

Abstract
Introduction
This article describes the protocol of an Ebola vaccine clinical trial which investigates the safety and immunogenicity of a two-dose prophylactic Ebola vaccine regimen comprised of two Ebola vaccines (Ad26.ZEBOV and MVA-BN-Filo) administered 56 days apart, followed by a booster vaccination with Ad26.ZEBOV offered at either 1 year or 2 years (randomisation 1:1) after the first dose. This clinical trial is part of the EBOVAC3 project (an Innovative Medicines Initiative 2 Joint Undertaking), and is the first to evaluate the safety and immunogenicity of two different booster vaccination arms in a large cohort of adults.
Methods and analysis
This study is an open-label, monocentric, phase 2, randomised vaccine trial. A total of 700 healthcare providers and frontliners are planned to be recruited from the Tshuapa province in the Democratic Republic of the Congo (DRC). The primary and secondary objectives of the study assess the immunogenicity of the first (Ad26.ZEBOV), second (MVA-BN-Filo) and booster (Ad26.ZEBOV) dose. Immunogenicity is assessed through the evaluation of EBOV glycoprotein binding antibody responses after vaccination. Safety is assessed through the collection of serious adverse events from the first dose until 6 months post booster vaccination and the collection of solicited and unsolicited adverse events for 1 week after the booster dose.
Ethics and dissemination
The protocol was approved by the National Ethics Committee of the Ministry of Health of the DRC (n°121/CNES/BN/PMMF/2019). The clinical trial was registered on 4 December 2019 on ClinicalTrials.gov. Trial activities are planned to finish in October 2022. All participants are required to provide written informed consent and no study-related procedures will be performed until consent is obtained. The results of the trial will be added on ClinicalTrials.gov, published in peer-reviewed journals and presented at international conferences.
Trial registration number
NCT04186000; Pre-results.
Keywords: protocols & guidelines, health & safety, immunology
Strengths and limitations of this study.
With this randomised vaccine trial, being the first to evaluate the safety and immunogenicity in two different booster vaccine arms 1 or 2 years after the prime dose, new contributions will be added to already existing safety and immunogenicity data. Additionally, it is the first trial to assess the antibody response and (serious) adverse event occurrence of two different booster arms in a large adult cohort.
Vaccination of healthcare providers and frontliners can potentially help protect a community which is at risk for future outbreaks.
Innovative use of iris scanning biometric material to identify participants enrolled in the trial.
This study takes place in a resource poor setting, impacting logistical set-up of the trial.
Long duration of the trial (2.5 years) may lead to considerable loss to follow-up.
Introduction
Ebolaviruses (negative stranded RNA viruses) belong to the Filoviridae family and cause Ebola virus disease (EVD), which often leads to severe haemorrhagic fever in humans and non-human primates.1 Contact with infected wild animals (such as fruit bats, gorillas, apes and monkeys) is often reported as the source of animal-to-human transmission2–4 and once among humans, these public health pathogens spread via direct (body fluids) or indirect (contaminated surfaces) human-to-human contact.2 3 While they do not spread via air or water,3 Ebolaviruses bring along a severe public health burden with case fatality rates that can range up to 90%.5 Since the discovery of the Ebolaviruses in 1976,6 more than 20 outbreaks (most are endemic to regions in equatorial and western Africa) have taken place.7 To date, the Democratic Republic of the Congo (DRC) has been the most affected country with its 12th outbreak taking place between February and May 2021.8 However, it is only recently that the search for a safe and effective vaccine against Ebola was accelerated when the epidemic potential of Ebolaviruses, and more specifically the species Zaïre Ebolavirus (virus name: Ebola virus; abbreviation: EBOV9), became clear through the West African Ebola epidemic (2013–2016; 28 616 cases with 11 310 deaths10).
One of the initiatives to develop such a vaccine came from an international consortium, funded by the Innovative Medicines Initiative 2 (IMI2) Joint Undertaking, dedicated to evaluate a prophylactic Ebola vaccine regimen comprised of two candidate Ebola vaccines (Ad26.ZEBOV and MVA-BN-Filo) and aiming to bring this prophylactic Ebola vaccine to licensure.11 Ad26.ZEBOV is a monovalent vaccine developed to provide active EBOV-specific immunity. MVA-BN-Filo, which is administered 56 days after the Ad26.ZEBOV vaccine, is a multivalent vaccine developed to establish active immunity to EBOV, Sudan Ebolavirus, Taï Forest Ebolavirus and the Marburg virus (also part of the Filoviridae family). In July 2020, the two-dose prophylactic vaccine regimen was granted market authorisation by the European Commission.12
Several projects (PREVAC, EBOVAC1, EBOVAC2 and EBOVAC3) within the IMI2 Joint Undertaking, of which the first in-human clinical trials started in 2014, were at the basis of this successful authorisation.11 Within these projects, multiple clinical trials assessed or are assessing the timing, tolerability, safety and immunogenicity of different Ad26.ZEBOV and MVA-BN-Filo vaccine regimen in healthy adults (≥18 years old) and children/adolescents (1–17 years old) via phase 1, 2, 2B and 3 studies. Initial trials showed that healthy adult participants had higher geometric mean concentrations of binding antibodies for the regimen where Ad26.ZEBOV vaccination was followed by an MVA-BN-Filo vaccination 56 days later. Moreover, 100% of participants had detectable Ebola glycoprotein-specific Immunoglobulin G (IgG) antibodies up to at least 240 days after vaccination.13–15 Even though some local (erythema, swelling and pain at injection site) and systemic (headache, nausea, fever, myalgia and fatigue) solicited adverse events (AEs) were recorded, the vaccine regimen was generally well tolerated across studies.13–17
While it is of utmost importance that the two-dose prophylactic vaccine regimen is safe and leads to an immune response, it is also crucial to find out whether or not this regimen can lead to induced immune memory at the time of imminent risk (ie, an outbreak) through a booster vaccination. To evaluate this induced immune memory response, three previous studies within EBOVAC projects have administered a booster vaccine with Ad26.ZEBOV at either 1 year (NCT02325050; NCT02564523) or 2 years (NCT02509494) post dose 1. Results from the NCT02325050 trial have already shown that an immunological memory was rapidly induced via booster vaccination with Ad26.ZEBOV, indicating that booster vaccination can be considered for at risk individuals (eg, when an outbreak occurs) that were previously vaccinated with the two-dose heterologous prophylactic regimen.18 However, these trials only evaluated booster vaccination in a small amount of participants (n≤39) and it still has to be explored whether the induced immune memory response differs depending on the timing of the booster dose (ie, 1 or 2 years after dose 1).
