

SUMMARY
The power of Drosophila melanogaster as a model system relies on tractable germline genetic manipulations. Despite Drosophila’s expansive genetics toolbox, such manipulations are still accomplished one change at a time and depend predominantly on phenotypic screening. We describe a drug-based genetic platform consisting of four selection and two counterselection markers, eliminating the need to screen for modified progeny. These markers work reliably individually or in combination to produce specific genetic outcomes. We demonstrate three example applications of multiplexed drug-based genetics by generating (1) transgenic animals, expressing both components of binary overexpression systems in a single transgenesis step; (2) dual selectable and counterselectable balancer chromosomes; and (3) selectable, fluorescently tagged P[acman] bacterial artificial chromosome (BAC) strains. We perform immunoprecipitation followed by proteomic analysis on one tagged BAC line, demonstrating our platform’s applicability to biological discovery. Lastly, we provide a plasmid library resource to facilitate custom transgene design and technology transfer to other model systems.
Graphical Abstract
In brief
Matinyan et al. describe a set of four drug selection and two drug counterselection markers for use in Drosophila genetics. The orthogonal markers can be used individually or combined for complex, multiplex genetic manipulations. Use of drug selection and counterselection eliminates screening efforts for modified animals, saving time and cost.
INTRODUCTION
Genetic engineering technologies in Drosophila melanogaster have greatly advanced the study of basic biology and human disease (Bier, 2005; Venken and Bellen, 2007, 2014; Venken et al., 2016; Bellen et al., 2019; Link and Bellen, 2020). Historically, germline genetic manipulations in fruit flies, e.g., insertional mutagenesis and transgenesis, almost exclusively relied on P element transposons (Ryder and Russell, 2003). They were either remobilized from one location in the genome to another (Cooley et al., 1988) or jumped into the genome from a microinjected plasmid (Rubin and Spradling, 1982). Transposon-based mutagenesis and transgenesis have largely been replaced by CRISPR-Cas9-mediated genome editing (Gratz et al., 2014; Bier et al., 2018) and ΦC31-mediated, site-specific integration (Groth et al., 2004; Bischof et al., 2007), respectively.
Introducing tractable genetic modifications into flies starts with microinjecting a plasmid (Venken and Bellen, 2007). This plasmid carrying a transgene coupled to a dominant physical marker is injected into early-stage, syncytial embryos targeting the future germline in a genetic background deficient for the marker (Venken and Bellen, 2007). Adult flies that survive the injection process may have transformed germ cells that upon crossing will produce offspring identifiable by marker expression (Figure S1A). The most commonly used dominant markers are the eye color markers, white+ and vermillion+, and the body color marker, yellow+ (Venken and Bellen, 2007). Screening genetically modified progeny can be time consuming and laborious (Venken et al., 2006, 2009, 2010; Venken and Bellen, 2007; Dahmann, 2008; Beumer and Carroll, 2014; Bier et al., 2018).
To eliminate the workload of screening, a drug-based selection approach can be used instead (Figure S1B; Steller and Pirrotta, 1985; Handler and O’Brochta, 1991; Semple et al., 2010; Giordano-Santini and Dupuy, 2011; Kandul et al., 2020). Selection markers confer resistance to universally toxic antibiotics, killing non-transgenic progeny but allowing transgenic, drug-resistant animals to survive (Steller and Pirrotta, 1985). As these markers are exogenous to the fly, drug-based selection can be used in any genetic background (Steller and Pirrotta, 1985), in contrast to many physical screening markers that require marker null allele genotypes (Venken and Bellen, 2007). Fluorescent markers are also exogenous and dominantly expressed, but they still require screening and can interfere with downstream analyses (e.g., fluorescence microscopy; Horn et al., 2002; Venken and Bellen, 2007). Selectable markers have been most successfully used in the nematode Caenorhabditis elegans, where several exogenous drug-resistance markers have been found effective (Giordano-Santini et al., 2010; Semple et al., 2010; Radman et al., 2013; Kim et al., 2014). In the fruit fly, drug-based selection was implemented shortly after the advent of P-element-based transgenesis (Rubin and Spradling, 1982; Steller and Pirrotta, 1985) but was largely abandoned due to variability of marker-conferred drug resistance, likely due to the inherent randomness of P element transposon integration (Steller and Pirrotta, 1985; Handler and O’Brochta, 1991). Selection strategies using orthogonal selectable marker genes would greatly expand the utility of this technology and provide opportunities for multiplexed selectable genetic engineering applications.
Similarly, counterscreening against an undesired outcome of a genetic manipulation depends on dominant physical or fluorescent markers. These are coupled to an unwanted genotype, and counterscreening against them can be equally laborious as screening (Figure S2A). Drug sensitivity markers eliminate counterscreening by counterselecting against those genotypes using the relevant counterselection agent instead (Figure S2B). Drug-sensitized animals will not survive, whereas non-sensitive progeny will be unaffected by drug exposure. In vivo animal counterselection, akin to counterselection in bacteria and cell culture, has been primarily used for efforts to control disease vector and agricultural pest insect populations by biasing genetic sexing (i.e., generation of purely male populations; Thomas et al., 2000; Flores and O’Neill, 2018; Kandul et al., 2020). Of the three most notable examples, two of the approaches use a two-component tetracycline-repressible expression system to drive female-specific expression of the proapoptotic gene head involution defective (hid) (Heinrich and Scott, 2000; Thomas et al., 2000). A third approach uses female-specific expression of the bacterial suicide gene cytosine deaminase (codA), sensitizing females to the nucleoside analog 5-fluorocytosine (Markaki et al., 2004). However, in vivo counterselection is not yet part of the basic genetic toolbox in Drosophila. Previous efforts have shown the efficacy of counterselectable chromosomes by coupling the cell death gene reaper (rpr) to a UAS element, which if crossed to a GAL4 source will result in the death of the resulting progeny (McMahan et al., 2013). However, this system precludes the use of the GAL4 binary expression system for other applications. Counterselection strategies using orthogonal counterselectable marker genes would further expand the utility of this application and make multiplex counterselectable genetic manipulations possible.
We describe a drug-based selection and counterselection platform for multiplexed genetic manipulations in Drosophila melanogaster that is readily transferrable to other model systems. We designed a compact expression cassette to test five drug-resistance and two drug-sensitivity markers. We successfully select drug-resistant transgenic animals with four of the five resistance markers using the drugs G418 sulfate, puromycin, blasticidin S, and hygromycin B. We also demonstrate effective counterselection against drug-sensitized animals using either ganciclovir or 5-fluorocytosine. By showing that marker-conferred drug resistance or sensitivity is specific to the corresponding drug, we combine multiple drugs in single, multiplexed genetic manipulations to perform co-selection, combination selection and counterselection, and co-counterselection, generating distinct genotypes. We also apply this platform to make double-transgenic animals in a single step, generate selectable and counterselectable balancer chromosomes, and make fluorescently tagged selectable P[acman] transgenics. We use one of the P[acman] transgenics to establish an unbiased interactome, demonstrating applicability of our platform for biological discovery. Finally, we provide a vector library resource to allow easy adoption of our selection and counterselection genetic platform, facilitating designs for custom applications as well as for adaptation to other model and non-model species.
RESULTS
Determining effective drug concentrations and specificity of selection and counterselection markers
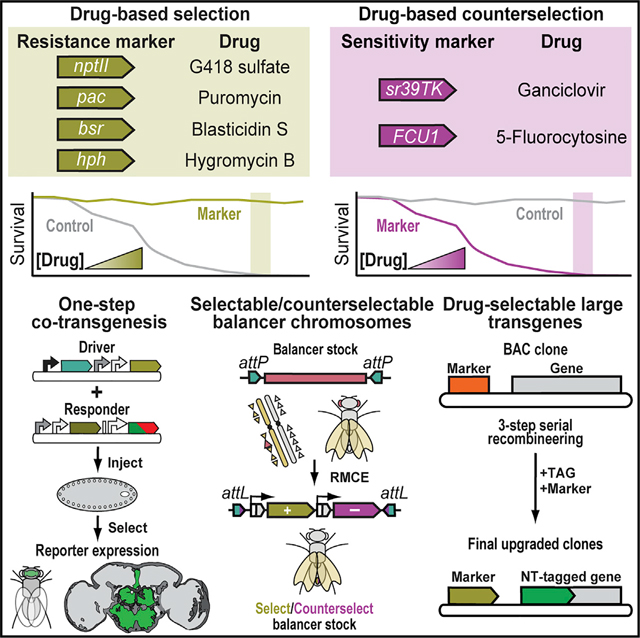
We developed a compact expression cassette using the same marker expression in bacteria for molecular cloning and in Drosophila for transgenesis (Figure 1A; see STAR Methods for details). We cloned five selection markers, two counterselection markers, and an EGFP control marker into our cassette (Table 1; Figure 1B). The five selection markers were neomycin phosphotransferase II (nptII), puromycin N-acetyltransferase (pac), blasticidin S-resistance (bsr), hygromycin B phosphotransferase (hph), and bleomycin resistance protein (ble). Their corresponding drugs are G418 sulfate, puromycin HCl, blasticidin S, hygromycin B, and zeocin or phleomycin. The two counterselection markers were thymidine kinase (sr39TK) and fusion protein of cytosine deaminase and uracilphosphoribosyltransferase (FCU1), used with ganciclovir and 5-fluorocytosine, respectively. We made transgenic fly strains for each marker, resulting in G418R, PuroR, BlastR, HygroR, ZeoR, GCVS, 5FCS, and EGFP flies, all inserted into the same genomic locus (Table 1; Figure 1B).
Figure 1. Determining effective drug concentrations and specificity of selection and counterselection markers.
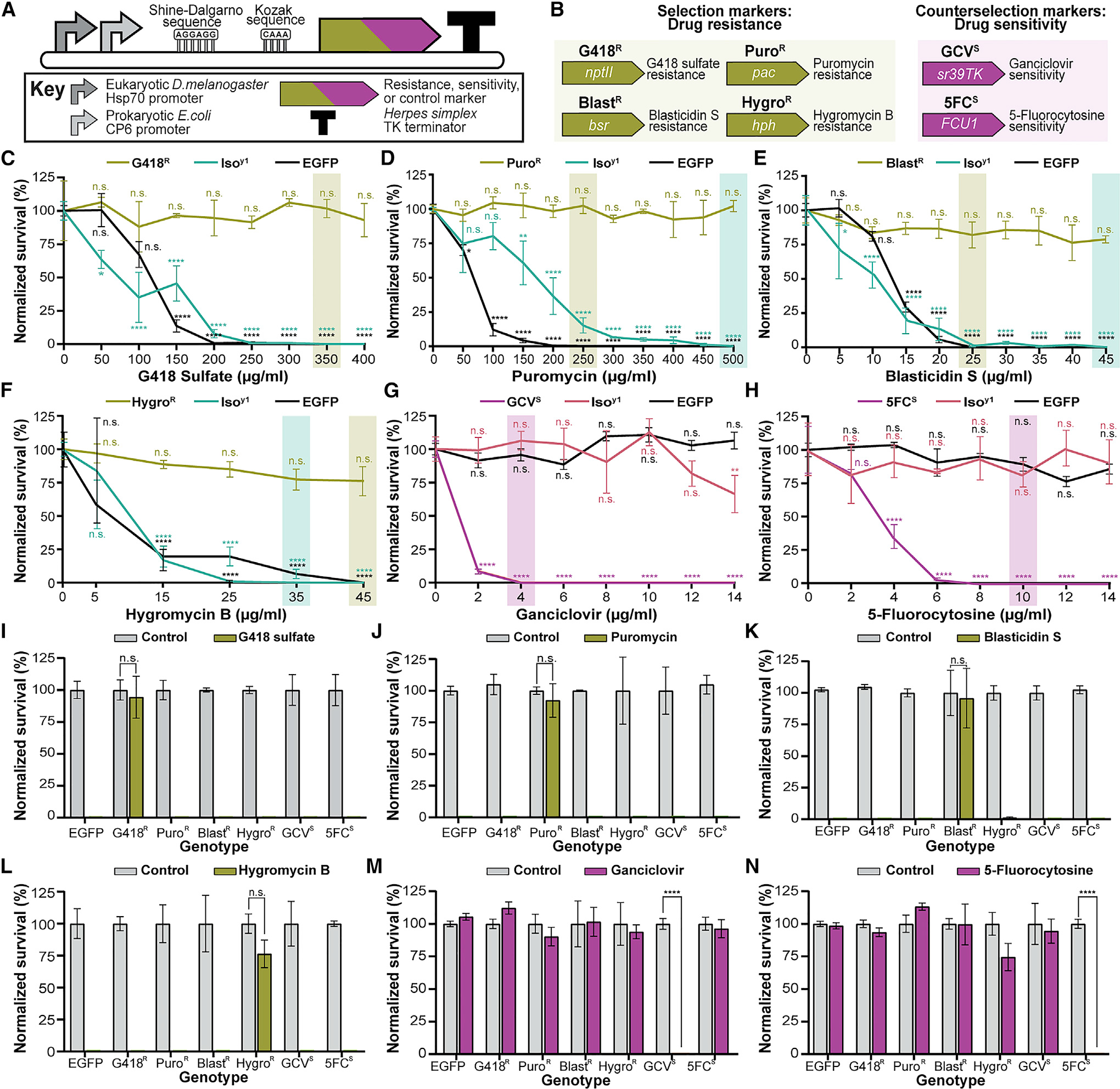
(A) Schematic of the marker expression cassette. The fusion promoter works in bacteria and flies. Marker expression can be enhanced in flies via heat shock. Expression-enhancing elements are indicated: a bacterial Shine-Dalgarno; a consensus Drosophila Kozak/Cavener sequence; and a TK poly(A) terminator.
(B) Schematic summary of markers (italics) and their corresponding drug resistance or sensitivity and strains (bold).
(C–H) Determining effective selection and counterselection concentrations for drug-based selection and counterselection in two genetic backgrounds.
(C–F) The effective selection concentration (ESC) is the concentration of drug at which the control, EGFP-expressing strain is eliminated while the drug-resistant strain is unaffected. ESC for G418 sulfate is 350 μg/mL (G418R) (C), 250 or 500 μg/mL for puromycin (PuroR) (D), 25 or 45 μg/mL for blasticidin S (BlastR) (E), and 65 or 45 μg/mL for hygromycin B (HygroR) (F).
(G and H) The effective counterselection concentration (ECC) is the drug concentration at which the sensitivity-marker-expressing strain is eliminated while the control EGFP-expressing fly strain survival is unaffected versus vehicle control. ECC for ganciclovir is 4 μg/mL (GCVS) (G) and 10 μg/mL for 5-fluorocytosine (5FCS) (H).
(I–L) Only correspondingly resistant fly strains survive drug treatment at determined ESC (see C–F) for G418 sulfate (I), puromycin (J), blasticidin S (K), and hygromycin B (L).
(M and N) Only the corresponding sensitive strain survival is affected by treatment with ganciclovir (M) or 5-fluorocytosine (N) at their respective ECC (Table 1).
Statistical significance for the survival curves was determined via two-way ANOVA using Dunnett’s multiple comparisons test for each strain on each drug compared to vehicle survival. Statistical significance of marker orthogonality was determined via multiple t test between untreated and treated vials of the same strain for each drug using the Holm-Sidak method. For both methods, α = 0.05, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, n.s. is non-significant, and data shown represent mean (Dunnett) or average (Holm-Sidak) and SEM for at least three biological replicates per condition. See also Figures S1–S3.
Table 1.
Summary of selection and counterselection markers
| Marker | Species origin | Encoded protein | Reference | Marker size (bp) | Drug(s)a | Solvent | Bacterial selection (μg/mL) | Drosophila size ESC/ECC (μg/mL) | Cost ($/vial) |
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| Resistance markers | |||||||||
| nptII | Klebsiella pneumoniae | neomycin phosphotransferase II | Davies and Smith, 1978 | 795 | kanamycin (bacteria), G418 sulfate (geneticin) | MQ H2O | 30 | 350 | 0.07 |
| pac | Streptomyces alboniger | puromycin N-acetyltransferase | Vara et al., 1985 | 600 | puromycin HCl | MQ H2O | 100 | 250–500 | 2.32– 4.64 |
| bsr b | Bacillus cereus | blasticidin S-resistance | Itaya et al., 1990 | 423 | blasticidin S | MQ H2O | 100 | 25–45 | 0.41– 0.73 |
| hph c | Escherichia coli | hygromycin B phosphotransferase | Gritz and Davies, 1983 | 1,026 | hygromycin B | MQ H2O | 75 | 35–45 | 0.04– 0.05 |
| ble d | Streptoalleteichus hindustanus | bleomycin resistance protein | Oliva-Trastoy et al., 2005 | 375 | zeocin or phleomycin | 1M HEPES | 25 or – | 350 | – or 9.35 |
| Sensitivity markers | |||||||||
| sr39TK e | herpes simplex virus-1 | thymidine kinase | Black et al., 2001 | 1,131 | ganciclovir | 0.1N NaOH | – | 4 | 0.01– 0.04 |
| FCU1 e | Saccharomyces cerevisiae | FCU1 | Erbs et al., 2000 | 1,122 | 5-fluorocytosine | 1×PBS | – | 10–15 | <0.01 |
The efficacy of seven different markers (five selection and two counterselection) were tested for in vivo selection and counterselection in Drosophila melanogaster. For each marker, we established an effective selection and counterselection concentration (ESC and ECC) defined as the minimal amount of drug required to either eliminate all non-resistant flies without significantly affecting resistance marker expressing fly viability or to eliminate all sensitivity-marker-expressing flies without significantly affecting non-sensitive flies. For the five selection markers, selection concentrations were also determined for use in bacteria.
Drugs used to determine cost were G418 sulfate (VWR 97063-060), puromycin HCl (VWR 97064-280), blasticidin S (VWR 71002-676), hygromycin B (VWR AAJ6068103), phleomycin (VWR AAJ67027-8EQ), ganciclovir (TCI America 50-155-694), and 5-fluorocytosine (TCI America 50-014-34810).
Although basal marker expression is enough to confer effective drug resistance when expressed from tested genomic docking sites, heat-shockenhanced expression is required when inserted into low-expressing genomic loci. Lower marker expression is also associated with blasticidin-S-related toxicity in resistant animals at higher concentrations (≥50 μg/mL).
