, reaction a). Different allyl-substituted carbazoles 308 were synthesized in good yields and complete regioselectivity.234 Noteworthy, when related cross-coupling reaction was performed in the presence of a second allenic unit 309, pharmacologically attractive 3-(buta-1,3-dienyl) carbazoles 310 were obtained (Scheme 52, reaction b). Yields were moderate to good and a wide number of allenols bearing a different pattern of substitution were reactive under those conditions. In addition, the transformation took place in a complete chemo- regio- and stereoselective manner, showing a previously unreported cross-coupling reaction of two allenic moieties including a carbocyclization process. Interestingly, two allenol units 300 and 309 showing a different chemical behavior can be found in this reaction. Indole-tethered allenols 300 behaved as π-activated alcohols where the late C–OH cleavage leads to aromatic rings through dehydration. On the other hand, acetyl protected allenols 309 behaved as activated alcohols where nucleophilic addition toward the central allenic carbon leads to the diene skeleton, as previously illustrated in prior sections. The authors proposed a mechanistic pathway starting from coordination of the palladium ion to the terminal allenic double bond to give complex 311, followed by 6-endo carbopalladation generating vinyl palladium species 312. HCl extrusion and dehydration would provide palladacarbazole 313. Then cross-coupling reaction toward the central allenic carbon of the acetyl-protected allenol 309 would lead to intermediate 310. Observed butadienyl carbazoles 310 could be eventually obtained by deacetoxy palladation of intermediates 314, regenerating the catalytic species after loss of one molecule of AcOH.235
Scheme 52. Palladium-Catalyzed Cross-Coupling Reactions of Indole-Tethered Allenols and Allyl Bromides or Acetyl Protected Allenols.

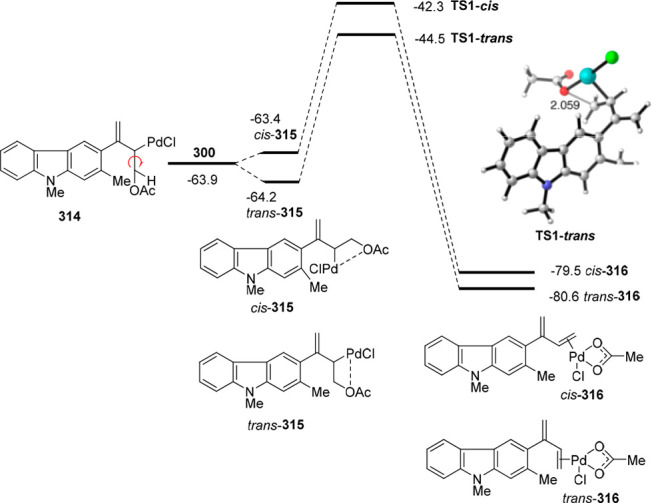
DFT calculations revealed a computed carbocyclization reaction profile notably lower in energy compared to the oxycyclization process from indole-tethered allenols 300, supporting the chemoselectivity observed in the first step of the tandem reaction. Also, the complete stereoselectivity observed in the diene generation can be explained considering the computed results for the depalladation step (Scheme 53). Free rotation along the C–C single bond in intermediate 314 could lead to both cis-315 or the more stabilized trans-315 complex. Demetalation step is calculated to proceed through a lower energy barrier from trans-315 adduct, yielding the also more favored trans-316 coordination complex. Thus, the more plausible reaction pathway is the kinetically and thermodynamically controlled trans-deacetoxypalladation process via transition state TS1-trans.
Scheme 53. DFT Computed Reaction Profile for Deacetoxypalladation Step in Allenol–Allenol Cross-Coupling Reaction.
Relative free energies are given in kcal mol–1.
3-Halo-(indol-2-yl)-α-allenols 317 revealed an intriguing reactivity pattern, showing divergent behavior depending on the halide substitution. 3-Chloro- and 3-bromo-indoles reacted with gold salts to yield dienes 318 via a 1,3-hydroxyl migration in complex reaction mixtures (Scheme 54, top left). Also, traces of oxycyclization products were observed. Interestingly, palladium catalysis only provided dihydrofuran systems 319 in low yields when 3-bromo-(indol-2-yl)-α-allenols 317 were employed (Scheme 54, bottom left). Noteworthy, when iodine-substituted indoles 317 were submitted to gold-catalyzed conditions a different reactivity was observed, obtaining mixtures of the previously reported carbazole structures 301 along with novel iodocarbazole compounds 320 (Scheme 54, top right). Complete selectivity toward the iodocarbazole skeleton was achieved under palladium conditions, yielding structures 320 with moderate to good yields (Scheme 54, bottom right). Opposite to normal metal catalyzed reactions from aryl halides where the halogen atom is lost during the reaction course, the observed iodine reincorporation into the final skeleton means an atom-economic improvement and unravels an unreported reaction mechanism.236
Scheme 54. Divergent Reactivity on 3-Halo-(indol-2-yl)-α-allenols under Metal Catalysis.

DFT calculations supported a 1,3-intramolecular iodine migration from dihydrocarbazole intermediate 321 to generate the corresponding iododihydrocarbazole 322. Iodonium cation 322 is proposed as the most favorable intermediate to achieve this transformation (Scheme 55, top). Also, computed reaction profile comparison of the migratory ability of chlorine, bromine, and iodine derivatives 321 supported the observed results. Activation barriers for the intramolecular 1,3-migration process are much higher for Br and Cl-substituted indoles (TS2-Br and TS2-Cl, respectively), leading therefore to diene adducts 318 or dihydrofurans 319. On the other hand, a lower energy barrier for the 1,3-iodine migration through transition state TS2-I facilitates the halogen recycling toward iodocarbazoles 320 (Scheme 55, bottom).
Scheme 55. DFT Computed Reaction Profile for the 1,3-Halogen Migration Step in Metal-Catalyzed Iodocarbazole Synthesis.
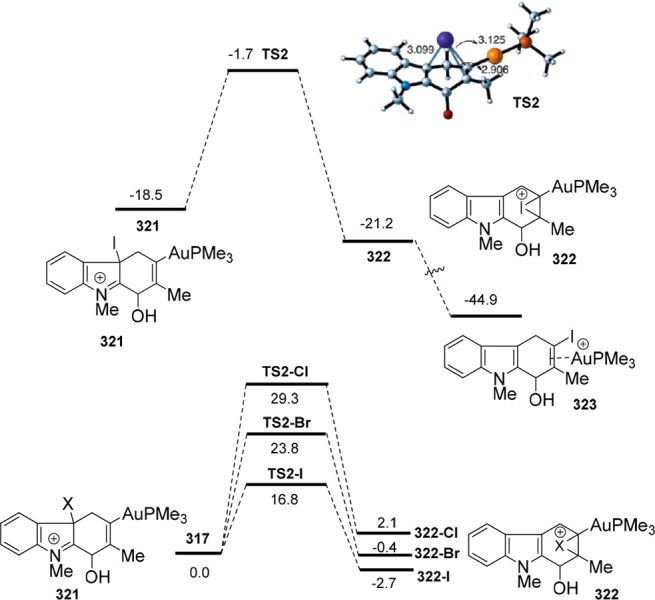
Relative free energies are given in kcal mol–1.
In a different approach, a wide family of naphthopyrans exhibiting large π-conjugation have been prepared. Naphthol (325) and related polyaromatic compounds reacted with conveniently substituted alkoxyallenes 324 in the presence of acid catalysts through a cascade process, providing naphthopyrans 326 in moderate to excellent yields. The reaction sequence includes a first allylation step to generate allyl naphthols 327, followed by oxycyclization, loss of one molecule of HOBn to build intermediate 315, and final dehydration to give the observed polyaromatic structures 326. Late C–OH cleavage inducing the extended conjugation in systems 326 is assumed as the driving force of the overall transformation (Scheme 56).237,238
Scheme 56. Acid-Catalyzed Synthesis of Naphthols.

3.2. Allenols as Bidentate Nucleophiles–Electrophiles
The inherent reactivity of hydroxyl groups and activated allene moieties as nucleophiles and electrophiles respectively, has prompted the oxycyclization reaction as one of the most extensively and traditionally reported transformation. The 5-Endo-trig cyclization leading to the dihydrofuran skeleton, 6-endo-trig processes providing dihydropyran motifs and cocyclization processes such as cyclocarbonilations leading to lactones have been widely employed in organic synthesis, exhibiting diverse applications in catalysis or natural products preparation.239−250 During the past decade the investigations in this field have been focused on developing new and more selective strategies for the oxycyclization reaction, greener and more economic procedures, and more sophisticated transformations for the synthesis of challenging molecular targets through tandem processes. Opposite to the π-activated alcohol reactivity discussed in the previous section, bidentate reactivity is not limited to α-allenol systems. Although less frequently reported, reactivity from β-, γ-, and δ-allenols will be also discussed. In addition, the oxycyclization of allenols has been largely employed as model reaction for the design of new catalysts with improved reactivity.
Alcaide and Almendros research group has devised a dual selectivity strategy for the oxycylization of α-allenols based both on the metal catalyst and on the allene substitution. To achieve a rare 4-exo-dig vs the most common 5-endo-trig cyclization, aryl-substituted allenes 330 were synthesized to induce an extra stabilization on the η2 gold intermediate complexes and to promote the nucleophlic attack toward the central allenic carbon. Interestingly, the selectivity toward the oxetene adducts 331 was improved by raising the temperature, indicating thermodynamic control over the 4-exo-trig products 331 (Scheme 57, top right). DFT calculations also supported this result, pointing to a reaction mechanism proceeding through a zwitterionic oxetene gold complex 335, which after loss of HCl and 1,3-gold migration could provide the rearranged neutral oxetane 337. A rare β-hydride elimination in gold catalysis could explain the observed oxetene adducts 331 (Scheme 57, bottom right).251
Scheme 57. Metal-Catalyzed 4-exo-dig versus 5-endo-dig Oxycyclizations of α-Allenols.

The use of platinum salts in substrates 330 revealed a divergent behavior toward the cycloetherification process. While PtCl2 cleanly provided the expected dihydrofuran systems 332, the addition of AgOTf promoted a dramatic change in the reactivity yielding exclusively substituted enones 333 (Scheme 57, top left). Supported on the precedents reported by the same group, the authors proposed a reaction mechanism passing by similar oxetene intermediates 335. A ring opening process instead of metal-migration to yield complex 338, followed by deprotopalladation would furnish the observed enones 333 (Scheme 57, bottom left). In addition, control experiments indicated the active role of silver ions in the reaction mechanism, through a yet not fully understood bimetallic catalytic species.252
Cycloetherification versus carbocyclization/dehydration has also been recently studied as a substrate-dependent methodology in metal-catalyzed experiments. α-Aryl-α′-hydroxyallenic esters 339 undergo 5-endo oxycyclization to yield aryl-substituted dihydrofurans 340 when the aryl moiety is decorated with electron withdrawing or mild electron donating groups (Scheme 58, reaction a, right).253 Nevertheless, and under similar reaction conditions, allenoates 339 were previously reported to provide functionalized naphthalene derivatives 341 when the aryl unit bears strong electron donating substituents (Scheme 58, reaction a, left). In this case, the enhanced nucleophilicity of the aromatic ring seems to be responsible for the reactivity switch of hydroxyallenic esters 339 toward a sequential carbocyclization/dehydratation, which is also favored by the extra stability of final aromatic compounds 341.254 A related approach from novel (indol-3-yl)-α-allenols 342 on the selective oxycyclization versus carbocyclization/aromatization process which can be easily modulated by changing the substitution on the pyrrolic nitrogen in 342 has been described. Thus, deactivated indoles yielded dihydrofuran derivatives 343 by gold-catalyzed cycloetherification processes with moderate to excellent yields, also allowing a wide functional group compatibility (Scheme 58, reaction b, right). On the other hand, NH-indoles 342 provided the carbazole skeleton 344 through a previously described tandem carbocyclization/dehydratation reaction. Moreover, cross-coupling reaction in the presence of allyl bromides and palladium catalysts generated the corresponding allyl-carbazoles 345 in good yields and in a regio- and chemo-selective fashion (Scheme 58, reaction b, left).255
Scheme 58. Cycloetherification vs Carbocyclization under Gold and Palladium Catalysis.

Allenyl acetates have also been used as competent substrates in cyclization reactions. Thus, Zhang reported a gold-catalyzed formal [3 + 3] benzannulation strategy for the preparation of polysubstituted benzyloxy arenes from 4-(benzyloxy)hexa-1,4,5-trien-3-yl acetates,256 while Mukai described a protocol for the formation of indole-2,3-quinodimethanes utilizing a potassium carbonate-promoted aminocyclization of 2-(2-((tert-butoxycarbonyl)amino)aryl)buta-2,3-dien-1-yl acetates with concomitant acetic acid release257 In an earlier report258 Cha prepared ethynyl-substituted ciclopropanes through the reaction of β-allenyl tosylates by basic treatment with LDA, in a cyclization which is supposed to proceed by sequential deprotonation of the internal allene hydrogen and cyclization with concurrent 4-methylbenzenesulfonic acid loss.
Different reports on the oxycyclization reactions selectivity including a competitive ring expansion versus cycloetherification in 3-allenyl-3-hydroxyindolones,259 counterion-controlled double bond isomerization in the oxycyclization of δ-allenols,260 or substrate-dependent 5-endo- versus 6-endo- in phosphorus-based allenols have appeared.261
Much effort has also been invested in developing different procedures to achieve the cycloetherification reaction from more economic or greener perspectives. In this regard, mercury salts were found to catalyze the oxycyclization of α-allenols 346 in a cheaper approach, compared to the most frequent precious metal-based methodologies. Thus, inexpensive and water-tolerant Hg(ClO4)2·3H2O provided the dihydrofuran skeleton 347 in good yields and wide scope, as it happens in sterically hindered tertiary allenols (Scheme 59, reaction a). In addition, complete selectivity toward the oxycyclization was observed even when electron rich aryl allenols 346a were employed, which normally led to mixtures 348a/347a under gold catalysis (Scheme 59, reaction b).262
Scheme 59. Hg-Catalyzed Cycloetherification of α-Allenols.

Recent metal-based alternatives to the classic cycloetherification reaction include the use of stoichiometric amounts of copper carboxylates 350 for the dioxygenation of allenols 349. Dihydrofuran systems 351 decorated with the vinyl carboxylate ester functionality were obtained through a 5-exo-trig cyclization of γ-allenols (Scheme 60, reaction a).263 Exocyclic γ-allenol 352 has been also described to react through a 5-exo-trig cyclization path in a Pd(0) catalyzed reaction (Scheme 60, reaction b).264 On the other hand, silver fluoride has been effectively used for the 5-endo-trig oxycyclization of highly substituted and sterically encumbered α-allenols 354, leading to dihydrofurans 355 exhibiting excellent yields and wide group compatibility (Scheme 60, reaction c).265
Scheme 60. Metal-Mediated Cycloetherification of Diverse Allenols.

Palladium nanoparticles (PdNPs) have shown high efficiency catalyzing the oxycyclization of differently substituted α-allenols 356a and 356b. Preformed nanoparticles using PdCl2 as metal source, K2CO3 as reducing agent and TBAB as stabilizer led to a wide family of dihydrofuran systems 357a and 357b in similar yields as the ones reported through the classic precious-metal approaches in homogeneous conditions (Scheme 61). Interestingly, phenols were needed as additives to achieve higher yields and conversions. TEM analysis showed an average particle size of 2.2 nm, and recycling experiments indicated a slight loss of the catalytic activity of solely 8% after four cycles, pointing to a low grade of bleaching in the catalytic system. Although higher temperatures are required compared to analogous homogeneous strategies, the lower catalyst loading of 1 mol %, the higher recycling performance, and the use of water as solvent establish the PdNP methodology as an effective greener procedure.266
Scheme 61. Heterogeneous Palladium-Catalyzed Oxycyclization of α-Allenols.

An alternative strategy for the catalyst recycling has been recently reported by Krause and collaborators, based on the use of gold catalysis in ionic liquids. Different trifluoromethylated allenols 358 were selected as model substrates for the 5-endo-trig cyclization reaction using both cationic and neutral gold species (Scheme 62). 1-Butyl-3-methylimidazolium hexafluorophosphate [BMIM][PF6] was selected as the best ionic media, allowing full conversions in most of the cases. Allenols 358 formed droplets when added to [BMIM][PF6], creating a heterogeneous system and allowing the gold catalyst recovering. A low decline of the yield in just 8% after 5 runs shows the practicality of the procedure. Also, mechanistic studies revealed a remarkable kinetic change when allenols bearing a R4 = CF3 substituent were submitted to Au-cycloeteherification conditions. In this case, formation of the π-complex was identified as the rate-determining step, opposite to the most habitual protodeauration reaction as the regulating step in gold-catalyzed oxycyclizations. This change is probably due to the stronger deactivating effect of the CF3 group when located at the terminus of the allenic moiety.267
Scheme 62. Cycloetherification of Trifluoromethylated Allenols in Ionic Liquids.

Gold catalyzed-cycloetherification has also been involved in the preparation of different compounds exhibiting an added value as molecular materials or naturally occurring alkaloid fragments. The 5-endo-dig cyclization of α-allenols 363 has been employed as one of the key steps en route to cyclophanes 365. Thus, double oxycyclization of allenols 363 led to bis(dihydrofurans) 364. Ruthenium-mediated ring closing metathesis yielded the expected aromatic, sugar- or beta lactam-based cyclophanes 365 (Scheme 63, reaction a).268
Scheme 63. Gold-Catalyzed Oxycyclization of Allenols for the Synthesis of Cyclophanes and Tetracyclic Indolines.

In a different approach, the tetracyclic indoline skeleton 368 was synthesized by a gold catalyzed cascade reaction, including a propargyl migration in aniline based compounds 366 to yield allenic indole intermediates 367. Further cyclization and rearrangement provided the observed bridged indolines 368. Challenging three-dimensional polycyclic skeletons are furnished, building three rings and four C–C bonds in one sole operational step (Scheme 63, reaction b).269
A metal-free oxycyclization of allenic hydroxyketones 369 in aqueous media has been reported. Inexpensive NaOH promotes the nucleophilic attack of the OH group toward the central allenic carbon through oxa-Michael-type reaction, yielding 3(2H)-furanones 370 with good yields (Scheme 64, reaction a). Moreover, cyclization may be achieved spontaneously by TBAF-mediated deprotection of silyl allenic ethers 371 (Scheme 64, reaction b). Noteworthy, gold catalysis failed promoting the oxycyclization of allenic hydroxyketones 372, stablishing the reported metal-free strategy as an alternative for the most common catalytic procedures.270
Scheme 64. Metal-Free Conversion of Allenic Hydroxyketones into 3(2H)-Furanones.

Despite that most of the transformations involving nucleophilic hydroxyls embedded in allenol moieties lead to cyclic final structures, mainly 5- or 6-membered oxacycles, some reports have appeared presenting the synthesis of open-chain products. Alcaide and Almendros research group have envisioned a change on the selectivity of the nucleophilic attack of allenols to rhodium carbenoids derived from triazoles, depending on the heterocycle substitution. Reaction of allenols 356 with 4-aryl-substituted triazoles 374a yielded pirrolines 373,271 while 4-acetyl-substituted triazoles 374b provided diketones 375, under otherwise identical reaction conditions (Scheme 65, top).272 Thus, when 1-tosyl-1,2,3-triazoles 374a presented an aryl group in C4, allenols 356 behaved as C-nucleophiles, generating intermediates 378 as the product of the nucleophilic addition of the central allenic carbon onto the rhodium carbenoid in intermediates 377. Further aza-cyclization of 378 yielded the experimentally observed pirrolines 373 (Scheme 65, bottom, left). Nonetheless, when 4-acetyl-1-tosyl-1,2,3-triazoles 374b were employed, allenols 356 selectively behaved as O-nucleophiles, leading to species 379 as reaction intermediates. Then, regeneration of the ruthenium species and protonation would provide allenyl vinyl compounds 380, which spontaneously evolve through a Claisen-type rearrangement to the observed final structures 375, exhibiting both the interesting 1,2-diketone and Z-1,3-diene frameworks (Scheme 65, bottom, right).
Scheme 65. Rhodium-Catalyzed Nucleophilic Addition of Allenols to 4-Substituted-1-tosyl-1,2,3-triazoles.

Zimmer, Reissing, and collaborators accessed the 1,2-diketone framework from α-allenols through a different pathway. Reaction of α-hydroxy methoxyallenes 381 with mCPBA yielded acyloxy-substituted 1,2-diketones 382 in reasonable yields. A mechanism rationale for this transformation would start from selective epoxidation of the proximal allenic double bond in 381 to generate intermediate 383. Acid-promoted ring opening and tautomerization would then lead to dicarbonyl 385, which may suffer intramolecular nucleophilic attack of the hydroxyl group of the former allenol moiety providing oxiranium 386. Nucleophilic attack of 3-chlorobenzoate would promote the ring opening of cationic intermediates 386 generating the observed diketones 382 after loss of methanol (Scheme 66).273
Scheme 66. mCPBA-Mediated Synthesis of 1,2-Diketones from α-Hydroxyl Methoxyallenes.

Enallenols, multifunctional molecular targets bearing an alkene and allene moieties along with a hydroxyl group, have attracted recent interest due to their divergent and intricate reactivity. Alkenol versus allenol selective reactivity has been encountered in metal-catalyzed reactions of different enallenols such as β-lactam-based compounds 387 or acyclic derivatives 390. Noteworthy, FeCl3 was found to exclusively provide alkenol oxycyclization adducts 388 and 391, leading therefore to the tetrahydrofuran skeleton (Scheme 67, right). On the other hand, gold and platinum salts selectively catalyzed the allenol 5-endo-trig oxycyclization of 387 and 390 generating the dihydrofuran motifs 389 and 392 in a chemoselective manner (Scheme 67, left).
Scheme 67. Allenol vs Alkenol Reactivity in Metal Catalyzed Reactions of Enallenols.

Density functional theory calculations showed coincident results according to the experimental observations. Thus, in both gold-based alkenol (reaction profile from 393) and allenol (reaction profile from 394) cyclizations, protodemetalation was identified as the bottleneck step of the whole process, finding a much lower energy barrier for the allenol cyclization resulting in the formation of dihydrofuran 396 (Scheme 68, left). Nevertheless, activation of the hydroxyl unit in substrates 397 should be the starting point of the catalytic cycle when iron salts are present, triggered by their strong Lewis acidity. In this case, alkenol cyclization pass by a notably lower energy barrier to yield the tetrahydrofuran skeleton 395 under kinetic control (Scheme 68, right).274
Scheme 68. Computed Reaction Profile for Allenol vs Alkenol Cyclization of Enallenols.

Relative free energy data are given in kcal mol–1. HCl-mediated oxycyclization is taken as model reaction for the Lewis acid-FeCl3 alkenol oxycyclization reaction.
Different reports on enallenol chemistry have stated the importance of designing the appropriate enallenol skeleton to modulate its reactivity. Bäckvall and co-workers have explored enallenol cyclization reactions under palladium catalysis in the presence of different cocyclization partners. In both substrates 398 and 403, where the previously mentioned allenol versus alkenol competitive cyclization is not feasible, palladium catalyzed cocyclization reaction should start by dual coordination of the metal ion with both allene and alkene moieties (coordination intermediates 400 and 406). Thus, when enallenols 398 were submitted to Pd(OAc)2 tretament under CO atmosphere, spirolactones 399 were obtained as sole reaction products in moderate to good yields. The process includes a cascade oxidative carbonylation-olefin insertion to form intermediates 402, followed by a second CO insertion-lactonization sequence providing spirocycles 399 (Scheme 69a).275 Noteworthy, three C–C single bonds, one C–O bond, and an all-carbon quaternary center are generated in one single operational step. On the other hand, when enallenols 403 were treated with palladium catalysts in the presence of terminal alkynes 405 as cocyclization partners, substituted furans 404 were obtained with good to excellent yields. In this case, a heterogeneous palladium catalyst was employed, based on an aminopropyl-decorated siliceous mesocellular foam which hosts palladium nanoparticles, exhibiting great performance and high recyclability. Again, palladium insertion into the central allenic carbon would yield intermediate 407, which may evolve through alkyne insertion to adduct 408. Nucleophilic attack of the hydroxyl unit to the Pd-activated alkyne and further isomerization would furnish the observed furans 404 (Scheme 69b).276
Scheme 69. Palladium-Catalyzed Cascade Processes of Enallenols.

Co-cyclization of allenols with aldehydes through Prins-type processes have also been reported, providing the synthesis of oxacycles with different ring sizes. β-Allenols 409 react with aromatic aldehydes in the presence of Bi(OTf)3 as Lewis acid catalyst to yield the dihydropyran skeleton 410 in moderate yields (Scheme 70, reaction a).277 When allenols 409 were treated with In(OTf)3 as Lewis acid catalyst, major efficiency in terms of catalyst loading toward the cocyclization adducts 410 was found (Scheme 70, reaction b).278 Interestingly, reaction of 5,5-dimethyl substituted β-allenol 409a and aldehydes under In(OTf)3 catalysis exhibited a special behavior. After the expected Prins-type cocyclization of 409a and the corresponding aldehydes, indium-mediated ring opening and further rearrangement took place, generating the alternative dihydropyran structures 411 in practical yields (Scheme 70, reaction c).278 In addition, tetrahydrofuran-based compounds 413a were achieved by reaction of α-hydroxy allenylsilane 412a with α,β-unsaturated aldehydes under acid catalysis. Also, larger ring sizes were accessible through this methodology in excellent yields, such as 3,4-dimethylidene oxepanes 413b obtained from the reaction of γ-allenol 412b using both aromatic and aliphatic aldehydes (Scheme 70, reaction d).279
Scheme 70. Prins-type Co-cyclization of Allenols and Aldehydes.

One recurrent strategy to achieve the synthesis of poly substituted furans or dihydrofurans lays on the metal-catalyzed oxycyclization of α-allenols, followed by a cross-coupling process using diverse reagents such as aryl halides. Taking advantage of the low redox potential of palladium, many reports have appeared describing cascade processes of allenols promoted by palladium salts.280−288 Thus, a multicomponent reaction of α-allenols 414 with aryl iodides 415, aliphatic alcohols 416 and carbon monoxide led to tetrasubstituted furans 417 through an oxidative addition/carbonylation reaction sequence (Scheme 71, reaction a).289 In a similar approach, the presence of tertiary amines 418 instead alcohols 416 in the reaction media provided the corresponding methylene acetamide-decorated furans 419 (Scheme 71, reaction b).290 Gong and collaborators have reported a related methodology using allenol 414a, aryl iodides 415 and imines 420 as reaction partners also under palladium catalysis to provide an extense family of oxazolidine derivatives 421. In this case, the proposed reaction mechanism includes carbopalladation onto the central carbon atom of the allene moiety, nucleophilic attack of the oxygen onto the C–C double bond of the imine, and latter ring closing step through nucleophilic addition of the nitrogen onto the inner π-allylic carbon atom (Scheme 71, reaction c).291
Scheme 71. Palladium-Catalyzed Multicomponent Synthesis of Tetrasubstituted Furans and Oxazolidine Derivatives.

Palladium species have also been found to be useful catalyzing homodimerization and heterodimerization processes of allenols. PdCl2 in the presence of NaI as additive has shown great activity promoting the tandem oxycyclization–cross-coupling reaction of 2-substituted allenols 422a, yielding 4-(1′,3′-dien-2′-yl)-2,5-dihydrofurans 423 (Scheme 72, reaction a).292 Interestingly, the reaction of two different allenic species under similar reaction conditions allowed the synthesis of substituted dihydrofurans 424, as a result of the chemoselective oxycyclization of 2-substituted allenols 422b, followed by cross-coupling reaction with 2-unsubstituted allenols 422c (Scheme 72, reaction b).293
Scheme 72. Palladium-Catalyzed Homo- And Heterodimeric Cross-Coupling Reaction of Allenols.

Besides the classical palladium-cross-coupling strategies, the past decade has started to witness the use of different metals to improve the efficiency and expand the scope of the methodology. Rhodium catalysis has been used to promote an oxycyclization–cross-coupling reaction of allenols with diverse benzamides including a challenging arene C–H bond insertion. Reaction of α-allenols 425 with Rh(III) species in the presence of N-methoxybenzamides 426 smoothly generated substituted dihydrofurans 427 with moderate to good yields. Notably, a wide substitution pattern on the allene moiety is tolerated, including sterically hindered substrates and tertiary alcohols. The most plausible reaction pathway would start by rhodation of benzamides 426 through the C–H bond, facilitated by coordination with the amide unit in complex 428. Further coordination with the allene moiety of 425 and oxyrhodation would afford intermediate 429, which could easily undergo reductive elimination to yield dihydrofurans 427 and liberate Rh(I) species. Atmospheric oxygen was used to reoxidize the catalytic species, returning the Rh(III) to the cycle (Scheme 73).294 A mechanistically different arene functionalization dealing with the intermolecular cyclization reaction of β-allenols in the presence of indoles catalyzed by a platinum salt, has been reported to afford C3-substituted indole derivatives with a tetrahydro-2H-pyran ring.295
Scheme 73. Rhodium-Catalyzed Oxycyclization/Cross-Coupling Reaction of α-Allenols and N-Methoxybenzamides.

Opposite to palladium or rhodium species, the high redox potential of gold makes the oxidative addition/reductive elimination steps hard to perform on gold-mediated transformations, and consequently inadequate for cross-coupling reactions. On the other hand, the well-known ability of gold salts to catalyze allenol oxycyclizations, frequently the first step in cross-coupling processes, has prompted different research groups to find solutions to circumvent this problem. Thus, photoredox catalysis has been successfully applied to achieve a gold-mediated cross-coupling reaction of allenols and diazonium salts. Allenols 430 reacted with aryldiazonium salts 431 in the presence of AuClPPh3 and [Ru(bpy)3][PF6]2 as photoactive catalyst under visible light. The reaction provided a wide family of 2,3,4-trisubstituted dihydrofurans 432 in a regioselective manner (Scheme 74). Yields were moderate to excellent, finding the best results when deactivated aryldiazonium salts 431 were employed. In addition, diverse functionalities were well tolerated, such as CF3, Br or OMe. A mechanistic proposal may start from oxidative arylation of the gold species promoted by single electron transfer from photoactivated ruthenium complex, generating Au(III) species 434. Coordination of 434 onto the allenic moiety of 430 resulted in complex 435, which would induce the oxycyclization step, generating intermediate 436. Reductive elimination would then recover the Au(I) species to the catalytic cycle and explain the formation of the observed 4-aryl-dihydrofurans 432 after deprotonation.296,297
Scheme 74. Gold-Catalyzed Photoredox Cross-Coupling of α-Allenols and Diazonium Salts.

The reactivity of allenols has also been used in catalysis as a platform for testing the versatility of recently developed metal complexes. The allenol oxycyclization process has been applied as a model reaction to investigate new catalytic pathways along with the design and tuning of novel catalysts. In this context, Rueping and collaborators have informed of a rare metal–ligand dual catalysis for the cycloisomerization of β-allenols. Allenes 437 bearing a β-hydroxyl unit reacted with catalytic amounts of iron cyclopentadienone complex 438, yielding 3,4-dihydro-2H-pyrans 439, through a selective 6-endo-trig oxycyclization/double bond isomerization process (Scheme 75, reaction a). The methodology was extended to both aromatic and aliphatic substituted allenols, providing the pyran skeleton in good yields. In addition, benzoxepine structure 441 was also accessible under similar reaction conditions through a more challenging 7-endo-trig heterocyclization (Scheme 75, reaction b).298 The cooperative metal–ligand catalysis strategy was further applied to the synthesis of dihydrofuran systems 443 through a 5-endo-trig cycloetherification of α-allenols 442 (Scheme 75, reaction c).299 Experimental and computational investigations revealed the role of the cyclopentadienone ligand as proton scavenger in a doubly activated intermediate complex 444. Also, it is proposed to act as proton shuttle/proton acceptor facilitating the 1,2-H shift to overcome the eventual double bond isomerization.
Scheme 75. Metal–Ligand Cooperative Catalysis in Cycloetherification of α-, β-, and γ-Allenols.