Healthcare settings play an important role in the control of EVD and therefore healthcare providers (HCP) and frontliners, due to occupational exposure, are not only more at risk of disease acquisition but also facilitate the spread of the virus.19–22 Knowing that outbreaks of EVD often occur in regions where there is already a shortage of HCP and frontliners, this further depletes a weak healthcare system and the quality of care. Consequently, the Strategic Advisory Group of Experts stated in 2018 that for preventive strategies, vaccination of HCP as part of an emergency preparedness plan has significant potential of reducing the scale and duration of outbreaks.23
This phase 2 randomised clinical trial aims to determine the safety and immunogenicity of the two-dose heterologous vaccine regimen with Ad26.ZEBOV followed by MVA-BN-Filo 56 days later. Additionally, this trial aims to assess the safety and immunogenicity of a booster Ad26.ZEBOV vaccine administered either 1 or 2 years post first dose and to compare the induced immune memory response between both booster arms. The trial is conducted in a cohort of HCP and frontliners from the Boende health district in DRC, a well-known population at risk from clinical and epidemiological perspective.
Methods
Study design and setting
This study is an open-label, monocentric, phase 2, randomised trial to evaluate the immunogenicity and safety of Ad26.ZEBOV (5×1010 viral particles) as first dose and MVA-BN-Filo (1×108 infectious units) as second dose vaccination at a 56-day interval in HCP and frontliners who may be exposed to Ebola in the event of a future Ebola outbreak in DRC. Additionally, after randomisation (1:1) a booster of Ad26.ZEBOV (5×1010 viral particles) will be offered at either 1 year or 2 years after the first dose (figure 1).
Figure 1.
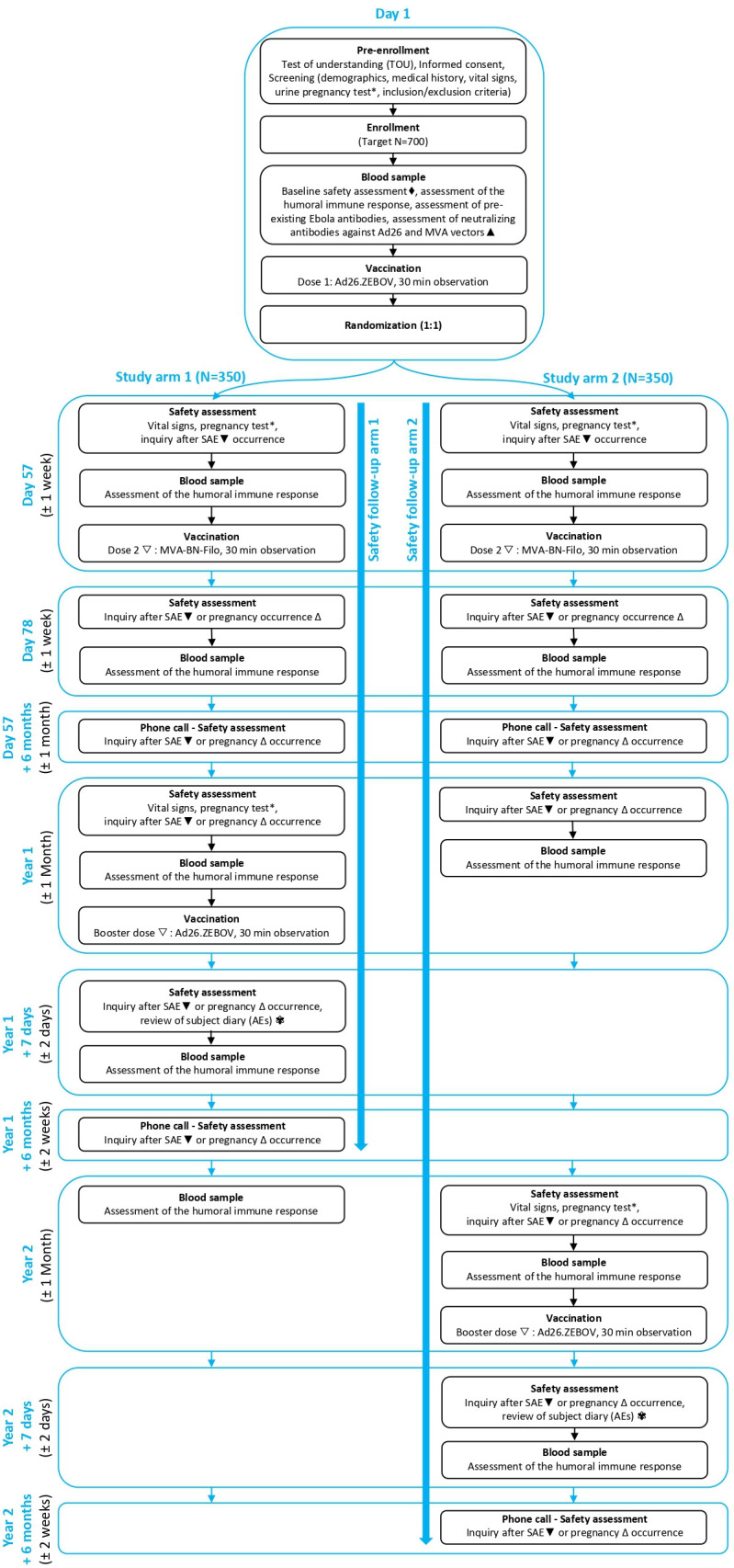
Study time and events overview. * Only for female participants of childbearing potential; ♦ Abnormal results will not exclude a participant, as results will not be reviewed prior to enrolment; ▲ Only the first 100 participants enrolled will be tested for neutralising antibody response against Ad26 virus neutralising assay and MVA vectors. Other blood analyses are for all 700 participants; ▼ Concomitant therapies given in conjunction with a serious adverse event (SAE) should be recorded from signing of the Informed Consent Form onwards until 6 months post booster; ▽ The investigator may withhold the second vaccine or booster dose if a participant’s clinical status changes prior to vaccination. The participant should continue to be followed for safety and immunogenicity according to the protocol; ∆ Only for female participants; ✾ Solicited and unsolicited adverse events (AEs) will be collected in a participant diary during 1 week post booster vaccination.
