

Abstract
Motivation
The absorption, distribution, metabolism, excretion, and toxicity (ADMET) of drugs plays a key role in determining which among the potential candidates are to be prioritized. In silico approaches based on machine learning methods are becoming increasing popular, but are nonetheless limited by the availability of data. With a view to making both data and models available to the scientific community, we have developed FPADMET which is a repository of molecular fingerprint-based predictive models for ADMET properties.
Summary
In this article, we have examined the efficacy of fingerprint-based machine learning models for a large number of ADMET-related properties. The predictive ability of a set of 20 different binary fingerprints (based on substructure keys, atom pairs, local path environments, as well as custom fingerprints such as all-shortest paths) for over 50 ADMET and ADMET-related endpoints have been evaluated as part of the study. We find that for a majority of the properties, fingerprint-based random forest models yield comparable or better performance compared with traditional 2D/3D molecular descriptors.
Availability
The models are made available as part of open access software that can be downloaded from https://gitlab.com/vishsoft/fpadmet.
Supplementary Information
The online version contains supplementary material available at 10.1186/s13321-021-00557-5.
Keywords: ADMET, Machine learning, Molecular fingerprints
Introduction
Properties such as absorption, distribution, metabolism, excretion and toxicity (ADMET), are an important component of pharmaceutical drug design. It is often reported that the failure to meet requisite ADMET criteria are a common cause for the high attrition rates of drug candidates [1]. Early ADMET profiling is indeed desirable so as to mitigate the risk of attrition. Various medium and high-throughput in vitro ADMET screens have therefore been developed, that have contributed to the available experimental data. These are nonetheless quite expensive especially when thousands of compounds are involved. Furthermore, reducing animal testing has now become a priority.
With the aim of facilitating rapid and inexpensive means of ADMET profiling, various in silico tools have been developed [2]. Using databases of experimentally measured ADMET properties [3], various quantitative structure-activity/property relationship (QSAR/QSPR) models have been generated that can predict a range of ADMET properties for novel chemical entities. Other efforts have made use of ADMET predictions to evaluate drug-likeness of a compound [4, 5]. While some of the models are available as part of commercial software packages based on proprietary datasets, there has been a significant push for open source software and web services [6–12].
Among the popular services, ADMETLab [12] offers 53 prediction models that are calculated using a multi-task graph attention network and operates on graph-structured data. The method is able to generate customized fingerprints from the general features for a specific task. Another web tool, SwissADME [9] evaluates pharmacokinetics, drug-likeness of small molecules. The predictions are based on a combination of fragmental methods (for solubility), as well as machine-learning based binary classification methods for other ADMET properties (cytochrome-P450 inhibitor, P-glycoprotein substrate). In ADMETSar [11], models for applications in both drug discovery and environmental risk assessment are built using MACCS and Morgan fingerprints. The toxicity models used in ProTox [13] are developed based on chemical similarities between compounds with known toxic effects and the presence of toxic fragments. Other models for hepatotoxicity, cytotoxicity, mutagenicity, and carcinogenicity rely on fingerprints (MACCS/Morgan). Extended connectivity fingerprints form the basis for the prediction of 15 ADMET properties in the vNN server [10] where models are trained using variable nearest neighbourhood method. pkCSM [6], on the other hand, uses graph-based signatures to develop predictive models of central ADMET properties. Other software such as MDCKPred [14], CarcinoPred-EL [15], CapsCarcino [16] focus on a single property such as the prediction of permeability coefficient and carcinogenic compounds. Overall, the molecular representations underlying these models include various molecular and physicochemical descriptors such as fingerprints, graph signatures, and other 2D/3D indices [17, 18]. Among these, fingerprint representations which are seen as an alternative to descriptors for QSPR studies, have been quite popular given their ease of computation and predictive value.
A number of fingerprints ranging from substructure/path to feature-class/circular have been proposed many of which are used in similarity searching [19, 20]. For ADMET studies however, the fingerprints studied so far have largely been restricted to a select few. In this study, we have evaluated the predictive efficacy of 20 different fingerprints ranging from substructure and extended/functional connectivity fingerprints to various path based encodings (depth-first search, shortest path, local path environments) [21]. The fingerprint-based regression/classification models were calculated for over 50 ADMET and ADMET-related endpoints (using data collated from various literature sources) and is to our knowledge one of the most comprehensive compilations analysed. For a majority of the endpoints, the prediction results were found to be comparable with more sophisticated descriptor formulations. Although the pharmacophore fingerprints yielded consistently poor results, others such as the PUBCHEM, MACCS and ECFP/FCFP encodings were found to yield the best results for most properties. The models and related software have been bundled into a downloadable package and is released under the GNU license.
Approach
Molecular representation
In this study, we have examined 20 different fingerprints (see Table 1) that are routinely used as similarity search tools in drug discovery. The ECFP- and FCFP-class fingerprints are circular topological fingerprints, where the former focuses on the atom properties (e.g. atomic number, charge, hydrogen count), whereas in the functional connectivity FPs, the emphasis is on properties that relate to ligand binding (e.g. hydrogen donor/acceptor, polarity, aromaticity). MACCS and PUBCHEM fingerprints are substructure fingerprints that cover a wide range of features such as element counts and ring systems, atom pairing, or atom environment etc. Other fingerprints include path based fingerprints such as the depth-first search fingerprints (DFS), all-shortest path encoding (ASP), radial fingerprints (Molprint2D), topological atom pairs (AP2D) and triplets (AT2D), pharmacophore pair and triplet encodings as well as local path environments [21]. Fingerprint calculations were performed using in-house code written in Java and makes use of the Chemistry Development Kit library [22]. The software merges existing fingerprints in the library with those calculated by the software jCompoundMapper [21].
Table 1.
Fingerprints used in this study to model different ADMET related properties
| Fingerprint | Size |
|---|---|
| MACCS | 166 |
| PUBCHEM | 881 |
| Klekota-Roth (KR) | 4860 |
| MOLPRINT (RAD2D) | 4096 |
| Atom pair (AP), atom triplet (AT) | 4096 |
| Local path environments (LSTAR) | 4096 |
| All-shortest path (ASP) | 4096 |
| Depth first search (DFS) | 4096 |
| Extended conectivity (ECFP: 0, 2, 4, 6) | 1024 |
| Functional class (FCFP: 0, 2, 4, 6) | 1024 |
| Pharmacophore: 2PPHAR/3PPHAR (2/3 point) | 4096 |
| ESTATE | 79 |
Descriptions and implementation details of the different fingerprints are provided in the article by Hinselmann et al [21] and the references therein
Data curation
Data for different endpoints were collected from previously published articles and databases with a primary source being the Online Chemical Database (OCHEM) [3]. The molecules were subsequently cleaned and duplicates (where present) were removed. Tables 2 and 3 lists the various endpoints and associated data sources considered in this study. Brief descriptions of the endpoints and the results from previous modelling efforts are provided in Additional file 1. Since, early identification of severe toxicity is a key requirement for the safety evaluation of drug candidates, we have evaluated a number of toxicity models covering a range of endpoints such as cardiac, hepatotoxicity, endocrine, urinary tract, carcinogenicity and cytotoxicity. While a majority of the models are binary classification models, for some endpoints such the metabolic intrinsic clearance, acute oral toxicity in rats, plasma protein binding and elimination half-life, multiclass models are proposed.
Table 2.
