

Abstract
Alterations in α-synuclein dosage lead to familial Parkinson’s disease (PD), and its accumulation results in synucleinopathies that include PD, dementia with Lewy bodies (DLB) and multiple system atrophy (MSA). Furthermore, α-synuclein contributes to the fibrilization of amyloid-β and tau, two key proteins in Alzheimer’s disease, which suggests a central role for α-synuclein toxicity in neurodegeneration. Recent studies of factors contributing to α-synuclein toxicity and its disruption of downstream cellular pathways have expanded our understanding of disease pathogenesis in synucleinopathies. In this Review, we discuss these emerging themes, including the contributions of aging, selective vulnerability and non-cell-autonomous factors such as α-synuclein cell-to-cell propagation and neuroinflammation. Finally, we summarize recent efforts toward the development of targeted therapies for PD and related synucleinopathies.
α-synuclein (SNCA) is a 14-kDa protein that forms a major component of abnormal neuronal aggregates known as Lewy bodies (LBs)1. It is highly soluble and enriched at presynaptic terminals, where it binds lipids and regulates the release of synaptic vesicles2,3. N-terminal point mutations in α-synuclein (A30P, E46K, H50Q, G51D, A53E and A53T)4–10 and genomic duplications or triplications that contain the α-synuclein locus result in autosomal dominant forms of familial PD11,12. In addition, multiple genome-wide association studies (GWASs) have identified single-nucleotide polymorphisms (SNPs) in α-synuclein as risk factors that increase susceptibility to sporadic PD13,14. Among these SNPs is a risk variant in a noncoding distal enhancer element of SNCA that leads to increased α-synuclein expression15. This genetic evidence helps to further highlight a crucial link between increased α-synuclein levels and PD pathogenesis.
α-synuclein also accumulates in other synucleinopathies, including dementia with Lewy bodies (DLB), multiple system atrophy (MSA)16,17 and various lysosomal-storage disorders, such as Gaucher’s disease18 (Table 1). Importantly, α-synuclein regulates the fibrilization of both amyloid-β (aβ) and tau, two key proteins in Alzheimer’s disease (AD) pathophysiology19–23. Thus, delineation of the downstream effects and factors that contribute to α-synuclein toxicity will be crucial for assessing potential therapeutics for both synucleinopathies and AD.
Table 1.
Summary of diseases associated with α-synuclein toxicity
| Disease | Genes associated | Symptoms | Pathology |
|---|---|---|---|
| Parkinson’s disease (PD) |
|
||
| Parkinson’s disease with dementia (PDD) or dementia with Lewy bodies (DLB) | N/A |
|
|
| Multiple system atrophy (MSA) |
|
|
|
| Gaucher’s disease (GD) |
|
|
|
| Additional lysosomal storage disorders (LSDs) |
|
|
|
| Neurodegeneration with brain iron accumulation (NBIA) |
|
|
|
| Alzheimer’s disease (AD) |
|
|
|
In this Review, we present evidence for various routes to cellular dysfunction caused by α-synuclein toxicity, including synaptic dysfunction, mitochondrial impairment, defective endoplasmic reticulum (ER) function and autophagy–lysosomal pathway and nuclear dysfunction. We discuss additional factors that contribute to α-synuclein toxicity, including the role of aging, selective neuronal vulnerability, non-cell-autonomous factors such as α-synuclein propagation and the role of glia and neuroinflammation, and the interplay of α-synuclein with aβ and tau. Finally, we compare therapeutic strategies currently being developed to target α-synuclein and minimize its toxicity in disease.
α-synuclein in neurodegenerative diseases
α-synuclein has been implicated in several diseases, which we discuss here to give an impression of the broad reach of its pathogenesis.
In PD, dopaminergic neurons in the substantia nigra pars compacta (SNc) degenerate, which results in dopamine loss in the basal ganglia, an area of the brain responsible for coordinating fine motor control, which ultimately leads to the onset of clinical Parkinson’s symptoms such as bradykinesia, muscular rigidity, resting tremors and postural instability24. α-synuclein accumulates in sporadic PD in neuronal cell bodies and processes to form Lewy bodies and Lewy neurites in the brain, spinal cord and peripheral nervous system24. However, Lewy pathology is also observed in a subset of neurologically healthy patients (in a condition called incidental Lewy body disease), which indicates that additional factors other than Lewy pathology alone might be required for α-synuclein’s toxicity in patients25. α-synuclein locus duplication results in autosomal dominant forms of PD with late onset (~60 years), typical of patients with sporadic PD. By contrast, α-synuclein locus triplication leads to early PD onset (<40 years)11, demonstrating that increased α-synuclein levels accelerate PD pathogenesis. Of note, patients with familial PD who have mutations in other PD-associated genes, such as leucine-rich repeat kinase 2 (LRRK2), can also develop Lewy body pathology26–28. LRRK2 overexpression was initially found to accelerate neuropathology progression in α-synuclein A53T mice (tetO-A53T), whereas LRRK2 depletion delayed its progression29. Subsequent studies demonstrated that LRRK2 knockout or inhibition prevented α-synuclein-induced neurodegeneration in animal models30,31, and mutated LRRK2 increases α-synuclein levels and its recruitment into neuronal inclusions32, which further suggests potential functional interplays between α-synuclein toxicity and other PD-associated genes.
Patients with MSA develop autonomic failure, as well as parkinsonism and cerebellar ataxia, as a result of respective striatonigral and olivopontocerebellar degeneration33. In contrast to individuals with PD, patients with MSA develop cytoplasmic α-synuclein inclusions in oligodendrocytes. Notably, various α-synuclein mutations result in both Parkinson’s and MSA symptoms33.
Parkinson’s disease with dementia (PDD) and DLB occurs in patients who first present with parkinsonism or dementia, respectively, which is followed by onset of other symptoms34. A subset of patients with PDD or DLB develop α-synuclein Lewy pathology and amyloid plaques, although a greater plaque load has been reported in DLB than in PDD34, which suggests an intimate link between α-synuclein and aβ toxicity. This contributes to a disease spectrum that ranges from PD with α-synuclein pathology to PDD and DLB with mixed pathology and AD with aβ pathology.
AD is the leading cause of dementia and involves progressive memory loss and cognitive impairment coupled with neurodegeneration of various brain regions, including the hippocampus35. In patients with AD, the microtubule-binding protein tau is hyperphosphorylated and accumulates into intracellular tangles, whereas aβ, an extracellular fragment cleaved from amyloid precursor protein (APP), accumulates into extracellular plaques35. Notably, elevated soluble α-synuclein levels have been observed in the AD-afflicted brain and are correlated with cognitive decline36.
Lysosomal storage disorders (LSD) such as Gaucher’s disease have also been linked to α-synuclein toxicity. Gaucher’s disease is caused by mutations in GBA1, which encodes glucocerebrosidase (GCase), a lysosomal hydrolase that converts glucosylceramide into ceramide and glucose37. Patients with Gaucher’s disease develop thrombocytopenia, anemia, hepatosplenomegaly and bone pain, and seizures and cognitive impairment occur in more severe neuropathic forms. Importantly, some patients develop parkinsonism coincident with α-synuclein Lewy body pathology37, and mutations in GBA1 are a risk factor for the development of PD38. Recently, several other LSDs, including Sanfilippo syndrome, GM2 gangliosidosis and Niemann–Pick type C, have also been shown to demonstrate α-synuclein pathology18.
Finally, neurodegeneration with brain iron accumulation (NBIA) is a collection of genetic diseases involving iron accumulation in the globus pallidus and substantia nigra, resulting in parkinsonism, spasticity and dystonia. To date, patients with mutations in the mitochondrial membrane protein C19orf12 and the A2 phospholipase PLA2G6 have confirmed Lewy body and neurites. In addition, mutations in ATP13A2 (also known as PARK9), a lysosomal ATPase present in Lewy bodies, also contribute to NBIA, juvenile onset parkinsonism, dementia and neuronal ceroid lipofuscinosis (NCL)39.
Pathways implicated in α-synuclein toxicity
α-synuclein is intrinsically disordered40 and forms multiple conformations, including amyloidogenic oligomers41. α-synuclein contains three distinct regions3: an amino-terminal lipid-binding region, a recently crystalized central NAC (non-amyloid-β component) hydrophobic region42, which contributes to its oligomerization, and an intrinsically disordered carboxy-terminal (Box 1).
Box 1. Understanding the many conformations of α-synuclein.
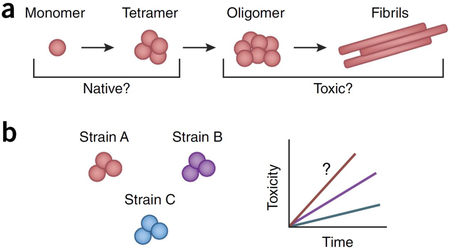
Native conformations: monomer and tetramer.
α-synuclein is able to transition between multiple different conformations, including monomers, tetramers, higher-level oligomers (soluble conformations), fibrils (highly ordered insoluble conformations characterized by β-sheet conformation) and aggregates shown in a. Early work demonstrated that α-synuclein natively exists as a monomer40, and recent studies have used α-synuclein purified from mouse brain198 and analysis of mammalian cells199 to demonstrate a compact monomeric state for native α-synuclein, which helps to shield its non-amyloid-β component (NAC) region from spontaneous aggregation. In addition, α-synuclein has also been observed to exist as both metastable conformers and stable monomers200, as well as to form tetramers201,202 mediated by its KTKEGV repeats203. PD-linked α-synuclein mutations, including A53T and E46K, decrease its tetrameric and increase its monomeric conformation, which suggests that the unfolded monomer might be a source of α-synuclein toxicity185. Thus, multiple forms of native α-synuclein might exist physiologically, depending on its cellular localization and membrane interactions.
Forming toxic conformations: oligomers and fibrils.
Multiple studies have examined the factors that promote α-synuclein’s initial oligomerization. Polyunsaturated fatty acids increase α-synuclein oligomer levels, whereas saturated fatty acids decrease them204. In addition, a low amount of negatively charged lipids205, mildly acidic environments such as endosomes and lysosomes206, and lipid vesicles all promote α-synuclein oligomerization207. Upon formation, α-synuclein oligomers subsequently undergo conformational changes to become more stable and compact proteinase-K-resistant oligomers, which induce higher oxidative stress, before becoming fibrils208.
Several studies have now begun to investigate whether oligomers or downstream fibrils are the more toxic conformation. Some reports have suggested that oligomers are more toxic, because α-synuclein transgenic mice and PD- and DLB-afflicted patient brains showed increased levels of soluble, lipid-dependent α-synuclein oligomers when compared to controls204. α-synuclein PD-linked A53T and A30P mutations also accelerate oligomerization but not fibrilization209, and injection of α-synuclein variants promoting oligomer rather than fibril formation into the rat brain causes more severe dopaminergic loss210.
By contrast, recent work has suggested that α-synuclein fibrils might be 1,000-fold more toxic than their precursors211, and injection of different human α-synuclein assemblies into rat SNc demonstrated that fibrils rather than ribbons or oligomers induced the greatest amount of motor impairment, dopaminergic cell loss and synaptic impairment140. Thus, further studying the role of different α-synuclein conformations, such as oligomers and fibrils, in disease will be helpful for understanding α-synuclein toxicity.