The study site is located in Boende, Tshuapa province, DRC (figure 2), at approximately 750 km north-west of Kinshasa. Study participants will be enrolled at the General Reference Hospital in Boende.
Figure 2.

Study site location. On the left, the Democratic Republic of Congo (DRC) is highlighted on a map of the African continent. On the right, the study site location (Boende, Tshuapa province) is marked on a map of DRC indicating its provinces.35
Objective
The primary, secondary and exploratory objectives and endpoints of this study are described in table 1.
Table 1.
Objectives and endpoints
| Objectives | Endpoints |
| Primary | |
|
|
| Secondary | |
|
|
|
|
| Exploratory | |
|
|
|
|
|
|
|
|
EBOV, Ebola virus; ELISA, enzyme-linked immunosorbent assay; FANG, Filovirus Animal Non-Clinical Group; GP, glycoprotein; NP, nucleoprotein; PRNT, plaque reduction neutralisation test; VNA, virus neutralising assay.
Participant population and sample size
A total of 700 registered HCPs and frontliners in DRC (working in the Boende General Reference Hospital, Health Centres or Health Posts in the Boende health district) are planned to be recruited from the Tshuapa province. This assessment was based on information obtained from an ongoing (monkeypox) vaccine trial in the same area at the time the protocol was being written.24 From discussions with the monkeypox research group, it became clear that a high enrolment rate and retention rate (>90% after 2 years) could be expected among HCPs and frontliners in the Boende health district. Based on this ongoing monkeypox trial, it was estimated that enrolling approximately 50% of the HCPs and frontliners working in the Boende health district would be feasible. The participant population is thus a convenience sample and the sample size is defined on the feasibility of recruitment of HCP and frontliners in the region.
However, to determine whether it would be possible to compare the induced immune responses of the two booster arms, a power analysis was performed. A power of 0.99 was calculated based on the following parameters: two-sided t-test, equal samples of 350 participants, significance level of 0.05, an effect size of 0.49 in antibody response. The effect size was calculated based on trial data (NCT02564523 and NCT02509494) available in the first edition of the combined investigator’s brochure of the vaccines with samples from 64 participants vaccinated either 1 year or 2 years after the first dose.25 To obtain the effect size, the difference in geometrical mean concentrations (log scale) of the EBOV glycoprotein (GP)-specific antibody responses between the two groups was divided by the pooled standard deviations.26 With a power of 0.99 it will thus be possible to perform a formal comparative analysis of the induced immune memory response of the two booster arms.
Unfortunately no power analysis could be performed to determine whether the sample size is sufficiently large to perform a formal statistical comparison of safety response (AEs and serious AEs (SAEs)) from both arms. In the current combined investigator’s brochure of the vaccines,25 safety information is pooled for all booster doses independent of the timing of its administration (1 year or 2 years post dose 1) and thus no effect size can be calculated until the unpooled data from the different trials is obtained.
Inclusion and exclusion criteria that determine the eligibility of participants are reported in box 1.
Box 1. Inclusion and exclusion criteria.
Inclusion criteria
Each potential participant must satisfy all of the following criteria to be enrolled in the study:
1. The participant must pass the test of understanding.
Note: If the participant fails the test of understanding on the first attempt, he/she must be retrained on the purpose of the study and must take the test again (two repeats are allowed). If participants fail on the third attempt, they should not continue with screening or consenting procedures.
2. Each participant must sign an informed consent form indicating that he or she understands the purpose of, and procedures required for, the study and is willing to participate in the study. In case the participant cannot read or write, the procedures must be explained and informed consent must be witnessed by a trusted literate third party not involved with the conduct of the study.
3. The participant must be a man or women aged 18 years or older.
4. The participant must be a documented healthcare provider in Democratic Republic of the Congo.
5. The participant must be healthy in the investigator’s clinical judgement and on the basis of vital signs assessed at day 1 screening.
Note: Subjects who are HIV-positive can be enrolled as long as their general condition is good, that is, they are on antiretroviral treatment or have no signs or symptoms of immunodepression, diagnosed on the basis of physical examination, medical history and the investigator’s clinical judgement.
6. Before vaccination, a woman must be either:
Of childbearing potential and practicing (or intending to practice) a highly effective method of birth control consistent with local regulations and/or local culture regarding the use of birth control methods for participants in clinical studies, beginning at least 28 days prior to vaccination and during the study up to at least 3 months after the first (or only) vaccination (Ad26.ZEBOV) and 1 month after the MVA-BN-Filo vaccination (if applicable); and then starting again 14 days before the booster vaccination until 3 months after the booster vaccination. The sponsor considers the following methods of birth control to be highly effective: established use of oral, injected or implanted hormonal methods of contraception; placement of an intrauterine device or intrauterine system; barrier methods: condom or occlusive cap (diaphragm or cervical/vault caps) with or without spermicidal foam/gel/film/cream/suppository; male partner sterilisation (the vasectomised partner should be the sole partner for that participant); true abstinence (when this is in line with the preferred and usual lifestyle of the participant); OR
Not of childbearing potential: postmenopausal (amenorrhoea for at least 12 months without alternative medical cause); permanently sterilised (eg, bilateral tubal occlusion (which includes tubal ligation procedures as consistent with local regulations), hysterectomy, bilateral salpingectomy, bilateral oophorectomy); or otherwise be incapable of pregnancy.
Note: If the social situation of a woman of childbearing potential changes (eg, woman who is not heterosexually active becomes active), she must begin a highly effective method of birth control, as described above.
7. Woman of childbearing potential must have a negative urine β-human chorionic gonadotropin pregnancy test immediately prior to each study vaccine administration.