Effective hygromycin B resistance requires heat-shock-enhanced expression in all tested contexts.
Zeocin was found ineffective at selecting flies. Phleomycin is a poorly effective selection agent, with high-ESC, batch-specific variability.
At higher concentrations, both tested counterselection drugs show general toxicity, with ganciclovir exhibiting toxicity above 15 μg/mL and 5-fluorocytosine above 50 μg/mL.
We next examined the survival of resistant, sensitive, and control marker strain flies on food treated with varying drug concentrations to determine their respective effective selection concentration (ESC) or effective counterselection concentration (ECC) (Figures 1C–1H). The ESC is the lowest drug concentration that eliminates all non-resistant flies without significantly affecting survival of the drug-resistant flies. Conversely, the ECC is the lowest drug concentration that eliminates all sensitized animals without significantly affecting control background fly survival. We carried out similar dose-response experiments using an isogenized fly strain (IsoY1) to determine the ESC and ECC dependency on genetic background (Figures 1C–1H). Except for the ble marker (ZeoR; Figure S3), we successfully determined an ESC or ECC for all drugs in both genetic backgrounds, but the effective concentrations varied somewhat between strains (Table 1). Hygromycin B was the only drug that required heat shocking during development to provide sufficient drug resistance (Figure 1F). Blasticidin S was toxic in the resistant strain at high concentrations, and both counterselection agents, ganciclovir and 5-fluorocytosine, also showed general toxicity above a certain concentration (Table 1). Importantly, drug exposure is limited to a single generation during selection or counterselection for the desired genetic event, after which animal maintenance occurs on regular food. We never observed defects in marker-expressing strains, their fecundity, or overall health on regular or drug-treated food. Although there does remain the potential for mutagenicity, the widespread and continued use of these drugs in mammalian cell culture without reported genetic defects (Eglitis, 1991; Mortensen and Kingston, 2009) bodes well for their use in Drosophila and other organisms.
To evaluate whether different markers can be combined, we determined the specificity of marker-conferred drug resistance and sensitivity by testing each marker-expressing strain and the EGFP control on each of the functional drugs and found that all markers are drug specific (Figures 1I–1N).
Multiplexed co-selection and/or co-counterselection produces genotypically pure populations
We next tested the robustness of combining multiple markers for co-selection, combination selection and counterselection, and co-counterselection (Figures 2A–2D, left, and S4). For each pair of marker heterozygotes, we set up crosses under four different drug conditions corresponding to the markers involved in the cross: vehicle control; drug 1; drug 2; and both drugs together. Each cross was set up twice, varying the sex of each marker heterozygote strain to account for potential parental effects on drug resistance or sensitivity (Figure S4). In each cross, there are four possible genotypes, whose proportions are genetically determined and modified by drug conditions (Figures 2A–2D, right, and S5).
Figure 2. Multiplexed co-selection and/or co-counterselection produces genotypically pure populations.
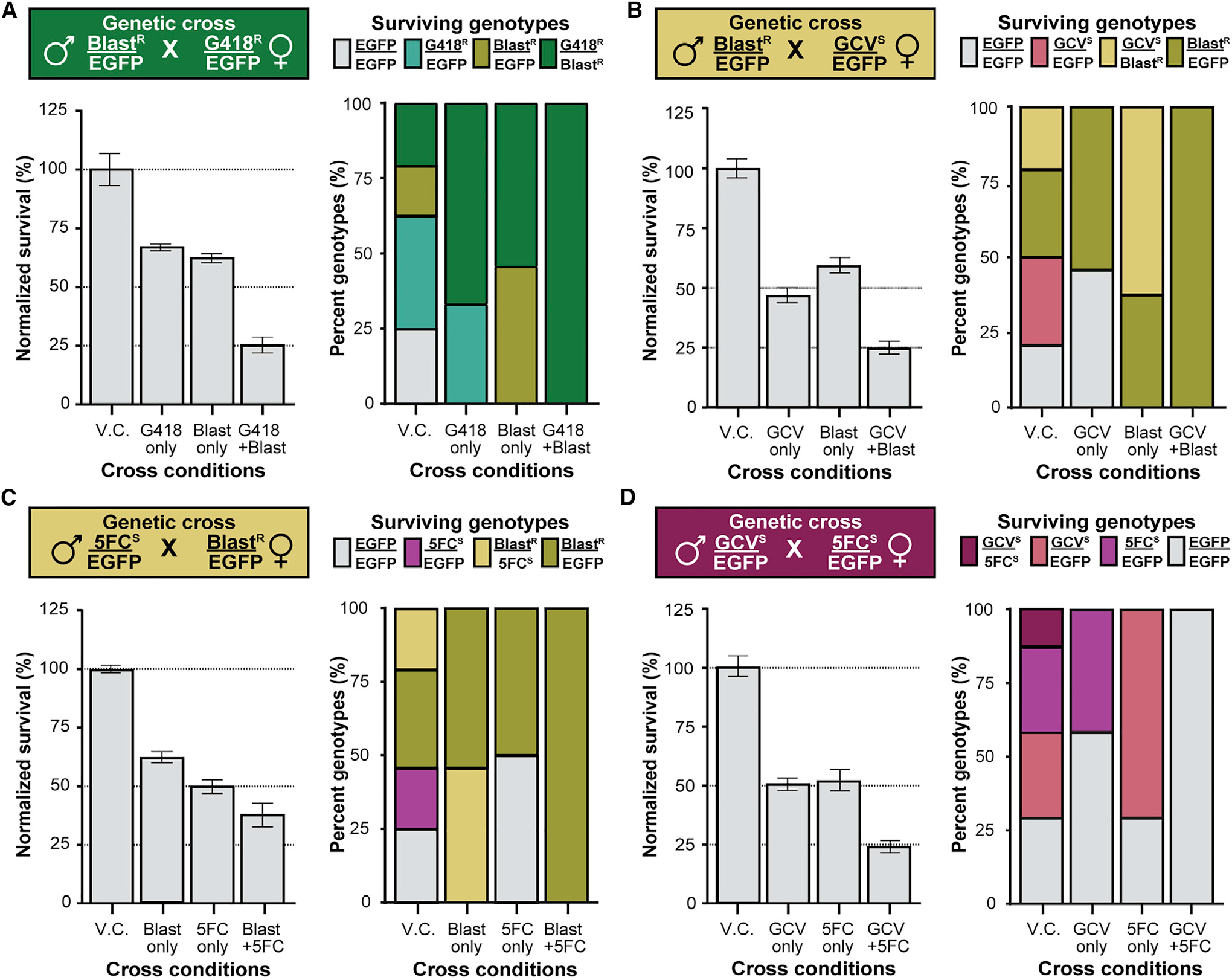
Heterozygous drug-resistant and/or drug-sensitive fly strains: BlastR and G418R (A); BlastR and GCVS (B); 5FCS and BlastR (C); and GCVS and 5FCS (D) were crossed together under four drug conditions. Fly crosses were tested on food with vehicle control (VC), drug A, drug B, or both drugs (A+B). Survival data matched expected frequencies, with selection or counterselection reducing normalized percent survival by 50% and dual-drug treatment by 75% versus vehicle control. For each cross, 24 flies from each of the four drug conditions were collected and individually genotyped. Genotyping produced expected genotypes for each drug condition in each individual cross. Selection with a single drug results in survival of only the corresponding resistant genotypes (A–C). Conversely, single-drug counterselection eliminates only the relevant sensitized strains (B–D). Finally, dual-drug co-selection produced only the dually resistant genotype (A), only BlastR heterozygotes survived combination selection and counterselection (A–C), and co-counterselection resulted in only EGFP homozygotes, sensitive to neither drug, surviving treatment (D). See also Figures S4–S6.
Co-selection of the G418R and BlastR heterozygote cross in vials treated with vehicle control, G418 sulfate, blasticidin S, or both drugs at their respective ESCs produced the expected genotype frequencies in each drug condition (Figures 2A and S4A). Compared to vehicle control, normalized survival decreased by about 50% in the single-drug conditions and 75% when co-selected on both drugs, as expected. Genotyping confirmed that all four genotypes were represented in roughly equal proportion in control vials, whereas in single-drug-treated vials, only the expected resistant heterozygous (teal or olive bars) and dually resistant transheterozygote genotypes (green bars) survived drug treatment. In vials drugged with both G418 sulfate and blasticidin S, only the transheterozygous animals expressing both markers survived co-selection (green bars, Figure S5A). We observed similarly robust results for the other two selection marker cross combinations (Figures S4B, S4C, S5B, and S5C) and for the combination selection and counterselection crosses (Figures 2B, 2C, S4D, S4E, S5D, and S5E), as well as the co-counterselection crosses between GCVS and 5FCS heterozygotes (Figures 2D, S4F, and S5F). In BlastR crosses where females conferred the blasticidin resistance marker, we found higher than expected survival frequencies, suggesting a maternal selection effect, as genotyping confirmed that all surviving progeny were BlastR positive (Figures S4 and S5).
Multiplexed drug-based selection and/or counterselection is analogous to the computer science concept of binary logic gates in which one or more inputs are computed into a single binary output using Boolean logic (Singh, 2014; Manzoni et al., 2016). Of the sixteen possible two-input binary Boolean logic gates, four commonly used gates can be directly represented using combinations of markers (AND, NOR, and both versions of NOT), whereas the other four commonly used gates (OR, NAND, XOR, and XNOR) can be generated indirectly by combining differentially selected and counterselected populations (Figure S6). Our experiments show that combining selection and/or counterselection drugs can generate a diverse set of genotypically pure populations in one or two generations, going beyond sex-specific selection of flies with a genetically engineered circuit coupled to G418 or puromycin resistance (Kandul et al., 2020). This combinatorial drug flexibility opens up opportunities for sophisticated genetic manipulations, crossing schemes, as well as population-control approaches.
Single-step co-transgenesis via dual drug co-selection
We next explored whether we could apply combinatorial drug selection to create genetically pure populations with direct practical significance. A simple but powerful demonstration of co-selection would be to make double-transgenic flies that carry both components of a binary expression system in a single-transgenic event. The original GAL4/UAS binary expression system and its later variants LexA/LexAOp and QF/QUAS have been powerful tools for functional analysis in Drosophila (Brand and Perrimon, 1993; Lai and Lee, 2006; Pfeiffer et al., 2010; Potter et al., 2010). However, generating such tools requires two separate transgenesis steps, each containing one half of the binary expression system (e.g., GAL4 and UAS). The resulting transgenics must then be crossed together, resulting in a strain expressing both elements of the system (e.g., GAL4/UAS). Instead, we generated GAL4/UAS and LexA/LexAOp double-transgenic animals expressing the complete binary expression system in a single transgenesis step using dual drug co-selection. This eliminates both the need to screen transgenic progeny and the required crosses, simplifying generation of these tools. We first generated ΦC31 transgenesis-compatible GAL4/LexA driver and UAS/LexAOp response vectors. Driver vectors confer G418 resistance, and response vectors provide blasticidin S resistance (Figure 3A). We made different driver vectors under the control of enhancer elements from the FlyLight enhancer collection producing pR76H03-GAL4G418R, pR20A02-GAL4G418R, and pR70B04-LexAG418R (Jenett et al., 2012). The FlyLight collection is well characterized and therefore serves as a good control for the resulting expression patterns of our binary expression system vectors (Jenett et al., 2012). We cloned response vectors each composed of a synthetic 5xUAS (Sarrion-Perdigones et al., 2013) or 12xLexAop element (Table S1) driving expression of a fluorescent protein, either superfolder GFP (sfGFP) (Pédelacq et al., 2006) or monomeric Cherry (mCherry) (Shaner et al., 2004), resulting in vectors p5xUAS-sfGFPBlastR, p5xUAS-mCherryBlastR, and p12xLexAOp-sfGFPBlastR (Table S2). All driver and response vectors also contain a physical eye marker with a 5xGMR enhancer element (Hay et al., 1994) driving expression of the white gene coding sequence or a synthetic mini-white eye marker (Figure 3A; Pirrotta, 1988). Driver-response vector pairs were injected into a double docking site (attP) fly strain (Table S3), and progeny were selected on G418 and blasticidin S dual-treated food. We successfully obtained double-transgenic flies for pR76H03-GAL4G418R–p5xUAS-mCherryBlastR, pR20A02-GAL4G418R–p5xUAS-sfGFPBlastR, and pR70B04-LexAG418R-p12xLexAOp-sfGFPBlastR driver-response element pairs.
Figure 3. Single-step co-transgenesis via dual-drug co-selection.
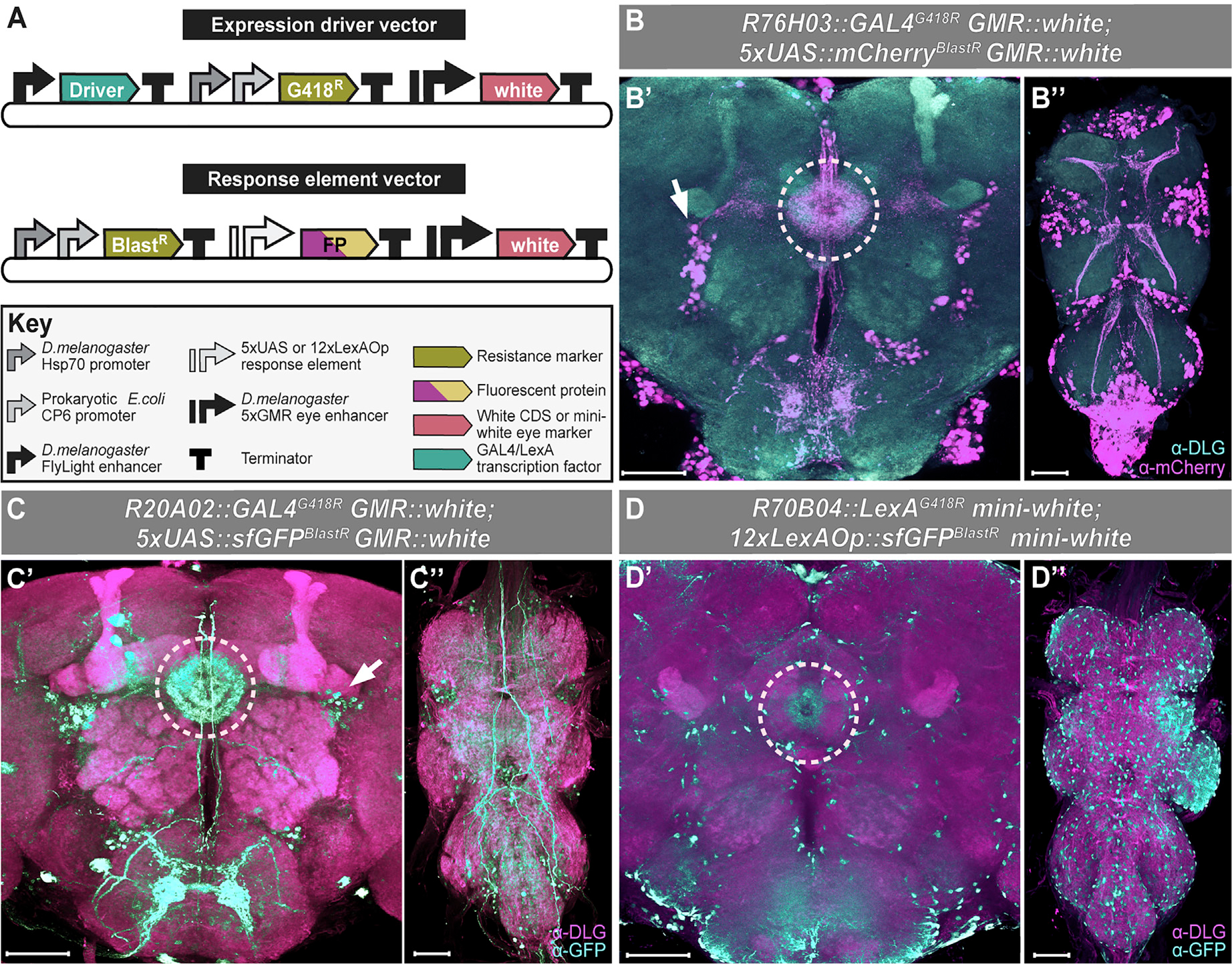
(A) Schematic of the drug-selectable driver-response vector pairs for either the GAL4/UAS or the LexA/LexAOp binary expression systems. Driver vectors consist of a FlyLight library genomic enhancer element regulating GAL4 or LexA transcription factor expression, a G418 resistance maker, and a visual eye marker. Response vectors contain a 5xUAS or a 12xLexAOp driver binding DNA motif upstream of a fluorescent protein (mCherry or sfGFP), a blasticidin resistance marker, and visual eye marker. Transgenics were generated via co-injections of driver-response vector pairs into a double-docking site fly line using ΦC31 integrase.
(B–D) Results were visualized via immunofluorescent staining for the respective fluorescent proteins.
(B) Staining for R76H03::GAL4-driven mCherry in the central complex of the adult fly brain showed expression in the ellipsoid body (dotted circle) and innervating R4 cells (arrow; B′), although in the ventral nerve cord, staining revealed an X-shaped pattern similar to FlyLight data (B″).
(C) Staining for GFP in R20A02::GAL4; UAS-GFP adult fly brains labels the ellipsoid body (dashed circle) and R4 cells (arrow) in the brain, similar to previously reported expression of this enhancer (C′). Expression in the ventral nerve cord (VNC) shows less similarity to the previously reported pattern (C″).
(D) Staining for R70B04::LexA-driven sfGFP showed only very faint expression in the ellipsoid body within the central complex compared to the described enhancer expression from FlyLight (D′), although in the VNC, expression was very similar to FlyLight expression for this enhancer (D″). Staining for Disc Large (DLG) was used as a counterstain.
Scale bars represent 50 μm. See also Figure S7.