Summary of the ADMET endpoints studied
| Endpoint | Model | #Compounds | Group | Data source |
|---|---|---|---|---|
| Blood brain barrier | BC | 7236 | Distribution | [3, 31] |
| Oral bioavailability | BC | 1822 | Absorption | [3, 32] |
| Anticommensal effect | BC | 1181 | Toxicity | [33, 34] |
| CYP450 (1A2) inhibition | BC | 17119 | Metabolism | [35] |
| CYP450 (2C19) inhibition | BC | 17119 | Metabolism | [35] |
| CYP450 (2C9) inhibition | BC | 17119 | Metabolism | [35] |
| CYP450 (2D6) inhibition | BC | 17119 | Metabolism | [35] |
| CYP450 (3A4) inhibition | BC | 17119 | Metabolism | [35] |
| CYP450 (2C8) inhibition | BC | 533 | Metabolism | [36] |
| HIA | BC | 1516 | Absorption | [3, 37] |
| BCRP inhibition | BC | 2799 | Metabolism | [38] |
| Metabolic intrinsic clearance | MC | 5278 | Excretion | [39] |
| Human liver microsomal stability | BC | 3654 | [40] | |
| PGP inhibitor | BC | 2930 | Distribution | [3, 41] |
| PGP substrate | BC | 2198 | Distribution | [3, 41] |
| DMSO solubility | BC | 59047 | [42] | |
| Phosphate buffer solubility | BC | 57584 | [43] | |
| Skin sensitization (LLNA) | BC | 1033 | Toxicity | [44] |
| Skin sensitization (KeratinSens) | BC | 190 | Toxicity | [44] |
| Skin sensitization (HRIPT) | BC | 138 | Toxicity | [44] |
| Skin sensitization (h-CLAT) | BC | 160 | Toxicity | [44] |
| Skin sensitization (DPRA) | BC | 194 | Toxicity | [44] |
| Rat acute oral toxicity () | MC | 11363 | Toxicity | [3, 45] |
| AMES mutagenecity | BC | 7950 | Toxicity | [46] |
| Cytotoxicity (HepG2) | BC | 6081 | Toxicity | [10] |
| Cytotoxicity (CRL-7250 cell line) | BC | 5241 | Toxicity | [47] |
| Cytotoxicity (HACAT cell line) | BC | 5241 | Toxicity | [47] |
| Cytotoxicity (HEK cell line) | BC | 5241 | Toxicity | [47] |
| Cytotoxicity (NIK cell line) | BC | 5241 | Toxicity | [47] |
| DILI | BC | 2478 | Toxicity | [48] |
| Hemolytic toxicity (saponins) | BC | 452 | Toxicity | [49] |
| hERG cardiotoxicity | BC | 7889 | Toxicity | [50] |
| hERG liability | BC | 9204 | [51] | |
| Mitochondrial toxicity | BC | 6467 | Toxicity | [52] |
| Urinary tract toxicity | BC | 213 | Toxicity | [53, 54] |
| Phototoxicity | BC | 516 | Toxicity | [55] |
| Phototoxicity | BC | 1419 | Toxicity | [55] |
| Toxic myopathy | BC | 232 | Toxicity | [56] |
| Myelotoxicity | BC | 907 | Toxicity | [57] |
| Phospholipidosis | BC | 1719 | Toxicity | [58] |
| Choleostasis | BC | 1926 | Toxicity | [59] |
| Rhabdomyolysis | BC | 1504 | Toxicity | [60] |
| Respiratory toxicity | BC | 1241 | Toxicity | [61] |
| Ototoxicity | BC | 2612 | Toxicity | [62] |
| MATE1 inhibition | BC | 853 | Metabolism | [63] |
| Hepatic steatosis | BC | 512 | Toxicity | [64] |
| Carcinogenecity | BC | 1003 | Toxicity | [15] |
| OATP1B1 inhibition | BC | 1339 | Metabolism | [65] |
| OATP2B1 inhibition | BC | 230 | Metabolism | [65] |
| OATP1B3 inhibition | BC | 1249 | Metabolism | [65] |
| BSEP inhibition | BC | 1634 | Metabolism | [66] |
| OCT2 inhibition | BC | 907 | Metabolism | [67] |
| PPB | MC | 8103 | Distribution | [3, 68] |
| Elimination half-life Human | MC | 2127 | Excretion | [69] |
| Elimination half-life Mouse | MC | 808 | Excretion | [69] |
| Elimination half-life Rat | MC | 1308 | Excretion | [69] |
Here BC and MC refer to binary and multiclass classification respectively
OATP organic anion transporting polypeptide, CYP-450 cytochrome-P450, BCRP breast cancer resistance protein, BSEP bile salt export pump, DILI drug-induced liver injury, OCT organic cation transporter 2, MATE1 multidrug toxin extrusion transporter, hERG human Ether-á-go-go-related gene, HIA human intestinal absorption, PPB plasma protein binding, PGP p-glycoprotein, LLNA local lymph node assay, DPRA direct peptide reactivity assay, h-CLAT human cell line activation, HRIPT human repeat insult patch test, HEK 293 human embryonic kidney 293 cell, MATE1 multidrug and toxin extrusion transporter 1
Table 3.
Summary of the ADMET and other endpoints for which fingerprint-based regression models were evaluated
| Endpoint | #Compounds | Group | Data source |
|---|---|---|---|
| Aqueous solubility (S) | 9982 | [70] | |
| Intrinsic clearance () | 244 | Excretion | [71] |
| Skin penetration () | 211 | Toxicity | [72] |
| Human serum albumin | 198 | [73, 74] | |
| Human placenta barrier (clearance index) | 88 | Distribution | [75] |
| Cancer potency in mouse () | 402 | Toxicity | [76] |
| Cancer potency in rat () | 511 | Toxicity | [76] |
| Steady state volume distribution () | 1951 | Distribution | [3, 77] |
| Distribution coefficient ( D) | 7321 | [3, 78] | |
| Fraction unbound in human plasma | 2319 | Distribution | [79] |
| Fraction unbound in the brain | 253 | Distribution | [80] |
| Human liver microsomal clearance | 5348 | Excretion | [30] |
| Rat liver microsomal clearance | 2166 | Excretion | [30] |
| Mouse liver microsomal clearance | 790 | Excretion | [30] |
| CACO-2 permeability | 2578 | Absorption | [30] |
| 11041 | [81, 82] | ||
| MDCK cell line permeability | 701 | Absorption | [3] |
| Human renal clearance () | 636 | Excretion | [83] |
| Hemolytic toxicity () | 875 | Toxicity | [84] |
MDCK Madin-Darby canine kidney
For other endpoints, regression models have been evaluated (see Table 3). These include the CACO-2 permeability which is commonly used to predict the absorption of orally administered drugs and other xenobiotics, the fraction of unbound drug in plasma, the liver microsomal clearance (typically used to predict hepatic clearance in humans), in vitro human skin permeability and the cancer potency. Models for other ADMET-related properties have also been studied. For instance, properties such as the dissociation constant () affect solubility ( S), permeability, distribution coefficient ( D) and oral absorption. These in turn along with other properties such as the human serum albumin (HSA) binding impact pharmacokinetic behaviour and drug bioavailability.
Modelling
In order to build the models, the Random Forest algorithm [23] was chosen which is an ensemble learning method for both classification and regression. The algorithm makes use of bagging and feature randomness to build multiple decision trees (each trained on a random subset of data) and merges them together. The models were trained using the ranger [24] library in the statistical computing environment R [25]. The number of trees used to compute the final average predicted value was set to 500. For each endpoint, the data was split randomly into separate training (80%) and test (20%) sets. A fivefold cross-validation was used to identify the best performing model. In order to rule out any selection bias, we repeated random splitting 3 times and the results were averaged to gain an understanding of the variability. Furthermore, y-randomization tests were conducted to assess the robustness of the final model. To address the problem with unequal distribution of samples between classes, data augmentation of the minority class was carried out using the synthetic minority oversampling technique (SMOTE) [26].