8. Participant must be available and willing to participate for the duration of the study.
9. Participant must be willing and able to comply with protocol requirements (including certain prohibitions and restrictions such as the use of anticonception and the discouragement of concomitant treatment that may alter the immune response).
10. Participant must be willing to provide verifiable identification.
11. Participant must have a means to be contacted.
Exclusion criteria
Participants will be excluded from study participation in case the following criteria apply:
1. The participant has a known history of Ebola virus disease.
2. The participant has received any experimental candidate Ebola vaccine less than 3 months prior to the first study visit.
3. The participant has received any experimental candidate Ad26-vaccine in the past.
Note: Receipt of any approved or experimental vaccinia/smallpox vaccine or experimental Ad-vector vaccine other than Ad26 prior to study entry is allowed.
4. The participant has a known allergy or history of anaphylaxis or other serious adverse reactions to vaccines or vaccine products (including any of the constituents of the study vaccines (eg, polysorbate 80, ethylenediaminetetraacetic acid or L-histidine for Ad26.ZEBOV vaccine; and tris (hydroxymethyl)-amino methane for MVA BN-Filo vaccine), including known allergy to egg, egg products and aminoglycosides.
5. The participant has an acute illness (this does not include minor illnesses such as mild diarrhoea or mild upper respiratory tract infection) or temperature ≥38°C on day 1. Participants with such symptoms will be excluded from enrolment at that time, but may be rescheduled for enrolment at a later date if feasible.
6. The participant is a pregnant or breastfeeding women, or women planning to become pregnant while enrolled in this study until at least 3 months after the Ad26.ZEBOV vaccination or 1 month after MVA-BM-Filo.
7. The participant has significant conditions or clinically significant findings at screening or vital signs for which, in the opinion of the investigator, participation would not be in the best interest of the participant (eg, compromise the safety or well-being) or that could prevent, limit or confound the protocol-specified assessments.
Note: Participants who have recently received treatment for acute, uncomplicated malaria are eligible for participation if at least 3 days have elapsed from the conclusion of a standard, recommended course of therapy for malaria; participants who are acutely ill with malaria at the time of screening should complete therapy and wait an additional 3 days after completion before screening for the study.
Note: Participants with sickle cell trait can be included.
8. The participant had major surgery (per the investigator’s judgement) within the 4 weeks prior to screening, or has planned major surgery during the study (from the start of screening onwards).
9. The participant had a post-organ and/or stem cell transplant whether or not with chronic immunosuppressive therapy.
10. The participant received an investigational drug or investigational vaccines or used an invasive investigational medical device within 3 months prior to screening, or current or planned participation in another clinical study during the study.
Note: Participation in an observational clinical study is allowed.
11. The participant has a history of chronic urticaria (recurrent hives).
Randomisation procedure
The study randomisation list will be developed using an algorithm in the Statistical Analysis System software. This algorithm will randomly assign a treatment group (1:1) to a sequential randomisation number. Once established, the list will be shared with the principal investigator (University of Kinshasa), who is in charge of creating sealed envelopes under sponsor (University of Antwerp) supervision. A total of at least 700 randomisation envelopes will be created. Thirty envelopes will be grouped into one larger envelope, referred to as a ‘booklet’. The booklets and envelopes will be numbered sequentially by a unique sequence of numbers. The booklets will be labelled in a sequential order (ie, 01–24) and the envelopes will be labelled with the study number ‘VAC52150-EBL-2007’ and a sequential randomisation number (ie, 001–700) to which a treatment group is linked via the algorithm. The staff delegated to make the envelopes will use the Envelope Assembly Record Worksheet, on which the randomisation number, initials of the assembler, date on which the assembly took place and the initials of the staff member(s) that performed the quality control are collected. The randomisation booklets with envelopes will be stored and used in the study clinic.
Delegated site staff will assign and open booklets and envelopes in sequential order during study visits. Each envelope will contain two stickers. The first will contain space for writing the subject ID and participant’s initials, the second will contain the randomisation number and treatment description (preprinted based on the study randomisation list). On opening the sealed envelope, the subject ID and initials must be written in the space provided on the first sticker and the subject ID sticker must be placed on the outside of the envelope. To ensure proper source documentation, the sticker with the treatment information must be placed on the corresponding randomisation worksheet. Thereafter, the empty envelope, with the subject ID sticker on the outside, must be placed back in the booklet. These booklets are to be stored by the principal investigator.
Study procedures
Figure 1 provides a visual overview of the study procedures. At day 1, interested participants are informed about the study and are required to pass a test of understanding before providing written consent. No study activities are performed before the participant has signed the informed consent form. Afterwards, the study medical doctor evaluates his/her general health based on the inclusion criteria, vital signs (blood pressure, pulse/heart rate (both at rest) and body temperature) are collected and a urine pregnancy test for women of childbearing potential is performed. Further during this first visit, a blood sample is taken for baseline testing of binding antibody level (ie, humoral immune response) against EBOV GP using Filovirus Animal Non-Clinical Group (FANG) Ebola virus ELISA and the presence of pre-existing human anti-EBOV GP IgG and anti-EBOV nucleoprotein (NP) IgG using LUMINEX assay. For day 1 samples, both FANG ELISA and LUMINEX assay will be carried out. FANG ELISA is performed for all EBOVAC trials in the same laboratory (for consistency and comparability) and LUMINEX assay will provide a more detailed array of IgG antibodies that are not obtained via FANG ELISA. For the first 100 enrolled participants an additional test on the collected serum is performed to measure the neutralising antibody level against Ad26 and MVA vectors using the Ad26 virus neutralising assay (VNA) and MVA plaque reduction immunogenicity test (PRNT), respectively. Subsequently, a blood sample for baseline safety assessment is collected to test haemoglobin, haematocrit, blood cell count (white and red), platelet count, urea, creatinine and transaminases. Then, participants are vaccinated with the first dose (Ad26.ZEBOV) and they are given instructions to contact the study team for any occurring SAEs, or in case of pregnancy of a participant during the study. After vaccination, participants remain at the study site for an observation period of 30 min to make sure no SAEs occur. SAEs are collected from first dose vaccination until 6 months post booster. Lastly on day 1, randomisation is performed to determine the timing of the booster vaccine at either 1 year or 2 years after the first dose. Contact information is verified, an appointment for the second dose on day 57 is arranged and a participant card is printed. Innovatively, next to a participant card, a biometric identification tool via iris scanning is foreseen to ensure correct identification of the participants during all study related visits.