The expression patterns in these strains are similar to the original FlyLight documented expression patterns (Figures 3B–3D; Jenett et al., 2012). Staining for the mCherry marker revealed expression in the central complex of adult brains of R76H03::GAL4 animals marking the ellipsoid body and innervating R4 cells (Figure 3B′). Staining in the adult ventral nerve cord showed a similar X-shaped expression pattern as seen in the FlyLight collection (Figure 3B″). R20A02-driven GAL4/UAS binary expression labels the ellipsoid body and R4 cells in the adult brain similar to previously reported expression of this enhancer (Figure 3C′), although GFP expression in the ventral nerve cord (VNC) was less similar to the previously reported pattern (Figure 3C″). We observed only faint sfGFP expression in the ellipsoid body within the central complex in the adult brains of R70B04::LexA-driven sfGFP (Figure 3D′). Staining in the adult VNC showed a similar expression pattern as the equivalent GAL4 enhancer line from the FlyLight collection (Figure 3D″). Differences between the observed expression patterns and those described in FlyLight are likely due to the use of different docking sites and differences in our cloned response elements affecting expression level. Despite these differences, our binary system reporter line shares significant similarities with previously reported expression patterns (Jenett et al., 2012). We also generated single GAL4 driver transgenics for these enhancers and crossed them to a strong UAS reporter line (Pfeiffer et al., 2012). The resulting expression patterns were even more similar to those previously described, confirming that expression differences likely result from design and insertion site variations (Figures S7A–S7C; Jenett et al., 2012). Together, these experiments show that making double-transgenic flies in a single step could accelerate the generation of split-GAL4 reagents or orthogonal binary expression drivers and repressors among other alternative applications.
Selection and counterselection to simplify balancer chromosome crosses
The ability to select for or against balancer chromosomes instead of screening and counterscreening them is useful when balancing transgenes or when only non-balancer offspring are needed for further analysis. Some dominant markers present on balancers or their dominantly marked counterparts are difficult to score, e.g., AntpHu (Humeral [Hu]) on the third chromosome balancer TM6B (Miller et al., 2016) or snaSco (Scutoid [Sco]) on the second balancer counterpart chromosome (Miller et al., 2018). Screening for the correct progeny in such crosses can be slow and tedious. Via recombinase-mediated cassette exchange (RMCE) (Bateman et al., 2006), we upgraded an existing third chromosome balancer (TM6B [Tb]), containing a P element insertion of a recombinase exchangeable cassette (Sun et al., 2012). We generated two selectable and counterselectable balancer chromosomes by combining either a blasticidin S or G418 resistance marker with a 5-fluorocytosine sensitivity marker expression cassette via synthetic assembly and then replacing the original P element insertion with our dual-marker cassette via RMCE (Figure 4A). The resulting transgenic strains have constitutive whole-body-marker expression that can be enhanced with heat shock treatment (Figures 4B and 4C).
Figure 4. Selection and counterselection to simplify balancer chromosome crosses.
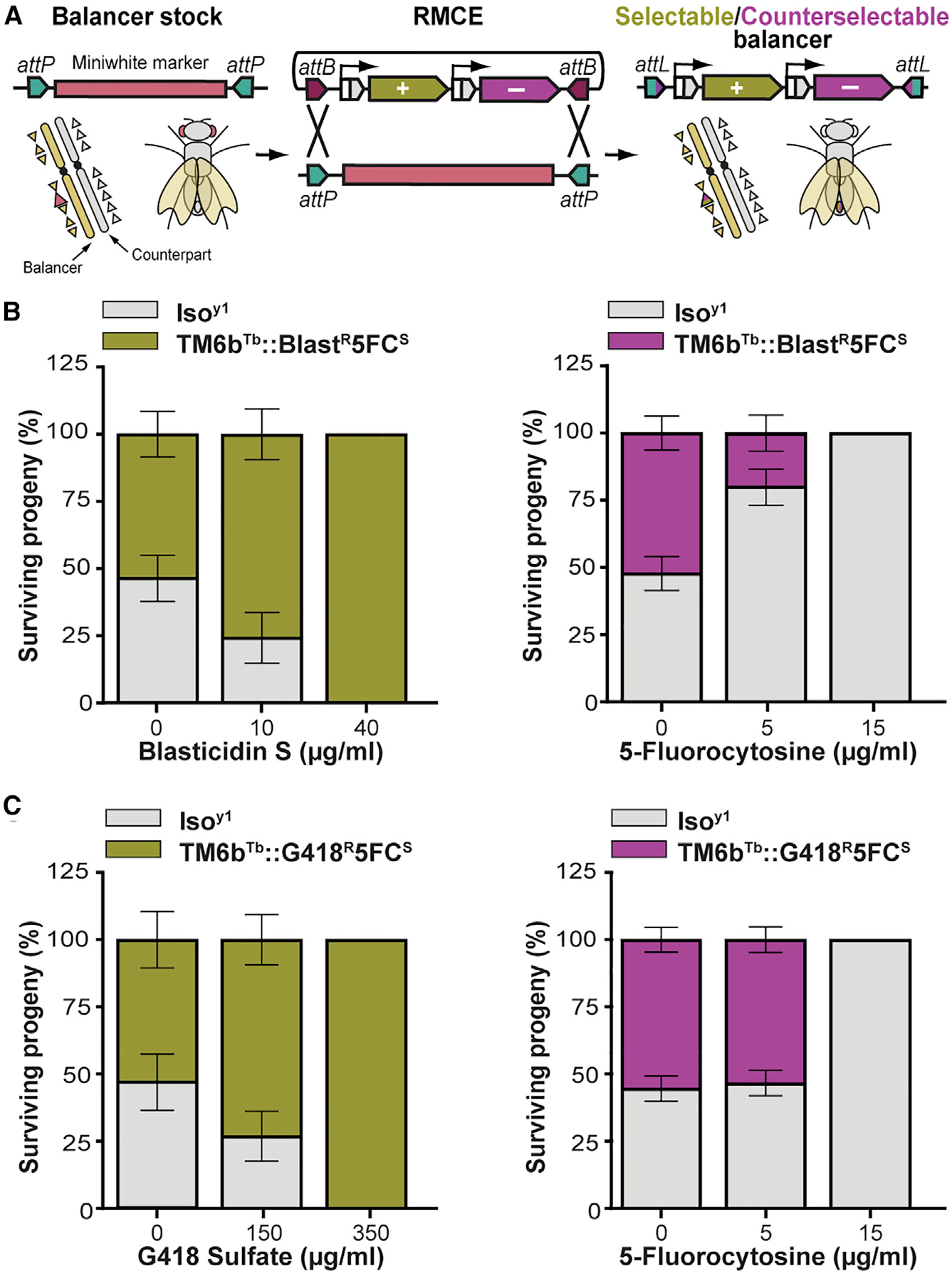
(A) Balancer chromosome fly stocks were generated via recombinase-mediated cassette exchange onto a TM6B, Tb[1] third chromosome balancer containing a double inverted attP docking site.
(B and C) Two different versions of the balancer stock were generated resistant to blasticidin S or G418 with both sensitive to 5-fluorocytosine and abbreviated as TM6bTb::BlastR5FCS (B) and TM6BTb::G418R5FCS (C). An equal number of non-virgin IsoY1 and balancer stock females, TM6bTb::BlastR5FCS (B) or TM6BTb::G418R5FCS (C), were mixed together in vials with varying concentrations of the appropriate drug. Populations selected with antibiotic produced only drug-resistant balancer animals at the respective drug ESCs, whereas concentration of 5-fluorocytosine had to be increased to 15 μg/mL for effective counterselection, likely due to lower basal marker expression in this docking site.
We tested the selectable and counterselectable balancers by mixing equal numbers of non-virgin isogenized control (IsoY1) and balancer strain females on vials with varying concentrations of the corresponding drugs. By exposing developing larvae to either blasticidin S (Figure 4B, left) or G418 (Figure 4C, left), we effectively selected for balancer flies. Conversely, we selected against the balancer strain via exposure to 5-fluorocytosine (Figures 4B and 4C, right). Basal marker expression was sufficient to confer G418 resistance, but heat shock treatment was required for complete blasticidin resistance, likely due to lower marker expression from this particular genomic locus (Figure 4B). The ECC for 5-fluorocytosine at 15 μg/mL was higher than in previous experiments, also likely due to position effects on marker expression. Heat shock induction would possibly reduce the amount of 5-fluorocytosine required for effective counterselection, though it is not required. A full set of selectable and counterselectable balancers as well as similar chromosomes for the Y and the 4th chromosomes would also be useful to simplify some fly genetics experiments. Our experiments demonstrate the feasibility of such a resource.
Drug-based selection of large fluorescently tagged P[acman] BAC transgenics to recapitulate endogenous expression patterns and identify protein complexes
Large transgenes (>20 kb), such as P[acman] bacterial artificial chromosome (BAC) clones (Venken et al., 2006, 2009), are an important tool in Drosophila genetics, as they provide endogenous genomic context for genes of interest. They are the gold standard for null allele rescue experiments (Venken et al., 2006, 2009, 2010) and demonstration of expression and localization of a tagged gene product covered by the genomic clone (Venken et al., 2008, 2009). However, the large size of the genomic clones results in lower transgenesis efficiency, requiring more extensive screening compared to smaller transgenes (Venken et al., 2006, 2009, 2010). Drug-based selection eliminates the need to screen, providing a simplified method for isolating rare transgenic events. We generated two transgenic strains with drug selectable and fluorescently protein-tagged P[acman] BAC library genomic clones using a 3-step serial recombineering strategy for N-terminal (Figure 5A) and C-terminal tagging (Figure 5B; see also Figure S8). The P[acman] BAC clone CH322–06D09 covering cysteine string protein (Csp) (38 kb) was upgraded with an N-terminal EGFP tag and a G418 resistance marker, although clone CH322–154P15 covering neurexin IV (NrxIV) (>35 kb) was upgraded with a C-terminal mCherry tag and a blasticidin S resistance marker.
Figure 5. Drug-based selection of large fluorescently tagged P[acman] BAC transgenics to recapitulate endogenous expression patterns and identify protein complexes.

(A and B) Using a 3-step serial recombineering strategy, two genomic P[acman] clones were upgraded with a fluorescent tag, either on the N (A) or C terminus (B), and a selection marker expression cassette conferring resistance to G418 or blasticidin S. The bacterial ampicillin resistance marker (AmpR) was used to select successful recombineering reactions in the first step. This marker is removed via Cre-recombinase-mediated reduction in the third step. A short, flexible peptide sequence links the fluorescent tag in frame to the encoded gene (Figure S8).
(C and D) Resulting selected flies were verified via immunofluorescent staining for tagged P[acman] BAC products.
(C) Staining data for N-terminal EGFP tagged cystein string protein (Csp) shows strong neuropil expression throughout the adult fly brain (C′). In the larval brain, staining against tagged Csp shows clear neuropil expression in the visual system, including the optic lobes (OLs) as well as expression in the VNC (C″).
(D) Staining for C terminus mCherry-tagged Neurexin IV (NrxIV) shows brain surface glial expression in the adult fly brain (D′). Similarly, staining shows glial expression in the larval brain with clear labeling of septate junctions formed at the borders of the surface subperineural glial (arrows) in the cerebral hemispheres and VNC (D″). Scale bars represent 50 μm.
(E) Schematic of the workflow of the co-immunoprecipitation of GFP-tagged Csp from isolated fly heads followed by proteomic analysis.
(F) Volcano plot shows differentially enriched proteins (by NCBI gene symbol) between tagged Csp and GFP control pull-downs. Colored dots represent peptides (291/624) that were found to be significantly different between the two groups at a false discovery rate (FDR) < 0.05 and enrichment of ≥4 fold.
(G) Interactome analysis was performed with STRING. Shown is a selected interactome focusing on Csp and putative connections to vesicle recycling (purple) and other neuronal proteins (teal). Proteins in olive indicate known protein-protein interactions with Csp.
See also Figures S8 and S9.
We stained adult fly brains for each tagged protein using antibodies against GFP (Figure 5C) or mCherry (Figure 5D). Consistent with the role of Csp in presynaptic vesicle exocytosis, we observed strong neuropil staining in the adult brain (Figure 4C′; Bronk et al., 2005). We also observed neuropil staining in the larval brain corresponding to previous reports (Figure 5C″; Eberle et al., 1998). Staining for mCherry-tagged NrxIV showed localization on the surface of the brain, corresponding to its known glial expression in the formation of septate junctions, in adult and larval brains (Figures 5D′ and 5C″; Baumgartner et al., 1996; Banerjee et al., 2008). In particular, staining in the larval brain clearly marks septate-junction-rich lateral borders of subperineural glia in the cerebral hemispheres and VNC (Figure 5D″; Carlson et al., 2000). These staining data show that we are able to modify BAC clones to be both selectable and fluorescently tagged.
We next assessed the applicability of our selection-based genetics platform to biologically relevant questions by performing immunoprecipitation followed by unbiased interactome analysis for one of our tagged-BAC clone transgenic strains. We chose to focus our analysis on N-terminally tagged Csp due to its role in synaptic vesicle exocytosis (Bronk et al., 2005). We pulled down EGFP-tagged Csp protein from fly heads and assessed the identities of co-immunoprecipitated proteins using mass spectrometry (Figure 5E). We identified 943 distinct peptides from our sample (Table S4), of which 624 could be 100% identified in either the tagged Csp or EGFP control groups. Of the 624 identified proteins, 291 exhibit significant differential enrichment between Csp and control samples at a false discovery rate (FDR) < 0.05 and fold enrichment of >4 versus control (Figure 5F). We found high enrichment of most proteins of both ribosomal subunits, making up 57 of the top 100 identified peptides by p value, suggesting whole ribosomes were pulled down (Figure S9). We also identified proteins associated with synaptic vesicle recycling or trafficking, resulting in a highly significant STRING network (Szklarczyk et al., 2019; Figure 5G). We detected Hsc70 peptides, which are known Csp interactors (Eberle et al., 1998; Tobaben et al., 2001; Nosková et al., 2011), and identified shibire and Comatose, whose human orthologs (DNM1 and NSF, respectively) interact either physically or biochemically with the human Csp ortholog (CSPα; Zhang et al., 2012). We also identified histones and mitochondrial-associated peptides enriched in our tagged Csp sample versus control (Figure S9). Further experiments are necessary to confirm the biological significance of these interactions.
A selection and counterselection vector library resource for iterative synthetic assembly
To make our selection and counterselection genetics platform easy to implement, we developed a vector library resource using GoldenBraid 2.0 cloning (GB2.0). GB2.0 is a multipartite, one-pot reaction synthetic assembly method that uses type IIs restriction enzymes, BsaI and Esp3I (isoschizomer of BsmBI), which cut away from their binding site to produce programmable four nucleotide overhangs (Engler et al., 2008; Sarrion-Perdigones et al., 2011, 2013). These overhangs produce a “grammar” that governs part assembly order in the reaction (Figure 6A). GB2.0 simplifies assembly of complex, multigenic constructs (Sarrion-Perdigones et al., 2013). Multiple compatible DNA elements can assemble in a single cloning reaction into a destination vector, either pColE1_Alpha1 or pColE1_Alpha2, via a BsaI digest, resulting in a functional transcription unit plasmid (pTU) (Figures 6B, S10A, and S10B). GB2.0 is iterative, using the products of one assembly step as reagents in the next, allowing further assembly of paired pTUs into a second set of destination vectors, either pColE1_Omega1 or pColE1_Omega2, via an Esp3I reaction, resulting in a multigenic genetic circuit (GC) (Figure S10C; Sarrion-Perdigones et al., 2013). GB2.0 allows further assembly of paired GC products back into alpha destination vectors, the products of which are reagents for additional assembly steps in an iterative cloning process, resulting in ever more complex constructs (Figure 6B). GB2.0 construction schemes to generate complex constructs needed for co-selection (Figure 3) and combined selection and counterselection (Figure 4) demonstrate overall feasibility, although selectable P[acman] BAC transgenics (Figure 5) demonstrate the compatibility of our DNA library with existing DNA resources (Venken et al., 2009). Altogether, these examples illustrate the versatility and usefulness of this vector library resource for a broad range of potential applications (Figure 6C).
Figure 6. A selection and counterselection vector library resource for iterative synthetic assembly.

(A) Iterative synthetic assembly with Goldenbraid 2.0 (GB2.0) cloning uses a predefined “grammar” of four nucleotide overhangs that determine DNA element identity and assembly order during cloning.
(B) GB2.0 uses a one-pot assembly of multiple DNA parts into one of two alpha-level destination vectors resulting in a transcription unit (TU). Paired TUs can be further assembled into an omega-level destination vector producing a multigenic genetic circuit (GC). These can then be further assembled back into an alpha-level vector to produce ever more complex constructs.
(C) A number of pre-built selectable and/or counterselectable vectors are provided for a variety of applications as well as a library of GB2.0-compatible DNA elements totaling 121 plasmids. Each of these vectors was itself cloned from smaller elements using GB2.0 synthetic assembly. The vectors require minimal modification using either GB2.0 or traditional cut-and-paste cloning for a particular application. More experienced users can generate fully customized vectors appropriate to their needs using GB2.0 cloning. Three examples of vector resources and their cloning assemblies are shown. All plasmids are publicly available via Addgene (https://www.addgene.org/), and a full list can be found in Tables S1 and S2.
See also Figure S10.
GB2.0 requires the use of compatible DNA elements with matching cloning overhangs and restrictions enzyme sites conforming to the established assembly grammar (Figure 6A; Sarrion-Perdigones et al., 2013). Here, we provide a collection of over 120 GB2.0-compatible vectors consisting of 64 DNA elements and 57 transgenesis ready vectors. Our DNA library consists of a variety of Drosophila melanogaster and non-Drosophila promoters, enhancers, markers (selection and counterselection, fluorescent, and physical), peptide linkers, tags, poly(A) terminators, and many other elements (Table S1). The provided toolkit of transgenesis-ready, drug-selectable and/or counterselectable vectors is compatible with GB2.0 and, in some cases, traditional restriction endonuclease cloning (Table S2). Included in the toolkit are (1) vectors for in vivo selection using one of four antibiotics (e.g., G418, puromycin, blasticidin S, and hygromycin B); (2) plasmids for generating drug-selectable GAL4/UAS, LexA/LexAOp, and QF/QUAS driver-response binary expression system vector pairs; (3) selectable and counterselectable RMCE-compatible vectors resistant to one of three drugs (G418, puromycin, or blasticidin S) and sensitive to 5-flurocytosine; and (4) selection and tagging cassettes for recombineering-based upgrading of genomic BAC clones (Table S2). The vector toolkit also contains all of the basic GB2.0 cloning vectors used in this work needed to adopt this cloning method. Because many of our vectors contain attB sites for ΦC31-integrase-mediated transgenesis, they can readily be transferred to additional insects, including other Drosophila and mosquito species (Holtzman et al., 2010; Labbé et al., 2010; Meredith et al., 2013; Pondeville et al., 2014; Volohonsky et al., 2015; Kudo et al., 2018), as well as vertebrates, such as fish and mice (Tasic et al., 2011; Kirchmaier et al., 2013; Mosimann et al., 2013; Roberts et al., 2014), that already have attP integration sites available.