For regression models, the performance was assessed using the squared regression coefficient () for the correlation between experimental and predicted values. the root mean squared error (RMSE) and the mean absolute error (MAE). For classification models, metrics that are sensitive to the class imbalance have been used. These include the balanced accuracy (BACC) given by:
| 1 |
where is the number of correct predictions in class i, m is the number of classes and is the number of examples in class i. In addition, other metrics such as the overall accuracy, the sensitivity (the true positive rate—TPR) and specificity (the true negative rate—TNR) and the area under the curve (AUC) are also reported (see Additional file 1).
Every model has a finite applicability domain (AD) within which its predictions can be trusted. For regression models, we quantify the prediction intervals (95%) using the quantile regression forests approach [27]. Here, a shorter prediction interval indicates the higher stability of prediction. In the case of classification, two values: confidence and credibility are associated with the predicted label based on the conformal prediction framework [28, 29]. While the confidence provides a measure of how likely a prediction is compared to all other possible classifications, the credibility measure (equal to the highest p-value of any one of the possible classifications being the true label) provides an indication of how good the training set is for classifying the given example.
Results and discussion
For the various endpoints, the relevant performance metrics associated with the best fingerprint-based models are summarized in Tables 4 (for classification models) and 5 (for regression models). The complete performance summary for the training and validations sets is listed in Additional file 1: Tables S1 and S2. For all cases, permutation tests confirmed (p-values < 0.001) that the probability that the model was obtained by chance is quite low. Overall, high classification accuracies () are obtained for the blood brain barrier permeability, plasma protein binding, CYP450 inhibition (3A4/2C19/1A2/2C9/2C8 isoforms), human intestinal absorption, breast cancer resistance protein inhibition, p-glycoprotein inhibitor/substrate and hemolytic/respiratory toxicity. For some of the other endpoints such as the mitochondrial/urinary tract toxicity, human liver microsomal stability, metabolic intrinsic clearance, AMES mutagenecity, cytotoxicity (multiple cell lines), hERG cardiotoxicity/liability, drug induced liver injury, myelotoxicity, phospholipidosis, rhabdomyolysis, OATP1B1/OATP1B3 inhibition, BSEP and OCT2 inhibition, moderate () performances were observed. Properties such as skin sensitization, acute oral toxicity, phototoxicity in humans, ototoxicity, choleostasis, hepatic steatosis, and carcinogenecity yielded somewhat average results. In the case of regression models, performances were largely on the poorer side with the exception of , S, D, human serum albumin and skin penetration, .
Table 4.
Performance metrics for the best performing fingerprint-based classification models
| Endpoint | FP | Calibration | Validation | ||
|---|---|---|---|---|---|
| BACC | AUC | BACC | AUC | ||
| Blood brain barrier | PUBCHEM | 0.82 | 0.90 | 0.81 | 0.92 |
| Oral bioavailability | PUBCHEM | 0.71 | 0.77 | 0.71 | 0.78 |
| Anticommensal effect | PUBCHEM | 0.76 | 0.82 | 0.74 | 0.81 |
| CYP450 (1A2) | PUBCHEM | 0.85 | 0.93 | 0.85 | 0.93 |
| CYP450 (2C19) | ECFP4 | 0.81 | 0.88 | 0.81 | 0.89 |
| CYP450 (2C9) | PUBCHEM | 0.78 | 0.88 | 0.79 | 0.89 |
| CYP450 (2D6) | FCFP4 | 0.73 | 0.86 | 0.73 | 0.87 |
| CYP450 (3A4) | FCFP6 | 0.80 | 0.89 | 0.80 | 0.90 |
| CYP450 (2C8) | PUBCHEM | 0.79 | 0.89 | 0.77 | 0.90 |
| HIA | MACCS | 0.84 | 0.89 | 0.83 | 0.89 |
| BCRP inhibition | FCFP4 | 0.89 | 0.95 | 0.90 | 0.96 |
| Metabolic intrinsic clearance | FCFP4 | 0.74 | 0.82 | 0.74 | 0.84 |
| Human liver microsomal stability | AT2D | 0.77 | 0.83 | 0.77 | 0.84 |
| PGP inhibitor | PUBCHEM | 0.84 | 0.91 | 0.85 | 0.92 |
| PGP substrate | ASP | 0.80 | 0.87 | 0.80 | 0.88 |
| DMSO solubility | ECFP2 | 0.72 | 0.78 | 0.73 | 0.80 |
| Phosphate buffer solubility | PUBCHEM | 0.79 | 0.87 | 0.79 | 0.87 |
| Skin sensitization (LLNA) | PUBCHEM | 0.69 | 0.76 | 0.67 | 0.74 |
| Skin sensitization (KeratinSens) | LSTAR | 0.64 | 0.65 | 0.57 | 0.60 |
| Skin sensitization (HRIPT) | ECFP0 | 0.70 | 0.74 | 0.67 | 0.72 |
| Skin sensitization (hCLAT) | MACCS | 0.65 | 0.70 | 0.61 | 0.68 |
| Skin sensitization (DPRA) | FCFP4 | 0.68 | 0.72 | 0.68 | 0.72 |
| Rat acute oral toxicity () | PUBCHEM | 0.69 | 0.78 | 0.68 | 0.81 |
| AMES mutagenecity | PUBCHEM | 0.79 | 0.86 | 0.79 | 0.87 |
| Cytotoxicity (HepG2) | AT2D | 0.78 | 0.85 | 0.78 | 0.85 |
| Cytotoxicity (CRL-7250 cell line) | AT2D | 0.79 | 0.87 | 0.78 | 0.86 |
| Cytotoxicity (HACAT cell line) | AT2D | 0.77 | 0.85 | 0.77 | 0.85 |
| Cytotoxicity (HEK cell line) | PUBCHEM | 0.77 | 0.87 | 0.76 | 0.86 |
| Cytotoxicity (NIK cell line) | PUBCHEM | 0.78 | 0.87 | 0.78 | 0.87 |
| DILI | PUBCHEM | 0.78 | 0.86 | 0.79 | 0.88 |
| Hemolytic toxicity (saponins) | FCFP6 | 0.84 | 0.88 | 0.85 | 0.90 |
| hERG cardiotoxicity | FCFP6 | 0.79 | 0.86 | 0.80 | 0.88 |
| hERG liability | PUBCHEM | 0.76 | 0.87 | 0.76 | 0.88 |
| Mitochondrial toxicity | PUBCHEM | 0.79 | 0.90 | 0.77 | 0.90 |
| Urinary tract toxicity | FCFP4 | 0.71 | 0.77 | 0.70 | 0.73 |
| Phototoxicity in vitro | KR | 0.70 | 0.76 | 0.69 | 0.80 |
| Phototoxicity human | PUBCHEM | 0.69 | 0.75 | 0.67 | 0.75 |
| Toxic myopathy | DFS | 0.68 | 0.74 | 0.63 | 0.74 |
| Myelotoxicity | FCFP4 | 0.72 | 0.79 | 0.71 | 0.80 |
| phospholipidosis | FCFP2 | 0.78 | 0.86 | 0.77 | 0.88 |
| Cholestasis | RAD2D | 0.67 | 0.73 | 0.67 | 0.74 |
| Rhabdomyolysis | MACCS | 0.71 | 0.80 | 0.70 | 0.83 |
| Respiratory toxicity | MACCS | 0.82 | 0.88 | 0.82 | 0.89 |
| Ototoxicity | PUBCHEM | 0.69 | 0.74 | 0.67 | 0.72 |
| MATE1 | DFS | 0.64 | 0.67 | 0.65 | 0.65 |
| Hepatic steatosis | MACCS | 0.63 | 0.67 | 0.59 | 0.68 |
| Carcinogenecity | PUBCHEM | 0.67 | 0.71 | 0.68 | 0.75 |
| OATP1B1 inhibition | ECFP6 | 0.72 | 0.80 | 0.73 | 0.82 |
| OATP2B1 inhibition | ECFP6 | 0.67 | 0.68 | 0.65 | 0.70 |
| OATP1B3 inhibition | PUBCHEM | 0.74 | 0.83 | 0.77 | 0.87 |
| BSEP inhibition | ECFP4 | 0.85 | 0.93 | 0.88 | 0.95 |
| OCT2 inhibition | PUBCHEM | 0.73 | 0.81 | 0.73 | 0.79 |
| PPB | PUBCHEM | 0.82 | 0.92 | 0.84 | 0.92 |
| Elimination half-life Human | ASP | 0.75 | 0.86 | 0.76 | 0.88 |
| Elimination half-life Mouse | ECFP2 | 0.74 | 0.86 | 0.72 | 0.84 |
| Elimination half-life Rat | KR | 0.74 | 0.86 | 0.74 | 0.83 |
The values reported are the balanced accuracies (BACC) and area under the ROC curve (AUC) (average of 3 independent runs) for the calibration/validation sets
Table 5.