At day 57, participants return to the study site for urine pregnancy testing (for women of childbearing potential), vital signs measurement, assessment of safety (SAEs), a blood sample for immunogenicity assessment (the binding antibody levels against EBOV GP using FANG ELISA) and afterwards administration of the second vaccine (MVA-BN-Filo). After an observation period of 30 min, participants are reminded to contact the study team for any SAEs that occurs, or in case of pregnancy of a participant during the study. Contact information is verified and an appointment for the 21-day post dose 2 visit (day 78) is arranged.
At 21 days post dose 2 (day 78), all participants return to the study site for a safety assessment (SAEs) and for the collection of a blood sample for immunogenicity assessment. Contact information is reverified and they are reminded to contact the study team in case of SAE occurrence, or in case of pregnancy of a participant.
To make sure no valuable information is missed, participants are contacted by phone to inquire about any occurrence of pregnancies (female participants) and SAEs at approximately 6 months post dose 2 vaccination.
At 1 year and 2 years after the first vaccine, when all participants return to the site, the clinical trial staff inquires after the occurrence of SAEs and a blood sample is collected for immunogenicity assessment of all participants (where applicable before administration of the booster dose). Depending on the study arm, a booster vaccination with Ad26.ZEBOV is given either 1 or 2 years after the first dose. Prior to vaccination, the general well-being of the participant is evaluated and urine pregnancy testing (for women of childbearing potential), as well as a vital signs measurement are performed. After vaccination, participants remain at the study site for a 30 min observation period. Participants are asked to collect solicited and unsolicited AEs in a participant diary starting on the day of the vaccination and continuing for the subsequent 7 days.
At day 8 post booster the safety data including solicited and unsolicited AEs is reviewed and a blood sample for immunogenicity assessment is taken to document the immune response. Should any solicited AEs persist at day 8 post booster, participants are asked to continue monitoring these in their participant diary. Once the solicited AEs have resolved, they are asked to make an unscheduled visit at the site so this information can be reported.
At 6 months post booster, all participants are contacted by phone and questioned about any SAEs or pregnancies (female participants) that have occurred since the last vaccination.
The total duration of the study is 2 years and 6 months post first dose. The study is considered completed when the last participant has been contacted for the 6 months post booster phone call or has left the study.
Study intervention
According to the predefined schedule (figure 1), participants receive a 0.5 mL intramuscular injection into the deltoid muscle of the upper arm with Ad26.ZEBOV or MVA-BN-Filo. The injection site should be free from any injury, local skin conditions or other issues that might interfere with the evaluation of local reactions. Every vaccination is given in the opposite arm of the previous vaccination (unless the opposite arm has a condition that prevents evaluating the arm after injection). No local or topical anaesthetic is used prior to the injection.
The second or booster vaccination is not administered if any of the following events occur at any time after the first dose vaccination:
A participant experiences anaphylaxis clearly attributable to vaccination with the study vaccine; OR
A participant experiences generalised urticaria within 72 hours of vaccination considered to be related to study vaccine; OR
A participant experiences a SAE considered to be related to the study vaccine; OR
A participant experiences injection site ulceration, abscess or necrosis considered to be related to the study vaccine; OR
A participant has confirmed EVD; OR
A female participant of childbearing potential has a positive urine β-human chorionic gonadotropin (β-HCG) pregnancy test before vaccination (on Day 57, Year 1 or Year 2 (depending on the randomisation group)); OR
A female participant of childbearing potential has a positive urine β-HCG pregnancy test between dose 2 and the booster dose and is still pregnant or breast feeding at the time of the booster dose; OR
A participant takes a concomitant treatment with drugs that may alter the immune response; OR
The principal investigator believes that for safety reasons it is in the best interest of a participant to discontinue the study intervention.
Participants experiencing any of the events described above are still followed up for safety and immunogenicity according to the protocol. The decision to discontinue the study intervention is at the discretion of the principal investigator (University of Kinshasa) and after consultation with the sponsor (University of Antwerp) for any of the events described above.
Patient and public involvement
Difficulties were expected when setting up a clinical trial in Boende, a remote and resource-limited area of DRC. However, to avoid and anticipate some of these challenges and in order to support vaccination compliance, a collaboration is established between the study team and the provincial division of health. Throughout the trial, workshops are organised for HCP in the health district of Boende to sensitise and inform about EVD and other relevant medical topics. These gatherings should not only facilitate enrolment in the trial but also increase the engagement of participants by enhancing their understanding of the clinical trial and the importance of adherence. During these workshops time is available for questions and discussions. In addition to these gatherings for trial participants, community engagement activities and the training and capacity building of the local clinical trial team that is executing the trial (under supervision of the University of Kinshasa as principal investigator) are organised for the duration of the trial.
Each participant receives an individual visit schedule on enrolment in the trial and when participants miss a planned study visit, community health workers of the Ministry of Health trace the individual participant. Consent is asked in the informed consent form for this mode of contact.
Furthermore, prior to the start of the clinical trial, a pilot study was performed whereby potential participants were interviewed on the feasibility and acceptability of the use of a biometric iris scanning tool for participant identification during the trial and the use of telephone messaging with visit reminders for participant adherence.27
Data management
All information is collected during study visits on source documents by study staff. These source documents with confidential information are transcribed into the electronic clinical database by site data managers. To make sure that all entered data (collected in DFexplore V.5.2.1) is correct, the principal investigator reviews each source document and confirms its correct transcription in the database. Additionally, the sponsor performs quality checks of the entered data in the database and, during monitoring visits, source data verification is performed.
Statistical analysis
A differentiation in analysis will be made according to: (1) the full analysis set (FAS; all participants who received at least one dose, regardless of the occurrence of protocol deviations), (2) per-protocol set for primary vaccination series (all vaccinated participants, who received both dose 1 and dose 2 (administered within the protocol-defined visit window) vaccinations, have at least one post-vaccination (ie, after the date of dose 1) evaluable immunogenicity sample and have no major protocol deviations influencing the immune response) and (3) per-protocol set for the booster vaccination (includes all participants in the per-protocol set for the primary vaccination series who received a booster dose and have at least one post booster vaccination evaluable immunogenicity sample, and have no major protocol deviations influencing the immune response).
