

SUMMARY
Addiction is a disorder of behavioral symptoms including enhanced incentive salience of drug-associated cues, but also a negative affective state. Cocaine-evoked synaptic plasticity in the reward system, particularly the nucleus accumbens (NAc), drives drug-adaptive behavior. However, how information is integrated downstream of the NAc remains unclear. Here, we identify the ventral pallidum (VP) as a site of convergence of medium spiny neurons expressing dopamine (DA) receptor type 1 (D1-MSNs) and type 2 (D2-MSNs) of the NAc. Repeated in vivo cocaine exposure potentiated output of D1-MSNs, but weakened output of D2-MSNs, occluding LTP and LTD at these synapses, respectively. Selectively restoring basal transmission at D1-MSN-to-VP synapses abolished locomotor sensitization, whereas restoring transmission at D2-MSN-to-VP synapses normalized motivational deficits. Our results support a model by which drug-evoked synaptic plasticity in the VP mediates opposing behavioral symptoms; targeting the VP may provide novel therapeutic strategies for addictive disorders.
In Brief
Creed et al. identify distinct forms of plasticity between D1- and D2-MSNs and the ventral pallidum. Cocaine exposure occluded activity-dependent LTP and LTD at D1-VP and D2-VP synapses. Transmission at these synapses mediates distinct behavioral symptoms of addiction.
INTRODUCTION
With repeated cocaine consumption, positive and negative symptoms accumulate to form a syndrome that is eventually diagnosed as addiction. The acute reinforcing effects of addictive drugs are mediated by dopamine (DA) release from the ventral tegmental area (VTA) to the nucleus accumbens (NAc) (Di Chiara and Imperato, 1988; Pascoli et al., 2015). The addicted state is defined by compulsive drug taking, driven by craving and the emergence of a negative affective state (Le Moal and Koob, 2007). Drug craving is a pathologically strong drug-associated memory that is precipitated by exposure to drug-associated cues and contexts and drives drug taking even after long periods of abstinence (Kalivas et al., 1998; Robbins et al., 1997). The emergence of a negative affective state after prolonged withdrawal also drives drug taking, which can relieve this aversive state through negative reinforcement. One prominent feature of this state is anhedonia (Le Moal and Koob, 2007).
Since all addictive drugs increase extracellular DA in the NAc (Di Chiara and Imperato, 1988; Koob and Bloom, 1988), which induces characteristic forms of plasticity (Lüscher, 2016; Wolf, 2016), we hypothesized that drug-evoked plasticity in the NAc may contribute to both the positive and negative symptoms of addiction. Considerable evidence implicates cocaine-evoked synaptic plasticity on medium spiny neurons that express D1 receptors (D1-MSNs) in drug reinforcement (Creed et al., 2015; Lobo and Nestler, 2011; Pascoli et al., 2014). Potentiation of inputs onto D1-MSNs has been implicated in the development of sensitization to addictive drugs (Pascoli et al., 2011; Robinson and Berridge, 2003) and in cue-induced drug seeking (Kalivas et al., 1998; Pascoli et al., 2014, 2015). Conversely, activity of the indirect pathway, which is modulated by DA acting on cells that express D2 receptors (D2-MSNs), is linked to aversion (Danjo et al., 2014; Hikida et al., 2010). Adaptations in D2-MSNs have been described in cocaine-addicted patients (Volkow et al., 1993) and animal models (Chandra et al., 2015; Lobo et al., 2010; Lobo and Nestler, 2011). While adaptations in D2-MSNs in the NAc have been implicated in anhedonic states arising from chronic pain (Ren et al., 2016; Schwartz et al., 2014) or stress (Francis et al., 2015; Khibnik et al., 2016), the involvement of D2-MSNs in anhedonia arising after cocaine withdrawal is undetermined. While there is evidence to support divergent roles of D1- and D2-MSNs in cocaine-induced behaviors, the synaptic basis of these adaptations is not known, nor is it clear which downstream target(s) of MSNs are implicated in maladaptive behaviors.
The ventral pallidum (VP) is a major target of the NAc (Heimer and Wilson, 1975), receiving innervation from D1- and D2-MSNs (Kupchik et al., 2015). VP neurons robustly respond to reward and reward-predictive cues (Tindell et al., 2005), while VP lesions reduce hedonic impact and motivation for reward (Cromwell and Berridge, 1993). The VP has also been directly implicated in drug-adaptive behavior (Robledo and Koob, 1993; Root et al., 2013); a subset of VP neurons project to the VTA, where they modulate activity of VTA and GABA neurons and mediate drug seeking (Mahler et al., 2014).
Despite evidence implicating the NAc and VP in reward-related behavior, how addictive drugs affect plasticity between the NAc and VP, and how this plasticity contributes to drug-adaptive behavior, remains largely unknown. Here, we propose a model by which cocaine potentiates and depresses D1- and D2-MSN inputs from the NAc to VP, respectively. We characterize divergent forms of homosynaptic plasticity at D1- and D2-MSN-to-VP synapses, which are presynaptically expressed and occluded by cocaine treatment. We selectively normalize transmission at these synapses to establish causal links between D1 projections and behavioral sensitization to cocaine, and between D2 projections and the emergence of anhedonia. Together, our data support a model whereby the VP integrates rewarding and aversive signals from the NAc that govern distinct behavioral symptoms of addictive disorders.
RESULTS
Cocaine Increases Inhibitory Transmission in the NAc-to-VP Pathway
To determine the effect of cocaine on inhibitory transmission in the VP, we used whole-cell patch-clamp electrophysiology in acute brain slices from adult mice. We first recorded electrically evoked inhibitory postsynaptic currents (IPSCs) from VP cells from mice that received five injections of saline or cocaine (20 mg/kg, i.p. [intraperitoneally]), followed by 10 days of withdrawal (Figure S1A, available online). Here, we focused exclusively on the ventromedial (vm) subdivision of the VP, which is innervated by MSNs from the shell of the NAc and sends projections to the VTA (Haber et al., 1985). We tested the ability of high-frequency stimulation (HFS) to induce long-term plasticity of inhibitory transmission onto VP neurons. Consistent with previous reports (Kupchik et al., 2014), HFS (100 pulses at 100 Hz, 20 s ISI) induced long-term potentiation (LTP) of IPSCs in saline-treated mice, whereas HFS failed to induce LTP in slices from cocaine-treated mice (Figures S1B and S1C). This failure to induce LTP was also apparent in mice that had self-administered cocaine, indicating that this plasticity is evoked by contingent or non-contingent exposure (Supplemental Experimental Procedures).
The NAc is considered the predominant source of inhibitory input to the VP (Heimer and Wilson, 1975). To resolve the neurons that contribute to the potentiation of inhibitory transmission in the VP after cocaine exposure, we first characterized the nature of the NAc-to-VP projection with optogenetic-assisted circuit mapping.
Accumbal D1- and D2-MSNs Both Project to the VP
In the dorsal striatum, D1- and D2-MSNs have largely segregated projection patterns, with D1-MSNs projecting to the substantia nigra and D2-MSNs projecting to the internal segment of the globus pallidus (Alexander et al., 1990). However, this segregation is not necessarily observed at the level of the NAc (Kupchik et al., 2015; Zahm and Heimer, 1990), and the proportion of D1- and D2-MSNs from the NAc shell to vmVP is not known. To address this, we injected the retrograde tracer cholera toxin subunit B (CTB) into the vmVP of bacterial artificial chromosome (BAC) transgenic mice expressing a reporter protein under the control of the D1R or D2R promoter and analyzed sections of the NAc shell (Figures 1A–1C). In D1-To (D1-Tomato) mice, of 500 MSNs positive for CTB, 282 were also positive for the D1-To reporter protein, and in D2-eGFP mice, 229/500 CTB-positive MSNs also expressed the D2-eGPF reporter (Figure 1D), indicating that D1- and D2-MSNs from the NAc shell equally project to the vmVP. We assayed the functional connectivity of D1- and D2-MSNs to the vmVP by injecting floxed ChR2 into the NAc shell of D1R- or D2R-Cre mice and recording light-evoked responses in vmVP cells (Figure 1E). We found that 92.8% of vmVP cells received innervation from D1-MSNs, while 75.0% of vmVP cells were innervated by D2-MSNs. Currents were GABAA mediated, as they were blocked by picrotoxin, with no differences in the release probability of D1- versus D2-MSN synapses (Figures 1F and 1G). Together, these experiments indicate that D1- and D2-MSNs in the NAc shell project in equal proportion to the vmVP, where they innervate large, overlapping populations of postsynaptic neurons.
Figure 1. D1- and D2-MSN Projection to the VP.