Performance metrics for the best performing fingerprint-based regression models
| Endpoint | FP | Calibration | Validation | ||||
|---|---|---|---|---|---|---|---|
| R2 | RMSE | MAE | R2 | RMSE | MAE | ||
| PUBCHEM | 0.77 | 1.15 | 0.81 | 0.78 | 1.12 | 0.78 | |
| Intrinsic clearance () | RAD2D | 0.48 | 0.83 | 0.65 | 0.29 | 1.02 | 0.82 |
| Skin penetration () | PUBCHEM | 0.73 | 0.60 | 0.48 | 0.75 | 0.56 | 0.43 |
| Human serum albumin | AP2D | 0.71 | 0.33 | 0.23 | 0.69 | 0.39 | 0.26 |
| Human placenta barrier | KR | 0.41 | 0.24 | 0.20 | 0.24 | 0.32 | 0.22 |
| Cancer potency in mouse () | AT2D | 0.33 | 0.98 | 0.75 | 0.27 | 0.96 | 0.72 |
| Cancer potency in rat () | AT2D | 0.41 | 1.08 | 0.83 | 0.35 | 1.14 | 0.87 |
| Steady state volume distribution () | ASP | 0.58 | 0.44 | 0.29 | 0.45 | 0.51 | 0.32 |
| Distribution coefficient () | PUBCHEM | 0.76 | 0.73 | 0.53 | 0.77 | 0.71 | 0.50 |
| Fraction unbound in human plasma | PUBCHEM | 0.60 | 0.46 | 0.35 | 0.63 | 0.44 | 0.34 |
| Fraction unbound in the brain | PUBCHEM | 0.48 | 0.58 | 0.46 | 0.56 | 0.56 | 0.45 |
| Human liver microsomal clearance | KR | 0.51 | 1.08 | 0.80 | 0.56 | 1.05 | 0.79 |
| Mouse liver microsomal clearance | AT2D | 0.52 | 1.21 | 0.92 | 0.53 | 1.16 | 0.88 |
| Rat liver microsomal clearance | KR | 0.64 | 1.08 | 0.83 | 0.67 | 1.01 | 0.76 |
| CACO-2 permeability | FCFP4 | 0.44 | 0.68 | 0.46 | 0.42 | 0.69 | 0.46 |
| ECFP2 | 0.71 | 1.85 | 1.15 | 0.74 | 1.78 | 1.11 | |
| MDCK cell line permeability | ECFP4 | 0.62 | 0.61 | 0.44 | 0.68 | 0.56 | 0.39 |
| Human renal clearance | MACCS | 0.25 | 0.54 | 0.43 | 0.27 | 0.53 | 0.42 |
| Hemolytic toxicity () | ASP | 0.68 | 0.47 | 0.35 | 0.68 | 0.44 | 0.34 |
The values reported are the squared correlation (), RMSE and MAE (average of 3 independent runs) for the calibration/validation sets
To identify which of the fingerprints perform well on the different datasets, we plotted heatmaps (see Figs. 1 and 2) of the balanced accuracies (for classification models) and squared correlations (in the case of regression) obtained for the different endpoints. While the pharmacaphore fingerprints (2PPHAR/3PPHAR) perform poorly on all datasets, fingerprints based on substructure keys (PUBCHEM, MACCS, KR) show moderate to high accuracies for a majority of the modelled endpoints. Although the performances for regression models are somewhat less encouraging, here too the for PUBCHEM, ECFP4, and ASP fingerprints yield better models than the other fingerprints tested.
Fig. 1.
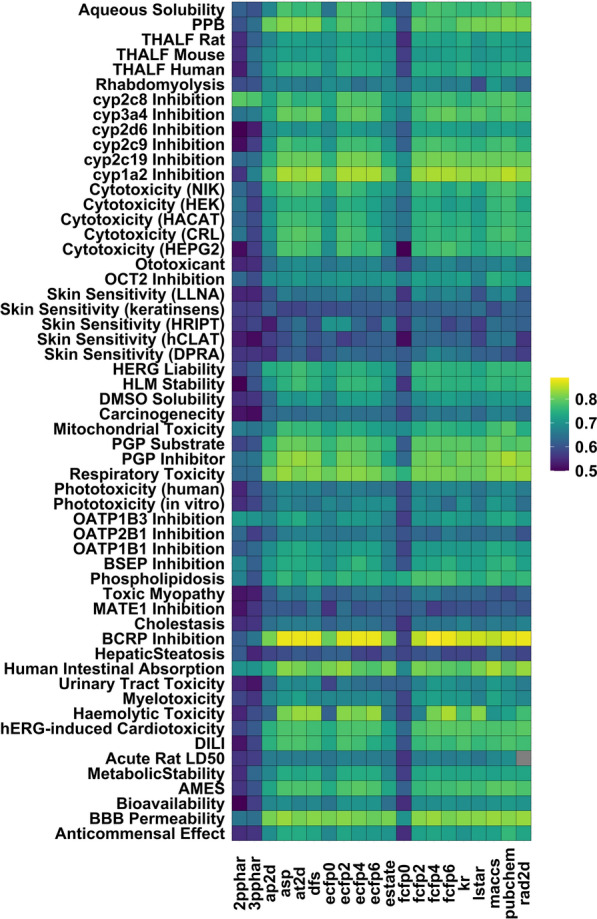
Heatmap showing the cross-validated balanced accuracies (average of 3 independent runs) achieved by different fingerprint-based models for the endpoints studied
Fig. 2.

Heatmap showing the cross-validated correlation coefficients (average of 3 independent runs) achieved by different fingerprint-based models for the endpoints studied
We further compared the performances achieved by the fingerprint models with those obtained for the 2D/3D descriptor based approaches. The barplots in Fig. 3 compare the accuracies achieved by the fingerprint models with values reported by the models published earlier. While results for most properties are comparable, for some endpoints such as myelotoxicity, ototoxicity, myopathy accuracies obtained using 2D/3D descriptors are only marginally better. Indeed better results are obtained for rhabdomyolysis, phospholipidosis, phototoxicity with other descriptor based models. For phototoxicity in particular, quantum chemistry-based 3D descriptors are used which can add to the time taken. It must however be pointed out that some of the better performing models take advantage of deep learning. Attempts to improve results for selected properties were carried out using support vector machines. However, the models were not always found to improve on the random forest approach.