Participant information (ie, demographics and baseline characteristics, disposition information, treatment compliance, extent of exposure, protocol deviations and concomitant medications) is planned to be tabulated and summarised with descriptive statistics for all participants. For continuous data, such as age, the mean and SD will be provided if applicable, otherwise the geometric means, related SD or median and IQRs will be used.
For the immunogenicity analysis, two per-protocol sets will be used, that is, the per-protocol set for primary vaccination series and the per-protocol set for the booster. If more than 10% of participants from the FAS are excluded from the per-protocol immunogenicity set, the immunogenicity analysis will be repeated on the FAS to evaluate the robustness of the analysis results. A subgroup analysis of the immune response at different time points will be performed stratified by age (18–40, 40–60 and >60), gender, prior vaccinia/smallpox vaccination, pre-existing Ebola antibodies (positive or negative for pre-existing human anti-EBOV GP IgG and anti-EBOV NP IgG and for both), baseline immunogenicity (positivity vs negativity for antibody levels against EBOV GP using FANG ELISA) and the presence of neutralising antibody levels against Ad26 and MVA vectors using Ad26 VNA and MVA PRNT assays (only the first 100 enrolled participants), respectively. For these planned subgroup analyses, N (%), geometric mean concentrations and 95% CIs will be provided as appropriate. Finally, a formal comparative analysis of the induced immune memory response between the two booster arms will be performed.
Safety analyses include SAEs collected during the whole study and solicited and unsolicited AEs for 1 week post booster vaccination. The analysis of SAEs will be performed using the FAS and the solicited and unsolicited AEs will be analysed for the participants who received the booster vaccination. Continuous variables will be summarised using the following statistics: number of observations; arithmetic or geometric mean/median (if applicable) with their related measures of dispersion (95% CI for the mean, SD or IQR (Q1–Q3)). Minimum and maximum frequencies and percentages (one decimal place) will be generated for categorical variables. If the unpooled safety data from the NCT02564523 and NCT02509494 studies can be obtained, a power analysis will be performed to assess whether the safety data of the two booster arms can potentially be compared through formal statistical analysis.
The primary endpoint analysis is planned to be performed when all participants have completed the 21-day post dose 2 visit (day 78) or discontinued earlier. This analysis includes all available immunogenicity and safety data up to this point (date cut-off). Additional interim analyses may be performed during the study for the purpose of informing future vaccine-related decisions in a timely manner.
The final analysis will be performed when all participants have completed the last study-related phone call (6 months post booster) or left the study.
Discussion
The aim of this phase 2 trial is to obtain further safety and immunogenicity data on the two-dose prophylactic heterologous Ebola vaccine regimen and to assess the safety and immunogenicity of a booster dose with Ad26.ZEBOV administered either 1 or 2 years post first dose in a large cohort of HCPs and frontliners. By doing so, this study will feed the immunogenicity and safety databases of the Ad26.ZEBOV and MVA-BN-Filo vaccines. It will also be the first study to compare the induced immune memory response between two different booster arms in a large cohort of adults.
Boende (the capital city of Tshuapa province) was selected as the trial site for several reasons. First, the Tshuapa province recovered from an EBOV outbreak that occurred in the Boende Health District in 2014.21 Following this outbreak, a study (n=565) conducted in the Tshuapa region in 2015 found that 41.4% of the tested HCPs were seroreactive to at least one EBOV protein and 2.8% of the HCPs showed a neutralising capacity while never having developed EVD symptoms.20 This observation suggests a possible endemic EBOV exposure in the Tshuapa province of DRC. These are interesting observations for future ecological research as the ecology and reservoir(s) of EBOV and other filoviruses remain largely unknown.28 29 Second, in addition to the previous outbreak of EVD, Boende was chosen to perform the current clinical trial as there was expertise available after carrying out a phase 3 monkeypox vaccine trial that took place in 2017.24
Some limitations are present in the current set-up of the trial. First, by focussing on occupation (registered HCPs and frontliners) rather than age and gender, in the inclusion and exclusion criteria, the aim is to easily reach the target of 700 participants. However, a recent review by Flanagan et al has shown that immune responses to vaccination can differ based on gender and age.30 To take this limitation into account, stratification for age and gender has been foreseen during statistical analysis. Second, while HIV-positive participants can participate in this trial if their general condition is good, it is not possible to be certain of the HIV-status of all participants as no routine checks prior to enrolment or during the course of the trial are foreseen. It is possible that some participants either are unwilling to share their HIV-positive status as a consequence of the stigma that is often linked to it31 or are simply unaware of their positive status (eg, during an asymptomatic phase of the disease32). However, due to the low prevalence (0.6%) of HIV-positive people in the province of the trial,31 it was chosen not to perform routine checks and to trust the willingness of a participant to share his/her status as it is not considered an exclusion criterium for the trial. Finally, at the start of the project the protocol initially only included a vaccination strategy with the two-dose heterologous vaccine regimen (Ad26.ZEBOV followed by MVA-BN-Filo 56 days later) and was later adapted to include a booster vaccination at the request of the vaccine producer. The purpose of the initial observational trial was, next to obtaining additional immunogenicity data, a way to see if performing a vaccination trial in a remote area of DRC was feasible and accepted by the population. While writing the protocol however, administering a booster dose in this large cohort was added as a novel aspect and thus this was entered as a secondary objective/endpoint. Currently it is unknown whether this booster dose will be required or not at the moment of an outbreak and what its protective effect would be. However, to explore its safety and immunogenicity, this study protocol was transformed and became a randomised clinical trial. Unfortunately, as the comparison of the two booster arm induced immune responses is not required for approval of the licensure of the two-dose heterologous vaccine regimen and the booster dose was added as a second stage to the study design, no sample size calculations were initially performed for this trial and sample size was selected based on available information from a previous monkeypox vaccine trial in the same area. While this trial thus mainly has a descriptive set-up, scientifically it is interesting to learn if there is a significant difference in the induced immune memory response of the two booster arms. For this reason, a power analysis was retrospectively performed to determine whether it would be possible to compare the induced immune memory response of the two arms. Fortunately, this will be possible as a power of 0.99 was calculated and a formal statistical comparison of the induced immune memory responses of the two booster arms has now been foreseen in the statistical analysis plan. It is however important to take into account that a varying antibody response after booster vaccination is not necessarily directly correlated with protective vaccine efficacy33 and that a high power (99% for this study) can lead to significant differences, even if the difference between both groups is small. Prudent and careful interpretation of the results will thus be crucial.34
In conclusion, because EVD remains a very deadly disease, effective and safe preventive measures will play a crucial role to protect vulnerable communities. While the prophylactic heterologous two-dose regimen was recently granted market authorisation by the European Commission, further research into the safety and immunogenicity of the two-dose regimen is still required to obtain worldwide licensure of the regimen. Furthermore, limited data has previously been collected on the safety and immunogenicity of a booster dose with Ad26.ZEBOV. This is the first large, randomised vaccine trial that assesses the safety and and compares the immunogenicity of two different booster arms in a large cohort.