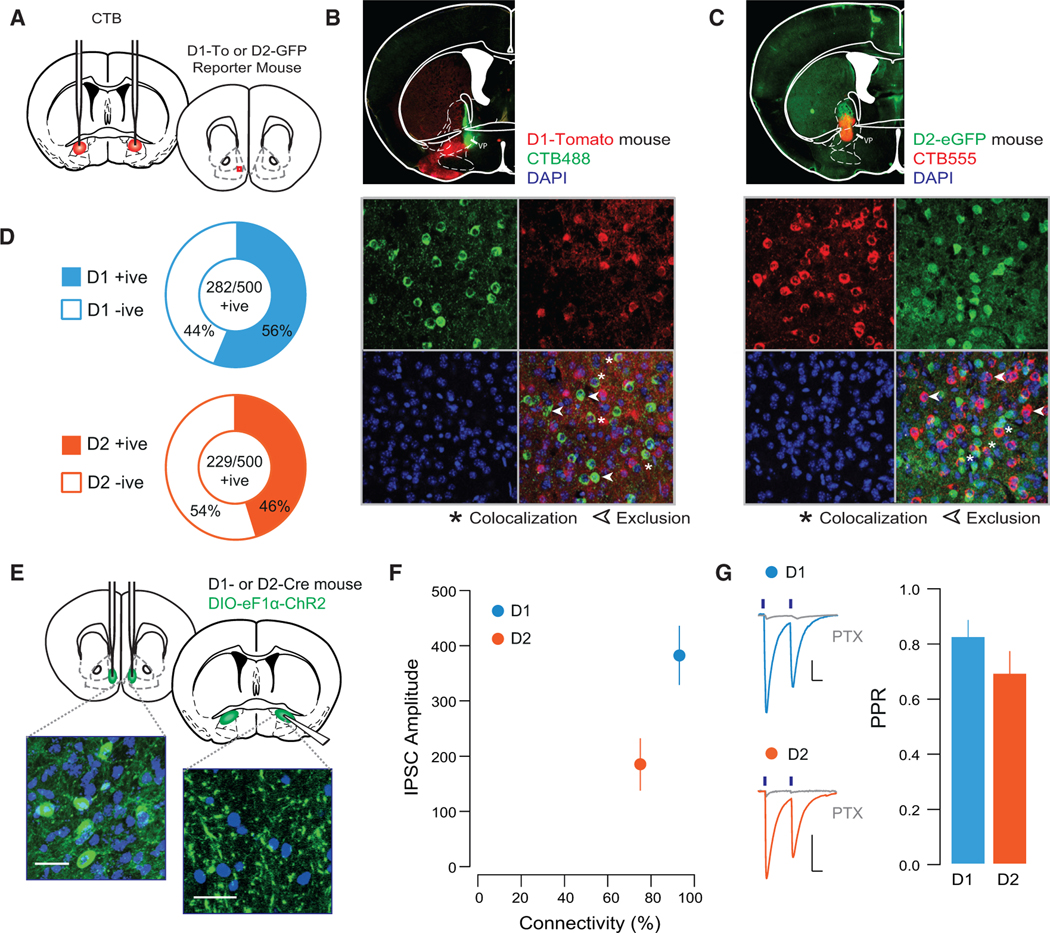
(A) Schematic of experiment.
(B) Representative injection site of CTB488 into the VP (top) and 40× confocal images of sections of the NAc shell of D1-TdTomato mice.
(C) Injection site of CTB555 into the VP (top) and 40× confocal images of sections of the NAc shell of D2-eGFP mice (n = 250 cells from 2 mice of each genotype).
(D) Quantification of CTB-positive cells based on their co-expression with reporter protein; in D1-TdTomato mice, 56% of CTB-positive cells (five sections/mouse from two mice) co-localized with TdTomato, while in D2-eGFP mice, 46% of CTB-positive cells co-localized with the D2-eGFP protein (five sections/mouse from two mice).
(E) Schematic and 40× confocal images of injection site in the NAc shell floxed-channelrhodpsin, and terminal fields in the VP (scale bars, 50 μM).
(F) Connectivity plot summarizing optogenetic circuit mapping. In the VP, 92.8% of neurons were innervated by D1-MSNs (83 cells from 4 mice; averageconnectivity strength, 382.5 ± 53.7 pA), whereas 75.0% received innervation from D2-MSNs (56 cells from 4 mice; average connectivity strength, 184.9 ± 47.2 pA).
(G) Currents blocked by picrotoxin (PTX; 20 μM). There was no difference in the PPR between D1- and D2-VP synapses (D1, 0.75 ± 0.05; D2, 0.82 ± 0.09; t = 0.655, p = 0.514).
Cocaine Occludes Bidirectional Plasticity at NAc-to-VP Synapses
Since D1- and D2-MSNs undergo distinct adaptations following cocaine (Bertran-Gonzalez et al., 2008; Lobo et al., 2010; Pascoli et al., 2014), we determined how cocaine affects transmission at both of these outputs to the VP. We selectively transfected the two classes of MSNs by injecting a floxed chETA construct into the NAc shell of D1- or D2-Cre mice. The VP is composed of interneurons and projection neurons with varied targets (Root et al., 2015; Zahm and Heimer, 1990); VTA-projecting VP neurons have been implicated in cocaine-seeking behavior (Mahler et al., 2014). We therefore selectively recorded from this population, which was identified by injecting the retrograde tracer CTB555 into the VTA (Figures 2A and 2B). Connectivity rates between MSNs and this subpopulation of VP neurons were similar to connectivity rates observed in the entire VP (connectivity, 222/256 D1-Cre and 263/329 D2-Cre mice).
Figure 2. Cocaine Occludes Bidirectional Plasticity at NAc-to-VP Synapses.

(A) Schematic of experiment.
(B) Representative section of ChR2 infection site in the NAc (left), and CTB seeding in the VTA (middle). Section (40×) of the VP showing labeled fibers opposing CTB-positive VP cells (left).
(C) IPSC recorded in VP cell during 100 Hz light stimulation.
(D) In saline-treated D1-Cre mice, HFS induced an LTP at NAc-VP synapses. HFS failed to induce LTP in cocaine-treated mice (saline [SAL], 181.16% ± 30.19%, 4 cells from 2 mice; cocaine [COC], 100.35% ± 18.53%, 8 cells from 3 mice). Representative traces (baseline, light blue; post-LTP, dark blue), single-cell examples (top), and group data (bottom) are shown.
(E) In saline-treated D2-Cre mice, HFS induced an LTD at NAc-VP synapses. HFS failed to induce LTD in cocaine-treated mice (SAL, 62.95% ± 11.61%, 10 cells from 3 mice; COC, 100.05% ± 5.95%, 10 cells from 3 mice). Representative examples of single-cell experiments (top) and group data (bottom) are shown.
(F) PPR was not different between D1- and D2-MSN-to-VP synapses in saline-treated mice (D1-MSNs, 0.733 ± 0.057, 10 cells from 3 mice; D2-MSNs, 0.708 ± 0.148, 10 cells from 3 mice). Relative to saline-treated mice, PPR was decreased in D1-MSNs (D1-MSNs, 0.530 ± 0.043, 10 cells from 3 mice; t = 2.84, p = 0.011) and increased in D2-MSNs (D2-MSNs, 1.207% ± 0.188%, 10 cells from 3 mice; t = 2.09, p = 0.051) in cocaine-treated mice.
*p < 0.05; all plots mean ± SEM. For representative traces, baseline (20 trials) and final 20 sweeps of recording are shown. All scale bars, 50 pA, 20 ms. Related to Figures S1–S3.
In saline-treated mice, HFS (Figure 2C) induced LTP of inhibitory transmission at D1-MSN-to-VP synapses (Figure 2D), analogous to what we have previously observed at D1-MSN synapses onto GABAergic neurons of the VTA (Bocklisch et al., 2013). Conversely, the same protocol applied at D2-MSN-to-VP synapses induced a long-term depression (LTD) of inhibitory transmission (Figure 2E). In cocaine-treated mice, HFS failed to alter the amplitude of evoked inhibitory transmission at either D1- or D2-MSN-to-VP synapses (Figures 2D and 2E). Cocaine treatment was associated with a presynaptic facilitation of release probability at D1-MSN synapses and a presynaptic depression of release probability of D2-MSN synapses (Figure 2F). This cocaine-evoked plasticity was not apparent on the first day of withdrawal from cocaine (Figure S2), suggesting that an incubation period is required for the occlusion of activity-dependent plasticity at NAc-to-VP synapses. In further support of an occlusion mechanism, we performed recordings of light-evoked currents in strontium, an established method to desynchronize release and reveal quantal events (Patton et al., 2016; Xu-Friedman and Regehr, 1999), (Figures S3A and S3B). There were no differences in event amplitude between saline- or cocaine-treated mice (Figure S3C). However, in D1-Cre mice, cocaine treatment increased the average number of events per light pulse, suggesting an increase in release sites. Conversely, the average number of events per light pulse was decreased at D2-MSN-to-VP synapses in cocaine-treated mice (Figure S3D).