Fig. 3.

Comparison of the accuracies achieved by the fingerprint based models in this study (“Current”) with those created using standard molecular graph based descriptors (“Original”) published in the literature. For OATP inhibtion, descriptors consist of constitutional, geometrical, electrostatic, and physicochemical indices. For phototoxicity, descriptors contain HOMO-LUMO gaps, spectral integrals, ionization potential, electron affinity and CATS descriptors. For properties such as toxic myopathy and MATE1 inhibition, the values compared are the accuracies and AUCs respectively
For the regression models calculated for selected properties: , S, D, skin penetration, human serum albumin, MDCK permeability , we assessed the prediction reliability based on the prediction intervals. Plots of the prediction intervals with respect to the observed response values for the test sets (see Additional file 1: Figure S1) showed that most of the samples lie within the 95% prediction interval which indicates that the constructed prediction intervals are reliable. For classification models, we focused on excluding compounds whose labels are predicted with low confidence and credibility. Thus, different thresholds for p-values (0.5, 0.6, 0.7, 0.8, 0.9) were applied and the corresponding fraction of molecules that would be withheld from further testing was recorded. A plot of the overall error rates and the percentage reduction in compounds excluded from further processing (see Additional file 1: Figure S2) shows that for many of the endpoints modelled, the predictive performance is not significantly impacted even at cutoffs of 0.50. Such a strategy that allows for compound selection based on static thresholds for the confidence/credibility offer a way to reduce the number of compounds that typically undergo experimental testing.
Software usage
FP-ADMET is available as open access software (GNU GPL v3.0) and can be downloaded from https://gitlab.com/vishsoft/fpadmet. Use of FP-ADMET proceeds in two steps (i) fingerprint calculation followed by (ii) predicting the ADMET endpoint of interest. The software is command line driven and is governed by a shell script (runadmet.sh) that can be run as:
bash runadmet.sh -f molecule.smi -p ## -a
The input to the script is a file (molecule.smi) containing SMILES strings. The ## is a number between 1 (predict Anticommensal Effect) and 56 (predict skin penetration) and corresponds to the prediction task. The results are written to a text file where each line contains molecule name and the predicted response. The “-a” option allows for the calculation of prediction intervals (in the case of regression) and confidence (for classification). For classification, conformal prediction is used to calculate a confidence (how certain the model is that the prediction is a singleton) and a credibility. For example, predicting AMES mutagenecity (task number 4) for a series of molecules produces the following results (see Table 6). The label “inactive” for compound G00001 suggests that the compound is predicted to be non-mutagenic. A confidence value of 0.95 suggests that the classifier is quite certain that the prediction is likely to be a single label. A relatively low value of credibility (0.57) suggests that the compounds like G00001 are not sufficiently represented in the training set and that the user needs to treat the prediction with caution. In the case of regression, a 95% prediction interval (predictions at the 0.025 and 97.5 percentiles for ) is calculated and provides a range for the predictions on an individual observation. Narrow prediction intervals indicate a lower uncertainty associated with the prediction.
Table 6.
Example showing the property ( and anticommensal effect) predictions and associated uncertainties for 3 molecules
| Name | Anticommensal effect | Confidence | Credibility | Q = 0.025 | Q = 0.975 | |
|---|---|---|---|---|---|---|
| G00001 | Inactive | 0.95 | 0.57 | 9.62 | 4.89 | 11.49 |
| G00002 | Active | 0.95 | 0.51 | 4.41 | − 1.60 | 13.06 |
| G00003 | Inactive | 0.95 | 0.57 | 3.37 | 1.66 | 6.10 |
and are the predictions calculated at percentiles 0.025 and 0.975 and allow for 95% prediction intervals
Conclusion
In this article, we have evaluated the performance of various molecular fingerprints for predicting a number of ADMET and ADMET-related endpoints. A total of 1500 models were analysed spanning 75 responses and 20 fingerprints. The results show that the machine learning performance using the different fingerprint encodings rival those of traditional descriptor-based methods. Future work will focus on combining different data sets in a multitask modeling approach which has been shown to yield statistically superior results compared with single-task models [12, 30]. In order to facilitate ADMET evaluation, the best performing models have been compiled into an open access software package called FPADMET that can be downloaded from https://gitlab.com/vishsoft/fpadmet.
Supplementary Information
Additional file 1. File contains brief descriptions of the properties modelled, additional performance statistics and figures referred to in the text.
Acknowledgements
The author thanks Dr. Amitava Roy (NIH) and Assoc. Prof. Travis Wheeler (University of Montana) for fruitful discussions.
Authors’ contributions
VV conceived and designed the study, performed the data analysis and wrote the paper. The author read and approved the final manuscript.
Funding
This work was supported through a grant (Grant No. 262152) from the Research Council of Norway.
Declarations
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Ferreira LLG, Andricopulo AD. ADMET modeling approaches in drug discovery. Drug Discov Today. 2019;24(5):1157–1165. doi: 10.1016/j.drudis.2019.03.015. [DOI] [PubMed] [Google Scholar]
- 2.Kar S, Leszczynski J. Open access in silico tools to predict the ADMET profiling of drug candidates. Expert Opin Drug Discov. 2020;15(12):1473–1487. doi: 10.1080/17460441.2020.1798926. [DOI] [PubMed] [Google Scholar]
- 3.Sushko I, Novotarskyi S, Körner R, Pandey AK, Rupp M, Teetz W, Brandmaier S, Abdelaziz A, Prokopenko VV, Tanchuk VY, Todeschini R, Varnek A, Marcou G, Ertl P, Potemkin V, Grishina M, Gasteiger J, Schwab C, Baskin II, Palyulin VA, Radchenko EV, Welsh WJ, Kholodovych V, Chekmarev D, Cherkasov A, Aires-de-Sousa J, Zhang Q-Y, Bender A, Nigsch F, Patiny L, Williams A, Tkachenko V, Tetko IV. Online chemical modeling environment (OCHEM): web platform for data storage, model development and publishing of chemical information. J Comput Aided Mol Des. 2011;25(6):533–554. doi: 10.1007/s10822-011-9440-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guan L, Yang H, Cai Y, Sun L, Di P, Li W, Liu G, Tang Y. ADMET-score—a comprehensive scoring function for evaluation of chemical drug-likeness. MedChemComm. 2019;10(1):148–157. doi: 10.1039/c8md00472b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jia C-Y, Li J-Y, Hao G-F, Yang G-F. A drug-likeness toolbox facilitates ADMET study in drug discovery. Drug Discov Today. 2020;25(1):248–258. doi: 10.1016/j.drudis.2019.10.014. [DOI] [PubMed] [Google Scholar]