Widenhoefer and co-workers have selected the oxycyclization of 2,2-diphenyl-4,5-hexadien-1-ol (445) furnishing vinyl tetrahydrofuran 449 to perform the first mechanistic investigation on gold-catalyzed hydroalkoxylations. Experimental observations gave light to two major significant conclusions: (i) reversibility across the C–O bond formation step, and (ii) the presence of outer sphere bis(gold) vinyl species 447 acting as catalyst reservoir. Intermediate 447 was characterized from in-solution samples after treatment of stable vinyl gold complex 446 with (L)AuOTs at low temperature (Scheme 76, top). Then NMR monitoring experiments of the catalytic hydroalkoxylation of allenols 445 indicated the uninterrupted presence of bis(gold) vinyl species 447 during the whole reaction time. According to the results obtained from kinetic and deuteration experiments, the proposed catalytic cycle would start with a reversible C–O bond formation to generate mono(gold) vinyl intermediate 446 and HOTs. Aggregation should take place at this point to produce bis(gold) intermediate 447 in an outer-sphere equilibrium with mono(gold) species 446. Also, kinetic experiments pointed the protodeauration of intermediate 446 as the rate-determining step, excluding a possible disproportionation of bis(gold) vinyl intermediate 447 as an alternative to yield the final tetrahydrofuran 449 (Scheme 76, bottom).300
Scheme 76. Mechanistic Pathway for the Gold-Catalyzed Hydroalkoxylation of Allenols.

Related bis-benzylic β-allenol 450a undergoes elimination in the presence of the major part of gold catalysts to yield conjugated vinyl allenes 451a instead of the oxycyclization product 452a (Scheme 77, reaction a). Same result has been observed on similar acid-sensitive allenols 450, due to the acidic nature of gold salts and complexes. Lacôte et al. have envisioned a gold self-buffering catalytic system to solve this problem and achieve the cycloetherification products 452 in substrates were dehydratation competes. Polyoxometalate (POM)-based catalyst 453 were designed and synthesized, using organotin-substituted polyoxotungstate [P2W17O16{Sn-(CH2)2–CO}]6– as POM surface and ω-amino gold phosphine complexes to provide the active metal site. Thus, acid-sensitive allenols 450 were treated with a catalytic amount of 453 and AgSbF6 to induce the activation of the gold specie as cationic gold. Conversions of allenols 450 into dihydropyran structures 452 were complete in all cases, with moderate to excellent yields and no elimination byproducts identified (Scheme 77, reaction b). Interestingly, mechanistic insights revealed a multiple role from the POM-Au catalyst: Coordination of the cationic gold to the surface may stabilize the intermediate 454, reducing catalytic activity at the same time it improves catalyst recyclability. The POM surface should be also acting as proton catcher, buffering the reaction media and facilitating the formation of intermediate 456. Nevertheless, eventual protodeauration step to yield final adducts 452 would need a disfavored proton release from the POM skeleton, explaining the low reaction rates (up to 5 days to completion) compared to regular homogeneous gold-catalyzed hydroalkoxylations. Finally, special solubility of POM systems provides catalyst recycling, constituting this approach as a greener and more economic strategy.301
Scheme 77. POM-Au Catalyzed Hydroalkoxylation of Acid-Sensitive Allenols.

During the past decade, different contributions on gold catalysis have appeared describing both a rationale design as well as synthesis and applications of novel catalysts that could be able to provide higher efficiency and greener procedures. Noteworthy, the hydroalkoxylation of allenols has been frequently chosen as model reaction. Hilvert’s research group has described the synthesis of thiazolium gold(I) carbenes 457, as a greener alternative to the well-known imidazolium analogous. The higher hydrophilicity of thiamine units, together with the presence of a pyrophosphate group, improves the catalyst stability, and allows the use of aqueous media in hydroalkoxylation reactions. Thus, γ-allenol 458 could be successfully transformed (up to 98% conversion) into tetrahydrofuran 459 under mild reaction conditions and open-air experiments (Scheme 78).302
Scheme 78. Thiazolium Gold(I) Carbene-Catalyzed Hydroalkoxylation of Allenols.

In the pursuit of more economic and environmentally friendly strategies including precious metal catalysis, Lipshutz and co-workers have recently reported the use of gold(I) salts in micellar systems. Surfactant Nok (SPGS-550-M) was employed in combination with the newly synthesized gold(I) salt 460, showing an improved lipophilicity (Scheme 79). The micellar cavities based on aggregation of Nok molecules in aqueous media behaved as organic-based nanoreactors, encapsulating the hydrophobic reagents and therefore increasing their effective concentration. Thus, both uses of solvent and catalyst could be minimized, resulting in high conversions with catalyst loadings of 0.1 mol %. Also, an E factor of 7.6 for the oxycyclization of allenols 462 into spirocyclic systems 463 indicates the promising greener advantages of the micellar-based strategy.303
Scheme 79. Micellar-Supported Gold(I) Catalyzed Hydroalkoxylation of Allenols.

Bergman, Raymond, Toste, and collaborators have introduced supramolecular chemistry in gold catalysis from a different perspective. Gallium-based tetrahedral macromolecule 464 has been used as a supramolecular host for cationic gold species, acting as an enzyme-mimic catalyst. Again, hydroalkoxylation of allenol 458 has been selected as model transformation, to study the catalytic activity of Au-464 species (Scheme 80). Noteworthy, encapsulation of the gold salt induces ionic bond dissociation, resulting in more active “naked” cationic gold species. Thus, the catalytic activity is 8-fold increased compared to regular homogeneous cationic gold procedures, and up to 67 catalytic turnovers were observed.304
Scheme 80. Supramolecular Gold GaL6-Hosted Catalyst and Its Application to Hydroalkoxylation Reaction.

Following a similar concept, Reek et al. have envisioned a gold(I) system bearing supramolecular ligands, as inductor of selectivity in the oxycyclization reaction of allenols. Pyridyl-decorated phosphoramidite ligands 465 were used to both bonding to the active gold ion and to link Zn template 466 through the pyridyl nitrogen atom, generating the supramolecular structure 467. Although lower conversions were achieved compared to AuCl(L) complexes under similar reaction conditions, complete selectivity toward 5-exo-trig cyclization of allenol 468 was observed, yielding vinyl tetrahydrofuran 469 as sole reaction product (Scheme 81).305
Scheme 81. Supramolecular Gold(I) Catalysis in Selective Oxycyclization of γ-Allenols.

3.3. Allenols as Allenes Showing an Extra Coordination Site
Besides the allenol transformations where the hydroxyl group behaves as a leaving group or as a nucleophile, seen in previous sections 3.1 and 3.2 respectively, the recent literature has also provided several examples of alternative hydroxyl-assisted reactions. This third class of allenol reactivity includes both metal-catalyzed processes where the M–O coordination is crucial for a specific transformation as well as bond migration reactions promoted by the hydroxyl lone electron pairs. In those cases, the hydroxyl unit is retained unaltered in the final products or oxidized into a carbonyl group.
Araki and co-workers reported one early example of OH-assisted allylindation of allenols. Hydroxyl-chelated bicyclic species are reported as the most plausible transition states for this transformation.306
Gong and collaborators have reported a three-component methodology for the synthesis of 3,3′-disubstituted allylic alcohols under palladium catalysis. Treatment of allenic alcohol 471 with aryl iodides, catalytic amounts of a Pd(0) complex and the adequate pro-nucleophile 472 or 473, provided allylic alcohols 474 and 475 with moderate to good yields and complete Z-selectivity (Scheme 82, reaction a). The hydroxyl group in the allenol skeleton 471 is proposed to perform a double role: (i) enhancement of the reactivity by palladium-coordination in metallacycle intermediate 481, and (ii) both regio- and stereodirection for the addition of the aryl and nucleophile moieties. A mechanistic proposal for this transformation would start with the oxidative addition of the Pd(0) catalyst to the corresponding aryl iodide to generate Pd(II) intermediate 479. Coordination and carbopalladation would furnish the above-mentioned cyclic intermediate 481. Reaction with the in situ generated nucleophile followed by reductive elimination could lead to the observed allylic alcohols 474 or 475 (Scheme 81, bottom). Interestingly, allene 476 lacking hydroxyl unit, failed to yield the corresponding substituted alkene 477, supporting the proposed mechanistic pathway and the coordinative role of the OH group in the reported transformation (Scheme 82, reaction b).307−309
Scheme 82. Pd-Mediated Synthesis of Allylic Alcohols.

Zhang’s research group has reported a three-component palladium-promoted variant for the synthesis of allylic alcohols from allenol 471, aryl iodides, and alcohols as O-nucleophiles. In this case, a cooperative borane-palladium catalyzed strategy was developed. Triethyl borane was used as coordinating agent, directing the addition of alcohols 483 toward the inner allenic carbon through intermediate 485. The methodology was extended to a wide variety of substituted aryl iodides and both aromatic and aliphatic alcohols 483, providing allylic alcohols 484 in moderate to good yields (Scheme 83).310
Scheme 83. Cooperative Pd/B-Catalyzed Transformation of Buta-2,3-dien-1-ol into Allylic Alcohols.

Recent examples on allenol reactions triggered by metal–oxygen coordination includes Shi’s contribution on Ru-catalyzed oxidative isomerization of vinylidene cyclopropanes 486 to aldehydes 487 trough intermediate 488 (Scheme 83, reaction a).311 Also, Lu and collaborators have reported the reaction of aromatic amides 490 and allenols 491 in the presence of catalytic amounts of Rh(III) to yield γ-lactams 492 (Scheme 84, reaction b). The authors stated the significant role of the hydroxyl group in controlling both the regio- and the stereochemical outcome, probably through coordination with the rhodium atom in intermediate 493. A control experiment from allene 494 lacking hydroxyl group, which lead to complex reaction mixtures supported the Rh–O coordination hypothesis (Scheme 84, reaction c).312
Scheme 84. Allenol Transformations Promoted by Previous Metal-Hydroxyl Previous Coordination.

The cyclopentenone skeleton is a recurring target in organic synthesis, exhibiting an extense range of biological activities and synthetic applications. Besides the well-known Pauson-Khand and Nazarov cyclizations, metal-catalyzed cycloisomerizations have recently appeared as synthetic strategies to achieve the cyclopentenone motif.313−316 In this context, Cha and co-workers have described an allenol-based ring expansion process involving a ruthenium–oxygen coordination to provide the cyclopentenone system. Thus, allenyl cyclopropanols 495 were treated with ruthenium complex 496 and In(OTf)3 as additive, generating cyclopentenones 497 in moderate to good yields (Scheme 85, reaction a, left). The mechanistic pathway may start from dual coordination of the metal in alcoholate 498. Ring opening of the strained three-membered ring would then provide intermediate 499, which might evolve through a migratory insertion to cyclic intermediate 501. Eventual ligand exchange would explain the observed cyclopentenones 497 and return the active metal species to the cycle (Scheme 85, reaction a, right).317,318 Alcaide and Almendros research group has also contributed to the allenol/cyclopentenone transformation in the context of a cooperative bimetallic catalysis. In this case, 2-iodoaryl allenols 503 and 505 were treated with [(PPh3)2PdCl2] and CuI as bimetallic pair, yielding differently substituted fused cyclopentenones 504 and 506 in good yields (Scheme 85, reaction b), through a proposed intramolecular Heck-type coupling reaction mechanism.319
Scheme 85. Synthesis of Cyclopentenones by Metal-Catalyzed Rearrangements of Allenols.

Hydroxyl-assisted bond migrations in allenol systems have been frequently reported by different research groups. Ma and collaborators have pioneered halogen-promoted 1,2-aryl shift, and 1,2-H shift in allenol skeletons.320,321 Thus, secondary and tertiary allenols were reported to smoothly generate 3-halo-3-alkenals and 2-halo-2-alkenyl ketones respectively, under halogenating reagents such as Br2, I2, NIS, or NBS. During the past decade, related halogen-promoted 1,2-bond migrations in allenol systems have been employed in ring expansion processes. Moreover, selenating reagents were found to induce an intriguing selectivity on this transformation. Thus, 2-azetidinone-tethered allenols 507 were reported to undergo a selective 1,2 C–C bond migration in the presence of NBS, providing tetramic acids 510 (Scheme 86, reaction a). In contratst, the use of N-phenylselenophthalimide as electrophile promoted the oxycyclization process yielding spirocyclic seleno-β-lactams 511, under otherwise similar reaction conditions.322 Species 508 and 509, formed by coordination of the electrophile to the proximal and distal allene double bond respectively, are proposed as raisonable intermediates for the divergent transformation (Scheme 86, reaction a). In addition, 2-indolinone-tethered allenols 512 under NBS conditions provided the corresponding quinolone skeletons 513 and 514 through a related ring expansion process (Scheme 86, reaction b, right).323 Noteworthy, mixtures of two regioisomers were frequently found, quinolone-2,3-diones 513 as major products from a C3–C4 bond cleavage, together with quinoline-2,4-diones 514 from a less favorable C2–C3 bond breakage. Interestingly, selenating reagents improved the divergency of the process, finding selenoquinoline-2,3-diones 515 as major or sole reaction products in the presence of N-phenylselenophthalimide (NPSP) (Scheme 86, reaction b, top left). Spirocyclic selenolactams 516 where achieved when phenylselenyl bromide was used. Also, AuCl3–NPSP cocatalyzed reaction of allenols 512 favored the formation of the spirocyclic products 516, probably due to the gold ability in promoting oxycyclization transformations in allenols (Scheme 86, reaction b, bottom left).324
Scheme 86. Halogen- and Selenium-Mediated Ring Expansion Reactions of Allenols.

Toste’s research group has presented a photoredox-catalyzed ring expansion methodology in ciclopropane-linked allenol systems. Compounds 517 undergo ring expansion and oxidative arylation processes in the presence of electrophilic gold(III)-aryl complex 520 which is generated in situ from benzenediazonium salt 518. Coordination of the gold complex to the proximal allenic double bond in allenols 517 to give intermdediates 521 would promote the oxidative ring expansion process toward intermediates 522. The observed four-membered cyclic ketones 519 are obtained in practical yields from reductive elimination of intermediates 522 (Scheme 87).325 On the same basis of photoredox catalysis, Almendros and Luna et al. have recently reported the synthesis of 3-(arylsulfonyl)but-3-enals 525 from allenols 523, sulfur dioxide, and arenediazonium salts 524 under visible light. The proposed mechanistic pathway includes a C–C bond migration facilitated by the latter oxidation of the hydroxyl group to the corresponding aldehyde in compounds 525 (Scheme 87, reaction b).326
Scheme 87. Dual Gold-Photoredox Catalyzed Arylative Rearrengement of Allenols.

Liu and co-workers have reported a methoxy-assisted allene migration to explain the synthesis of pyrroles 530 and pyrrolo[1,2-a]quinoline derivatives 531 from 4-methoxy-1,2-dienyl-5-ynes 526 with anthranil (527). Anthranil is proposed to attack the π-activated alkyne moiety in complex 528 (formed in the presence of gold salts), generating α-imino gold carbene intermediate 529. 1,2-Allene migration and further gold-mediated aza-cyclization would furnish pyrroles 530. When allene ester systems are employed, subsequent aldol reaction would explain evolution toward the polycyclic structures 531 (Scheme 88, top).327 On the other hand, reaction of 4-methoxy-1,2-dienyl-5-ynes 526 with isoxazole (532) as nucleophile provided the indolizine skeleton 536 and 537 under identical reaction conditions, unravelling a different mechanistic pathway. In this case, alkyne attack to the π-activated allene moiety in gold complex 533 would produce cyclic intermediate 534, which afer methoxy-assisted ring-opening and isomerization could lead to vinyl gold carbene 535. Nucleophilic attack of the isoxazole unit to the gold carbene carbon followed by a cascade azacyclization/rearrangement and aromatization would explain the observed indolizine compounds 536 and 537 (Scheme 88, bottom).328 Selenium-based π-acid-type catalysis has been used for the preparation of α,β-unsaturated α′-alkoxy ketones from alkoxy-allenes through alkoxy migration.329
Scheme 88. Reaction of Methoxyallenes with Anthraniland Isoxazole through Gold-Carbene Intermediates.

3.4. Allenols in Chirality Transfer Processes
The development of new strategies to provide enantioenriched molecules constitutes one of the principal interests of the chemistry community. During the past decade, allenol-based reactions have also provided a notorious and increasing number of chirality transfer methodologies to get access to a wide family of enantioenriched structures. Chirality transfer from both central and axially chiral allenols has been reported. Also, it has been described the use of racemic allenols as precursors for the obtention of enantioenriched final compounds employing enzymatic catalysis, optically pure ligands in metal catalysis, and hybrid methodologies.
3.4.1. Central-to-Central Chirality Transfer
The Alcaide and Almendros research group has described the synthesis of optically pure dihydropyran, tetrahydrofuran, and tetrahydrooxepine skeletons from enantiopure β,γ- and γ,δ-allendiols. Also, furan systems were synthesized. The methodology revealed an intriguing selectivity toward the oxycyclization reaction of secondary hydroxyls versus primary hydroxyls in both β,γ- and γ,δ-allendiols. The choice of the metal catalyst and the appropriate substituent was found to be crucial to achieve the optimal regioselectivity. Thus, β,γ-allendiols 538 reacted with gold(III) salts to produce the dihydropyran skeleton 539 through a 6-endo cycloisomerization process involving the secondary hydroxyl group and the terminal allenic carbon (Scheme 89, reaction a, top right). Interestingly, allendiol 538 provided carbaldehyde 540 in the presence of platinum salts, from a similar 6-endo cycloetherification reaction followed by a subsequent oxidation (Scheme 89, reaction a, bottom right). In contrast, furan 541 was obtained when β,γ-allendiol 538 was exposed to a lanthanide complex under catalytic conditions, through a 5-exo cyclization toward the central allenic carbon (Scheme 89, reaction a, top left). Although palladium salts failed in promoting an effective cycloetherification to yield dihydropyrans 539 from allendiols 538, the use of a Pd(II) catalysts and allyl bromide as coupling counterpart smoothly led to substituted dihydropyrans 542 (Scheme 89, reaction a, bottom left). This transformation revealed a selective 6-endo cyclization/cross-coupling cascade reaction toward the terminal allenic carbon. On the other hand, benzyloxy homologous γ,δ-allendiols 543a provided dihydrofurans 544a under gold(III) catalytic conditions (Scheme 89, reaction b, top right). Noteworthy, diastereomer 543b provided the corresponding 5-membered oxacycle 544b through a related 5-exo-trig cyclization, but exhibiting a decrease in the diastereoselectivity (Scheme 89, reaction b, top left). Although platinum catalysts did not show efficiency in the conversion of γ,δ-allendiols into oxacycle systems, lanthanide complexes provided dihydrofuran 544a in similar yields as gold salts (Scheme 89, reaction b, right). More interestingly, palladium-catalyzed cross-coupling reaction conditions unravelled a dramatic change in the regiochemistry of the oxycyclization reaction depending on the absolute configuration of the starting material. Thus, γ,δ-allendiols 543b provided the substituted tetrahydrooxepine 545 through a rare 7-endo-trig cyclization, while diastereomer 543a yielded tetrahydrofuran 545 through a 5-exo-trig cyclization toward the inner allenic carbon (Scheme 89, reaction b, bottom).330
Scheme 89. Synthesis of Enantiopure Tetrahydrofurans, Dihydropyrans, and Tetrahydrooxepines through Metal-Catalyzed Cyclization of Optically Pure β,γ- and γ,δ-Allendiols.

More recently, the same research group has reported the metal-based chemoselective aza- versus oxy-cyclization reaction of enantiopure α-amino-β-hydroxyallenes. Gold salts were reported to selectively promote the 5-endo azacyclization toward the synthesis of optically pure 2,5-dihydro-1H-pyrroles, while the palladium cyclization/cross-coupling cascade strategy yielded 6-dihydro-2H-pyrans through a 6-endo cycloetherification reaction.331 Also, optically pure β-lactam-tethered allenols were employed as chirality transfer reagents for the preparation of a wide variety of enantiopure polycyclic structures, such as morpholines, oxocines, dioxonines,332 and the already mentioned tetramic acids 510 and spirolactams 511 (see Scheme 86),322 through halogen or selenium-promoted reactions, respectively.
Ma and collaborators have described the synthesis of enantiopure oxacycles from optically pure allenols presenting a tetrahedral chiral carbon through metal-catalyzed cascade processes involving oxycyclization steps. Highly substituted 2(5H)-furanones 548a/548b were obtained by treatment of allenols 547a/547b with Grignard reagents and CO2 atmosphere (Scheme 90, reaction a). The proposed reaction mechanism starts with the insertion of the organometallic reagent into the terminal allenic double bond, generating the cyclic intermediate 549 by Mg–O interaction. Reaction with CO2 would produce the corresponding γ-hydroxy-Z-alkenoic carboxylic acid 550, which could undergo lactonization to yield the observed butenolides 548 without racemization.333
Scheme 90. Metal-Catalyzed Cascade Oxycyclization from Enantiopure Allenols.

In a different approach, oxa-bridged benzocycloheptanes 551a/551b were synthesized by reaction between optically pure allenols 547 and iodobenzaldehyde in the presence of palladium salts and a base (Scheme 90, reaction b). In this case, initial oxidative addition of Pd(0) species into the aryl halide would provide intermediate 552. Then, carbopalladation of the corresponding allenol 547 (547b in Scheme 90, reaction b, bottom) would produce both π-allyl intermediates anti-553 and syn-553, being the latter the most sterically favored. Intramolecular attack of the alkoxide to the carbaldehyde group in syn-553 would lead to diastereomers 554 and 555. The observed diasteroselectivity of the overall process could be explained by selective oxycyclization toward the allylic position in 554, providing the less sterically hindered oxa-bridged benzocycloheptanes 551b from allenes 547b.334
Taking advantage of the versatile chemical behavior of the enallenol skeleton, Bäckvall research group has reported the preparation of a wide variety of molecules exhibiting high structural diversity and up to two sterogenic centers in optically pure form. Enantioenriched enallenol 556 reacted with Pd(TFA)2 under CO atmosphere to yield bicyclic lactone 557 with complete enantioretention (Scheme 91, reaction a).335 The reaction mechanism to explain this transformation would include a palladium-mediated allene-alkene carbocyclization reaction, followed by a sequential carbonylation/lactonization step, resembling previous palladium-lactonization of enallenols (see Scheme 69, section 3.2). Homologous enallenol 558 provided cyclohexanol skeleton 559 in good yield in the presence of B2pin2, exhibiting also full enantioretention (Scheme 91, reaction b). Homogeneous Pd(OAc)2 catalysis was employed to achieve this transformation, which should imply a related allene-alkene carbocyclization followed by ligand exchange with the borane reagent and reductive elimination.336 Noteworthy, coordination of the hydroxyl unit with the metal ion at the first stages of the reaction mechanism is proposed to be crucial to explain formation of compounds 557 and 559.
Scheme 91. Enantioenriched Enallenols as Chirality Transfer Reagents in Palladium-Catalyzed Cyclization Reactions.

In contrast, enallenols 560 were transformed into oxaboroles 561 by reaction with B2pin2 under heterogeneous palladium catalysis (Scheme 91, reaction c, right). Enantioenriched allenols 560 were prepared through kinetic resolution of the racemic mixture using Candida Antarctica Lipase B, showing >99% ee. Interestingly, despite of the palladium ability to induce racemization in allylic alcohols, no loss of optical purity was detected.210 The same catalytic system provided γ-lactones 562 in optically pure manner from reaction of allenols 560 under CO atmosphere. Moderate to good yields were achieved with full retention of the enantiopurity in final compounds 562 (Scheme 91, reaction c, left).337 Amino-supported heterogeneous palladium has also been recently used by Bäckvall et al. in a domino reaction from related enallenols with alkynes. The chelating activity of the hydroxyl group is responsible of the observed diastereoselectivity.338
More particular methodologies for the synthesis of stereodefined oxacycles have been recently reported. Anderson et al. have described the reaction of enantiopure cyclic alkynyl carbonates with palladium catalysts to yield alkynyl tetrahydrofuran systems through in situ generated allenol-palladium intermediates.339 Guinchard and collaborators have employed gold-catalyzed cyclizations of tetrahydro β-carboline structures decorated with enallenol motifs for the synthesis of optically pure decahydrofuro[2,3-f]indolo-[2,3-a]quinolizines. Gold(I) salts have been reported to exhibit optimal reaction conversions providing full enantioretention and good control on the enantioselectivity of the newly formed sterogenic centers.340
Opposite to the most commonly reported cyclization reactions, intermolecular processes involving enantioenriched allenols in asymmetric synthesis are scarce. Taking advantage of the chelating effect of the hydroxyl group, as explored in section 3.3, palladium-catalyzed multicomponent reactions of different allenols 563, aryl iodides 564 and benzylamine proceeded with no loss of enantiopurity, even in the absence of enantiopure phosphine ligands. Interestingly, a change on the allenol substitution provided a dramatic change on the regioselectivity. Thus, phenyl-substituted allenols yielded homoalylic alcohols 565, while alkyl-substituted substrates provided allylic alcohols 566, through the attack of the amine to the terminal allenic carbon (Scheme 92).341
Scheme 92. Asymmetric Multicomponent Reaction of Enantioenriched Allenols, Aryl Iodides, and Amines under Palladium Catalysis.

3.4.2. Axial-to-Central Chirality Transfer
Opposite to central-to-central chirality transfer methodologies from enantioenriched allenols, reports on axial-to-central chirality transfer using axially chiral enantioenriched allenols are rare. One of the principal challenges to circumvent is the ease of racemization of the allene moiety under metal catalyzed conditions. Normally, metal activation of the allene unit starts with π-coordination of one of the double allenic bonds with the metal ion in a η2 complex 567 (Scheme 93). When such intermediates are in equilibrium with the corresponding π-allyl cations 568, free bond rotation falls into loss of the optical purity of the allene moiety. Avoiding the above-mentioned equilibrium by stabilizing the η2 complex constitutes one of the most recurrent strategies to achieve axial chirality transfer reactions in allenes. The choice of the metal catalyst and the appropriate substituents on the allene core are the principal tools to achieve a successful chirality transfer transformation.
Scheme 93. Racemization of Axially Chiral Allenes by Metal-Coordination.

In the context of allenol-based reactions, Lalic and co-workers have reported the first synthesis of teytrahydrofuran and tetrahydropyran systems bearing a tetrasubstituted stereogenic center. Substituted β- and γ-allenols 570 were submitted to gold catalysis providing the expected oxycyclization products 571 with good to excellent yields and a chirality transfer of up to 99% (Scheme 94). Gold(I)-based salts bearing a tosylate group together with electron-rich sterically hindered phosphine ligands were found to provide the best results, also promoting complete E-selectivity in the double bond formation in compounds 571. A plausible reaction mechanism would start from coordination of the metal species to the proximal double bond in intermediate 572, followed by carbometalation to furnish vinyl gold intermediate 573 without the occurrence of allene racemization. Opposite to Widenhoefer’s statement about digold alkenyl intermediates as catalyst resting states,300 monogold(I) intermediate 573 is herein presented as the most likely resting state due to the weakly electrophilicity of the gold complex, and the highly coordinating character of the counterion.342
Scheme 94. Chirality Transfer in Gold-Catalyzed Cycloisomerization Reactions of γ- and δ-Allenols.

Similarly, Yin’s research group has reported the synthesis of substituted dihydrofurans 575 showing up to 20:1 dr through a gold-catalyzed oxycyclization process. Allenols 574, exhibiting both axial and central chirality, have been prepared according to the previously mentioned asymmetric Cu-catalyzed alkynylogous aldol reaction (Scheme 22, reaction a), and successfully converted into cyclic structures 575, current motifs in anti-Alzheimer and Down’s Syndrome drugs (Scheme 95).145
Scheme 95. Synthesis of Optically Active Dihydrofurans through Gold-Catalyzed Oxycyclization of Enantioenriched Allenols.

A different approach based on gold catalysis has been reported by Krause and Lipshutz et al., following their interest in discovering new micellar systems to provide greener methodologies, high catalyst efficiency and recyclability.281 It has been reported that micellar catalysis in aqueous media was also effective for the synthesis of enantioenriched structures from α-allenols, employing poly(oxyethyl)-α-tocopheryl sebacate (PTS, 578) as amphiphile and AuBr3 as metal source. Dihydrofuran structures 577 bearing two stereogenic centers were achieved, exhibiting good to excellent yields and complete chirality transfer (Scheme 96).343
Scheme 96. Chirality Transfer in Micellar-Supported Gold-Catalyzed Oxycyclization Reactions of Allenols.

Ma and collaborators have synthesized optically pure lactones 581a and 581b from N-methoxybenzamide 580 and enantioenriched substituted allenols 579a and 579b under rhodium catalysis conditions (Scheme 97). Axial chirality in starting allenols 579 was fully transferred into lactones 581 exhibiting a stereogenic center with moderate yields. The mechanistic proposal would start with rhodation of the N-methoxybenzamide unit 580 to yield cyclic intermediate 582 (Scheme 97, bottom). Coordination of the metal ion to the less substituted allenic bond would then lead to complex 583, in equilibrium with the less favored intermediate complex 584. Carbometalation of the allenic double bond from the less sterically hindered adduct 583 could explain the E-selectivity observed, furnishing vinyl rhodium compound 585, which may suffer protonolysis to give 586. Eventual lactonization would explain the experimentally observed cyclic structures 581.344
Scheme 97. Chirality Transfer on Rhodium-Mediated C–H Insertion/Lactonization Reaction of N-Methoxybenzamide with Allenols.

Metal-free strategies regarding axial-to-central chirality transfer are scarce. Zhang and Bao and collaborators have recently reported the NIS-promoted allenol oxycyclization to yield dihydropyran systems showing central chirality.345 Sakaguchi and Ohfune and co-workers have described the enantiomeric version of the Prins-type reaction of allenols with carbonyls. β-Allenols 587 bearing a terminal silyl group were selected as chirality transfer agents, reacting through an uncommon 5-endo-trig cyclization in the presence of TMSOTf as Lewis acid catalyst and both aldehydes and ketones as reaction counterparts. Silylalkynyl-decorated tetrahydrofurans 589 were obtained as single diastereomers, exhibiting two stereogenic centers (Scheme 98, reaction a). To test the axial-to-central chirality transfer efficiency, enantioenriched allenol 587a was synthesized with a 92% ee. Reaction of 587a with benzaldehyde (588a) and TMSOTf as acid catalyst provided the expected oxycyclization product 589a showing an enantiomeric excess of 78%. Increasing the Lewis acid load up to 1.1 equiv allowed an 85% ee in final tetrahydrofuran 589a, showing solely slight racemization during the reaction (Scheme 98, reaction b).346
Scheme 98. Tetrahydrofuran Synthesis through Prins-type Cyclization of β-Allenols.

3.4.3. Metal-Based Catalysts as Chirality Transfer Agents
Metal catalysis offers a wide variety of alternatives to achieve an efficient chirality transfer with associated formation of enantioenriched molecules. The most traditional and extended strategy is based in the joined use of the metal along with a chiral phosphine ligand which induces enantioselectivity during the reaction. Michelet and Scalone and collaborators have synthesized an unprecedented bromine-decorated diphosphine ligand (S)-591 showing chiral atropisomerism. The catalytic system Ag/591 was applied for the synthesis of vinyl tetrahydrofurans 592 through a 5-exo-trig cyclization of γ-allenols 590. Yields were moderate to excellent, and enantiomeric ratios up to 91.5:8.5 were stated (Scheme 99, reaction a).347 Ma’s research group has devised an enantioselective palladium-catalyzed cross-coupling reaction of γ-allenols 590 with aryl iodides 594 yielding styryl tetrahydrofuran systems 577 through a similar 5-exo-trig cyclization. Optically pure diphophine (R,R)-591 was employed, finding enantioinversion in vinyl-substituted tetrahydrofuran structures 595 with respect to systems 592. Good yields and up to 92% ee were stated (Scheme 99, reaction b).348
Scheme 99. Synthesis of Enantioenriched Vinyl Tetrahydrofuran Systems through Metal-Catalyzed/Phosphine Ligand-Mediated Oxycyclization.