Occlusion of activity-dependent plasticity by cocaine suggests that these forms of plasticity share common underlying mechanisms. We therefore sought to determine the expression mechanisms underlying these two forms of activity-dependent plasticity at NAc-to-VP synapses.
Cocaine Occludes Activity-Dependent Plasticity at D1-MSN NAc-to-VP Synapses
We selectively targeted D1-MSNs with chETA and measured light-evoked currents in VTA-projecting VP cells. We confirmed that HFS induced an LTP, which was insensitive to inclusion of the calcium chelator 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA) in the patch pipette (Figure 3A). In further support of a presynaptic mechanism of this homosynaptic plasticity, we observed that HFS induced a decrease in the failure rate and paired-pulse ratio (PPR), and increase in the variance of inhibitory currents (Figures 3B and 3C). This is reminiscent of a form of inhibitory plasticity at D1-MSN-to-VTA GABA neuron synapses that is presynaptically mediated, protein kinase A (PKA) and D1R dependent, and also occluded by cocaine treatment (Bocklisch et al., 2013). We confirmed that at D1-MSN-to-VP synapses, HFS-induced LTP was prevented by incubation in the D1 antagonist SCH23390 or the PKA activator forskolin (FSK; Figure 3D). In support of an occlusion mechanism by cocaine, FSK application induced an LTP of inhibitory transmission that was completely absent in cocaine-treated mice (Figure 3E). Application of FSK also induced an LTP of transmission at D2-MSN-toVP synapses (Figure 3F). These results confirm that HFS-induced LTPatD1-MSN-to-VP synapses is presynaptic, and D1R and PKA mediated, whereas HFS differentially affects plasticity at D1- and D2-MSN-to-VP synapses through divergent mechanisms.
Figure 3. HFS-Induced LTP at D1-VP Synapses Is Presynaptic and PKA Dependent.

(A) HFS-induced LTP at D1-VP synapses was insensitive to postsynaptic BAPTA (mock HFS, 100.98% ± 16.51%, 6 cells from 3 mice; HFS, 188.63% ± 32.94%,11 cells from 4 mice; HFS with BAPTA, 170.09% ± 18.09%, 5 cells from 2 mice).
(B) HFS-induced LTP was associated with a decrease in the PPR, decrease in failure rate (FR), and no change in 1/CV2 (paired t test PPR, t = 2.27, p = 0.046; FR, t = 2.34, p = 0.042; 1/CV2, t = 0.863, p = 0.407, 11 cells from 4 mice).
(C) Twenty individual traces pre- and post-HFS protocol.
(D) HFS-induced LTP was prevented by incubation in SCH23390 of FSK (control [CTRL], 223.91% ± 16.09%, 4 cells from 2 mice; SCH23390, 92.69% ± 12.67%,9 cells from 3 mice; FSK, 114.3% ± 30.13%, 7 cells from 3 mice).
(E) Application of FSK induced an LTP at D1-VP synapses, which was occluded in cocaine-treated mice (SAL, 220.7% ± 21.72%, 10 cells from 3 mice; COC, 97.46% ± 9.06%, 6 cells from 2 mice).
(F) Application of FSK induced an LTP at D2-VP synapses (CTRL, 92.51% ± 9.11%, 4 cells from 3 mice; FSK, 175.5% ± 33.62%, 4 cells from 3 mice).
*p < 0.05; all plots mean ± SEM. For representative traces, first and last 20 trials of recording in light and dark blue, respectively. Scale bars, 20 pA, 20 ms.
Cocaine Occludes Activity-Dependent Plasticity at D2-MSN NAc-to-VP Synapses
We confirmed that HFS induced an LTD of transmission at D2-MSN-to-VP synapses. This plasticity was insensitive to postsynaptic BAPTA (Figure 4A) and was associated with an increase in the failure rate and PPR (Figures 4B and 4C). How can a single stimulation protocol induce divergent forms of plasticity at D1- and D2-MSN-to-VP synapses? In addition to the DA receptors, the two populations of MSNs differ in their expression of endogenous opioids and receptors. Specifically, D1-MSNs release dynorphin (an endogenous κ-ligand) and express μ-opioid receptors, whereas D2-MSNs release enkephalin (an endogenous δ-ligand) and express both the δ- and κ-opioid receptors (Gerfen et al., 1990; Gerfen and Young, 1988). δ-opioid receptors (DORs) on D2R-MSNs may thus act as autoreceptors when opioids are released by high-frequency activity. Through this mechanism, enkephalin decreases the probability of transmitter release from the presynaptic terminal, which may be long lasting (Patton et al., 2016; Spanagel et al., 1990). Application of the selective DOR agonist, DPDPE, induced an LTD of D2-MSN-to-VP transmission. The long-lasting nature of this plasticity was confirmed by chasing DPDPE with the DOR antagonist naltrindol (NAL). DPDPE failed to induce an LTD at D2-MSN-to-VP synapses in cocaine-treated mice, supporting an occlusion scenario (Figure 4D). Further implicating DORs in HFS-induced LTD, we applied HFS following incubation with NAL; in this condition of DOR antagonism, HFS failed to induce an LTD (Figure 4E). Previous studies have reported that enkephalin levels in the reward system are sensitive to drug administration (Crespo et al., 2001). To test the possibility that HFS failed to induce an LTD after cocaine due to a change in tonic levels of enkephalin, we quantified levels of enkephalin in the NAc and VP (Supplemental Experimental Procedures) but found no evidence for a decrease in enkephalin levels in the VP following cocaine treatment (Figures S4A and S4B).
Figure 4. HFS-Induced LTD at D2-VP Synapses Is Presynaptic and DOR Mediated.

(A) HFS-induced LTD at D2-VP synapses was insensitive to postsynaptic BAPTA (CTRL, 103.54% ± 17.57%, 5 cells from 2 mice; HFS, 61.43% ± 9.92%, 10 cells from 4 mice; HFS with BAPTA, 60.62% ± 6.42%, 6 slices from 3 mice).
(B) HFS-LTD was associated with an increase in the PPR and failure rate and a decrease in C1/V12 (paired t test PPR, t = 2.45, p = 0.035; FR, t = 2.79, p = 0.019; 1/CV2, t = 1.079, p = 0.306, 11 cells from 6 mice).
(C) Twenty individual IPSC traces pre- and post-HFS protocol.
(D) DPDPE induced an LTD in saline-, but not cocaine-treated, mice (SAL, 59.33% ± 7.12%, 10 cells from 3 mice; COC, 97.994% ± 8.14%, 8 cells from 3 mice). Naltrindole (NAL) applied after DPDPE-LTD did not reverse the plasticity (52.92% ± 8.5%, 5 cells from 2 mice).
(E) Incubation in NAL prevented the HFS-induced LTD (CTRL, 61.43% ± 9.92%, 10 cells from 4 mice; NAL, 130.4% ± 8.84%, 9 cells from 5 mice).
(F) DPDPE did not affect transmission at D1-VP synapses (CTRL, 111.85% ± 18.49%, 3 cells from 3 mice; DPDE, 106.99% ± 6.49%, 11 cells from 4 mice); DAMGO (1 μM) induced a depression of IPSCs (61.33% ± 6.08%, 6 cells from 3 mice).
*p < 0.05, all plots mean ± SEM. For representative traces, first and last 20 trials of recording in light and dark orange, respectively. Scale bars, 20 pA, 20 ms. Related to Figure S4.
Finally, this mechanism was unique to D2-MSN-to-VP synapses, since DPDPE had no effect on inhibitory transmission at D1-MSN-to-VP synapses. Given that D1-MSNs express the μ-opioid receptor, we confirmed that D1-MSN-to-VP synapses were depressed by application of the selective μ-opioid receptor agonist, DAMGO (Figure 4F). These results suggest that activity-dependent LTD occurs at D2-MSN-, but not D1-MSN-to-VP synapses, due to enkephalin co-release. This plasticity is presynaptically expressed, DOR mediated, and occluded by cocaine treatment.
Diverse Symptoms of Addiction Emerge after Withdrawal from Cocaine
Addiction is characterized by positive and negative symptoms, which are reflected as drug-evoked craving and emergence of a negative affective state, respectively. Behavioral sensitization to cocaine is the phenomenon by which repeated exposure to an identical concentration of drug induces a progressively enhanced locomotor response. This response reflects drug-evoked plasticity since it persists for months after cocaine withdrawal, and models features of reinstatement of drug-seeking behavior (Robinson and Berridge, 1993; Steketee and Kalivas, 2011). Over five daily exposures to cocaine i.p., the locomotor response to cocaine was steadily increased and remained elevated 10 days following the last cocaine exposure (Creed et al., 2015; Pascoli et al., 2011). This time point is critical, as it also reflects the emergence of a negative affective state following cocaine withdrawal (Le Moal and Koob, 2007; Meye et al., 2015). One feature of this negative affective state is a decreased motivation for natural reward and anhedonia (Barnea-Ygael et al., 2016; Barr and Phillips, 1999).
