

Abstract
Proteolysis targeting chimeras (PROTACs) represent a new class of promising therapeutic modalities. PROTACs hijack E3 ligases and the ubiquitin-proteasome system (UPS), leading to selective degradation of the target proteins. However, only a very limited number of E3 ligases have been leveraged to generate effective PROTACs. Herein, we report that the KEAP1 E3 ligase can be harnessed for targeted protein degradation utilizing a highly selective, noncovalent small-molecule KEAP1 binder. We generated a proof-of-concept PROTAC, MS83, by linking the KEAP1 ligand to a BRD4/3/2 binder. MS83 effectively reduces protein levels of BRD4 and BRD3, but not BRD2, in cells in a concentration-, time-, KEAP1- and UPS-dependent manner. Interestingly, MS83 degrades BRD4/3 more durably than the CRBN-recruiting PROTAC dBET1 in MDA-MB-468 cells and selectively degrades BRD4 short isoform over long isoform in MDA-MB-231 cells. It also displays improved antiproliferative activity than dBET1. Overall, our study expands the limited toolbox for targeted protein degradation.
Graphical Abstract
1. INTRODUCTION
Proteolysis targeting chimeras (PROTACs) are heterobifunctional small molecules, composed of three components: a small molecule that binds to the protein of interest (POI), an E3 ligase-recruiting small-molecule ligand, and a linker that ties these two moieties together.1,2 Within the last 15 years, PROTAC technology has been effectively applied to degrade a great variety of proteins.3–6 However, a key issue in the PROTAC field is that only a distinct few E3 ligases (e.g., CRBN, VHL, MDM2, cIAP1, DCAF15) have been harnessed to generate effective PROTACs, with CRBN and VHL being utilized most extensively.4,5 Recently, E3 ligases RNF4, RNF114, and DCAF16 have been leveraged for targeted protein degradation (TPD) via using covalent ligands of these E3 ligases, but without widespread use thus far.5 As a result of the paucity of TPD-harnessed E3 ligases, many POIs cannot be effectively degraded by PROTACs, partially due to the fact that the targeted E3 ligase has low expression levels or functional activity in the target tissues or cell types. Another important factor for building effective PROTACs is the generation of a sufficiently stable and productive POI–PROTAC–E3 ligase ternary complex.6 The great promise of PROTACs derives from the versatility and diversity of individual E3 ligases and substrate pairings and thus the ability to target a broad range of proteins previously considered undruggable. Yet, to date, considering that there are approximately 600 human E3 ligases, most have not been leveraged for TPD. This presents tremendous potential to generate PROTACs targeting oncoproteins conceivably with tumor and tissue selectivity. Here, we report kelch-like ECH-associated protein-1 (KEAP1, also known as KLHL19) as another E3 ligase to be exploited for generating effective PROTACs.
KEAP1 functions as a substrate adaptor protein for cullin 3 (CUL3) E3 ligase complexes. These complexes are typically consist of three modules: a CUL3 scaffold protein, RING-box protein 1 (RBX1), and a bric-a-brac tramtrack broad (BTB) protein such as KEAP1. Distinct from other cullin-RING E3 ligases (CRLs), within the CUL3 complex, a single BTB protein carries out a dual adaptor and substrate recognition functionality, as opposed to separating these functions into individual subunits.7 KEAP1, the best-characterized kelch-like family member, manipulates the levels of the transcription factor Nrf2 (nuclear factor erythroid 2-related factor 2), which in turn regulates the oxidative stress response.8,9 Under normal conditions, Nrf2 is sequestered by dimeric KEAP1/Cullin 3 complex (CRL3KEAP1) and polyubiquitinated for proteasomal degradation. However, oxidative stress inhibits the ability of the Cul3-KAP11-E3 ligase complex to ubiquitinate Nrf2, due to chemical modifications to cysteine residues of KEAP1. These modifications of cysteine residues could lead to dissociation of Cul3 as well as other conformational changes to KEAP1.8,9 Analysis of KEAP1 protein expression levels in human tissues shows that KEAP1 has distinctive tissue distributions compared to other E3 ligases such as CRBN, VHL, RNF4, and RNF114 (Figure S1). In addition, KEAP1 is highly expressed in human cancers, with similar expression levels as VHL but higher than CRBN in general (Figure S2). In particular, expression levels of KEAP1 are the highest in lung adenocarcinoma, kidney renal clear cell carcinoma, breast invasive carcinoma, prostate adenocarcinoma, and glioblastoma, compared with that of VHL and CRBN (Figure S3).
Given the importance of Nrf2 in cellular processes, small molecules that interrupt the KEAP1-Nrf2 protein–protein interaction in a covalent or noncovalent manner have been developed.10–12 For example, several electrophilic small molecules including bardoxolone (CDDO) and its derivatives covalently modify the cysteine residues of KEAP1 that sense oxidative stress.13 Nrf2 binds two kelch domains in the KEAP1 dimer through its high-affinity ETGE and low-affinity DLG motifs.14 A number of noncovalent small-molecule inhibitors binding the KEAP1 kelch domain at the Nrf2 binding site, mimicking the ETGE motif, have also been identified.10
A peptide-based KEAP1-recruiting degrader was recently reported and used for degradation of intracellular Tau.15 This Nrf2 ETGE motif-based peptidic degrader induces KEAP1-dependent Tau degradation; it binds tightly to KEAP1 and Tau in vitro and exhibits enhanced Tau ubiquitination. However, peptide-based degraders have limited utilities due to their poor cell permeability and poor pharmacokinetic properties. Very recently, a nonpeptidic degrader utilizing bardoxolone (CDDO) as a covalent ligand of KEAP1 and JQ1 as the ligand to target BRD4 was also reported.16 While degradation of BRD4 was demonstrated, it should be noted that CDDO, the KEAP1 ligand utilized in this study, is not selective for KEAP1.16 For example, CDDO and its derivatives interact with other protein targets, such as mTOR.16,17 It was acknowledged that the degradation effect observed for the aforementioned heterobifunctional degrader may not be solely due to recruiting KEAP1, as CDDO is a nonselective and reactive ligand and may covalently modify other E3 ligases.16 In light of these limitations, a more general approach using a selective small-molecule ligand of KEAP1 to harness KEAP1 for PROTAC development is highly desirable.
In this proof-of-concept study, we utilize a highly potent, selective, and noncovalent small-molecule KEAP1 ligand, KEAP1-L (also known as KI696) (Figure 1A)18 which disrupts the KEAP1 kelch–Nrf2 interaction by binding the Nrf2 binding pocket of KEAP1, as the ligand for our KEAP1-recruiting PROTAC approach.18,19 We show that bromodomain and extra-terminal (BET) family proteins BRD4 and BRD3 can be effectively degraded using KEAP1-L as the E3 ligase recruiting ligand, and JQ1 as the BRD4/3/2 ligand. The discovery and functional characterization of our KEAP1-recruiting BRD4/3 PROTAC, MS83, and its negative control have demonstrated for the first time that the E3 ligase KEAP1 can be harnessed for TPD using a selective small-molecule KEAP1 ligand, thus significantly enhancing the general applicability of the KEAP1-based PROTAC approach.
Figure 1.
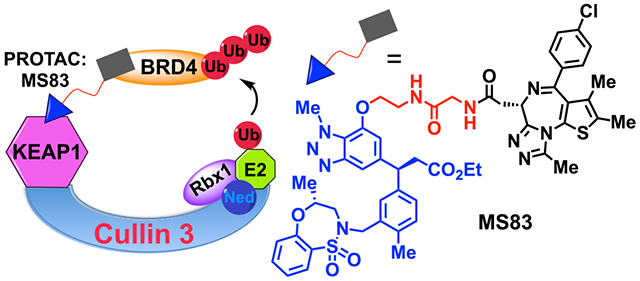
Design and biophysical characterizations of KEAP1-recruiting BRD4 PROTAC degraders MS83 and MS83A and the negative control MS83N1. (A) Chemical structures of KEAP1 kelch domain binding ligand, KEAP1-L and its ethyl ester, KEAP1-L-OEt. (B) Cocrystal structure (PDB code: 5FNU) of the KEAP1 kelch domain (shown in gray) in complex with KEAP1-L (blue). The methoxy group, highlighted by the dashed black circle, reaches out to the binding pocket. The key hydrogen-bonding interactions of KEAP1-L (blue) with the KEAP1 residues (gray sticks) and water molecules (red spheres) are represented by yellow dashed lines. (C) Chemical structures of KEAP1-recruiting BRD4 PROTAC degraders MS83 and MS83A and their negative control, MS83N1. (D) Binding affinity of KEAP1-L (left), MS83A (middle), and MS83N1 (right) to the KEAP1 kelch domain as measured by ITC. The calculated values represent the mean ± SD from two independent experiments. First injection was removed from the fitting.