Ethics and dissemination
This protocol was submitted and approved by the National Ethics Committee of the Ministry of Health of the DRC (approval number: n°121/CNES/BN/PMMF/2019). Prior to being enrolled in the trial, all participants are required to provide written informed consent by signing the informed consent form after having performed a test of understanding. If the participant is unable to read or write, an impartial witness should be present for the entire informed consent process (which includes reading and explaining all written information) and should personally date and sign the informed consent form after the oral consent of the participant is obtained. No study-related procedures are performed until the participant has signed the informed consent form.
The trial was registered on ClinicalTrials.gov on 4 December 2019 and recruitment started on 18 December 2019. All participants were recruited by 8 February 2020 and the study is planned to finish in October 2022. Results of the trial will be entered on ClinicalTrials.gov, published in peer-reviewed journals and presented at international conferences.
Supplementary Material
Acknowledgments
We acknowledge Janssen Vaccines & Prevention B.V. (in collaboration with Bavarian Nordic GmbH), the London School of Hygiene and Tropical Medicine (LSHTM), the Institut National de la Santé et de la Recherche Médicale (INSERM) and the College of Medicine and Allied Health Sciences (COMAHS) for their contribution in the EBOVAC3 project. We are grateful to the Division Provinciale de la Santé and, the political-administrative authorities of the Tshuapa province for a trustful collaboration. We acknowledge the reliability and motivation of the study site team and are grateful.
Footnotes
Contributors: YL wrote the manuscript. TZ, ES, YL, VM, JM, PM, HM-M, J-PVG and PVD wrote the initial English protocol on which this manuscript is based. TZ, VM, PM, JM and HM-M translated the English protocol into French for submission to the National Ethics Committee and the ‘Direction de la Pharmacie et des Médicaments’ of the Ministry of Health of the Democratic Republic of Congo as well as the National Scientific committee against Ebola. All authors (YL, TZ, ES, JDB, VM, JM, PM, HM-M, J-PVG and PVD) reviewed and contributed to the final manuscript.
Funding: The EBOVAC3 project has received funding from the IMI2 Joint Undertaking under grant agreement No 800 176 (IMI-EU). This Joint Undertaking receives support from the European Union’s Horizon 2020 research and innovation programme, European Federation of Pharmaceutical Industries and Associations (EFPIA) and the Coalition for Epidemic Preparedness Innovations (CEPI). For this trial, the Contract Research Organisation and part of the FANG ELISA analyses are funded by the CEPI. All other trial activities are funded by the IMI2 Joint Undertaking grant. All vaccines and neutralising antibody level analyses against Ad26 at the first visit are provided by Janssen Vaccines & Prevention B.V.
Map disclaimer: The depiction of boundaries on this map does not imply the expression of any opinion whatsoever on the part of BMJ (or any member of its group) concerning the legal status of any country, territory, jurisdiction or area or of its authorities. This map is provided without any warranty of any kind, either express or implied.
Competing interests: None declared.
Patient and public involvement: Patients and/or the public were involved in the design, or conduct, or reporting, or dissemination plans of this research. Refer to the Methods section for further details.
Provenance and peer review: Not commissioned; externally peer reviewed.
Ethics statements
Patient consent for publication
Not applicable.
References
- 1.Baseler L, Chertow DS, Johnson KM, et al. The pathogenesis of Ebola virus disease. Annu Rev Pathol 2017;12:387–418. 10.1146/annurev-pathol-052016-100506 [DOI] [PubMed] [Google Scholar]
- 2.Muyembe-Tamfum JJ, Mulangu S, Masumu J, et al. Ebola virus outbreaks in Africa: past and present. Onderstepoort J Vet Res 2012;79:451. 10.4102/ojvr.v79i2.451 [DOI] [PubMed] [Google Scholar]
- 3.Rewar S, Mirdha D. Transmission of ebola virus disease: an overview. Ann Glob Health 2014;80:444–51. 10.1016/j.aogh.2015.02.005 [DOI] [PubMed] [Google Scholar]
- 4.Rouquet P, Froment J-M, Bermejo M, et al. Wild animal mortality monitoring and human Ebola outbreaks, Gabon and Republic of Congo, 2001-2003. Emerg Infect Dis 2005;11:283–90. 10.3201/eid1102.040533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.World Health Organization . Ebola virus disease. Fact sheet N 103, 2014. [Google Scholar]
- 6.Report of an International Commission . Ebola haemorrhagic fever in Zaire, 1976. Bull World Health Organ 1978;56:271–93. [PMC free article] [PubMed] [Google Scholar]
- 7.Malvy D, McElroy AK, de Clerck H, et al. Ebola virus disease. The Lancet 2019;393:936–48. 10.1016/S0140-6736(18)33132-5 [DOI] [PubMed] [Google Scholar]
- 8.World health Organization . Ebola - North Kivu, Democratic Republic of the Congo, 2021, 2021. Available: https://www.who.int/emergencies/situations/ebola-2021-north-kivu
- 9.International Committee on Taxonomy . Virus metadata Repository: version may 1, 2020; MSL35, 2020. [Google Scholar]
- 10.World Health Organization . Situation report Ebola virus disease. Available: http://apps.who.int/ebola/ebola-situation-reports [Accessed 10 Jun 2016].