Zhang’s research group has envisioned an accelerative gold catalysis by ligand–metal coordination to γ-allenols 590 providing vinyl tetrahydrofuran skeletons 595 (Scheme 100, reaction a). Opposite to classic enantiopure ligand approaches, where chiral induction is frequently based in sterically hindered ligands which normally show a decelerating effect in the reaction rate, Zhang’s ligand (R)-596 bearing an amide remote group exhibited an 88-fold rate increase compared with unsubstituted bisphenyl- or bisnaphthyl phosphines. Simoultaneous metal-allene and carbonyl-hydroxyl coordination in intermediate 597 explains the enhancement of the catalytic activity, yielding tetrahydrofurans 595 with good to excellent yields and enantioselectivities up to >99% ee. Also, catalyst loadings as low as 200 ppm were allowed.349 A similar strategy was applied to the synthesis of enantioenriched dihydrofuran structures from in situ generated α-allenol systems.350 The 5-exo-trig oxycyclization of γ-allenol 598 has also been in the focus of an intriguing case of enantioinversion in final vinyl tetrahydrofuran molecules (S)-592a and (R)-592a (Scheme 98, reaction b). Fürstner et al. have found that even under the same optically pure gold catalyst 599, formed from AuCl and phosphoramidite (S,S,S,S)-599, enantioselection could be achieved by changing the solvent, temperature, and counterion. Moreover, a synergistic effect between the three parameters could be perform, achiving enantiomeric excesses from 97% ee in (R)-595a (Scheme 100, reaction b, right) to 68% ee in (S)-592a (Scheme 100, reaction b, left). Experimental and computational studies revealed that a change in the rate determining step lays on the base of the change in the sterochemical outcome. Protic solvents and coordinating counterions supported an additive-assisted reaction mechanism favoring the (R)-595a isomer. Also, temperature has a dramatic entropic effect, promoting the additive-assisted mechanism on cryogenic conditions providing (R)-579a isomer as major compound.351,352
Scheme 100. Synthesis of Enantiopure Vinyl Tetrahydrofurans through Chirality Transfer from Gold-Based Catalytic Systems.

Related enantiopure oxaphosphorous ligands 602 and 603 have been used in the context on an unprecedented enantioselective silver-catalyzed oxycyclization of allenols. Reaction of Ag2CO3 salts with the corresponding phosphoric acid yielded catalyst complexes Ag-602 and Ag-603. Treatment of different allenols 600 with the preprepared catalytic species allowed the synthesis of vinyl tetrahydrofuran and tetrahydropyran compounds 601 (Scheme 101). Also, furanones were obtained starting from the corresponding allenic carboxilyc acids. DFT calculations on this transformation pointed to ionic interactions between ligand and substrate as the major forces contributing to the enantioselectivity of the process.353
Scheme 101. Silver-Catalyzed Enantioselective Oxycyclization of Allenols.

Chemical desymmetrization of prochiral allendiols has been accomplished using palladium catalysts and enantiopure phosphoric acids as ligands. α,α′-Allendiols 604 were submitted to Pd(OAc)2 catalysis in the presence of catalytic amounts of chiral ligand (R)-605, generating dihydrofurans 606 in good to excellent yields, and practical enantiomeric excesses up to 85% ee (Scheme 102, reaction a). Adduct 607 was proposed as the stereodetermining intermediate, showing a dual interaction of the catalytic system and the allenol unit through both metal-coordination to the central allenic carbon and hydrogen bonding with the hydroxyl group.354 In a similar but conceptually different approach, γ,γ′-allendiols 609 were converted into enantioenriched tetrahydrofuran derivatives 611 through a gold-catalyzed oxycyclization (Scheme 102, reaction b). In this case, enantiopure phosphoric silver salts (R)-609 were introduced in the reaction system as chiral counterions, while achiral phosphine ligands such as 610 are linked to the reactive gold ion and are used as enantioselectivity modulators. This methodology allowed the generation of two stereogenic centers in one single operational step, furnishing oxacycles 611 with good to excellent yields and up to 93% ee.355
Scheme 102. Metal-Catalyzed Desymmetrization of Allendiols.

Different authors have investigated the synergistic effect of both enantiopure chiral ligands and enantiopure counterions to achieve higher enantioselectivities without loss of catalytic activity. Regarding allenol oxycyclizations, Mikami et al. have described the use of neutral dinuclear gold complexes 613 as catalytic species with improved activity for the 5-exo-trig cyclization of substituted γ-allenols (Scheme 103, reaction a). The optimal reaction conditions were found when same catalytic amount of 613 and the silver salt Ag-614 bearing an enantiopure chiral phosphoric acid counterion were added. Vinyl tetrahydrofurans 615 were obtained with good to excellent yields and up to 95% ee. According to experimental studies, it has been proposed species (R)-616 from ligand exchange with one equivalent of silver complex Ag-614 as the catalytically active system. Complex (R)-613 in the absence of silver salts was ineffective for the indicated transformation, while complex (R)-617 from the addition of two equivalents of Ag-614 showed lower yields and enantioselectivity (Scheme 103, reaction a).356 Toste and co-workers described the enantioselective aza-cyclization/halogenation tandem reaction of allenamides 618 with brominating reagent 619 to yield bromovivnyl pyrrolidine structures 621 under gold catalysis in the presence of BINAP-type ligands (Scheme 103, reaction b). The methodology also included one example of 5-exo-trig oxycyclization of γ-allenol 622 to produce bromovinyl tetrahydrofuran 625 (Scheme 103, reaction c). This transformation promoted by gold catalyst 623 provided a poor enantioselectivity (25% ee) of the indicated structure 625. Interestingly, the combined use of 623 and silver salt Ag-624 bearing a chiral counterion provoked a remarkable impact, both increasing the selectivity up to 86% ee and reverting the enantioselectivity compared to adducts 621.357
Scheme 103. Chirality Transfer from Enantiopure Chiral Ligands and Chiral Counterions on Gold-Catalyzed Oxycyclization of γ-Allenols.

Despite the widespread presence of carbocations in organic synthesis, its utilization as reaction intermediates in asymmetric synthesis is still scarcely described. The challenging facial discrimination in planar carbocation species has been mainly limited to diastereoselective-substrate control, or to the addition of an enantiopure chiral anion forming ion pairs with the carbocationic molecule. Carreira research group has envisioned an alternative approach for developing asymmetric SN1-type reactions in α-allenols and related systems, based in η2 coordination complex 633 (Scheme 104, bottom). Species 633 formed from interaction of iridium salts bearing an enantiopure chiral phosphine and the distal double bond of the allene moiety can be considered as mimics of diastereoselective-control substrates and constitutes one rare example of enantioselectivity in allenol transformations apart from oxycyclization processes. Thus, racemic Boc-protected α-allenols 626 reacted with organozinc nucleophiles 628 in the presence of iridium catalysts Ir-(R)-627 to provide compounds 629 in practical yields and excellent enantioselectivities, through an asymmetric allene-transfer reaction (Scheme 104, reaction a).358 In addition, related methodology has been applied to describe the first example of an enantioselective reductive deoxygenation of tertiary alcohols. Thus, racemic α-allenols 630 were submitted to catalytic system Ir-(S)-627 in the presence of Hantzsch ester analogues 631 as hydride source, providing enantioenriched allenes 632. Kinetic and computational insights revealed the presence of η2 carbocationic complexes as the most plausible reaction intermediates (Scheme 104, reaction b).359
Scheme 104. Enantioselective SN1-type Reactions Involving Iridium-Based Allenic Carbocations.

Different approaches to achieve asymmetric synthesis from racemic allenols and enantiopure catalysts also includes denitrogenative annulation of 1,2,3-benzotriazin-4(3H)-ones with allenes under nickel catalysis,360 synthesis of 1H-isochromene structures through copper catalyzed oxycupration/allylation reaction using optically pure phosphine ligands,361 the enantioselective synthesis of spiropentanes from hydroxymethylallenes catalyzed by Zn,362 the asymmetric palladium-catalyzed homoallenilation of amines,363 or the synthesis of cyclodextrin-tethered gold(I) carbene complexes as water-soluble and recyclable catalysts for several transformations including oxycyclizations of α- and γ-allenols.364
Racemic 2-(2′,3′-alkadienyl)malonates were obtained through the Pd(PPh3)4-catalyzed alkylation reaction of allenyl acetates with malonates365 while racemic allenes bearing a quaternary carbon center α to the cumullene were prepared by [Ir(cod)Cl]2/dppe-catalyzed allylic alkylation of 1,1-disubstituted-2,3-butadienyl acetates with malonates.366 A smart approach for obtaining optically active allenes is the direct preparation from achiral allenyl acetates, allenyl phosphonates, and allenyl carbamates via π-allylmetal intermediates, taking advantage of the great leaving aptitude of the acetate, phosphonate, and carbamate moieties. Imada, Murahashi, and Naota achieved the metal-catalyzed synthesis of enantioneriched α-allenamines 635 by asymmetric amination of allenyl phosphonates 634 using Pd2(dba)3CHCl3 as the palladium source and (R)-SEGPHOS as the ligand (Scheme 105, reaction a).367 Starting from precursors 1, the same research group did also reported the asymmetric alkylation with 2-acetamidomalonate.368 Trost and co-workers accomplished the asymmetric synthesis of allenes (S)-635 and (S)-637 from allenyl acetates 636 by palladium-catalyzed dynamic kinetic reactions with both malonates and amines involving α-methylidene π-allylpalladium species (Scheme 105, reaction b).369 The optimized reaction conditions require the use of Pd2dba3 (2.5 mol %), phosphine 104 (7.5 mol %), THACl (tetrahexylammonium chloride) (5 mol %), and a base in THF. Hamada performed the same palladium-catalyzed reaction between allenyl acetates 636 and malonates but replacing Trost ligand with (S,RP)-DIAPHOXs, a chiral nonracemic diaminophosphine oxide, which results in the formation of axially chiral allenes (R)-637 in good yields with up to 99% ee.370 The above-mentioned palladium-catalyzed amination of allenyl phosphonates,371 and the addition of malonates to allenyl acetates,372,373 have also been explored by Ma and collaborators in the context of achieving central as much as axial chirality from racemic allenes. The same research group has deeply investigated different allenyl esters in the presence of various nucleophiles yielding substituted allenes exhibiting both types of chirality. Thus, racemic allenyl acetates 638 have been reported to undergo a SN2′-type oxidative addition in the presence of palladium complexes and enantiopure (R)-DTBM-SEGPHOS as ligand. Echoing effect between the central and axial chirality is stated, providing enantioenriched allenes 640 showing both axial and central chirality (Scheme 105, recation c).374 Asymmetric allenylation of malonates have also been achieved using racemic allenyl carbonates. Selectivity between mono- and bis-allenylation is reported using Pd2(dba)3/(R)-DTBM-SEGPHOS as catalytic pair.375 In a related work, a smart approach to allenylamines exhibiting axial chirality has been accomplished through the palladium-catalyzed decarboxylative amination of allenyl carbamates 641. Pd2(dba)3/(S)-DTBM-SEGPHOS catalytic pair promotes the loss of CO2 providing the corresponding π-allylpalladium intermediates. Further nucleophilic attack of the in situ generated amide ion yields the observed allenylamines 635a exhibiting up to 99% ee and good to excellent yields (Scheme 105, reaction d).376
Scheme 105. Palladium-Catalyzed Enantioselective Synthesis of Functionalized Allenes from Allenyl Esters.

3.4.4. Chirality Transfer in Enzymatic Catalysis
Although enzymatic systems have been largely employed as biocatalysts for KR and DKR in allenol synthesis, strategies for the preparation of enantiopure compounds from racemic allenols including enzymatic resolution are almost unexplored. Bäckvall research group has contributed to develop this methodology with the synthesis of dihydrofuran and cyclobutanol skeletons with excellent enantiomeric excesses. During attempts to achieve an efficient DKR of allenols, Shvo catalyst (645) was used in combination with Candida Antarctica lipase B (CAlB) to promote the expected consecutive recemization and selective acetylation of α-allenols 642. Despite the well-known activity of ruthenium catalyst 645 for the racemization of secondary alcohols, oxycyclization products 643 were found instead, along with the corresponding acetylated allenols 644 (Scheme 106, reaction a). Noteworthy, enantioselectivities in dihydrofuran skeletons 643 were remarkably higher in comparaison with any other standard oxycyclization procedure. In addition, mechanistic insights pointed to ruthenium carbene species 647 as reaction intermediates, explaining the eventual double bond isomerization found in final adducts 643 through a 1,2-H shift.377 Alternatively, a cheaper and less toxic approach based on iron catalyst 646 and same enzymatic system allowed the preparation of dydrofurans 643 through milder reaction conditions (Scheme 106, reaction b).378
Scheme 106. Hybrid Enzymatic/Transition Metal Catalyzed Synthesis of Dihydrofuran Compounds.

The same research group has also taken advantage of the fruitful reactivity of enallenol skeletons under palladium catalysis, previously mentioned in section 3.2, to perform the synthesis of cyclobutenol structures. Reaction of racemic enalenols 648 and boronic esters 649 in the presence of palladium nanoparticles yielded the four-membered ring systems 650 trough a tandem carbocyclization/borylation reaction. Moderate to good yields and high diastereoselectivities were observed. In addition, palladium nanoparticles were suspended on amino-decorated mesocellular foam (Pd-Amp-MCF), providing high recyclability and efficiency (Scheme 101, reaction a). In combination with biocatalyst CAlB, cyclobutenol structure (1S,4S)-650a was obtained with a good 83% yield from enantioenriched enallenol (S)-648a, and a high 95% ee (Scheme 107, reaction b). From the mechanistic point of view, hydroxyl group is proposed to perform a multiple role, promoting the carbocyclization process by coordination with the metal unit, and directing the stereoselectivity on final adducts 630 through intermediates 651.379
Scheme 107. Hybrid Enzymatic/Transition Metal Catalyzed Synthesis of Enantioenriched Cyclobutenols.

Following their interest in supramolecular host–guest catalyzed reactions (see Scheme 80, ref (282), Bergman, Raymond, and Toste and collaborators have extended the applications of Ga4L6-encapsulated gold ions 464 in tandem reactions with biocatalysts. Nonencapsulated metal catalyst, especially gold species, can partially poison biocatalysts by binding amino-acid groups from the protein skeleton. This fact may incur in a loss of catalytic activity, unless great excess of enzyme is used. In addition, the use of supramolecular hosts exhibited many other advantages such as catalyst stabilization or aqueous media allowance. Thus, racemic acetylated γ-allenols 652 were reported to undergo KR in the presence of different enzymes such as Amano lipase PS providing enantioenriched allenols 653, which after oxycyclization in the presence of supramolecular catalyst 464 yielded dihydrofurans 654 showing up to 96% ee. Notably, the supramolecular host–guest catalytic system 464 allowed a decrease in the enzymatic loading to six units, compared with the 25 units needed when naked Me3PAuCl complex was used as catalyst. Kinetic experiments supported the hypothesis of the enzyme poisoning from free metal salts, revealing no interference between encapsulated gold systems 464 and lipase enzymes (Scheme 108).380
Scheme 108. Hybrid Enzymatic/Transition Metal Catalyzed Oxycyclization of Allenols Using Supramolecular Hosts.

4. Allenols in Natural Products
The diverse reactivity of allenols under different reaction conditions has been applied to the synthesis of a wide family of natural and pharmaceutically attractive products. During the past decade, several reports have appeared describing the total synthesis of naturally occurring structures incorporating allenol chemistry, normally as a key step in the whole reaction sequence. Also, natural products have been characterized and synthesized exhibiting the allenol motif in the final structure. In the first part of this section, the most significant and recent uses of allenols as key intermediates in total synthesis will be described. In a second part, allenol synthesis strategies applied to the preparation of natural product bearing an allenol unit will be detailed.
4.1. Allenols as Key Intermediates in Natural Product Synthesis
The great ability of allenes to undergo carbo- and heterocyclization reactions has been largely employed for the synthesis of the cyclic core of different naturally occurring compounds. Concretely, oxycyclization of allenols has been one the most recurring tools to get access to natural products containing 5- and 6-membered oxacycles. Silver catalysis has been frequently described to provide tetrahydrofuran systems from ennartioenriched allenols without racemization. In this context, Ballereau and co-workers have reported one short synthetic route to the natural product Jaspine B (655), a cytotoxic marine compound consisting in a trisubstituted tetrahydrofuran skeleton. The heterocyclic core was obtained through a silver-catalyzed 5-endo-trig cyclization of α-allenol 657, obtained from the enantioselective Crabbé-type reaction of propargylic alcohol (655) with aldehyde 656 using optically pure (R)-α,α-diphenylprolinol as secondary amine (Scheme 109, reaction a). Axial-to-central chirality transfer from enantioenriched allenol 657 allowed the full retention of the enantiopurity in dihydrofuran 658. Further transformations included epoxidation, ring opening reaction in the presence of sodium azide and reduction to yield Jaspine B (659) in 12% overall yield through a six-step sequence.381
Scheme 109. Silver-Catalyzed Oxycyclization of α-Allenols in Natural Product Synthesis.

Deska et al. reported the desymmetrization of prochiral allendiol 660 using enzymatic catalysis providing enantioenriched allenol 661 exhibiting 99% ee (Scheme 109, reaction b). Silver-catalyzed 5-endo-trig cyclization followed by enzymatic ester hydrolysis generated the corresponding dihydrofuran skeleton in product 662, with no racemization observed. Compound 662 was used as precursor for the synthesis of diastereomers Hyperione A (663) and Hyperione B (664), secondary metabolites found in the leaves of Hypericum Chinese, showing pharmaceutical activity.382
(+)-Sylvone A (669) is a highly functionalized tetrahydrofuran metabolite principally extracted from the seeds and fruits of piper sylvaticum and piper logum plants. Yu’s research group has envisioned a synthetic sequence to yield (+)-Sylvone A, based also on a silver-catalyzed 5-endo-trig cyclization of enantioenriched α-allenols. In this case, optically pure borane 666 was used as chiral inductor in the enantioselective aldol-type reaction of allenoate 665 with 3,4-dimethoxybenzaldehyde to yielded enantioenriched allenol 667 exhibiting 93% ee (Scheme 109, reaction c). Silver nitrate was then employed as the most convenient metal catalyst to promote the dihydrofuran generation from 667 to compound 668 avoiding racemization processes. Further transformations including a Michael addition provided the expected natural product (+)-Sylvone A (669) through a short 5-step reaction sequence.383
Metal-catalyzed oxycyclization of α-allenols have also been involved in longer synthethic pathways toward the synthesis of natural products exhibiting higher structural complexity. Carter et al. have reported the total synthesis of macrolide 670, a naturally occurring product found in Amphidinium sp. organisms. Compound 670 shows one of the most densely functionalized structures among the family of amphidinolides, and 11 stereogenic centers, endowing both a synthethic and analytical challenge. Also, two trans-disposed tetrahydrofuran units are present in the skeleton of compound 670, both synthesized from optically pure alkynol 671 (Scheme 110, reaction a). In the presence of AgBF4 as metal salt, alkynol 671 rearranges to produce α-allenol 672 as nonisolable reaction intermediate. In situ oxycyclization of 672 generates dihydrofuran 673 in 85% yield and dr >20:1. The complete synthesis of macrolide 670 comprises 34 steps in the longest linear sequence, and its synthesis has helped to elucidate the absolute configuration of the whole structure, unresolved since its first isolation more than two decades ago.384
Scheme 110. Silver-Mediated Oxycyclization of α-Allenols in the Total Synthesis of Amphidinolide F and Leiodolide B.

Leiodolide B metabolite (674) is a natural product isolated from marine sponges, showing a challenging tetrahydrofuran unit bearing four stereogenic centers on its northern fragment (Scheme 110, reaction b). Fürstner and collaborators have envisioned a total synthesis of compound 674 through a 26-step reaction sequence. To achieve the tetrahydrofuran structure with the appropriate stereochemistry, a silver-promoted 5-endo-trig oxycyclization of enantioenriched α-allenol 675 was proposed. In this manner, dihydrofuran 676 was therefore achieved in high 91% yield and further converted into tetrahydrofuran 677. Further transformations allowed the preparation of macrolide 674, although full charecterization and interpretation of the naturally isolated analogous remains unresolved, leaving the quest for the Leiodolide B absolute configuration still open.385
The γ-butyrolactone scaffold is ubiquitous in nature and present in different biologically active alkaloids. Stenine (578) and Stemoamide (579), two naturally occurring heterocycles from the stemona alkaloid family, exhibit a γ-butyrolactone unit which has been achieved trough a ruthenium-catalyzed carbonylation of allenols. In both cases, related exocyclic allenols 580 and 583 were synthesized and submitted to ruthenium catalysis under CO atmosphere to yield butenolide systems 581 and 584 (Scheme 111). In the final steps of the synthesis of the stenine and stemoamide cores, double bond reduction by treatment with Mg/MeOH and nickel boride, respectively, led to the desired compounds 682 and 679.386,387
Scheme 111. Ruthenium-Catalyzed Carbonylation of α-Allenols toward the Synthesis of the γ-Butyrolactone Scaffold in Natural Products.

In a recent total synthesis of (+)-Xilogyblactone A (685), an alternative gold-based methodology has been introduced to access the butanolide motif. tButyl allenoate 686 reacted through an asymmetric aldol-type transformation with aldehyde 687 in the presence of enantiopure organoboron reagent 666. Enantioenriched α-allenol 688 was therefore synthesized exhibiting >99% ee. Interestingly, gold treatment of allenol 688 yielded the butenolide skeleton from selective nucleophilic attack of the carboxylic oxygen to the allene moiety. (+)-Xilogyblactone (685) was obtained after acidic hydrolysis of the TBSO group through a short three-step sequence and a 41% overall yield (Scheme 112).388 A related gold-catalyzed cycloisomerization of allenyl carboxylates has also provided the γ-butyrolactone unit in the total synthesis of Xestospongienes E, F, G, and H.389
Scheme 112. Synthesis of (+)-Xylogiblactone A through Gold-Catalyzed Cycloisomerization of Enantioenriched Allenols.

Dihydropyran and tetrahydropyran fragments found in natural products have been accessed from oxycyclization of β-allenols through diverse strategies. The dihydropyran C1–C15 subunit 689 of Sorangicin A, a potent antibiotic isolated from Sorangium Cellulosumi bacteria, has been synthesized through the gold-catalyzed 6-endo-trig cyclization of enantioenriched β-allenol 692a, prepared from oxidation and further asymmetric reduction of the diastereomeric mixture 692a/692b (Scheme 113, reaction a).390
Scheme 113. Synthesis of the Dihydropyran and Tetrahydropyran Fragments of Sorangicin A, Eribulin, and Halichondrin.

Prins-type cyclization of β-allenol 695 with dimethyl acetals 696 and 697 followed by Tsuji reduction led to the C14–C35 fragment of Eribulin (693) and the C14–C38 fragment of Halichondrin (694), respectively, two macrolides of marine origin exhibiting potent antitumor activity. Chirality transfer from enantioenriched allenols and acetals led to the stereocontrolled generation of the C27 sterocenter, and further Tsuji reduction under palladium conditions provided the stereodefined C25 center in both fragments 693 and 694 (Scheme 113, reaction b).391
A more intrincate reaction mechanism was envisioned for the synthesis of (−)-Gilbertine natural product (680), a member of the uleine alkaloid family. Allenyl azide 678 undergoes photoinduced azacyclization to yield indolidene intermediate 679, which after 6-exo-trig oxycyclization reaction from the hydroxyl group yields the fused tetrahydropyran skeleton, favored by the formation of the more stable indol aromatic ring in compound 680 (Scheme 114).392
Scheme 114. Synthesis of (−)-Gilbertine through Photoinduced Cyclization of Azido-allenols.

In the context of allenol oxycyclization reactions toward the synthesis of naturally occurring products, related allenyl hydroxylamines have been described to undergo in situ cycloisomerization to yield polycyclic oxazines en route to the total synthesis of Casuarine, Australine, and diverse non-natural derivatives.393
Breit and co-workers have recently reported a diasteroselective synthesis of dihydropyrans through the rhodium-catalyzed oxycyclization of both terminal and internal allenols, using dppf as ligand. The methodology has been successfully applied to the synthesis of (−)-centrolobine (683) through a six-steps reaction sequence and 20% overall yield (Scheme 115).394
Scheme 115. Synthesis of (−)-Centrolobine Natural Product from an Enantioenriched Internal Allenol.

The β-allenol scaffold has been also presented as precursor of acyclic fragments. Syn-1,3-diol is a common motif in every compound of the statin family. Breit and collaborators have developed the diasteroselective synthesis of syn-dioxanes 690 from in situ generated allenyl hemiacetals 689 as syn-1,3-diol precursors (Scheme 116). The asymmetric version of this transformation was achieved using acetylsultam borylenolate 686 as chiral inductor. Coupling reaction of compound 686 with allenyl carbaldehyde 687 followed by amide hydrolysis led to enantiopure β-allenol 688. Further coupling reaction of syn-dioxanes 690 with the appropriate phosphoryl compound 691 and acetal hydrolysis allowed the total synthesis of Rosuvastatin (684) and Pitavastatin (685).395
Scheme 116. Synthesis of Rosuvastatin and Pitavastatin Natural Products from an Enantiopure β-Allenol.

As previously mentioned in section 3.1.2, tandem carbocyclization/dehydratation reactions of allenols constitute straightforward procedures for the preparation of aromatic and heteroaromatic polycyclic structures. Ma and co-workers have employed this strategy for the synthesis of a wide variety of alkaloids from the carbazole family, starting from readily available methoxypropadiene (693) and indole-2-carbaldehydes 694. Treatment of methoxypropadiene (693) with nBuLi in the presence of indole-2- carbaldehydes 694 led to indole-tethered allenols 695 (Scheme 117). Carbocyclization of compounds 695 under precious metal catalyzed conditions followed by in situ dehydratation provided 2-methoxy-3-methylcarbazoles 696 in high yields. Carbazoles 696 were employed as key intermediates for the synthesis through short reaction sequences of natural occurring alkaloids such as Siamenol (697) or the Clausine family drugs (698–700), both exhibiting promising anti-HIV activities and commonly used in traditional medicine. Also, Girinimbine (701), Murrayacine (702), or Mukoenine-type structures (703, 704), showing cytotoxic activity against a wide variety of cell lines were obtained.232
Scheme 117. Synthesis of Carbazole Alkaloids from Indole-Tethered Allenols.

Cycloaddition reactions involving allenol molecules en route to natural products and fragments have also been described. Early examples deal with the Diels-Alder of allenols and methyl propiolate in the total synthesis of Quassin.396 More recent advances include the intramolecular (5 + 2) cycloaddition of allenol 705 for the synthesis of the tetracyclic core of Bufogargarizin C (707) (Scheme 118, reaction a),397 or the tandem Diels-Alder/carbonyl-ene reaction from allenol 708 to provide the Chloropupukeananin D analogous 710 (Scheme 118, reaction b).398 The first and asymmetric total synthesis of the bioactive bufospirostenin A, an unusual spirostanol natural product, has been accomplished taking advantage of the intramolecular allenic Pauson–Khand reaction of an alkyne-tethered allenol for the construction of a tetracyclic skeleton.399
Scheme 118. Synthesis of Natural Products from Allenol Cycloaddition Processes.

Jogyamicin (712) is an aminocyclopentitol-based natural product recently isolated from Streptomyces culture broth. Its potent antiprotozoal activity along with its challenging structure has attracted the interest of diverse research groups. One recent approach to the five-membered core of Jogyamicin starts from enantioenriched β-allenol 713, which after protection as allenic sulfamate 714, followed by oxidative allene amination under rhodium catalysis led to cyclic sulfamate 716 through aziridine-intermediate 715 (Scheme 119). A 15-step reaction sequence from sulfamate 716 provided the pentacyclic structure 717 in a 6% overall yield, a known key intermediate in the total synthesis of Jogyamicin.400
Scheme 119. Formal Synthesis of Jogyamycin through Rhodium-Catalyzed Azacyclization of a β-Allenol.

Palladium-catalyzed additions and hydroborations of allenes has been applied to the preparation of different natural products. Yoshida’s research group has described the synthesis of sesquiterpenes (−)-HM-3 and (−)-HM-4 based on the palladium-catalyzed addition of boronic acids to α-allenols,401 and the synthesis of enokipodins A and B, two sesquiterpenoids from the α-cuparenone family exhibiting antimicrobial activity through a similar strategy.402 Roulland and co-workers have reported the total synthesis of the antibiotic Tiacumicin B incorporating a palladium-mediated cross-coupling reaction of alkynes and allenols.403 Hong and collaborators have envisioned a total synthesis of Lasonolide A (718), a natural product from marine origin and promising activity in pancreatic cancer therapies. The proposed retrosynthesis disconnects the macrolide product in fragments 719 and 720, prepared from the hydroboration of both allenes (+)-721-Ac and (−)-721, after a 12- and 11-step sequence, respectively. Enantiopure allenes (+)-721-Ac and (−)-721 have been prepared taking advantage of the enzymatic resolution of racemic allenol 721. Julia-type olefination of fragments 719 and 720 followed by Yamaguchi macrolactonization and total desilylation provided the expected structure of the Lasonolide A polyketide (Scheme 120).404
Scheme 120. Total Synthesis of Lasonolide A from Enantioenriched α-Allenols.

Recently, the ability of allenyl carbamates, readily available from the corresponding allenols, to generate dienes as reaction intermediates has been employed in the total synthesis of trachelanthamidine and supinidine through a (4 + 1) ring closing process. Thus, allenyl carbamates 725 reacted under phosphine-promoted conditions to yield pyrrolines 727, key reaction intermediates in the total synthesis of pyrrolizidine alkaloids 728 and 729 (Scheme 121).405
Scheme 121. Total Synthesis of Trachelanthamidine and Supinidine.

4.2. Natural Products Bearing the Allenol Motif
Once considered chemical curiosities and extremely reactive compounds, allenes are currently found in more than 150 thermally and photochemically stable natural products.406 Despite the allenol system is infrequent in naturally occurring systems, and linear allenes commonly show chemical instability, some simple linear molecules bearing the allenol motif have exhibited important antibiotic activity, such as the diyonic compounds marasin (730),407 and 07F275 molecule (731),408 both described in the late 80s (Scheme 122).
Scheme 122. Linear Allenols Exhibiting Antibiotic Properties.

During the past decade, some examples describing naturally occurring linear allenes have been reported. Ma’s research group has applied their chiral amine enantioselective allenation of terminal alkynes for the one-step synthesis of (R)-8-hydroxyocta-5,6-dienoate (735), a potent antifungal and antibiotic molecule extracted from the Japanese tallow tree Sapium japonicum. Reaction of propargylic alcohol (732) and methyl-5-oxopentanoate 733 in the presence of (S)-diphenyl(pyrrolidin-2-yl)methanol 734 and CuBr2 as metal catalyst led to the expected 1,3-disubstituted allene moiety in compound 735 (Scheme 123). Efficient chirality transfer from optically pure secondary amine 734 allowed the obtention of allenol 735 in 94% ee.389
Scheme 123. Enantioselective Allenation of Terminal Alkynes for the Synthesis of Naturally Occurring Allenol Structure.

Thomas and collaborators have reported the total synthesis of the allenol-based natural product Puna’auic acid (736), a fatty acid isolated from marine cyanobacterium. The allenol motif is generated in one of the latter steps of the reaction sequence, through a copper-catalyzed conjugated hydride addition to enantioenriched epoxy alkyne 737, following Krause’s procedure.409 Although the allene biosynthethic origins are not yet fully understood, the discovery of minor alkyne metabolites related to structure 736 from the same natural sources point to a similar conjugated hydride addition as the most plausible biosynthethic route (Scheme 124).410
Scheme 124. Conjugated Hydride Addition to Epoxy Alkynes Towards the Total Synthesis of Allenol-Based Puna’auic Fatty Acid.