Pharmacological manipulation of GABAergic and δ-opioid signaling in the VP has profound effects on motivation for and hedonic impact of natural reward (Inui and Shimura, 2014; Johnson and Stellar, 1994); we confirmed that following withdrawal from repeated cocaine injections, motivation for and hedonic impact of sucrose reward were decreased. Mice were first trained on an operant task, where a lever press was required to earn access to a 10% sucrose reward (Figure S5A). Motivation for sucrose was then tested in a progressive ratio (PR) task. Cocaine-treated mice exhibited a significant decrease in break point during the PR task relative to baseline; there was no change in breakpoint in saline-treated mice (Figure S5B). Sucrose consumption was also assessed in a free access task. Mice explored an open arena with a reward zone containing 10% sucrose. During this task, the orofacial reactions of mice to sucrose were quantified (Figure S6A). In response to palatable tastes, mice exhibit stereotyped tongue protrusions emulating licking, which reflect the hedonic value of the reward (Berridge, 2000; Steiner et al., 2001). We first validated that hedonic tongue protrusions followed a typical dose-response curve in response to increasing concentrations of sucrose (Figure S6B). We found that cocaine treatment did not affect the amount of entries made into the reward zone during the task, but significantly reduced the amount of time spent in the reward zone and the number of hedonic tongue protrusions (Figures S6C–S6E). These findings confirm that cocaine decreases both motivation and hedonic impact for natural reward, which reflect the emergence of a negative affective state.
We then hypothesized that cocaine-evoked plasticity at D1and D2-MSN-to-VP synapses may differentially contribute to these behavioral adaptations.
Validation of Rescue Stimulation Protocols
Given the opposing effects of cocaine at D1- and D2-MSN-to-VP synapses, we sought to develop stimulation protocols that could selectively decrease transmission at D1-MSN synapses and potentiate transmission at D2-MSN synapses in cocaine-treated mice.
First, we applied protocols ex vivo on VP slices obtained from cocaine-treated mice (Figure 5A). At D1-MSN-to-VP synapses, intermittent low-frequency stimulation (trains of five pulses at 1 Hz, with 10 s between trains, applied for 10 min) induced an LTD of inhibitory transmission (Figure 5B). At D2-MSN-to-VP synapses, bursts of 10 Hz stimulation (trains of ten pulses at 10 Hz, with 10 s between trains, applied for 10 min) induced an LTP of inhibitory transmission (Figure 5C). Next, we validated these protocols in vivo; optic fibers were implanted over the VP and rescue protocols were applied 24 hr prior to preparing slices of the VP and carrying out whole-cell recordings (Figure 5D). In cocaine-treated D1-Cre mice, the rescue protocol restored the ability of HFS to induce an LTP at D1-MSN-to-VP synapses, without altering the magnitude of HFS-induced LTP in saline-treated mice (Figure 5E). In cocaine-treated D2-Cre mice, application of the D2-selective rescue protocol restored the ability of HFS to induce an LTD at D2-MSN-to-VP synapses (Figure 5F). We observed that in saline-treated mice that had undergone the in vivo rescue protocol, the HFS-induced LTD was more pronounced than in naive saline-treated mice, suggesting that the protocol may have induced a slight potentiation relative to baseline levels.
Figure 5. Validation of Stimulation Protocols to Normalize Transmission at D1- or D2-MSN-to-VP Synapses.

(A) Schematic of experiments.
(B) In cocaine-treated mice, trains of five pulses at 1 Hz, ITI of 10 s applied for 10 min induced an LTD at D1-MSN-to-VP synapses (49.41% ± 5.93%, 5 cells from 2 mice).
(C) In cocaine-treated mice, trains of ten pulses at 10 Hz, ITI of 10 s applied for 10 min, induced an LTP at D2-MSN-to-VP synapses (217.48% ± 51.38%, 6 cells from 3 mice).
(D) Schematic of experiments.
(E) In cocaine-treated mice that had undergone in vivo stimulation (protocol as in B), HFS induced an LTP at D1-MSN-to-VP synapses (175.5% ± 33.62%, 4 cells from 3 mice). Control data from Figure 2D are plotted for comparison.
(F) In cocaine-treated mice that had undergone in vivo stimulation (protocol as in C), HFS induced an LTP at D1-MSN-to-VP synapses (48.89% ± 14.29%, 9 cells from 2 mice). Control data from Figure 2E are plotted here for comparison.
*p < 0.05; all plots mean ± SEM. For representative traces, first and last 20 trials of recording are shown. Scale bars, 20 pA, 20 ms.
Having validated that these protocols normalized transmission selectively at D1- or D2-MSN-to-VP synapses, we applied these protocols in vivo (Figures S7A and S7B) to determine the role of the plasticity at each synapse in drug-adaptive behavior.
Potentiation of D1-MSN-to-VP Synapses Drives Behavioral Sensitization to Cocaine
Our previous work has implicated potentiation of excitatory inputs onto D1-MSNs in behavioral sensitization to cocaine; depotentiating excitatory inputs onto D1-MSNs abolished the sensitized locomotor response to cocaine (Creed et al., 2015; Pascoli et al., 2011). However, how cocaine-induced changes in output of the D1-MSNs contribute to behavioral sensitization is not known. We therefore tested the hypothesis that normalizing the output of these neurons would also abolish locomotor sensitization. A single dose of cocaine (20 mg/kg, i.p.) induced an increase in locomotor activity, which was further enhanced with subsequent doses of cocaine, and persisted for 10 days after withdrawal (Figures 6A–6C). In vivo depotentiation of D1-MSN output to the VP abolished the sensitized locomotor response to cocaine, but had no effect on the acute locomotor response (Figure 6B). Conversely, rescuing D2-MSN-to-VP transmission had no effect on either the sensitized or acute locomotor response to cocaine (Figure 6C). These findings further implicate synaptic adaptations selectively in D1-MSNs in drug-adaptive behaviors modeling craving and relapse, the so-called positive symptoms of addiction.
Figure 6. Selectively Normalizing Transmission at D1- or D2-MSN-to-VP Synapses Differentially Affects Drug-Adaptive Behavior.

(A–C) Locomotor sensitization experiments.
(A) Schematic of experiment.
(B) In vivo rescue of D1 transmission abolished the sensitized response to cocaine (COC CTRL, 1,578.0 ± 248.8, n = 7; COC with D1 rescue, 1,026.1 ± 133.08,n = 12; t = 2.087, p = 0.05), but had no effect on acute response to cocaine (SAL CTRL, 878.83 ± 227.41, n = 6; SAL with D1 rescue, 860.3 ± 134.6, n = 8; t = 0.07, p = 0.94).
(C) Rescue of D2-MSN VP transmission had no effect on cocaine response (COC CTRL, 1,735.86 ± 281.74, n = 7; COC with D2 rescue, 1,513.58 ± 161.20, n = 12; t = 0.21, p = 0.83; SAL CTRL, 1,042.17 ± 151.81, n = 6; SAL with D2 rescue, 1,151.25 ± 66.55, n = 8; t = 0.70, p = 0.50).
(D) Schematic of sucrose-related experiments.
(E and F) Operant task (E) raster plots of first 30 min of PR test in saline- (top) and cocaine-treated mouse (bottom). (F) Cocaine-treated mice had lower break points during the test relative to baseline (SAL CTRL, 1.10 ± 0.17, n = 9, t = 0.594, p = 0.56; COC CTRL, 0.597 ± 0.18, n = 14, t = 2.24, p = 0.034) and performance was impaired in cocaine-treated mice following D1 rescue (0.49 ± 0.06, n = 8, t = 8.67, p < 0.001), while D2 rescue normalized the break point (1.05 ± 0.18, n = 9, t = 0.26, p = 0.801).
(G–J) Free access task (G) track plots of a saline- (top) and cocaine-treated mouse (bottom) during the free access task. (H) There was no effect of cocaine or intervention on reward zone entries. (I) Cocaine-treated mice spent less time in the reward zone during the test relative to baseline (SAL CTRL, 1.00 ± 0.097, n = 9, t = 0.01, p = 0.99; COC CTRL, 0.79 ± 0.06, n = 14, t = 3.47, p = 0.002) and performance was impaired following D1 rescue (SAL, 0.72 ± 0.11, n = 6, t = 2.47, p = 0.032; COC, 0.65 ± 0.13, n = 8, t = 2.69, p = 0.0176), while D2 rescue normalized the break point in cocaine-treated mice (1.07 ± 0.18, n = 9, t = 0.38, p = 0.71).
(J) Hedonic tongue protrusions were reduced relative to baseline in cocaine-treated mice (0.582 ± 0.091, n = 14, t = 4.59, p < 0.001) and mice that had D1 rescue protocol (0.627 ± 0.15, n = 8, t = 2.85, p = 0.013); mice with the D2 rescue protocol were not impaired relative to baseline (0.92 ± 0.22, n = 9, t = 0.36, p = 0.727). *p < 0.05, **p < 0.01, ***p < 0.001 by t test comparing locomotor response to cocaine challenge (B and C) or normalized response to null hypothesis (F and H–J). All plots are mean ± SEM. Related to Figures S5–S8 and Table S1.