2. RESULTS AND DISCUSSION
2.1. Design and Biophysical Characterization of the KEAP1-Recruiting PROTAC MS83.
To generate effective KEAP1-recruiting PROTAC degraders, we first determined a suitable linker attachment point by analyzing the cocrystal structure of KEAP1-L in complex with the KEAP1 kelch domain (Figure 1B).19 KEAP1-L fits into the Nrf2 binding pocket very efficiently, with a significant proportion of the molecule engaging directly with the Nrf2 binding site. In addition, the carboxylic acid of KEAP1-L mimicks the Nrf2 ETGE motif and thus creates a critical interaction with the kelch domain of KEAP1. On the other hand, the protrusion of the methoxy group at the benzotriazole 7-position out of the binding pocket presents a very suitable handle and, therefore, is used for the attachment of a linker (Figure 1B, black dotted circle). To show that KEAP1-recruiting PROTACs can be used for effective degradation of target proteins, we chose to target the transcription coactivator, BET family protein BRD4. BRD4 was chosen not only due to extensive studies and successes of diverse BRD4 degraders4 but also due to its critical role in driving both hematological and solid tumor cancers.20–22 Thus, we next designed and synthesized novel putative degraders 1–9, which connect the KEAP1 ligand to the BET bromodomain inhibitor (+)-JQ1 via a variety of linkers (Figure S4A and Scheme S1). To enhance cell permeability, we prepared an ester derivative, KEAP1-L-OEt (Figure 1A, R = Et), of the KEAP1-L as a pro-drug, and utilized it for the synthesis of initial putative degraders. We used exactly the same attachment point for JQ1 as the first-generation BET degrader, dBET1 that was designed to recruit cereblon (CRBN) E3 ligase to degrade BET domain proteins. Through immunoblotting analysis of these initial compounds (Figure S4B), we identified MS83 (Figure 1C) as an effective degrader of BRD4. We also designed and synthesized MS83A (Figure 1C and Scheme S1), which contains a carboxylic acid group instead of the ethyl ester group in MS83. Furthermore, we designed a negative control, MS83N1 with a highly diminished binding affinity to the KEAP1 kelch domain (Figure 1C). This KEAP1 inactive ligand was designed by converting the key carboxylic acid functional group to a methylketone and also by moving the N-methyl group from the 1-position to the 3-position of the triazole ring, in turn blocking crucial hydrogen-bonding interactions. Using isothermal titration calorimetry (ITC), we assessed binding affinities of KEAP1-L, MS83A, and MS83N1 to the KEAP1 kelch domain. Compared to KEAP1-L (Kd = 6 ± 6 nM), MS83A (Kd = 16 ± 5 nM) maintained similar binding affinity to KEAP1, validating our design hypothesis (Figure 1D, left and middle). Furthermore, as expected, MS83N1, featuring a KEAP1 inactive ligand, did not show any appreciable binding (Figure 1D, right).
2.2. Degradation of BET Proteins by the KEAP1-Recruiting PROTAC MS83 in Cells.
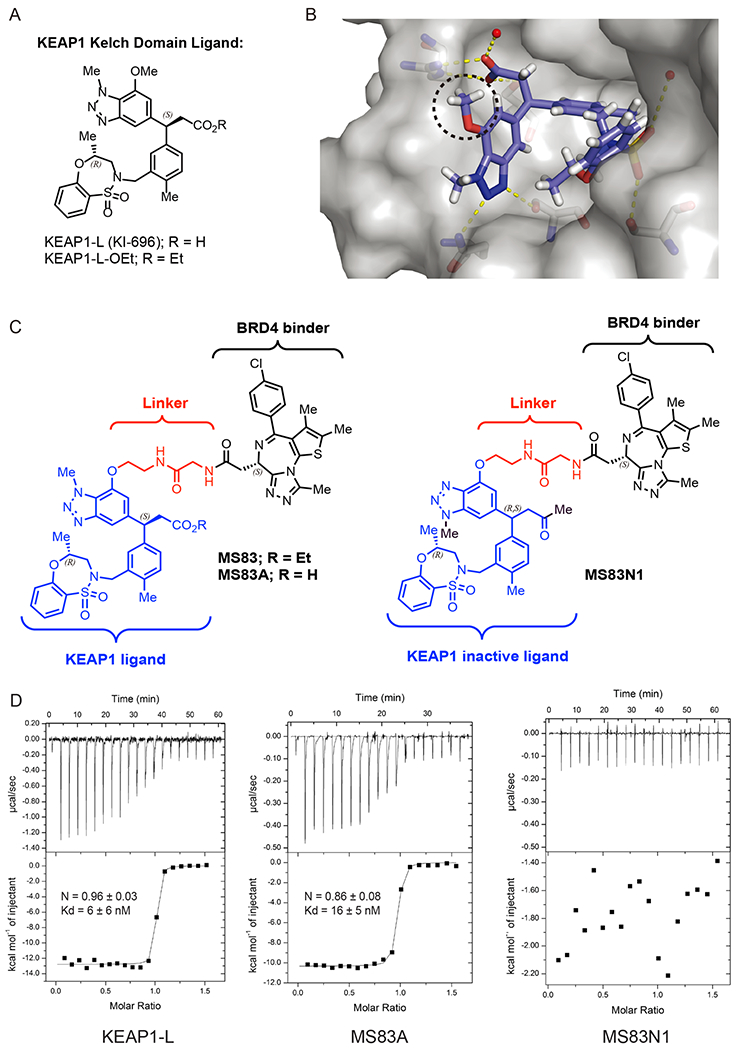
To assess degradation effects of KEAP1-recruiting PROTACs MS83 and MS83A, we performed Western blotting analysis in the triple-negative breast cancer (TNBC) cell line MDA-MB-468. This cell line was chosen based on the reported BRD4-c-MYC axis in TNBC23 and our determination of sufficient KEAP1 protein expression levels by Western blotting (Figure S5). We used the well-known CRBN-recruiting BET PROTAC degrader dBET1 as a positive control in our experiments. As mentioned above, MS83A featured the KEAP1-L with the carboxylic acid group, while MS83 was designed and synthesized as the pro-drug by using the ethyl ester derivative, KEAP1-L-OEt, to enhance cell permeability of this compound (Figure 1A,C). We separately treated MDA-MB-468 cells with 0.5, 1, and 5 μM of dBET1, MS83A, or MS83 and monitored the change of BRD4 as well as BRD3 and BRD2 protein levels at different time points (Figure 2A and Figure S6). Protein levels of both BRD4 long (L) and short (S) isoforms were reduced significantly when treated with dBET1 for 12 h; however, the protein levels rebounded remarkably, almost to their original levels within a 36 h treatment. On the other hand, BRD4 (both L and S isoforms) was robustly degraded by MS83A treatment (at 5 μM) for 12 h, and degradation of BRD4 persisted for 72 h, while lower concentrations (0.5 and 1 μM) of MS83A did not show obvious degradation of BRD4. MS83 displayed slower degradation of BRD4 (L and S isoforms) compared with MS83A and dBET1, with moderate reductions of BRD4 (L and S) protein levels after 24 treatment with 0.5 μM of MS83. Similar to MS83A, the degradation effect of MS83 was durable for 72 h. Notably, we observed a “hook effect” in cells treated with MS83 at the highest concentration (5 μM).24 While MS83A requires higher concentrations to degrade BRD4, presumably due to its lower cell permeability, MS83 effectively degraded BRD4 (both L and S isoforms) at lower concentrations, but requires longer time to do so, likely due to that the in-cell hydrolysis of its ethyl ester group is required to release the carboxylic acid group. As mentioned above, this carboxylic acid group is crucial for effective binding to KEAP1. In addition, we examined the effect of these compounds on degrading BRD2 and BRD3. BRD2 and BRD3 protein levels were also reduced by dBET1, with robust degradation within a 12 h treatment followed by significant rebounding of the protein levels within a 72 h treatment. Interestingly, 5 μM of MS83A or 0.5 μM of MS83 induced obvious degradation of BRD3, but not BRD2 with a 36 h treatment, and the degradation effect on BRD3 was durable for 72 h. In addition, we pretreated MDA-MB-468 cells with 50 μg/mL of cycloheximide (CHX) and then chased with 0.5 μM of MS83 at 0, 12, 24, and 36 h time points (Figure S7). While the DMSO treatment did not cause obvious changes in the BRD4 (S) protein level at these time points, the MS83 treatment induced degradation of BRD4 (S) very clearly at 24 and 36 h (Figure S7). Collectively, our results indicate that (1) MS83 and MS83A effectively degrade BRD4 (both L and S isoforms); (2) MS83 is more effective than MS83A at lower concentrations to induce degradation of BRD4; (3) MS83 and MS83A effectively degrade BRD3 in addition to BRD4, but not BRD2, while in comparison dBET1 does not exhibit any selectivity; and (4) MS83 is more effective in degrading BRD4 over BRD3. Based on these results, we advanced MS83 for the following studies.