Differential strains.
Recent studies on α-synuclein have identified the existence of different strains, shown in b defined as conformational variants of α-synuclein—that exhibit distinct properties such as differences in structure and toxicity and ability to seed, propagate108 and cross-seed tau fibrilization20. These strain differences can occur both naturally or result from nuances in experimental preparation of higher-level α-synuclein conformations (i.e., oligomers and/or fibrils). Differences in α-synuclein strains have also been observed across different species (such as human and mouse); human α-synuclein but not mouse α-synuclein fibril inoculation into wild-type mice decreased Lewy body formation and was more effective in α-synuclein knockout mice as compared to wild-type mice, which suggests that efficient spreading might be attenuated when fibril and host monomers are from different species212,213.
In addition, differences in α-synuclein strains also exist between synucleinopathies, such as PD and MSA. Interestingly, brain extracts from patients with MSA but not those with PD accelerated neurodegeneration when injected into transgenic mice, suggesting that MSA-derived strains of α-synuclein might be more toxic214. Furthermore, brain extracts from different patients with MSA showed variable rates of α-synuclein transmission215, which suggests the presence of patient-to-patient strain variability even within MSA. Importantly, α-synuclein strain differences among patients may contribute to patient variability such as age of onset and rate of disease progression.
Although α-synuclein normally localizes to the presynaptic terminal, its oligomers and aggregates localize throughout the cell body and neurites, which suggests that α-synuclein might disrupt cellular function beyond the presynaptic terminal. Indeed, multiple organelles are implicated in α-synuclein toxicity, including synaptic vesicles, mitochondria, ER and Golgi, lysosomes and autophagosomes and the nucleus (Fig. 1). Moreover, interorganelle contacts and organelle axonal transport are also disrupted by α-synuclein toxicity.
Figure 1.
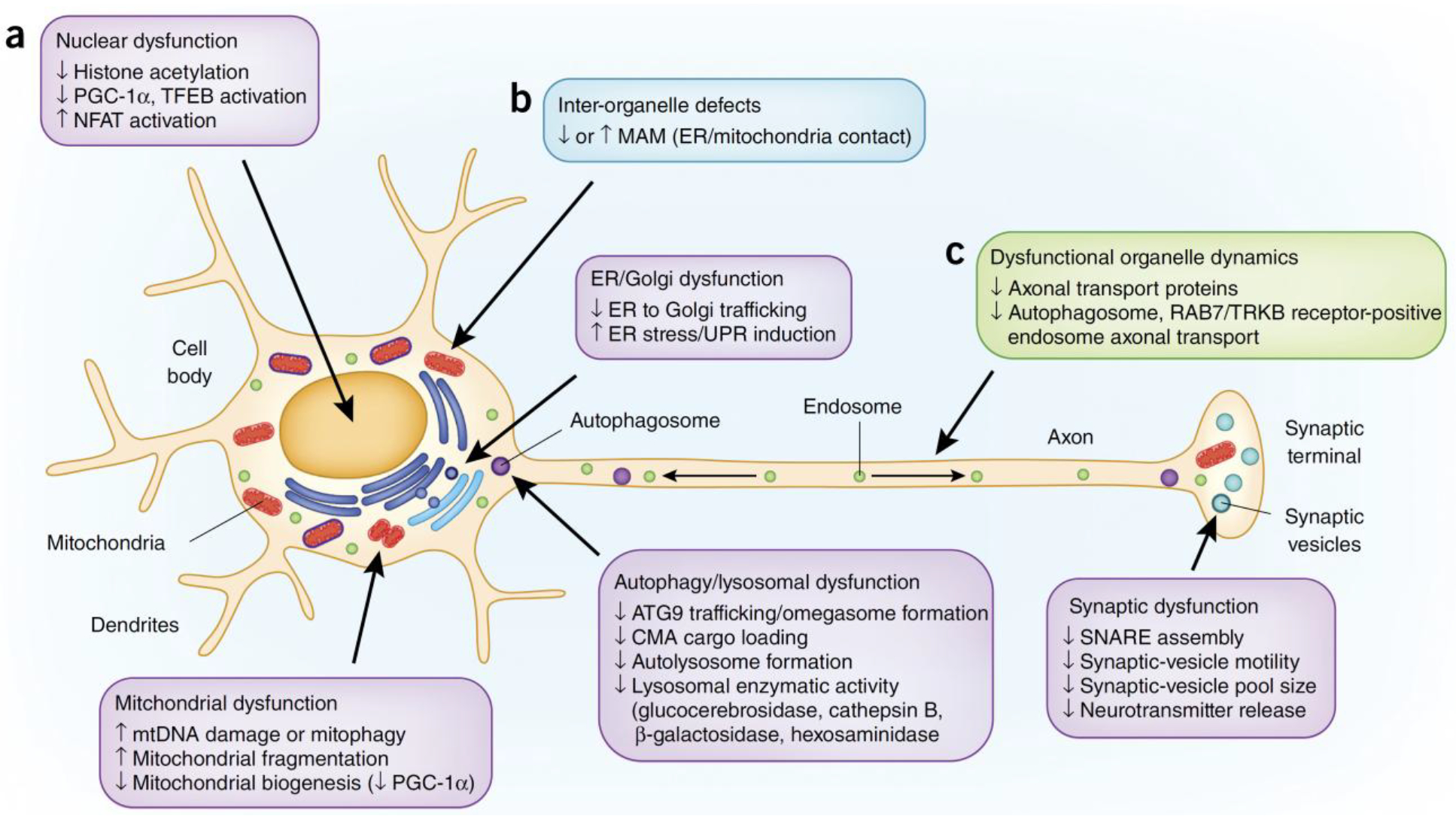
Pathways implicated in α-synuclein toxicity. (a–c) Organelle dysfunction (a, purple boxes), defects in inter-organelle contacts (b, blue box) and dysfunctional organelle dynamics (c, green box) have all been implicated in α-synuclein toxicity.
Synaptic-vesicle trafficking.
α-synuclein normally localizes to the presynaptic terminal, where it associates with synaptic vesicles43,44, binds membranes45 and induces membrane curvature46. α-synuclein regulates soluble NSF attachment protein receptor (SNARE) complex assembly47 by binding the SNARE protein synaptobrevin-2/vesicle-associated membrane protein 2 (VAMP2) to promote synaptic-vesicle fusion at the presynaptic terminal2. It also potentially regulates additional steps during synaptic-vesicle trafficking48–52.
Large α-synuclein oligomers preferentially bind VAMP2 and disrupt SNARE complex formation, dopamine release53 and synaptic-vesicle motility54, and increased α-synuclein levels disrupt neurotransmitter release via decreased synaptic-vesicle recycling-pool size55 and mobility50. Thus, it has been proposed that α-synuclein’s normal function might be disrupted in synucleinopathies. By contrast, other studies have suggested that α-synuclein’s physiological role at the synapse is not altered in disease, and have found that α-synuclein oligomers actually promoted SNARE assembly56, and that PD-linked mutations (except the lipid-binding-deficient A30P mutation) does not disrupt SNARE assembly57 or synaptic-vesicle clustering51.
Elevated levels of α-synuclein might also disrupt dopamine neurotransmission. Mice lacking α-synuclein show increased dopamine release from nigrostriatal terminals58, although α-synuclein deletion does not affect cytosolic dopamine levels59. Conversely, transgenic mice overexpressing human α-synuclein show dopaminergic terminal loss60, deficient dopamine release and altered synaptic-vesicle distribution61. Increased α-synuclein expression has also been linked to a reduction in dopamine reuptake and defective dopamine transporter (DAT) function62, which suggests several potential mechanisms through which α-synuclein might disrupt dopamine turnover.
Mitochondrial function.
Mitochondria are crucial for ATP synthesis, calcium storage, lipid metabolism and neuronal survival63. α-synuclein toxicity might directly disrupt mitochondrial homeostasis, given that mice with A53T α-synuclein mutations have increased mitochondrial DNA damage64 and mitophagy65,66, and increased α-synuclein levels promote dynamin related protein 1 (DRP1)-independent mitochondrial fission in cell lines and mouse models overexpressing α-synuclein67,68. Interestingly, in mice lacking α-synuclein, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced degeneration of dopaminergic neurons is prevented69, potentially because α-synuclein oligomers promote mitochondrial dysfunction via increased calcium uptake70. Post-translationally modified species of α-synuclein have also recently been suggested to disrupt mitochondrial function by impairing mitochondrial protein import71. By contrast, mitochondrial dysfunction might also be induced indirectly by α-synuclein toxicity via decreased levels of the mitochondrial biogenesis factor PGC-1α. This has been observed in cell models that express oligomeric α-synuclein, mice that express A30P α-synuclein72 and the brains of patients with PD, whereas PGC-1α activation rescued neuronal loss induced by mutant α-synuclein73. Dopaminergic α-synuclein induced pluripotent stem cell (iPSC) models of PD also display inhibition of the MEF2C–PGC-1α mitochondrial transcription network via increased S-nitrosylation of transcription factor MEF2C (ref. 74).
Endoplasmic reticulum and Golgi function, and the endocytic pathway.
The endoplasmic reticulum (ER) is essential for protein folding, trafficking to the Golgi, calcium buffering and the unfolded protein response (UPR). Both wild-type and mutant α-synuclein disrupt ER to Golgi trafficking in yeast75 and induce ER stress and early secretory-pathway dysfunction, which is rescued by certain Rab GTPases such as RAB1, RAB3A or RAB8A (refs. 76,77). Increased α-synuclein expression also disrupts endosomal transport events via the E3 ubiquitin ligase yeast RSP5 and its mammalian homologue NEDD4 and endosomal transport can be rescued by the drug N-aryl benzimidazole78,79. Moreover, α-synuclein accumulation disrupts GCase trafficking in PD iPSCs and patient brains; this leads to reduced GCase lysosomal activity80, which in turn might be mediated by the aberrant association of α-synuclein with cis-Golgi–tethering factor GM130 and RAB1A mislocalization81 or by inhibiting ER to Golgi formation via the R-SNARE ykt6 (ref. 82). Notably, DAT trafficking is also disrupted upon α-synuclein overexpression83. Increased α-synuclein levels also raise cytoplasmic calcium levels, which leads to the activation of a toxic calmodulin–calcineurin cascade84,85 and suggests that calcium buffering in the ER might be disrupted by α-synuclein. Finally, α-synuclein has been proposed to bind the ER chaperone GRP78 (also known as BIP)86 and interfere with ER folding machinery, which is ameliorated in α-synuclein transgenic mice by decreasing ER stress87,88.
Autophagy or lysosomal pathway.
Autophagy is a dynamic pathway involved in the degradation of damaged organelles and protein aggregates89. α-synuclein overexpression disrupts ER to Golgi trafficking of the autophagic transmembrane protein ATG9 and decreases the formation of omegasomes, a precursor for autophagosome biogenesis90. In chaperone-mediated autophagy (CMA), A53T and A30P α-synuclein bind the lysosomal receptor LAMP2A more tightly than wild-type α-synuclein, which prevents their own degradation and cargo loading of other CMA substrates into lysosomes91. In addition, dopamine-modified α-synuclein also blocks CMA, which might contribute to selective dopaminergic vulnerability in PD92. In neuronal models incubated with α-synuclein preformed fibrils (pffs), autophagosomes formed normally but had defective lysosomal fusion and autophagic cargo accumulation93, potentially owing to defective autophagosome axonal transport94. Finally, because efficient autophagic degradation relies on lysosomal enzymatic activity, the lysosomal activity of multiple enzymes, including GCase, cathepsin B, β-galactosidase and hexosaminidase, is reduced in α-synuclein triplication PD iPSCs as compared to control iPSCs as a result of defective ER-to-Golgi trafficking80,81,95.