- 11.Ebovac . EBOVAC3, 2020. Available: https://www.ebovac.org/ebovac-3/
- 12.European Commission . Vaccine against Ebola: Commission grants new market authorisation, 2020. Available: https://ec.europa.eu/commission/presscorner/detail/en/ip_20_1248
- 13.Anywaine Z, Whitworth H, Kaleebu P, et al. Safety and immunogenicity of a 2-Dose heterologous vaccination regimen with Ad26.ZEBOV and MVA-BN-Filo Ebola vaccines: 12-month data from a phase 1 randomized clinical trial in Uganda and Tanzania. J Infect Dis 2019;220:46–56. 10.1093/infdis/jiz070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Milligan ID, Gibani MM, Sewell R, et al. Safety and immunogenicity of novel adenovirus type 26- and modified vaccinia Ankara-Vectored Ebola vaccines: a randomized clinical trial. JAMA 2016;315:1610–23. 10.1001/jama.2016.4218 [DOI] [PubMed] [Google Scholar]
- 15.Mutua G, Anzala O, Luhn K, et al. Safety and immunogenicity of a 2-Dose heterologous vaccine regimen with Ad26.ZEBOV and MVA-BN-Filo Ebola vaccines: 12-month data from a phase 1 randomized clinical trial in Nairobi, Kenya. J Infect Dis 2019;220:57–67. 10.1093/infdis/jiz071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shukarev G, Callendret B, Luhn K, et al. A two-dose heterologous prime-boost vaccine regimen eliciting sustained immune responses to Ebola Zaire could support a preventive strategy for future outbreaks. Hum Vaccin Immunother 2017;13:266–70. 10.1080/21645515.2017.1264755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Safety and immunogenicity of a two-dose heterologous Ad26 . ZEBOV and MVA-BN®-Filo Ebola vaccine regimen: a phase 2 randomised clinical study in Europe (EBOVAC2. Amsterdam, Netherlands: 29th ECCMID, 2019. [Google Scholar]
- 18.Goldstein N, Bockstal V, Bart S. Safety and immunogenicity of heterologous and homologous 2-Dose regimens of adenovirus serotype 26– and modified vaccinia Ankara–Vectored Ebola vaccines: a randomized, controlled phase 1 study. J Infect Dis 2020;4. 10.1093/infdis/jiaa586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Evans DK, Goldstein M, Popova A. Health-care worker mortality and the legacy of the Ebola epidemic. Lancet Glob Health 2015;3:e439–40. 10.1016/S2214-109X(15)00065-0 [DOI] [PubMed] [Google Scholar]
- 20.Hoff NA, Mukadi P, Doshi RH, et al. Serologic markers for ebolavirus among healthcare workers in the Democratic Republic of the Congo. J Infect Dis 2019;219:517–25. 10.1093/infdis/jiy499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maganga GD, Kapetshi J, Berthet N, et al. Ebola virus disease in the Democratic Republic of Congo. N Engl J Med 2014;371:2083–91. 10.1056/NEJMoa1411099 [DOI] [PubMed] [Google Scholar]
- 22.Nanclares C, Kapetshi J, Lionetto F, et al. Ebola virus disease, Democratic Republic of the Congo, 2014. Emerg Infect Dis 2016;22:1579–86. 10.3201/eid2209.160354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.World Health Organization . Meeting of the strategic Advisory group of experts on immunization, October 2018–Conclusions and recommendations.. Weekly Epidemiological Record 2018;93:661–79. [Google Scholar]
- 24.Petersen BW, Kabamba J, McCollum AM, et al. Vaccinating against monkeypox in the Democratic Republic of the Congo. Antiviral Res 2019;162:171–7. 10.1016/j.antiviral.2018.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vaccines J, Prevention BV. Investigator’s Brochure VAC52150 (Ad26.ZEBOV, MVA-BN-Filo [MVA-mBN226B. 1 ed, 2020: 1–163. [Google Scholar]
- 26.Kabacoff RI. Power analysis, 2017. Available: https://www.statmethods.net/stats/power.html
- 27.Zola Matuvanga T, Johnson G, Larivière Y, et al. Use of iris scanning for biometric recognition of healthy adults participating in an Ebola vaccine trial in the Democratic Republic of the Congo: mixed methods study. J Med Internet Res 2021;23:e28573. 10.2196/28573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gryseels S, Mbala-Kingebeni P, Akonda I, et al. Role of wildlife in emergence of Ebola virus in Kaigbono (Likati), Democratic Republic of the Congo, 2017. Emerg Infect Dis 2020;26:2205–9. 10.3201/eid2609.191552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marí Saéz A, Weiss S, Nowak K, et al. Investigating the zoonotic origin of the West African Ebola epidemic. EMBO Mol Med 2015;7:17–23. 10.15252/emmm.201404792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Flanagan KL, Fink AL, Plebanski M, et al. Sex and gender differences in the outcomes of vaccination over the life course. Annu Rev Cell Dev Biol 2017;33:577–99. 10.1146/annurev-cellbio-100616-060718 [DOI] [PubMed] [Google Scholar]
- 31.UNICEF . Deuxième enquête démographique et de santé (EDS-RDC II 2013-2014, 2014. [Google Scholar]
- 32.Moir S, Chun T-W, Fauci AS. Pathogenic mechanisms of HIV disease. Annu Rev Pathol 2011;6:223–48. 10.1146/annurev-pathol-011110-130254 [DOI] [PubMed] [Google Scholar]
- 33.Meyer M, Malherbe DC, Bukreyev A. Can Ebola virus vaccines have universal immune correlates of protection? Trends Microbiol 2019;27:8–16. 10.1016/j.tim.2018.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Colquhoun D. The reproducibility of research and the misinterpretation of p-values. R Soc Open Sci 2017;4:171085. 10.1098/rsos.171085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.d-maps.com . cartographer Map DR of the Congo: boundaries, provinces. Available: https://d-maps.com/carte.php?num_car=4886&lang=en2020
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.