Figure 2.
KEAP1-recruiting PROTACs MS83 and MS83A induce degradation of BRD4 and BRD3, but not BRD2, in TNBC cells. (A) MDA-MB-468 cells were treated with 0.1% DMSO, dBET1, MS83A, or MS83 at the indicated concentrations for 12, 24, 36, 48, and 72 h. The indicated protein levels were determined by Western blotting and as the loading control α-tubulin was used. (B) MDA-MB-468 cells were treated with DMSO or MS83 at the indicated concentrations for 48 h. The indicated protein levels were determined by Western blotting and normalized with β-actin. Protein intensity relative to DMSO control for each concentration of MS83 is shown under the indicated protein band. (C) MDA-MB-468 cells were treated with 0.1% DMSO, dBET1, MS83N1, or MS83 at the indicated concentrations for 48 h. The indicated protein levels were determined by Western blotting and as the loading control α-tubulin was used. (D) MDA-MB-231 cells were treated with DMSO or MS83 at the indicated concentrations for 48 h. The indicated protein levels were determined by Western blotting and as the loading control α-tubulin was used. WB results are representative of at least two independent experiments.
To further evaluate the degradation potency of MS83, we treated MDA-MB-468 cells with MS83 for 48 h at concentrations ranging from 25 nM to 10 μM in a concentration response experiment (Figure 2B and Figure S8). Both BRD4 (L) and BRD4 (S) were degraded by MS83 starting at 100 nM, 80% maximum degradation was reached at 0.5 μM, and a “hook effect” was observed starting at 2.5 μM. Furthermore, BRD3 was degraded by 50% when cells were treated with 0.25 μM or 0.5 μM of MS83, and an obvious hook effect was observed starting at 1 μM. On the other hand, no degradation of BRD2 was observed at any concentrations tested. It has been frequently reported that JQ1 treatment in tumor models suppresses transcription of oncogene MYC, 2,21,25 thus reducing its post-transcriptional level. Indeed, we found the c-MYC protein level was significantly reduced by MS83 treatment at 0.25 μM or above in a concentration-dependent manner.
To clearly show that the degradation of BRD4 induced by MS83 is mediated via recruitment of KEAP1, we tested the effect of MS83N1, the negative control of MS83, on BET protein levels via Western blotting (Figure 2C). As stated before, the key interacting motifs of MS83 were altered in MS83N1 to prevent its effective binding to KEAP1 (Figure 1C, right), and MS83N1 indeed does not bind KEAP1 effectively (Figure 1D, right). As shown in Figure 2C, in contrast to MS83, MS83N1 was not able to degrade BRD4 (L) and (S), BRD3, and BRD2 when MDA-MB-468 cells were incubated with 0.5 or 1 μM of MS83N1 for 48 h. We also compared the effect of MS83 on downregulating c-MYC with that of dBET1, MS83N1, and JQ1 in MDA-MB-468 cells (48 h treatment) (Figure 2C and Figure S9). While dBET1 did not display an obvious effect in this experimental setting (Figure 2C), MS83 exhibited a similar effect as JQ1 at 0.5 μM, but was more effective than JQ1 at 2.5 μM in downregulating c-MYC (Figure S9). As expected, MS83 was more effective than the negative control MS83N1 in downregulating c-MYC (Figure 2C and Figure S9).
To assess whether MS83 affects the transcription level of BRD4, we performed a qRT-PCR study to evaluate the BRD4 mRNA level in MDA-MB-468 cells treated with 0.5 μM of JQ1, MS83N1, or MS83 for 48 h (Figure S10A). Interestingly, we found that BRD4 transcription was suppressed by JQ1 and MS83N1, but not MS83 (Figure S10A), which explains the observation of a slight reduction of the BRD4 (S) protein level by 5 μM of MS83N1 (Figure 2C). These qRT-PCR data, together with the Western blotting results (Figure 2C), suggest that the reduction of BRD4 protein levels induced by MS83 occurs post-translationally, thus confirming that MS83 is a PROTAC degrader of BRD4 (L and S isoforms). Moreover, we evaluated the mRNA levels of c-MYC and CDKN1A (encodes p21CIP1/WAF1), which are involved in cell proliferation and senescence, oppositely regulated by BRD4 (c-Myc downregulation and p21 upregulation).21,23 Indeed, all three compounds significantly suppressed MYC transcription (Figure S10B), consistent with the Western blotting results (Figure 2C), and remarkably induced CDKN1A transcription (Figure S10C), suggesting secondary effects on effector transcription. The effect of the negative control MS83N1 on MYC and CDKN1A transcriptions is likely due to that MS83N1 retains the BRD4 inhibitory activity as it contains the JQ1 moiety.
We further extended our investigation to another TNBC cell line, MDA-MB-231, which also expresses a sufficient level of the KEAP1 protein (Figure S5). Interestingly, in MDA-MB-231 cells treated with MS83 at concentrations ranging from 25 nM to 10 μM for 48 h, we observed potent degradation of BRD4 (S) and BRD3 without an obvious hook effect, while no degradation of BRD4 (L) and BRD2 was observed with treatment of MS83 even at 10 μM (Figure 2D). It has been reported recently that the BRD4 short isoform is an important oncoprotein, while the BRD4 long isoform may have tumor suppressor functions.26,27 Therefore, the selectivity of MS83 for BRD4 (S) over BRD4 (L) in MDA-MB-231 cells could be potentially important and is worth further investigation in the future. Additionally, downregulation of c-MYC by MS83 at 0.25 μM or above was also observed in MDA-MB-231 cells (Figure 2D). Taken together, these results show that our KEAP1-recruiting PROTAC MS83 can effectively degrade BET proteins in cells with distinct selectivity depending on cellular contexts and with different degradation kinetics compared with the CRBN-recruiting PROTAC dBET1.
2.3. Mechanism of Action of BRD4 Degradation Induced by MS83.
To establish the dependency of MS83-induced BRD4 degradation on the ubiquitin proteasome system, we conducted a set of rescue experiments in MDA-MB-468 cells (Figure 3A). A 2 h pretreatment with either JQ1 (1, 3, or 10 μM), which competes with MS83 for binding BRD4, or with the KEAP1 ligand KEAP1-L-OEt (1, 3, or 10 μM), which competes with MS83 for binding KEAP1, significantly rescued the reduction of BRD4 (L) and (S) protein levels induced by a 24 h treatment with 0.5 μM of MS83. Moreover, pretreatment with MLN4924 (1, 3, or 5 μM), which inhibits NEDD8-activiting enzyme (NAE) responsible for neddylation of cullin-RING E3 ubiquitin ligase activation, was also effective in blocking MS83-induced BRD4 degradation. To further confirm the MS83-induced BRD4 depends on the KEAP1 E3 ligase, we conducted a transient KEAP1 knockdown with shRNA in MDA-MB-468 cells. After confirmation of the effectiveness of KEAP1 shRNA via Western blotting (Figure 3B, right), we treated the KEAP1 knockdown and control cells with MS83. Upon treatment with MS83, BRD4 (S) and (L) isoforms were effectively degraded in control shRNA knockdown cells, while the BRD4 degradation was significantly diminished in the KEAP1 knockdown cells (Figure 3B, left). Furthermore, we conducted endogenous immunoprecipitation (IP) experiments to determine whether the interaction between KEAP1 and BRD4 can be observed in the presence of MS83 in MDA-MB-468 cells. As expected, the strong interaction between KEAP1 and BRD4 was induced by MS83, but no interaction was induced by DMSO or the negative control MS83N1 (Figure 3C). The results of the knockdown and co-IP experiments, together with the inability of the negative control MS83N1 to reduce BRD4 protein levels, clearly indicate that the effect of MS83 on degrading BRD4 is mediated by hijacking the KEAP1 E3 ligase.