Nuclear function.
Although α-synuclein was also originally localized to the nucleus96, this finding has since been up for debate, potentially because of the use of different antibodies against cleaved forms of α-synuclein (Box 1). The targeting of α-synuclein to the nucleus has been proposed to be regulated by the nuclear protein TRIM28 (ref. 97) and to inhibit histone acetylation98. Moreover, PD-associated mutations (A30P, A53T and G51D) in α-synuclein demonstrate increased nuclear localization as compared to wild-type α-synuclein98,99. Altered activation of various transcription factors has also been observed, including decreased activation of the mitochondrial biogenesis factor PGC-1α in α-synuclein A53T PD-patient-derived iPSC neurons and PD-afflicted substantia nigra72–74; decreased activation of the autophagy–lysosomal pathway transcription factor TFEB in rats overexpressing α-synuclein via adeno-associated virus (AAV)100; and increased activation of nuclear factor of activated T cells (NFAT) via calcineurin activation in cell lines that overexpress wild-type or A53T α-synuclein, in dopaminergic neurons from α-synuclein transgenic mice and in brains of patients with PD or DLB84,85.
Disruption of inter-organelle contacts.
In the past few years, multiple inter-organelle contacts have emerged as sites of cellular homeostatic regulation. One such site is the mitochondria-associated ER membrane (MAM), a subdomain of the ER tethered to mitochondria via a group of adaptor proteins, which serves as a critical site for autophagosome biogenesis, mitochondrial fission, calcium homeostasis, phospholipid transport and fatty acid and cholesterol transfer101. Two recent studies have implicated the MAM in α-synuclein toxicity with opposing conclusions; one of these studies found increased numbers of MAM contact sites, resulting in increased mitochondrial calcium uptake from the ER upon α-synuclein overexpression102, whereas the other study identified α-synuclein in MAM fractions and saw decreased numbers of MAM contact sites upon wild-type, A53T or A30P α-synuclein overexpression103. However, in both reports, mitochondrial fragmentation was observed. Thus, further studies to reconcile these differences, as well as experiments in neurons, will be crucial for understanding the interaction of α-synuclein with MAMs, as well as the role of other inter-organelle contacts in the context of α-synuclein toxicity.
Misregulation of organelle dynamics.
The majority of studies on α-synuclein cellular toxicity have been performed at steady state. However, organelles are highly dynamic and undergo axonal transport, fission and fusion events and maturation, and are intricately regulated by numerous signaling pathways mediated by phosphorylation, Rab GTPase activity, calcium signaling and electrical activity. Recently, α-synuclein fibrils were found to impair the axonal transport of autophagosomes and RAB7- and TrkB-receptor-positive endosomes, but not the transport of synaptophysin or mitochondria, which suggests that α-synuclein does not cause a generalized defect in axonal transport94. This may be partially due to decreased levels of axonal transport proteins in patients with sporadic PD when compared to age-matched controls104 or decreased microtubule stability and kinesin-dependent cargo mobility, as observed in cellular models expressing α-synuclein oligomers105. Transport defects might also be mediated by interactions of α-synuclein with tau, a microtubule-binding protein that stabilizes and promotes microtubule assembly20,23. Neuronal exposure to extracellular α-synuclein also disrupts actin turnover and actin waves along axons, owing to cofilin inactivation106. By contrast, very little work has examined α-synuclein’s regulation of vesicle fission and fusion and maturation dynamics in neurons, which suggests that further investigation of these processes might advance our understanding of α-synuclein’s role in neurodegeneration.
Understanding cellular dysfunction in α-synuclein toxicity
Numerous reports have described multiple pathways of cellular dysfunction in models of α-synuclein toxicity. Why have these different pathways been implicated thus far in α-synuclein toxicity?
One possibility is that distinct pathways might be affected in different synucleinopathies. The pathways affected in familial PD caused by α-synuclein mutations might not be identical to those disrupted by other PD-associated genes or in sporadic PD, and may be different from those involved in MSA or LBD. These differences might be exacerbated by differences in α-synuclein strain characteristics, specific cell types affected and diverse protein interactions.
Moreover, multiple pathways could be affected in synucleinopathies at different time points of disease progression. Some pathways may be disrupted early in disease (presymptomatic) versus late stage (post-symptomatic), whereas others might be compensating for dysfunction in other pathways. In addition, different pathways might have different rates of dysfunction, which may initially be below detection until the cell has already degenerated. Furthermore, additional factors, such as aging and genetic variability, might also affect when and which pathways become dysfunctional.
Finally, the differences in cellular-pathway defects observed across studies might also be in part due to differences in the experimental model of α-synuclein toxicity. α-synuclein toxicity is commonly modeled by the overexpression of wild-type α-synuclein, expression of PD-linked α-synuclein mutations, injection or incubation with α-synuclein preformed oligomers or fibrils or targeted expression of α-synuclein using AAV. These studies are further complicated by differences in the choice of cell type (non-neuronal versus neuronal versus glial), animal model (human versus mouse versus rat) and time point of analysis (2 d versus 200 d versus 2 years). Indeed, using different models of α-synuclein toxicity, different steps in the autophagic pathway have been found to be disrupted, including autophagosome formation90, cargo loading in CMA91, autophagosome fusion with lysosomes and axonal transport93,94, and lysosomal degradative function78,80,107. Moreover, small changes in experimental preparation of α-synuclein oligomers or fibrils can lead to strain differences (Box 1) with distinct seeding, propagation and toxicity-inducing capabilities20,108, which further contributes to potential differences in observed cellular dysfunction. Mouse models of α-synuclein toxicity enable analysis of functional neuronal circuitry, but they do not exhibit accumulation of neuromelanin, a dark pigment that consists of oxidized catechols such as dopamine, which is a key feature of human SNc dopaminergic neurons. By contrast, dopaminergic neurons differentiated from human iPSCs allow for the study, over hundreds of days, of patient-derived cells with endogenous levels of α-synuclein, but lack the complex connections of an intact basal ganglia circuitry. Thus, understanding the capabilities of each model and characteristics such as which synucleinopathy and which stage of the disease it most accurately reflects will be crucial for designing and interpreting future studies on α-synuclein toxicity.
Steps to α-synuclein toxicity
Although multiple cellular pathways have been implicated downstream of α-synuclein toxicity, several contributing factors might be crucial for the spread, extent and onset of α-synuclein toxicity. Indeed, human postmortem studies have shown that a subset of the population (10–20%) exhibits incidental Lewy body disease but presents as neurologically normal25, so the contributing factors that we discuss below might be crucial for determining which patients will become symptomatic, as well as influence their age of disease onset and rate of disease progression. Of note, different conformations of α-synuclein, as well as differences between α-synuclein strains, might further influence the toxicity of α-synuclein (Box 1). Finally, additional pathways have also been implicated in the augmentation of α-synuclein toxicity in Drosophila and mouse models, including loss of the molecular chaperone HSP70 (ref. 109) and presynaptic scaffold protein septin 4 (ref. 110), histone deacetylase sirtuin 2 activation111 and α-synuclein S129 phosphorylation112.
Aging.
Aging is the greatest risk factor for multiple neurodegenerative diseases, partially owing to decreased organelle function. Substantia nigra neurons demonstrate higher levels of mitochondrial DNA (mtDNA) deletion with age, which causes mitochondrial dysfunction via respiratory-chain deficiency113. Proteasome dysfunction in multiple cell types, including neurons, also increases with age owing to disassembly of proteasomes and decreased proteasome subunit expression114, and autophagy is less efficient with age because of decreased levels of autophagy proteins such as beclin 1, ATG5 and ATG7 in human brains89. These defects might further contribute to α-synuclein toxicity because defective protein degradation accelerates α-synuclein accumulation. Indeed, α-synuclein levels increase with age in the human substantia nigra115,116.
Oxidative stress also increases with age117, which contributes to pathogenic modifications of α-synuclein, such as nitration of tyrosine residues, which have been observed in the brains of patients afflicted with PD, DLB and MSA118. Importantly, α-synuclein nitration promotes its aggregation119 and decreases its lipid-binding ability120. In dopaminergic neurons, autophagic vesicles containing lipofuscin and neuromelanin, generated by iron-catalyzed oxidation and oxidized catecholamines, respectively, accumulate with age121. α-synuclein has been proposed to associate with the lipid component of neuromelanin in SNc dopaminergic neurons to promote its own aggregation122. However, given that neuromelanin may be neuroprotective by sequestering oxidized catecholamines, it remains unclear whether α-synuclein’s interaction with neuromelanin is beneficial.
Selective vulnerability.
Synucleinopathies are a diverse group of diseases with disparate affected cell types, including SNc dopaminergic neurons in PD, oligodendrocytes in MSA and cortical neurons in LBD. This suggests that cell-autonomous factors might further augment α-synuclein toxicity in different populations. Identifying these factors for different cellular populations could thus facilitate targeted treatment development for specific synucleinopathies.
In PD, multiple cellular populations are lost, including neuromelanin-positive catecholamine neurons in the SNc and locus coeruleus, dorsal motor nucleus of the vagus neurons in the majority of patients and norepinephrine and dopaminergic neurons in the enteric nervous system123. Lewy body pathology is present but not limited to these regions, and has been proposed to progress through the brain in a caudo-rostral fashion124. Factors that might contribute to the susceptibility of these populations include the use of a monomamine neurotransmitter, highly branched axons with multiple release sites and increased excitability owing to autonomous pacemaking with low intrinsic calcium buffering abilities123.
Specifically, dopamine might also enhance α-synuclein toxicity, because dopamine adducts stabilize α-synuclein protofibrils125 by inhibiting α-synuclein aggregation and promoting oligomer formation126,127. Moreover, lowering cytosolic dopamine in cultured midbrain neurons is neuroprotective, and dopaminergic neurons lacking α-synuclein are resistant to L-DOPA-induced cell death59. Additionally, neuromelanin, a downstream product of oxidized dopamine, might be toxic, given that neuromelanin-containing neurons are lost in PD128. Neuromelanin is a homeostatic byproduct of increased oxidation and catecholamine adducts. However, in cases of excessive oxidation, elevated levels of neuromelanin accumulation in neurons might also contribute to neurodegeneration.
In addition, given that L-DOPA increases dopamine levels and is used to treat PD, and that A10 dopaminergic neurons from the ventral tegmental are spared in PD, dopamine alone might not be sufficient to cause degeneration in the context of α-synuclein toxicity during PD. SNc A9 dopaminergic neurons lost in PD might be more susceptible than A10 neurons owing to their increased cytosolic calcium levels59, which drive dopamine metabolism and increase cellular metabolism. Of note, SNc degeneration in PD might also be mediated by non-cell-autonomous mechanisms, such as the expression of cytokines (e.g., interferon (IFN)-γ)129.