(+)-Iso-A82775C natural product (739) has been isolated from the fermentation culture of fungus Pestalotiopsis fici, and it has been proposed as a biosynthethic intermediate of the previously mentioned chloropupukeananin family. Compound 739 is a polysubstituted cyclohexane ring bearing an exocyclic allene. Its structure and complex stereochemistry have attracted the interest of different research groups, reporting alternative synthethic strategies. Suzuki and Tanino and co-workers have proposed a Seyferth–Gilbert homologation of carbaldehyde 740 to generate ethynyl cyclohexane structure 742 (Scheme 125, reaction a). Epoxidation of the endocyclic olefin followed by Cu-mediated anti-SN2′ reaction of the chloroalkyne with isopropenyl magnesium bromide provided adduct 743 exhibiting the allene moiety with the adequate stereochemistry. Natural product 739 was obtained after desilylation of protected alcohols with TBAF.411 Han et al. have envisioned a different approach to get access to the allene moiety in 739. Stille coupling of iodo vinyl derivative 744 and organostannane 745 led to the dienyne 746 (Scheme 125, reaction b). Selective endocyclic double bond reduction with K-selectride yielded cyclohexanone 747, which suffered enyne-enallene isomerization in the presence of catalytic amounts of triethylamine. Total synthesis of 739 was completed with further carbonyl reduction and desylylation steps.412
Scheme 125. Alternative Approaches to the Allenic Moiety Generation in (+)-Iso-A82775C Total Synthesis.

Carotenoids represent the largest group of natural products exhibiting the allenol motif, being the Grasshopper ketone (749) one of the most commonly reported. First synthesis and isolation date to the late 60s,413 pointing to a dietary metabolism of larger carotenoids as the most plausible biological origin of compound 749. Eugster et al. described a total synthesis of allenol 749 based on a SN2′ hydride addition onto propargylic oxirane precursor 750, followed by selective oxidation under MnO2 conditions (Scheme 126).414
Scheme 126. Synthesis of the Grasshopper Ketone.

A related exocyclic allene unit is present in the wide family of xanthophyll norcarotenoids, naturally occurring compounds isolated from marine microalgae. Great effort has been made during the last years to provide an efficient synthethic route to some of these carotenoid molecules such as the most abundant Peridinin (752a), Fucoxanthin (752c), the biosynthetic intermediate Paracentrone (752d), and diverse natural and non-natural derivatives (Scheme 127).
Scheme 127. Allenol-Based Natural Products from the Norcarotenoid Family.

Álvarez and de Lera et al. have proposed a retrosynthethic analysis for the total synthesis of Peridinin based on Julia-Kocienski olefinations, and a Stille coupling reaction to incorporate the allenic moiety from fragment 753 (Scheme 128, path a). Also, in depth investigations on the stereoselective oxidative addition or SN2′ substitution of palladium reagents to iodoallene derivatives 753 were reported. Iodoallene 753 was prepared from the corresponding alkyne 755.415−417 Burke’s research group has devised a Suzuki coupling of iodoallene 753 and the corresponding boronic acids 756 to incorporate the allenic fragment utilizing the same disconnection strategy (Scheme 128, path b).418,419 A different retrosynthetic approach for the total synthesis of analogous deoxy-Peridinin (752b) has been proposed by Sakaguchi and Katsumara and collaborators. Despite this strategy being Suzuki-based, the novelty lays on a different disconnection, which results in allenic fragment 757 and boronic acids 758 (Scheme 128, path c).420 Wittig olefination from previously known enallenal 759 provided the dienallenyl iodide 757.421
Scheme 128. Retrosynthethic Strategies for the Synthesis of Peridinin and Deoxy-Peridinin Natural Products.

Katsumara’s research group has proposed a total synthesis for both Fucoxanthin (752c) and Paracentrone (752d) natural products following similar strategies. Sonogashira coupling of ethynyl epoxide 760 and iodo triene 761 yielded trienynyl carboxylate 762 (Scheme 129). Next, treatment with DIBAL-H as hydride source smoothly rearranged the ethynyl epoxide moiety to generate the allenol motif and also reduced the carboxylate unit to the corresponding terminal alcohol building compound 763. Further Dess-Martin oxidation followed by Wittig olefination in the presence of phosphonium salt 765 provided the Paracentrone skeleton (752d) (Scheme 129, left).422 A Suzuki alternative for the Wittig olefination in the last steps of the synthesis has also been stated,423 also allowing the synthesis of related 19-hexanoyloxyparacentrone 3-acetate.424 Likewise, Fucoxanthin (752c) total synthesis has been achieved through a Julia-type olefination of the allenic fragment 764 with hydroxysulfone 766 (Scheme 129, right).425
Scheme 129. Synthesis of Paracentrone and Fucoxanthin Natural Products.

Besides the above-mentioned synthethic approaches to naturally occurring targets, the determination of both structure and absolute configuration of allenol-containing natural products have been reported. Thus, Maoka et al. have stated the absolute configuration of minor carotenoid 4-Ketodeepoxyneoxanthin (767) in basis of NMR investigations.426 Che’s and Souto’s research groups have respectively proposed the full structure and stereochemistry of Chloropestolide metabolite 768,427 and Marilzabicycloallene A (769), exhibiting an unusual bromoallene motif (Scheme 130).428
Scheme 130. Proposed Structure of Allenol-Containing Natural Products As Determined by NMR Investigations.

The diverse and intriguing biological properties of naturally occurring allenols described so far, have paved the way to the synthesis of non-natural analogous and the examination of their pharmaceutical activities. Zemlicka has recently reported a critical comparison of the antiviral properties of a wide family of lipophilic nucleoside analogous and their phosphoramidates, including several examples of allenol-containing systems.429 For instance, adenosine- and cytosine-based compounds 770 and 771 have been prepared through basic equilibration from the corresponding alkynol precursors. Both molecules exhibit potent cytotoxic and antiviral activities. Interestingly, anti-HIV properties of 770 and 771 were found to be in close dependency of the absolute configuration of the allene moiety, being the (R)-isomer obtained through enzymatic resolution the active species (Scheme 131).430,431
Scheme 131. Allenol Motifs in Nucleoside Analogous with Pharmaceutical Activities.