Figure 3.

The BRD4 degradation induced by MS83 is mediated by the KEAP1 E3 ligase. (A) MDA-MB-468 cells were pretreated with the BET inhibitor JQ1 (1, 3, or 10 μM), KEAP1 ligand KEAP1-L-OEt (1, 3, or 10 μM), NAE inhibitor MLN4924 (1, 3, or 5 μM), or 0.1% DMSO for 2 h, followed by 24 h treatment with 0.5 μM of MS83. The indicated protein levels were determined by Western blotting and as the loading control α-tubulin was used. (B) The shRNA-mediated KEAP1 knockdown experiments were conducted in MDA-MB-468 cells, followed by a 24 h treatment with 0.5 μM of MS83. The knockdown efficiency of KEAP1 is shown in the right panel. The indicated protein levels were determined by Western blotting and as the loading control α-tubulin was used. (C) Endogenous IP of the long BRD4 isoform in MDA-MB-468 cells after an 18 h treatment with 0.5 μM of MS83N1, MS83, or 0.1% DMSO. The indicated protein levels were determined by Western blotting. The input is shown in the left panel, and the IP result is shown in the right panel. WB results are representative of at least two independent experiments.
In addition, we synthesized a JQ1 nonbinding version of MS83, which is named MS83N2, as a negative control using (−)-JQ1 instead of (+)-JQ1 (Scheme S2).28 We also synthesized MS83N2A, which is the carboxylic acid version of MS83N2 (Scheme S2). In contrast to MS83, we found that MS83N2 (at 0.5 μM) did not degrade BRD4 (L) and BRD4 (S) in MDA-MB-468 cells after 48 h treatment (Figure S11). We next assessed binding of MS83N2A to BRD4-BD1 and BRD4-BD2 in comparison with MS83A and MS83N1 using AlphaScreen assays and found that MS83N2A was indeed unable to bind effectively either bromodomain of BRD4, while MS83A and MS83N1 displayed comparable binding affinity to both bromodomains of BRD4 similar to JQ1 (Figure S12A–D). These results further support that MS83 is a BRD4 PROTAC.
2.4. Selectivity of MS83.
We next performed unbiased tandem mass tag (TMT)-based global proteomic profiling studies in MDA-MB-468 cells to further evaluate the selectivity of MS83. Over 7000 proteins were quantified in the protein samples from MDA-MB-468 cells that were treated with MS83 (0.5 μM), MS83N1 (0.5 μM), or DMSO for 48 h. 47 out of 7094 (0.66%) proteins were downregulated by the MS83 treatment compared with DMSO (Figure 4A and Table S1), and 4 out of 7094 (0.06%) proteins were downregulated by the MS83 treatment compared with the MS83N1 treatment (Figure 4B and Table S2). Notably, MS83 induced a significant reduction in BRD4 and BRD3 protein levels (Figure 4), consistent with the Western blotting results above (Figure 2A–C). Interestingly, the MS83 treatment did not lead to a significant change in protein levels of several other bromodomain-containing proteins such as BRD1, BRD2, BRD7, BRD8 and BRD9 (Figure 4). This BRD2 result is also consistent with our Western blotting results (Figure 2A–C). We next determined binding affinity of MS83A to BRD2-BD1, BRD2-BD2, BRD3-BD1 and BRD3-BD2 in addition to BRD4-BD1 and BRD4-BD2 using AlphaScreen assays (Figure S12A–B). As expected, MS83A effectively binds all of these bromodomains with similar affinity as JQ1 (Figure S12A–B). These results suggest that MS83’s degradation selectivity for BRD4 and BRD3 over BRD2 is not due to the lack of binding to BRD2, but likely due to unfavorable BRD2-MS83A-KEAP1 ternary complex formation. This warrants further investigation. Overall, we confirmed that MS83 is a BRD4 and BRD3 degrader using a complementary method and show that MS83 is relatively selective for BRD4 and BRD3 in unbiased global proteomic studies.
Figure 4.
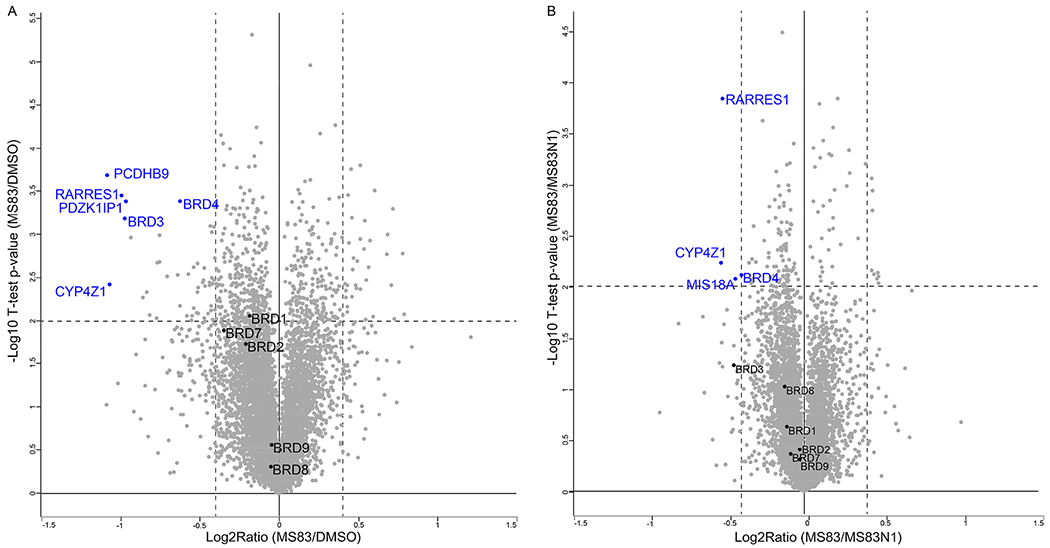
Quantitative MS-based proteomic analysis indicates that MS83 selectively degrades BRD4 and BRD3 over other BRDs in MDA-MB-468 cells. MDA-MB-468 cells were treated with 0.1% DMSO, 0.5 μM of MS83, or 0.5 μM of MS83N1 for 48 h before they were harvested for TMT-based mass spectrometry analysis. Volcano plots of the −log 10 (p value) versus the log 2-fold change are displayed for (A) MS83 versus DMSO and (B) MS83 versus MS83N1. Several proteins that were significantly downregulated by the MS83 treatment are labeled in blue (FDR = 0.01). P values were calculated from the data of two technical replicates with two-sample t test statistics.