- 6.Pires DEV, Blundell TL, Ascher DB. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem. 2015;58(9):4066–4072. doi: 10.1021/acs.jmedchem.5b00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clark AM, Dole K, Coulon-Spektor A, McNutt A, Grass G, Freundlich JS, Reynolds RC, Ekins S. Open source Bayesian models. 1. Application to ADME/Tox and drug discovery datasets. J Chem Inf Model. 2015;55(6):1231–1245. doi: 10.1021/acs.jcim.5b00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lagorce D, Bouslama L, Becot J, Miteva MA, Villoutreix BO. FAF-drugs4: free ADME-Tox filtering computations for chemical biology and early stages drug discovery. Bioinformatics. 2017;33(22):3658–3660. doi: 10.1093/bioinformatics/btx491. [DOI] [PubMed] [Google Scholar]
- 9.Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017 doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schyman P, Liu R, Desai V, Wallqvist A. vNN web server for ADMET predictions. Front Pharmacol. 2017 doi: 10.3389/fphar.2017.00889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang H, Lou C, Sun L, Li J, Cai Y, Wang Z, Li W, Liu G, Tang Y. admetSAR 2.0: web-service for prediction and optimization of chemical ADMET properties. Bioinformatics. 2018;35(6):1067–1069. doi: 10.1093/bioinformatics/bty707. [DOI] [PubMed] [Google Scholar]
- 12.Xiong G, Wu Z, Yi J, Fu L, Yang Z, Hsieh C, Yin M, Zeng X, Wu C, Lu A, Chen X, Hou T, Cao D. ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic Acids Res. 2021;49(W1):5–14. doi: 10.1093/nar/gkab255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Banerjee P, Eckert AO, Schrey AK, Preissner R. ProTox-II: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018;46(W1):257–263. doi: 10.1093/nar/gky318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patel RD, Kumar SP, Pandya HA, Solanki HA. MDCKpred: a web-tool to calculate MDCK permeability coefficient of small molecule using membrane-interaction chemical features. Toxicol Mech Methods. 2018;28(9):685–698. doi: 10.1080/15376516.2018.1499840. [DOI] [PubMed] [Google Scholar]
- 15.Zhang L, Ai H, Chen W, Yin Z, Hu H, Zhu J, Zhao J, Zhao Q, Liu H. CarcinoPred-EL: novel models for predicting the carcinogenicity of chemicals using molecular fingerprints and ensemble learning methods. Sci Rep. 2017 doi: 10.1038/s41598-017-02365-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y-W, Huang L, Jiang S-W, Li K, Zou J, Yang S-Y. CapsCarcino: a novel sparse data deep learning tool for predicting carcinogens. Food Chem Toxicol. 2020;135:110921. doi: 10.1016/j.fct.2019.110921. [DOI] [PubMed] [Google Scholar]
- 17.Yap CW. PaDEL-descriptor: an open source software to calculate molecular descriptors and fingerprints. J Comp Chem. 2010;32(7):1466–1474. doi: 10.1002/jcc.21707. [DOI] [PubMed] [Google Scholar]
- 18.Venkatraman V, Alsberg BK. KRAKENX: software for the generation of alignment-independent 3D descriptors. J Mol Model. 2016 doi: 10.1007/s00894-016-2957-5. [DOI] [PubMed] [Google Scholar]
- 19.Muegge I, Mukherjee P. An overview of molecular fingerprint similarity search in virtual screening. Expert Opin Drug Discov. 2015;11(2):137–148. doi: 10.1517/17460441.2016.1117070. [DOI] [PubMed] [Google Scholar]
- 20.Cereto-Massagué A, Ojeda MJ, Valls C, Mulero M, Garcia-Vallvé S, Pujadas G. Molecular fingerprint similarity search in virtual screening. Methods. 2015;71:58–63. doi: 10.1016/j.ymeth.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 21.Hinselmann G, Rosenbaum L, Jahn A, Fechner N, Zell A. jCompoundMapper: an open source java library and command-line tool for chemical fingerprints. J Cheminf. 2011 doi: 10.1186/1758-2946-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Willighagen EL, Mayfield JW, Alvarsson J, Berg A, Carlsson L, Jeliazkova N, Kuhn S, Pluskal T, Rojas-Chertó M, Spjuth O, Torrance G, Evelo CT, Guha R, Steinbeck C. The chemistry development kit (CDK) v2.0: atom typing, depiction, molecular formulas, and substructure searching. J Cheminf. 2017 doi: 10.1186/s13321-017-0220-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Breiman L. Random forests. Mach Learn. 2001;45(1):5–32. doi: 10.1023/a:1010933404324. [DOI] [Google Scholar]
- 24.Wright MN, Ziegler A. ranger: a fast implementation of random forests for high dimensional data in C++ and R. J Stat Soft. 2017;77(1):1–17. doi: 10.18637/jss.v077.i01. [DOI] [Google Scholar]
- 25.R Core Team (2020) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. R Foundation for Statistical Computing. https://www.R-project.org/
- 26.Chawla NV, Bowyer KW, Hall LO, Kegelmeyer WP. SMOTE synthetic minority over-sampling technique. J Artif Intell Res. 2002;16:321–357. doi: 10.1613/jair.953. [DOI] [Google Scholar]
- 27.Meinshausen N. Quantile regression forests. J Mach Learn Res. 2006;7(35):983–999. [Google Scholar]
- 28.Papadopoulos H. Chap. 18. Inductive conformal prediction: theory and application to neural networks. In: Fritzsche P, editor. Tools in artificial intelligence. Rijeka: IntechOpen; 2008. [Google Scholar]
- 29.Ahlberg E, Hammar O, Bendtsen C, Carlsson L. Current application of conformal prediction in drug discovery. Ann Math Artif Intell. 2017;81(1–2):145–154. doi: 10.1007/s10472-017-9550-1. [DOI] [Google Scholar]
- 30.Wenzel J, Matter H, Schmidt F. Predictive multitask deep neural network models for ADME-Tox properties: learning from large data sets. J Chem Inf Model. 2019;59(3):1253–1268. doi: 10.1021/acs.jcim.8b00785. [DOI] [PubMed] [Google Scholar]
- 31.Shaker B, Yu M-S, Song JS, Ahn S, Ryu JY, Oh K-S, Na D. LightBBB: computational prediction model of blood–brain-barrier penetration based on LightGBM. Bioinformatics. 2020 doi: 10.1093/bioinformatics/btaa918. [DOI] [PubMed] [Google Scholar]
- 32.Falcón-Cano G, Molina C, Cabrera-Pérez MÁ. ADME prediction with KNIME: development and validation of a publicly available workflow for the prediction of human oral bioavailability. J Chem Inf Model. 2020;60(6):2660–2667. doi: 10.1021/acs.jcim.0c00019. [DOI] [PubMed] [Google Scholar]
- 33.Maier L, Pruteanu M, Kuhn M, Zeller G, Telzerow A, Anderson EE, Brochado AR, Fernandez KC, Dose H, Mori H, Patil KR, Bork P, Typas A. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature. 2018;555(7698):623–628. doi: 10.1038/nature25979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zheng S, Chang W, Liu W, Liang G, Xu Y, Lin F. Computational prediction of a new ADMET endpoint for small molecules: anticommensal effect on human gut microbiota. J Chem Inf Model. 2018;59(3):1215–1220. doi: 10.1021/acs.jcim.8b00600. [DOI] [PubMed] [Google Scholar]