In MSA, α-synuclein preferentially accumulates in oligodendrocytes, but whether this occurs in a cell-autonomous manner or results from neuronal release and subsequent oligodendrocyte uptake remains unclear. Transgenic mice overexpressing α-synuclein in oligodendrocytes undergo degeneration and myelin autophagocytosis130. Selective vulnerability of oligodendrocytes in MSA might be mediated by the accumulation and binding of p25α, an oligodendrocyte-specific protein, to aggregated α-synuclein131, or by decreased glial-derived neurotrophic factor (GDNF) levels132.
Cell-to-cell spreading of α-synuclein.
α-synuclein spreading was observed initially from host human PD-afflicted brains into grafted nigral neurons over a decade after transplantation133,134. Subsequently, α-synuclein transmission via endocytosis to neighboring neurons was observed in cell culture and mouse models135. Exogenous introduction of human α-synuclein fibrils recruited endogenous α-synuclein to form Lewy body–like pathology in human cell lines136 and mouse neurons137. Studies in vivo subsequently found that mouse α-synuclein fibril inoculation into wild-type mouse brains caused Lewy pathology propagation and progressive dopaminergic SNc loss138. α-synuclein intramuscular injection also induced Lewy pathology139 and demonstrated efficient crossing of the blood–brain barrier140, and injection into the olfactory bulb also mimicked the sequential progression of Lewy-body-like pathology141.
One potential mechanism for α-synuclein secretion is exosome release142, which is increased upon the expression of ATP13A2 via increased exosome biogenesis143,144; RAB11 expression145; and in a calcium-dependent manner upon thapsigargin treatment146. α-synuclein spreading might also involve clathrin-mediated endocytosis147, lymphocyte-activation gene 3 (LAG3)-mediated endocytosis148 or lysosomal vesicles traveling through tunneling nanotubes149. Importantly, protein spreading might occur in multiple neurodegenerative diseases because neurons are particularly vulnerable to protein propagation owing to their extensive anatomical connections150. However, various issues remain unaddressed, including whether the spreading of Lewy body pathology is required for clinical symptoms, the relevance of using elevated fibril concentrations to induce spreading in various models and the heterogeneity of Lewy body pathology across PD cases that do not always follow the stereotypical caudo-rostral progression151. Thus, the role, timing and mechanisms of α-synuclein propagation remain to be further investigated in the context of cellular pathogenesis.
Glia and neuroinflammation.
The role of glia (oligodendrocytes, astrocytes and microglia) has been increasingly studied in the context of neurodegeneration in recent years. However, although α-synuclein accumulates in oligodendrocytes in MSA, whether oligodendrocyte dysfunction occurs in other synucleinopathies has not been well characterized. Similarly, astrocyte involvement in α-synuclein toxicity has not been clearly elucidated, although astrocytes can take up neuronally released α-synuclein via endocytosis, which leads to gene expression changes that are indicative of an inflammatory response152.
Microglial activation has been observed both in patients with PD153 and those with MSA154, which suggests that neuroinflammation might contribute to the pathogenesis of α-synuclein-mediated toxicity. Indeed, neuroinflammation has been observed in multiple animal models of α-synuclein toxicity155–158 and might be mediated by microglial expression of the major histocompatibility complex (MHC) II, a key regulator of the immune response, given that MHCII depletion reduces microglial activation and dopaminergic neurodegeneration in mouse models of α-synuclein toxicity159. Increased α-synuclein expression also increases toll-like receptor (TLR)-4 immunoreactivity in mouse models of MSA154.
Various mechanisms have been proposed through which α-synuclein might induce a neuroinflammatory response. Extracellular α-synuclein released from neurons has been found to be an endogenous agonist for TLR2, leading to microglial activation160, whereas oligomeric α-synuclein has been proposed to directly bind the heterodimer TLR1/2 on the cell membrane to induce a proinflammatory response dependent on myeloid differentiation primary response gene 88 (MyD88) (ref. 161). α-synuclein has also been suggested to be a chemoattractant that promotes microglial migration162, and proinflammatory responses to α-synuclein toxicity might be additionally mediated by the microRNA-155 (miR-155)163. Finally, dopaminergic neurons in PD might be particularly susceptible to immunomodulation, because mice lacking cytokine IFN-β develop spontaneous neurodegeneration of dopaminergic neurons, motor and cognitive impairments and Lewy body pathology164, and genetic polymorphisms in the locus of HLA-DR—a component of MHCII—have been linked to late-onset sporadic PD165. Further studies on the role of neuroinflammation and microglial activation in α-synuclein toxicity will thus be important for understanding α-synuclein pathophysiology and might additionally contribute to the development of future therapeutics for synucleinopathies.
Interplay of α-synuclein with aβ and tau.
Lewy body pathology is observed in about half of patients with AD, and soluble α-synuclein levels are increased in AD-afflicted brains and correlated with cognitive decline36, which suggests that α-synuclein might contribute to hippocampal and cholinergic neurodegeneration in AD. Early studies using doubly transgenic mouse models found that aβ enhanced α-synuclein fibrilization both in vitro and in vivo22, and that α-synuclein promoted aβ 1–38 aggregation upon coincubation in vitro21. By contrast, injection of α-synuclein fibrils into transgenic mouse models of AD failed to cross-seed aβ in vivo, and mice co-expressing α-synuclein A30P actually inhibited plaque formation in transgenic mutant amyloid precursor protein (APP) and presenilin 1 (PS1) mice19. These intriguing findings suggest that in contrast to α-synuclein’s ability to cross-seed both itself and tau, α-synuclein suppresses aβ deposition and reduces plaque formation in vivo. Further studies will thus be crucial for examining whether α-synuclein directly interacts with aβ and whether preventing aβ aggregation increases the levels of toxic aβ oligomers and contributes to neuronal dysfunction, despite a reduction in plaque formation.
Interestingly, tau has previously been identified as a PD risk factor13. α-synuclein and tau cross-seed one another, and their coincubation leads to the fibrilization of both proteins20,23. Misexpression of α-synuclein and tau in Drosophila leads to neuronal dysfunction and axonal transport defects166, and α-synuclein oligomers potentially impair microtubule assembly105. However, the precise role of the interplay between tau and α-synuclein in disease and the downstream cellular dysfunction that may occur remain to be further investigated.
Future questions in α-synuclein research
Multiple pathways and contributing factors have been investigated in the context of α-synuclein toxicity. However, several crucial questions remain unanswered in this field:
What are the precise mechanisms through which α-synuclein disrupts cellular pathways?
At the synapse, the pathological role of α-synuclein still remains controversial as to whether α-synuclein toxicity involves a loss of its physiological function at the synapse. Furthermore, the exact steps of synaptic-vesicle trafficking that are disrupted in disease, whether synaptic defects preferentially affect specific neuronal subpopulations and how pathogenic forms of α-synuclein interact with synaptic-vesicle machinery still remain unclear. In addition, whether α-synuclein affects mitochondrial function directly by interacting with mitochondrial lipids and proteins or indirectly through other organelles is also unknown. Similarly, α-synuclein has been shown to disrupt multiple ER functions, including Golgi trafficking and calcium buffering, but the precise mechanisms through which α-synuclein interacts with ER proteins to induce these defects also remain unclear. Finally, as previously mentioned, few studies have examined the effect of α-synuclein toxicity on inter-organelle contacts or the live-cell dynamics of organelles. Thus, studies investigating α-synuclein’s cellular localization, dynamics and interaction partners will be important for advancing our understanding of the basic cellular mechanisms through which α-synuclein disrupts organelle function.
Which cellular pathways are disrupted earliest in synucleinopathies?
This might depend on both the type of synucleinopathy and on genetic heterogeneity between patients, which could leave different pathways more susceptible to dysfunction. PD-linked mutations and risk factors cluster predominantly around endolysosomal proteins (LRRK2, ATP13A2 and GCase) and mitochondrial proteins (parkin, PINK1 and DJ-1), which suggests that α-synuclein might mediate the convergence between endolysosomal and mitochondrial pathway dysfunction in PD, and that these pathways might be disrupted early on in disease progression. Importantly, identifying which pathways are affected earliest in disease will be highly valuable for therapeutic development. For example, if decreased mitochondrial biogenesis due to decreased PGC-1α activation is an early step in α-synuclein-induced toxicity, activators of PGC-1α might be important for reducing cellular dysfunction. By contrast, if decreased mitochondrial biogenesis occurs as a compensatory response to inefficient lysosomal degradation, the activation of PGC-1α could potentially exacerbate α-synuclein toxicity at later stages. Thus, future studies examining organelle dynamics and function in living cells over extended periods of time will be crucial to confirm the order of cellular pathway dysfunction in PD and other synucleinopathies.
Which factors contribute to α-synuclein accumulation?
Genetic forms of PD exemplify the crucial role of α-synuclein dosage on its toxicity, because both α-synuclein locus duplication and triplication lead to familial PD11,12. In these cases, α-synuclein accumulation is a known starting cause of pathophysiology, and targeting α-synuclein directly or potential downstream pathways might be beneficial. By contrast, in sporadic forms of PD and other synucleinopathies, α-synuclein ultimately accumulates into Lewy bodies even without mutations or increased dosage levels of α-synuclein, which suggests that upstream factors, such as oxidation, nitration, decreased proteasomal and/or lysosomal function, increased dopamine adducts or interplay with aβ and tau might contribute to α-synuclein accumulation. Furthermore cell-to-cell spreading involving neurons and/or glia might further contribute to the accumulation of α-synuclein in specific cellular subtypes, such as SNc dopaminergic neurons in PD or oligodendrocytes in MSA. Importantly, different factors might be more important in different synucleinopathies, or a combination of factors might be necessary to trigger initial α-synuclein accumulation. In these cases, targeting upstream factors that lead to α-synuclein accumulation would be most effective, and could even be preventive if targeted during prodromal stages before clinical onset. Finally, a subset of familial forms of PD linked to genes other than that encoding α-synuclein also results in α-synuclein accumulation, which suggests that mutations in these genes might exacerbate the role of the upstream factors previously mentioned, or alternatively, might lead to α-synuclein accumulation through an independent pathway specific to that gene. Thus, understanding the nuances and the role that each factor has in different synucleinopathies and across patient populations will be crucial for informing our knowledge of α-synuclein toxicity in disease.
Therapeutically targeting α-synuclein in neurodegeneration
Various studies have targeted α-synuclein directly at various stages of its synthesis and action as a potential therapeutic intervention (Fig. 2). In addition, there are other therapeutic approaches currently in clinical trials that target contributing factors to α-synuclein toxicity, such as the selective vulnerability of A9 SNc dopaminergic neurons in PD using the calcium-channel blocker isradipine, which may also prove to be successful for treating synucleinopathies.
Figure 2.