2.5. Antiproliferative Activity of MS83 in TNBC Cells.
The Western blotting (Figure 2C) and qRT-PCR (Figure S2) results suggest that MS83 could suppress cell proliferation. Therefore, we next evaluated the antiproliferative activity of MS83 in MDA-MB-468 and MDAMB-231 cells using the WST-8 cell viability assay (Figure 5A,B). Indeed, MS83 displayed a potent antiproliferation effect in both cell lines in a concentration-dependent manner (GI50 = 280 ± 48 nM in MDA-MB-468 cells; GI50 = 130 ± 30 nM in MDA-MB-231 cells). It exhibited more than 3-fold improved potency over MS83N1 (GI50 = 1000 ± 74 nM in MDA-MB-468 cells; GI50 = 490 ± 30 nM in MDA-MB-231 cells). MS83 also showed 3- and 17-fold higher potency than dBET1 (GI50 = 1200 ± 150 nM in MDA-MB-468 cells; GI50 = 2300 ± 210 nM in MDA-MB-231 cells) in the two cell lines, respectively. Among the compounds tested, JQ1, which is a small-molecule inhibitor of BET proteins, was the most potent in inhibiting the growth in both cell lines (GI50 = 250 ± 48 nM in MDA-MB-468 cells; GI50 = 8 ± 3 nM in MDA-MB-231 cells), likely due to that it has better cell permeability than MS83 and MS83N1. Compared with MS83, MS83A was much less potent in suppressing cell proliferation in both cell lines, consistent with its weak degradation effect of BRD4. The KEAP1 ligand KEAP1-L-OEt had no effect on cell proliferation, suggesting that the KEAP1 binding portion of MS83 does not contribute to the antiproliferative activity of MS83. In addition, we found that MS83N2 displayed little or no effect on suppressing cell proliferation (Figure S13), further suggesting that the antiproliferative activity of MS83 is not due to its KEAP1 ligand and linker portions. We next extended the compound treatment time to 8 days to observe the effect of MS83 on cell colony formation using crystal violet staining (Figure 5C,D). Consistent with the results from the cell viability assay, MS83 showed a much more potent inhibitory effect than the negative control MS83N1 and dBET1 on the formation of cell colonies in both MDA-MB-468 and MDA-MB-231 cells. These results demonstrate that the antiproliferative effect of MS83 is largely due to its ability to degrade, but not its ability to inhibit, BRD4, as MS83 and MS83N1 have similar BRD4 inhibitory activity due to the same JQ1 portion of both molecules. In summary, these results establish the capability of the KEAP1-recruiting BRD4/3 degrader MS83 to inhibit TNBC cell proliferation effectively and its superior potency compared to the CRBN-recruiting BRD4/3/2 degrader dBET1.
Figure 5.
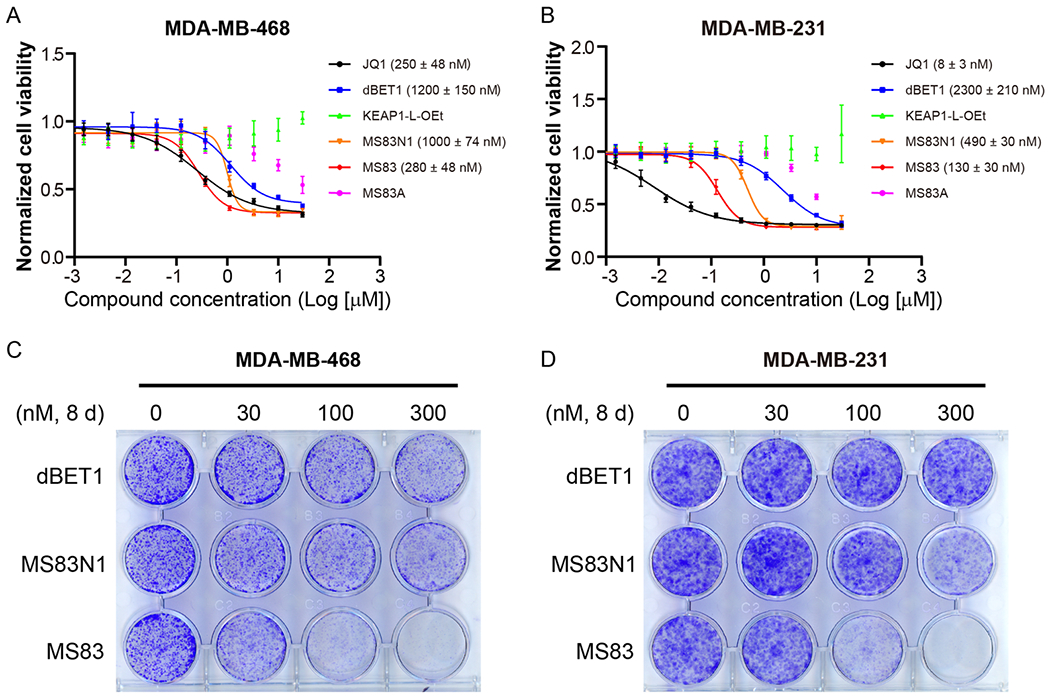
MS83 effectively suppresses proliferation of TNBC cells. Cell viability assays using the WST-8 reagent were conducted in MDA-MB-468 (A) and MDA-MB-231 (B) cells after a 3 d treatment with DMSO or indicated compounds with 3-fold dilutions. GraphPad Prism 8 was used in analysis of raw data. For each concentration point, in triplicate, mean value ± SD from three independent experiments is shown in the curves. GI50 values of the indicated compounds are shown in the plots. Clonogenic assays were conducted in MDA-MB-468 (C) and MDA-MB-231 (D) cells treated for 8 d with DMSO or 30, 100, or 300 nM of indicated compounds. Cells were then fixed and stained with crystal violet. The images are representatives of three independent experiments.
3. CONCLUSION
In this study, we demonstrated that the E3 ligase KEAP1 can be harnessed for PROTAC development using a highly selective and noncovalent ligand of KEAP1. We discovered a proof-of-concept small-molecule PROTAC degrader, MS83, which is a novel KEAP1-recruiting PROTAC based on a previously reported KEAP1 small-molecule ligand that selectively binds the KEAP1 kelch domain. MS83 hijacks the KEAP1-dependent CUL3 ligase complex (CRL3KEAP1), leading to polyubiquitination and subsequent degradation of BRD4 and BRD3, but not BRD2, by the 26S proteasome. In particular, we show that MS83 can effectively reduce the protein levels of BRD4 long and short isoforms in MDA-MB-468 cells in a concentration-, time-, KEAP1-, and UPS-dependent manner. We also show that MS83 degrades BRD4 and BRD3 more durably than the CRBN-recruiting BRD4/3/2 PROTAC dBET1 in MDA-MB-468 cells. As our MS-based quantitative global proteomics analysis demonstrates, MS83 is selective for BRD4 and BRD3 over several other bromodomain-containing proteins such as BRD1, BRD2, BRD7, BRD8, and BRD9. MS83 is also effective in degrading BRD4 and BRD3, but not BRD2, in MDA-MB-231 cells. Interestingly, in MDA-MB-231 cells, MS83 selectively degrades the BRD4 short isoform over the BRD4 long isoform. As the BRD4 short isoform is an important oncoprotein, while the BRD4 long isoform may have tumor suppressor functions, this unprecedented selectivity will likely be important and warrants further investigation. Furthermore, we generated two negative control compounds with structural similarity: MS83N1, which exhibits highly diminished KEAP1 binding but binds BRD4 effectively, and MS83N2, which retains the same KEAP1 binding moiety and linker but does not bind BRD4 effectively. We show that MS83N1 and MS83N2 did not induce significant degradation of BRD4 and BRD3. Moreover, our KEAP1-recuiting BRD4/3 PROTAC MS83 effectively inhibited TNBC cell proliferation and was more potent than the CRBN-recruiting BRD4/3/2 PROTAC dBET1. This could be due to that MS83 degrades BRD4 and BRD3 more durably than dBET1. Taken together, our results show that the KEAP1 E3 ligase can be exploited for developing effective PROTACs using a selective small-molecule KEAP1 ligand, which significantly enhances general applicability of the KEAP1-based PROTAC approach. This study expands the toolbox for targeted protein degradation.
4. EXPERIMENTAL METHODS
4.1. Compound Synthesis.
Synthesis and characterization of MS83, MS83A, MS83N1, MS83N2, and compounds 2–9 as well as the ligand KEAP1-L-OEt and intermediates are detailed in the Supporting Information.
4.2. Cell Culture.
MDA-MB-468 and MDA-MB-231 cells29 were cultivated in DMEM medium supplemented with 10% FBS, 10 μg/mL of streptomycin, and 100 units/mL of penicillin.