DISCUSSION
In this study, we developed a multiplex drug-based selection and counterselection strategy to simplify genetic manipulations in Drosophila melanogaster. Selection- and counterselection-based genetics eliminates the need to screen for or counterscreen against modified progeny via visual markers, significantly reducing workload. We generated four selection and two counterselection markers with broad applicability. Although previous efforts to adapt selection and counterselection genetics to Drosophila have had only limited success (Steller and Pirrotta, 1985; Handler and O’Brochta, 1991), our work demonstrates effective drug-based selection and counterselection to make genetically pure populations of animals through multiplexed marker co-selection, combination selection and counterselection, and co-counterselection. To demonstrate the power of multiplexing selection and counterselection drugs, we generated GAL4/UAS and LexA/LexAop double-transgenic animals in a single co-transgenesis step and made selectable and counterselectable balancer chromosomes. Generating transgenic fly lines for an experiment often takes 3 to 4 months to create and bring together multiple genetic elements into a single strain. Especially if using visible markers, this can involve time-consuming screening to identify progeny with the correct physical makers. Both applications eliminate the need for screening and complex cross schemes, making these genetic manipulations simpler and faster.
We also selected single transgenics for two different N- or C-terminally fluorescently tagged P[acman] BAC genomic library clones using either G418 or blasticidin S. We then analyzed the interactome of Csp by performing mass spectrometry on immunoprecipitated GFP-tagged Csp from fly heads and identified Hsc70–3, Hsc70–4, Shi, and Comt, known interactors of Csp or orthologs of interactors of human Cspα (Eberle et al., 1998; Nosková et al., 2011; Tobaben et al., 2001; Zhang et al., 2012). The co-immunoprecipitation of a large number of ribosomal subunit proteins suggests that Csp may also interact with the ribosome. Whether this interaction represents a role of Csp at the synapse or is due to aggregation because of modestly elevated Csp expression levels will require further investigation to validate its biological significance.
Choosing the right marker for a given application depends foremost on the site of insertion, as degree of resistance varies with marker expression. Although we designed our expression cassette to maximize marker expression, the genomic context of an insertion site does affect marker effectiveness. Expression can be enhanced with heat shock treatments but in our hands is not usually necessary. Our experience shows that the nptII marker is the most robust and should be the primary choice for most applications. The second preferred marker depends on application: for insertions into a well characterized, high expression docking site, the bsr marker (blasticidin S) is a good choice, but for insertions into a poorly expressing site, the pac marker is likely better, as puromycin does not seem to be toxic to resistant animals even at high concentrations (700 μg/mL), unlike blasticidin S. However, the high cost per vial for puromycin may be prohibitive for large-scale use. Finally, the hph marker is the least preferred marker, as heat shock is required for effective drug resistance.
Similar considerations apply to using multiple counterselectable drugs simultaneously. Although cost per vial is negligible for the counterselection markers, sr39TK or FCU1, degree of marker-conferred sensitivity also varies with insertion-site expression level. Importantly, for all counterselection drugs tested, there is a maximal concentration, beyond which even non-sensitized animals exhibit drug toxicity. The negligible cost of the counterselectable drugs makes their corresponding markers useful for RMCE-based (Bateman et al., 2006) upgrading of existing transposon insertions (e.g., MiMIC; Venken et al., 2011) or targeted CRISPR alleles (Zhang et al., 2014; Li-Kroeger et al., 2018; Kanca et al., 2019), after appropriately modifying them with a counterselectable marker.
We recommend testing innate drug resistance and sensitivity of a particular strain prior to using any of the markers (Matinyan et al., 2021), similar to establishing lethality curves in mammalian cell culture. Once established in a particular background, markers and their corresponding drugs can be used without further troubleshooting.
We modified a pair of balancer chromosomes by upgrading them with a selection and counterselection cassette. Although we only provide upgraded balancers for the third chromosome, multiple balancers exist for each of the three main Drosophila chromosomes and are often used in conjunction during complex crossing schemes. In the future, it may be possible to use multiple selectable balancers for different chromosomes.
Some small changes could further improve our drug-based genetics system. Substituting the Hsp70 promoter for promoters from constitutively expressed genes, such as α1-tubulin (Angelichio et al., 1991; Zhang et al., 2013), polyubiquitin (Handler and Harrell, 2001), actin 5C (Angelichio et al., 1991), or armadillo (Vincent and Girdham, 1997), may provide higher and more consistent expression and may decrease variability between different genomic locations. Substituting the minimal HSV-TKpA transcriptional terminator (Steller and Pirrotta, 1985) for other polyadenylation signals, e.g., SV40 (Angelichio et al., 1991), may do the same. These modifications may improve the usefulness of the hygromycin B resistance marker especially.
To make our platform broadly useful, we generated a library of DNA elements and a transgenesis ready vector toolkit compatible with GB2.0 cloning for selection and counterselection genetics. This vector resource will make our next-generation transgenesis system immediately useful not just for the fly community but also for other model systems. The large library of DNA elements makes it possible for other labs to quickly build their own constructs and tools based on their needs. As all the markers are wholly exogenous to insects, selection and counterselection genetics can be readily applied to other drosophilids (Holtzman et al., 2010; Kudo et al., 2018) and other insect species like mosquitoes (Labbé et al., 2010; Meredith et al., 2013; Pondeville et al., 2014; Volohonsky et al., 2015; Matthews and Vosshall, 2020), provided careful selection of well-expressing promoters to ensure effective drug resistance and sensitivity. Obviously, the use of these markers in other species will have to be adapted to the particularities of their life cycles, life stages, and culture conditions. Robust selection and counterselection will likely be very valuable in species currently lacking available genetic tools and where transgenesis is not as efficient as in the fruit fly.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Polyclonal rabbit GFP | Invitrogen/Thermo Fisher | Cat# A11122, RRID:AB_221569 |
| Mouse monoclonal anti-Dlg | Developmental Studies Hybridoma Bank | Cat# 4F3, RRID:AB_528203 |
| Rat monoclonal mCherry | Invitrogen/Thermo Fisher | Cat# M11217, RRID:AB_2536611 |
| Mouse monoclonal dCsp | Developmental Studies Hybridoma Bank | Cat# Ab49, RRID:AB_2307340 |
| Chicken anti-rabbit AlexaFluor 488 | Invitrogen/Thermo-Fisher | Cat# A-21441, RRID:AB_2535859 |
| Goat anti-mouse AlexaFluor 568 | Invitrogen/Thermo-Fisher | Cat# A11004, RRID:AB_2534072 |
| Goat anti-rat AlexaFluor 568 | Invitrogen/Thermo-Fisher | Cat# A-11077, RRID:AB_141874 |
| Chicken anti-mouse AlexaFluor 568 | Invitrogen/Thermo-Fisher | Cat# A21200, RRID:AB_2535786 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| Chemocompetent Escherichia coli strain DH10B | Invitrogen/Thermo Fisher Scientific | Cat# EC0113 |
| Transformax EPI300 electrocompetent strain cells | Epicenter/Lucigen | Cat# EC300110, |
| EL350 recombineering strain bacterial cells | Kind gift from Donald Court, National Cancer Institute | n/a |
| EC100D pir-116 | Epicenter/Lucigen | Cat# EC6P095H |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| G418 disulfate | VWR | Cat# 97063–060 |
| Puromycin dihydrochloride | VWR | Cat# 97064–280 |
| Blasticidin S hydrochloride | VWR | Cat# 71002–676 |
| Hygromycin B | VWR | Cat# AAJ6068103 |
| Ganciclovir | TCI America | Cat# G0315 |
| 5-Fluorocytosine | TCI America | Cat# F0321 |
| BsaI-HFv2 | New England Biolabs | Cat# R3733 |
| Esp3I | New England Biolabs | Cat# R0734 |
| T4 DNA Ligase | Promega | Cat# M1801 |
| iProof High-Fidelity DNA Polymerase | Biorad | Cat# 1725301 |
| Phusion High-Fidelity DNA Polymerase | New England Biolabs | Cat# M0530 |
| Q5 High-Fidelity DNA Polymerase | New England Biolabs | Cat# M0491 |
| BioReady rTaq DNA polymerase | Bulldog Bio | Cat# BSAX050 |
| Zeocin | Alfa Aesar | Cat# J67140 |
| Phleomycin | VWR | Cat# AAJ67027–8EQ |
| BbsI-HF | New England Biolabs | Cat# R3539L |
| CopyControl Fosmid Autoinduction Solution | Epicenter/Lucigen | Cat# CCIS125 |
| Trypsin protease | Gendepot | Cat# T9600 |
| Ammonium bicarbonate | Fisher Scientific | Cat# 3003–1 |
| LC-MS methanol | Fisher Scientific | Cat# A456 |
| LC-MS water | J.T.Baker | Cat# JT9831–3 |
| LC-MS acetonitrile | Fisher Scientific | Cat# A955 |
| Formic Acid | Fisher Scientific | Cat# A117 |
|
| ||
| Critical commercial assays | ||
|
| ||
| QIAquick PCR Purification Kit | QIAGEN | Cat# 28106 |
| QIAquick Gel Extraction Kit | QIAGEN | Cat# 28506 |
| QIAprep Spin Miniprep Kit | QIAGEN | Cat# 27106 |
| ChargeSwitch-Pro plasmid Miniprep kit | Invitrogen/Thermo Fisher Scientific | Cat# CS30250 |
| ZR-96 Quick-gDNA kit | Zymo Research | Cat# D3010 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Drosophila melanogaster: EGFP: y[1]w[1118]; PBac{y[+mDint2] w[+mC] = P[acman]-attB-Hsp70-CP6-EGFP-TKpA} VK00033 | This work, available from the BDSC | Cat# 92331, RRID:BDSC_92331 |
| Drosophila melanogaster: G418R: y[1]w[1118]; PBac{y[+mDint2] w[+mC] = P[acman]-attB-Hsp70-CP6-nptII-TKpA} VK00033 | This work, available from the BDSC | Cat# 92332, RRID:BDSC_92332 |
| Drosophila melanogaster: PuroR:y[1]w[1118]; PBac{y[+mDint2] w[+mC] = P[acman]-attB-Hsp70-CP6-pac-TKpA} VK00033 | This work, available from the BDSC | Cat# 92333, RRID:BDSC_92333 |
| Drosophila melanogaster: BlastR: y[1]w[1118]; PBac{y[+mDint2] w[+mC] = P[acman]-attB-Hsp70-CP6-bsd-TKpA} VK00033/TM6B, Tb[1] | This work, available from the BDSC | Cat# 92334, RRID:BDSC_92334 |
| Drosophila melanogaster: HygroR: y[1]w[1118]; PBac{y[+mDint2] w[+mC] = P[acman]-attB-Hsp70-CP6-hph-TKpA} VK00033 | This work, available from the BDSC | Cat# 92335, RRID:BDSC_92335 |
| Drosophila melanogaster: ZeoR: y[1]w[1118]; PBac{y[+mDint2] w[+mC] = P[acman]-attB-Hsp70-CP6-bsr-TKpA} VK00033 | This work, available from the BDSC | Cat# 92336, RRID:BDSC_92336 |
| Drosophila melanogaster: GCVS:y[1]w[1118]; PBac{y[+mDint2] w[+mC] = P[acman]-attB-Hsp70-CP6-sr39TK-TKpA} VK00033 | This work, available from the BDSC | Cat# 92337, RRID:BDSC_92337 |
| Drosophila melanogaster: 5FCS: y[1]w[1118]; PBac{y[+mDint2] w[+mC] = P[acman]-attB-Hsp70-CP6-FCU1-TKpA} VK00033 | This work, available from the BDSC | Cat# 92338, RRID:BDSC_92338 |
| Drosophila melanogaster: FM7h-25C-1B: FM7h-RMCE-w[+mC]-25C-1B | This work, available from the BDSC | Cat# 92339, RRID:BDSC_92339 |
| Drosophila melanogaster: CyO-25C-4A: y[1]w[1118]/Dp(1;y)y[+];CyO-RMCE-25C-4A(w[+mC])/L | This work, available from the BDSC | Cat# 92340, RRID:BDSC_92340 |
| Drosophila melanogaster: CyO-52D-3A: y[1]w[1118]/Dp(1;y)y[+];CyO-RMCE-52D-3A(w[+mC])/L | This work, available from the BDSC | Cat# 92341, RRID:BDSC_92341 |
| Drosophila melanogaster: TM6B-25C-5A: y[1]w[1118]/Dp(1;y)y[+];TM6b-RMCE-25C-5A(w[+mC])/D | This work, available from the BDSC | Cat# 92342, RRID:BDSC_92342 |
| Drosophila melanogaster: TM6B-52D-1A: y[1]w[1118]/Dp(1;y)y[+];TM6b-RMCE-52D-1A(w[+mC])/D | This work, available from the BDSC | Cat# 92343, RRID:BDSC_92343 |
| Drosophila melanogaster: TM6bTb::BlastR5FCS: y[1]w[1118]/Dp(1;y)y[+];TM6b-RMCE{BlastR-5FCS-w[5xGMR-CDS]}-25C-5A(w[+mC])/D | This work, available from the BDSC | Cat# 92344, RRID:BDSC_92344 |
| Drosophila melanogaster: TM6bTb::G418R5FCS: y[1]w[1118]/Dp(1;y)y[+];TM6b-RMCE{G418R-5FCS-w[5xGMR-CDS]}-25C-5A(w[+mC])/D | This work, available from the BDSC | Cat# 92345, RRID:BDSC_92345 |
| Drosophila melanogaster: 2xattP::VK00033;VK00020:y[1] M{RFP[3xP3.PB] GFP[E.3xP3] = vas-int.B}ZH-2A w[*]; PBac{y[+]-attP-3B}VK00033, PBac{y[+]-attP-9A}VK00020 | This work, available from the BDSC | Cat# 92346, RRID:BDSC_92346 |
| Drosophila melanogaster: N-EGFP-Csp: y[1]w[1118]; PBac{y[+mDint2] w[+mC] = P[acman]-attB-CH322-06D09-N-EGFP-Csp-G418}VK00033 | This work, available from the BDSC | Cat# 92347, RRID:BDSC_92347 |
| Drosophila melanogaster: NrxIV-CmCherry: y[1]w[1118]; PBac{y[+mDint2] w[+mC] = P[acman]-attB-CH322-154P15-NrxIV-C-Cherry-Blast}VK00033 | This work, available from the BDSC | Cat# 92348, RRID:BDSC_92348 |
| Drosophila melanogaster: R20A02::GAL4; w[5xGMR-CDS] = pR20A02-GAL4-G418R } VK000XX, w[5xGMR-CDS] = p5xUASsfGFP-BlastR }VK000XX 5xUAS::sfGFP: | This work, available from the BDSC | Cat# 92764, RRID:BDSC_92364 |
| Drosophila melanogaster: R76H03::GAL4; 5xUAS::mCherry: w[5xGMR-CDS] = pR76H03-GAL4-G418R }VK000XX, w[5xGMR-CDS] = p5xUAS-mCherry-BlastR } VK000XX | This work, available from the BDSC | Cat# 92765, RRID:BDSC_92365 |
| Drosophila melanogaster: R70B04::LexA; 12xLexAOp::sfGFP: w[+mC] = pR70B04-LexA-G418R }VK000XX, w[+mC] = p12xLexAOp-sfGFP-BlastR }VK000XX | This work, available from the BDSC | Cat# 92766, RRID:BDSC_92366 |
| Drosophila melanogaster: IsoY1: y[1] w[67c23] | This work, available from the BDSC | Cat# 92349, RRID:BDSC_92349 |
|
| ||
| Oligonucleotides | ||
|
| ||
| TKpA-F | This work | N/A |
| TKpA-R | This work | N/A |
| attB-FOR | This work | N/A |
| attB-REV | This work | N/A |
| ModuleA-FOR | This work | N/A |
| ModuleA-REV | This work | N/A |
| ModuleF-FOR | This work | N/A |
| ModuleF-REV | This work | N/A |
| R6Kg-FOR | This work | N/A |
| R6Kg-REV | This work | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| p3xP3-Hsp70b | This work, available from AddGene | Cat# 165773 |
| pMCS-TCCC | This work, available from AddGene | Cat# 165774 |
| p5xGMR | This work, available from AddGene | Cat# 165775 |
| pdSCP | This work, available from AddGene | Cat# 165776 |
| pDmActin5c | This work, available from AddGene | Cat# 165777 |
| p3xP3 | This work, available from AddGene | Cat# 165779 |
| pHsp70b-TACT | This work, available from AddGene | Cat# 165780 |
| pDmAlfaTub84b | This work, available from AddGene | Cat# 165781 |
| pDmVasa | This work, available from AddGene | Cat# 165783 |
| pDmHsp70b | This work, available from AddGene | Cat# 165785 |
| pDmPolyUbiq | This work, available from AddGene | Cat# 165786 |
| pPromoterless-P2A | This work, available from AddGene | Cat# 165787 |
| Promoterless TP10 | This work, available from AddGene | Cat# 165790 |
| pR20A02 | This work, available from AddGene | Cat# 165791 |
| pR70B04 | This work, available from AddGene | Cat# 165792 |
| pR76H03 | This work, available from AddGene | Cat# 165793 |
| pCP6-Dros | This work, available from AddGene | Cat# 165794 |
| pCP6-STD | This work, available from AddGene | Cat# 165795 |
| pCP7-STD | This work, available from AddGene | Cat# 165796 |
| pTP10 | This work, available from AddGene | Cat# 165797 |
| pSV40L | This work, available from AddGene | Cat# 165798 |
| pAdh Short 3′UTR | This work, available from AddGene | Cat# 165800 |