More non-natural pharmacologically attractive allenols were described and reviewed during the 80s and 90s decades such as allenol-based prostaglandin and carbacyclin systems,432 allenic amino acids bearing hydroxyl groups,433 and allenic steroids.434
5. Conclusions
Allenol chemistry remains to be a hot research topic, which includes two main areas, namely the development of new methods for their synthesis and the discovery of novel and fascinating reactivity, which converts the allenol moiety in a powerful building block in the modern synthetic arsenal. Allenes decorated with hydroxyl units, namely allenols, exhibit unique and particular reactivity compared to the nonsubstituted analogous. The allenol reactivity could be classified in three main categories: (i) Allenols reacting as π-activated alcohols, where the hydroxyl group leaves the molecule or undergoes 1,3-migration rearrangement processes, leading to open-chained systems such as dienes or enynes, or to aromatic cyclic structures. (ii) Those reacting as bidentate nucleophiles-electrophiles, taking advantage of both the hydroxyl nucleophilicity as well as the allene electrophilicity when π-metal activation takes place. Oxacyclic structures such as furans or pyrans are accessed. (iii) Those where the hydroxyl group assists any kind of allene transformation, frequently by metal intermediate coordination. Also, the frequent use of allenols as key intermediates for the total synthesis of natural product deserves to be mentioned.
The extensive use of allenols as synthetic intermediates is associated with the implementation of an increasing number of methodologies for their preparation, both in racemic and enantiopure manner. Particularly attractive is the use of modern catalytic methods for the synthesis of enantioenriched allenols, which can display astonishing axial and central chirality. Last but not least, despite that the allenol scaffold is not commonly encountered in Nature, several allenol-based natural products have been isolated, characterized and synthesized. All contributions together support the widespread use and importance of the allenol functionality in current organic chemistry.
The intriguing reactivity so far exhibited by the allenol functional group, and the wide range of structures accessible from allenol starting materials will certainly inspire organic chemist to pursuit new advances and results that will be shortly coming in this area. In one hand, allenol-containing molecules constitute an ideal playground to continue the development of modern synthetic methodologies, as it has been recently illustrated by the recent micellar catalysis or the gold-based supramolecular catalysis, both of them based on allenol oxycyclization reactions. The field is dominated by the use of catalysts derived from expensive transition metals such as gold and palladium, with punctual incorporation of other metals. It should be desirable the widespread use of inexpensive and more environmentally friendly metals such as iron, copper, etc. Besides, despite the appearance of several catalytic protocols in heterogeneous phase through the use of metal nanoparticles, the more of the reactions are performed in homogeneous conditions. On the other hand, covalent–organic frameworks (COFs) and metal–organic frameworks (MOFs) have attracted considerable interest in recent years, but its application in allenol chemistry remains elusive. Consequently, more sustainable processes are desirable. In this context, the incorporation of recent progresses in photochemical methods and modern electrochemistry persist as a challenge. The application of photochemistry in allenol chemistry is restricted to a couple of isolated reports dealing with photoredox catalysis while there is absence of information concerning electrochemical methods. The use of enzymes in allenol chemistry is limited to the classical use for the resolution of racemic mixtures, but an efficient use of bioengineering advances should be taken into account. Besides, ongoing endeavors to discover competent asymmetric routes are largely based on designing and building new and exotic chiral nonracemic ligands or catalysts; however, the recognition of conveniently activated allenol precursors to enlarge catalytic effectiveness is critical too. On the other hand, the potential axial chirality of the allene motif is still unexploited, being the axial-to-central chirality transfer processes from axially enantioenriched 1,2-dienes one of the most notable challenges regarding the chemistry of allenes. Also, the inexhaustible search of new and more potent drugs, and the synthesis and characterization of yet unreported natural products often bearing oxacyclic moieties, will be certainly supported by the rich and efficient chemistry of the allenol system.
Acknowledgments
This work was supported in part by AEI (MICIU) and FEDER (Project PGC2018-095025–B-I00). We thank Prof. Benito Alcaide and Prof. Pilar M. Fresneda for their mentorship in past years.
Glossary
Abbreviations
- Acac
acetyl acetonate
- BINAP
2,2′-bis(diphenylphosphino)-1,1′-binaphthalene
- Bn
benzyl
- Boc
tert-butyloxycarbonyl
- Bpy
2,2′-bipyridine
- BPS
4,4′-sulfonyldiphenol
- BQ
benzoquinone
- BT
benzothiazole
- Bz
benzoyl
- Cbz
benzyloxycarbonyl
- Cod
cyclooctadiene
- CPME
cyclopentyl methyl ether
- CpMe4
tetramethylciclopentadienyl
- Cy
cyclohexyl
- DABCO
1,4-diazabicyclo[2.2.2]octane
- Dba
dibenzylacetone
- DCE
dichloroethane
- DCM
dichloromethane
- DDQ
2,3-dichloro-5,6-dicyano-1,4-benzoquinone
- DEAD
diethyl azodicarboxylate
- DIBAL-H
diisobutylaluminum hydride
- DMAP
4-dimethylaminopyridine
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- DPEphos
bis[(2-diphenylphosphino)phenyl] ether
- Dppf
1,1′-Ferrocenediyl-bis(diphenylphosphine
- DTBM-SEGPHOS
5,5′-bis[di(3,5-di- tert-butyl-4-methoxyphenyl)phosphino]-4,4′-bi-1,3-benzodioxole
- HMDS
hexamethyldisilazane
- HMPA
hexamethylphosphoramide
- LDA
lithium diisopropyl amine
- m-CPBA
meta-chloroperbenzoic acid
- MOM
methoxymethyl
- Ms
methanesulfonyl chloride
- NBS
N-bromosuccinimide
- NCS
N-chlorosuccinimide
- NFSI
N-fluorobenzenesulfonimide
- NP
nanoparticles
- NPSP
N-(phenylseleno)phthalimide
- PCC
pyridinium chlorochromate
- Pin
pinacol
- PINAP
(2-phosphino-1-naphthyl)phthalazinamine
- Piv
pivaloyl
- PMP
p-methoxyphenyl
- PNB
p-nitrobenzoyl
- PPL
lipase porcine pancreas
- PTSA
p-toluenesulfonic acid
- Py
pyridine
- Rh2(S-DOSP)4
dirhodium tetrakis((S)-N-(dodecylbenzenesulfonyl)prolinate)
- Sia2BH
disiamylborane
- TBAB
tetrabutylammonium bromide
- TBAI
tetrabutylammonium iodide
- TBDPS
tert-butyldiphenylsilyl
- TBHP
tert-butyl hydroperoxide
- TBS
tert-butyl dimethylsilyl
- TDMPP
tris(2,6-dimethoxyphenyl)phosphine
- TEA
triethylamine
- TEMPO
(2,2,6,6-tetramethylpiperidin-1-yl)oxyl
- TES
triethylsilyl
- Tf
trifluoromethane sulfonyl
- TFA
trifluoroacetic acid
- TMANO
trimethylamine N-oxide
- TMEDA
tetramethylethylenediamine
- TMS
trimethylsilyl
- Ts
tosyl
- 9-BBN
9-borabicyclo[3.3.1]nonane
Biographies
José Miguel Alonso (Madrid, 1975) received his B.S. degree (1998) and his Ph.D. degree (2003) from Universidad Complutense de Madrid (Prof. Benito Alcaide and Prof. Pedro Almendros). Besides a diverse experience in the industry (environmental science, polymer chemistry), he has been working as a postdoctoral researcher at Universidade de Santiago de Compostela (Santiago de Compostela, Spain), Universidad Complutense de Madrid (Madrid, Spain), University of East Anglia (Norwich, UK), and IQOG-CSIC (Madrid, Spain). He has held an Assistant Professor position in the Organic and Inorganic Chemistry Department at Universidad de Alcalá (Madrid, Spain), and he is currently Assistant Professor in the Organic Chemistry Department at Universidad Complutense de Madrid. His research interests include allene and aryne chemistry, metal-promoted heterocyclizations, and homogeneous and heterogeneous catalysis.
Pedro Almendros (Albacete, 1966) earned his Ph.D. in chemistry from Universidad de Murcia (1994, Profs. Molina and Fresneda). He was a Spanish MEC and European Marie Curie Postdoctoral Programs Fellow (1995-1998, University of Manchester, Prof. Eric J. Thomas). He joined Universidad Complutense de Madrid (UCM) in 1998 as Associate Researcher (Prof. Benito Alcaide). Subsequent appointments have included Assistant Professor at the UCM (2000–2002), Científico Titular “Tenured Scientist” (2002–2007), and Investigador Científico “Researcher Scientist“ (2007–2016) at the Instituto de Química Orgánica General, CSIC, Madrid. In 2016, he was promoted to Profesor de Investigación “Researcher Professor” at the IQOG-CSIC, Madrid. His research interests include allene and alkyne reactivity, C–C coupling reactions, and triflone chemistry.
The authors declare no competing financial interest.
References
- Van’t Hoff J. H.La Chimie dans L’Espace; Bazendijk: Rotterdam, 1875. [Google Scholar]
- Hoffmann-Roder A.; Krause N. Synthesis and Properties of Allenic Natural Products and Pharmaceuticals. Angew. Chem., Int. Ed. 2004, 43, 1196–1216. 10.1002/anie.200300628. [DOI] [PubMed] [Google Scholar]
- Alcaide B.; Almendros P. Allenyl-β-lactams: Versatile Scaffolds for the Synthesis of Heterocycles. Chem. Rec. 2011, 11, 311–330. 10.1002/tcr.201100011. [DOI] [PubMed] [Google Scholar]
- Yu S.; Ma S. Allenes in Catalytic Asymmetric Synthesis and Natural Product Syntheses. Angew. Chem., Int. Ed. 2012, 51, 3074–3112. 10.1002/anie.201101460. [DOI] [PubMed] [Google Scholar]
- Aso M.; Kanematsu K. Allenes as Versatile Synthons. Trends in Organic Chemistry 1995, 5, 157–169. [Google Scholar]
- Allenes and Cumulenes. Comprehensive Organic Functional Group Transformations; Bruneau C., Dixneuf P. H., Katritzky A. R., Meth-Cohn O., Rees C. W., Eds., 1995; Vol. 1, pp 953–995. [Google Scholar]
- Hashmi A. S. K. New and Selective Transition Metal Catalyzed Reactions of Allenes. Angew. Chem., Int. Ed. 2000, 39, 3590–3593. . [DOI] [PubMed] [Google Scholar]
- Alcaide B.; Almendros P. Progress in Allene Chemistry. Chem. Soc. Rev. 2014, 43, 2879–3205. 10.1039/c4cs90020k. [DOI] [PubMed] [Google Scholar]
- Alcaide B.; Almendros P.; Aragoncillo C. Cyclization Reactions of Bis(allenes) for the Synthesis of Polycarbo(hetero)cycles. Chem. Soc. Rev. 2014, 43, 3106–3135. 10.1039/C3CS60462D. [DOI] [PubMed] [Google Scholar]
- Krause N.; Winter C. Gold-Catalyzed Nucleophilic Cyclization of Functionalized Allenes: A Powerful Access to Carbo- and Heterocycles. Chem. Rev. 2011, 111, 1994–2009. 10.1021/cr1004088. [DOI] [PubMed] [Google Scholar]
- Aubert C.; Fensterbank L.; Garcia P.; Malacria M.; Simonneau A. Transition Metal Catalyzed Cycloisomerizations of 1,n–Allenynes and – Allenenes. Chem. Rev. 2011, 111, 1954–1993. 10.1021/cr100376w. [DOI] [PubMed] [Google Scholar]
- Zimmer R.; Dinesh C. U.; Nandanon E.; Khan F. A. Palladium-Catalyzed Reactions of Allenes. Chem. Rev. 2000, 100, 3067–3126. 10.1021/cr9902796. [DOI] [PubMed] [Google Scholar]
- Alonso J. M.; Muñoz M. P. When Indoles Meet Allene and its Derivatives. Eur. J. Org. Chem. 2020, 2020, 7197. 10.1002/ejoc.202001269. [DOI] [Google Scholar]
- Ma S. Electrophilic Addition and Cyclization Reactions of Allenes. Acc. Chem. Res. 2009, 42, 1679–1688. 10.1021/ar900153r. [DOI] [PubMed] [Google Scholar]
- Ma S. Control of Regio- and Stereoselectivity in Electrophilic Addition Reactions of Allenes. Pure Appl. Chem. 2007, 79, 261–267. 10.1351/pac200779020261. [DOI] [Google Scholar]
- Ma S.Ionic Additions to Allenes. In Modern Allene Chemistry; Krause N., Hashmi A. S. K., Eds.; Wiley: Weinheim, 2004; Vol. 2, pp 595–699. [Google Scholar]
- Chen W.-Q.; Ma J.-A.; Knochel P.; Molander G. A. Stabilized Nucleophiles with Electron Deficient Alkenes, Alkynes, Allenes. Comprehensive Organic Synthesis 2014, 4, 1–85. 10.1016/B978-0-08-097742-3.00401-8. [DOI] [Google Scholar]
- Mascareñas J. L.; Varela I.; López F. Allenes and Derivatives in Gold(I)- and Platinum(II)-Catalyzed Formal Cycloadditions. Acc. Chem. Res. 2019, 52, 465–479. 10.1021/acs.accounts.8b00567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lledó A.; Pla-Quintana A.; Roglans A. Allenes, Versatile Unsaturated Motifs in Transition-Metal-Catalysed [2 + 2+2] Cycloaddition Reactions. Chem. Soc. Rev. 2016, 45, 2010–2023. 10.1039/C5CS00535C. [DOI] [PubMed] [Google Scholar]
- Wang L.-J.; Tang Y.. Intermolecular 1,3-Dipolar Cycloadditions of Alkenes, Alkynes, and Allenes. In Comprehensive Organic Synthesis, 2nd ed.; Knochel P., Molander G. A., Eds.; Elsevier: Amsterdam, 2014; Vol. 4, pp 1342–1383. [Google Scholar]
- Alcaide B.; Almendros P.; Aragoncillo C. Exploiting [2 + 2] Cycloaddition Chemistry: Achievements with Allenes. Chem. Soc. Rev. 2010, 39, 783–816. [DOI] [PubMed] [Google Scholar]
- Pan F.; Fu C.-L.; Ma S. Radical Reactions of Allenes. Chin. J. Org. Chem. 2004, 24, 1168–1190. [Google Scholar]
- Hartung J.; Kopf T.. Fundamentals and Application of Free Radical Addition to Allenes. In Modern Allene Chemistry; Krause N.; Hashmi A. S. K., Eds.; Wiley: Weinheim, 2004; Vol. 2, pp 701–726. [Google Scholar]
- Muzart J. Gold-Catalyzed Reactions of Alcohols: Isomerization, Inter- and Intramolecular Reactions leading to C-C and C-Heteroatom Bonds. Tetrahedron 2008, 64, 5815–5849. 10.1016/j.tet.2008.04.018. [DOI] [Google Scholar]
- Brasholz M.; Reissig H.-U.; Zimmer R. Sugars, Alkaloids, and Heteroaromatics: Exploring Heterocyclic Chemistry with Alkoxyallenes. Acc. Chem. Res. 2009, 42, 45–56. 10.1021/ar800011h. [DOI] [PubMed] [Google Scholar]
- Alcaide B.; Almendros P. Gold-Catalyzed Cyclization Reactions of Allenol and Alkynol Derivatives. Acc. Chem. Res. 2014, 47, 939–952. 10.1021/ar4002558. [DOI] [PubMed] [Google Scholar]
- Lu Z.; Ma S. Metal-Catalyzed Enantioselective Allylation in Asymmetric Synthesis. Angew. Chem., Int. Ed. 2008, 47, 258–297. 10.1002/anie.200605113. [DOI] [PubMed] [Google Scholar]
- Cowen B. J.; Miller S. J. Enantioselective Catalysis and Complexity Generation from Allenoates. Chem. Soc. Rev. 2009, 38, 3102–3116. 10.1039/b816700c. [DOI] [PubMed] [Google Scholar]
- Alcaide B.; Almendros P.; Martínez del Campo T. Cross-Coupling/Cyclization Reactions of Two Different Allenic Moieties. Chem. - Eur. J. 2010, 16, 5836–5842. 10.1002/chem.201000550. [DOI] [PubMed] [Google Scholar]
- Le Bras J.; Muzart J. Palladium-catalysed Inter- and Intramolecular Formation of C–O Bonds from Allenes. Chem. Soc. Rev. 2014, 43, 3003–3040. 10.1039/C3CS60379B. [DOI] [PubMed] [Google Scholar]
- Barreiro E. M.; Adrio L. A.; Hii K. K.; Brazier J. B. Coinage Metal Catalysts for the Addition of O–H to C = C Bonds. Eur. J. Org. Chem. 2013, 2013, 1027–1039. 10.1002/ejoc.201201441. [DOI] [Google Scholar]
- Yang B.; Qiu Y.; Bäckvall J.-E. Control of Selectivity in Palladium(II)-Catalyzed Oxidative Transformations of Allenes. Acc. Chem. Res. 2018, 51, 1520–1531. 10.1021/acs.accounts.8b00138. [DOI] [PubMed] [Google Scholar]
- Muñoz M. P. Silver and Platinum-catalysed Addition of O–H and N–H Bonds to Allenes. Chem. Soc. Rev. 2014, 43, 3164–3183. 10.1039/c3cs60408j. [DOI] [PubMed] [Google Scholar]
- Ma S. Control of Regio- and Stereoselectivity in Electrophilic Addition Reactions of Allenes. Pure Appl. Chem. 2007, 79, 261–267. 10.1351/pac200779020261. [DOI] [Google Scholar]
- Fan X.; He Y.; Zhang X. Recent Advances in the Reactions of 1,2-Allenic Ketones and a-Allenic Alcohols. Chem. Rec. 2016, 16, 1635–1646. 10.1002/tcr.201500301. [DOI] [PubMed] [Google Scholar]
- Alcaide B.; Almendros P.; Martínez del Campo T.; Soriano E.; Marco-Contelles J. Heterocyclization of Allenes Catalyzed by Late Transition Metals: Mechanisms and Regioselectivity. Top. Curr. Chem. 2011, 302, 183–224. 10.1007/128_2010_120. [DOI] [PubMed] [Google Scholar]
- Crabbé P.; André D.; Fillion H. Synthesis of Homodinordrin, Allenyl A-nor and Dinor-steroids. Tetrahedron Lett. 1979, 20, 893–896. 10.1016/S0040-4039(01)93581-6. [DOI] [Google Scholar]
- Crabbé P.; Fillion H.; André D.; Luche J. Efficient Homologation of Acetylenes to Allenes. J. Chem. Soc., Chem. Commun. 1979, 859–860. 10.1039/C39790000859. [DOI] [Google Scholar]
- Searles S.; Li Y.; Nassim B.; Lopes M. R.; Tran P. T.; Crabbé P. Observation on the Synthesis of Allenes by Homologation of Alk-1-ynes. J. Chem. Soc., Perkin Trans. 1 1984, 1, 747–751. 10.1039/P19840000747. [DOI] [Google Scholar]
- For a review, see:; Huang X.; Ma S. Allenation of Terminal Alkynes with Aldehydes and Ketones. Acc. Chem. Res. 2019, 52, 1301–1312. 10.1021/acs.accounts.9b00023. [DOI] [PubMed] [Google Scholar]
- Kuang J.; Ma S. An Efficient Synthesis of Terminal Allenes from Terminal 1-Alkynes. J. Org. Chem. 2009, 74, 1763–1765. 10.1021/jo802391x. [DOI] [PubMed] [Google Scholar]
- Chen B.; Wang N.; Fan W.; Ma S. Efficient Synthesis of N-(buta-2,3-dienyl) Amides from Terminal N-propargyl Amides and Their Synthetic Potential Towards Oxazoline Derivatives. Org. Biomol. Chem. 2012, 10, 8465–8470. 10.1039/c2ob26291f. [DOI] [PubMed] [Google Scholar]
- Luo H.; Yang Z.; Lin W.; Zheng Y.; Ma S. A Catalytic Highly Enantioselective Allene Approach to Oxazolines. Chem. Sci. 2018, 9, 1964–1969. 10.1039/C7SC04079B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N.; Chen B.; Ma S. Studies on Electrophilic Cyclization of N-(Buta-2,3- dienyl)amides with N-Bromosuccinimide and its Applications. Adv. Synth. Catal. 2014, 356, 485–492. 10.1002/adsc.201300959. [DOI] [Google Scholar]
- Kuang J.; Xie X.; Ma S. A General Approach to Terminal Allenols. Synthesis 2013, 45, 592–595. 10.1055/s-0032-1317949. [DOI] [Google Scholar]
- Luo H.; Ma S. CuI-Catalyzed Synthesis of Functionalized Terminal Allenes from 1-Alkynes. Eur. J. Org. Chem. 2013, 2013, 3041–3048. 10.1002/ejoc.201201696. [DOI] [Google Scholar]
- Kuang J.; Ma S. One-Pot Synthesis of 1,3-Disubstituted Allenes from 1-Alkynes, Aldehydes, and Morpholine. J. Am. Chem. Soc. 2010, 132, 1786–1787. 10.1021/ja910503k. [DOI] [PubMed] [Google Scholar]
- Kuang J.; Luo H.; Ma S. Copper (I) Iodide-Catalyzed One-Step Preparation of Functionalized Allenes from Terminal Alkynes: Amine Effect. Adv. Synth. Catal. 2012, 354, 933–944. 10.1002/adsc.201100772. [DOI] [Google Scholar]
- Tang X.; Zhu C.; Cao T.; Kuang J.; Lin W.; Ni S.; Zhang J.; Ma S. Cadmium Iodide-Mediated Allenylation of Terminal Alkynes with Ketones. Nat. Commun. 2013, 4, 1–7. 10.1038/ncomms3450. [DOI] [PubMed] [Google Scholar]
- Tang X.; Kuang J.; Ma S. CuBr for KA2 reaction: En route to Propargylic Amines Bearing a Quaternary Carbon Center. Chem. Commun. 2013, 49, 8976–8978. 10.1039/c3cc45301d. [DOI] [PubMed] [Google Scholar]
- Kuang J.; Tang X.; Ma S. Zinc Diiodide-promoted Synthesis of Trisubstituted Allenes from Propargylic Amines. Org. Chem. Front. 2015, 2, 470–475. 10.1039/C5QO00047E. [DOI] [Google Scholar]
- Xiao Q.; Xia Y.; Li H.; Zhang Y.; Wang J. Coumpling of N-Tosylhydrazones with Terminal Alkynes Catalyzed by Copper(I): Synthesis of Trisubstituted Allenes. Angew. Chem., Int. Ed. 2011, 50, 1114–1117. 10.1002/anie.201005741. [DOI] [PubMed] [Google Scholar]
- Poh J.-S.; Tran D. N.; Battilocchio C.; Hawkins J. M.; Ley S. V. A Versatile Room-Temperature Route to Di- and Trisubstituted Allenes Using Flow-Generated Diazo Compounds. Angew. Chem., Int. Ed. 2015, 54, 7920–7923. 10.1002/anie.201501538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabbasani V.; Mamidipalli P.; Lu H.; Xia Y.; Lee D. Subtle Electronic Effects in Metal-Free Rearrangement of Allenic Alcohols. Org. Lett. 2013, 15 (7), 1552–1555. 10.1021/ol400331r. [DOI] [PubMed] [Google Scholar]
- Xu S.; Chen R.; Fu Z.; Gao Y.; Wang J. Cu (I)-Catalyzed Coupling of Bis(trimethylsilyl)diazomethane with Terminal Alkynes: A Synthesis of 1,2-Disilyl Allenes. J. Org. Chem. 2018, 83 (11), 6189–6192. 10.1021/acs.joc.8b00651. [DOI] [PubMed] [Google Scholar]
- Wu C.; Hu F.; Liu Z.; Deng G.; Ye F.; Zhang Y.; Wang J. Cu (I)-Catalyzed Coupling of Diaryldiazomethanes with Terminal Alkynes: An Efficient Synthesis of Tri-aryl-substituted Allenes. Tetrahedron 2015, 71, 9196–9201. 10.1016/j.tet.2015.10.043. [DOI] [Google Scholar]
- Masuyama Y.; Ito A.; Fukuzawa M.; Terada K.; Kusuru Y. Carbonyl Propargylation or Allenylation by 3-Haloprop-1-yne with Tin(II) Halides and Tetrabutylammonium Halides. Chem. Commun. 1998, 2025–2026. 10.1039/a806206d. [DOI] [Google Scholar]
- Masuyama Y.; Watabe A.; Ito A.; Kuturu Y. Carbonyl Propargylation by 1-Substituted 2-Propynyl Mesylates and Carbonyl Allenylation by 3-Substituted 2-Propynyl Mesylates with Tin(II) Iodide and Tetrabutylammonium Iodide. Chem. Commun. 2000, 2009–2010. 10.1039/b006646j. [DOI] [Google Scholar]
- Savall B.; Powell N.; Roush W. Highly Diastereoselective Synthesis of Propargylic 1,2-anti-Diol Derivatives Using α-Alkoxypropargylstannanes. Org. Lett. 2001, 3, 3057–3060. 10.1021/ol016536m. [DOI] [PubMed] [Google Scholar]
- Chemla F.; Bernard N.; Ferreira F.; Normant J. F. Preparation of Propargylic Carbenoids and Reactions with Carbonyl Compounds – A Stereoselective Synthesis of Propargylic Halohydrins and Oxiranes. Eur. J. Org. Chem. 2001, 2001, 3295–3300. . [DOI] [Google Scholar]
- Tamaru Y.; Goto S.; Tanaka A.; Shimizu M.; Kimura M. Propargylation of Carbonyl Compounds by Umpolung of Propargylpalladium Complexes with Diethylzinc. Angew. Chem., Int. Ed. Engl. 1996, 35, 879–880. 10.1002/anie.199608781. [DOI] [Google Scholar]
- The reaction of formyl N-methyliminoiacetyl boronate with propargylzinc reagents, which were generated in situ from 1-bromobut-2-yne or (3-bromoprop-1-yn-1-yl)benzene and Zn in the presence of Cp2TiCl2 proceeded to led the corresponding boronate-allenol derivatives in low yields.; Ivon Y. M.; Mazurenko I. V.; Kuchkovska Y. O.; Voitenko Z. V.; Grygorenko O. O. Formyl MIDA Boronate: C1 Building Block Enables Straightforward Access to α-Functionalized Organoboron Derivatives. Angew. Chem., Int. Ed. 2020, 59, 18016–18022. 10.1002/anie.202007651. [DOI] [PubMed] [Google Scholar]
- Yi X.-H.; Meng Y.; Hua X.-G.; Li C.-J. Regio- and Diastereoselective Allenylation of Aldehydes in Aqueous Media: Total Synthesis of (+)-Goniofufurone. J. Org. Chem. 1998, 63, 7472–7480. 10.1021/jo9815610. [DOI] [PubMed] [Google Scholar]
- Isaac M.; Chan T.-K. Indium-Mediated Coupling of Aldehydes with Prop-2-ynyl Bromides in Aqueous Media. J. Chem. Soc., Chem. Commun. 1995, 1003–1004. 10.1039/c39950001003. [DOI] [Google Scholar]
- Alcaide B.; Almendros P.; Aragoncillo C. Regio- and Stereocontrolled Metal-Mediated Carbonyl Propargylation or Allenylation of Enantiomerically Pure Azetidine-2,3-diones: Synthesis of Highly Functionalized 3-Substituted 3-Hydroxy- β-lactams. Org. Lett. 2000, 2, 1411–1414. 10.1021/ol005736f. [DOI] [PubMed] [Google Scholar]
- Alcaide B.; Almendros P.; Rodríguez-Acebes R. Metal–Mediated Entry to Functionalized 3-Substituted 3-Hydroxyindolin-2-ones via Regiocontrolled Carbonyl-Allylation, Bromoallylation, 1,3-Butadien-2-ylation, Propargylation, or Allenylation Reactions of Isatins in Aqueous Media. J. Org. Chem. 2005, 70, 3198–3204. 10.1021/jo050130w. [DOI] [PubMed] [Google Scholar]
- Park C.; Lee P. H. Indium-Mediated Regio- and Chemoselective Synthesis of α-Hydroxyalkyl Allenic Esters and Gold-Catalyzed Cyclizations to Ethyl 2-Naphthoate Derivatives. Org. Lett. 2008, 10, 3359–3362. 10.1021/ol801196g. [DOI] [PubMed] [Google Scholar]
- Fandrick D. R.; Fandrick K. R.; Reeves J. T.; Tan Z.; Tang W.; Capacci A. G.; Rodriguez S.; Song J. J.; Lee H.; Yee N. K.; Senanayake C. H. Copper Catalyzed Asymmetric Propargylation of Aldehydes. J. Am. Chem. Soc. 2010, 132, 7600–7601. 10.1021/ja103312x. [DOI] [PubMed] [Google Scholar]
- Zou Y.; Gutierrez O.; Sader A. C.; Patel N. D.; Fandrick D. R.; Busacca C. A.; Fandrick K. R.; Kozlowski M.; Senanayake C. H. A Computational Investigation of the Ligand-Controlled Cu-Catalyzed Site-Selective Propargylation and Allenylation of Carbonyl Compounds. Org. Lett. 2017, 19, 6064–6067. 10.1021/acs.orglett.7b02845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan Y.; Cai Z.; Li X.; Ramella D.; Miao Z.; Wang W. An Efficient Nozaki–Hiyama Allenylation Promoted by the Acid Derived MIL-101 MOF.. RSC Adv. 2019, 9, 7479–7484. 10.1039/C8RA09600G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita Y.; Cui Y.; Xie P.; Kobayashi S. Zinc Amide Catalyzed Regioselective Allenylation and Propargylation of Ketones with Allenyl Boronate. Org. Lett. 2015, 17, 6042–6045. 10.1021/acs.orglett.5b03045. [DOI] [PubMed] [Google Scholar]
- Bhowmick M.; Lepore S. D. Manganese η2-Complexes as Auxiliaries for Stereoselective Aldol Synthesis of Allenyl Carbinols. Org. Lett. 2010, 12, 5078–5080. 10.1021/ol1021096. [DOI] [PubMed] [Google Scholar]
- Kim H. Y.; Li J.-Y.; Oh K. A Soft Vinyl Enolization Approach to α-Acylvinyl Anions: Direct Aldol/Aldol Condensation Reactions of (E)-β-Chlorovinyl Ketones. Angew. Chem., Int. Ed. 2013, 52, 3736–3740. 10.1002/anie.201209876. [DOI] [PubMed] [Google Scholar]
- Zhao G.-L.; Shi M. Baylis–Hillman Reactions of N-Tosyl Aldimines and Aryl Aldehydes with 3-Methylpenta-3,4-dien-2-one. Org. Biomol. Chem. 2005, 3, 3686–3694. 10.1039/b510572b. [DOI] [PubMed] [Google Scholar]
- Selig P.; Turockin A.; Raven W. Guanidine-Catalyzed γ-Selective Morita–Baylis–Hillman Reactions on α,γ-Dialkyl-Allenoates: Access to Densely Substituted Heterocycles. Synlett 2013, 24, 2535–2539. 10.1055/s-0033-1339471. [DOI] [Google Scholar]
- Maity P.; Lepore S. D. Anion-Catalyzed Addition of γ-Silylallenyl Esters to Aldehydes: A New Entry into [3.2.1] Bicyclic Natural Products. J. Am. Chem. Soc. 2009, 131, 4196–4197. 10.1021/ja810136m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tata R. R.; Hampton C. S.; Altenhofer E. F.; Topinka M.; Ying W.; Gao X.; Harmata M. New Synthesis of α’-Hydroxydienones. Chem. - Eur. J. 2014, 20, 13547–13550. 10.1002/chem.201404638. [DOI] [PubMed] [Google Scholar]
- Kessler S. N.; Hundemer F.; Bäckvall J. E. A Synthesis of Substituted α-Allenols via Iron-Catalyzed Cross-Coupling of Propargyl Carboxylates with Grignard Reagents. ACS Catal. 2016, 6, 7448–7451. 10.1021/acscatal.6b02114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green N. J.; Willis A. C.; Sherburn M. S. Direct Cross-Couplings of Propargylic Diols. Angew. Chem., Int. Ed. 2016, 55, 9244–9248. 10.1002/anie.201604527. [DOI] [PubMed] [Google Scholar]
- Xing J.; Zhu Y.; Lin X.; Liu N.; Shen Y.; Lu T.; Dou X. Rhodium-Catalyzed Arylative Transformations of Propargylic Diols: Dual Role of the Rhodium Catalyst. Adv. Synth. Catal. 2018, 360, 1595–1599. 10.1002/adsc.201800048. [DOI] [Google Scholar]
- Marshall J. A.; Pinney K. G. Stereoselective Synthesis of 2,5-Dihydrofurans by Sequential SN2′ Cleavage of Alkynyloxiranes and Ag+-Catalyzed Cyclization of the Allenylcarbinol Products. J. Org. Chem. 1993, 58, 7180–7184. 10.1021/jo00077a048. [DOI] [Google Scholar]
- Morita N.; Krause N. Gold Catalysis in Organic Synthesis: Efficient Cycloisomerization of α-Aminoallenes to 3-Pyrrolines. Org. Lett. 2004, 6, 4121–4123. 10.1021/ol0481838. [DOI] [PubMed] [Google Scholar]
- Yoshida M.; Ueda H.; Ihara M. Palladium-Catalyzed Coupling Reaction of Propargylic Oxiranes with Arylboronic Acids in Aqueous Media. Tetrahedron Lett. 2005, 46, 6705–6708. 10.1016/j.tetlet.2005.07.134. [DOI] [Google Scholar]
- Miura T.; Shimada M.; Ku S.-Y.; Tamai T.; Murakami M. Stereoselective Synthesis of α-Allenols by Rhodium-Catalyzed Reaction of Alkynyl Oxiranes with Arylboronic Acids. Angew. Chem., Int. Ed. 2007, 46, 7101–7103. 10.1002/anie.200701505. [DOI] [PubMed] [Google Scholar]
- Yoshida M.; Hayashi M.; Shishido K. Palladium-Catalyzed Diastereoselective Coupling of Propargylic Oxiranes with Terminal Alkynes. Org. Lett. 2007, 9, 1643–1646. 10.1021/ol070224n. [DOI] [PubMed] [Google Scholar]
- Miura T.; Shimada M.; de Mendoza P.; Deutsch C.; Krause N.; Murakami M. Stereoselective Synthesis of syn-Configured α-Allenols by Rhodium-Catalyzed Reaction of Alkynyl Oxiranes with Arylboronic Acids. J. Org. Chem. 2009, 74, 6050–6054. 10.1021/jo900987w. [DOI] [PubMed] [Google Scholar]
- Alexakis A.; Marek I.; Mangeney P.; Normant J. F. Diastereoselective Syn or Anti Opening of Propargylic Epoxides. Synthesis of α-Allenic Alcohols. Tetrahedron 1991, 47, 1677–1696. 10.1016/S0040-4020(01)96911-X. [DOI] [Google Scholar]
- Alexakis A.; Marek I.; Mangeney P.; Normant J. F. Diastereoselective Synthesis of α-Allenic Alcohols from Propargylic Epoxides. Tetrahedron Lett. 1989, 30, 2387–2390. 10.1016/S0040-4039(01)80406-8. [DOI] [Google Scholar]
- Bertozzi F.; Crotti P.; Macchia F.; Pineschi M.; Arnold A.; Feringa B. L. A New Diastereo- and Enantioselective Copper-Catalyzed Conversion of Alkynyl Epoxides into α-Allenic Alcohols. Tetrahedron Lett. 1999, 40, 4893–4896. 10.1016/S0040-4039(99)00905-3. [DOI] [Google Scholar]
- For a recent example on heteronucleophile addition to related propargyl carbonates, see:; Guo K.; Kleij A. W. Cu-Catalyzed Synthesis of Tetrasubstituted 2,3-Allenols through Decarboxylative Silylation of Alkyne-Substituted Cyclic Carbonates. Org. Lett. 2020, 22, 3942–3945. 10.1021/acs.orglett.0c01222. [DOI] [PubMed] [Google Scholar]