4.3. Western Blotting.
Cells were collected and lysed on ice for 30 min in lysis buffer (20 mM HEPES, pH 7.4, 150 mM NaCl, 2 mM EDTA, and 1% Triton X-100), supplemented with EDTA-free phosphatase and protease inhibitor. The samples then were centrifuged at 14,000g for 10 min at 4 °C to get the supernatant as cell lysate. The Pierce rapid gold BCA protein assay kit was used for the measurement of protein concentrations. The primary antibodies used were BRD2 (5848S, CST), BRD3 (ab50818, Abcam), BRD4 (ab128874, Abcam), c-MYC (5605S, CST), KEAP1 (sc-365626, Santa Cruz), β-actin (A4700, Sigma-Aldrich), and α-tubulin (T6074, Sigma-Aldrich). The secondary antibodies were fluorescence-labeled IRDye 800CW goat antirabbit IgG (926-32211, LI-COR) and IRDye 680CW donkey antimouse IgG (926-68072, LI-COR). Protein signals were detected by OdysseyCLx imaging system (LI-COR) and then analyzed by Image Studio Lite software (LI-COR).
4.4. KEAP1 Protein Purification.
KEAP1 kelch domain (residues 321–609) was synthesized into pET15b vector with a N-terminal His tag by Genscript. The synthesized plasmid was expressed in E. coli RIPL cells with induction by 0.5 mM IPTG at 16 °C for 18 h. Induced cultures were harvested, and the pellets were resuspended in cold lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 25 mM imidazole, 0.001% IGEPAL, 7 mM β- mercaptoethanol and 1 mM AEBSF). Cells were lysed using a sonicator, and the lysate was clarified by centrifugation at 18,000g at 4 °C for 60 min. The supernatant was filtered with 0.45 μm filter and then loaded onto a 5 mL HisTrap HP column (GE Healthcare), equilibrated with the wash buffer (same buffer as lysis buffer, without AEBSF). The column was then washed with wash buffer, and protein was eluted using a 30–300 mM imidazole gradient. The eluted fractions were analyzed using SDS-PAGE, pooled, concentrated, and further purified by size exclusion chromatography using a 320 mL HiLoad 26/600 Superdex 200 pg column (GE Healthcare) under the buffer condition (50 mM HEPES pH 7.5, 150 mM NaCl, and 1 mM TCEP). The eluted fractions were first analyzed using SDS-PAGE, pooled accordingly, concentrated to 69 mg/mL, and stored in aliquots in −80 °C freezer for further usage.
4.5. Isothermal Titration Calorimetry.
ITC experiments were performed on MicroCal iTC200 (Malvern Instruments Ltd.) at 25 °C in 50 mM HEPES pH 7.5, 150 mM NaCl and 1 mM TCEP, 5% DMSO. Lyophilized compounds (KEAP1-L, MS83A, MS83N1) were dissolved in DMSO as a stock solution and then diluted to the appropriate concentrations using the same buffer, matching the DMSO concentrations between the protein and the compound samples. The solution of KEAP1 (25 μM) was titrated with the test compound (250 μM) under 19 injections (2 μL per injection) at a 3 min interval, stirring at 600 rpm. Data analysis was performed using Microcal Origin 7.0 (Malvern). The reported values represent the mean ± SD from two independent measurements.
4.6. qRT-PCR.
RNA was isolated from MDA-MB-468 cells after compound treatment using RNeasy Mini Kit (74104, Qiagen). A 1.5–2 μg of RNA for each sample was used for reverse transcription using SuperScript Reverse Transcriptase III (18080-051, Life technologies). cDNA was diluted 1:50, and then 1 μL used as template for qRT-PCR in 10 μL of reaction system with Power SYBR Green PCR Master Mix (4367659, Applied Biosystems) in triplicate. GraphPad Prism 8 was used for the analysis of quantification from the data of three independent experiments. The following primers were used:
GAPDH (F): GCGAGATCCCTCCAAAATCAA
GAPDH (R): GTTCACACCCATGACGAACAT
BRD4 (F): AGCGCTATGTCACCTCCTGT
BRD4 (R): GTTTCGGAGTCTTCGCTGTC
c-MYC (F): GCTGCTTAGACGCTGGATTT
c-MYC (R): CACCGAGTCGTAGTCGAGGT
CDKN1A (F): TGGACCTGTCACTGTCTTG
CDKN1A (R): GGCTTCCTCTTGGAGAAGAT
4.7. shRNA-Mediated KEAP1 Knockdown.
HEK293T cells were used to transfect the plasmids (PMD(VSVG)/pCMVΔ8.2/pLKO.1) for lentivirus packaging. 48 h later, viruses were harvested and filtered with 0.45 μm membrane filter. Then appropriate volumes of viruses with 10 μ/mL of Polybrene (TR-1003, Sigma-Aldrich) were incubated with MDA-MB-468 cells. After 24 h transduction, the medium was changed with fresh full-medium containing 2 μ/mL of puromycin (P8833, Sigma-Aldrich) for 48 h selection, and then cells expressing shControl or shKEAP1 (TRCN0000156676, Sigma-Aldrich) were digested and seeded into 6-well plates. After 20 h, 0.1% of DMSO or 0.5 μM of MS83 was added into the medium for another 24 h. Then cells were collected for Western blotting analysis.
4.8. Endogenous Immunoprecipitation.
2 ×106 of MDA-MB-468 cells were seeded in 10 cm dish. After 20–24 h, cells were treated with 0.1% DMSO, 0.5 μM of MS83N1, or 0.5 μM of MS83 for 18 h. Cells were then washed with ice-cold PBS twice and centrifuged at 200g, 3 min to get cell pellets which were treated with 500 μL of lysis buffer (20 mM HEPES, pH 7.4, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, and EDTA-free phosphotase and protease inhibitor) for 30 min on ice, then centrifuged at 14,000g, 10 min to get supernatant as cell lysate. 100 μL of lysates was used as input, then the lysates remnant were divided into two equal aliquots. Each 200 μL of lysate was incubated with 3 μg of BRD4 antibody (A301-985A100, Bethyl Laboratories) or control rabbit IgG (2729S, CST, 1 mg/mL) at 4 °C overnight followed by incubation with 30 μL of Dynabeads Protein G (10003D, Thermo Fisher Scientific) for 3 h. After washed 3 times with cell lysis buffer containing 0.05% NP-40, immunoprecipitates were resuspended in 60 μL of 1 × SDS-PAGE Laemmli buffer for boil. The protein samples were then tested by Western blotting.
4.9. Proteomics Studies.
4.9.1. Reagents.
All chemicals used were HPLC-grade unless otherwise indicated. Trypsin was purchased from Promega. TMT10plex Isobaric Label Reagent was purchased from Thermo fisher (cat. 90110).
4.9.2. TMT-Based Proteomics Global Profiling Sample Preparation.
The cell pellets were resuspended in 8 M urea, 50 mM Tris-HCl pH 8.0, followed by 30 min, room temperature reduction with dithiothreitol (5 mM final), and a 45 min, room temperature alkylation with iodoacetamide (15 mM final) in the dark. The 4-fold dilution of samples in 25 mM Tris-HCl pH 8.0, 1 mM CaCl2 was then digested at room temperature overnight, with trypsin at a 1:100 (w/w, trypsin: protein) ratio. Peptides were desalted on homemade C18 stagetips. TMT reagent was used to label 100 μg of each peptide sample following manufacturer’s protocol. Next, this labeled peptide mixture was desalted and fractionated into 24 fractions in 10 mM trimethylammonium bicarbonate buffer containing 5–40% acetonitrile.
4.9.3. Mass Spectrometry Analysis.
A mixture of 0.1% formic acid and 2% acetonitrile was used to dissolve dried peptides. Peptide concentration was measured for global profiling samples, utilizing the Pierce Quantitative Colorimetric Peptide Assay (Thermofisher). Analysis of 0.5 μg of each fraction was performed on a Q-Exactive HF-X coupled with an Easy nanoLC 1200 (Thermo Fisher Scientific, San Jose, CA). Peptides were loaded on to a nanoEase MZ HSS T3 column (100 Å, 1.8 μm, 75 μm × 150 mm, Waters). A 100 min gradient was used to achieve analytical separation of all peptides. A linear gradient (over 5 min) of 5–10% buffer B, (over 70 min), of 10–31% buffer B, and (over 15 min) of 31–75% buffer B was executed at a 300 nL/min flow rate followed a ramp in 1 min to 100% B and 9 min wash with 100% B (buffer A: aqueous 0.1% formic acid, and buffer B: 80% acetonitrile and 0.1% formic acid). LC-MS experiments were also carried out in a data-dependent mode with full MS (externally calibrated to a mass accuracy of <5 ppm and a resolution of 120,000 for TMT-labeled samples at m/z 200) followed by high-energy collision-activated dissociation MS/MS (resolution of 45,000 for TMT-labeled global samples at m/z 200). Peptides were dissociated at a normalized collision energy of 32 eV (for TMT-labeled sample) in the presence of nitrogen bath gas atoms by using high-energy collision-activated dissociation MS/MS. Dynamic exclusion was 45 or 20 s.