| pHSV-TK | This work, available from AddGene | Cat# 165801 |
| prrnBT1_STD | This work, available from AddGene | Cat# 165803 |
| prrnBT2_STD | This work, available from AddGene | Cat# 165804 |
| pL3S2P21_STD | This work, available from AddGene | Cat# 165805 |
| pspy_STD | This work, available from AddGene | Cat# 165806 |
| pGGGSx4 | This work, available from AddGene | Cat# 165807 |
| pT2A | This work, available from AddGene | Cat# 165809 |
| pGGSx4 NT | This work, available from AddGene | Cat# 165810 |
| pGGSx4 CT | This work, available from AddGene | Cat# 165811 |
| p5xUAS-A1 | This work, available from AddGene | Cat# 165812 |
| 4xLexAOp-A1 | This work, available from AddGene | Cat# 165814 |
| 4xLexAOp-A2 | This work, available from AddGene | Cat# 165815 |
| 4xLexAOp-O1 | This work, available from AddGene | Cat# 165816 |
| 4xLexAOp-O2 | This work, available from AddGene | Cat# 165817 |
| 5xQUAS | This work, available from AddGene | Cat# 165818 |
| pGal4BD 1–147 | This work, available from AddGene | Cat# 165819 |
| pGAL4AD 768–881 | This work, available from AddGene | Cat# 165820 |
| pLexA BD | This work, available from AddGene | Cat# 165821 |
| pVP16 AD | This work, available from AddGene | Cat# 165822 |
| pQFBD | This work, available from AddGene | Cat# 165823 |
| pQFAD | This work, available from AddGene | Cat# 165824 |
| pGAL80 | This work, available from AddGene | Cat# 165825 |
| pQS | This work, available from AddGene | Cat# 165826 |
| pmCherry | This work, available from AddGene | Cat# 165828 |
| psfGFP | This work, available from AddGene | Cat# 165829 |
| pEGFP | This work, available from AddGene | Cat# 165830 |
| pEBFP2 | This work, available from AddGene | Cat# 165831 |
| pmCherry TL1 | This work, available from AddGene | Cat# 165832 |
| pmCherry TL5 | This work, available from AddGene | Cat# 165833 |
| pEGFP TL1 | This work, available from AddGene | Cat# 165834 |
| pEGFP TL5 | This work, available from AddGene | Cat# 165835 |
| pNPTII | This work, available from AddGene | Cat# 165836 |
| pPAC | This work, available from AddGene | Cat# 165837 |
| pBSR | This work, available from AddGene | Cat# 165838 |
| pHPH | This work, available from AddGene | Cat# 165839 |
| pBLE | This work, available from AddGene | Cat# 165840 |
| pTK | This work, available from AddGene | Cat# 165841 |
| pFCU1 | This work, available from AddGene | Cat# 165842 |
| pFC31 attB O1 | This work, available from AddGene | Cat# 165843 |
| pFC31 attB O2 | This work, available from AddGene | Cat# 165844 |
| pFC31 | This work, available from AddGene | Cat# 165845 |
| pWhite_CDS | This work, available from AddGene | Cat# 165846 |
| pUPD2 | This work, available from AddGene | Cat# 165856 |
| pVD2_purple | This work, available from AddGene | Cat# 165857 |
| P[acman]-A1 | This work, available from AddGene | Cat# 165858 |
| P[acman]-A2 | This work, available from AddGene | Cat# 165859 |
| P[acman]-O1 | This work, available from AddGene | Cat# 165860 |
| P[acman]-O2 | This work, available from AddGene | Cat# 165861 |
| pR6Kg-A1 | This work, available from AddGene | Cat# 165862 |
| pR6Kg-A2 | This work, available from AddGene | Cat# 165863 |
| pR6Kg-O1 | This work, available from AddGene | Cat# 165864 |
| pR6Kg-O2 | This work, available from AddGene | Cat# 165865 |
| pR6Kg-A1spm | This work, available from AddGene | Cat# 165866 |
| pColE1-AlphaOmega | This work, available from AddGene | Cat# 165867 |
| pEGFP-NT-Tagging | This work, available from AddGene | Cat# 165868 |
| pEGFP-CT-Tagging | This work, available from AddGene | Cat# 165869 |
| pmCherry-NT-Tagging | This work, available from AddGene | Cat# 165870 |
| pmCherry-CT-Tagging | This work, available from AddGene | Cat# 165871 |
| pMiniwhiteA1 | This work, available from AddGene | Cat# 165872 |
| pMiniWhiteA2 | This work, available from AddGene | Cat# 165873 |
| pGMR-WhiteA2 | This work, available from AddGene | Cat# 165874 |
| p3XP3-WhiteA2 | This work, available from AddGene | Cat# 165875 |
| pHSP70B-CP6-nptII | This work, available from AddGene | Cat# 165876 |
| pHSP70B-CP6-pac | This work, available from AddGene | Cat# 165877 |
| pHSP70B-CP6-bsr | This work, available from AddGene | Cat# 165878 |
| pHSP70B-CP6-hph | This work, available from AddGene | Cat# 165879 |
| pHSP70B-CP6-ble | This work, available from AddGene | Cat# 165880 |
| pHSP70B-CP6-TK | This work, available from AddGene | Cat# 165881 |
| pHSP70B-CP6-FCU1 | This work, available from AddGene | Cat# 165882 |
| pG418R-GMRWhite | This work, available from AddGene | Cat# 165883 |
| pPuroR-GMRWhite | This work, available from AddGene | Cat# 165884 |
| pBlastR-GMRWhite | This work, available from AddGene | Cat# 165885 |
| pHygroR-GMRWhite | This work, available from AddGene | Cat# 165886 |
| pRMCE{G418R-5FCS-GMRWhite}-VasaphiC31 | This work, available from AddGene | Cat# 165887 |
| pRMCE{PuroR-5FCS-GMRWhite}-VasaphiC31 | This work, available from AddGene | Cat# 165888 |
| pRMCE{BlastR-5FCS-GMRWhite}-VasaphiC31 | This work, available from AddGene | Cat# 165889 |
| pG418R-MCS-GAL4-MiniWhite | This work, available from AddGene | Cat# 165890 |
| pPuroR-MCS-GAL4-MiniWhite | This work, available from AddGene | Cat# 165892 |
| pBlastR-MCS-GAL4-MiniWhite | This work, available from AddGene | Cat# 165894 |
| pG418R-MCS-LexA-VP16-GMRWhite | This work, available from AddGene | Cat# 165895 |
| pG418R-MCS-LexA-VP16-MiniWhite | This work, available from AddGene | Cat# 165896 |
| pPuroR-MCS-LexA-VP16-GMRWhite | This work, available from AddGene | Cat# 165897 |
| pPuroR-MCS-LexA-VP16-MiniWhite | This work, available from AddGene | Cat# 165898 |
| pBlastR-MCS-LexA-VP16-MiniWhite | This work, available from AddGene | Cat# 165900 |
| pG418R-MCS-QF-MiniWhite | This work, available from AddGene | Cat# 165902 |
| pBlastR-MCS-QF-MiniWhite | This work, available from AddGene | Cat# 165906 |
| pBlastR-5xUAS-sfGFP-GMRWhite | This work, available from AddGene | Cat# 165907 |
| pBlastR-5xUAS-mCherry-GMRWhite | This work, available from AddGene | Cat# 165908 |
| pBlastR-5xUAS-sfGFP-MiniWhite | This work, available from AddGene | Cat# 165910 |
| pBlastR-5xUAS-mCherry-MiniWhite | This work, available from AddGene | Cat# 165911 |
| pBlastR-5xUAS-EBFP2-MiniWhite | This work, available from AddGene | Cat# 165912 |
| pBlastR-12xLexAOp-sfGFP-MiniWhite | This work, available from AddGene | Cat# 165916 |
| pBlastR-12xLexAOp-mCherry-MiniWhite | This work, available from AddGene | Cat# 165917 |
| pBlastR-12xLexAOp-EBFP2-MiniWhite | This work, available from AddGene | Cat# 165918 |
| pBlastR-5xQUAS-mCherry-GMRWhite | This work, available from AddGene | Cat# 165920 |
| pBlastR-5xQUAS-EBFP2-GMRWhite | This work, available from AddGene | Cat# 165921 |
| pBlastR-5xQUAS-sfGFP-MiniWhite | This work, available from AddGene | Cat# 165922 |
| pBlastR-5xQUAS-mCherry-MiniWhite | This work, available from AddGene | Cat# 165923 |
| pBlastR-5xQUAS-EBFP2-MiniWhite | This work, available from AddGene | Cat# 165924 |
|
| ||
| Software and algorithms | ||
|
| ||
| Prism 7 software v9.1.1 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| Adobe Illustrator Creative Cloud | Adobe | https://www.adobe.com/creativecloud.html |
| Adobe Photoshop Creative Cloud | Adobe | https://www.adobe.com/creativecloud.html |
| Search Tool for the Retrieval of Interacting Genes/Proteins | STRING (https://www.string-db.org) | Szklarczyk et al., 2019 |
| Cytoscape v3.8.2 | https://cytoscape.org | Shannon et al., 2003 |
| Zen Software Blue Version 2.3 pro HWL | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html |
| Codon Optimization Tool | IDT | http://www.idtdna.com/pages/tools/codon-optimization-tool?returnurl=%2FCodonOpt |
| Splice Site Prediction by Neural Network | Berkley Drosophila Genome Project | https://www.fruitfly.org/seq_tools/splice.html |
| SnapGene v4.2.3 | GSL Biotech LLC | https://www.snapgene.com:443/products/snapgene/ |
|
| ||
| Other | ||
|
| ||
| ChromoTek GFP-Trap Dynabeads | ChromoTek | Cat# gtd-10, RRID:AB_2827592 |
| NuPAGE 10%, Bis-Tris protein gel | Thermo Fisher Scientific | NP0315BOX |
| Reprosil-Pur Basic C18 beads | Dr.Maisch GmbH, Germany | R119.b9.3 |
| Fused silica tubing | IDEX Health | FS-110 |
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Koen Venken; Email: koen.j.t.venken@gmail.com, Phone: 1-713-798-3698.
Materials availability
There are no restrictions on material availability of any reagent produced in this work. All plasmids generated in this study are made available through Addgene (http://www.addgene.org/). Generated plasmid materials and distinct identifiers are summarized in Tables S1 and S2 and the Key resources table. All fly stocks generated in this study (Table S3; Key resources table) are made available through the Bloomington Drosophila Stock Center (https://bdsc.indiana.edu/). Fly stocks, primer sequences used in this study and stock numbers are summarized in the Key resources table.
Data and code availability
Data for Figure 5F is available as Table S4. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD026579. This paper does not report original code. Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Animals used in this study: Drosophila melanogaster, males and females, aged 2–10 days. Drosophila melanogaster strains used and developed in this study are listed in Table S3 and the Key resources table.
Bacteria
Microbial strains used in this study: Chemocompetent Escherichia coli strain DH10B (Invitrogen/Thermo Fisher Scientific), Transformax EPI300 (Epicenter/Lucigen), strain EL350 recombineering bacterial cells (kind gift from Donald Court, National Cancer Institute), and Transformax EC100D pir-116 (Epicenter/Lucigen).
METHOD DETAILS
Molecular biology enzymes
Restriction enzyme cloning was accomplished using: BsaI-HFv2 (New England Biolabs, #R3733), Esp3I (New England Biolabs, R0734) and T4 Ligase (Promega, M1801). PCR was performed using proofreading enzymes iProof High-Fidelity DNA Polymerase (Biorad, #1725301), Phusion High-Fidelity DNA Polymerase (New England Biolabs, M0530), or Q5 High-Fidelity DNA Polymerase (New England Biolabs, M0491). Molecular biology experiments, including in silico designs and experimentation, as well as plasmid maps, were designed using SnapGene software (https://www.snapgene.com:443/products/snapgene/) (GSL Biotech LLC). All plasmids developed in this project are listed in Tables S1 and S2 and the Key resources table.
General molecular biology
Molecular biology cloning was confirmed by agarose gel DNA electrophoresis after restriction enzyme digestion to expose diagnostic DNA bands of specific lengths, as well as control uncut plasmid to eliminate unwanted multimeric assemblies. All end products were verified by Sanger sequencing using GeneWiz (https://www.genewiz.com/) or Eurofins genomics (https://eurofinsgenomics.com/). PCR purifications, gel extractions and all plasmid preparations, excluding all bacterial artificial chromosome (BAC) backbone vectors, were performed using the QIAquick PCR Purification Kit (QIAGEN, #28106), QIAquick Gel Extraction Kit (QIAGEN, #28506), and QIAprep Spin Miniprep Kit (QIAGEN, #27106), respectively according to manufacturer’s instruction. All BAC plasmids were prepared using the ChargeSwitch-Pro plasmid miniprep kit (Invitrogen/Thermo Fisher Scientific, CS30250) according to manufacturer’s instruction. Primers were obtained from MilliporeSigma.
Chemical reagents
The following drugs were used: G418 sulfate (VWR 97063–060), puromycin HCl (VWR 97064–280), blasticidin S (VWR 71002–676), hygromycin B (VWR AAJ6068103), zeocin (Alfa Aesar J67140), phleomycin (VWR AAJ67027–8EQ), ganciclovir (TCI America 50–155-694), and 5-fluorocytosine (TCI America 50–014-34810).
Antibodies for immunofluorescence
The following primary antibodies were used for immunofluorescence: polyclonal rabbit GFP (Invitrogen/Thermo Fisher Cat# A11122, RRID:AB_221569, 1/500), mouse monoclonal anti-Dlg (Developmental Studies Hybridoma Bank Cat# 4F3, RRID:AB_528203, 1/100), rat monoclonal mCherry (Invitrogen/Thermo Fisher Cat# M11217, RRID:AB_2536611, 1/500), and mouse monoclonal dCsp (Developmental Studies Hybridoma Bank Cat# Ab49, RRID:AB_2307340, 1/100). Secondary antibodies were obtained from Invitrogen and were used at a final concentration of 1/500: chicken anti-rabbit AlexaFluor 488 (Invitrogen/Thermo-Fisher, Cat# A-21441, RRID:AB_2535859), goat anti-mouse AlexaFluor 568 (Invitrogen/Thermo-Fisher, Cat# A11004, RRID:AB_2534072), goat anti-Rat AlexaFluor 568 (Invitrogen/Thermo-Fisher, Cat# A-11077, RRID:AB_141874), and chicken anti-mouse AlexaFluor 568 (Invitrogen/Thermo-Fisher, Cat# A21200, RRID:AB_2535786). Images were captured with Zen Software (Blue Version 2.3 pro HWL, Zeiss).
Computational resources
All synthesized coding DNA sequences were first codon optimized for expression in Drosophila melanogaster using an online tool (http://www.idtdna.com/pages/tools/codon-optimization-tool?returnurl=%2FCodonOpt, IDT), and then analyzed for splice acceptor and donor sites using a splice site prediction online tool (https://www.fruitfly.org/seq_tools/splice.html, BDGP) which were then manually removed.
Dual-kingdom (counter)selection markers
We generated an expression cassette for the selection marker Neomycin phosphotransferase II/nptII (G418R) in the plasmid pBluescript II KS (+) (pBS-KS) (Stratagene/Agilent). The Hsp70 promoter was amplified from plasmid template pCasper-HS, obtained from the Drosophila Genomics Resource Center (1215) (Steller and Pirrotta, 1986), using primers Hsp70-F and Hsp70-CP6-R. The CP6 promoter was amplified from genomic DNA isolated from the TP977 strain (kind gift from Anthony Poteete, University of Massachusetts Medical School) (Poteete et al., 2006) using primers Hsp70-CP6-F and CP6-Neo-R (for primers see Table S5 and the Key resources table). The open reading frame for a non-synthetic codon optimized version of G418 was amplified from plasmid template pEGFP-N1 (Clontech/Takara Bio) using primers CP6-G418-F and HSVTK-R1. Next, a secondary overlap extension PCR (Horton et al., 1989) was performed using purified products of all three primary PCR products (Hsp70 promoter, CP6 promoter and G418R open reading frame) as templates, and primers Hsp70-F and HSVTK-R2, resulting in the Hsp70-CP6-G418R-HSV-TKpA fragment. This fragment was cloned as a NotI cut fragment in a NotI linearized pBS-KS plasmid, resulting in pBS-KS-Hsp70-CP6-G418-HSV-TKpA maintained in the DH10B bacterial strain (Thermo Fisher Scientific) that can grow on LB plates with 100 μg/ml Ampicillin and 30 μg/ml Kanamycin, demonstrating functionality of the hybrid Hsp70/CP6 promoter in bacteria.
A cloning cassette compatible with downstream sequence and ligation independent cloning (SLIC) (Li and Elledge, 2007), as well as chew-back, anneal and repair (CBAR) cloning, now commonly known as Gibson cloning (Gibson et al., 2009), was cloned in attB-Pacman-ApR (Venken et al., 2006). The Gibson cloning cassette consisting of the dual-kingdom promoter, Hsp70-CP6, separated from the HSV-TKpA transcriptional terminator, by a distinct NheI restriction enzyme site, was obtained by primary PCR amplification of the Hsp70-CP6 part using template pBS-KS-Hsp70-CP6-G418-HSV-TKpA and primers Hsp70-Ascl-F and Hsp70-CP6-TKpA·R1, followed by a secondary PCR using the primary template as PCR and primers Hsp70-Ascl-F and Hsp70CP6-TKpA-R2-Pacl, and cloned as an AscI/PacI cut fragment into an AscI/PacI linearized attB-P[acman]-ApR plasmid resulting in plasmid P[acman]-attB-Hsp70CP6-NheI-TKpA. Plasmid copy number induction was performed using fosmid autoinduction solution performed as described (Epicenter/Lucigen).