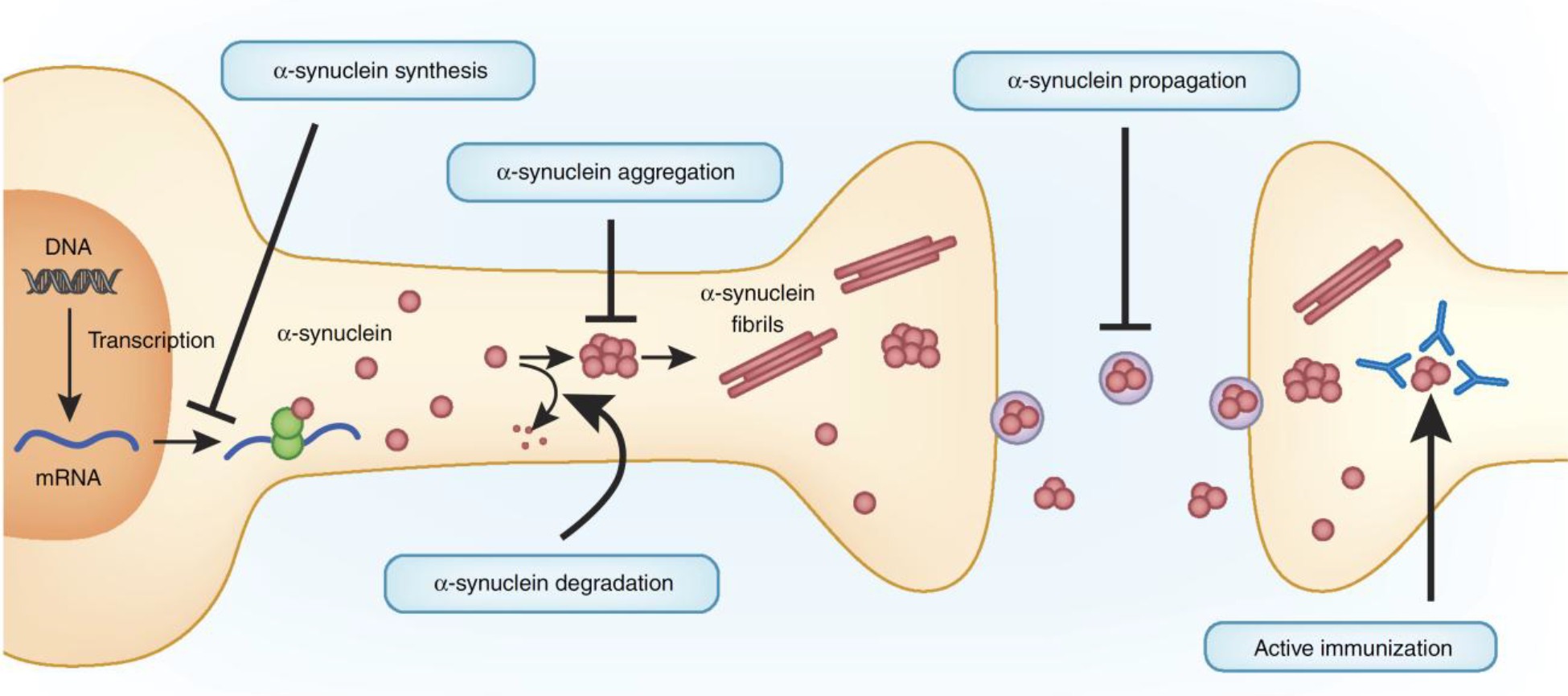
Therapeutically targeting α-synuclein toxicity. Various pathways have been manipulated to decrease α-synuclein toxicity, largely in mouse models of α-synuclein toxicity. These include (i) reducing α-synuclein synthesis with siRNAs, (ii) increasing α-synuclein degradation, (iii) reducing α-synuclein aggregation, (iv) blocking α-synuclein propagation and (v) active immunization of α-synuclein. Drug development to increase lysosomal activity via glucocerebrosidase (GCase) to accelerate α-synuclein degradation, as well as clinical trials using both passive and active immunization against α-synuclein, are currently under way.
Reducing α-synuclein synthesis.
Given that increased α-synuclein expression causes familial PD, several studies have aimed to decrease its synthesis by using siRNA that targets α-synuclein mRNA. In mice, direct siRNA infusion decreases hippocampal and cortical α-synuclein levels for a week after infusion167, whereas in mice expressing a human form of α-synuclein, injecting siRNA-containing exosomes decreases protein aggregation in the SNc168. Given that antisense oligonucleotide (ASO)-mediated therapies are being tested in clinical trials currently as potential therapeutics for other neurodegenerative diseases, ASO targeting of α-synuclein may also be effective.
Increasing α-synuclein protein degradation.
Another mechanism for targeting α-synuclein involves increasing its lysosomal and/or autophagic degradation. Indeed, α-synuclein degradation is regulated by SIAH169 and NEDD4 (ref. 170) ubiquitin ligases, S129 phosphorylation by polo-like kinase 2 (PLK2)171 and activation of lysosomal cysteine cathepsins172. Overexpression of the lysosomal transcription factor TFEB in rats expressing α-synuclein decreases α-synuclein oligomer levels and prevents lysosomal dysfunction decline and neurodegeneration100. Passive immunization of mice overexpressing α-synuclein by using antibodies against α-synuclein also promote its lysosomal clearance173 or target it into microglia, which rescues α-synuclein-induced neurodegeneration and behavioral deficits174.
Another important therapeutic target that has emerged for α-synuclein degradation is GCase, because increased GCase levels enhance α-synuclein degradation in human neurons80. Importantly expression of GCase in the central nervous system reduces α-synuclein aggregation in a presymptomatic mouse model of Gaucher’s-related synucleinopathy mice; decreases soluble α-synuclein in mice expressing mutant human A53T α-synuclein175; and reduces α-synuclein accumulation and dopaminergic neurodegeneration in several rodent models of α-synuclein toxicity176,177. Recently, two noninhibitory GCase modulators (NCGC00188758 and NCGC607) were also found to increase GCase activity and decrease α-synuclein accumulation and toxicity in human neurons derived from iPSCs107,178. Furthermore, patients with Gaucher’s disease who receive GCase enzyme replacement therapy (ERT) for more than 5 years have reduced oligomeric α-synuclein levels when compared with those who did not receive therapy179, which further confirms the validity of increasing GCase activity as a therapeutic approach. Finally, increased expression of other lysosomal proteins, including LIMP2 (ref. 180) and ATP13A2 (refs. 181,182), also promote α-synuclein degradation in various models, presenting additional targets for the acceleration of α-synuclein degradation.
Reducing α-synuclein aggregation.
Given that α-synuclein oligomers and fibrils are implicated in α-synuclein toxicity, several strategies have been attempted to reduce their formation. The porphyrin phtalocyanine tetrasulfonate binds and stabilizes vesicle-associated α-synuclein, which thus delays its misfolding and aggregation183. Passive immunization using a protofibril-selective antibody decreases soluble and membrane-bound α-synuclein protofibrils in the spinal cord and decreases motor dysfunction in mice expressing PD-associated mutant A30P α-synuclein184. In addition, if native α-synuclein exists predominantly as a tetramer, compounds that stabilize α-synuclein in this conformation—as has been applied for transthyretin, which misfolds in the multisystem disorder transthyretin-related amyloidosis—might be effective at combating α-synuclein toxicity185. Several clinical trials are currently using small molecules186 to inhibit either α-synuclein aggregation (glycerol phenylbutyrate (University of Colorado, Denver); nilotinib (Georgetown University)) or α-synuclein oligomer formation (EGCG (University of Munich)).
Blocking α-synuclein propagation.
Because α-synuclein propagation may contribute to the spreading of α-synuclein toxicity, passive immunization studies to block spreading have also been performed. Antibodies against C-terminal truncated α-synuclein decrease its propagation in vitro and rescue motor and memory impairments in an α-synuclein mouse model187, and monoclonal α-synuclein antibodies prevent propagation and uptake of α-synuclein and rescue dopaminergic neuron loss and motor deficits in mice injected with α-synuclein pffs188. Importantly, understanding the role of propagation in disease will help to clarify whether these therapies will be successful for patients.
Active immunization.
Multiple studies have used active immunization to target α-synuclein. The first of these studies vaccinated human α-synuclein transgenic mice using human α-synuclein, which cleared α-synuclein aggregates, potentially via lysosomal pathways189. Subsequent studies have treated different α-synuclein transgenic mice with various forms of human α-synuclein antigen, with similar reductions in aggregates, improved motor and memory functions and reduced dopaminergic degeneration and dopamine loss186. Current PD and MSA clinical trials directly targeting α-synuclein involve active immunization with the vaccines PD01A and PD03A (AFFiRiS) for α-synuclein186 or passive immunization with antibodies against α-synuclein190, including PRX002 (Prothena) and BIIB054 (Biogen).
Targeted drug development
Previous clinical trials for PD have focused predominantly on improving general cellular pathways to slow disease progression, such as correcting mitochondrial dysfunction and oxidative damage with coenzyme Q10 (ref. 191). However, these studies have been unsuccessful at slowing disease progression, potentially because of the absence of defined therapeutic molecular pathways and targets, heterogeneity within patient populations and a lack of relevant biomarkers as clear readouts for drug efficacy. Consequently, future clinical studies with well-defined therapeutic targets backed by an improved understanding of the basic cellular mechanisms behind disease pathophysiology and the pathways affected early in disease will be crucial for improving clinical trial outcomes, both in PD and other synucleinopathies.
Therapeutically targeting α-synuclein via GCase is one example of a well-defined molecular target; multiple studies have now shown that GCase activation promotes α-synuclein degradation both in vitro and in vivo192. Moreover, because GCase is an enzyme with well-characterized substrates, drugs that target GCase can be examined early on during the clinical trial for their efficacy by measuring GCase enzyme activity levels in plasma and cerebrospinal fluid (CSF) to show effective target engagement, as well as α-synuclein plasma and CSF levels as clear readouts of α-synuclein degradation rates and potential biomarkers for disease risk and progression193. Finally, further stratification of patient populations by well-characterized biomarkers, genetic screening and consistent clinical diagnostics before trial enrollment will also be important for producing a more homogenous patient population. Importantly, future work advancing our knowledge of α-synuclein toxicity as it relates to factors such as strain differences, propagation and interaction with other proteins such as aβ and tau may also influence future drug development and clinical trial design.
Targeting the immune system.
Finally, the role of neuroinflammation in α-synuclein toxicity has also received much attention recently, and multiple studies are now examining the effect of manipulating the immune system to reduce α-synuclein toxicity. Treatment with the hypertension drug Candesartan cilexetil inhibited TLR2 expression and reduced the proinflammatory response in vitro in microglia treated with oligomeric α-synuclein161. In rat models of α-synuclein toxicity, treatment with either the immunosuppressant FK506 or AZD1480, a JAK1/2 inhibitor of the JAK–STAT pathway important for microglial activation and cytokine expression, reduced dopaminergic neurodegeneration194,195. In addition, treatment with potentially anti-inflammatory drugs, including hypoestoxide (a NF-κB modulator) or lenalidomide (an NF-κB signaling inhibitor) reduced motor-behavior defects and microgliosis in α-synuclein transgenic mice196,197. Thus, immunomodulation might be an important regulator of α-synuclein toxicity and a potential therapeutic target in combination with direct targeting of α-synuclein.
Conclusions and future directions
Two decades ago, α-synuclein was identified as the main component of Lewy bodies1 and genetically linked to familial PD4. Since then, numerous studies have implicated multiple dysfunctional pathways in its toxicity, as well as contributing factors, including its propagation, strain differences and glial activation. Importantly, these findings have identified various routes by which to therapeutically target α-synuclein, ranging from active and passive immunization to the upregulation of lysosomal machinery.