- Zhao J.; Szabó K. J. Catalytic Borylative Opening of Propargyl Cyclopropane, Epoxide, Aziridine, and Oxetane Substrates: Ligand Controlled Synthesis of Allenyl Boronates and Alkenyl Diboronates. Angew. Chem., Int. Ed. 2016, 55, 1502–1506. 10.1002/anie.201510132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjellgren J.; Sundén H.; Szabó K. J. Palladium Pincer Complex Catalyzed Stannyl and Silyl Transfer to Propargylic Substrates: Synthetic Scope and Mechanism. J. Am. Chem. Soc. 2005, 127, 1787–1796. 10.1021/ja043951b. [DOI] [PubMed] [Google Scholar]
- Shen R.; Yang J.; Zhao H.; Feng Y.; Zhang L.; Han L.-B. Cu-Catalyzed Hydrophosphorylative Ring Opening of Propargyl Epoxides: Highly Selective Access to 4-Phosphoryl 2,3-Allenols. Chem. Commun. 2016, 52, 11959–11962. 10.1039/C6CC05428E. [DOI] [PubMed] [Google Scholar]
- Shen T.; Yang J.; Zhang M.; Han L.-B. Silver-Catalyzed Atom-Economic Hydrophosphorylation of Propargyl Epoxides: An Access to 4-Phosphoryl 2,3-Allenols and Stereodefined 1-Phosphoryl 1,3-Dienes. Adv. Synth. Catal. 2017, 359, 3626–3637. 10.1002/adsc.201700421. [DOI] [Google Scholar]
- For a related approach using cyclopropyl alkynols, see:; Watson H. A.; Manaviazar S.; Steeds H. G.; Hale K. J. Fast Ring-Opening of an Intermediary α-Stannyl-β-Cyclopropylvinyl Radical Does Not Support Formation of an α-Stannylvinyl Cation in the O-Directed Free Radical Hydrostannation of Dialkyl Acetylenes. Chem. Commun. 2019, 55, 14454–14457. 10.1039/C9CC05492H. [DOI] [PubMed] [Google Scholar]
- Liepouri F.; Bernasconi G.; Petasis N. A. Component-Selective and Stereocontrolled One-Step Three-Component Reaction among Aldehydes, Amines, and Allenyl Boronic Acids or Allenyl Pinacolboronates. Org. Lett. 2015, 17, 1628–1631. 10.1021/acs.orglett.5b00024. [DOI] [PubMed] [Google Scholar]
- Mundal D. A.; Lutz K. E.; Thomson R. J. A Direct Synthesis of Allenes by a Traceless Petasis Reaction. J. Am. Chem. Soc. 2012, 134, 5782–5785. 10.1021/ja301489n. [DOI] [PubMed] [Google Scholar]
- Deng Y.; Bäckvall J. E. Palladium-Catalyzed Oxidative Acyloxylation/Carbocyclization of Allenynes. Angew. Chem., Int. Ed. 2013, 52, 3217–3221. 10.1002/anie.201208718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padial N. M.; Hernández-Cervantes C.; Muñoz-Bascón J.; Roldán-Molina E.; García-Martínez M.; Ruiz-Muelle A. B.; Rosales A.; Álvarez-Corral M.; Muñoz-Dorado M.; Rodríguez-García I.; et al. Ti-Catalyzed Synthesis of Exocyclic Allenes on Oxygen Heterocycles. Eur. J. Org. Chem. 2017, 2017, 639–645. 10.1002/ejoc.201601444. [DOI] [Google Scholar]
- Roldán-Molina E.; Nievas M. M.; Navarro J. A. R.; Oltra J. E. CpTiCl2-Catalyzed Cross-Coupling between Internal Alkynes and Ketones: A Novel Concept in the Synthesis of Halogenated, Conjugated Dienes. Chem. - Eur. J. 2017, 26, 8296–8301. 10.1002/chem.202001023. [DOI] [PubMed] [Google Scholar]
- Jiang H.; Wang W.; Yin B.; Liu W. Facile Synthesis of Trisubstituted Allenynes by Phosphane-Mediated Deoxygenation of 2,4-Pentadiyn-1-ol. Eur. J. Org. Chem. 2010, 4450–4453. 10.1002/ejoc.201000483. [DOI] [Google Scholar]
- De Nisi A.; Sierra S.; Ferrara M.; Monari M.; Bandini M. TBAF Catalyzed One-pot Synthesis of Allenyl-indoles. Org. Chem. Front. 2017, 4, 1849–1853. 10.1039/C7QO00414A. [DOI] [Google Scholar]
- Huang X.; Bugarin A. Direct Synthesis of α-Allenols from TMS-Protected Alkynes and Aldehydes Mediated by Tetrabutylammonium Fluoride. Chem. - Eur. J. 2016, 22, 12696–12700. 10.1002/chem.201603250. [DOI] [PubMed] [Google Scholar]
- Ye C.; Li Y.; Zhu X.; Hu S.; Yuan D.; Bao H. Copper-Catalyzed 1,4-Alkylarylation of 1,3-Enynes with Masked Alkyl Electrophiles. Chem. Sci. 2019, 10, 3632–3636. 10.1039/C8SC05689G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B.; Li Y.; Pang J. H.; Watanabe K.; Takita R.; Chiba S. Hydromagnesiation of 1,3-Enynes by Magnesium Hydride for Synthesis of Tri- and Tetra-substituted Allenes. Angew. Chem. 2021, 133, 219–223. 10.1002/ange.202012027. [DOI] [PubMed] [Google Scholar]
- Huang X.; Cao T.; Han Y.; Jiang X.; Lin W.; Zhang J.; Ma S. General CuBr2-Catalyzed Highly Enantioselective Approach for Optically Active Allenols from Terminal Alkynols. Chem. Commun. 2015, 51, 6956–6959. 10.1039/C5CC00697J. [DOI] [PubMed] [Google Scholar]
- Ye J.; Fan W.; Ma S. tert-Butyldimethylsilyl-Directed Highly Enantioselective Approach to Axially Chiral α-Allenols. Chem. - Eur. J. 2013, 19, 716–720. 10.1002/chem.201201948. [DOI] [PubMed] [Google Scholar]
- Ma D.; Duan X.; Fu C.; Huang X.; Ma S. Dimethylprolinol Versus Diphenylprolinol in CuBr2-Catalyzed Enantioselective Allenylation of Terminal Alkynols. Synthesis 2018, 50, 2533–2545. 10.1055/s-0036-1592007. [DOI] [Google Scholar]
- Bang J.; Oh C.; Lee H.; Jeong H.; Lee J.; Ryu J. Y.; Kim J.; Yu C.-M. A Regulation of Regiodivergent Routes for Enantioselective Aldol Addition of 2-Alkyl Allenoates with Aldehydes: α-Addition versus γ-Addition. Org. Lett. 2018, 20, 1521–1525. 10.1021/acs.orglett.8b00219. [DOI] [PubMed] [Google Scholar]
- Pak G.; Park E.; Park S.; Kim J. Synthesis of (+)-Hypoxylactone through Allenoate γ-Addition: Revision of Stereochemistry. J. Org. Chem. 2020, 85, 14246–14252. 10.1021/acs.joc.0c02194. [DOI] [PubMed] [Google Scholar]
- For α-selective versus γ-selective addition through the same methodology, see:; Bang J.; Kim H.; Kim J.; Yu C.-M. Asymmetric Aldol Reaction of Allenoates: Regulation for the Selective Formation of Isomeric Allenyl or Alkynyl Aldol Adduct. Org. Lett. 2015, 17, 1573–1576. 10.1021/acs.orglett.5b00454. [DOI] [PubMed] [Google Scholar]
- For a recent asymmetric aldol-type addition of aldehydes to enantioenriched alkynes, see:; Sechi M. L.; Andrade M.; Foy H.; Pilkington M.; Dudding T.; Metallinos C. Stereoselective Synthesis of N-Propargyl Alkynes and Axial Chiral N-Allenes with Epimeric Pyrroloimidazolone Auxiliaries. Adv. Synth. Catal. 2020, 362, 4397. 10.1002/adsc.202000574. [DOI] [Google Scholar]
- Spino C.; Fréchette S. Synthesis of Non-Racemic Unsymmetrical Tetrasubstituted Vinylallenes. Tetrahedron Lett. 2000, 41, 8033–8036. 10.1016/S0040-4039(00)01406-4. [DOI] [Google Scholar]
- Fürstner A.; Méndez M. Iron-Catalyzed Cross-Coupling Reactions: Efficient Synthesis of 2,3-Allenol Derivatives. Angew. Chem., Int. Ed. 2003, 42, 5355–5357. 10.1002/anie.200352441. [DOI] [PubMed] [Google Scholar]
- Jiang H.; Holub N.; Paixao M. W.; Tiberi C.; Falcicchio A.; Jørgensen K. A. Iminium-Ion Activation as an Efficient Strategy for Divergent Synthesis of Optically Active Propargylic, Homopropargylic, and Allenic Compounds. Chem. - Eur. J. 2009, 15, 9638–9641. 10.1002/chem.200901921. [DOI] [PubMed] [Google Scholar]
- Jarava-Barrera C.; Parra A.; Amenos L.; Arroyo A.; Tortosa M. Stereoselective Traceless Borylation–Allenation of Propargylic Epoxides: Dual Role of the Copper Catalyst. Chem. - Eur. J. 2017, 23, 17478–17481. 10.1002/chem.201705019. [DOI] [PubMed] [Google Scholar]
- Ziyanak F.; Kus M.; Alkan-Karadeniz L.; Artok L. Palladium-Catalysed Reactions of Conjugated Enyne Oxiranes with Organoborons: A Diastereoselective Method of the Synthesis of 2,4,5-Trienol Derivatives. Tetrahedron 2018, 74, 3652–3662. 10.1016/j.tet.2018.05.030. [DOI] [Google Scholar]
- Domin S.; Kedzierski J.; Zambroń B. K. Remote 1,5-Stereoselectivity Control by an N-Ligand Switch in the Pd(0)/InI-Promoted Reactions of 4-Ethynyl-β-lactams with Aldehydes. Org. Lett. 2019, 21, 3904–3908. 10.1021/acs.orglett.9b00891. [DOI] [PubMed] [Google Scholar]
- Jiang X.; Fu C.; Ma S. Highly Stereoselective Iodolactonization of 4,5-Allenoic Acids—An Efficient Synthesis of 5-(1’-Iodo-1’(Z)-alkenyl)-4,5-dihydro-2(3H)-furanones. Chem. - Eur. J. 2008, 14, 9656–9664. 10.1002/chem.200801363. [DOI] [PubMed] [Google Scholar]
- Jiang X.; Fu C.; Ma S. A Concise Synthesis of (−)- and (+)-trans-Whisky Lactones. Eur. J. Org. Chem. 2010, 2010, 687–693. 10.1002/ejoc.200901058. [DOI] [Google Scholar]
- Lü B.; Jiang X.; Fu C.; Ma S. Highly Regio- and Stereoselective Cyclic Iodoetherification of 4,5-Alkadienols. An Efficient Preparation of 2-(1′(Z)-Iodoalkenyl)tetrahydrofurans. J. Org. Chem. 2009, 74, 438–441. 10.1021/jo802079b. [DOI] [PubMed] [Google Scholar]
- Zheng W.-F.; Zhang W.; Huang C.; Wu P.; Qian H.; Wang L.; Guo Y.-L.; Ma S. Tetrasubstituted Allenes via the Palladium-Catalysed Kinetic Resolution of Propargylic Alcohols using a Supporting Ligand. Nat. Catal. 2019, 2, 997–1005. 10.1038/s41929-019-0346-z. [DOI] [Google Scholar]
- Yin S. H.; Ma S.; Jiao N.; Tao F.-G. Dramatic Solvent Effect in the Reduction of 2,3-Allenoic Acid Esters. A Simple Synthesis of 2,3-Allenols from 2,3-Allenoates. Chin. J. Chem. 2002, 20, 707–710. 10.1002/cjoc.20020200717. [DOI] [Google Scholar]
- Liu Z.; Gu P.; Shi M. Asymmetric Addition of Arylboronic Acids to Cumulene Derivatives Catalyzed by Axially Chiral N-Heterocyclic Carbene–Pd2+ Complexes. Chem. - Eur. J. 2011, 17, 5796–5799. 10.1002/chem.201100703. [DOI] [PubMed] [Google Scholar]
- Chen B.; Lu Z.; Chai G.; Fu C.; Ma S. An Efficient Double 1,2-Addition Reaction of 2,3-Allenoates with Allyl Magnesium Chloride. J. Org. Chem. 2008, 73, 9486–9489. 10.1021/jo801809j. [DOI] [PubMed] [Google Scholar]
- Griebenow N.; Cumine F.; Dilmac A. M. Investigations on the [2,3]-Wittig Rearrangement of Chiral 4-Aminopropargyloxy Acetates to Novel α-Hydroxy-γ-amino Acids Containing an Allene Unit. Synlett 2015, 26, 1702–1704. 10.1055/s-0034-1380740. [DOI] [Google Scholar]
- Rodríguez R. I.; Ramírez E.; Fernández-Salas J. A.; Sánchez-Obregón R.; Yuste F.; Alemán J. Asymmetric [2,3]-Wittig Rearrangement: Synthesis of Homoallylic, Allenylic, and Enynyl α-Benzyl Alcohols. Org. Lett. 2018, 20, 8047–8051. 10.1021/acs.orglett.8b03659. [DOI] [PubMed] [Google Scholar]
- Zamani F.; Pyne S. G.; Hyland C. J. T. Oxazolidinones and 2,5-Dihydrofurans via Zinc-Catalyzed Regioselective Allenylation Reactions of L-α-Amino Aldehydes. J. Org. Chem. 2017, 82, 6819–6830. 10.1021/acs.joc.7b00969. [DOI] [PubMed] [Google Scholar]
- Hernandez E.; Soderquist J. A. Nonracemic α-Allenyl Carbinols from Asymmetric Propargylation with the 10-Trimethylsilyl-9-borabicyclo[3.3.2]decanes. Org. Lett. 2005, 24, 5397–5400. 10.1021/ol051886k. [DOI] [PubMed] [Google Scholar]
- Barluenga J.; Suero M. G.; De la Campa R.; Flórez J. Enantioselective Synthesis of 4-Hydroxy-2-cyclohexenones through a Multicomponent Cyclization. Angew. Chem., Int. Ed. 2010, 49, 9720–9724. 10.1002/anie.201004413. [DOI] [PubMed] [Google Scholar]
- Carballeira J. D.; Krumlinde P.; Bocola M.; Vogel A.; Reetz M. T.; Bäckvall J.-E. Directed Evolution and Axial Chirality: Optimization of the Enantioselectivity of Pseudomonas Aeruginosa Lipase towards the Kinetic Resolution of a Racemic Allene. Chem. Commun. 2007, 19, 1913–1915. 10.1039/B700849J. [DOI] [PubMed] [Google Scholar]
- Deska J.; Bäckvall J.-E. Enzymatic Kinetic Resolution of Primary Allenic Alcohols. Application to the Total Synthesis and Stereochemical Assignment of Striatisporolide A. Org. Biomol. Chem. 2009, 7, 3379–3381. 10.1039/b912128p. [DOI] [PubMed] [Google Scholar]
- Horváth A.; Bäckvall J.-E. Catalyzed Racemization of Allenes. Chem. Commun. 2004, 964–965. 10.1039/B316482A. [DOI] [PubMed] [Google Scholar]
- For a review including enzymatic approaches to chiral allenols, see:; Skrobo B.; Rolfes J. D.; Deska J. Enzymatic Approaches for the Preparation of Optically Active Non-Centrochiral Compounds. Tetrahedron 2016, 72, 1257–1275. 10.1016/j.tet.2016.01.026. [DOI] [Google Scholar]
- Deska J.; del Pozo Ochoa C.; Bäckvall J.-E. Chemoenzymatic Dynamic Kinetic Resolution of Axially Chiral Allenes. Chem. - Eur. J. 2010, 16, 4447–4451. 10.1002/chem.201000301. [DOI] [PubMed] [Google Scholar]
- Manzuna Sapu C.; Backvall J.-E.; Deska J. Enantioselective Enzymatic Desymmetrization of Prochiral Allenic Diols. Angew. Chem., Int. Ed. 2011, 50, 9731–9734. 10.1002/anie.201103227. [DOI] [PubMed] [Google Scholar]
- Xu D.; Li Z.; Ma S. Efficient Preparation of Highly Optically Active (S)-(−)-2,3-Allenols and (R)-(+)-2,3-Allenyl Acetates by a Clean Novozym-435-Catalyzed Enzymatic Separation of Racemic 2,3-Allenols. Chem. - Eur. J. 2002, 8, 5012–5018. . [DOI] [PubMed] [Google Scholar]
- Li W.; Lin Z.; Chen L.; Tian X.; Wang Y.; Huang S.-H.; Hong R. Highly Stereoselective Kinetic Resolution of α-Allenic Alcohols: An Enzymatic Approach. Tetrahedron Lett. 2016, 57, 603–606. 10.1016/j.tetlet.2015.12.098. [DOI] [Google Scholar]
- Wang Y.; Zheng K.; Hong R. Chiral Silver Phosphate-Catalyzed Cycloisomeric Kinetic Resolution of α-Allenic Alcohols. J. Am. Chem. Soc. 2012, 134, 4096–4099. 10.1021/ja300453u. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Hoen R.; Hong R. Development of a Nonenzymatic Kinetic Resolution of α-Allenic Alcohols. Synlett 2012, 23, 2729–2734. 10.1055/s-0032-1317566. [DOI] [Google Scholar]
- Ye J.; Li S.; Chen B.; Fan W.; Kuang J.; Liu J.; Liu Y.; Miao B.; Wan B.; Wang Y.; et al. Catalytic Asymmetric Synthesis of Optically Active Allenes from Terminal Alkynes. Org. Lett. 2012, 14, 1346. 10.1021/ol300296k. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Ye J.; Ma S. Harmony of CdI2 with CuBr for the One-Pot Synthesis of Optically Active α-Allenols. Org. Biomol. Chem. 2015, 13, 4080–4089. 10.1039/C4OB02673J. [DOI] [PubMed] [Google Scholar]
- Liu Q.; Cao T.; Han Y.; Jiang X.; Tang Y.; Zhai Y.; Ma S. Copper(I) Iodide-Catalyzed Asymmetric Synthesis of Optically Active Tertiary α-Allenols. Synlett 2019, 30, 477–482. 10.1055/s-0037-1611641. [DOI] [Google Scholar]
- Zhong F.; Xue Q.-Y.; Yin L. Construction of Chiral 2,3-Allenols through Copper(I)-Catalyzed Asymmetric Direct Alkynylogous Aldol Reaction. Angew. Chem., Int. Ed. 2020, 59, 1562–1566. 10.1002/anie.201912140. [DOI] [PubMed] [Google Scholar]
- For a related recent example of asymmetric synthesis of allenols through copper-catalyzed aldol-type reaction, see:; Kondo Y.; Nagao K.; Ohmiya H. Reductive Umpolung for Asymmetric Synthesis of Chiral α-Allenic Alcohols. Chem. Commun. 2020, 56, 7471–7474. 10.1039/D0CC02619K. [DOI] [PubMed] [Google Scholar]
- Wang G.; Liu X.; Chen Y.; Yang J.; Li J.; Lin L.; Feng X. Diastereoselective and Enantioselective Alleno-aldol Reaction of Allenoates with Isatins to Synthesis of Carbinol Allenoates Catalyzed by Gold. ACS Catal. 2016, 6, 2482–2486. 10.1021/acscatal.6b00294. [DOI] [Google Scholar]
- Trost B. M.; Schultz J. E.; Chang T.; Maduabum M. R. Chemo-, Regio-, Diastereo-, and Enantioselective Palladium Allylic Alkylation of 1,3-Dioxaboroles as Synthetic Equivalents of α-Hydroxyketones. J. Am. Chem. Soc. 2019, 141, 9521–9526. 10.1021/jacs.9b04658. [DOI] [PubMed] [Google Scholar]
- Liao Y.; Yin X.; Wang X.; Yu W.; Fang D.; Hu L.; Wang M.; et al. Enantioselective Synthesis of Multisubstituted Allenes by Cooperative Cu/Pd-Catalyzed 1,4-Arylboration of 1,3-Enynes. Angew. Chem., Int. Ed. 2020, 59, 1176–1180. 10.1002/anie.201912703. [DOI] [PubMed] [Google Scholar]
- Yang C.; Liu Z.-L.; Dai D.-T.; Li Q.; Ma W.-W.; Zhao M.; Xu Y.-H. Catalytic Asymmetric Conjugate Protosilylation and Protoborylation of 2-Trifluoromethyl Enynes for Synthesis of Functionalized Allenes. Org. Lett. 2020, 22, 1360–1367. 10.1021/acs.orglett.9b04647. [DOI] [PubMed] [Google Scholar]
- Bayeh-Romero L.; Buchwald S. L. Copper Hydride Catalyzed Enantioselective Synthesis of Axially Chiral 1,3-Disubstituted Allenes. J. Am. Chem. Soc. 2019, 141, 13788–13794. 10.1021/jacs.9b07582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aparece M. D.; Hu W.; Morken J. P. Catalytic Enantioselective Synthesis of β-Allenyl Boronic Esters by Conjunctive Cross-Coupling with Propargylic Carbonates. ACS Catal. 2019, 9, 11381–11385. 10.1021/acscatal.9b04453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J. T.; Yun J. Asymmetric Synthesis of α-Chiral β-Hydroxy Allenes: Copper-Catalyzed C-Selective Borylative Coupling of Vinyl Arenes and Propargyl Phosphates. Chem. Commun. 2019, 55, 9813–9816. 10.1039/C9CC04165F. [DOI] [PubMed] [Google Scholar]
- Li Z.; Boyarskikh V.; Hansen J. H.; Autschbach J.; Musaev D. J.; Davies H. M. L. Scope and Mechanistic Analysis of the Enantioselective Synthesis of Allenes by Rhodium-Catalyzed Tandem Ylide Formation/[2,3]-Sigmatropic Rearrangement between Donor/Acceptor Carbenoids and Propargylic Alcohols. J. Am. Chem. Soc. 2012, 134, 15497–15504. 10.1021/ja3061529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X.; Zhang J.; Dong S.; Lin L.; Lin X.; Liu X.; Feng X. Nickel(II)-Catalyzed Asymmetric Propargyl [2,3] Wittig Rearrangement of Oxindole Derivatives: A Chiral Amplification Effect. Angew. Chem., Int. Ed. 2018, 57, 8734–8738. 10.1002/anie.201804080. [DOI] [PubMed] [Google Scholar]
- Teng S.; Jiao J.; Chi Y. R.; Zhou J. S. Asymmetric Wacker-Type Oxyallenylation and Azaallenylation of Cyclic Alkenes. Angew. Chem., Int. Ed. 2020, 59, 2246–2250. 10.1002/anie.201911961. [DOI] [PubMed] [Google Scholar]
- Durán-Galván M.; Connell B. T. Asymmetric Synthesis of (1,3-Butadien-2-yl)methanols from Aldehydes via [1-(Silylmethyl)allenyl]methanols. Eur. J. Org. Chem. 2010, 2010, 2445–2448. 10.1002/ejoc.201000199. [DOI] [Google Scholar]
- Lee K. A.; Silverio D. L.; Torker S.; Robbins D. W.; Haeffner F.; van der Mei F. W.; Hoveyda A. H. Catalytic Enantioselective Addition of Organoboron Reagents to Fluoroketones Controlled by Electrostatic Interactions. Nat. Chem. 2016, 8, 768–777. 10.1038/nchem.2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M.; Khan S.; Miliordos E.; Chen M. Enantioselective Allenylation of Aldehydes via Brønsted Acid Catalysis. Adv. Synth. Catal. 2018, 360, 4634–4639. 10.1002/adsc.201801080. [DOI] [Google Scholar]
- Jiang Y.; Diagne A. B.; Thomson R. J.; Schaus S. E. Enantioselective Synthesis of Allenes by Catalytic Traceless Petasis Reactions. J. Am. Chem. Soc. 2017, 139, 1998–2005. 10.1021/jacs.6b11937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tap A.; Blond A.; Wakchaure V. N.; List B. Chiral Allenes via Alkynylogous Mukaiyama Aldol Reaction. Angew. Chem., Int. Ed. 2016, 55, 8962–8965. 10.1002/anie.201603649. [DOI] [PubMed] [Google Scholar]
- Okamoto S.; Sato H.; Sato F. Highly Efficient Synthesis of Alka-1,3-dien-2-yltitanium Compounds from Alka-2,3-dienyl Carbonates. A New, Practical Synthesis of 1,3-Dienes and 2-Iodo-1,3-dienes. Tetrahedron Lett. 1996, 37, 8865–8868. 10.1016/S0040-4039(96)02126-0. [DOI] [Google Scholar]
- Horváth A.; Bäckvall J.-E. Palladium(II)-Catalyzed SN2′ Reactions of α-Allenic Acetates. Stereoconvergent Synthesis of (Z,E)-2-Bromo-1,3-dienes. J. Org. Chem. 2001, 66, 8120–8126. 10.1021/jo015950x. [DOI] [PubMed] [Google Scholar]
- Ma S.; Gu Z. PdCl2-Catalyzed Two-Component Cross-Coupling Cyclization of 2,3-Allenoic Acids with 2,3-Allenols. An Efficient Synthesis of 4-(1’,3′-Dien-2′-yl)-2(5H)-furanone Derivatives. J. Am. Chem. Soc. 2005, 127, 6182–6183. 10.1021/ja0500815. [DOI] [PubMed] [Google Scholar]
- Schneekloth J. S. Jr.; Pucheault M.; Crews C. M. Construction of Highly Substituted Stereodefined Dienes by Cross-Coupling of α-Allenic Acetates. Eur. J. Org. Chem. 2007, 2007, 40–43. 10.1002/ejoc.200600721. [DOI] [Google Scholar]
- Shimp H. L.; Micalizio G. C. An Alkoxide-Directed Alkyne–Allene Cross-Coupling for Stereoselective Synthesis of 1,4-Dienes. Chem. Commun. 2007, 4531–4533. 10.1039/b708256h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzas A. K.; Istrate F. M.; Gagosz F. Gold(I)-Catalyzed Isomerization of Allenyl Carbinol Esters: An Efficient Access to Functionalized 1,3-Butadien-2-ol Esters. Org. Lett. 2007, 9, 985–988. 10.1021/ol063031t. [DOI] [PubMed] [Google Scholar]
- Deng Y.; Jin X.; Ma S. Studies on Highly Stereoselective Addition-Elimination Reactions of 3-(Methoxycarbonyl)-1,2-allen-4-ols with MX. An Efficient Synthesis of 3-(Methoxycarbonyl)-2-halo-1,3(Z)-dienes. J. Org. Chem. 2007, 72, 5901–5904. 10.1021/jo070725m. [DOI] [PubMed] [Google Scholar]
- McLaughlin M.; Shimp H. L.; Navarro R.; Micalizio G. C. Regio- and Stereoselective Direct Cross-Coupling of Aromatic Imines with Allenic Alcohols. Synlett 2008, 2008, 735–738. 10.1055/s-2008-1042808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlan A. U.; Micalizio G. C. The Regio- and Stereochemical Course of Reductive Cross-Coupling Reactions between 1,3-Disubstituted Allenes and Vinylsilanes: Synthesis of (Z)-Dienes. Tetrahedron 2010, 66, 4775–4783. 10.1016/j.tet.2010.02.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eom D.; Kim S. H.; Lee P. H. Efficient Synthetic Method of Z-Selective 2-Halo-1,3-dienes from Reactions of Allenols Possessing Ethoxycarbonyl and Vinyl Group with Indium Trihalide. Bull. Korean Chem. Soc. 2010, 31, 645–649. 10.5012/bkcs.2010.31.03.645. [DOI] [Google Scholar]
- Zhang X.; Fu C.; Ma S. Highly Stereoselective Facile Synthesis of 2-Acetoxy-1,3(E)-alkadienes via a Rh(I)-Catalyzed Isomerization of 2,3-Allenyl Carboxylates. Org. Lett. 2011, 13, 1920–1923. 10.1021/ol200198z. [DOI] [PubMed] [Google Scholar]
- Wang H.; Beiring B.; Yu D.-G.; Collins K. D.; Glorius F. [3]Dendralene Synthesis: Rhodium(III)-Catalyzed Alkenyl C–H Activation and Coupling Reaction with Allenyl Carbinol Carbonate. Angew. Chem., Int. Ed. 2013, 52, 12430–12434. 10.1002/anie.201306754. [DOI] [PubMed] [Google Scholar]
- Liu T.; Dong J.; Cao S.-J.; Guo L.-C.; Wu L. Suzuki–Miyaura Coupling of Phosphinoyl-α-Allenic Alcohols with Arylboronic Acids Catalyzed by a Palladium Complex “on Water”: An Efficient Method to Generate Phosphinoyl 1,3-Butadienes and Derivatives. RSC Adv. 2014, 4, 61722–61726. 10.1039/C4RA12251H. [DOI] [Google Scholar]
- Webster S.; Young P. C.; Barker G.; Rosair G. M.; Lee A.-L. Dehydrative Thiolation of Allenols: Indium vs Gold Catalysis. J. Org. Chem. 2015, 80, 1703–1718. 10.1021/jo502648w. [DOI] [PubMed] [Google Scholar]
- Lan Y.; Hammond G. B. Functionalization of Monofluoroallene and the Synthesis of Aryl-Substituted Conjugated Fluorodienes. Org. Lett. 2002, 4, 2437–2439. 10.1021/ol026196k. [DOI] [PubMed] [Google Scholar]
- Yoshida M.; Gotou T.; Ihara M. Palladium-catalysed coupling reaction of allenic alcohols with aryl- and alkenylboronic acids. Chem. Commun. 2004, 1124–1125. 10.1039/b402047b. [DOI] [PubMed] [Google Scholar]
- Shimizu I.; Sugiura T.; Tsuji J. Facile Synthesis of ß-Aryl- or ß-Alkenyl-ß-methyl-α,ß-unsaturated Carbonyl Compounds by Palladium-Catalyzed Reaction of l,2-Dien-4-ols with Aryl or Alkenyl Halides. J. Org. Chem. 1985, 50, 537–539. 10.1021/jo00204a024. [DOI] [Google Scholar]
- Choi S. Y.; Chung Y. K. N-Heterocyclic Carbene (NHC)-Rhodium-Catalyzed Carbonylative C–C Bond Formation of Allenols with Arylboronic Acids under Carbon Monoxide. Adv. Synth. Catal. 2011, 353, 2609–2613. 10.1002/adsc.201100309. [DOI] [Google Scholar]
- Fan C.; Cazes B. Nucleophilic Substitution of α-Allenic Alcohols via the Murahashi Method: 1,3-Dienes Synthesis. Tetrahedron Lett. 1988, 29, 1701–1704. 10.1016/S0040-4039(00)82022-5. [DOI] [Google Scholar]
- Piotti M. E.; Alper H. Regioselective and in Some Cases Stereoselective Carbonylation of α-Allenic Alcohols to α-Vinylacrylic Acids Catalyzed by a Cationic Palladium Complex. J. Org. Chem. 1994, 59, 1956–1957. 10.1021/jo00087a002. [DOI] [Google Scholar]
- Ma S.; Wang G. Unexpected SN2′-Type Addition–Elimination Reactions of 1-Aryl-2,3-allenols with LiX. Synthesis and Synthetic Application of 1-Aryl-3-halo-1,3-dienes. Tetrahedron Lett. 2002, 43, 5723–5726. 10.1016/S0040-4039(02)01207-8. [DOI] [Google Scholar]
- Wang B.; Li Y.; Pang J. H.; Watanabe K.; Takita R.; Chiba S. Hydromagnesiation of 1,3-Enynes by Magnesium Hydride for Synthesis of Tri- and Tetra-substituted Allenes. Angew. Chem., Int. Ed. 2021, 60, 217–221. 10.1002/anie.202012027. [DOI] [PubMed] [Google Scholar]
- Shimp H. L.; Hare A.; McLaughlin M.; Micalizio G. C. Allene–Alkyne Cross-Coupling for Stereoselective Synthesis of Substituted 1,4-Dienes and Cross-Conjugated Trienes. Tetrahedron 2008, 64, 6831–6837. 10.1016/j.tet.2008.03.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazmaier U.; Lucas S.; Klein M. Syntheses and Synthetic Applications of Stannylated Allylic Alcohols. J. Org. Chem. 2006, 71, 2429–2433. 10.1021/jo052611l. [DOI] [PubMed] [Google Scholar]
- Alcaide B.; Almendros P.; Aragoncillo C.; Redondo M. C. Stereoselective Synthesis of 1,2,3-Trisubstituted 1,3-Dienes through Novel [3,3]-Sigmatropic Rearrangements in α-Allenic Methanesulfonates: Application to the Preparation of Fused Tricyclic Systems by Tandem Rearrangement/Diels-Alder Reaction. Eur. J. Org. Chem. 2005, 2005, 98–106. 10.1002/ejoc.200400527. [DOI] [Google Scholar]
- Alcaide B.; Almendros P.; Aragoncillo C.; Fernández I.; Gómez-Campillos G. Metal-Free Allene-Based Synthesis of Enantiopure Fused Polycyclic Sultones. Chem. - Eur. J. 2016, 22, 285–294. 10.1002/chem.201504045. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Wang Y.; Gu Z.; Wang Z. [3,3]-Sigmatropic Rearrangement of Allenic Alcohols: Stereoselective Synthesis of 1,3-Diene-2-ol Sulfonates. Org. Biomol. Chem. 2017, 15, 4014–4021. 10.1039/C7OB00578D. [DOI] [PubMed] [Google Scholar]
- Sabbasani V. R.; Mamidipalli P.; Lu H.; Xia Y.; Lee D. Subtle Electronic Effects in Metal-Free Rearrangement of Allenic Alcohols. Org. Lett. 2013, 15, 1552–1555. 10.1021/ol400331r. [DOI] [PubMed] [Google Scholar]
- Wu L.; Huang H.; Liang Y.; Cheng P. Regio- and Stereoselective Synthesis of 2-Amino-dienes via Decarboxylative Amination of 4-(Ethoxycarbonyl)-2,3-allenols by TsNCO. J. Org. Chem. 2014, 79, 11264–11269. 10.1021/jo502098b. [DOI] [PubMed] [Google Scholar]
- Matsumoto K.; Mizushina N.; Yoshida M.; Shindo M. Stereocontrolled Synthesis of Multisubstituted 1,3-Dienes via an Allene–Claisen Rearrangement. Synlett 2017, 28, 2340–2344. 10.1055/s-0036-1590970. [DOI] [Google Scholar]
- Rinaolo V. J.; Robinson E. E.; Diagne A. B.; Schaus S. B.; Thomson R. J. Diene Synthesis by the Reductive Transposition of 1,2-Allenols. Synlett 2019, 30, 2073–2076. 10.1055/s-0039-1690692. [DOI] [Google Scholar]
- Liu T.; Xia Y.-T.; Zhu J.; Lu A.-M.; Wu L. Metal-free Synthesis of Chlorinated and Brominated Phosphinoyl 1,3-Butadiene Derivatives and its Synthetic Applications. Tetrahedron Lett. 2015, 56, 6508–6512. 10.1016/j.tetlet.2015.10.029. [DOI] [Google Scholar]
- For a recently published synthesis of phosphinoylbutadienes through an alternative procedure, see:; Hu S.; Sun W.; Chen J.; Li S.; Zhao R.; Xu P.; Gao Y.; Zhao Y. Palladium-Catalyzed C–P Cross-Coupling of Allenic Alcohols with H-Phosphonates Leading to 2-Phosphinoyl-1,3-butadienes. Chem. Commun. 2021, 57, 339–342. 10.1039/D0CC07022J. [DOI] [PubMed] [Google Scholar]
- Ni C.; Zhang Y.; Hou Y.; Tong X. Access to Thiopyrano[2,3-b]indole via Tertiary Amine-catalyzed Formal (3 + 3) Annulations of β’-Acetoxy Allenoates with Indoline-2-thiones. Chem. Commun. 2017, 53, 2567–2570. 10.1039/C6CC09788J. [DOI] [PubMed] [Google Scholar]
- For DABCO-promoted isomerization of allenols to enones, see:; Dong W.; Hu P.; Hu J.; Tong X. Lewis Base-Catalyzed Divergent Isomerizations of 5-Hydroxyl-2,3-dienoate. Tetrahedron Lett. 2014, 55, 1682–1685. 10.1016/j.tetlet.2013.10.059. [DOI] [Google Scholar]
- Shukla R. K.; Nair A. M.; Khan S.; Volla C. M. R. Cobalt-Catalyzed C8-Dienylation of Quinoline-N-Oxides. Angew. Chem., Int. Ed. 2020, 59, 17042–17048. 10.1002/anie.202003216. [DOI] [PubMed] [Google Scholar]
- Alcaide B.; Almendros P.; Luna A.; Prieto N. Direct FeX3-Based Stereocontrolled Access to (Z)-3-Alkenyl-oxindoles from Allenols. J. Org. Chem. 2012, 77, 11388–11392. 10.1021/jo302296r. [DOI] [PubMed] [Google Scholar]
- Lin M.-H.; Li Y.-H.; Kuo C.-K.; Chen C.-H.; Huang Y.-C.; Liang K.-Y.; Chen Y.-C.; Tsai C.-H.; Chuang T.-H. Synthesis of Hexahydro-1H-isoindole Derivatives from Arylacyl Bromides via Homoallenic Bromohydrins. J. Org. Chem. 2015, 80, 2462–2466. 10.1021/acs.joc.5b00072. [DOI] [PubMed] [Google Scholar]
- Shen R.; Yang J.; Zhang M.; Han L.-B. Silver-Catalyzed Atom-Economic Hydrophosphorylation of Propargyl Epoxides: An Access to 4-Phosphoryl 2,3-Allenols and Stereodefined 1-Phosphoryl 1,3-Dienes. Adv. Synth. Catal. 2017, 359, 3626–3637. 10.1002/adsc.201700421. [DOI] [Google Scholar]
- Yoshida M.; Matsuda K.; Shoji Y.; Gotou T.; Ihara M.; Shishido K. Regiocontrolled Addition of Arylboronic Acids to Allenes Using Palladium and Platinum Catalysts. Org. Lett. 2008, 22, 5183–5185. 10.1021/ol8021484. [DOI] [PubMed] [Google Scholar]
- Miura T.; Shimizu H.; Igarashi T.; Murakami M. Stereoselective Synthesis of Vinyl-substituted (Z)-Stilbenes by Rhodium-catalysed Addition of Arylboronic Acids to Allenic Alcohols. Org. Biomol. Chem. 2010, 8, 4074–4076. 10.1039/c0ob00163e. [DOI] [PubMed] [Google Scholar]
- Chen D.; Chen X.; Lu Z.; Cai H.; Shen J.; Zhu G. Palladium-Catalyzed Dienylation of Haloalkynes using 2,3-Butadienyl Acetates: A Facile Access to (1Z)-1,2-Dihalo-3-vinyl-1,3-dienes. Adv. Synth. Catal. 2011, 353, 1474–1478. 10.1002/adsc.201100113. [DOI] [Google Scholar]
- Alcaide B.; Almendros P.; Cembellín S.; Martínez del Campo T. Acid-Catalyzed Synthesis of α,β-Disubstituted Conjugated Enones by a Meyer–Schuster-Type Rearrangement in Allenols. Adv. Synth. Catal. 2015, 357, 1070–1078. 10.1002/adsc.201400928. [DOI] [Google Scholar]