4.9.4. Raw Proteomics Data Processing and Analysis.
The MaxQuant software version 1.6.10.43 (Max Planck Institute, Germany) was used for mass spectra processing and peptide identification. All protein database searches were performed against the UniProt human protein sequence database (UP000005640). A false discovery rate (FDR) for both peptide-spectrum match and protein assignment were set at 1%. Search parameters included protein N-terminal acetylation as a dynamic modification and up to two missed cleavages at Lys/Arg on the sequence as well as oxidation of methionine. Carbamidomethylation of cysteine residues was considered as a static modification. Peptide identifications are reported by filtering of reverse and contaminant entries and assigning to their leading razor protein. The TMT reporter intensity found in MaxQuant was for quantitation. Perseus (Version 1.6.10.50) was used to process the data and perform statistical analyses. TMT reporter intensity from MaxQuant was used to perform protein quantitation. A statistically significant protein abundance fold-change (30%) was determined with a two-sample t test statistics on two technical replicates with a p-value of 1% to report.
4.10. Cell Viability Assay.
MDA-MB-468 or MDA-MB-231 cells (1500 or 2000 cells per well) were seeded into 96-well microplates. After 20 h, cells were treated with 3-fold serially diluted compounds in triplicate for 3 d. Cell viability was evaluated using WST-8 reagent (CK04, Dojindo). Absorbance signals were obtained with Infinite F PLEX plate reader (TECAN, Morrisville, NC) at 450 nm with 690 nm as reference wavelength after 3 h incubation at 37 °C. GraphPad Prism 8 was used in the analysis of GI50 values from the data of three independent experiments.
4.11. Clonogenic Assay.
MDA-MB-468 or MDA-MB-231 cells (2500 cells per well) were seeded into 12-well tissue culture plates. After 20 h, cells were treated with 0.1% DMSO or diluted concentrations of indicated compounds for 8 d. Cell medium was changed with fresh full medium containing indicated compounds every 3 d. The plates were then washed with PBS and stained with the solution containing 0.5% (w/v) crystal violet and 6% (v/v) glutaraldehyde for 30 min. The plates were then washed with running water until cell colonies were clear without background color and dried at rt. Epson Perfection V600 Photo was used for the acquisition of the images.
4.12. AlphaScreen Assay.
Eight μL/well of assay buffer (50 mM HEPES, pH 7.5, 100 mM NaCl, 0.05% CHAPS, 0.1% BSA, 0.9% DMSO) was delivered into columns 1 and 13 without BRDs as control. With same volume, the assay buffer containing BRDs were delivered into columns 2–12 and 14–24. Using an Echo liquid handler (Beckman Coulter, USA), compounds were serially diluted as 3-fold from 10 μM to 0.17 nM followed by spin and shake and preincubated for 30 min with gentle mixing. Then, substrate solution containing histone H4 peptide (1–21) K5/8/12/16Ac-biotin-OH was delivered into all wells as 4 μL/well. After incubation at room temperature for 30 min with gentle mixing under dark conditions, 4 μL of assay buffer containing AlphaScreen Ni-acceptor beads (final 20 μg/mL) and SA-donor beads (final 20 μg/mL) was dispensed into every well. Finally, the reaction mixture was incubated for 1 h at room temperature and then read by EnSpire (PerkinElmer, USA) using AlphaScreen mode.
4.13. Statistic Methods.
For all biological studies, at least two independent experiments were performed. The number of biologically independent experiments and technical replicates as well as error bars and P values for all biological data are described in the figure legends. Two-tailed Student’s t tests were utilized for the indicated analyses, respectively, with P ≥ 0.05, ns; 0.01 ≤ P < 0.05, *; 0.001 ≤ P < 0.01, **; P < 0.001, ***.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by the US National Institutes of Health grants R01CA218600, R01CA230854, R01CA260666, R01GM122749, and P30CA196521 and an endowed professorship from the Icahn School of Medicine at Mount Sinai to J.J. This work utilized the AVANCE NEO 600 MHz NMR spectrometer system that was upgraded with funding from a National Institutes of Health SIG grant 1S10OD025132-01A1. We thank Meng Cheng for helpful discussions on the TCGA analysis.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c04841.
The synthetic procedures and the characterization of all novel compounds; 1H NMR and 13C NMR spectra of MS83, MS83A, MS83N1, MS83N2, MS83N2A, and KEAP1-L-OEt; supplemental Figures S1–S13, Tables S1 and S2, and Schemes S1, S2, and S3 are included (PDF)
The authors declare the following competing financial interest(s): The Jin laboratory received research funds from Celgene Corporation, Levo Therapeutics, and Cullgen, Inc. J.J. is a co-founder, equity shareholder, and consultant of Cullgen, Inc.
Contributor Information
Jieli Wei, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Fanye Meng, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Kwang-Su Park, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Hyerin Yim, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Julia Velez, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Prashasti Kumar, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Li Wang, Department of Biochemistry and Biophysics, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
Ling Xie, Department of Biochemistry and Biophysics, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
He Chen, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Yudao Shen, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Emily Teichman, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Dongxu Li, Department of Biochemistry and Biophysics, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States; Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
Gang Greg Wang, Department of Biochemistry and Biophysics, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States; Lineberger Comprehensive Cancer Center, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
Xian Chen, Department of Biochemistry and Biophysics, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
H. Ümit Kaniskan, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Jian Jin, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
REFERENCES
- (1).Bondeson DP; Mares A; Smith IE; Ko E; Campos S; Miah AH; Mulholland KE; Routly N; Buckley DL; Gustafson JL; Zinn N; Grandi P; Shimamura S; Bergamini G; Faelth-Savitski M; Bantscheff M; Cox C; Gordon DA; Willard RR; Flanagan JJ; Casillas LN; Votta BJ; den Besten W; Famm K; Kruidenier L; Carter PS; Harling JD; Churcher I; Crews CM Catalytic in vivo Protein Knockdown by Small-Molecule PROTACs. Nat. Chem. Biol 2015, 11 (8), 611–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Winter GE; Buckley DL; Paulk J; Roberts JM; Souza A; Dhe-Paganon S; Bradner JE Phthalimide Conjugation as A Strategy for in vivo Target Protein Degradation. Science 2015, 348 (6241), 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Bondeson DP; Smith BE; Burslem GM; Buhimschi AD; Hines J; Jaime-Figueroa S; Wang J; Hamman BD; Ishchenko A; Crews CM Lessons in PROTAC Design from Selective Degradation with A Promiscuous Warhead. Cell Chem. Biol 2018, 25 (1), 78–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Schapira M; Calabrese MF; Bullock AN; Crews CM Targeted Protein Degradation: Expanding the Toolbox. Nat. Rev. Drug Discovery 2019, 18 (12), 949–963. [DOI] [PubMed] [Google Scholar]