- 35.Veith H, Southall N, Huang R, James T, Fayne D, Artemenko N, Shen M, Inglese J, Austin CP, Lloyd DG, Auld DS. Comprehensive characterization of cytochrome p450 isozyme selectivity across chemical libraries. Nat Biotechnol. 2009;27(11):1050–1055. doi: 10.1038/nbt.1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang X, Zhao P, Wang Z, Xu X, Liu G, Tang Y, Li W. In silico prediction of CYP2c8 inhibition with machine-learning methods. Chem Res Toxicol. 2021;34(8):1850–1859. doi: 10.1021/acs.chemrestox.1c00078. [DOI] [PubMed] [Google Scholar]
- 37.Wang N-N, Huang C, Dong J, Yao Z-J, Zhu M-F, Deng Z-K, Lv B, Lu A-P, Chen AF, Cao D-S. Predicting human intestinal absorption with modified random forest approach: a comprehensive evaluation of molecular representation, unbalanced data, and applicability domain issues. RSC Adv. 2017;7(31):19007–19018. doi: 10.1039/c6ra28442f. [DOI] [Google Scholar]
- 38.Jiang D, Lei T, Wang Z, Shen C, Cao D, Hou T. ADMET evaluation in drug discovery. 20. Prediction of breast cancer resistance protein inhibition through machine learning. J Cheminf. 2020 doi: 10.1186/s13321-020-00421-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Esaki T, Watanabe R, Kawashima H, Ohashi R, Natsume-Kitatani Y, Nagao C, Mizuguchi K. Data curation can improve the prediction accuracy of metabolic intrinsic clearance. Mol Inf. 2018;38(1–2):1800086. doi: 10.1002/minf.201800086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu R, Schyman P, Wallqvist A. Critically assessing the predictive power of QSAR models for human liver microsomal stability. J Chem Inf Model. 2015;55(8):1566–1575. doi: 10.1021/acs.jcim.5b00255. [DOI] [PubMed] [Google Scholar]
- 41.Wang P-H, Tu Y-S, Tseng YJ. PgpRules: a decision tree based prediction server for p-glycoprotein substrates and inhibitors. Bioinformatics. 2019;35(20):4193–4195. doi: 10.1093/bioinformatics/btz213. [DOI] [PubMed] [Google Scholar]
- 42.Tetko IV, Novotarskyi S, Sushko I, Ivanov V, Petrenko AE, Dieden R, Lebon F, Mathieu B. Development of dimethyl sulfoxide solubility models using 163000 molecules: using a domain applicability metric to select more reliable predictions. J Chem Inf Model. 2013;53(8):1990–2000. doi: 10.1021/ci400213d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perryman AL, Inoyama D, Patel JS, Ekins S, Freundlich JS. Pruned machine learning models to predict aqueous solubility. ACS Omega. 2020;5(27):16562–16567. doi: 10.1021/acsomega.0c01251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Borba JVB, Braga RC, Alves VM, Muratov EN, Kleinstreuer N, Tropsha A, Andrade CH. Pred-skin: a web portal for accurate prediction of human skin sensitizers. Chem Res Toxicol. 2020 doi: 10.1021/acs.chemrestox.0c00186. [DOI] [PubMed] [Google Scholar]
- 45.Gadaleta D, Vuković K, Toma C, Lavado GJ, Karmaus AL, Mansouri K, Kleinstreuer NC, Benfenati E, Roncaglioni A. SAR and QSAR modeling of a large collection of LD50 rat acute oral toxicity data. J Cheminf. 2019 doi: 10.1186/s13321-019-0383-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu C, Cheng F, Chen L, Du Z, Li W, Liu G, Lee PW, Tang Y. In silico prediction of chemical Ames mutagenicity. J Chem Inf Model. 2012;52(11):2840–2847. doi: 10.1021/ci300400a. [DOI] [PubMed] [Google Scholar]
- 47.Sun H, Wang Y, Cheff DM, Hall MD, Shen M. Predictive models for estimating cytotoxicity on the basis of chemical structures. Bioorg Med Chem. 2020;28(10):115422. doi: 10.1016/j.bmc.2020.115422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mora JR, Marrero-Ponce Y, García-Jacas CR, Causado AS. Ensemble models based on QuBiLS-MAS features and shallow learning for the prediction of drug-induced liver toxicity: improving deep learning and traditional approaches. Chem Res Toxicol. 2020;33(7):1855–1873. doi: 10.1021/acs.chemrestox.0c00030. [DOI] [PubMed] [Google Scholar]
- 49.Zheng S, Wang Y, Liu W, Chang W, Liang G, Xu Y, Lin F. In silico prediction of hemolytic toxicity on the human erythrocytes for small molecules by machine-learning and genetic algorithm. J Med Chem. 2019;63(12):6499–6512. doi: 10.1021/acs.jmedchem.9b00853. [DOI] [PubMed] [Google Scholar]
- 50.Cai C, Guo P, Zhou Y, Zhou J, Wang Q, Zhang F, Fang J, Cheng F. Deep learning-based prediction of drug-induced cardiotoxicity. J Chem Inf Model. 2019;59(3):1073–1084. doi: 10.1021/acs.jcim.8b00769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Siramshetty VB, Nguyen D-T, Martinez NJ, Southall NT, Simeonov A, Zakharov AV. Critical assessment of artificial intelligence methods for prediction of hERG channel inhibition in the "big data" era. J Chem Inf Model. 2020;60(12):6007–6019. doi: 10.1021/acs.jcim.0c00884. [DOI] [PubMed] [Google Scholar]
- 52.Hemmerich J, Troger F, Füzi B, Ecker FG. Using machine learning methods and structural alerts for prediction of mitochondrial toxicity. Mol Inf. 2020;39(5):2000005. doi: 10.1002/minf.202000005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lei T, Sun H, Kang Y, Zhu F, Liu H, Zhou W, Wang Z, Li D, Li Y, Hou T. ADMET evaluation in drug discovery. 18. Reliable prediction of chemical-induced urinary tract toxicity by boosting machine learning approaches. Mol Pharm. 2017;14(11):3935–3953. doi: 10.1021/acs.molpharmaceut.7b00631. [DOI] [PubMed] [Google Scholar]
- 54.Zhang H, Ren J-X, Ma J-X, Ding L. Development of an in silico prediction model for chemical-induced urinary tract toxicity by using Naïve Bayes classifier. Mol Divers. 2018;23(2):381–392. doi: 10.1007/s11030-018-9882-8. [DOI] [PubMed] [Google Scholar]
- 55.Schmidt F, Wenzel J, Halland N, Güssregen S, Delafoy L, Czich A. Computational investigation of drug phototoxicity: photosafety assessment, photo-toxophore identification, and machine learning. Chem Res Toxicol. 2019;32(11):2338–2352. doi: 10.1021/acs.chemrestox.9b00338. [DOI] [PubMed] [Google Scholar]
- 56.Hu X, Yan A. In silico models to discriminate compounds inducing and noninducing toxic myopathy. Mol Inf. 2011;31(1):27–39. doi: 10.1002/minf.201100067. [DOI] [PubMed] [Google Scholar]
- 57.Zhang H, Yu P, Zhang T-G, Kang Y-L, Zhao X, Li Y-Y, He J-H, Zhang J. In silico prediction of drug-induced myelotoxicity by using Naïve Bayes method. Mol Divers. 2015;19(4):945–953. doi: 10.1007/s11030-015-9613-3. [DOI] [PubMed] [Google Scholar]
- 58.Fusani L, Brown M, Chen H, Ahlberg E, Noeske T. Predicting the risk of phospholipidosis with in silico models and an image-based in vitro screen. Mol Pharm. 2017;14(12):4346–4352. doi: 10.1021/acs.molpharmaceut.7b00388. [DOI] [PubMed] [Google Scholar]
- 59.Kotsampasakou E, Ecker GF. Predicting drug-induced cholestasis with the help of hepatic transporters—an in silico modeling approach. J Chem Inf Model. 2017;57(3):608–615. doi: 10.1021/acs.jcim.6b00518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cui X, Liu J, Zhang J, Wu Q, Li X. In silico prediction of drug-induced rhabdomyolysis with machine-learning models and structural alerts. J Appl Toxicol. 2019;39(8):1224–1232. doi: 10.1002/jat.3808. [DOI] [PubMed] [Google Scholar]