Selection/counterselection markers were commercially synthesized as Drosophila codon optimized fragments (Thermo Fisher Scientific) and amplified using SLIC and CBAR/Gibson compatible primers as follows: EGFP-SLIC was amplified from the synthetic construct encoding the MiMIC transposable element (Venken et al., 2011) with EGFP-F and EGFP-R; G418-SLIC was amplified from the G418 synthetic fragment (Davies and Smith, 1978) with G418-F and G418-R; Puro-SLIC was amplified from the Puro synthetic fragment (Vara et al., 1985) with Puro-F and Puro-R; Blast-SLIC was amplified from the Blast synthetic fragment (Itaya et al., 1990) with Blast-F and Blast-R; Hygro-SLIC was amplified from the Hygro synthetic fragment (Gritz and Davies, 1983) with Hygro-F and Hygro-R; Zeo-SLIC was amplified from the Zeo synthetic fragment (Oliva-Trastoy et al., 2005) with Zeo-F and Zeo-R; GCV-SLIC was amplified from the GCV synthetic fragment (Black, Kokoris and Sabo, 2001) with GCV-F and GCV-R; and 5FC-SLIC was amplified from the 5FC synthetic fragment (Erbs et al., 2000) with 5FC-F and 5FC-R. PCR amplified SLIC-fragments were subcloned in NheI linearized F-2-5-attB-Hsp70-CP6-TKpA as previously described (Li and Elledge, 2007), and annealing reactions transformed in home-made chemocompetent EPI300 cells (Epicenter/Lucigen). Plasmid copy number induction was performed using fosmid autoinduction solution performed as described (Epicenter/Lucigen), resulting in plasmids: P[acman]-attB-Hsp70-CP6-EGFP-TKpA, P[acman]-attB-Hsp70-CP6-G418-TKpA, P[acman]-attB-Hsp70-CP6-Puro-TKpA, P[acman]-attB-Hsp70-CP6-Blast-TKpA, P[acman]-attB-Hsp70-CP6-Hygro-TKpA, P[acman]-attB-Hsp70-CP6-Zeo-TKpA, P[acman]-attB-Hsp70-CP6-GCV-TKpA, and P[acman]-attB-Hsp70-CP6-5FC-TKpA.
Transgenic selectable/counterselectable flies
Plasmids P[acman]-attB-Hsp70-CP6-EGFP-TKpA, P[acman]-attB-Hsp70-CP6-G418-TKpA, P[acman]-attB-Hsp70-CP6-Puro-TKpA, P[acman]-attB-Hsp70-CP6-Blast-TKpA, P[acman]-attB-Hsp70-CP6-Hygro-TKpA, P[acman]-attB-Hsp70-CP6-Zeo-TKpA, P[acman]-attB-Hsp70-CP6-GCV-TKpA, and P[acman]-attB-Hsp70-CP6-5FC-TKpA were prepared for microinjection using the QIAprep Spin Miniprep Kit (QIAGEN) according to the handbook and concentrations adjusted to 150 ng/μl. Transgenic flies were generated by microinjection using a custom injection stock, containing a germline driven ΦC31 source on the X (Bischof et al., 2007) (kind gift from Johannes Bischof and Konrad Basler, University of Zurich), and an attP docking site on the third chromosome (Venken et al., 2006): y[1] M{RFP[3xP3.PB] GFP[E.3xP3] = vas-int.B}ZH-2A w[*]; PBac{y[+]-attP-3B}VK00033 with putative integration events (identified by mini-white expression) balanced and correct integration events verified molecularly as previously described (Venken et al., 2009, 2010). The resulting strains are listed in Table S3 and the Key resources table.
Preparation of drug containing fly food
Fly food was made in a large kettle with mixer with the following ingredients per 1 l water: 6.4 g agar (Genesee Scientific, 66–103), 30 g yeast (Red Star, Webstaurant), 70 g cornmeal (Luby’s Cafeteria), 55 g dextrose (VWR, JT1910–5), 30 g sucrose (VWR, BDH9308), 4 mL 20% tegosept dissolved in Ethanol (Genesee Scientific, 20–258), and 4 mL propionic acid (Sigma-Aldrich, P1386). Agar is dissolved in heating water first, followed by adding yeast, cornmeal, dextrose and sucrose. When everything is dissolved, heat is turned down, fly food allowed to cool to 70°C, and tegosept and propionic acid carefully added to avoid bubbling.
A piston-driven food dispenser, DAB-8–4 (Filamatic) was used to precisely dispense 8 mL of food into each vial. Food was allowed to cool and air dry overnight in covered but unplugged vials. 20 holes were then poked three-fourths of the way down into the food using a home-made hole puncher. Appropriately solubilized drug was added to the fly food at 50X concentration, i.e., 160 μl for 8 ml. Drug was allowed to permeate into the food for 48 hours prior to introducing adult flies. Drugs were dissolved in MilliQ water (G418 sulfate, puromycin, blasticidin S, and hygromycin B), pH7.0 HEPES buffer (Zeocin and Phleomycin), 0.1N NaOH (ganciclovir) or 1x PBS (5-fluorocytosine).
Generation of the isogenized IsoY1 strain
Fly strains with the following genotypes, y[1] w[67c23] (BDSC #6599) and P{w[+mW.Scer\FRT.hs] = RS3}l(1)CB-6411-3[1], w[1118]/FM7h (BDSC #6878), were used in conjunction with a fly stock double balanced for the second and third chromosomes, to generate a fly stock isogenized for all three major chromosomes: X (y[1] w[67c23] from BDSC #6599), 2 and 3 (both from BDSC #6878). This strain is abbreviated to IsoY1.
Effective drug concentration determination
For each drug titration, self-crosses were set up between the relevant drug resistant or sensitive strain (G418R, PuroR, BlastR, HygroR, ZeoR, GCVS, or 5FCS), or the control strain (EGFP or IsoY1) with 3 mating pairs per vial to determine the effective selection/counterselection concentration (ESC or ECC). For each drug, four vials were set up per drug concentration per fly strain tested on said drug. Flies were allowed to mate and lay eggs for one week at 25°C in an acclimated incubator before being removed. Larvae were left to develop at 25°C for an additional two weeks. Surviving adult flies in each vial at the end of three weeks, if any, were counted, and results normalized to vehicle control treated vials and reported as percent survival ± SEM. We averaged at least three replicates per drug concentration per drug as occasionally a vial would not result in a fertile cross. Heat shock induction of marker expression was performed as follows: crosses with 3 pairs per vial were setup on vials with drug and allowed to lay overnight at 25°C. After 24 hours, developing eggs and larvae were heat shocked by placing vials into a 37°C water bath for 30 min. Care was taken to ensure vials were only 3/4th submerged in water to improve adult fly survival while still providing heat shock to eggs and larvae in the food. After 30 min, vials were removed, dried, and placed back into the incubator. Heat shock was repeated every 2 days for a week before adults were discarded and remaining larvae allowed to develop at 25°C for an additional 2 weeks as above. This frequency balances marker expression with overall survival of heat shock treatment. Significance of change in percent survival per strain per drug concentration was determined via 2-way ANOVA using Dunnett’s multiple comparisons test at α = 0.05. A detailed protocol for determining effective drug concentrations is provided elsewhere (Matinyan et al., 2021).
Specificity of marker/drug combinations
Drug specificity for each selection and counterselection marker was characterized by exposing all marker expressing strains, except ZeoR (G418R, PuroR, BlastR, HygroR, GCVS, or 5FCS) as well as the control strain (EGFP) to each of the six drugs (G418, puromycin, blasticidin S, hygromycin B, ganciclovir, or 5-fluorocytosine) at their determined ESC or ECC (Figures 1C–1H). Strain self-crosses were setup as described above with four crosses per strain on food containing drug or vehicle control, added as described above. Results were averaged with at least three replicates per strain per drug and normalized to vehicle control treated vials and reported as percent survival. Statistical significance was determined via multiple t test using the Holms-Sidack method with an α = 0.05.
Robustness of marker multiplexing
Homozygous drug resistant and drug sensitive fly strains were crossed to homozygous control (EGFP) flies to generate marker/EGFP heterozygotes: G418R/EGFP, PuroR/EGFP, BlastR/EGFP, GCVS/EGFP and 5FCS/EGFP. Heterozygous fly strains were then crossed to each other as follows: G418R/EGFP to BlastR/EGFP, PuroR/EGFP to G418R/EGFP, BlastR/EGFP to PuroR/EGFP, BlastR/EGFP to GCVS/EGFP, BlastR/EGFP to 5FCS/EGFP, and 5FCS/EGFP to GCVS/EGFP. All six cross schemes were set up with reciprocal male and female parents. Each cross was further subdivided into four crosses under different drug conditions: vehicle control, one of the drugs, the other drug, or both drugs. Food was drugged by adding either 160 μl of a single drug at 50X its ESC/ECC or 80 μl of both drugs at 100X ESC/ECC when two drugs were used. Vehicle control vials were made with 160 μl of vehicle control if both drugs shared the same solvent or with 80 μl of each different solvent if the two drugs were in different solvents. Fly crosses were setup as described above with four replicates per drug conditions per experiment. The number of surviving adult flies was normalized to vehicle control in each experiment and results were reported as percent survival averaged from at least three fertile replicates per cross condition per experiment. On occasion one of four crosses per condition would not produce progeny and was excluded from analysis.
Genotyping dually (counter)selected flies
From each of the crosses described above, 24 flies were collected randomly for each of the four drug conditions. Single flies were put into each well of a 96-well plate along with a 5 mm stainless steel ball and 500 μL of genomic lysis buffer of the ZR-96 Quick-gDNA kit (Zymo Research, D3010). Plates were sealed and placed into a tissue homogenizer, 1600 MiniG (SPEX SamplePrep). Flies were homogenized for 45 s at 1000 rpm. DNA was extracted using the ZR-96 Quick-gDNA kit according to the manufacturer’s instructions (Zymo Research). Flies were genotyped using a set of three PCRs per extracted DNA plate each using BioReady rTaq DNA polymerase (Bulldog Bio, BSAX050) and primer pairs specific to one of the three potential markers (Table S5; Key resources table).
Generation of a double-docking site fly stock
A double attP docking site chromosome was generated by crossing y[1] M{RFP[3xP3.PB] GFP[E.3xP3] = vas-int.B}ZH-2A w[*]; PBac{y[+]-attP-3B}VK00033 males (Venken et al., 2009, 2010) to y[1] M{RFP[3xP3.PB] GFP[E.3xP3] = vas-int.B}ZH-2A w[*]; PBac{y[+]-attP-9A}VK00020 females (Venken et al., 2009, 2010), resulting in transheterozygous female progeny with the genotype, y[1] M{RFP[3xP3.PB] GFP[E.3xP3] = vas-int.B}ZH-2A w[*]; PBac{y[+]-attP-3B}VK00033/PBac{y[+]-attP-9A}VK00020 that were backcrossed to double balanced males, w[*] dlg1[14] P{w[+mW.hs] = FRT(w[hs])}101/FM7a; PBac{w[+mC] = PB}CG11583[c01124] P{ry[+t7.2] = neoFRT}80B/TM3, Sb[1] (BDSC #36283). Single double balanced flies, containing y[1] M{RFP[3xP3.PB] GFP[E.3xP3] = vas-int.B}ZH-2A w[*] and putatively a recombinant double attP docking site chromosome, PBac{y[+]-attP-3B}VK00033, PBac{y[+]-attP-9A}VK00020, were backcrossed to double balanced flies, whose double balanced progeny was self-crossed and homozygosed, generating the y[1] M{RFP[3xP3.PB] GFP[E.3xP3] = vas-int.B}ZH-2A w[*]; PBac{y[+]-attP-3B}VK00033, PBac{y[+]-attP-9A}VK00020 stock. Presence of both docking sites was confirmed by PCR amplification from genomic DNA from putative 2x docking site stain flies using primers VK33_RIGHT_F, VK33_RIGHT_R, VK33_LEFT_F, and VK33_LEFT_R for VK00033 and VK20_RIGHT_F, VK20_RIGHT_R, VK20_LEFT_F and VK20_LEFT_R for VK00020 (see Table S5 and the Key resources table for primer sequences). Flies are available from the Bloomington Drosophila Stock Center (https://bdsc.indiana.edu/; see Table S3 and the Key resources table).
GoldenBraid 2.0 cloning reactions
GoldenBraid 2.0 cloning (GB2.0) was used to generate and assemble all DNA parts and vector toolkit elements. GB2.0 cloning is an assembly method where all part vectors, destination vector, restriction enzyme, and ligase are mixed together in a single reaction (Sarrion-Perdigones et al., 2011, 2013). Briefly, 40 ng of destination vector and 40 ng of each donor-part vector are mixed with 1 μL of the appropriate restriction enzyme (BsaI-HFv2 or Esp3I, New England BioLabs), 1 μL of T4 Ligase (Promega), and 2 μL of the Ligase 10x Buffer (Promega) in a final volume of 20 μL. Reactions were set up in a standard thermocycler (Applied Biosystems) with at least 25 cycles of digestion/ligation reactions (2′ at 37°C, 5′ at 16°C with a final extension step of 10’ at 72°C). More cycles are recommended for complex assemblies of more than 3 parts or involving parts with large size differences (> 2kb) up to 50 cycles.
GB2.0 cloning depends on an extensive library of compatible DNA elements as reagents for assembly steps (Sarrion-Perdigones et al., 2011, 2013). DNA parts are incorporated into the system, or domesticated, by addition of appropriate GB2.0 cloning overhangs (the grammar of which determines the order of part assembly) via PCR amplification or commercial synthesis of compatible fragments (Figure S10A; Engler et al., 2008; Sarrion-Perdigones et al., 2013). Fragments are then cloned into one of three universal part domesticator (pUPD) vector backbone via Esp3I enzyme digestion: pUPD (kind gift from Diego Orsaez, The Institute for Plant Molecular and Cellular Biology), pUPD2 (this work), and pUPD3 (Addgene, #118043) (Sarrion-Perdigones et al., 2019), followed by transformation of 2 μl of reaction product into chemocompetent E.coli DH10B cells (Epicenter/Lucigen; Sarrion-Perdigones et al., 2013, 2019). All domesticated parts were confirmed via restriction endonuclease fingerprinting (New England BioLabs), agarose gel electrophoresis visualization and sequence verified by Sanger sequencing (GeneWiz, Eurofins Genomics). GB2.0 vectors and parts construction are described below.
Cloning of selectable GAL4/UAS vector pairs
GAL4/UAS and LexA/LexAop binary systems driver and response vectors were cloned using GB2.0. GAL4 or LexA driver vector assembly began with cloning of a G418 resistance cassette, as described above, with one of three GAL4- or LexA-expressing cassettes, each driven by distinct DNA enhancer elements, previously characterized by the FlyLight consortium to produce intermediates pR76H03-GAL4G418R, pR20A02-GAL4G418R, and pR70B04-LexAG418R (Jenett et al., 2012). Expression cassettes consist of GB2.0 compatible enhancer element fragments, R76H03, R20A02, or R70B04 (Jenett et al., 2012) were assembled with the Drosophila synthetic core promoter dSCP fragment (Pfeiffer et al., 2008), GAL4 or LexA DNA binding domain fragment, GAL4BD1−147 or LexABD, (Luan et al., 2006) flexible peptide linker fragment, GGGSx4 (4 repeats of gly-gly-gly-ser; Venken et al., 2011), GAL4 (GAL4AD768−881) or VP16 (VP16AD) activation domain fragment (Luan et al., 2006), and the late transcription terminator of simian virus 40, SV40L (Angelichio et al., 1991). Intermediates were then further assembled with a ΦC31 attB site fragment (Venken et al., 2006) as described above, to produce intermediates pattB-R76H03-GAL4G418R, pattB-R20A02-GAL4G418R, pattB-R70B04-GAL4G418R, and pattB-R70B04-LexAG418R. A final assembly with a visible eye marker cassette, as described above, produced the final driver vectors, pattB-R76H03-GAL4G418R-5xGMRwhite, pattB-R20A02-GAL4G418R-5xGMRwhite, pattB-R70B04-GAL4G418R-5xGMRwhite, and pattB-R70B04-LexAG418R-Mini-white (Table S2; Key resources table). UAS/LexAop response vectors began by assembly of a blasticidin S resistance cassette, as described above, with one of three UAS reporter cassettes consisting of a synthetic 5xUAS or a 4xLexAOp element (Sarrion-Perdigones et al., 2013), dSCP part (Pfeiffer et al., 2008), Drosophila codon optimized coding sequence parts for one of two fluorescent proteins, sfGFP (Pédelacq et al., 2006) or mCherry (Shaner et al., 2004), and SV40L (Angelichio et al., 1991). Assembly produced intermediates pUAS-sfGFPBlastR, pUAS-mCherryBlastR, and pLexAOp-sfGFPBlastR. These were then further assembled with a ΦC31 attB site (Groth et al., 2004) to produce pattB-UAS-sfGFPBlastR, pattB-UAS-mCherryBlastR, pattB-LexAOp-sfGFPBlastR. Final assembly with a visible eye marker cassette, as described above, produced the final UAS/LexAop response vectors, pattB-UAS-sfGFPBlastR-GMRwhite, pattB-UAS-mCherryBlastR-GMRwhite, pattB-LexAOp-sfGFPBlastR-Miniwhite (Table S2; Key resources table).
Single-step, dual selection co-transgenesis
Plasmids pairs pR76H03-GAL4G418R-5xGMRwhite and pUAS-mCherryBlastR-5xGMRwhite, pR20A02-GAL4G418R-5xGMRwhite and pUAS-sfGFPBlastR-5xGMRwhite, and pR70B04-LexAG418R–Mini-white and pLexAOp-sfGFPBlastR–Mini-white were mixed together in a 1:1 ratio by weight (ng) at a final concentration of ~500 ng/ul, and co-injected into early stage embryos of the y[1] M{RFP[3xP3.PB] GFP[E.3xP3] = vas-int.B}ZH-2A w[*]; PBac{y[+]-attP-3B}VK00033, PBac{y[+]-attP-9A}VK00020 double attP docking site fly strain, abbreviated J15; VK00033, VK00020. Surviving adults were individually crossed to IsoY1 flies of opposite sex on food with G418 sulfate (300 μg/ml) and Blasticidin S (35 μg/ml) as previously described. Transgenic offspring were then individually balanced with a third chromosome balancer line (w1118; Sb/TM6b) on regular food. Resulting balanced progeny were then self-crossed to generate third chromosome isogenized, double transgenic GAL4/UAS or LexA/LexAop stocks. Flies are available through the Bloomington Drosophila Stock Center (https://bdsc.indiana.edu/; Table S3; Key resources table).
Testing synthetic GAL4 driver functionality
Prior to generating dual transgenic binary expression system animals, we first tested the expression and function of our generated GAL4 driver vectors. Transgenic animals were generated as described above and selected for and visually confirmed by screening for expression of 5xGMR white, producing a strong red eye phenotype, in selected animals. Transgenic animals were then crossed to a previously described strong UAS reporter line P{10XUAS-IVS-GFP}attP2 (BDSC #32201) (Pfeiffer et al., 2010). Adult brains from the resulting progeny were dissected and immunostained for GFP expression as described below (Figure S7).