- (5).Ishida T; Ciulli A E3 Ligase Ligands for PROTACs: How They Were Found and How to Discover New Ones. SLAS Discovery 2021, 26 (4), 484–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Dale B; Cheng M; Park K-S; Kaniskan HÜ; Xiong Y; Jin J Advancing Targeted Protein Degradation for Cancer Therapy. Nat. Rev. Cancer 2021. DOI: 10.1038/s41568-021-00365-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Genschik P; Sumara I; Lechner E The Emerging Family of CULLIN3-RING Ubiquitin Ligases (CRL3s): Cellular Functions and Disease Implications. EMBO J. 2013, 32 (17), 2307–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Zhang DD; Lo SC; Cross JV; Templeton DJ; Hannink M Keap1 is A Redox-Regulated Substrate Adaptor Protein for A Cul3-Dependent Ubiquitin Ligase Complex. Mol. Cell. Biol 2004, 24 (24), 10941–10953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Lee S; Hu L Nrf2 Activation through the Inhibition of Keap1-Nrf2 Protein-Protein Interaction. Med. Chem. Res 2020, 29 (5), 846–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Pallesen JS; Tran KT; Bach A Non-covalent Small-Molecule Kelch-like ECH-Associated Protein 1-Nuclear Factor Erythroid 2-Related Factor 2 (Keap1-Nrf2) Inhibitors and Their Potential for Targeting Central Nervous System Diseases. J. Med. Chem 2018, 61 (18), 8088–8103. [DOI] [PubMed] [Google Scholar]
- (11).Cleasby A; Yon J; Day PJ; Richardson C; Tickle IJ; Williams PA; Callahan JF; Carr R; Concha N; Kerns JK; Qi H; Sweitzer T; Ward P; Davies TG Structure of the BTB Domain of Keap1 and its Interaction with the Triterpenoid Antagonist CDDO. PLoS One 2014, 9 (6), No. e98896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Honda T; Rounds BV; Gribble GW; Suh N; Wang Y; Sporn MB Design and Synthesis of 2-Cyano-3,12-dioxoolean-1,9-dien-28-oic Acid, A Novel and Highly Active Inhibitor of Nitric Oxide Production in Mouse Macrophages. Bioorg. Med. Chem. Lett 1998, 8 (19), 2711–2714. [DOI] [PubMed] [Google Scholar]
- (13).Meng X; Waddington JC; Tailor A; Lister A; Hamlett J; Berry N; Park BK; Sporn MB CDDO-Imidazolide Targets Multiple Amino Acid Residues on the Nrf2 Adaptor, Keap1. J. Med. Chem 2020, 63 (17), 9965–9976. [DOI] [PubMed] [Google Scholar]
- (14).Tong KI; Katoh Y; Kusunoki H; Itoh K; Tanaka T; Yamamoto M Keap1 Recruits Neh2 through Binding to ETGE and DLG Motifs: Characterization of the Two-Site Molecular Recognition Model. Mol. Cell. Biol 2006, 26 (8), 2887–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Lu M; Liu T; Jiao Q; Ji J; Tao M; Liu Y; You Q; Jiang Z Discovery of A Keap1-Dependent Peptide PROTAC to Knockdown Tau by Ubiquitination-Proteasome Degradation Pathway. Eur. J. Med. Chem 2018, 146, 251–259. [DOI] [PubMed] [Google Scholar]
- (16).Tong B; Luo M; Xie Y; Spradlin JN; Tallarico JA; McKenna JM; Schirle M; Maimone TJ; Nomura DK Bardoxolone Conjugation Enables Targeted Protein Degradation of BRD4. Sci. Rep 2020, 10 (1), 15543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Yore MM; Kettenbach AN; Sporn MB; Gerber SA; Liby KT Proteomic Analysis Shows Synthetic Oleanane Triterpenoid Binds to mTOR. PLoS One 2011, 6 (7), No. e22862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Davies TG; Wixted WE; Coyle JE; Griffiths-Jones C; Hearn K; McMenamin R; Norton D; Rich SJ; Richardson C; Saxty G; Willems HM; Woolford AJ; Cottom JE; Kou JP; Yonchuk JG; Feldser HG; Sanchez Y; Foley JP; Bolognese BJ; Logan G; Podolin PL; Yan H; Callahan JF; Heightman TD; Kerns JK Monoacidic Inhibitors of the Kelch-Like ECH-Associated Protein 1: Nuclear Factor Erythroid 2-Related Factor 2 (KEAP1:NRF2) Protein-Protein Interaction with High Cell Potency Identified by Fragment-Based Discovery. J. Med. Chem 2016, 59 (8), 3991–4006. [DOI] [PubMed] [Google Scholar]
- (19).Heightman TD; Callahan JF; Chiarparin E; Coyle JE; Griffiths-Jones C; Lakdawala AS; McMenamin R; Mortenson PN; Norton D; Peakman TM; Rich SJ; Richardson C; Rumsey WL; Sanchez Y; Saxty G; Willems HMG; Wolfe L; Woolford AJA; Wu ZN; Yan HX; Kerns JK; Davies TG Structure-Activity and Structure-Conformation Relationships of Aryl Propionic Acid Inhibitors of the Kelch-like ECH-Associated Protein 1/Nuclear Factor Erythroid 2-Related Factor 2 (KEAP1/NRF2) Protein-Protein Interaction. J. Med. Chem 2019, 62 (9), 4683–4702. [DOI] [PubMed] [Google Scholar]
- (20).Chaidos A; Caputo V; Karadimitris A Inhibition of Bromodomain and Extra-Terminal Proteins (BET) as A Potential Therapeutic Approach in Haematological Malignancies: Emerging Preclinical and Clinical Evidence. Ther. Adv. Hematol 2015, 6 (3), 128–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Hines J; Lartigue S; Dong H; Qian Y; Crews CM MDM2-Recruiting PROTAC Offers Superior, Synergistic Antiproliferative Activity via Simultaneous Degradation of BRD4 and Stabilization of p53. Cancer Res. 2019, 79 (1), 251–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Donati B; Lorenzini E; Ciarrocchi A BRD4 and Cancer: Going Beyond Transcriptional Regulation. Mol. Cancer 2018, 17 (1), 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Bai L; Zhou B; Yang CY; Ji J; McEachern D; Przybranowski S; Jiang H; Hu J; Xu F; Zhao Y; Liu L; Fernandez-Salas E; Xu J; Dou Y; Wen B; Sun D; Meagher J; Stuckey J; Hayes DF; Li S; Ellis MJ; Wang S Targeted Degradation of BET Proteins in Triple-Negative Breast Cancer. Cancer Res. 2017, 77 (9), 2476–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Pettersson M; Crews CM Proteolysis TArgeting Chimeras (PROTACs) - Past, Present and Future. Drug Discovery Today: Technol. 2019, 31, 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Delmore JE; Issa GC; Lemieux ME; Rahl PB; Shi J; Jacobs HM; Kastritis E; Gilpatrick T; Paranal RM; Qi J; Chesi M; Schinzel AC; McKeown MR; Heffernan TP; Vakoc CR; Bergsagel PL; Ghobrial IM; Richardson PG; Young RA; Hahn WC; Anderson KC; Kung AL; Bradner JE; Mitsiades CS BET Bromodomain Inhibition as A Therapeutic Strategy to Target c-Myc. Cell 2011, 146 (6), 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wu SY; Lee CF; Lai HT; Yu CT; Lee JE; Zuo H; Tsai SY; Tsai MJ; Ge K; Wan Y; Chiang CM Opposing Functions of BRD4 Isoforms in Breast Cancer. Mol. Cell 2020, 78 (6), 1114–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Han X; Yu D; Gu R; Jia Y; Wang Q; Jaganathan A; Yang X; Yu M; Babault N; Zhao C; Yi H; Zhang Q; Zhou MM; Zeng L Roles of the BRD4 Short Isoform in Phase Separation and Active Gene Transcription. Nat. Struct. Mol. Biol 2020, 27 (4), 333–341. [DOI] [PubMed] [Google Scholar]
- (28).Filippakopoulos P; Qi J; Picaud S; Shen Y; Smith WB; Fedorov O; Morse EM; Keates T; Hickman TT; Felletar I; Philpott M; Munro S; McKeown MR; Wang Y; Christie AL; West N; Cameron MJ; Schwartz B; Heightman TD; La Thangue N; French CA; Wiest O; Kung AL; Knapp S; Bradner JE Selective Inhibition of BET Bromodomains. Nature 2010, 468 (7327), 1067–7103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Ma A; Stratikopoulos E; Park KS; Wei J; Martin TC; Yang X; Schwarz M; Leshchenko V; Rialdi A; Dale B; Lagana A; Guccione E; Parekh S; Parsons R; Jin J Discovery of A First-in-Class EZH2 Selective Degrader. Nat. Chem. Biol 2020, 16 (2), 214–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.