- Zhao R.; Huang X.; Wang M.; Hu S.; Gao Y.; Xu P.; Zhao Y. TfOH-Catalyzed Phosphinylation of 2,3-Allenols into γ-Ketophosphine Oxides. J. Org. Chem. 2020, 85, 8185–8195. 10.1021/acs.joc.0c00328. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Tracy J. S. Contemporaneous Dual Catalysis: Aldol Products from Non-Carbonyl Substrates. Chem. - Eur. J. 2015, 21, 15108–15112. 10.1002/chem.201502973. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Tracy J. S. Carbon–Nitrogen Bond Formation via the Vanadium Oxo Catalyzed Sigmatropic Functionalization of Allenols. Org. Lett. 2017, 19, 2630–2633. 10.1021/acs.orglett.7b00961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. B.; Newman J. J.; Taylor D. R. Ene Reactions of Allenes. Part 5. Regio- and Stereo-Selective Ene Reactions of a Trialkylallene. J. Chem. Soc., Perkin Trans. 1 1978, 10, 1161–1168. 10.1039/p19780001161. [DOI] [Google Scholar]
- Sabbasani V. R.; Huang G.; Xia Y.; Lee D. Facile Alder-Ene Reactions of Silylallenes Involving an Allenic C(sp2)-H Bond. Chem. - Eur. J. 2015, 21, 17210–17214. 10.1002/chem.201503243. [DOI] [PubMed] [Google Scholar]
- Trost B. M.; Tracy J. S.; Yusoontorn T. Vanadium-Catalyzed Coupling of Allenols with Electrophilic Halide Sources for the Formation of α-Halo-α′,β′-unsaturated Ketones. Org. Lett. 2019, 21, 1207–1211. 10.1021/acs.orglett.9b00195. [DOI] [PubMed] [Google Scholar]
- Ghotekar G. S.; Shirsath S. R.; Shaikh A. C.; Muthukrishnan M. 1,6-Conjugate Addition Initiated Formal [4 + 2] Annulation of p-Quinone Methides with Sulfonyl Allenols: A Unique Access to Spiro[5.5]undeca-1,4-dien-3-one Scaffolds. Chem. Commun. 2020, 56, 5022–5025. 10.1039/D0CC01005G. [DOI] [PubMed] [Google Scholar]
- Lázaro-Milla C.; Macicior J.; Yanai H.; Almendros P. Trifluorosulfonylation Cascade in Allenols: Stereocontrolled Synthesis of Bis(triflyl)enones. Chem. - Eur. J. 2020, 23, 8983. 10.1002/chem.202001236. [DOI] [PubMed] [Google Scholar]
- Alcaide B.; Almendros P.; Herrera F.; Luna A.; de Orbe M. E.; Torres M. R. Gallium-Catalyzed Domino Arylation/Oxycyclization of Allenes with Phenols. J. Org. Chem. 2015, 80, 4157–4163. 10.1021/acs.joc.5b00106. [DOI] [PubMed] [Google Scholar]
- Deng Y.; Jin X.; Fu C.; Ma S. Efficient Highly Selective Synthesis of Methyl 2-(Ethynyl)alk-2(E)-enoates and 2-(1′-Chlorovinyl)alk-2(Z)-enoates from 2-(Methoxycarbonyl)-2,3-allenols. Org. Lett. 2009, 11, 2169–2172. 10.1021/ol9004273. [DOI] [PubMed] [Google Scholar]
- Alcaide B.; Almendros P.; Martínez del Campo T. Direct Allenol-Based Stereocontrolled Access to Substituted (E)-1,3-Enynes. Org. Biomol. Chem. 2012, 10, 7603–7609. 10.1039/c2ob26085a. [DOI] [PubMed] [Google Scholar]
- Alcaide B.; Almendros P.; Martínez del Campo T.; Torres M. R. Synthesis of Fused-β-Lactams through Selective Gold-Catalyzed Oxycyclization of Dioxolane-Tethered Enynes. J. Org. Chem. 2013, 78, 8956–8965. 10.1021/jo401390k. [DOI] [PubMed] [Google Scholar]
- Alcaide B.; Almendros P.; Martínez del Campo T.; Naranjo T. Gold-Catalyzed Preparation of Anellated 2-Azetidinones via Divergent Heterocyclization of Enyne-Tethered Oxazolidines. Org. Chem. Front. 2018, 5, 817–821. 10.1039/C7QO00950J. [DOI] [Google Scholar]
- Asai S.; Kato M.; Monguchi Y.; Sajiki H.; Sawama Y. Phosphate-Mediated Enyne Synthesis from Allenols. ChemistrySelect 2017, 2, 876–878. 10.1002/slct.201601789. [DOI] [Google Scholar]
- Choe Y.; Lee P. H. Stereoselective DABCO-Catalyzed Synthesis of (E)-α-Ethynyl-α,β-Unsaturated Esters from Allenyl Acetates. Org. Lett. 2009, 11, 1445–1448. 10.1021/ol9001703. [DOI] [PubMed] [Google Scholar]
- Sai M.; Matsubara S. Copper-Catalyzed Direct and Stereoselective Synthesis of Conjugated Enynes from α-Allenols. Adv. Synth. Catal. 2019, 361, 39–43. 10.1002/adsc.201801211. [DOI] [Google Scholar]
- Xu H.; Zhang X.; He Y.; Guo S.; Fan X. Tandem Reaction of 3-Hydroxyhexa-4,5-Allenic Esters: A Novel Access to Diversely Substituted 2H-Pyran-2-ones and Indenes. Chem. Commun. 2012, 48, 2121–3123. 10.1039/c2cc30247k. [DOI] [PubMed] [Google Scholar]
- He Y.; Zhang X.-Y.; Cui L.-Y.; Fan X.-S. Iodine-Initiated Domino Reaction of Hepta-1,2-dien-6-yn-4-ols and Brønsted Acid Promoted Cyclization of Hepta-1,2,6-trien-4-ols Leading to Functionalized Benzenes. Chem. - Asian J. 2013, 8, 717–722. 10.1002/asia.201201151. [DOI] [PubMed] [Google Scholar]
- Ma J.; Peng L.; Zhang X.; Zhang Z.; Campbell M.; Wang J. InI3- or ZnI2-Catalyzed Reaction of Hydroxylated 1,5-Allenynes with Thiols: A New Access to 3,5-Disubstituted Toluene Derivatives. Chem. - Asian J. 2010, 5, 2214–2220. 10.1002/asia.201000267. [DOI] [PubMed] [Google Scholar]
- Guo B.; Huang X.; Fu C.; Ma S. Carbazoles from the [4C+2C] Reaction of 2,3-Allenols with Indoles. Chem. - Eur. J. 2016, 22, 18343–18348. 10.1002/chem.201604317. [DOI] [PubMed] [Google Scholar]
- Luna A.; Herrera F.; Higuera S.; Murillo A.; Fernández I.; Almendros P. AgNO3·SiO2: Convenient AgNPs Source for the Sustainable Hydrofunctionalization of Allenyl-Indoles using Heterogeneous Catalysis. J. Catal. 2020, 389, 432–439. 10.1016/j.jcat.2020.06.002. [DOI] [Google Scholar]
- Tsukamoto H.; Ito K.; Doi T. Synthesis of Multi-Substituted Dihydrofurans via Palladium-Catalysed Coupling Between 2,3-Alkadienols and Pronucleophiles. Chem. Commun. 2018, 54, 5102–5105. 10.1039/C8CC02589D. [DOI] [PubMed] [Google Scholar]
- Sai M.; Yorimitsu H.; Oshima K. Allyl-, Allenyl-, and Propargyl-Transfer Reactions through Cleavage of C-C Bonds Catalyzed by an N-Heterocyclic Carbene/Copper Complex: Synthesis of Multisubstituted Pyrroles. Angew. Chem., Int. Ed. 2011, 50, 3294–3298. 10.1002/anie.201100631. [DOI] [PubMed] [Google Scholar]
- For a palladium-mediated synthesis of N-hydroxyaminoallenes by allene transfer reaction from 2,3-butadienols and hydroxylamines, see:; Sharma P.; Liu R.-S. Cu-Catalyzed Aerobic Oxidative Cyclizations of 3-N-Hydroxyamino-1,2-propadienes with Alcohols, Thiols, and Amines to Form a -O-, S-, and N-Substituted 4-Methylquinoline Derivatives. Chem. - Eur. J. 2015, 21, 4590–4594. 10.1002/chem.201406317. [DOI] [PubMed] [Google Scholar]
- Kong W.; Fu C.; Ma S. An Efficient Synthesis of Carbazoles from PtCl2-Catalyzed Cyclization of 1-(Indol-2-yl)-2,3-allenols. Chem. Commun. 2009, 4572–4574. 10.1039/b909649c. [DOI] [PubMed] [Google Scholar]
- Barluenga J.; Piedrafita M.; Ballesteros A.; Suárez-Sobrino A. L.; González J. M. Gold-Catalyzed Annulations of 1-(2,3-Butadienyl)-1H-Indole Derivatives. Chem. - Eur. J. 2010, 16, 11827–11831. 10.1002/chem.201001754. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Liu C.; Kinder R. E.; Han X.; Quian H.; Widenhoefer R. A. Highly Active Au(I) Catalyst for the Intramolecular exo-Hydrofunctionalization of Allenes with Carbon, Nitrogen, and Oxygen Nucleophiles. J. Am. Chem. Soc. 2006, 128, 9066–9073. 10.1021/ja062045r. [DOI] [PubMed] [Google Scholar]
- Dai J.; Ma D.; Fu C.; Ma S. Gram Scale Total Synthesis of 2-Hydroxy-3-methylcarbazole, Pyrano[3,2-a]carbazole and Prenylcarbazole Alkaloids. Eur. J. Org. Chem. 2015, 2015, 5655–5662. 10.1002/ejoc.201500783. [DOI] [Google Scholar]
- Kong W.; Fu C.; Ma S. General Au-Catalyzed Benzannulation Towards Naturally Occurring Carbazole Alkaloids from Methoxypropadiene. Chem. - Eur. J. 2011, 17, 13134–13137. 10.1002/chem.201102251. [DOI] [PubMed] [Google Scholar]
- Alcaide B.; Almendros P.; Alonso J. M.; Quirós M. T.; Gadzinski P. Gold- or Palladium-Catalyzed Allene Carbocyclization/Functionalization: Simple and Efficient Synthesis of Carbazoles. Adv. Synth. Catal. 2011, 353, 1871–1876. 10.1002/adsc.201100209. [DOI] [Google Scholar]
- Alcaide B.; Almendros P.; Alonso J. M.; Fernández I. Palladium-Catalyzed Carbocyclization–Cross-Coupling Reactions of Two Different Allenic Moieties: Synthesis of 3-(Buta-1,3-dienyl) Carbazoles and Mechanistic Insights. Chem. Commun. 2012, 48, 6604–6606. 10.1039/c2cc32015k. [DOI] [PubMed] [Google Scholar]
- Alcaide B.; Almendros P.; Alonso J. M.; Cembellín S.; Fernández I.; Martínez del Campo T.; Torres M. R. Iodine Recycling via 1,3-Migration in Iodoindoles under Metal Catalysis. Chem. Commun. 2013, 49, 7779–7781. 10.1039/c3cc44073g. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Zhu L.; Shen K.; Yang H.; Hang X.-C.; Jiang G. Brønsted Acid-Catalyzed Aromatic Annulation of Alkoxyallenes with Naphthols: A Reaction Sequence to Larger π-Conjugated Naphthopyrans with Aggregation-Induced Emission Characters. Chem. Sci. 2019, 10, 1070–1074. 10.1039/C8SC03837F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a review on the synthetic utility of alkoxyallenes, including α-hydroxy-substituted alkoxyallenes, see:; Zimmer R.; Reissig H.-U. Alkoxyallenes as building blocks for organic synthesis. Chem. Soc. Rev. 2014, 43, 2888–2903. 10.1039/C3CS60429B. [DOI] [PubMed] [Google Scholar]
- Olsson L. I.; Claesson A. Allenes and acetylenes. Synthesis of 2,5-Dihydrofurans and 5,6-Dihydro-2H-pyrans by Silver(I)-Catalyzed Cyclization of Allenic Alcohols. Synthesis 1979, 9, 743–745. 10.1055/s-1979-28825. [DOI] [Google Scholar]
- Beaulieu P. L.; Morisset V. M.; Garratt D. G. The Synthesis of 2,5-Dihydrofurans from α-Allenic Alcohols. Tetrahedron Lett. 1980, 21, 129–132. 10.1016/S0040-4039(00)71393-1. [DOI] [Google Scholar]
- Gill G. B.; Idris M. S. H. Synthesis of α,β-Unsaturated Butenolides from Allenic Acids. Tetrahedron Lett. 1985, 26, 4811–4814. 10.1016/S0040-4039(00)94958-X. [DOI] [Google Scholar]
- Hoffmann-Roeder A.; Krause N. Gold(III) Chloride Catalyzed Cyclization of α-Hydroxyallenes to 2,5-Hihydrofurans. Org. Lett. 2001, 3, 2537–2538. 10.1021/ol016205+. [DOI] [PubMed] [Google Scholar]
- Alcaide B.; Almendros P.; Martínez del Campo T. Metal-Catalyzed Regiodivergent Cyclization of γ-Allenols: Tetrahydrofurans versus Oxepanes. Angew. Chem., Int. Ed. 2007, 46, 6684–6687. 10.1002/anie.200701611. [DOI] [PubMed] [Google Scholar]
- Yu X.; Seo S.; Marks T. J. Effective, Selective Hydroalkoxylation/Cyclization of Alkynyl and Allenyl Alcohols Mediated by Lanthanide Catalysts. J. Am. Chem. Soc. 2007, 129, 7244–7245. 10.1021/ja071707p. [DOI] [PubMed] [Google Scholar]
- Kim S.; Lee P. H. Cyclization of Allenyne-1,6-diols Catalyzed by Gold and Silver Salts: An Efficient Selective Synthesis of Dihydrofuran and Furan Derivatives. Adv. Synth. Catal. 2008, 350, 547–551. 10.1002/adsc.200700471. [DOI] [Google Scholar]
- Alcaide B.; Almendros P.; Martínez del Campo T.; Carrascosa R. Metal-Catalyzed Cycloetherification Reactions of β,γ- and γ, δ-Allendiols. Chemo-, Regio- and Stereocontrol in the Synthesis of Oxacycles. Chem. - Eur. J. 2010, 16, 13243–13252. 10.1002/chem.201001520. [DOI] [PubMed] [Google Scholar]
- Alcaide B.; Almendros P. Gold-Catalyzed Cyclization Reactions of Allenol and Alkynol Derivatives. Acc. Chem. Res. 2014, 47, 939–952. 10.1021/ar4002558. [DOI] [PubMed] [Google Scholar]
- Barreiro E. M.; Adrio L. A.; Hii K. K.; Brazie J. B. Coinage Metal Catalysts for the Addition of O–H to C = C Bonds. Eur. J. Org. Chem. 2013, 2013, 1027–1039. 10.1002/ejoc.201201441. [DOI] [Google Scholar]
- Alcaide B.; Almendros P.; Martínez del Campo T.. Metal-Mediated Synthesis of Nonaromatic Oxacycles from Allenols. In Advances in Transition-Metal Mediated Heterocyclic Synthesis; Solé D., Fernández I., Eds.; Elsevier: Oxford, UK, 2018; pp 1–31. [Google Scholar]
- Yoneda E.; Zhang S.-W.; Zhou D.-Y.; Onitsuka K.; Takahashi S. Ruthenium-Catalyzed Cyclocarbonylation of Allenyl Alcohols and Amines: Selective Synthesis of Lactones and Lactams. J. Org. Chem. 2003, 68, 8571–8576. 10.1021/jo0350615. [DOI] [PubMed] [Google Scholar]
- Alcaide B.; Almendros P.; Martínez del Campo T.; Fernández I. Fascinating Reactivity in Gold Catalysis: Synthesis of Oxetenes Through Rare 4-exo-dig Allene Cyclization and Infrequent β-Hydride Elimination. Chem. Commun. 2011, 47, 9054–9056. 10.1039/c1cc13212a. [DOI] [PubMed] [Google Scholar]
- Alcaide B.; Almendros P.; Fernández I.; Martínez del Campo T.; Naranjo T. Platinum-Catalyzed Divergent Reactivity of α-Hydroxyallenes: Synthesis of Dihydrofurans and α,β-Unsaturated Ketones. Adv. Synth. Catal. 2013, 355, 2681–2685. 10.1002/adsc.201300413. [DOI] [Google Scholar]
- Eom D.; Kang D.; Lee P. H. Synthesis of 2-Alkyl- and Aryl-3-ethoxycarbonyl-2,5-dihydrofurans through Gold-Catalyzed Intramolecular Hydroalkoxylation. J. Org. Chem. 2010, 75, 7447–7450. 10.1021/jo101474s. [DOI] [PubMed] [Google Scholar]
- Park C.; Lee P. H. Indium-Mediated Regio- and Chemoselective Synthesis of α-Hydroxyalkyl Allenic Esters and Gold-Catalyzed Cyclizations to Ethyl 2-Naphthoate Derivatives. Org. Lett. 2008, 10, 3359–3362. 10.1021/ol801196g. [DOI] [PubMed] [Google Scholar]
- Alcaide B.; Almendros P.; Alonso J. M.; Fernández I. Carbocyclization versus Oxycyclization on the Metal-Catalyzed Reactions of Oxyallenyl C3-Linked Indoles. J. Org. Chem. 2013, 78, 6688–6701. 10.1021/jo401013d. [DOI] [PubMed] [Google Scholar]
- Huang X.; Zhang L. AuCl-Catalyzed Synthesis of Benzyl-Protected Substituted Phenols: A Formal [3 + 3] Approach. Org. Lett. 2007, 9, 4627–4630. 10.1021/ol7021356. [DOI] [PubMed] [Google Scholar]
- Kuroda N.; Takahashi Y.; Yoshinaga K.; Mukai C. A Novel Generation of Indole-2,3-quinodimethanes. Org. Lett. 2006, 8, 1843–1845. 10.1021/ol060418n. [DOI] [PubMed] [Google Scholar]
- Pyo S.; Skowron J. F.; Cha J. K. An Improved Synthesis of Cyclopropanes from Homoallenic Alcohols. Tetrahedron Lett. 1992, 33, 4703–4706. 10.1016/S0040-4039(00)61263-7. [DOI] [Google Scholar]
- Alcaide B.; Almendros P.; Luna A.; Prieto N. Metal-catalyzed Rearrangements of 3-Allenyl 3-hydroxyindolin-2-ones in the Presence of Halogenated Reagents. Org. Biomol. Chem. 2013, 11, 1216–1225. 10.1039/c2ob27359d. [DOI] [PubMed] [Google Scholar]
- Jung M. S.; Kim W. S.; Shin Y. H.; Jin H. J.; Kim Y. S.; Kang E. J. Chemoselective Activities of Fe(III) Catalysts in the Hydrofunctionalization of Allenes. Org. Lett. 2012, 14, 6262–6265. 10.1021/ol303021d. [DOI] [PubMed] [Google Scholar]
- Sajna K. V.; Swamy K. C. K. Allenylphosphonates/Allenylphosphine Oxides as Intermediates/Precursors for Intramolecular Cyclization Leading to Phosphorus-Based Indenes, Indenones, Benzofurans, and Isochromenes. J. Org. Chem. 2012, 77, 5345–5356. 10.1021/jo300705f. [DOI] [PubMed] [Google Scholar]
- Alcaide B.; Almendros P.; Luna A.; Soriano E. An Alternative to Precious Metals: Hg(ClO4)2·3H2O as a Cheap and Water-Tolerant Catalyst for the Cycloisomerization of Allenols. J. Org. Chem. 2015, 80, 7050–7057. 10.1021/acs.joc.5b00887. [DOI] [PubMed] [Google Scholar]
- Casavant B. J.; Khoder Z. M.; Berhane I. A.; Chemler S. R. Copper(II)-Promoted Cyclization/Difunctionalization of Allenols and Allenylsulfonamides: Synthesis of Heterocycle-Functionalized Vinyl Carboxylate Esters. Org. Lett. 2015, 17, 5958–5961. 10.1021/acs.orglett.5b02833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Ready J. M. Cyclocondensation of Amino-propargyl Silanes. Org. Lett. 2012, 14, 2308–2311. 10.1021/ol300724c. [DOI] [PubMed] [Google Scholar]
- Tata R. R.; Harmata M. Silver-Catalyzed Cyclization of Sulfonyl Allenes to Dihydrofurans. Org. Lett. 2016, 18, 5684–5687. 10.1021/acs.orglett.6b02917. [DOI] [PubMed] [Google Scholar]
- Alcaide B.; Almendros P.; González A. M.; Luna A.; Martínez-Ramírez S. Palladium Nanoparticles in Water: A Reusable Catalytic System for the Cycloetherification or Benzannulation of α-Allenols. Adv. Synth. Catal. 2016, 358, 2000–2006. 10.1002/adsc.201501132. [DOI] [Google Scholar]
- Lempke L.; Sak H.; Kubicki M.; Krause N. Gold-Catalyzed Cycloisomerization of Trifluoromethylated Allenols: Sustainability and Mechanistic Studies. Org. Chem. Front. 2016, 3, 1514–1519. 10.1039/C6QO00423G. [DOI] [Google Scholar]
- Alcaide B.; Almendros P.; Quirós M. T.; Lázaro C.; Torres M. R. Three-Step Metal-Promoted Allene-Based Preparation of Bis(heterocyclic) Cyclophanes from Carbonyl Compounds. J. Org. Chem. 2014, 79, 6244–6255. 10.1021/jo500993x. [DOI] [PubMed] [Google Scholar]
- Tokimizu Y.; Oishi S.; Fujii N.; Ohno H. Gold-Catalyzed Cascade Cyclization of 2-Alkynyl-N-Propargylanilines by Rearrangement of a Propargyl Group. Angew. Chem., Int. Ed. 2015, 54, 7862–7866. 10.1002/anie.201502256. [DOI] [PubMed] [Google Scholar]
- Poonoth M.; Krause N. Cycloisomerization of Bifunctionalized Allenes: Synthesis of 3(2H)-Furanones in Water. J. Org. Chem. 2011, 76, 1934–1936. 10.1021/jo102416e. [DOI] [PubMed] [Google Scholar]
- Alcaide B.; Almendros P.; Cembellín S.; Martínez del Campo T.; Palop G. Allenols versus Allenones: Rhodium-Catalyzed Regiodivergent and Tunable Allene Reactivity with Triazoles. Chem. - Eur. J. 2017, 23, 13754–13759. 10.1002/chem.201702468. [DOI] [PubMed] [Google Scholar]
- Alcaide B.; Almendros P.; Fernández I.; Martínez del Campo T.; Palop G.; Toledano-Pinedo M.; Delgado-Martínez P. Chemoselectivity Switching in the Rhodium-Catalyzed Reactions of 4-Substituted-1-sulfonyl-1,2,3-triazoles with Allenols: Noticeable Differences between 4-Acyl- and 4-Aryl-Triazoles. Adv. Synth. Catal. 2019, 361, 1160–1165. 10.1002/adsc.201801424. [DOI] [Google Scholar]
- Klemme R.; Bentz C.; Zukowski T.; Schefzig L.; Lentz D.; Reissig H.-U.; Zimmer R. Percarboxylic Acid Oxidation of α-Hydroxy-Substituted Alkoxyallenes: The Unexpected Formation of Acyloxy-Substituted 1,2-Diketones and the Synthesis of Functionalized Quinoxalines. Synthesis 2016, 48, 1491–1501. 10.1055/s-0035-1561751. [DOI] [Google Scholar]
- Alcaide B.; Almendros P.; Martínez del Campo T.; Redondo M. C.; Fernández I. Striking Alkenol Versus Allenol Reactivity: Metal-Catalyzed Chemodifferentiating Oxycyclization of Enallenols. Chem. - Eur. J. 2011, 17, 15005–15013. 10.1002/chem.201102100. [DOI] [PubMed] [Google Scholar]
- Qiu Y.; Yang B.; Jiang T.; Zhu C.; Bäckvall J.-E. Palladium-Catalyzed Oxidative Cascade Carbonylative Spirolactonization of Enallenols. Angew. Chem., Int. Ed. 2017, 56, 3221–3225. 10.1002/anie.201612384. [DOI] [PubMed] [Google Scholar]
- Li M.-B.; Posevins D.; Geoffroy A.; Zhu C.; Bäckvall J.-E. Efficient Heterogeneous Palladium-Catalyzed Oxidative Cascade Reactions of Enallenols to Furan and Oxaborole Derivatives. Angew. Chem., Int. Ed. 2020, 59, 1992–1996. 10.1002/anie.201911462. [DOI] [PubMed] [Google Scholar]
- Devi N. R.; Sultana S.; Borah M.; Saikia A. K. Regio- and Diastereoselective Synthesis of Dihydropyrans and Pyranopyrans via Oxonium–Ene Reaction of β-Allenols and Aldehydes. J. Org. Chem. 2018, 83, 14987–14998. 10.1021/acs.joc.8b02244. [DOI] [PubMed] [Google Scholar]
- Han Y.; Tang X.; Cheng J.; Ma S. Indium(III) Triflate-Catalyzed Efficient Prins-Type Cyclization of β-Allenols and Aldehydes. Adv. Synth. Catal. 2016, 358, 4019–4029. 10.1002/adsc.201600588. [DOI] [Google Scholar]
- Ullapu P. R.; Kim Y. S.; Lee J. K.; Pae A. N.; Kim Y.; Min S. J.; Cho Y. S. Synthesis of 3,4-Dimethylidene Oxacycles through Prins-Type Cyclization of Allenylmethylsilanes. Chem. - Asian J. 2011, 6, 2092–2100. 10.1002/asia.201100162. [DOI] [PubMed] [Google Scholar]
- Ma S.; Zhao S. Pd(0)-Catalyzed Insertion-Cyclization Reaction of 2,3-Allenols with Aryl or Alkenyl Halides. Diastereoselective Synthesis of Highly Optically Active trans-2,3-Disubstituted Vinylic Oxiranes. J. Am. Chem. Soc. 1999, 121, 7943–7944. 10.1021/ja9904888. [DOI] [Google Scholar]
- Ma S.; Gao W. Efficient Synthesis of 4-(2‘-Alkenyl)-2,5-dihydrofurans and 5,6-Dihydro-2H-pyrans via the Pd-Catalyzed Cyclizative Coupling Reaction of 2,3- or 3,4-Allenols with Allylic Halides. J. Org. Chem. 2002, 67, 6104–6112. 10.1021/jo0163997. [DOI] [PubMed] [Google Scholar]
- Alcaide B.; Almendros P.; Rodríguez-Acebes R. Pd-Cu Bimetallic Catalyzed Domino Cyclization of α-Allenols–Coupling Reactions. New Sequence Leading to Functionalized Spirolactams. Chem. - Eur. J. 2005, 11, 5708–5712. 10.1002/chem.200500228. [DOI] [PubMed] [Google Scholar]
- Alcaide B.; Almendros P.; Martínez del Campo T. Reaction of Two Different α-Allenols in a Heterocyclization Cross-Coupling Sequence: Convenient Access to Functionalized Buta-1,3-dienyl Dihydrofurans. Angew. Chem., Int. Ed. 2006, 45, 4501–4504. 10.1002/anie.200601055. [DOI] [PubMed] [Google Scholar]
- For a review on Pd-catalyzed cross-coupling reactions of allenol-based systems, see:; Alcaide B.; Almendros P.; Martínez del Campo T. Cross-Coupling/Cyclization Reactions of Two Different Allenic Moieties. Chem. - Eur. J. 2010, 16, 5836–5842. 10.1002/chem.201000550. [DOI] [PubMed] [Google Scholar]
- Ma S.; Gao W. Efficient Synthesis of 4-(2′-Alkenyl)-2,5-dihydrofurans via PdCl2-Catalyzed Coupling–Cyclization Reaction of 2,3-Allenols with Allylic Halides. Tetrahedron Lett. 2000, 41, 8933–8936. 10.1016/S0040-4039(00)01585-9. [DOI] [Google Scholar]
- Miao P.; Wang H.; Liu L.; Chang W.; Li J. Palladium-Catalyzed Highly Chemoselective Cascade Coupling-Cyclization of Allenol Derivatives and Aryl Halides for the Construction of Dihydrobenzofuranols or Chromanols and Indolinols. Asian J. Org. Chem. 2015, 4, 1050–1054. 10.1002/ajoc.201500263. [DOI] [Google Scholar]
- Kang S.-K.; Yamaguchi T.; Pyung S.-J.; Lee Y.-T.; et al. Palladium-Catalyzed Arylation of α-Allenic Alcohols with Hypervalent lodonium Salts: Synthesis of Epoxides and Diol Cyclic Carbonates. Tetrahedron Lett. 1998, 39, 2127–2130. 10.1016/S0040-4039(98)00076-8. [DOI] [Google Scholar]
- Naapuri J. M.; Aberg G. A.; Palomo J. M.; Deska J. Arylative Allenol Cyclization via Sequential One-pot Enzyme & Palladium Catalysis. ChemCatChem 2021, 12, 763–769. 10.1002/cctc.202001619. [DOI] [Google Scholar]
- He Y.; Zhang X.; Fan X. Synthesis of Diversely Substituted 2-(Furan-3-yl)acetates from Allenols through Cascade Carbonylations. Chem. Commun. 2015, 51, 16263–16266. 10.1039/C5CC06150D. [DOI] [PubMed] [Google Scholar]
- He Y.; Zheng Z.; Liu Q.; Song G.; Sun N.; Chai X. Tunable Synthesis of 2-Ene-1,4-diones, 4-Hydroxycyclopent-2-en-1-ones, and 2-(Furan-3-yl)acetamides via Palladium-Catalyzed Cascade Reactions of Allenols. J. Org. Chem. 2018, 83, 12514–12526. 10.1021/acs.joc.8b01753. [DOI] [PubMed] [Google Scholar]
- Hou Y.; Qin M.; Yang X.; Shen Q.; Zhao Y.; Liu Y.; Gong P. Palladium-Catalyzed Three-Component Tandem Cyclization of Buta-2,3-dien-1-ol, Aryl Iodides, and Imines: An Efficient Protocol for the Synthesis of Oxazolidine Derivatives. RSC Adv. 2017, 7, 7401–7405. 10.1039/C6RA27993G. [DOI] [Google Scholar]
- Deng Y.; Yu Y.; Ma S. PdCl2/NaI-Catalyzed Homodimeric Coupling-Cyclization Reaction of 2,3-Allenols: An Efficient Synthesis of 4-(1′,3′-Dien-2′-yl)-2,5-dihydrofuran Derivatives. J. Org. Chem. 2008, 73, 585–589. 10.1021/jo702332m. [DOI] [PubMed] [Google Scholar]
- Deng Y.; Li J.; Ma S. PdI2-Catalyzed Coupling–Cyclization Reactions Involving Two Different 2,3-Allenols: An Efficient Synthesis of 4-(1’,3′-Dien-2′-yl)-2,5-dihydrofuran Derivatives. Chem. - Eur. J. 2008, 14, 4263–4266. 10.1002/chem.200800167. [DOI] [PubMed] [Google Scholar]
- Zeng R.; Ye J.; Fu C.; Ma S. Arene C-H Bond Functionalization Coupling with Cyclization of Allenes. Adv. Synth. Catal. 2013, 355, 1963–1970. 10.1002/adsc.201300408. [DOI] [Google Scholar]
- Kong W.; Cui J.; Yu Y.; Chen G.; Fu C.; Ma S. PtCl4-Catalyzed Cyclization Reaction of β-Allenols in the Presence of Indoles. Org. Lett. 2009, 11, 1213–1216. 10.1021/ol802838v. [DOI] [PubMed] [Google Scholar]
- Alcaide B.; Almendros P.; Aparicio B.; Lázaro-Milla C.; Luna A.; Nieto Faza O. Gold-Photoredox-Cocatalyzed Tandem Oxycyclization/Coupling Sequence of Allenols and Diazonium Salts with Visible Light Mediation. Adv. Synth. Catal. 2017, 359, 2789–2800. 10.1002/adsc.201700598. [DOI] [Google Scholar]
- Cyclopropane-linked allenols suffered arylative ring expansion under gold-catalyzed photoredox conditions:; Shu X.-Z.; Zhang M.; He Y.; Frei H.; Toste D. F. Dual Visible Light Photoredox and Gold-Catalyzed Arylative Ring Expansion. J. Am. Chem. Soc. 2014, 136, 5844–5847. 10.1021/ja500716j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sepelgy O.; Brzozowska A.; Azofra L. M.; Jang Y. K.; Cavallo L.; Rueping M. Experimental and Computational Study of an Unexpected Iron-Catalyzed Carboetherification by Cooperative Metal and Ligand Substrate Interaction and Proton Shuttling. Angew. Chem., Int. Ed. 2017, 56, 14863–14867. 10.1002/anie.201708240. [DOI] [PubMed] [Google Scholar]
- El-Sepelgy O.; Brzozowska A.; Sklyaruk J.; Jang Y. K.; Zubar V.; Rueping M. Cooperative Metal–Ligand Catalyzed Intramolecular Hydroamination and Hydroalkoxylation of Allenes Using a Stable Iron Catalyst. Org. Lett. 2018, 20, 696–699. 10.1021/acs.orglett.7b03828. [DOI] [PubMed] [Google Scholar]
- Brown T. J.; Weber D.; Gagné M. R.; Widenhoefer R. A. Mechanistic Analysis of Gold(I)-Catalyzed Intramolecular Allene Hydroalkoxylation Reveals an Off-Cycle Bis(gold) Vinyl Species and Reversible C–O Bond Formation. J. Am. Chem. Soc. 2012, 134, 9134–9137. 10.1021/ja303844h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupré N.; Brazel C.; Fensterbank L.; Malacria M.; Thorimbert S.; Hasenknopf B.; Lacôte E. Self-Buffering Hybrid Gold-Polyoxometalate Catalysts for the Catalytic Cyclization of Acid-Sensitive Substrates. Chem. - Eur. J. 2012, 18, 12962–12965. 10.1002/chem.201201007. [DOI] [PubMed] [Google Scholar]
- Dince C. C.; Meoded R. A.; Hilvert D. Synthesis and Characterization of Catalytically Active Thiazolium Gold(I)-carbenes. Chem. Commun. 2017, 53, 7585–7587. 10.1039/C7CC03791K. [DOI] [PubMed] [Google Scholar]
- Klumphu P.; Desfeux C.; Zhang Y.; Handa S.; Gallou F.; Lipshutz B. H. Micellar Catalysis-Enabled Sustainable ppm Au-catalyzed Reactions in Water at Room Temperature. Chem. Sci. 2017, 8, 6354–6358. 10.1039/C7SC02405C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z. J.; Brown C. J.; Bergman R. G.; Raymond K. N.; Toste F. D. Hydroalkoxylation Catalyzed by a Gold(I) Complex Encapsulated in a Supramolecular Host. J. Am. Chem. Soc. 2011, 133, 7358–7360. 10.1021/ja202055v. [DOI] [PubMed] [Google Scholar]
- Gramage-Doria R.; Bellini R.; Rintjema J.; Reek J. N. H. Supramolecular Ligands in Gold(I) Catalysis. ChemCatChem 2013, 5, 1084–1087. 10.1002/cctc.201200541. [DOI] [Google Scholar]
- Araki S.; Usui H.; Kato M.; Butsugan Y. Regio- and Stereoselective Allylindation of Allenols Regulated by a Hydroxyl-Chelated Bicyclic Transition State. J. Am. Chem. Soc. 1996, 118, 4699–4700. 10.1021/ja954318g. [DOI] [Google Scholar]
- Hou Y.; Shen Q.; Li Z.; Chen S.; Zhao Y.; Qin M.; Gong P. Palladium-Catalyzed Three-Component Tandem Reaction for One-Pot Highly Stereoselective Synthesis of (Z)-α-Hydroxymethyl Allylic Sulfones. Adv. Synth. Catal. 2018, 360, 631–636. 10.1002/adsc.201701011. [DOI] [Google Scholar]
- Hu G.; Wang J.; Li Z.; Liu Y.; Gong P. Palladium-Catalyzed Three-Component Reaction for the Synthesis of 3,3-Disubstituted Allylic Alcohols with Regio- and Stereoselectivity. New J. Chem. 2018, 42, 1736–1739. 10.1039/C7NJ04342B. [DOI] [Google Scholar]
- Hou Y.; Shen Q.; Zhu L.; Han Y.; Zhao Y.; Qin M.; Gong P. Palladium-Catalyzed Three-Component Tandem Reaction of Sulfonyl Hydrazones, Aryl Iodides and Allenes: Highly Stereoselective Synthesis of (Z)-α-Hydroxymethyl Allylic Sulfones. RSC Adv. 2017, 7, 50372–50377. 10.1039/C7RA08208H. [DOI] [Google Scholar]
- Li H.; Li T.; Hsueh Y. J.; Wu X.; Xu F.; Zhang Y. J. Tandem Arylation and Regioselective Allylic Etherification of 2,3-Allenol via Pd/B Cooperative Catalysis. Org. Biomol. Chem. 2019, 17, 8075–8078. 10.1039/C9OB01792E. [DOI] [PubMed] [Google Scholar]
- Lu B.-L.; Shi M. Oxidative Isomerization of Vinylidenecyclopropanes to Dimethylenecyclopropanes and Brønsted Acid-Catalyzed Further Transformation. Eur. J. Org. Chem. 2011, 2011, 243–248. 10.1002/ejoc.201001484. [DOI] [Google Scholar]
- Zhou Z.; Liu G.; Lu X. Regiocontrolled Coupling of Aromatic and Vinylic Amides with α-Allenols to Form γ-Lactams via Rhodium(III)-Catalyzed C–H Activation. Org. Lett. 2016, 18, 5668–5671. 10.1021/acs.orglett.6b02903. [DOI] [PubMed] [Google Scholar]
- Shi X.; Gorin D. J.; Toste F. D. Synthesis of 2-Cyclopentenones by Gold(I)-Catalyzed Rautenstrauch Rearrangement. J. Am. Chem. Soc. 2005, 127, 5802–4803. 10.1021/ja051689g. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Wang S. Efficient Synthesis of Cyclopentenones from Enynyl Acetates via Tandem Au(I)-Catalyzed 3,3-Rearrangement and the Nazarov Reaction. J. Am. Chem. Soc. 2006, 128, 1442–1443. 10.1021/ja057327q. [DOI] [PubMed] [Google Scholar]
- Brooks L. L.; Caruana P. E.; Frontier A. J. Conjugate Addition-Initiated Nazarov Cyclization. J. Am. Chem. Soc. 2011, 133, 12454–12457. 10.1021/ja205440x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a review, see:; Mack D. J.; Njardarson J. T. Recent Advances in the Metal-Catalyzed Ring Expansions of Three- and Four-Membered Rings. ACS Catal. 2013, 3, 272–286. 10.1021/cs300771d. [DOI] [Google Scholar]
- Gyanchander E.; Ydhyam S.; Tumma N.; Belmore K.; Cha J. K. Mechanism of Ru(II)-Catalyzed Rearrangements of Allenyl- and Alkynylcyclopropanols to Cyclopentenones. Org. Lett. 2016, 18, 6098–6101. 10.1021/acs.orglett.6b03088. [DOI] [PubMed] [Google Scholar]
- Tumma N.; Gyanchander E.; Cha J. K. J. J. Org. Chem. 2017, 82, 4379–4385. 10.1021/acs.joc.7b00432. [DOI] [PubMed] [Google Scholar]