Depression of D2-MSN-to-VP Synapses Drives Impaired Motivation and Hedonic Impact
Given the role of D2-MSNs in aversion and depressive symptoms (Kravitz et al., 2012; Lobo et al., 2010; Ren et al., 2016; Schwartz et al., 2014), we next determined whether plasticity at D2-MSN-to-VP synapses mediates symptoms of the negative affective state that emerges after cocaine withdrawal. Motivation for and hedonic reactions to sucrose were tested with a PR and free access task, respectively (Figure 6D). Cocaine-treated mice exhibited a decrease in break point in the PR task (Figures 6E and 6F). The application of the in vivo depression protocol at D1-MSN-to-VP synapses had no effect on PR break point. However, application of the in vivo potentiation protocol at D2-MSN-to-VP synapses normalized performance on the PR task in cocaine-treated mice (Figures 6F, S5D, and S5E). In the free access task, cocaine-treated mice made equal numbers of entries into the reward zone (Figures 6G and 6H) but spent less time in the reward zone and exhibited fewer hedonic tongue protrusions (Figures 6I and 6J). Neither rescue protocol affected the number of entries made into the reward zone (Figures 6H and S6F). While D1 rescue had no effect on the amount of time spent in the reward zone or the number of hedonic tongue protrusions, application of the D2 rescue protocol normalized both of these parameters to levels equal to saline-treated controls (Figures 6I, 6J, S6G, and S6H). These results strongly implicate D2-MSNto-VP synapses in cocaine-induced anhedonia and amotivation for natural reward.
DISCUSSION
We focus on the VP because it is the primary output of the NAc and is necessary for the expression of motivated behaviors (Root et al., 2015). Both D1- and D2-MSNs project from the NAc to VP neurons that then project to the VTA. Cocaine treatment potentiated transmission at D1-MSN-to-VP synapses and depressed transmission at D2-MSN-to-VP synapses. By selectively normalizing transmission at each output, we demonstrate a role for D1-MSN-to-VP synapses in behavioral sensitization, and D2-MSNto-VP synapses in the emergence of a cocaine-induced negative affective state.
Anatomical Integration of D1- and D2-MSNs in the VP
In the dorsal striatum, D1- or D2-MSN projections are largely segregated, with D1-MSNs projecting to the substantia nigra and D2-MSNs innervating the globus pallidus. This dissociation has been questioned in the VP, which anatomically is subdivided into the vm and dorsomedial (dm) portions (Root et al., 2015). Here, we focused on VTA-projecting VP neurons in the rostral subdivision of the VP, where we found that the majority of neurons were also innervated by both D1- and D2-MSNs from the NAc shell. D1-MSNs, also referred to as “direct pathway” neurons, have been linked to reward and positive affect, whereas the D2-MSNs, or “indirect” pathway neurons, are linked to aversive stimuli and negative affect. The anatomy of the VP thus suggests that aversive and reinforcing signals are integrated and synthesized in order to drive multiple components of drug-adaptive behavior.
Cocaine Occludes Bidirectional, Activity-Dependent Plasticity
How, then, does cocaine affect plasticity at these synapses, and how does this plasticity mediate different facets of cocaine-adaptive behavior? We characterized activity-dependent plasticity and its interaction with cocaine-evoked plasticity. D1-MSN outputs to the VTA exhibit a form of homosynaptic LTP, which is induced by high-frequency stimulation with concomitant DA signaling and expressed by an increase in presynaptic release probability (Bocklisch et al., 2013). We also observe this form of plasticity at D1-MSN terminals in the VP. Signaling through D1R is necessary for LTP expression since the D1 antagonist, SCH23390, blocked the potentiation. The DA transporter and DARs are expressed in the VP (Mengual and Pickel, 2004; Napier and Maslowski-Cobuzzi, 1994). Ambient signaling through D1 receptors is permissive for plasticity in other regions of the reward system (Bocklisch et al., 2013; Creed et al., 2015; Shen et al., 2008), and we propose that this mechanism is also implicated in the plasticity described here.
In contrast, the identical HFS protocol at D2-MSN-to-VP synapses induced a depression that is explained by co-released opioid peptides. Enkephalin is co-released with GABA when D2-MSNs are stimulated at high frequency, which acts on presynaptic DORs to decrease exocytosis through Gi (inhibitory regulative G-protein) signaling. This depression persists even in the absence of the ligand; enkephalin levels are normal after withdrawal from cocaine. Moreover, the DOR agonist DPDPE mimicked LTD at D2-MSNs but failed to do so at D1-MSN-to-VP synapses because DORs are not expressed on these terminals. Acutely, cocaine likely inhibits D2-MSNs via increase of DA levels. However, after cocaine withdrawal, potentiation of excitatory inputs onto these MSNs may enhance their activity, sufficient to drive co-release of enkephalin. This differential co-release and the expression of opioid receptors on MSN populations provide a substrate for divergent effects of cocaine on activity-dependent output of MSN pathways.
Importantly, neither LTP nor LTD could be induced after mice were exposed to cocaine, suggesting occlusion. This interpretation is supported by the changes of PPR and frequency of evoked unitary events. Thus, cocaine treatment potentiates transmission at D1-MSN-to-VP synapses and weakens transmission at D2-MSN-to-VP synapses. This is in line with previous studies showing distinct cocaine-induced adaptations in D1- and D2-MSNs. Cocaine treatment increases activity-dependent gene expression and basal calcium activity in D1-MSNs, with concomitant decreases in D2-MSNs (Calipari et al., 2016; Lobo et al., 2010, 2013). Differential expression of DARs has been implicated in distinct cocaine-induced adaptations, since plasticity in D1-MSNs is due to D1R activation (Bertran-Gonzalez et al., 2008; Pascoli et al., 2011), while D2R activation decreases excitability and GABA release from D2-MSNs (Ericsson et al., 2013; Perez et al., 2006). Our work posits an additional role of endogenous opioids in cocaine-evoked plasticity.
Transmission between D1-MSNs and the VP Mediates Behavioral Sensitization
We used insight into the expression mechanisms of cocaine-evoked plasticity to design cell-type-specific reversal protocols. At D1-MSNs, a low-frequency stimulation protocol depotentiated the synapse. The efficient protocol at the D2-MSN used an intermediate 10 Hz stimulation, presumably in the window of sufficient presynaptic calcium influx without evoking enkephalin release.
The selective normalization of transmission at D1-MSN-to-VP synapses abolished locomotor sensitization to cocaine. These results are consistent with literature suggesting that activation of D1-MSNs is reinforcing (Kravitz et al., 2012; Vicente et al., 2016) and mediates the association between cocaine and context (Calipari et al., 2016). Also, cocaine-evoked potentiation of excitatory inputs onto D1-MSNs drives locomotor sensitization to cocaine (Creed et al., 2015; Pascoli et al., 2011) and drug seeking (Pascoli et al., 2014). Here, we show that this signal in the accumbal D1-MSNs is transmitted downstream to the VP, and that interference at this node in the circuit is also sufficient to abolish sensitization to cocaine.
In contrast, normalizing transmission at D2-MSN-to-VP synapses had no effect on sensitization, in line with studies that fail to find a role for D2R contribution to locomotor sensitization with non-contingent cocaine (Terrier et al., 2016). Still, plasticity in D2-MSNs may be relevant to cocaine-seeking behavior. In cases where high doses of cocaine are self-administered, excitatory input from the basolateral amygdala to D2-MSNs is potentiated, which may contribute to reduced responding during initial drug seeking (Terrier et al., 2016). Consistent with this interpretation, chemogenetic activation of D2-MSNs promotes resistance to cocaine seeking (Bock et al., 2013). Future studies will be needed to parse the interplay of D1- and D2-MSNs in drug seeking, in which aversion and reward interact to drive behavior.
D2-MSN-to-VP Transmission Is Implicated in Decreased Motivation and Hedonic Impact of Sucrose
Our slice physiology experiments revealed a dysregulation of GABAergic and opioid transmission between D2-MSNs of the NAc and the VP after cocaine treatment. Manipulation of opioid and GABA transmission in the NAc and VP profoundly changes motivation “wanting” and hedonic impact “liking” of reward (Root et al., 2015). We identified synaptic depression at D2-MSN-to-VP synapses as a substrate of impaired motivation and hedonic impact of sucrose after cocaine exposure. The negative affective state associated with cocaine withdrawal also includes anxiety- and stress-related behavior. It is likely that altered activity in brain areas such as the lateral habenula (LHb), amygdala, and hippocampus contributes to these components of drug-induced negative affect (Le Moal and Koob, 2007).