Several key goals still remain, including the establishment of a timeline of cellular pathway dysfunction during the progression of different synucleinopathies, further characterization of toxic and native α-synuclein species in control and disease-afflicted human brains, the identification of the role of α-synuclein propagation versus cell-autonomous factors and investigation of the role of glia and neuroinflammation. Finally, because α-synuclein is intimately connected with the fibrilization of both aβ and tau—key proteins in AD pathogenesis—understanding the interplay between these three proteins in the context of propagation and cellular toxicity will be crucial for advancing our understanding of AD and synucleinopathies, and potentially, for identifying novel therapeutics that are relevant to both fields.
ACKNOWLEDGMENTS
This work was supported by NIH/NINDS R01 NS076054 (D.K.) and NIH/NINDS 5T32NS041234 (Y.C.W.).
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Spillantini MG et al. α-synuclein in Lewy bodies. Nature 388, 839–840 (1997). [DOI] [PubMed] [Google Scholar]
- 2.Burré J et al. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329, 1663–1667 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bendor JT, Logan TP & Edwards RH The function of α-synuclein. Neuron 79, 1044–1066 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Polymeropoulos MH et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047 (1997). [DOI] [PubMed] [Google Scholar]
- 5.Krüger R et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet 18, 106–108 (1998). [DOI] [PubMed] [Google Scholar]
- 6.Zarranz JJ et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol 55, 164–173 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Appel-Cresswell S et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov. Disord 28, 811–813 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Proukakis C et al. A novel α-synuclein missense mutation in Parkinson disease. Neurology 80, 1062–1064 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lesage S et al. G51D α-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol 73, 459–471 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Pasanen P et al. Novel alpha-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol. Aging 35, 2180e.1–2180.e5 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Singleton AB et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302, 841 (2003). [DOI] [PubMed] [Google Scholar]
- 12.Chartier-Harlin MC et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 364, 1167–1169 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Simón-Sánchez J et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet 41, 1308–1312 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nalls MA et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet 46, 989–993 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Soldner F et al. Parkinson-associated risk variant in distal enhancer of α-synuclein modulates target gene expression. Nature 533, 95–99 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spillantini MG et al. Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci. Lett 251, 205–208 (1998). [DOI] [PubMed] [Google Scholar]
- 17.Spillantini MG, Crowther RA, Jakes R, Hasegawa M & Goedert M alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. USA 95, 6469–6473 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shachar T et al. Lysosomal storage disorders and Parkinson’s disease: Gaucher disease and beyond. Mov. Disord 26, 1593–1604 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Bachhuber T et al. Inhibition of amyloid-β plaque formation by α-synuclein. Nat. Med 21, 802–807 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Guo JL et al. Distinct α-synuclein strains differentially promote tau inclusions in neurons. Cell 154, 103–117 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yoshimoto M et al. NACP, the precursor protein of the non-amyloid beta/A4 protein (A beta) component of Alzheimer disease amyloid, binds A beta and stimulates A beta aggregation. Proc. Natl. Acad. Sci. USA 92, 9141–9145 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Masliah E et al. beta-amyloid peptides enhance alpha-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer’s disease and Parkinson’s disease. Proc. Natl. Acad. Sci. USA 98, 12245–12250 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giasson BI et al. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science 300, 636–640 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Kalia LV & Lang AE Parkinson’s disease. Lancet 386, 896–912 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Frigerio R et al. Incidental Lewy body disease: do some cases represent a preclinical stage of dementia with Lewy bodies? Neurobiol. Aging 32, 857–863 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paisán-Ruíz C et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 44, 595–600 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Zimprich A et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44, 601–607 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Trinh J, Guella I & Farrer MJ Disease penetrance of late-onset parkinsonism: a meta-analysis. JAMA Neurol. 71, 1535–1539 (2014). [DOI] [PubMed] [Google Scholar]
- 29.Lin X et al. Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson’s-disease-related mutant alpha-synuclein. Neuron 64, 807–827 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Daher JP et al. Leucine-rich repeat kinase 2 (LRRK2) pharmacological inhibition abates α-synuclein gene-induced neurodegeneration. J. Biol. Chem 290, 19433–19444 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Daher JP, Volpicelli-Daley LA, Blackburn JP, Moehle MS & West AB Abrogation of α-synuclein-mediated dopaminergic neurodegeneration in LRRK2-deficient rats. Proc. Natl. Acad. Sci. USA 111, 9289–9294 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Volpicelli-Daley LA et al. G2019S-LRRK2 expression augments α-synuclein sequestration into inclusions in neurons. J. Neurosci 36, 7415–7427 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fanciulli A & Wenning GK Multiple-system atrophy. N. Engl. J. Med 372, 249–263 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Goldman JG, Williams-Gray C, Barker RA, Duda JE & Galvin JE The spectrum of cognitive impairment in Lewy body diseases. Mov. Disord 29, 608–621 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang Y & Mucke L Alzheimer mechanisms and therapeutic strategies. Cell 148, 1204–1222 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Larson ME et al. Soluble α-synuclein is a novel modulator of Alzheimer’s disease pathophysiology. J. Neurosci 32, 10253–10266 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37.Blanz J & Saftig P Parkinson’s disease: acid-glucocerebrosidase activity and alpha-synuclein clearance. J. Neurochem 139 (Suppl. 1), 198–215 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Sidransky E et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med 361, 1651–1661 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arber CE, Li A, Houlden H & Wray S Review: Insights into molecular mechanisms of disease in neurodegeneration with brain iron accumulation: unifying theories. Neuropathol. Appl. Neurobiol 42, 220–241 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weinreb PH, Zhen W, Poon AW, Conway KA & Lansbury PT Jr. NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry 35, 13709–13715 (1996). [DOI] [PubMed] [Google Scholar]
- 41.Iwai A, Yoshimoto M, Masliah E & Saitoh T Non-A beta component of Alzheimer’s disease amyloid (NAC) is amyloidogenic. Biochemistry 34, 10139–10145 (1995). [DOI] [PubMed] [Google Scholar]
- 42.Rodriguez JA et al. Structure of the toxic core of α-synuclein from invisible crystals. Nature 525, 486–490 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iwai A et al. The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 14, 467–475 (1995). [DOI] [PubMed] [Google Scholar]
- 44.Kahle PJ et al. Subcellular localization of wild-type and Parkinson’s disease-associated mutant alpha-synuclein in human and transgenic mouse brain. J. Neurosci 20, 6365–6373 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Davidson WS, Jonas A, Clayton DF & George JM Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem 273, 9443–9449 (1998). [DOI] [PubMed] [Google Scholar]
- 46.Varkey J et al. Membrane curvature induction and tubulation are common features of synucleins and apolipoproteins. J. Biol. Chem 285, 32486–32493 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chandra S, Gallardo G, Fernández-Chacón R, Schlüter OM & Südhof TC Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 123, 383–396 (2005). [DOI] [PubMed] [Google Scholar]
- 48.Cabin DE et al. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. J. Neurosci 22, 8797–8807 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murphy DD, Rueter SM, Trojanowski JQ & Lee VM Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J. Neurosci 20, 3214–3220 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Scott D & Roy S α-Synuclein inhibits intersynaptic vesicle mobility and maintains recycling-pool homeostasis. J. Neurosci 32, 10129–10135 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Diao J et al. Native α-synuclein induces clustering of synaptic-vesicle mimics via binding to phospholipids and synaptobrevin-2/VAMP2. eLife 2, e00592 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vargas KJ et al. Synucleins regulate the kinetics of synaptic vesicle endocytosis. J. Neurosci 34, 9364–9376 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Choi BK et al. Large α-synuclein oligomers inhibit neuronal SNARE-mediated vesicle docking. Proc. Natl. Acad. Sci. USA 110, 4087–4092 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang L et al. α-synuclein multimers cluster synaptic vesicles and attenuate recycling. Curr. Biol 24, 2319–2326 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nemani VM et al. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 65, 66–79 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Burré J, Sharma M & Südhof TC α-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc. Natl. Acad. Sci. USA 111, E4274–E4283 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Burré J, Sharma M & Südhof TC Systematic mutagenesis of α-synuclein reveals distinct sequence requirements for physiological and pathological activities. J. Neurosci 32, 15227–15242 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abeliovich A et al. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25, 239–252 (2000). [DOI] [PubMed] [Google Scholar]
- 59.Mosharov EV et al. Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron 62, 218–229 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Masliah E et al. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science 287, 1265–1269 (2000). [DOI] [PubMed] [Google Scholar]
- 61.Janezic S et al. Deficits in dopaminergic transmission precede neuron loss and dysfunction in a new Parkinson model. Proc. Natl. Acad. Sci. USA 110, E4016–E4025 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lundblad M, Decressac M, Mattsson B & Björklund A Impaired neurotransmission caused by overexpression of α-synuclein in nigral dopamine neurons. Proc. Natl. Acad. Sci. USA 109, 3213–3219 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nunnari J & Suomalainen A Mitochondria: in sickness and in health. Cell 148, 1145–1159 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Martin LJ et al. Parkinson’s disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J. Neurosci 26, 41–50 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Choubey V et al. Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy. J. Biol. Chem 286, 10814–10824 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen L, Xie Z, Turkson S & Zhuang X A53T human α-synuclein overexpression in transgenic mice induces pervasive mitochondria macroautophagy defects preceding dopamine neuron degeneration. J. Neurosci 35, 890–905 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kamp F et al. Inhibition of mitochondrial fusion by α-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J. 29, 3571–3589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nakamura K et al. Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J. Biol. Chem 286, 20710–20726 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dauer W et al. Resistance of alpha-synuclein null mice to the parkinsonian neurotoxin MPTP. Proc. Natl. Acad. Sci. USA 99, 14524–14529 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Luth ES, Stavrovskaya IG, Bartels T, Kristal BS & Selkoe DJ Soluble, prefibrillar α-synuclein oligomers promote complex I-dependent, Ca2+-induced mitochondrial dysfunction. J. Biol. Chem 289, 21490–21507 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Di Maio R et al. α-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med 8, 342ra78 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Eschbach J et al. Mutual exacerbation of peroxisome proliferator-activated receptor γ coactivator 1α deregulation and α-synuclein oligomerization. Ann. Neurol 77, 15–32 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zheng B et al. PGC-1α, a potential therapeutic target for early intervention in Parkinson’s disease. Sci. Transl. Med 2, 52ra73 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ryan SD et al. Isogenic human iPSC Parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1α transcription. Cell 155, 1351–1364 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Outeiro TF & Lindquist S Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science 302, 1772–1775 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cooper AA et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 313, 324–328 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gitler AD et al. The Parkinson’s disease protein alpha-synuclein disrupts cellular Rab homeostasis. Proc. Natl. Acad. Sci. USA 105, 145–150 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chung CY et al. Identification and rescue of α-synuclein toxicity in Parkinson patient-derived neurons. Science 342, 983–987 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tardiff DF et al. Yeast reveal a “druggable” Rsp5/Nedd4 network that ameliorates α-synuclein toxicity in neurons. Science 342, 979–983 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mazzulli JR et al. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146, 37–52 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mazzulli JR, Zunke F, Isacson O, Studer L & Krainc D α-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc. Natl. Acad. Sci. USA 113, 1931–1936 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Thayanidhi N et al. Alpha-synuclein delays endoplasmic reticulum (ER)-to-Golgi transport in mammalian cells by antagonizing ER/Golgi SNAREs. Mol. Biol. Cell 21, 1850–1863 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Oaks AW, Marsh-Armstrong N, Jones JM, Credle JJ & Sidhu A Synucleins antagonize endoplasmic reticulum function to modulate dopamine transporter trafficking. PLoS One 8, e70872 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Caraveo G et al. Calcineurin determines toxic versus beneficial responses to α-synuclein. Proc. Natl. Acad. Sci. USA 111, E3544–E3552 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Luo J et al. A calcineurin- and NFAT-dependent pathway is involved in α-synuclein-induced degeneration of midbrain dopaminergic neurons. Hum. Mol. Genet 23, 6567–6574 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bellucci A et al. Induction of the unfolded protein response by α-synuclein in experimental models of Parkinson’s disease. J. Neurochem 116, 588–605 (2011). [DOI] [PubMed] [Google Scholar]
- 87.Colla E et al. Endoplasmic reticulum stress is important for the manifestations of α-synucleinopathy in vivo. J. Neurosci 32, 3306–3320 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Colla E et al. Accumulation of toxic α-synuclein oligomer within endoplasmic reticulum occurs in α-synucleinopathy in vivo. J. Neurosci 32, 3301–3305 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wong YC & Holzbaur EL Autophagosome dynamics in neurodegeneration at a glance. J. Cell Sci 128, 1259–1267 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Winslow AR et al. α-Synuclein impairs macroautophagy: implications for Parkinson’s disease. J. Cell Biol 190, 1023–1037 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT & Sulzer D Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305, 1292–1295 (2004). [DOI] [PubMed] [Google Scholar]
- 92.Martinez-Vicente M et al. Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J. Clin. Invest 118, 777–788 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tanik SA, Schultheiss CE, Volpicelli-Daley LA, Brunden KR & Lee VM Lewy body-like α-synuclein aggregates resist degradation and impair macroautophagy. J. Biol. Chem 288, 15194–15210 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Volpicelli-Daley LA et al. Formation of α-synuclein Lewy neurite-like aggregates in axons impedes the transport of distinct endosomes. Mol. Biol. Cell 25, 4010–4023 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wong YC & Krainc D Lysosomal trafficking defects link Parkinson’s disease with Gaucher’s disease. Mov. Disord 31, 1610–1618 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Maroteaux L, Campanelli JT & Scheller RH Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci 8, 2804–2815 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rousseaux MW et al. TRIM28 regulates the nuclear accumulation and toxicity of both alpha-synuclein and tau. eLife 5, e19809 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kontopoulos E, Parvin JD & Feany MB Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum. Mol. Genet 15, 3012–3023 (2006). [DOI] [PubMed] [Google Scholar]
- 99.Fares MB et al. The novel Parkinson’s disease linked mutation G51D attenuates in vitro aggregation and membrane binding of α-synuclein, and enhances its secretion and nuclear localization in cells. Hum. Mol. Genet 23, 4491–4509 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Decressac M et al. TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity. Proc. Natl. Acad. Sci. USA 110, E1817–E1826 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Phillips MJ & Voeltz GK Structure and function of ER membrane contact sites with other organelles. Nat. Rev. Mol. Cell Biol 17, 69–82 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Calì T, Ottolini D, Negro A & Brini M α-Synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum-mitochondria interactions. J. Biol. Chem 287, 17914–17929 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Guardia-Laguarta C et al. α-Synuclein is localized to mitochondria-associated ER membranes. J. Neurosci 34, 249–259 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chu Y et al. Alterations in axonal transport motor proteins in sporadic and experimental Parkinson’s disease. Brain 135, 2058–2073 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Prots I et al. α-Synuclein oligomers impair neuronal microtubule-kinesin interplay. J. Biol. Chem 288, 21742–21754 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tilve S, Difato F & Chieregatti E Cofilin 1 activation prevents the defects in axon elongation and guidance induced by extracellular alpha-synuclein. Sci. Rep 5, 16524 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mazzulli JR et al. Activation of β-glucocerebrosidase reduces pathological α-synuclein and restores lysosomal function in Parkinson’s patient midbrain neurons. J. Neurosci 36, 7693–7706 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bousset L et al. Structural and functional characterization of two alpha-synuclein strains. Nat. Commun 4, 2575 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Auluck PK, Chan HY, Trojanowski JQ, Lee VM & Bonini NM Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science 295, 865–868 (2002). [DOI] [PubMed] [Google Scholar]
- 110.Ihara M et al. Sept4, a component of presynaptic scaffold and Lewy bodies, is required for the suppression of alpha-synuclein neurotoxicity. Neuron 53, 519–533 (2007). [DOI] [PubMed] [Google Scholar]
- 111.Outeiro TF et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science 317, 516–519 (2007). [DOI] [PubMed] [Google Scholar]
- 112.Chen L & Feany MB Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat. Neurosci 8, 657–663 (2005). [DOI] [PubMed] [Google Scholar]
- 113.Bender A et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet 38, 515–517 (2006). [DOI] [PubMed] [Google Scholar]
- 114.Vilchez D, Saez I & Dillin A The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat. Commun 5, 5659 (2014). [DOI] [PubMed] [Google Scholar]
- 115.Li W et al. Stabilization of alpha-synuclein protein with aging and familial parkinson’s disease-linked A53T mutation. J. Neurosci 24, 7400–7409 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Chu Y & Kordower JH Age-associated increases of alpha-synuclein in monkeys and humans are associated with nigrostriatal dopamine depletion: Is this the target for Parkinson’s disease? Neurobiol. Dis 25, 134–149 (2007). [DOI] [PubMed] [Google Scholar]
- 117.Finkel T & Holbrook NJ Oxidants, oxidative stress and the biology of ageing. Nature 408, 239–247 (2000). [DOI] [PubMed] [Google Scholar]
- 118.Giasson BI et al. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 290, 985–989 (2000). [DOI] [PubMed] [Google Scholar]
- 119.Paxinou E et al. Induction of alpha-synuclein aggregation by intracellular nitrative insult. J. Neurosci 21, 8053–8061 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hodara R et al. Functional consequences of alpha-synuclein tyrosine nitration: diminished binding to lipid vesicles and increased fibril formation. J. Biol. Chem 279, 47746–47753 (2004). [DOI] [PubMed] [Google Scholar]
- 121.Zecca L et al. Neuromelanin can protect against iron-mediated oxidative damage in system modeling iron overload of brain aging and Parkinson’s disease. J. Neurochem 106, 1866–1875 (2008). [DOI] [PubMed] [Google Scholar]
- 122.Halliday GM, Macdonald V & Henderson JM A comparison of degeneration in motor thalamus and cortex between progressive supranuclear palsy and Parkinson’s disease. Brain 128, 2272–2280 (2005). [DOI] [PubMed] [Google Scholar]
- 123.Sulzer D & Surmeier DJ Neuronal vulnerability, pathogenesis, and Parkinson’s disease. Mov. Disord 28, 41–50 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Braak H et al. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211 (2003). [DOI] [PubMed] [Google Scholar]
- 125.Conway KA, Rochet JC, Bieganski RM & Lansbury PT Jr. Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science 294, 1346–1349 (2001). [DOI] [PubMed] [Google Scholar]
- 126.Mazzulli JR, Armakola M, Dumoulin M, Parastatidis I & Ischiropoulos H Cellular oligomerization of alpha-synuclein is determined by the interaction of oxidized catechols with a C-terminal sequence. J. Biol. Chem 282, 31621–31630 (2007). [DOI] [PubMed] [Google Scholar]
- 127.Mazzulli JR et al. Cytosolic catechols inhibit alpha-synuclein aggregation and facilitate the formation of intracellular soluble oligomeric intermediates. J. Neurosci 26, 10068–10078 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hirsch E, Graybiel AM & Agid YA Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson’s disease. Nature 334, 345–348 (1988). [DOI] [PubMed] [Google Scholar]
- 129.Chakrabarty P et al. Interferon-γ induces progressive nigrostriatal degeneration and basal ganglia calcification. Nat. Neurosci 14, 694–696 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yazawa I et al. Mouse model of multiple system atrophy alpha-synuclein expression in oligodendrocytes causes glial and neuronal degeneration. Neuron 45, 847–859 (2005). [DOI] [PubMed] [Google Scholar]
- 131.Lindersson E et al. p25alpha Stimulates alpha-synuclein aggregation and is co-localized with aggregated alpha-synuclein in alpha-synucleinopathies. J. Biol. Chem 280, 5703–5715 (2005). [DOI] [PubMed] [Google Scholar]
- 132.Ubhi K et al. Neurodegeneration in a transgenic mouse model of multiple system atrophy is associated with altered expression of oligodendroglial-derived neurotrophic factors. J. Neurosci 30, 6236–6246 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kordower JH, Chu Y, Hauser RA, Freeman TB & Olanow CW Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat. Med 14, 504–506 (2008). [DOI] [PubMed] [Google Scholar]
- 134.Li JY et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med 14, 501–503 (2008). [DOI] [PubMed] [Google Scholar]
- 135.Desplats P et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc. Natl. Acad. Sci. USA 106, 13010–13015 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Luk KC et al. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc. Natl. Acad. Sci. USA 106, 20051–20056 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Volpicelli-Daley LA et al. Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72, 57–71 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Luk KC et al. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338, 949–953 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sacino AN et al. Intramuscular injection of α-synuclein induces CNS α-synuclein pathology and a rapid-onset motor phenotype in transgenic mice. Proc. Natl. Acad. Sci. USA 111, 10732–10737 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Peelaerts W et al. α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522, 340–344 (2015). [DOI] [PubMed] [Google Scholar]
- 141.Rey NL et al. Widespread transneuronal propagation of α-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson’s disease. J. Exp. Med 213, 1759–1778 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Danzer KM et al. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol. Neurodegener 7, 42 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Tsunemi T, Hamada K & Krainc D ATP13A2/PARK9 regulates secretion of exosomes and α-synuclein. J. Neurosci 34, 15281–15287 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kong SM et al. Parkinson’s disease-linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes α-Synuclein externalization via exosomes. Hum. Mol. Genet 23, 2816–2833 (2014). [DOI] [PubMed] [Google Scholar]
- 145.Chutna O et al. The small GTPase Rab11 co-localizes with α-synuclein in intracellular inclusions and modulates its aggregation, secretion and toxicity. Hum. Mol. Genet 23, 6732–6745 (2014). [DOI] [PubMed] [Google Scholar]