- Alcaide B.; Almendros P.; Alonso J. M.; Fernández I.; Khodabakhshi S. Synthesis of Fused Cyclopentenones through Palladium-Catalyzed Cyclization of 2-Iodoaryl Allenols. Adv. Synth. Catal. 2014, 356, 1370–1374. 10.1002/adsc.201301127. [DOI] [Google Scholar]
- Li J.; Fu C.; Chen G.; Chai G.; Ma S. Studies on Electrophilic Interaction of 2,3-Allenols with Electrophilic Halogen Reagents: Selective Synthesis of 2,5-Dihydrofurans, 3-Halo-3-alkenals, or 2-Halo-2-alkenyl Ketones. Adv. Synth. Catal. 2008, 350, 1376–1382. 10.1002/adsc.200800088. [DOI] [Google Scholar]
- Fu C.; Li J.; Ma S. Efficient Two-step Synthesis of 3-Halo-3-enals or 2-Halo-2-alkenyl Ketones from Propargylic Bromides via a Unique Cationic 1,2-Aryl or Proton Shift in Electrophilic Addition Reaction of 2,3-Allenols with X+. Chem. Commun. 2005, 4119–4121. 10.1039/b508069j. [DOI] [PubMed] [Google Scholar]
- Alcaide B.; Almendros P.; Luna A.; Torres M. R. Divergent Reactivity of 2-Azetidinone-Tethered Allenols with Electrophilic Reagents: Controlled Ring Expansion versus Spirocyclization. Adv. Synth. Catal. 2010, 352, 621–626. 10.1002/adsc.200900864. [DOI] [Google Scholar]
- Alcaide B.; Almendros P.; Luna A.; Cembellín S.; Arnó M.; Domingo L. R. Controlled Rearrangement of Lactam-Tethered Allenols with Brominating Reagents: A Combined Experimental and Theoretical Study on α- versus β-Keto Lactam Formation. Chem. - Eur. J. 2011, 17, 11559–11566. 10.1002/chem.201101160. [DOI] [PubMed] [Google Scholar]
- Alcaide B.; Almendros P.; Luna P.; Gómez-Campillos G.; Torres M. R. Ring Enlargement versus Selenoetherification on the Reaction of Allenyl Oxindoles with Selenenylating Reagents. J. Org. Chem. 2012, 77, 3549–3556. 10.1021/jo202495z. [DOI] [PubMed] [Google Scholar]
- Shu X.-Z.; Zhang M.; He Y.; Frei H.; Toste F. D. Dual Visible Light Photoredox and Gold-Catalyzed Arylative Ring Expansion. J. Am. Chem. Soc. 2014, 136, 5844–5847. 10.1021/ja500716j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera F.; Luna A.; Almendros P. Visible-Light-Mediated Ru-Catalyzed Synthesis of 3-(Arylsulfonyl)but-3-enals via Coupling of α-Allenols with Diazonium Salts and Sulfur Dioxide. Org. Lett. 2020, 22, 9490–9494. 10.1021/acs.orglett.0c03482. [DOI] [PubMed] [Google Scholar]
- Kulandai Raj A. S.; Tan K.-C.; Chen L.-Y.; Cheng M.-J.; Liu R.-S. Gold-Catalyzed Bicyclic Annulations of 4-Methoxy-1,2-dienyl-5-ynes with Isoxazoles to Form Indolizine Derivatives via an Au-π-allene Intermediate. Chem. Sci. 2019, 10, 6437–6442. 10.1039/C9SC00735K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh H. C.; Tan K.-C.; Raj A. S. K.; Liu R.-S. Gold-catalyzed [4 + 1]-Annulation Reactions Between Anthranils and 4-Methoxy-1,2-dienyl-5-ynes Involving a 1,2-Allene Shift. Chem. Commun. 2019, 55, 1979–1982. 10.1039/C8CC09082C. [DOI] [PubMed] [Google Scholar]
- Toledano-Pinedo M.; Martínez del Campo T.; Tiemblo M.; Fernández I.; Almendros P. Organoseleno-Catalyzed Synthesis of α,β-Unsaturated α’-Alkoxy Ketones from Allenes Enabled by Se···O interactions. Org. Lett. 2020, 22, 3979–3984. 10.1021/acs.orglett.0c01288. [DOI] [PubMed] [Google Scholar]
- Alcaide B.; Almendros P.; Carrascosa R.; Martínez del Campo T. Metal-Catalyzed Cycloetherification Reactions of β,γ- and γ,δ-Allendiols: Chemo-, Regio-, and Stereocontrol in the Synthesis of Oxacycles. Chem. - Eur. J. 2010, 16, 13243–13252. 10.1002/chem.201001520. [DOI] [PubMed] [Google Scholar]
- Alcaide B.; Almendros P.; Aragoncillo C.; Gómez-Campillos G.; Quirós M. T.; Soriano E. Tunable Metal-Catalyzed Heterocyclization Reactions of Allenic Amino Alcohols: An Experimental and Theoretical Study. J. Org. Chem. 2016, 81, 7362–7372. 10.1021/acs.joc.6b00934. [DOI] [PubMed] [Google Scholar]
- Alcaide B.; Almendros P.; Carrascosa R.; Casarrubios L.; Soriano E. A Versatile Synthesis of β-Lactam-Fused Oxacycles through the Palladium-Catalyzed Chemo-, Regio-, and Diastereoselective Cyclization of Allenic Diols. Chem. - Eur. J. 2015, 21, 2200–2213. 10.1002/chem.201405181. [DOI] [PubMed] [Google Scholar]
- Li S.; Miao B.; Yuan W.; Ma S. Carbometalation-Carboxylation of 2,3-Allenols with Carbon Dioxide: A Dramatic Effect of Halide Anion. Org. Lett. 2013, 15, 977–979. 10.1021/ol4000197. [DOI] [PubMed] [Google Scholar]
- Li Q.; Jiang X.; Fu C.; Ma S. One-Pot Construction of Aza- or Oxa-Bridged Benzocycloheptanes from Readily Available 2,3-Allenyl Malonates or 2,3-Allenols and o-Iodobenzaldehyde or Imine. Org. Lett. 2011, 13, 466–469. 10.1021/ol102811x. [DOI] [PubMed] [Google Scholar]
- Posevins D.; Li M.-B.; Grape E. S.; Inge A. K.; Qiu Y.; Bäckvall J.-E. Highly Diastereoselective Palladium-Catalyzed Oxidative Cascade Carbonylative Carbocyclization of Enallenols. Org. Lett. 2020, 22, 417–421. 10.1021/acs.orglett.9b04134. [DOI] [PubMed] [Google Scholar]
- Posevins D.; Qiu Y.; Bäckvall J.-E. Highly Diastereoselective Palladium-Catalyzed Oxidative Carbocyclization of Enallenes Assisted by a Weakly Coordinating Hydroxyl Group. J. Am. Chem. Soc. 2018, 140, 3210–3214. 10.1021/jacs.7b13563. [DOI] [PubMed] [Google Scholar]
- Li M.-B.; Inge A. K.; Posevins D.; Gustafson K. P. J.; Qiu Y.; Bäckvall J.-E. Chemodivergent and Diastereoselective Synthesis of γ-Lactones and γ-Lactams: A Heterogeneous Palladium-Catalyzed Oxidative Tandem Process. J. Am. Chem. Soc. 2018, 140, 14604–14608. 10.1021/jacs.8b09562. [DOI] [PubMed] [Google Scholar]
- Li M.-B.; Yang J.; Yang Y.; Xu G.-Y.; Luo G.; Yang J.; et al. Bäckvall, J.-E. Amino-Supported Solid Palladium Catalyst for Chemo- and Stereoselective Domino Reactions. Angew. Chem., Int. Ed. 2021, 60, 670–674. 10.1002/anie.202011708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels D. S. B.; Thompson A. L.; Anderson E. A. Palladium-Catalyzed Asymmetric Synthesis of 2-Alkynyl Oxacycles. Angew. Chem., Int. Ed. 2011, 50, 11506–11510. 10.1002/anie.201105720. [DOI] [PubMed] [Google Scholar]
- Gobé V.; Dousset M.; Retailleau P.; Gandon V.; Guinchard X. Dissecting the Gold(I)-Catalyzed Carboaminations of N-Allyl Tetrahydro-β-carbolines to Allenes. J. Org. Chem. 2018, 83, 898–912. 10.1021/acs.joc.7b02900. [DOI] [PubMed] [Google Scholar]
- Ma S.; Zhao S. Novel Substituent and Chelating Effects in the Pd-Catalyzed Reaction of 2,3-Allenols, Aryl Iodides, and Amines. Highly Regio- and Stereoselective Synthesis of 2-Amino-3-alken-1-ols or 4-Amino-2(E)-alken-1-ols. J. Am. Chem. Soc. 2001, 123, 5578–5579. 10.1021/ja0035553. [DOI] [PubMed] [Google Scholar]
- Cox N.; Uehling M. R.; Haelsig K. T.; Lalic G. Catalytic Asymmetric Synthesis of Cyclic Ethers Containing an α-Tetrasubstituted Stereocenter. Angew. Chem., Int. Ed. 2013, 52, 4878–4882. 10.1002/anie.201300174. [DOI] [PubMed] [Google Scholar]
- Minkler S. R. K.; Lipshutz B. H.; Krause N. K. Gold Catalysis in Micellar Systems. Angew. Chem., Int. Ed. 2011, 50, 7820–7823. 10.1002/anie.201101396. [DOI] [PubMed] [Google Scholar]
- Zeng R.; Fu C.; Ma S. Highly Selective Mild Stepwise Allylation of N-Methoxybenzamides with Allenes. J. Am. Chem. Soc. 2012, 134, 9597–9600. 10.1021/ja303790s. [DOI] [PubMed] [Google Scholar]
- Zeng Y.; Chiou M. F.; Zhu X.; Cao J.; Lv D.; Jian W.; Li Y.; Zhang X.; Bao H. Copper-Catalyzed Enantioselective Radical 1,4-Difunctionalization of 1,3-Enynes. J. Am. Chem. Soc. 2020, 142, 18014–18021. 10.1021/jacs.0c06177. [DOI] [PubMed] [Google Scholar]
- Okada T.; Shimoda A.; Shinada T.; Sakaguchi K.; Ohfune Y. Regioselective Prins Cyclization of Allenylsilanes. Stereoselective Formation of Multisubstituted Heterocyclic Compounds. Org. Lett. 2012, 14, 6130–6133. 10.1021/ol302669q. [DOI] [PubMed] [Google Scholar]
- Le Boucher d’Herouville F.; Millet A.; Scalone M.; Michelet V. Synthesis of Atropisomeric MeOBIPHEP Analogues and Their Application in Silver-Catalyzed Cycloisomerization of Allenols. Synthesis 2016, 48, 3309–3316. 10.1055/s-0035-1562448. [DOI] [Google Scholar]
- Xie X.; Ma S. Palladium-Catalyzed Asymmetric Coupling Cyclization of Terminal γ-Allenols with Aryl Iodides. Chem. Commun. 2013, 49, 5693–5695. 10.1039/c3cc40892b. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Nicolini C.; Hervieu C.; Wong Y.-F.; Zanoni G.; Zhang L. Remote Cooperative Group Strategy Enables Ligands for Accelerative Asymmetric Gold Catalysis. J. Am. Chem. Soc. 2017, 139, 16064–16067. 10.1021/jacs.7b09136. [DOI] [PubMed] [Google Scholar]
- Cheng X.; Wang Z.; Quintanilla C. D.; Zhang L. Chiral Bifunctional Phosphine Ligand Enabling Gold-Catalyzed Asymmetric Isomerization of Alkyne to Allene and Asymmetric Synthesis of 2,5-Dihydrofuran. J. Am. Chem. Soc. 2019, 141, 3787–3791. 10.1021/jacs.8b12833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilg M. K.; Wolf L. M.; Mantilli L.; Farès C.; Thiel W.; Fürstner A. A Striking Case of Enantioinversion in Gold Catalysis and Its Probable Origins. Chem. - Eur. J. 2015, 21, 12279–12284. 10.1002/chem.201502160. [DOI] [PubMed] [Google Scholar]
- Teller H.; Corbet M.; Mantilli L.; Gopakumar G.; Goddard R.; Thiel W.; Fürstner A. One-Point Binding Ligands for Asymmetric Gold Catalysis: Phosphoramidites with a TADDOL-Related but Acyclic Backbone. J. Am. Chem. Soc. 2012, 134, 15331–15342. 10.1021/ja303641p. [DOI] [PubMed] [Google Scholar]
- Arbour J. L.; Rzepa H. S.; Contreras-García J.; Adrio L. A.; Barreiro E. M.; Hii K. K. Silver-Catalysed Enantioselective Addition of O-H and N-H Bonds to Allenes: A New Model for Stereoselectivity Based on Noncovalent Interactions. Chem. - Eur. J. 2012, 18, 11317–11324. 10.1002/chem.201200547. [DOI] [PubMed] [Google Scholar]
- Cao K.-S.; Zheng W.-H. Enantioselective Desymmetrization of Prochiral Allenic Diols via Cooperative Catalysis of Pd(OAc)2 and a Chiral Phosphoric Acid. Tetrahedron: Asymmetry 2015, 26, 1150–1155. 10.1016/j.tetasy.2015.08.010. [DOI] [Google Scholar]
- Zi W.; Toste F. D. Gold(I)-Catalyzed Enantioselective Desymmetrization of 1,3-Diols through Intramolecular Hydroalkoxylation of Allenes. Angew. Chem., Int. Ed. 2015, 54, 14447–14451. 10.1002/anie.201508331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aikawa K.; Kojima M.; Mikami K. Synergistic Effect: Hydroalkoxylation of Allenes through Combination of Enantiopure BIPHEP-Gold Complexes and Chiral Anions. Adv. Synth. Catal. 2010, 352, 3131–3135. 10.1002/adsc.201000672. [DOI] [Google Scholar]
- Miles D. H.; Veguillas M.; Toste F. D. Gold(I)-catalyzed Enantioselective Bromocyclization Reactions of Allenes. Chem. Sci. 2013, 4, 3427–3431. 10.1039/c3sc50811k. [DOI] [Google Scholar]
- Petrone D. A.; Isomura M.; Franzoni I.; Rössler S. L.; Carreira E. M. Allenylic Carbonates in Enantioselective Iridium-Catalyzed Alkylations. J. Am. Chem. Soc. 2018, 140, 4697–4704. 10.1021/jacs.8b01416. [DOI] [PubMed] [Google Scholar]
- Isomura M.; Petrone D. A.; Carreira E. M. Coordination-Induced Stereocontrol over Carbocations: Asymmetric Reductive Deoxygenation of Racemic Tertiary Alcohols. J. Am. Chem. Soc. 2019, 141, 4738–4748. 10.1021/jacs.9b00862. [DOI] [PubMed] [Google Scholar]
- Yamauchi M.; Morimoto M.; Miura T.; Murakami M. Enantioselective Synthesis of 3,4-Dihydroisoquinolin-1(2H)-ones by Nickel-Catalyzed Denitrogenative Annulation of 1,2,3-Benzotriazin-4(3H)-ones with Allenes. J. Am. Chem. Soc. 2010, 132, 54–55. 10.1021/ja909603j. [DOI] [PubMed] [Google Scholar]
- Kawai J.; Chikkade P. K.; Shimizu Y.; Kanai M. In situ Catalytic Generation of Allylcopper Species for Asymmetric Allylation: Toward 1H-Isochromene Skeletons. Angew. Chem., Int. Ed. 2013, 52, 7177–7180. 10.1002/anie.201302027. [DOI] [PubMed] [Google Scholar]
- Charette A. B.; Jolicoeur E.; Bydlinski G. A. S. Enantioselective Synthesis of Spiropentanes from Hydroxymethylallenes. Org. Lett. 2001, 21, 3293–3295. 10.1021/ol0165140. [DOI] [PubMed] [Google Scholar]
- Imada Y.; Nishida M.; Naota T. Sequential Asymmetric Homoallenylation of Primary Amines with a Palladium Catalyst. Tetrahedron Lett. 2008, 49, 4915–4917. 10.1016/j.tetlet.2008.05.144. [DOI] [Google Scholar]
- Sak H.; Mawick M.; Krause N. Sustainable Gold Catalysis in Water Using Cyclodextrin-tagged NHC-Gold Complexes. ChemCatChem 2019, 11, 5821–5829. 10.1002/cctc.201901658. [DOI] [Google Scholar]
- Ma S.; Jiao N.; Yang Q.; Zheng Z. Highly Regio- and Stereoselective Synthesis of Polysubstituted Cyclopropane Compounds via the Pd(0)-Catalyzed Coupling-Cyclization Reaction of 2-(2′,3′-Allenyl)malonates with Organic Halides. J. Org. Chem. 2004, 69, 6463–6466. 10.1021/jo049323u. [DOI] [PubMed] [Google Scholar]
- Kezuka S.; Kanemoto K.; Takeuchi R. Iridium Complex-Catalyzed Method for the Construction of a Quaternary Carbon Center α to Allene. Tetrahedron Lett. 2004, 45, 6403–6406. 10.1016/j.tetlet.2004.07.002. [DOI] [Google Scholar]
- Imada Y.; Nishida M.; Kutsuwa K.; Murahashi S.-I.; Naota T. Palladium-Catalyzed Asymmetric Amination and Imidation of 2,3-Allenyl Phosphates. Org. Lett. 2005, 7, 5837–5839. 10.1021/ol0523502. [DOI] [PubMed] [Google Scholar]
- Imada Y.; Ueno K.; Kutsuwa K.; Murahashi S.-I. Palladium-Catalyzed Asymmetric Alkylation of 2,3-Alkadienyl Phosphates. Synthesis of Optically Active 2-(2,3-Alkadienyl)malonates. Chem. Lett. 2002, 31, 140–141. 10.1246/cl.2002.140. [DOI] [Google Scholar]
- Trost B. M.; Fandrick D. R.; Dinh D. C. Dynamic Kinetic Asymmetric Allylic Alkylations of Allenes. J. Am. Chem. Soc. 2005, 127, 14186–14187. 10.1021/ja0543705. [DOI] [PubMed] [Google Scholar]
- Nemoto T.; Kanematsu M.; Tamura S.; Hamada Y. Palladium-Catalyzed Asymmetric Allylic Alkylation of 2,3-Allenyl Acetates Using a Chiral Diaminophosphine Oxide. Adv. Synth. Catal. 2009, 351, 1773–1778. 10.1002/adsc.200900151. [DOI] [Google Scholar]
- Li Q.; Fu C.; Ma S. Palladium-Catalyzed Asymmetric Amination of Allenyl Phosphates: Enantioselective Synthesis of Allenes with an Additional Unsaturated Unit. Angew. Chem., Int. Ed. 2014, 53, 6511–6514. 10.1002/anie.201402647. [DOI] [PubMed] [Google Scholar]
- Li Q.; Fu C.; Ma S. Catalytic Asymmetric Allenylation of Malonates with the Generation of Central Chirality. Angew. Chem., Int. Ed. 2012, 51, 11783–11786. 10.1002/anie.201204346. [DOI] [PubMed] [Google Scholar]
- Song S.; Zhou J.; Fu C.; Ma S. Catalytic Enantioselective Construction of Axial Chirality in 1,3-Disubstituted Allenes. Nat. Commun. 2019, 10, 1–9. 10.1038/s41467-018-07908-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J.; Duan X.; Zhou J.; Fu C.; Ma S. Catalytic Enantioselective Simultaneous Control of Axial Chirality and Central Chirality in Allenes. Chin. J. Chem. 2018, 36, 387–391. 10.1002/cjoc.201800090. [DOI] [Google Scholar]
- Song S.; Zhou J.; Fu C.; Ma S. Highly Selective Nucleophilic 4-Aryl-2,3-allenylation of Malonates. Chin. J. Chem. 2020, 38, 1233–1238. 10.1002/cjoc.202000300. [DOI] [Google Scholar]
- Wan B.; Ma S. Enantioselective Decarboxylative Amination: Synthesis of Axially Chiral Allenyl Amines. Angew. Chem., Int. Ed. 2013, 52, 441–445. 10.1002/anie.201204796. [DOI] [PubMed] [Google Scholar]
- Yang B.; Zhu C.; Qiu Z.; Bäckvall J.-E. Enzyme- and Ruthenium-Catalyzed Enantioselective Transformation of α-Allenic Alcohols into 2,3-Dihydrofurans. Angew. Chem., Int. Ed. 2016, 55, 5568–5572. 10.1002/anie.201601505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guđmundsson A.; Gustafson K. P. J.; Mai B. K.; Yang B.; Himo F.; Bäckvall J.-E. Efficient Formation of 2,3-Dihydrofurans via Iron-Catalyzed Cycloisomerization of α-Allenols. ACS Catal. 2018, 8, 12–16. 10.1021/acscatal.7b03515. [DOI] [Google Scholar]
- Li M.-B.; Posevins D.; Gustafson K. P. J.; Tai C.-W.; Shchukarev A.; Qiu Y.; Bäckvall J.-E. Diastereoselective Cyclobutenol Synthesis: A Heterogeneous Palladium-Catalyzed Oxidative Carbocyclization-Borylation of Enallenols. Chem. - Eur. J. 2019, 25, 210–215. 10.1002/chem.201805118. [DOI] [PubMed] [Google Scholar]
- Wang Z. J.; Clary K. N.; Bergman R. G.; Raymond K. N.; Toste F. D. A Supramolecular Approach to Combining Enzymatic and Transition Metal Catalysis. Nat. Chem. 2013, 5, 100–103. 10.1038/nchem.1531. [DOI] [PubMed] [Google Scholar]
- Alnazer H.; Castellan T.; Salma Y.; Genisson Y.; Ballereau S. Enantioselective Stereodivergent Synthesis of Jaspine B and 4-epi-Jaspine B from Axially Chiral Allenols. Synlett 2019, 30, 185–188. 10.1055/s-0037-1610344. [DOI] [Google Scholar]
- Manzuna Sapu C.; Deska J. Chemoenzymatic Total Synthesis of Hyperione A and B. Org. Biomol. Chem. 2013, 11, 1376–1382. 10.1039/c2ob27073k. [DOI] [PubMed] [Google Scholar]
- Lee E.; Bang J.; Kwon J.; Yu C.-M. Enantioselective Synthesis of a Furan Lignan (+)-Sylvone. J. Org. Chem. 2015, 80, 10359–10363. 10.1021/acs.joc.5b01677. [DOI] [PubMed] [Google Scholar]
- Mahapatra S.; Carter R. G. Enantioselective Total Synthesis of Amphidinolide F. Angew. Chem., Int. Ed. 2012, 51, 7948–7951. 10.1002/anie.201203935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larivée A.; Unger J. B.; Thomas M.; Wirtz C.; Dubost C.; Handa S.; et al. Fürstner. The Leiodolide B Puzzle. Angew. Chem., Int. Ed. 2011, 50, 304–309. 10.1002/anie.201005850. [DOI] [PubMed] [Google Scholar]
- For the synthesis of the Stenine core, see:; Bates R. W.; Sridhar S. Synthesis of the Stenine Ring System from Pyrrole. J. Org. Chem. 2011, 76, 5026–5035. 10.1021/jo200699k. [DOI] [PubMed] [Google Scholar]
- For the synthesis of Stemoadine, see:; Wang Y.; Zhu L.; Zhang Y.; Hong R. Bioinspired and Concise Synthesis of (±)-Stemoamide. Angew. Chem., Int. Ed. 2011, 50, 2787–2790. 10.1002/anie.201005833. [DOI] [PubMed] [Google Scholar]
- Park S.; Pak G.; Oh C.; Lee J.; Kim J.; Yu C.-M. Kinetic Resolution of Racemic Aldehydes through Asymmetric Allenoate γ-Addition: Synthesis of (+)-Xylogiblactone A. Org. Lett. 2019, 21, 7660–7664. 10.1021/acs.orglett.9b02982. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Fu C.; Ma S. Gold-Catalyzed Stereoselective Cycloisomerization of Allenoic Acids for Two Types of Common Natural γ-Butyrolactones. Nat. Commun. 2018, 9, 1–10. 10.1038/s41467-018-03894-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavan S.; Nyalata S. Convergent Synthesis of the Dihydropyran Core Containing the C1–C15 Subunit of Sorangicin A Employing Gold(I)-Catalyzed Cyclization of an Allenic Alcohol. J. Org. Chem. 2016, 81, 10698–10706. 10.1021/acs.joc.6b01743. [DOI] [PubMed] [Google Scholar]
- Choi H.-W.; Fang F. G.; Fang H.; Kim D.-S.; Mathieu S. R.; Yu R. T. Prins Reaction of Homoallenyl Alcohols: Access to Substituted Pyrans in the Halichondrin Series. Org. Lett. 2017, 19, 6092–6095. 10.1021/acs.orglett.7b02934. [DOI] [PubMed] [Google Scholar]
- Feldman K. S.; Folda T. S. Studies on the Synthesis of the Alkaloid (−)-Gilbertine via Indolidene Chemistry. J. Org. Chem. 2016, 81, 4566–4575. 10.1021/acs.joc.6b00348. [DOI] [PubMed] [Google Scholar]
- Parmeggiani C.; Cardona F.; Giusti L.; Reissig H.-U.; Goti A. Stereocomplementary Routes to Hydroxylated Nitrogen Heterocycles: Total Syntheses of Casuarine, Australine, and 7-epi-Australine. Chem. - Eur. J. 2013, 19, 10595–10604. 10.1002/chem.201301320. [DOI] [PubMed] [Google Scholar]
- Schmidt J. P.; Breit B. Rhodium-Catalyzed Cyclization of Terminal and Internal Allenols: An Atom Economic and Highly Stereoselective Access Towards Tetrahydropyrans. Angew. Chem., Int. Ed. 2020, 59, 23485–23490. 10.1002/anie.202009166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spreider P. A.; Breit B. Palladium-Catalyzed Stereoselective Cyclization of in Situ Formed Allenyl Hemiacetals: Synthesis of Rosuvastatin and Pitavastatin. Org. Lett. 2018, 20, 3286–3290. 10.1021/acs.orglett.8b01156. [DOI] [PubMed] [Google Scholar]
- Dion A.; Dubé P.; Spino C. A Triple Diene-Transmissive Diels-Alder Strategy to Build the Quassinoid Framework. Org. Lett. 2005, 7, 5601–5604. 10.1021/ol0522236. [DOI] [PubMed] [Google Scholar]
- Fan J.-H.; Hu Y.-J.; Guo Q.; Li S.; Zhao J.; Li C.-C. Asymmetric Synthesis of the Tetracyclic Core of Bufogargarizin C by an Intramolecular [5 + 2] Cycloaddition. Org. Chem. Front. 2019, 6, 22–26. 10.1039/C8QO01089G. [DOI] [Google Scholar]
- Suzuki T.; Miyajima Y.; Suzuki K.; Iwakiri K.; Koshimizu M.; Hirai G.; Sodeoka M.; Kobayashi S. Unexpected Diels-Alder/Carbonyl-ene Cascade toward the Biomimetic Synthesis of Chloropupukeananin. Org. Lett. 2013, 15, 1748–1751. 10.1021/ol400549q. [DOI] [PubMed] [Google Scholar]
- Cheng M.-J.; Zhong L.-P.; Gu C.-C.; Zhu X.-J.; Chen B.; Liu J.-S.; Wang L.; Ye W.-C.; Li C.-C. Asymmetric Total Synthesis of Bufospirostenin A.. J. Am. Chem. Soc. 2020, 142 (29), 12602–12607. 10.1021/jacs.0c05479. [DOI] [PubMed] [Google Scholar]
- Gerstner N. C.; Adams C. S.; Grigg R. D.; Tretbar M.; Rigoli J. W.; Schomaker J. M. Diastereoselective Synthesis of the Aminocyclitol Core of Jogyamycin via an Allene Aziridination Strategy. Org. Lett. 2016, 18, 284–287. 10.1021/acs.orglett.5b03453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M.; Kasai T.; Mizoguchi T.; Namba K. Total Synthesis of (−)-HM-3 and (−)-HM-4 Utilizing a Palladium-Catalyzed Addition of an Arylboronic Acid to an Allenic Alcohol Followed by Eschenmoser-Claisen Rearrangement. Synlett 2014, 25, 1160–1162. 10.1055/s-0033-1341059. [DOI] [Google Scholar]
- Yoshida M.; Shoji Y.; Shishido K. Total Syntheses of Enokipodins A and B Utilizing Palladium-Catalyzed Addition of An Arylboronic Acid to An Allene. Org. Lett. 2009, 6, 1441–1443. 10.1021/ol9001637. [DOI] [PubMed] [Google Scholar]
- Jeanne-Julien L.; Masson G.; Astier E.; Genta-Jouve G.; Servajean V.; Beau J.-M.; Norsikian S.; Roulland E. Synthesis of a Tiacumicin B Protected Aglycone. Org. Lett. 2017, 19, 4006–4009. 10.1021/acs.orglett.7b01744. [DOI] [PubMed] [Google Scholar]
- Yang L.; Lin Z.; Shao S.; Zhao Q.; Hong R. An Enantioconvergent and Concise Synthesis of Lasonolide A. Angew. Chem., Int. Ed. 2018, 57, 16200–16204. 10.1002/anie.201811093. [DOI] [PubMed] [Google Scholar]
- Kwon O.; Blank B.; Andrews I. Phosphine-Catalyzed (4 + 1) Annulation: Rearrangement of Allenylic Carbamates to 3-Pyrrolines through Phosphonium Diene Intermediates. ChemCatChem 2020, 12, 4352–4372. 10.1002/cctc.202000626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a review on natural products showing the allene functional group, see:; Hoffmann-Röder A.; Krause N. Synthesis and Properties of Allenic Natural Products and Pharmaceuticals. Angew. Chem., Int. Ed. 2004, 43, 1196–1216. 10.1002/anie.200300628. [DOI] [PubMed] [Google Scholar]
- de Graaf W.; Smits A.; Boersma J.; van Koten G.; et al. Synthesis of Marasin and 9-Me-Marasin. Tetrahedron 1988, 44, 6699–6704. 10.1016/S0040-4020(01)90110-3. [DOI] [Google Scholar]
- Schlingmann G.; Milne L.; Pearce C. J.; Borders D. B.; Greenstein M.; Maiese W. M.; Carter G. T. Isolation, Characterisation and Structure of a New Allenic Polyine Antibiotic Produced by Fungus LL-07F275. J. Antibiot. 1995, 48, 375–379. 10.7164/antibiotics.48.375. [DOI] [PubMed] [Google Scholar]
- Deutsch C.; Lipshutz B. H.; Krause N. Small but Effective: Copper Hydride Catalyzed Synthesis of Alpha-Hydroxyallenes. Angew. Chem., Int. Ed. 2007, 46, 1650–1653. 10.1002/anie.200603739. [DOI] [PubMed] [Google Scholar]
- Roulland E.; Solanki H.; Calabro K.; Zubia M.; Genta-Jouve G.; Thomas O. P. Stereochemical Study of Puna’auic Acid, an Allenic Fatty Acid from the Eastern Indo-Pacific Cyanobacterium Pseudanabaena sp. Org. Lett. 2018, 20, 2311–2314. 10.1021/acs.orglett.8b00654. [DOI] [PubMed] [Google Scholar]
- Suzuki T.; Watanabe S.; Kobayashi S.; Tanino K. Enantioselective Total Synthesis of (+)-Iso-A82775C, a Proposed Biosynthetic Precursor of Chloropupukeananin. Org. Lett. 2017, 19, 922–925. 10.1021/acs.orglett.7b00085. [DOI] [PubMed] [Google Scholar]
- Kim G.; Kim T.; Han S. Total Synthesis of (+)-Pestalofone A and (+)-Iso-A82775C. J. Org. Chem. 2020, 85, 6815–6821. 10.1021/acs.joc.0c00770. [DOI] [PubMed] [Google Scholar]
- Meinwald J.; Erickson K.; Hartshorn M.; Meinwald Y. C.; Eisner T. Defensive mechanisms of arthropods. XXIII. An Allenic Sesquiterpenoid from the Grasshopper Romalea Microptera. Tetrahedron Lett. 1968, 9, 2959–2962. 10.1016/S0040-4039(00)89622-7. [DOI] [Google Scholar]
- Baumeler A.; Brade W.; Haag A.; Eugster C. H. Synthesis of Optically Pure Grasshopper Ketone, its Diastereoisomers, and Related Compounds. Helv. Chim. Acta 1990, 73, 700–715. 10.1002/hlca.19900730319. [DOI] [Google Scholar]
- Otero L.; Vaz B.; Álvarez R.; de Lera A. Total Synthesis of (8R,6’R)-Peridinin-5,8-Furanoxide. Chem. Commun. 2013, 49, 5043. 10.1039/c3cc41552j. [DOI] [PubMed] [Google Scholar]
- For previous work on steroselective Stille coupling reaction with iodoallene derivatives, see:; Vaz B.; Pereira R.; Pérez M.; Álvarez R.; de Lera A. J. Org. Chem. 2008, 73, 6534–6541. 10.1021/jo800756b. [DOI] [PubMed] [Google Scholar]
- For previous synthesis of related 6′-epi-Peridinin, see:; Vaz B.; Álvarez R.; Brückner; de Lera A. The Stille Reaction in the Synthesis of Carotenoid Butenolides: Synthesis of 6’-epi-Peridinin. Org. Lett. 2005, 7, 545–548. 10.1021/ol0478281. [DOI] [PubMed] [Google Scholar]
- Woerly E. M.; Cherney A. H.; Davis E. K.; Burke M. D. Stereoretentive Suzuki-Miyaura Coupling of Haloallenes Enables Fully Stereocontrolled Access to (−)-Peridinin. J. Am. Chem. Soc. 2010, 132, 6941–6943. 10.1021/ja102721p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haley H. M. S.; Hill A. G.; Greenwood A. I.; Woerly E. M.; Rienstra C. M.; Burke M. D. Peridinin Is an Exceptionally Potent and Membrane-Embedded Inhibitor of Bilayer Lipid Peroxidation. J. Am. Chem. Soc. 2018, 140, 15227–15240. 10.1021/jacs.8b06933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinashi N.; Katsumura S.; Shinada T.; Sakaguchi K. Stereocontrolled Synthesis of 19′-Deoxyperidinin. Org. Lett. 2018, 20, 582–585. 10.1021/acs.orglett.7b03695. [DOI] [PubMed] [Google Scholar]
- Yamano Y.; Tode C.; Ito M. Carotenoids and Related Polyenes. Part 3. First Total Synthesis of Fucoxanthin and Halocynthiaxanthin Using Oxo-metallic Catalyst. J. Chem. Soc., Perkin Trans. 1 1995, 1, 1895–1904. 10.1039/p19950001895. [DOI] [Google Scholar]
- Murakami Y.; Nakano M.; Shimofusa T.; Furuichi N.; Katsumura S. Total Synthesis of Paracentrone, C31-Allenic Apo-carotenoid. Org. Biomol. Chem. 2005, 3, 1372–1374. 10.1039/b500316d. [DOI] [PubMed] [Google Scholar]
- Nishioka Y.; Yano Y.; Kinashi N.; Oku N.; Toriyama Y.; Katsumura S.; Shinada T.; Sakaguchi K. Stereocontrolled Synthesis of Paracentrone. Synlett 2017, 28, 327–332. 10.1055/s-0036-1588906. [DOI] [Google Scholar]
- Sakaguchi K.; Nishioka Y.; Kinashi N.; Yukihira N.; Shinada T.; Nishimura T.; Hashimoto H.; Katsumura S. Synthesis of Allene-Containing Apocarotenoids by Cross-Coupling Strategy. Synthesis 2020, 52, 3007–3017. 10.1055/s-0040-1707906. [DOI] [Google Scholar]
- Kajikawa T.; Okumura S.; Iwashita T.; Kosumi T.; Hashimoto H.; Katsumura S. Stereocontrolled Total Synthesis of Fucoxanthin and Its Polyene Chain-Modified Derivative. Org. Lett. 2012, 14, 808–811. 10.1021/ol203344c. [DOI] [PubMed] [Google Scholar]
- Maoka T.; Akimoto N.; Terada Y.; Komemushi S.; Harada R.; Sameshima N.; Sakagami Y. Structure of Minor Carotenoids from the Crown-of-Thorns Starfish, Acanthaster planci. J. Nat. Prod. 2010, 73, 675–678. 10.1021/np100021u. [DOI] [PubMed] [Google Scholar]
- Liu L.; Li Y.; Li L.; Cao Y.; Guo L.; Liu G.; Che Y. Spiroketals of Pestalotiopsis fici Provide Evidence for a Biosynthetic Hypothesis Involving Diversified Diels–Alder Reaction Cascades. J. Org. Chem. 2013, 78, 2992–3000. 10.1021/jo302804h. [DOI] [PubMed] [Google Scholar]
- Gutiérrez-Cepeda A.; Fernández J. J.; Norte M.; Souto M. L. New Bicyclotridecane C15 Nonterpenoid Bromoallenes from Laurencia marilzae. Org. Lett. 2011, 13, 2690–2693. 10.1021/ol200792v. [DOI] [PubMed] [Google Scholar]
- Zemlicka J. Lipophilic Phospharamidates as Antiviral Pronucleotides. Biochim. Biophys. Acta, Mol. Basis Dis. 2002, 1587, 276–286. 10.1016/S0925-4439(02)00090-X. [DOI] [PubMed] [Google Scholar]
- Jones B. C. N. M.; Silverton J. V.; Simons C.; Megati S.; Nishimura H.; Maeda Y.; Mitsuya H.; Zemlicka Synthesis, Absolute Configuration, and Enantioselectivity of Antiretroviral Effect of (R)-(−)- and (S)-(+)-Cytallene. Lipase-Catalyzed Enantioselective Acylations of (±)-N4-Acylcytallenes J.. J. Med. Chem. 1995, 38, 1397–1405. 10.1021/jm00008a018. [DOI] [PubMed] [Google Scholar]
- Megati S.; Goren Z.; Silverton J. V.; Orlina J.; Nishimura H.; Shirasaki T.; Mitsuya H.; Zemlicka J. (R)-(−)- and (S)-(+)-Adenallene: Synthesis, Absolute Configuration, Enantioselectivity of Antiretroviral Effect, and Enzymic Deamination. J. Med. Chem. 1992, 35, 4098–4104. 10.1021/jm00100a016. [DOI] [PubMed] [Google Scholar]
- Collins P. W.; Djuric S. W. Synthesis of Therapeutically Useful Prostaglandin and Prostacyclin Analogs. Chem. Rev. 1993, 93, 1533–1564. 10.1021/cr00020a007. [DOI] [Google Scholar]
- Casara P.; Jund K.; Bey P. General Synthetic Access to α-Allenyl Amines and α-Allenyl-α-aminoacids as Potential Enzyme Activated Irreversible Inhibitors of PLP Dependent Enzymes. Tetrahedron Lett. 1984, 25, 1891–1894. 10.1016/S0040-4039(01)90068-1. [DOI] [Google Scholar]
- Burger A.; Roussel J.-P.; Hetru C.; Hoffmann J. A.; Luu B. Allenic Cholesteryl Derivatives as Inhibitors of Ecdysone Biosynthesis. Tetrahedron 1989, 45, 155–164. 10.1016/0040-4020(89)80042-0. [DOI] [Google Scholar]