Our focus on VTA-projecting VP neurons may reveal the circuit that underlies the negative affective state emerging after cocaine withdrawal. Plasticity in D2-MSNs may represent opponent processes that emerge to counter the activation caused by the rewarding properties of drugs of abuse (Le Moal and Koob, 2007; Solomon and Corbit, 1974). Since the VP has reciprocal connections with brain areas such as the LHb, amygdala, and hippocampus (Haber et al., 1985; Root et al., 2015), future work will be needed to determine how addictive drugs modify plasticity at other nodes between the VP and the larger limbic system.
Future studies may address how two inhibitory inputs to a largely overlapping population of VP neurons mediate distinct components of drug-adaptive behavior. It is possible that VP neurons innervated selectively by D1-MSNs may be particularly relevant for driving sensitization behavior, whereas VP neurons innervated exclusively by D2-MSNs modulate affective state. Alternatively, through impinging onto different compartments of the VP neuron, or being active at different times during behavior, D1- and D2-MSNs could differentially modulate VP neuron activity. There is precedent for different inputs to single neurons mediating distinct components of behavior. We previously reported that excitatory inputs from the medial prefrontal cortex (mPFC) and ventral hippocampus onto overlapping populations of D1-MSNs in the NAc mediate action-outcome associations and contextual information driving drug seeking, respectively (Pascoli et al., 2014). Here, we extend this circuit analysis by examining outputs of MSNs in the NAc shell and also find evidence for overlapping circuits mediating distinct components of drug-adaptive behavior. An additional question is whether the changes in D2-MSNs that mediate anhedonia contribute to cocaine seeking. This seems plausible as the opponent process theory proposes that the emergence of a negative affective state contributes to the maintenance of addiction (Le Moal and Koob, 2007).
Conclusion
Our results extend a circuit model of addiction (Lüscher, 2016) whereby cocaine induces divergent forms of plasticity at synapses between D1- and D2-MSNs of the NAc and the VP, which in turn mediate sensitization and impaired processing of natural reward, respectively. The VP thus acts as a hub, integrating information regarding the incentive salience of drug-associated cues as well as the induction of anhedonia. The adaptations that mediate different symptoms emerging after cocaine withdrawal may contribute to individual variability in susceptibility to addiction. Alternatively, the imbalance between direct and indirect circuits before the convergence in the VP may reflect specific experience and may predispose individuals to different symptoms of addiction. Targeting these pathways or their integration downstream in the VP may provide novel therapeutic strategies for addictive disorders.
EXPERIMENTAL PROCEDURES
Animals
All experiments were carried out in accordance with the Institutional Animal Care and Use Committee of the University of Geneva with permission of the cantonal authorities. Tracing experiments were done in heterozygous BAC transgenic mice (Drd1a-tdTomato from Jackson Laboratories, Drd2-eGFP from GENSAT; Mutant Mouse Regional Resource Center; Shuen et al., 2008). Behavior and electrophysiology experiments were performed on C57BL/6 mice or heterozygous Drd1-Cre and Drd2-Cre mice (Tg(Drd1a-cre) EY262Gsat/Mmucd, Tg(Drd2-cre)ER44Gsat/Mmucd), which were obtained from the Mutant Mouse Regional Resource Center, an NCRR-NIH-funded strain repository, and were originally donated to the MMRRC by the NINDS-funded GENSAT BAC transgenic project (Gerfen et al., 2013). Transgenic mice had been backcrossed in the C57BL/6 line for a minimum of four generations. For all experiments, males and females were used (Table S1). Mice were injected with cocaine (20 mg/kg, i.p.) or 0.9% saline, given daily for 5 days in a novel environment.
Stereotaxic Surgery
Anesthesia was induced and maintained with isoflurane at 5% and 2%, respectively. For optogenetic experiments, AAV2-EF1α-ChETA-EYFP (University of North Carolina Vector Core) was bilaterally injected in the NAc shell (coordinates relative to Bregma, anterior-posterior [AP], +1.7 mm; medial lateral [ML], ± 0.6 mm; dorsal ventral [DV], 4.5 mm). Injections were carried out using graduated pipettes with tip diameter of 10–15 μm, at 0.05 μL/min for a total of 250 nL; pipettes were left in situ for 8 min to allow for the diffusion. The minimum viral expression time was 21 days. For behavior experiments, optic fibers (constructed in house as described previously; Sparta et al., 2011) were implanted bilaterally over the VP and secured with three skull screws and dental cement (coordinates relative to Bregma, AP, +0.26 mm; ML, ± 1.5 mm; DV, 4.55 mm). Mice were allowed to recover for at least 7 days before behavioral training; only mice with fiber placements in the VP were included in the analyses (Figure S6).
Anatomical Tracing
For retrograde tracing of VP-projecting MSNs, CTB conjugated to Alexafluor 488 or Alexafluor 555 (150 nL, 1 mg/mL) in sterile PBS was injected into the VP of 8-week-old mice (coordinates from Bregma, AP, 0.3 mm; ML, ± 1.5 mm; DV, −4.6 mm). After 10 days, mice were perfused transcardially with 4% paraformaldehyde (PFA) in phosphate buffer (PBS). Brains were removed and left in 4% PFA overnight before being transferred into PBS. Coronal sections of the NAc were sliced at 40 μM, mounted with fluorashield media containing DAPI and imaged using a laser-scanning confocal microscope (Zeiss 700) with a 40× oil objective. For labeling of VTA-projecting VP cells, injections were performed as above (200 nL Alexafluor 555, coordinates from Bregma, AP, 3.4 mm; ML, ± 0.8 mm; DV, 4.3 mm) and slice experiments were performed 7–11 days after injection.
Whole-Cell Electrophysiology
Coronal slices of the VP were prepared using a vibratome in ice cold artificial cerebrospinal fluid (aCSF), continuously bubbled with 95% O2 and 5% CO2. Slices were superfused with aCSF at 30°C −32°C and visualized with a 40× objective lens. Neurons were recorded with borosilicate glass pipettes (6–9 MΩ) by means of whole-cell voltage-clamp at 60 mV. The internal solution contained 100 mM KCl, 30 mM potassium gluconate, 4 mM MgCl2, 10 mM sodium creatine phosphate, 3.4 mM Na2ATP, 0.1 mM Na3GTP, 1.1 mM EGTA, and 5 mM HEPES. Currents were amplified, filtered at 2 kHz, and digitized at 10 kHz. Access resistances were monitored by a hyperpolarizing step of −4 mV every 10 s; the experiment was discarded if the access resistance changed by more than 20%. ChETA was stimulated by flashing 470 nm light (4 ms) through the light path of the microscope using an LED powered by an LED driver. For plasticity experiments, light-evoked IPSCs were recorded in the presence of kynurenic acid (2 mM). PPRs were evoked at an interval of 50 ms. Example traces are an average of 30 consecutive IPSCs, taken from the last 5 min of the baseline and the last 5 min of the plasticity experiment. For strontium experiments, Ca2+ was replaced with 2 mM Sr2+ in the aCSF, and unitary events were recorded between 10 and 60 ms after the light pulse. DPDE, FSK, NAL, SCH23390, picrotoxin, and kyurenic acid were used where indicated; all agents were purchased from Sigma Biosciences.
Behavior
Locomotor Sensitization
Locomotor activity was measured for 60 min immediately after drug injection and reported as the number of quarter turns in a circular corridor (outer/inner diameter, 30/10 cm; video tracking system, Anymaze, Stoeling). After 3 days of habituation, mice underwent five daily injections of 20 mg/kg cocaine i.p. After 9 days of withdrawal, in vivo HFS through a fiber optic and connected solid-state laser was applied, and 24 hr later a challenge dose of cocaine was given.
Operant Training
Training was conducted in operant chambers (ENV-307A-CT, Med Associates). Two levers were present on either side of a port for liquid reward delivery and cue light. Each session started with the presentation of the levers into the operant chamber and was terminated when 60 rewards had been earned or after a maximum of 60 min. During initial training, a single active lever press resulted in a 20 s presentation of the cue light and liquid reward dispenser in the reward port. Sucrose solution (10%) was available for the entire 20 s, to a maximum volume of 0.1 mL. After the mouse had reached 5 days of stable responding on the FR1 (fixed ratio) schedule (<20% variability over 5 days), mice began 5 days of FR2 responding. Mice that did not learn discrimination of the active lever (active lever presses at least three times higher than inactive lever presses) or did not reach stable FR1 responding after 12 days were excluded.
PR Task
Mice underwent one PR session at baseline and at re-test. During the session, the number of active lever presses required to earn a reward increased, according to the schedule in Figure S5B. Reward delivery and cue light presentation were identical to operant training conditions. Sessions lasted 4 hr or were terminated if no responses occurred for a 20 min period.