Immunofluorescent staining
Staining and imaging was performed as previously described (Gnerer et al., 2015). Briefly, adult brains were dissected in ice-cold PBS and fixed in 4% PFA/PBS for one hour. Next, the brains were rinsed 3 times with PBS-0.5% Triton X-100 (PBT) and then washed three times for 20 minutes in PBT at room temperature. The brains were then blocked in 5% normal goat serum (NGS) in PBT for one hour at room temperature. Samples were incubated in 5% NGS/PBT with primary antibody for 48 hours at 4°C. After three short rinses and three 20 minute washes in PBT, brains were incubated in 5% NGS/PBT with secondary antibody for 48 hours at 4°C. Brains were then rinsed three times and washed three times for 20 minutes at room temperature and then for 2 days at 4°C. Finally, brains were mounted in SlowFade mounting medium (Invitrogen/Thermo Fisher Scientific) and covered with a no. 0 glass coverslip that was separated from the slide by two strips of scotch tape. Immunostained brains were imaged with an upright Zeiss fluorescence Microscope (Axio Imager M2) equipped with an ApoTome2 (Zeiss) and a Hamamatsu Flash 4.0 V3 sCMOS Camera (Hamamtsu Photonics).
Generation of upgradeable balancer stocks
Double attP docking site-containing balancer chromosomes for the 1st, 2nd, and 3rd chromosomes, FM7c, CyO, and TM6B, Tb[1], respectively, were generated through P element mobilization using the P transposase source Δ2–3 (Robertson et al., 1988), obtained from the Bloomington Drosophila Stock Center, y[1] w[1]; Ki[1] P{ry[+t7.2] = Delta2-3}99B (BDSC #4368), and available double attP docking sites previously generated for ΦC31 integrase-mediated cassette exchange, located on the second chromosome (25C and 52D) (kind gifs of Jack Bateman, Bowdoin College; Bateman et al., 2006). P element-mediated mobilization resulted in five double attP docking site-containing balancer chromosomes that are available from the Bloomington Drosophila Stock Center (https://bdsc.indiana.edu/), abbreviated to FM7c-25C-1B, CyO-25C-4A, CyO-52D-3A, TM6B-25C-5A, TM6B-52D-1A, respectively (see Table S3 and the Key resources table).
Selection & counterselection RMCE cassettes
GB2.0 cloning was used to generate three all-in-one recombinase mediated cassette exchange (RMCE) genetic circuits. Each vector consists of one of three drug resistance markers (conferring resistance to G418, blasticidin S, or puromycin) and the 5-fluorocytosine sensitivity marker along with a synthetic white coding sequence visual eye marker all flanked on either end by inverted attP sites. All vectors also express a germline ΦC31 integrase source (Bischof et al., 2007) in the backbone. Briefly, each of the three resistance marker cassettes was combined with the 5-fluorocytosine sensitivity cassette, resulting in cloning intermediates pG418R-5FCS, pBlastR-5FCS or pPuroR5FCS. Marker cassettes are GB2.0 compatible versions of our dual-kingdom expression cassette generated via GB2.0 assembly from basic DNA parts. Next, a ΦC31 attB site (Groth et al., 2004) fragment was added onto the 5′ end of each intermediate to generate pattB-G418R-5FCS, pattB-BlastR-5FCS or pattB-PuroR5FCS. These were then each assembled with a synthetic visual eye marker cassette to produce pattB-G418R-5FCS-5xGMRwhite, pattB-BlastR-5FCS-5xGMRwhite or pattB-PuroR5FCS-5xGMRwhite. A second inverted ΦC31 attB site (Groth et al., 2004) fragment was cloned onto the 3′ end of each intermediate to produce pattB-G418R-5FCS-5xGMRwhite-attB, pattB-BlastR-5FCS-5xGMRwhite-attB or pattB-PuroR5FCS-5xGMRwhite-attB. These were then cloned together with a germline expressing ΦC31 integrase cassette (Groth et al., 2004; Bischof et al., 2007) to generate the final all-in-one RMCE vectors: pRMCE{G418R-5FCS}, pRMCE{BlastR-5FCS}, and pRMCE{PuroR-5FCS} (Table S2; Key resources table).
Selection/counterselection balancer stocks
Transgenic flies were generated by microinjection of plasmids pRMCE{G418R-5FCS} and pRMCE{BlastR-5FCS} into a TM6B, Tb[1] RMCE balancer stock. Surviving adult flies were crossed to two Isoy1 strain animals of the opposite sex on untreated food as previously described. Transgenic progeny were screened based on the visual marker described above, then backcrossed to the original balancer strain and finally self-crossed. Stocks are available through the Bloomington Drosophila Stock Center (https://bdsc.indiana.edu/; Table S3; Key resources table).
Validating selection/counterselection stocks
Balancer chromosome stocks were validated by mixing equal numbers of non-virgin balancer females and non-virgin IsoY1 stock females in vials drugged with varying concentrations of G418, blasticidin S, or 5-fluorocytosine up to the previously determined ESC and ECC for IsoY1. Initial ECC experiments were repeated using higher 5-fluorocytosine concentrations as the original ECC was not fully effective at eliminating all balancer strain flies. Four replicates of two IsoY1 and two balancer chromosome stock females were set up per drug concentration per drug per balancer chromosome as described above. Surviving animals were visually sorted by genotype and counted. Results were reported as the average percentage of surviving flies of each genotype.
Selection/tagging cassettes for BAC upgrading
GB2.0 compatible G418 and blasticidin dual-kingdom marker expression cassettes were PCR amplified using overhang primers Marker-RECO1-F and Marker-RECO1-R to add 50 base pair arms on both 5′ and 3′ ends with homology to regions flanking the chloramphenicol bacterial resistance marker common to P[acman] BAC library genomic clones. Cassettes were then further amplified by secondary PCR using primers Marker-RECO2-F and Marker-RECO2-R to add GB2.0 cloning and XbaI restriction enzyme sites on both 5′ and 3′ ends (Table S5; Key resources table). Both cassettes were then cloned using GB2.0 into a compatible conditionally replicative vector backbone (Rakowski and Filutowicz, 2013), pGB2-R6Kγ-A1Spm, and sequence verified via commercial Sanger sequencing, resulting in plasmids, pG418R-RECO and pBlastR-RECO (Figure S8A). Similarly, a pair of fluorescent tag cassettes expressing either EGFP (Cormack et al., 1996) for N-terminal or mCherry (Shaner et al., 2004) for C-terminal protein tagging was cloned using GB2.0 methodology into an Alpha level GB2.0 vector as previously described (Sarrion-Perdigones et al., 2013). Cassettes consist of a human EF1a promoter piece (Kim et al., 1990) (cloning purposes only), an EGFP (Cormack et al., 1996) or mCherry coding sequence (Shaner et al., 2004), a loxP flanked ampicillin bacterial resistance marker, and an N- or C-terminal GGGSx4 peptide linker (Venken et al., 2011). Tagging cassettes were then PCR amplified using primers, Csp-RECO1-F and Csp-RECO1-R for the N-terminal EGFP cassette, and Nrx-RECO1-F and Nrx-RECO1-R for the C-terminal mCherry cassette, adding the 50 base pair of homologous sequence required for recombineering. In a secondary PCR, GB2.0 cloning and XhoI restriction cut sites were added using primers Csp-RECO2-F and Csp-RECO2-R for the N-terminal variant, and Nrx-RECO2-F and Nrx-RECO2-R for the C-terminal cassette. Both PCR fragments were then GB2.0 cloned into vector pGB2-R6Kγ-A1Spm and Sanger sequence verified resulting in plasmids pEGFPNT-RECO and pmCherryCT-RECO (Figure S8B).
Serial recombineering of P[acman] BAC clones
Genomic P[acman] BAC clones CH322–06D09 (Cysteine String Protein Csp), and CH322–154P15 (Neurexin IV protein, NrxIV) (Venken et al., 2009) were isolated as described above and electroporated into strain EL350 recombineering bacterial cells (kind gift from Donald Court, National Cancer Institute; Lee et al., 2001; Venken et al., 2009). The selection marker and tagging cassettes described above were linearized from their respective pGB2-R6Kγ-A1Spm vector backbones via XbaI or XhoI digestion, respectively. The genomic P[acman] BAC clones were upgraded through serial recombineering, using XhoI-released tagging cassettes during a first recombineering step, XbaI-released selection cassettes during a second recombineering step, and finally Cre-recombinase mediated reduction of the ampicillin resistance marker as previously described (Figure S8D; Venken et al., 2008). The resulting modified clones, CH322-06D09-N-EGFP-Csp-G418 and CH322-154P15-NrxIV-C-Cherry-Blast were isolated from EL350 cells, electroporated into, electrocompetent EPI300 cells (Epicenter/Lucigen), and DNA was isolated as previously described (Venken et al., 2006, 2009, 2010).
Selectable/tagged P[acman] BAC transgenics
Upgraded P[acman] BACs CH322-06D09-N-EGFP-Csp-G418, and CH322-154P15-NrxIV-C-Cherry-Blast were microinjected singly into a dual ΦC31 integrase source, single docking site fly stock, y[1] M{RFP[3xP3.PB] GFP[E.3xP3] = vas-int.B}ZH-2A w[*] v[1] P{y[+t7.7] = nos-ϕ31\int.NLS}X; PBac{y[+]-attP-3B}VK00033 (BDSC #32543) as previously described (Venken et al., 2009, 2010). Surviving adults were then crossed to Isoy1 flies on food with either G418 sulfate (350 μg/ml) or Blasticidin S (35 μg/ml) as previously described. Putative integration events were balanced as previously described via crossing to a third chromosome balancer strain (w1118; Sb/TM6b) on un-drugged food (Venken et al., 2006, 2009). Balanced transgenics were then isogenized via self-cross on untreated food to generate the final homozygous transgenic lines. Integration events were verified molecularly as previously described (Venken et al., 2009). The resulting flies are available through the Bloomington Drosophila Stock Center (https://bdsc.indiana.edu/; Table S3; Key resources table).
Co-immunoprecipitation of eGFP-tagged Csp
Equal number of adult male and female flies were frozen in liquid nitrogen. Approximately 4,500 fly heads were collected by sieving the frozen flies. The heads were homogenized in chilled NETN buffer (50mM Tris pH 7.3, 170mM NaCl, 1mM EDTA, 0.5% NP-40, and protease and phosphatase inhibitors) using a 1600 MiniG automated tissue homogenizer and cell lyser (SPEX SamplePrep, NJ, USA) at 1250 rpm for 20 s in 6 rounds with 30 s gaps between each round when samples were on ice. The lysates were centrifuged at 12,000 g for 5 minutes to pellet the exoskeleton and undissolved material. The lysates were then sonicated and ultracentrifuged at 100,000 g for 20 minutes at 4°C to clear any undissolved material, cellular polymers, or insoluble lipids. The clear supernatant from this step was used for the immunoprecipitation. The lysates were incubated with ChemoTek GFP-Trap Dynabeads (ChromoTek, Cat# gtd-10, RRID:AB_2827592) previously equilibrated with ice cold NETN buffer. Following the 1 hour of incubation period the beads were washed with ice cold NETN buffer. To elute the proteins, beads were boiled in 2X SDS-PAGE sample loading buffer and subjected to SDS-PAGE. The gels were analyzed by Coomassie staining prior to mass spectrometry.
Mass spectrometry analysis of Csp interactome
The immuno-precipitated samples were resolved on NuPAGE 10% Bis-Tris Gel (Life Technologies) and the gel pieces were processed for in-gel digestion using trypsin enzyme (Gendepot T9600). The tryptic peptides were analyzed on nano-LC 1000 system (Thermo Fisher Scientific, San Jose, CA) coupled to an Orbitrap Fusion (Thermo Fisher Scientific, San Jose, CA) mass spectrometer. The peptides were loaded on a two-column setup using a pre-column trap of 2cm × 100μm size (Reprosil-Pur Basic C18 1.9 μm, Dr. Maisch GmbH, Germany) and a 20cm × 75μm analytical column (Reprosil-Pur Basic C18 1.9 μm, Dr.Maisch GmbH, Germany). The peptides were resolved on a 110-minute gradient of 6%–30% acetonitrile/0.1% formic acid at a flow rate of 200nl/min. The eluted peptides were directly electro-sprayed into the mass spectrometer operated in the data-dependent acquisition (DDA) mode. The full MS scan was acquired in Orbitrap in the range of 300–1400 m/z at 120,000 resolution, followed by Top35 MS2 in an ion trap (HCD 30% collision energy), with 5 s dynamic exclusion time. The MS/MS spectra were searched using the Mascot algorithm (Mascot 2.4, Matrix Science) against the Drosophila melanogaster NCBI refseq protein database (01/14/2019 download) in the Proteome Discoverer (PD 2.1, Thermo Fisher) interface. The precursor mass tolerance was confined to 20 ppm, fragment mass tolerance of 0.5 Da, and a maximum of two missed cleavages was allowed. Dynamic modification of oxidation on methionine, protein N-terminal Acetylation and DeStreak on cysteine was allowed. The gene product inference and iBAQ-based label free quantification was performed by gpGrouper algorithm (Saltzman et al., 2018). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (Deutsch et al., 2020) via the PRIDE partner repository (Perez-Riverol et al., 2019) with the dataset identifier PXD026579.
Basic GoldenBraid 2.0 cloning vectors
We generated several basic GB2.0 cloning vectors with different backbone functionalities. Vectors were cloned using GB2.0 assembly via BbsI-HF (New England Biolabs, R3539L) mediated cloning reaction as previously described (Sarrion-Perdigones et al., 2013). Briefly, vector backbone elements were either commercially synthesized as GB2.0 cloning compatible fragments or PCR amplified from existing plasmids with GB2.0 compatible cloning overhangs (Figure 6A). The pUPD2 DNA element domestication vector was cloned by first synthesizing a LacZ expressing GB2.0 entry cassette (Sarrion-Perdigones et al., 2013) flanked on either side by pairs of inverted bacterial transcription terminator elements, rrnBT1 (Green et al., 1985), rrnBT2 (Green et al., 1985), L3S2P21 (Chen et al., 2013), and spy (Clarke et al., 2018), as described above with BsaI restriction sites on both 5′ and 3′ ends of the fragment. The synthesized element was cloned into a universal part domesticator vector, pVD2_purple, via BsaI cloning reaction described below. The pVD2_purple vector features a purple chromophore screening marker and is itself derived from a previously described pVD2 domesticator vector (Sarrion-Perdigones et al., 2013). All other backbone elements of pUPD2 were PCR amplified as a single BbsI restriction site flanked amplicon using primers pUPD_FOR and pUPD_REV using the pUPD vector as template (Sarrion-Perdigones et al., 2013). The amplified backbone elements were assembled and circularized with pUPD2 entry cassette via BbsI cloning reaction described below. The resultant vector was transformed into chemocompetent DH10B cells (Invitrogen/Thermo Fisher Scientific) and correct assemblies were identified using standard blue-white colony screening. Plasmid DNA was prepared using the QIAprep Spin Miniprep Kit (QIAGEN) and characterized via restriction enzyme digestion. Results were visualized via gel electrophoresis on 1% agarose gel. Correct clones were fully Sanger sequenced via commercial service (GeneWiz).
GB2.0 compatible P[acman] vectors were assembled by first amplifying backbone elements using the original P[acman] empty backbone vector (Venken et al., 2006) as the template and domesticating them into pVD2_purple via BsaI cloning. The ΦC31 attB site containing element was amplified using primers attB-FOR and attB-REV, an element containing the sopC and sopB genes was amplified using primers moduleA_FOR and moduleA_REV, the element containing sopA, incC and partial repE genes was commercially synthesized and domesticated into pVD2_purple, and the element containing the rest of repE along with the oriS and oriV replications of origin was amplified using primers moduleF_FOR and moduleF_REV. All domesticated elements were then assembled with one of four corresponding GB2.0 entry cassettes: Alpha1, Alpha2, Omega1 or Omega2, and either a kanamycin resistance bacterial marker in the case of Alpha level vectors or a chloramphenicol marker for Omega level vectors using BbsI mediated GB2.0 cloning. Reaction products were transformed, isolated, and analyzed as described above albeit using purple-white colony screening rather than traditional blue-white screening. The resulting P[acman] plasmids are totally free of P element transposon inverted transposon repeat sequences and lack a mini-white marker, reducing the vector size to just under 7 kb.
Conditionally replicative pGB2-R6Kγ GB2.0 compatible vectors were assembled by first PCR amplifying the R6K gamma replication of origin (Rossignol et al., 2001) using primers R6Kγ_FOR and R6Kg_REV from template vector MCS2-R6Kgamma-CORRECT-Kan-rc (unpublished data) and domestication of the amplicon into pVD2 (Sarrion-Perdigones et al., 2013) via a BsaI GB2.0 cloning reaction. pGB2-R6Kg vectors were assembled via a BbsI GB2.0 assembly reaction by combining the domesticated R6Kγ replication of origin with an appropriate GB2.0 entry cassette (Alpha1, Alpha2, Omega1, or Omega2) similar in design to the pUPD2 entry cassette, and either a domesticated kanamycin or chloramphenicol resistance marker for Alpha or Omega vectors respectively. A spectinomycin resistance variant of pGB2-R6Kγ-Alpha1 was also generated as described above but with a spectinomycin resistance marker instead. Resultant assembly products were transformed in EC100D pir-116 cells (Epicenter/Lucigen), isolated and analyzed as described above.