- 61.Zhang H, Ma J-X, Liu C-T, Ren J-X, Ding L. Development and evaluation of in silico prediction model for drug-induced respiratory toxicity by using Naïve Bayes classifier method. Food Chem Toxicol. 2018;121:593–603. doi: 10.1016/j.fct.2018.09.051. [DOI] [PubMed] [Google Scholar]
- 62.Zhang H, Liu C-T, Mao J, Shen C, Xie R-L, Mu B. Development of novel in silico prediction model for drug-induced ototoxicity by using Naïve Bayes classifier approach. Toxicol In Vitro. 2020;65:104812. doi: 10.1016/j.tiv.2020.104812. [DOI] [PubMed] [Google Scholar]
- 63.Wittwer MB, Zur AA, Khuri N, Kido Y, Kosaka A, Zhang X, Morrissey KM, Sali A, Huang Y, Giacomini KM. Discovery of potent, selective multidrug and toxin extrusion transporter 1 (MATE1, SLC47a1) inhibitors through prescription drug profiling and computational modeling. J Med Chem. 2013;56(3):781–795. doi: 10.1021/jm301302s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jain S, Norinder U, Escher SE, Zdrazil B. Combining in vivo data with in silico predictions for modeling hepatic steatosis by using stratified bagging and conformal prediction. Chem Res Toxicol. 2020 doi: 10.1021/acs.chemrestox.0c00511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Türková A, Jain S, Zdrazil B. Integrative data mining, scaffold analysis, and sequential binary classification models for exploring ligand profiles of hepatic organic anion transporting polypeptides. J Chem Inf Model. 2018;59(5):1811–1825. doi: 10.1021/acs.jcim.8b00466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McLoughlin KS, Jeong CG, Sweitzer TD, Minnich AJ, Tse MJ, Bennion BJ, Allen JE, Calad-Thomson S, Rush TS, Brase JM. Machine learning models to predict inhibition of the bile salt export pump. J Chem Inf Model. 2021;61(2):587–602. doi: 10.1021/acs.jcim.0c00950. [DOI] [PubMed] [Google Scholar]
- 67.Kido Y, Matsson P, Giacomini KM. Profiling of a prescription drug library for potential renal drug–drug interactions mediated by the organic cation transporter 2. J Med Chem. 2011;54(13):4548–4558. doi: 10.1021/jm2001629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yuan Y, Chang S, Zhang Z, Li Z, Li S, Xie P, Yau W-P, Lin H, Cai W, Zhang Y, Xiang X. A novel strategy for prediction of human plasma protein binding using machine learning techniques. Chemom Intell Lab Syst. 2020;199:103962. doi: 10.1016/j.chemolab.2020.103962. [DOI] [Google Scholar]
- 69.Podlewska S, Kafel R. MetStabOn—online platform for metabolic stability predictions. Int J Mol Sci. 2018;19(4):1040. doi: 10.3390/ijms19041040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sorkun MC, Khetan A, Er S. AqSolDB, a curated reference set of aqueous solubility and 2D descriptors for a diverse set of compounds. Sci Data. 2019 doi: 10.1038/s41597-019-0151-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hsiao Y-W, Fagerholm U, Norinder U. In silico categorization of in vivo intrinsic clearance using machine learning. Mol Pharm. 2013;10(4):1318–1321. doi: 10.1021/mp300484r. [DOI] [PubMed] [Google Scholar]
- 72.Lindh M, Karlén A, Norinder U. Predicting the rate of skin penetration using an aggregated conformal prediction framework. Mol Pharm. 2017;14(5):1571–1576. doi: 10.1021/acs.molpharmaceut.7b00007. [DOI] [PubMed] [Google Scholar]
- 73.Serra A, Önlü S, Coretto P, Greco D. An integrated quantitative structure and mechanism of action-activity relationship model of human serum albumin binding. J Cheminf. 2019 doi: 10.1186/s13321-019-0359-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ciura K, Ulenberg S, Kapica H, Kawczak P, Belka M, Bączek T. Drug affinity to human serum albumin prediction by retention of cetyltrimethylammonium bromide pseudostationary phase in micellar electrokinetic chromatography and chemically advanced template search descriptors. J Pharm Biomed. 2020;188:113423. doi: 10.1016/j.jpba.2020.113423. [DOI] [PubMed] [Google Scholar]
- 75.Giaginis C, Zira A, Theocharis S, Tsantili-Kakoulidou A. Application of quantitative structure activity relationships for modeling drug and chemical transport across the human placenta barrier: a multivariate data analysis approach. J Appl Toxicol. 2009;29(8):724–733. doi: 10.1002/jat.1466. [DOI] [PubMed] [Google Scholar]
- 76.Bercu JP, Morton SM, Deahl JT, Gombar VK, Callis CM, van Lier RBL. In silico approaches to predicting cancer potency for risk assessment of genotoxic impurities in drug substances. Regul Toxicol Pharmacol. 2010;57(2):300–306. doi: 10.1016/j.yrtph.2010.03.010. [DOI] [PubMed] [Google Scholar]
- 77.Simeon S, Montanari D, Gleeson MP. Investigation of factors affecting the performance of in silico volume distribution QSAR models for human, rat, mouse, dog & monkey. Mol Inf. 2019;38(10):1900059. doi: 10.1002/minf.201900059. [DOI] [PubMed] [Google Scholar]
- 78.Fu L, Liu L, Yang Z-J, Li P, Ding J-J, Yun Y-H, Lu A-P, Hou T-J, Cao D-S. Systematic modeling of based on ensemble machine learning, group contribution, and matched molecular pair analysis. J Chem Inf Model. 2019;60(1):63–76. doi: 10.1021/acs.jcim.9b00718. [DOI] [PubMed] [Google Scholar]
- 79.Watanabe R, Esaki T, Kawashima H, Natsume-Kitatani Y, Nagao C, Ohashi R, Mizuguchi K. Predicting fraction unbound in human plasma from chemical structure: improved accuracy in the low value ranges. Mol Pharm. 2018;15(11):5302–5311. doi: 10.1021/acs.molpharmaceut.8b00785. [DOI] [PubMed] [Google Scholar]
- 80.Esaki T, Ohashi R, Watanabe R, Natsume-Kitatani Y, Kawashima H, Nagao C, Mizuguchi K. Computational model to predict the fraction of unbound drug in the brain. J Chem Inf Model. 2019;59(7):3251–3261. doi: 10.1021/acs.jcim.9b00180. [DOI] [PubMed] [Google Scholar]
- 81.Lu Y, Anand S, Shirley W, Gedeck P, Kelley BP, Skolnik S, Rodde S, Nguyen M, Lindvall M, Jia W. Prediction of pKa using machine learning methods with rooted topological torsion fingerprints: application to aliphatic amines. J Chem Inf Model. 2019;59(11):4706–4719. doi: 10.1021/acs.jcim.9b00498. [DOI] [PubMed] [Google Scholar]
- 82.Mansouri K, Cariello NF, Korotcov A, Tkachenko V, Grulke CM, Sprankle CS, Allen D, Casey WM, Kleinstreuer NC, Williams AJ. Open-source QSAR models for pKa prediction using multiple machine learning approaches. J Cheminf. 2019 doi: 10.1186/s13321-019-0384-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen J, Yang H, Zhu L, Wu Z, Li W, Tang Y, Liu G. In silico prediction of human renal clearance of compounds using quantitative structure-pharmacokinetic relationship models. Chem Res Toxicol. 2020;33(2):640–650. doi: 10.1021/acs.chemrestox.9b00447. [DOI] [PubMed] [Google Scholar]
- 84.Zheng S, Xiong J, Wang Y, Liang G, Xu Y, Lin F. Quantitative prediction of hemolytic toxicity for small molecules and their potential hemolytic fragments by mach. learn. and recursive fragmentation methods. J Chem Inf Model. 2020;60(6):3231–3245. doi: 10.1021/acs.jcim.0c00102. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. File contains brief descriptions of the properties modelled, additional performance statistics and figures referred to in the text.