Free Access Task
Testing was conducted in a plexiglass arena (50 × 50 cm). Mice were given 2 days of 20 min habituation sessions to an empty arena. For the test, a glass well (7.5 cm in diameter) filled with sucrose solution (10%) was placed in the corner. The number of entries made into the reward zone and amount of time spent in the reward zone were quantified over three sessions (Anymaze, Steoling). Each session was 5 min, performed 60 min apart. No changes in locomotor activity were found (Figure S8).
Orofacial Taste Reactivity
During the first session of the free access task, mice were filmed at 240 frames/s in the reward zone. Hedonic tongue protrusions were scored offline and defined as any event where the tongue was extended out of the mouth but did not make contact with sucrose solution (Movie S1).
Statistics
Plasticity experiments were analyzed by comparing average current amplitudes at baseline (30 time points over 5 min) with the final 5 min of recording using paired Student’s t test. Behavioral experiments were analyzed with repeated-measures ANOVA, with genotype and treatment (i.e., cocaine or saline) as between-subject factors and test day as the within-subject factor. All analyses were performed in SPSS (version 17.0).
Supplementary Material
Highlights.
The ventral pallidum (VP) integrates inputs from D1- and D2-MSNs from the NAc
Cocaine exposure potentiates output of D1-MSNs and depresses output of D2-MSNs to the VP
Plasticity at D1-MSN-to-VP synapses is implicated in behavioral sensitization to cocaine
Plasticity at D2-MSN-to-VP synapses contributes to cocaine-induced negative affect
ACKNOWLEDGMENTS
We thank M. Mameli, A.V. Kravitz, and E.C. O’Connor for discussion and comments on the manuscript; C. Gerfen for providing Cre mouse lines through the MMRC repository; Iris Lindberg for radioimmunoassay reagents; and K. Deisseroth for optogenetic effectors. This work was financed by a grant from the Swiss National Science Foundation, the National Center of Competence in Research SYNAPSY-The Synaptic Bases of Mental Diseases, and an advanced grant from the European Research Council (MeSSI).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, eight figures, one table, and one movie and can be found with this article online at http://dx.doi.org/10.1016/j.neuron.2016.09.001.
REFERENCES
- Alexander GE, Crutcher MD, and DeLong MR (1990). Basal ganglia-thalamocortical circuits: parallel substrates for motor, oculomotor, “prefrontal” and “limbic” functions. Prog. Brain Res 85, 119–146. [PubMed] [Google Scholar]
- Barnea-Ygael N, Gal R, and Zangen A. (2016). Chronic cocaine administration induces long-term impairment in the drive to obtain natural reinforcers in high- but not low-demanding tasks. Addict. Biol 21, 294–303. [DOI] [PubMed] [Google Scholar]
- Barr AM, and Phillips AG (1999). Withdrawal following repeated exposure to d-amphetamine decreases responding for a sucrose solution as measured by a progressive ratio schedule of reinforcement. Psychopharmacology (Berl.) 141, 99–106. [DOI] [PubMed] [Google Scholar]
- Berridge KC (2000). Measuring hedonic impact in animals and infants: microstructure of affective taste reactivity patterns. Neurosci. Biobehav. Rev 24, 173–198. [DOI] [PubMed] [Google Scholar]
- Bertran-Gonzalez J, Bosch C, Maroteaux M, Matamales M, Hervé D, Valjent E, and Girault JA (2008). Opposing patterns of signaling activation in dopamine D1 and D2 receptor-expressing striatal neurons in response to cocaine and haloperidol. J. Neurosci 28, 5671–5685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock R, Shin JH, Kaplan AR, Dobi A, Markey E, Kramer PF, Gremel CM, Christensen CH, Adrover MF, and Alvarez VA (2013). Strengthening the accumbal indirect pathway promotes resilience to compulsive cocaine use. Nat. Neurosci 16, 632–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocklisch C, Pascoli V, Wong JC, House DR, Yvon C, de Roo M, Tan KR, and Lüscher C. (2013). Cocaine disinhibits dopamine neurons by potentiation of GABA transmission in the ventral tegmental area. Science 341, 1521–1525. [DOI] [PubMed] [Google Scholar]
- Calipari ES, Bagot RC, Purushothaman I, Davidson TJ, Yorgason JT, Peña CJ, Walker DM, Pirpinias ST, Guise KG, Ramakrishnan C, et al. (2016). In vivo imaging identifies temporal signature of D1 and D2 medium spiny neurons in cocaine reward. Proc. Natl. Acad. Sci. USA 113, 2726–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra R, Francis TC, Konkalmatt P, Amgalan A, Gancarz AM, Dietz DM, and Lobo MK (2015). Opposing role for Egr3 in nucleus accumbens cell subtypes in cocaine action. J. Neurosci 35, 7927–7937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creed M, Pascoli VJ, and Lüscher C. (2015). Addiction therapy. Refining deep brain stimulation to emulate optogenetic treatment of synaptic pathology. Science 347, 659–664. [DOI] [PubMed] [Google Scholar]
- Crespo JA, Manzanares J, Oliva JM, Corchero J, Palomo T, and Ambrosio E. (2001). Extinction of cocaine self-administration produces a differential time-related regulation of proenkephalin gene expression in rat brain. Neuropsychopharmacology 25, 185–194. [DOI] [PubMed] [Google Scholar]
- Cromwell HC, and Berridge KC (1993). Where does damage lead to enhanced food aversion: the ventral pallidum/substantia innominata or lateral hypothalamus? Brain Res. 624, 1–10. [DOI] [PubMed] [Google Scholar]
- Danjo T, Yoshimi K, Funabiki K, Yawata S, and Nakanishi S. (2014). Aversive behavior induced by optogenetic inactivation of ventral tegmental area dopamine neurons is mediated by dopamine D2 receptors in the nucleus accumbens. Proc. Natl. Acad. Sci. USA 111, 6455–6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Chiara G, and Imperato A. (1988). Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc. Natl. Acad. Sci. USA 85, 5274–5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ericsson J, Stephenson-Jones M, Pérez-Fernández J, Robertson B, Silberberg G, and Grillner S. (2013). Dopamine differentially modulates the excitability of striatal neurons of the direct and indirect pathways in lamprey. J. Neurosci 33, 8045–8054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis TC, Chandra R, Friend DM, Finkel E, Dayrit G, Miranda J, Brooks JM, Iñiguez SD, O’Donnell P, Kravitz A, and Lobo MK (2015). Nucleus accumbens medium spiny neuron subtypes mediate depression-related outcomes to social defeat stress. Biol. Psychiatry 77, 212–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, and Young WS 3rd (1988). Distribution of striatonigral and striatopallidal peptidergic neurons in both patch and matrix compartments: an in situ hybridization histochemistry and fluorescent retrograde tracing study. Brain Res. 460, 161–167. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Engber TM, Mahan LC, Susel Z, Chase TN, Monsma FJ Jr., and Sibley DR (1990). D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science 250, 1429–1432. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Paletzki R, and Heintz N. (2013). GENSAT BAC cre-recombinase driver lines to study the functional organization of cerebral cortical and basal ganglia circuits. Neuron 80, 1368–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber SN, Groenewegen HJ, Grove EA, and Nauta WJ (1985). Efferent connections of the ventral pallidum: evidence of a dual striato pallidofugal pathway. J. Comp. Neurol 235, 322–335. [DOI] [PubMed] [Google Scholar]
- Heimer L, and Wilson RD (1975). The subcortical projections of allocortex: similarities in the neural associations of the hippocampus, the piriform cortex and the neocortex. In Golgi Centennial Symposium Proceedings, Santini M, ed. (Raven Press; ), pp. 173–193. [Google Scholar]
- Hikida T, Kimura K, Wada N, Funabiki K, and Nakanishi S. (2010). Distinct roles of synaptic transmission in direct and indirect striatal pathways to reward and aversive behavior. Neuron 66, 896–907. [DOI] [PubMed] [Google Scholar]
- Inui T, and Shimura T. (2014). Delta-opioid receptor blockade in the ventral pallidum increases perceived palatability and consumption of saccharin solution in rats. Behav. Brain Res 269, 20–27. [DOI] [PubMed] [Google Scholar]
- Johnson PI, and Stellar JR (1994). Comparison of delta opiate receptor agonist induced reward and motor effects between the ventral pallidum and dorsal striatum. Neuropharmacology 33, 1171–1182. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Pierce RC, Cornish J, and Sorg BA (1998). A role for sensitization in craving and relapse in cocaine addiction. J. Psychopharmacol. (Oxford) 12, 49–53. [DOI] [PubMed] [Google Scholar]
- Khibnik LA, Beaumont M, Doyle M, Heshmati M, Slesinger PA, Nestler EJ, and Russo SJ (2016). Stress and cocaine trigger divergent and cell type-specific regulation of synaptic transmission at single spines in nucleus accumbens. Biol. Psychiatry 79, 898–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, and Bloom FE (1988). Cellular and molecular mechanisms of drug dependence. Science 242, 715–723. [DOI] [PubMed] [Google Scholar]