- 146.Emmanouilidou E et al. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci 30, 6838–6851 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Oh SH et al. Mesenchymal stem cells inhibit transmission of α-Synuclein by modulating clathrin-mediated endocytosis in a Parkinsonian model. Cell Rep. 14, 835–849 (2016). [DOI] [PubMed] [Google Scholar]
- 148.Mao X et al. Pathological α-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 353, aah3374 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Abounit S et al. Tunneling nanotubes spread fibrillar α-synuclein by intercellular trafficking of lysosomes. EMBO J. 35, 2120–2138 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Brettschneider J, Del Tredici K, Lee VM & Trojanowski JQ Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat. Rev. Neurosci 16, 109–120 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Walsh DM & Selkoe DJ A critical appraisal of the pathogenic protein spread hypothesis of neurodegeneration. Nat. Rev. Neurosci 17, 251–260 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lee HJ et al. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J. Biol. Chem 285, 9262–9272 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ouchi Y et al. Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann. Neurol 57, 168–175 (2005). [DOI] [PubMed] [Google Scholar]
- 154.Stefanova N et al. Microglial activation mediates neurodegeneration related to oligodendroglial alpha-synucleinopathy: implications for multiple system atrophy. Mov. Disord 22, 2196–2203 (2007). [DOI] [PubMed] [Google Scholar]
- 155.Chung CY, Koprich JB, Siddiqi H & Isacson O Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV alpha-synucleinopathy. J. Neurosci 29, 3365–3373 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Theodore S, Cao S, McLean PJ & Standaert DG Targeted overexpression of human alpha-synuclein triggers microglial activation and an adaptive immune response in a mouse model of Parkinson disease. J. Neuropathol. Exp. Neurol 67, 1149–1158 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lastres-Becker I et al. α-Synuclein expression and Nrf2 deficiency cooperate to aggravate protein aggregation, neuronal death and inflammation in early-stage Parkinson’s disease. Hum. Mol. Genet 21, 3173–3192 (2012). [DOI] [PubMed] [Google Scholar]
- 158.Gao HM et al. Neuroinflammation and oxidation/nitration of alpha-synuclein linked to dopaminergic neurodegeneration. J. Neurosci 28, 7687–7698 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Harms AS et al. MHCII is required for α-synuclein-induced activation of microglia, CD4 T cell proliferation, and dopaminergic neurodegeneration. J. Neurosci 33, 9592–9600 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kim C et al. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun 4, 1562 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Daniele SG et al. Activation of MyD88-dependent TLR1/2 signaling by misfolded α-synuclein, a protein linked to neurodegenerative disorders. Sci. Signal 8, ra45 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Wang S et al. α-Synuclein, a chemoattractant, directs microglial migration via H2O2-dependent Lyn phosphorylation. Proc. Natl. Acad. Sci. USA 112, E1926–E1935 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Thome AD, Harms AS, Volpicelli-Daley LA & Standaert DG microRNA-155 regulates alpha-synuclein-induced inflammatory responses in models of Parkinson disease. J. Neurosci 36, 2383–2390 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Ejlerskov P et al. Lack of neuronal IFN-β-IFNAR causes Lewy body- and Parkinson’s disease-like dementia. Cell 163, 324–339 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hamza TH et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat. Genet 42, 781–785 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Roy B & Jackson GR Interactions between Tau and α-synuclein augment neurotoxicity in a Drosophila model of Parkinson’s disease. Hum. Mol. Genet 23, 3008–3023 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Lewis J et al. In vivo silencing of alpha-synuclein using naked siRNA. Mol. Neurodegener 3, 19 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Cooper JM et al. Systemic exosomal siRNA delivery reduced alpha-synuclein aggregates in brains of transgenic mice. Mov. Disord 29, 1476–1485 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Rott R et al. α-Synuclein fate is determined by USP9X-regulated monoubiquitination. Proc. Natl. Acad. Sci. USA 108, 18666–18671 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Tofaris GK et al. Ubiquitin ligase Nedd4 promotes alpha-synuclein degradation by the endosomal-lysosomal pathway. Proc. Natl. Acad. Sci. USA 108, 17004–17009 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Oueslati A, Schneider BL, Aebischer P & Lashuel HA Polo-like kinase 2 regulates selective autophagic α-synuclein clearance and suppresses its toxicity in vivo. Proc. Natl. Acad. Sci. USA 110, E3945–E3954 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.McGlinchey RP & Lee JC Cysteine cathepsins are essential in lysosomal degradation of α-synuclein. Proc. Natl. Acad. Sci. USA 112, 9322–9327 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Masliah E et al. Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of Lewy body disease. PLoS One 6, e19338 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 174.Bae EJ et al. Antibody-aided clearance of extracellular α-synuclein prevents cell-to-cell aggregate transmission. J. Neurosci 32, 13454–13469 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Sardi SP et al. Augmenting CNS glucocerebrosidase activity as a therapeutic strategy for parkinsonism and other Gaucher-related synucleinopathies. Proc. Natl. Acad. Sci. USA 110, 3537–3542 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Rocha EM et al. Glucocerebrosidase gene therapy prevents α-synucleinopathy of midbrain dopamine neurons. Neurobiol. Dis 82, 495–503 (2015). [DOI] [PubMed] [Google Scholar]
- 177.Rockenstein E et al. Glucocerebrosidase modulates cognitive and motor activities in murine models of Parkinson’s disease. Hum. Mol. Genet 25, 2645–2660 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Aflaki E et al. A new glucocerebrosidase chaperone reduces α-synuclein and glycolipid levels in iPSC-derived dopaminergic neurons from patients with Gaucher disease and parkinsonism. J. Neurosci 36, 7441–7452 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Pchelina SN et al. Increased plasma oligomeric alpha-synuclein in patients with lysosomal storage diseases. Neurosci. Lett 583, 188–193 (2014). [DOI] [PubMed] [Google Scholar]
- 180.Rothaug M et al. LIMP-2 expression is critical for β-glucocerebrosidase activity and α-synuclein clearance. Proc. Natl. Acad. Sci. USA 111, 15573–15578 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Gitler AD et al. α-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat. Genet 41, 308–315 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Tsunemi T & Krainc D Zn2+ dyshomeostasis caused by loss of ATP13A2/PARK9 leads to lysosomal dysfunction and alpha-synuclein accumulation. Hum. Mol. Genet 23, 2791–2801 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Fonseca-Ornelas L et al. Small molecule-mediated stabilization of vesicle-associated helical α-synuclein inhibits pathogenic misfolding and aggregation. Nat. Commun 5, 5857 (2014). [DOI] [PubMed] [Google Scholar]
- 184.Lindström V et al. Immunotherapy targeting α-synuclein protofibrils reduced pathology in (Thy-1)-h[A30P] α-synuclein mice. Neurobiol. Dis 69, 134–143 (2014). [DOI] [PubMed] [Google Scholar]
- 185.Dettmer U et al. Parkinson-causing α-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation. Nat. Commun 6, 7314 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Schneeberger A, Tierney L & Mandler M Active immunization therapies for Parkinson’s disease and multiple system atrophy. Mov. Disord 31, 214–224 (2016). [DOI] [PubMed] [Google Scholar]
- 187.Games D et al. Reducing C-terminal-truncated alpha-synuclein by immunotherapy attenuates neurodegeneration and propagation in Parkinson’s disease-like models. J. Neurosci 34, 9441–9454 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Tran HT et al. A-synuclein immunotherapy blocks uptake and templated propagation of misfolded α-synuclein and neurodegeneration. Cell Rep. 7, 2054–2065 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Masliah E et al. Effects of alpha-synuclein immunization in a mouse model of Parkinson’s disease. Neuron 46, 857–868 (2005). [DOI] [PubMed] [Google Scholar]
- 190.Bergstrom AL, Kallunki P & Fog K Development of passive immunotherapies for synucleinopathies. Mov. Disord 31, 203–213 (2016). [DOI] [PubMed] [Google Scholar]
- 191.Beal MF et al. A randomized clinical trial of high-dosage coenzyme Q10 in early Parkinson disease: no evidence of benefit. JAMA Neurol. 71, 543–552 (2014). [DOI] [PubMed] [Google Scholar]
- 192.Sybertz E & Krainc D Development of targeted therapies for Parkinson’s disease and related synucleinopathies. J. Lipid Res 55, 1996–2003 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Chen-Plotkin AS Unbiased approaches to biomarker discovery in neurodegenerative diseases. Neuron 84, 594–607 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Van der Perren A et al. FK506 reduces neuroinflammation and dopaminergic neurodegeneration in an α-synuclein-based rat model for Parkinson’s disease. Neurobiol. Aging 36, 1559–1568 (2015). [DOI] [PubMed] [Google Scholar]
- 195.Qin H et al. Inhibition of the JAK/STAT pathway protects against α-synuclein-induced neuroinflammation and dopaminergic neurodegeneration. J. Neurosci 36, 5144–5159 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Valera E, Mante M, Anderson S, Rockenstein E & Masliah E Lenalidomide reduces microglial activation and behavioral deficits in a transgenic model of Parkinson’s disease. J. Neuroinflammation 12, 93 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Kim C et al. Hypoestoxide reduces neuroinflammation and α-synuclein accumulation in a mouse model of Parkinson’s disease. J. Neuroinflammation 12, 236 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Burre J et al. Properties of native brain α-synuclein. Nature 498, E4–E6, discussion E6–7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Theillet FX et al. Structural disorder of monomeric α-synuclein persists in mammalian cells. Nature 530, 45–50 (2016). [DOI] [PubMed] [Google Scholar]
- 200.Gould N et al. Evidence of native α-synuclein conformers in the human brain. J. Biol. Chem 289, 7929–7934 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Wang W et al. A soluble α-synuclein construct forms a dynamic tetramer. Proc. Natl. Acad. Sci. USA 108, 17797–17802 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Bartels T, Choi JG & Selkoe DJ α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 477, 107–110 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Dettmer U, Newman AJ, von Saucken VE, Bartels T & Selkoe D KTKEGV repeat motifs are key mediators of normal α-synuclein tetramerization: Their mutation causes excess monomers and neurotoxicity. Proc. Natl. Acad. Sci. USA 112, 9596–9601 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Sharon R et al. The formation of highly soluble oligomers of alpha-synuclein is regulated by fatty acids and enhanced in Parkinson’s disease. Neuron 37, 583–595 (2003). [DOI] [PubMed] [Google Scholar]
- 205.Ferreon AC, Gambin Y, Lemke EA & Deniz AA Interplay of alpha-synuclein binding and conformational switching probed by single-molecule fluorescence. Proc. Natl. Acad. Sci. USA 106, 5645–5650 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Buell AK et al. Solution conditions determine the relative importance of nucleation and growth processes in α-synuclein aggregation. Proc. Natl. Acad. Sci. USA 111, 7671–7676 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Galvagnion C et al. Lipid vesicles trigger α-synuclein aggregation by stimulating primary nucleation. Nat. Chem. Biol 11, 229–234 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Cremades N et al. Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell 149, 1048–1059 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Conway KA et al. Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. USA 97, 571–576 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Winner B et al. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. USA 108, 4194–4199 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Pieri L, Madiona K, Bousset L & Melki R Fibrillar α-synuclein and huntingtin exon 1 assemblies are toxic to the cells. Biophys. J 102, 2894–2905 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Fares MB et al. Induction of de novo α-synuclein fibrillization in a neuronal model for Parkinson’s disease. Proc. Natl. Acad. Sci. USA 113, E912–E921 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Luk KC et al. Molecular and biological compatibility with host alpha-synuclein influences fibril pathogenicity. Cell Rep. 16, 3373–3387 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Prusiner SB et al. Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc. Natl. Acad. Sci. USA 112, E5308–E5317 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Woerman AL et al. Propagation of prions causing synucleinopathies in cultured cells. Proc. Natl. Acad. Sci. USA 112, E4949–E4958 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]