- Kravitz AV, Tye LD, and Kreitzer AC (2012). Distinct roles for direct and indirect pathway striatal neurons in reinforcement. Nat. Neurosci 15, 816–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupchik YM, Scofield MD, Rice KC, Cheng K, Roques BP, and Kalivas PW (2014). Cocaine dysregulates opioid gating of GABA neurotransmission in the ventral pallidum. J. Neurosci 34, 1057–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupchik YM, Brown RM, Heinsbroek JA, Lobo MK, Schwartz DJ, and Kalivas PW (2015). Coding the direct/indirect pathways by D1 and D2 receptors is not valid for accumbens projections. Nat. Neurosci 18, 1230–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Moal M, and Koob GF (2007). Drug addiction: pathways to the disease and pathophysiological perspectives. Eur. Neuropsychopharmacol 17, 377–393. [DOI] [PubMed] [Google Scholar]
- Lobo MK, and Nestler EJ (2011). The striatal balancing act in drug addiction: distinct roles of direct and indirect pathway medium spiny neurons. Front. Neuroanat 5, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo MK, Covington HE 3rd, Chaudhury D, Friedman AK, Sun H, Damez-Werno D, Dietz DM, Zaman S, Koo JW, Kennedy PJ, et al. (2010). Cell type-specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science 330, 385–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo MK, Zaman S, Damez-Werno DM, Koo JW, Bagot RC, DiNieri JA, Nugent A, Finkel E, Chaudhury D, Chandra R, et al. (2013). DFosB induction in striatal medium spiny neuron subtypes in response to chronic pharmacological, emotional, and optogenetic stimuli. J. Neurosci 33, 18381–18395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C. (2016). The emergence of a circuit model for addiction. Annu. Rev. Neurosci 39, 257–276. [DOI] [PubMed] [Google Scholar]
- Mahler SV, Vazey EM, Beckley JT, Keistler CR, McGlinchey EM, Kaufling J, Wilson SP, Deisseroth K, Woodward JJ, and Aston-Jones G. (2014). Designer receptors show role for ventral pallidum input to ventral tegmental area in cocaine seeking. Nat. Neurosci 17, 577–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengual E, and Pickel VM (2004). Regional and subcellular compartmentation of the dopamine transporter and tyrosine hydroxylase in the rat ventral pallidum. J. Comp. Neurol 468, 395–409. [DOI] [PubMed] [Google Scholar]
- Meye FJ, Valentinova K, Lecca S, Marion-Poll L, Maroteaux MJ, Musardo S, Moutkine I, Gardoni F, Huganir RL, Georges F, and Mameli M. (2015). Cocaine-evoked negative symptoms require AMPA receptor trafficking in the lateral habenula. Nat. Neurosci 18, 376–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napier TC, and Maslowski-Cobuzzi RJ (1994). Electrophysiological verification of the presence of D1 and D2 dopamine receptors within the ventral pallidum. Synapse 17, 160–166. [DOI] [PubMed] [Google Scholar]
- Pascoli V, Turiault M, and Lüscher C. (2011). Reversal of cocaine-evoked synaptic potentiation resets drug-induced adaptive behaviour. Nature 481, 71–75. [DOI] [PubMed] [Google Scholar]
- Pascoli V, Terrier J, Espallergues J, Valjent E, O’Connor EC, and Lüscher C. (2014). Contrasting forms of cocaine-evoked plasticity control components of relapse. Nature 509, 459–464. [DOI] [PubMed] [Google Scholar]
- Pascoli V, Terrier J, Hiver A, and Lüscher C. (2015). Sufficiency of mesolimbic dopamine neuron stimulation for the progression to addiction. Neuron 88, 1054–1066. [DOI] [PubMed] [Google Scholar]
- Patton MH, Roberts BM, Lovinger DM, and Mathur BN (2016). Ethanol disinhibits dorsolateral striatal medium spiny neurons through activation of a presynaptic delta opioid receptor. Neuropsychopharm. 41, 1831–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez MF, White FJ, and Hu XT (2006). Dopamine D(2) receptor modulation of K(+) channel activity regulates excitability of nucleus accumbens neurons at different membrane potentials. J. Neurophysiol 96, 2217–2228. [DOI] [PubMed] [Google Scholar]
- Ren W, Centeno MV, Berger S, Wu Y, Na X, Liu X, Kondapalli J, Apkarian AV, Martina M, and Surmeier DJ (2016). The indirect pathway of the nucleus accumbens shell amplifies neuropathic pain. Nat. Neurosci 19, 220–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins SJ, Ehrman RN, Childress AR, and O’Brien CP (1997). Relationships among physiological and self-report responses produced by cocaine-related cues. Addict. Behav 22, 157–167. [DOI] [PubMed] [Google Scholar]
- Robinson TE, and Berridge KC (1993). The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res. Brain Res. Rev 18, 247–291. [DOI] [PubMed] [Google Scholar]
- Robinson TE, and Berridge KC (2003). Addiction. Annu. Rev. Psychol 54, 25–53. [DOI] [PubMed] [Google Scholar]
- Robledo P, and Koob GF (1993). Two discrete nucleus accumbens projection areas differentially mediate cocaine self-administration in the rat. Behav. Brain Res 55, 159–166. [DOI] [PubMed] [Google Scholar]
- Root DH, Ma S, Barker DJ, Megehee L, Striano BM, Ralston CM, Fabbricatore AT, and West MO (2013). Differential roles of ventral pallidum subregions during cocaine self-administration behaviors. J. Comp. Neurol 521, 558–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Root DH, Melendez RI, Zaborszky L, and Napier TC (2015). The ventral pallidum: Subregion-specific functional anatomy and roles in motivated behaviors. Prog. Neurobiol 130, 29–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz N, Temkin P, Jurado S, Lim BK, Heifets BD, Polepalli JS, and Malenka RC (2014). Chronic pain. Decreased motivation during chronic pain requires long-term depression in the nucleus accumbens. Science 345, 535–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Flajolet M, Greengard P, and Surmeier DJ (2008). Dichotomous dopaminergic control of striatal synaptic plasticity. Science 321, 848–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuen JA, Chen M, Gloss B, and Calakos N. (2008). Drd1a-tdTomato BAC transgenic mice for simultaneous visualization of medium spiny neurons in the direct and indirect pathways of the basal ganglia. J. Neurosci 28, 2681–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon RL, and Corbit JD (1974). An opponent-process theory of motivation. I. Temporal dynamics of affect. Psychol. Rev 81, 119–145. [DOI] [PubMed] [Google Scholar]
- Spanagel R, Herz A, and Shippenberg TS (1990). The effects of opioid peptides on dopamine release in the nucleus accumbens: an in vivo microdialysis study. J. Neurochem 55, 1734–1740. [DOI] [PubMed] [Google Scholar]
- Sparta DR, Stamatakis AM, Phillips JL, Hovelsø N, van Zessen R, and Stuber GD (2011). Construction of implantable optical fibers for long-term optogenetic manipulation of neural circuits. Nat. Protoc 7, 12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner JE, Glaser D, Hawilo ME, and Berridge KC (2001). Comparative expression of hedonic impact: affective reactions to taste by human infants and other primates. Neurosci. Biobehav. Rev 25, 53–74. [DOI] [PubMed] [Google Scholar]
- Steketee JD, and Kalivas PW (2011). Drug wanting: behavioral sensitization and relapse to drug-seeking behavior. Pharmacol. Rev 63, 348–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrier J, Lüscher C, and Pascoli V. (2016). Cell-type specific insertion of GluA2-lacking AMPARs with cocaine exposure leading to sensitization, cue-induced seeking, and incubation of craving. Neuropsychopharm. 41, 1779–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tindell AJ, Berridge KC, Zhang J, Peciña S, and Aldridge JW (2005). Ventral pallidal neurons code incentive motivation: amplification by mesolimbic sensitization and amphetamine. Eur. J. Neurosci 22, 2617–2634. [DOI] [PubMed] [Google Scholar]
- Vicente AM, Galvão-Ferreira P, Tecuapetla F, and Costa RM (2016). Direct and indirect dorsolateral striatum pathways reinforce different action strategies. Curr. Biol 26, R267–R269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Fowler JS, Wang GJ, Hitzemann R, Logan J, Schlyer DJ, Dewey SL, and Wolf AP (1993). Decreased dopamine D2 receptor availability is associated with reduced frontal metabolism in cocaine abusers. Synapse 14, 169–177. [DOI] [PubMed] [Google Scholar]
- Wolf ME (2016). Synaptic mechanisms underlying persistent cocaine craving. Nat. Rev. Neurosci 17, 351–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu-Friedman MA, and Regehr WG (1999). Presynaptic strontium dynamics and synaptic transmission. Biophys. J 76, 2029–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahm DS, and Heimer L. (1990). Two transpallidal pathways originating in the rat nucleus accumbens. J. Comp. Neurol 302, 437–446. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.