DNA part library and vector toolkit
GoldenBraid 2.0 (GB2.0) DNA parts were designed as follows (for DNA parts see Table S1 and the Key resources table): all non-coding sequences (e.g., promoters, poly(A) terminators, etc.) were synthesized as linear double stranded DNA with appropriate flanking GB2.0 cloning overhangs and restriction enzyme sites (IDT, Eurofins Genomics) or commercially cloned DNA fragments (Twist Bioscience). Internal GB2.0 cloning restriction enzyme sites (Esp3I and BsaI) were removed where necessary. All coding DNA sequences were first codon optimized for expression in Drosophila melanogaster using an online tool (http://www.idtdna.com/pages/tools/codon-optimization-tool?returnurl=%2FCodonOpt, IDT), then analyzed for splice acceptor and donor sites using a splice site prediction online tool (https://www.fruitfly.org/seq_tools/splice.html, BDGP (Reese et al., 1997), and finally any internal GB2.0 cloning enzyme sites were manually removed prior to commercial DNA synthesis. DNA fragments were domesticated as described above. Initially parts were domesticated into pUPD vector backbones (Sarrion-Perdigones et al., 2011). However, due to variable quality of prepared DNA, poor blue-white colony differentiation, and requirement of IPTG induction for strong blue color, we developed pUPD2 to overcome these limitations. In this vector, DNA parts are insulated from the rest of the vector backbone by flanking pairs of inverted bacterial poly(A) terminator sequences (see above) and LacZ expression is driven by a constitutive synthetic bacterial promoter, Em7 (Zhang et al., 1998). For DNA subcloning, such as when switching the grammar of an already domesticated element, we also developed a third domestication vector, pUPD3. This version also features a constitutively expressed LacZ blue-white screening marker and a chloramphenicol bacterial resistance marker to facilitate backbone switching from pUPD or pUPD2. Smaller parts up to 50 bp were synthesized as a pair of single stranded DNA oligos (Sigma-Aldrich), diluted to 10 μM, mixed in a 1:1 ratio by volume, and allowed to anneal at room temperature for at least an hour. Three μl of annealed oligos was then added to a domestication GB2.0 cloning reaction as described above.
Chemocompetent DH10B strain bacterial cells (Invitrogen/Thermo Fisher Scientific) were then transformed via heat shock with 2 μl of assembly product and plated onto solid media containing X-Gal and appropriate antibiotic. Assembly into the pGB2-R6Kγ vector backbones was accomplished via electroporation of 1 μl of assembly product into electrocompetent EC100D pir-116 cells (Epicenter/Lucigen). Assembly into GB2.0 compatible P[acman] vector backbones was accomplished by transforming 2 μl of assembly product into chemocompetent EPI300 cells (Epicenter/Lucigen). Correct assemblies were identified using standard blue-white colony screening. Plasmid DNA was prepared using the QIAprep Spin Miniprep Kit (QIAGEN) for regular high-copy plasmid backbones, and ChargeSwitch-Pro plasmid miniprep kit (Invitrogen/Thermo Fisher Scientific) for P[acman] vector backbones, after copy number induction by CopyControl Fosmid Autoinduction Solution (Epicenter/Lucigen, CCIS125). All elements of the DNA part library are summarized in Table S1 and the Key resources table. All primers used are summarized in Table S5 and the Key resources table.
All GB2.0 vectors were cloned using alternating BsaI and Esp3I mediated assembly steps as previously described (Sarrion-Perdigones et al., 2013, 2019). Briefly, domesticated parts are initially assembled into transcriptional units (TUs) consisting of, minimally, a promoter, a CDS, and a poly(A) terminator sequence into an Alpha level destination vector via BsaI-HFv2 mediated assembly as described above. More complex assemblies involving additional parts are possible. We are routinely able to assemble up to 7 different parts of varying size and complexity at high efficiency. A pair of Alpha level vectors can then be assembled further using GB2.0 grammar into an Omega level vector via an Esp3I mediated assembly reaction to form a pair of TUs. Paired Omega vector can then be assembled again back into an Alpha level vector via BsaI-HFv2 assembly to form a genetic circuit of multiple TUs. This process continues until the desired end product is reached. All cloning backbone vectors, domesticated parts, and assembled plasmids are available through Addgene (https://www.addgene.org/; Tables S1 and S2; Key resources table).
QUANTIFICATION AND STATISTICAL ANALYSIS
All data were analyzed using Excel (Microsoft Corporation), followed by Prism 7 software (GraphPad Software) for statistical analysis and graphing. Statistical parameters are indicated when applicable at the end of figure legends (Figures 1, S3, and S4). The resulting graphs were then edited for publication using Adobe Illustrator Creative Cloud (Adobe). Immunofluorescent images were obtained as described above and then edited for publication using Adobe Photoshop Creative Cloud and Adobe Illustrator Creative Cloud (Adobe). Statistical methods are described in relevant sections. Interactome analysis was performed and visualized using the biological database and web resource of known and predicted protein–protein interactions, called STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) (https://www.string-db.org; Szklarczyk et al., 2019). Synaptic specific network was further visualized using the open source bioinformatics software platform for visualizing molecular interaction networks, called Cytoscape v3.8.2 (https://cytoscape.org; Shannon et al., 2003).
Supplementary Material
Highlights.
In vivo analysis of four selection and two counterselection markers
Individual or multiplexed use of markers to produce complex genotypes
Demonstrating usefulness of markers through three separate applications
Provision of DNA and vector resources to facilitate future applications
ACKNOWLEDGMENTS
Stocks obtained from the Bloomington Drosophila Stock Center (supported by NIH grant P40ODO18537) were used in this study. We thank Konrad Basler (University of Zurich), Jack Bateman (Bowdoin College), Johannes Bischof (University of Zurich), Donald Court (National Cancer Institute), Diego Orsaez (The Institute for Plant Molecular and Cellular Biology), Anthony Poteete (University of Massachusetts Medical School), and the Drosophila Genomics Resource Center (Indiana University, supported by NIH grant 2P40OD010949) for plasmids or flies. We are grateful to Robert Harrell, David O’Brochta, and colleagues at the Insect Genetic Technologies Research Coordination Network (University of Maryland, supported by NSF grant 1352978) for help in training with the injection routine. This work was supported by start-up and additional seed funds provided by Baylor College of Medicine (K.J.T.V.), the McNair Medical Institute at The Robert and Janice McNair Foundation (K.J.T.V.), the Foundation for Angelman Syndrome Therapeutics grant FT2016-002 (K.J.T.V.), the Cancer Prevention and Research Institute of Texas grant R1313 (K.J.T.V.), and the National Institutes of Health grants 1R21HG006726 (K.J.T.V.), 1R21GM110190 (K.J.T.V.), 1R21OD022981 (K.J.T.V.), R01GM109938 (H.A.D.), and R01MH107474 (H.A.D.). The Dan L. Duncan Comprehensive Cancer Center and the BCM Mass Spectrometry Proteomics Core are supported by the National Institutes of Health (P30 CA125123). The BCM Mass Spectrometry Proteomics Core is additionally supported by a CPRIT Core Facility Award (RP170005). This work is dedicated to Dr. Alejandro Sarrion-Perdigones in memoriam.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109700.
SUPPORTING CITATIONS
The following references appear in the supplemental information: Ai et al. (2007); Donnelly et al. (2001); Ingolia et al. (1980); Jensen and Hammer (1998); Lee and Luo (1999); Riabinina et al. (2015); Sano et al. (2002).
REFERENCES
- Ai H, Shaner NC, Cheng Z, Tsien RY, and Campbell RE (2007). Exploration of new chromophore structures leads to the identification of improved blue fluorescent proteins. Biochemistry 46, 5904–5910. [DOI] [PubMed] [Google Scholar]
- Angelichio ML, Beck JA, Johansen H, and Ivey-Hoyle M (1991). Comparison of several promoters and polyadenylation signals for use in heterologous gene expression in cultured Drosophila cells. Nucleic Acids Res. 19, 5037–5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S, Bainton RJ, Mayer N, Beckstead R, and Bhat MA (2008). Septate junctions are required for ommatidial integrity and blood-eye barrier function in Drosophila. Dev. Biol 317, 585–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman JR, Lee AM, and Wu CT (2006). Site-specific transformation of Drosophila via phiC31 integrase-mediated cassette exchange. Genetics 173, 769–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgartner S, Littleton JT, Broadie K, Bhat MA, Harbecke R, Lengyel JA, Chiquet-Ehrismann R, Prokop A, and Bellen HJ (1996). A Drosophila neurexin is required for septate junction and blood-nerve barrier formation and function. Cell 87, 1059–1068. [DOI] [PubMed] [Google Scholar]
- Bellen HJ, Wangler MF, and Yamamoto S (2019). The fruit fly at the interface of diagnosis and pathogenic mechanisms of rare and common human diseases. Hum. Mol. Genet 28 (R2), R207–R214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beumer KJ, and Carroll D (2014). Targeted genome engineering techniques in Drosophila. Methods 68, 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bier E (2005). Drosophila, the golden bug, emerges as a tool for human genetics. Nat. Rev. Genet 6, 9–23. [DOI] [PubMed] [Google Scholar]
- Bier E, Harrison MM, O’Connor-Giles KM, and Wildonger J (2018). Advances in engineering the fly genome with the CRISPR-Cas system. Genetics 208, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischof J, Maeda RK, Hediger M, Karch F, and Basler K (2007). An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proc. Natl. Acad. Sci. USA 104, 3312–3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black ME, Kokoris MS, and Sabo P (2001). Herpes simplex virus-1 thymidine kinase mutants created by semi-random sequence mutagenesis improve prodrug-mediated tumor cell killing. Cancer Res. 61, 3022–3026. [PubMed] [Google Scholar]
- Brand AH, and Perrimon N (1993). Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118, 401–415. [DOI] [PubMed] [Google Scholar]
- Bronk P, Nie Z, Klose MK, Dawson-Scully K, Zhang J, Robertson RM, Atwood HL, and Zinsmaier KE (2005). The multiple functions of cysteine-string protein analyzed at Drosophila nerve terminals. J. Neurosci 25, 2204–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson SD, Juang JL, Hilgers SL, and Garment MB (2000). Blood barriers of the insect. Annu. Rev. Entomol 45, 151–174. [DOI] [PubMed] [Google Scholar]
- Chen Y-J, Liu P, Nielsen AA, Brophy JA, Clancy K, Peterson T, and Voigt CA (2013). Characterization of 582 natural and synthetic terminators and quantification of their design constraints. Nat. Methods 10, 659–664. [DOI] [PubMed] [Google Scholar]
- Clarke AM, Engel KL, Giles KE, Petit CM, and Schneider DA (2018). NETSeq reveals heterogeneous nucleotide incorporation by RNA polymerase I. Proc. Natl. Acad. Sci. USA 115, E11633–E11641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooley L, Kelley R, and Spradling A (1988). Insertional mutagenesis of the Drosophila genome with single P elements. Science 239, 1121–1128. [DOI] [PubMed] [Google Scholar]
- Cormack BP, Valdivia RH, and Falkow S (1996). FACS-optimized mutants of the green fluorescent protein (GFP). Gene 173, 33–38. [DOI] [PubMed] [Google Scholar]
- Dahmann C (2008). In Drosophila Methods and Protocols, Dahmann C, ed. (Humana; ). [Google Scholar]
- Davies J, and Smith DI (1978). Plasmid-determined resistance to antimicrobial agents. Annu. Rev. Microbiol 32, 469–518. [DOI] [PubMed] [Google Scholar]
- Deutsch EW, Bandeira N, Sharma V, Perez-Riverol Y, Carver JJ, Kundu DJ, García-Seisdedos D, Jarnuczak AF, Hewapathirana S, Pullman BS, et al. (2020). The ProteomeXchange consortium in 2020: enabling ‘big data’ approaches in proteomics. Nucleic Acids Res. 48 (D1), D1145–D1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly MLL, Hughes LE, Luke G, Mendoza H, Ten Dam E, Gani D, and Ryan MD (2001). The ‘cleavage’ activities of foot-and-mouth disease virus 2A site-directed mutants and naturally occurring ‘2A-like’ sequences. J. Gen. Virol 82, 1027–1041. [DOI] [PubMed] [Google Scholar]
- Eberle KK, Zinsmaier KE, Buchner S, Gruhn M, Jenni M, Arnold C, Leibold C, Reisch D, Walter N, Hafen E, et al. (1998). Wide distribution of the cysteine string proteins in Drosophila tissues revealed by targeted mutagenesis. Cell Tissue Res. 294, 203–217. [DOI] [PubMed] [Google Scholar]
- Eglitis MA (1991). Positive selectable markers for use with mammalian cells in culture. Hum. Gene Ther 2, 195–201. [DOI] [PubMed] [Google Scholar]
- Engler C, Kandzia R, and Marillonnet S (2008). A one pot, one step, precision cloning method with high throughput capability. PLoS One 3, e3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erbs P, Regulier E, Kintz J, Leroy P, Poitevin Y, Exinger F, Jund R, and Mehtali M (2000). In vivo cancer gene therapy by adenovirus-mediated transfer of a bifunctional yeast cytosine deaminase/uracil phosphoribosyltransferase fusion gene. Cancer Res. 60, 3813–3822. [PubMed] [Google Scholar]
- Flores HA, and O’Neill SL (2018). Controlling vector-borne diseases by releasing modified mosquitoes. Nat. Rev. Microbiol 16, 508–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA 3rd, and Smith HO (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345. [DOI] [PubMed] [Google Scholar]
- Giordano-Santini R, and Dupuy D (2011). Selectable genetic markers for nematode transgenesis. Cell. Mol. Life Sci 68, 1917–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano-Santini R, Milstein S, Svrzikapa N, Tu D, Johnsen R, Baillie D, Vidal M, and Dupuy D (2010). An antibiotic selection marker for nematode transgenesis. Nat. Methods 7, 721–723. [DOI] [PubMed] [Google Scholar]
- Gnerer JP, Venken KJT, and Dierick HA (2015). Gene-specific cell labeling using MiMIC transposons. Nucleic Acids Res. 43, e56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratz SJ, Ukken FP, Rubinstein CD, Thiede G, Donohue LK, Cummings AM, and O’Connor-Giles KM (2014). Highly specific and efficient CRISPR/Cas9-catalyzed homology-directed repair in Drosophila. Genetics 196, 961–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green CJ, Stewart GC, Hollis MA, Vold BS, and Bott KF (1985). Nucleotide sequence of the Bacillus subtilis ribosomal RNA operon, rrnB. Gene 37, 261–266. [DOI] [PubMed] [Google Scholar]
- Gritz L, and Davies J (1983). Plasmid-encoded hygromycin B resistance: the sequence of hygromycin B phosphotransferase gene and its expression in Escherichia coli and Saccharomyces cerevisiae. Gene 25, 179–188. [DOI] [PubMed] [Google Scholar]
- Groth AC, Fish M, Nusse R, and Calos MP (2004). Construction of transgenic Drosophila by using the site-specific integrase from phage phiC31. Genetics 166, 1775–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handler AM, and Harrell RA 2nd. (2001). Polyubiquitin-regulated DsRed marker for transgenic insects. Biotechniques 31, 820–824, 828. [PubMed] [Google Scholar]
- Handler AM, and O’Brochta DA (1991). Prospects for gene transformation in insects. Annu. Rev. Entomol 36, 159–183. [DOI] [PubMed] [Google Scholar]
- Hay BA, Wolff T, and Rubin GM (1994). Expression of baculovirus P35 prevents cell death in Drosophila. Development 120, 2121–2129. [DOI] [PubMed] [Google Scholar]
- Heinrich JC, and Scott MJ (2000). A repressible female-specific lethal genetic system for making transgenic insect strains suitable for a sterile-release program. Proc. Natl. Acad. Sci. USA 97, 8229–8232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman S, Miller D, Eisman R, Kuwayama H, Niimi T, and Kaufman T (2010). Transgenic tools for members of the genus Drosophila with sequenced genomes. Fly (Austin) 4, 349–362. [DOI] [PubMed] [Google Scholar]
- Horn C, Schmid BG, Pogoda FS, and Wimmer EA (2002). Fluorescent transformation markers for insect transgenesis. Insect Biochem. Mol. Biol 32, 1221–1235. [DOI] [PubMed] [Google Scholar]
- Horton RM, Hunt HD, Ho SN, Pullen JK, and Pease LR (1989). Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77, 61–68. [DOI] [PubMed] [Google Scholar]
- Ingolia TD, Craig EA, and McCarthy BJ (1980). Sequence of three copies of the gene for the major Drosophila heat shock induced protein and their flanking regions. Cell 21, 669–679. [DOI] [PubMed] [Google Scholar]
- Itaya M, Yamaguchi I, Kobayashi K, Endo T, and Tanaka T (1990). The blasticidin S resistance gene (bsr) selectable in a single copy state in the Bacillus subtilis chromosome. J. Biochem 107, 799–801. [DOI] [PubMed] [Google Scholar]
- Jenett A, Rubin GM, Ngo TT, Shepherd D, Murphy C, Dionne H, Pfeiffer BD, Cavallaro A, Hall D, Jeter J, et al. (2012). A GAL4-driver line resource for Drosophila neurobiology. Cell Rep. 2, 991–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen PR, and Hammer K (1998). The sequence of spacers between the consensus sequences modulates the strength of prokaryotic promoters. Appl. Environ. Microbiol 64, 82–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanca O, Zirin J, Garcia-Marques J, Knight SM, Yang-Zhou D, Amador G, Chung H, Zuo Z, Ma L, He Y, et al. (2019). An efficient CRISPR-based strategy to insert small and large fragments of DNA using short homology arms. eLife 8, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandul NP, Liu J, Hsu AD, Hay BA, and Akbari OS (2020). A drug-inducible sex-separation technique for insects. Nat. Commun 11, 2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DW, Uetsuki T, Kaziro Y, Yamaguchi N, and Sugano S (1990). Use of the human elongation factor 1 a promoter as a versatile and efficient expression system. Gene 91, 217–223. [DOI] [PubMed] [Google Scholar]
- Kim H, Ishidate T, Ghanta KS, Seth M, Conte D Jr., Shirayama M, and Mello CC (2014). A co-CRISPR strategy for efficient genome editing in Caenorhabditis elegans. Genetics 197, 1069–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchmaier S, Höckendorf B, Möller EK, Bornhorst D, Spitz F, and Wittbrodt J (2013). Efficient site-specific transgenesis and enhancer activity tests in medaka using PhiC31 integrase. Development 140, 4287–4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo A, Awasaki T, Ishikawa Y, and Matsuo T (2018). piggyBac- and phiC31 integrase-mediated transgenesis in Drosophila prolongata. Genes Genet. Syst 92, 277–285. [DOI] [PubMed] [Google Scholar]
- Labbé GMC, Nimmo DD, and Alphey L (2010). piggybac- and PhiC31-mediated genetic transformation of the Asian tiger mosquito, Aedes albopictus (Skuse). PLoS Negl. Trop. Dis 4, e788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai SL, and Lee T (2006). Genetic mosaic with dual binary transcriptional systems in Drosophila. Nat. Neurosci 9, 703–709. [DOI] [PubMed] [Google Scholar]
- Lee T, and Luo L (1999). Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron 22, 451–461. [DOI] [PubMed] [Google Scholar]
- Lee EC, Yu D, Martinez de Velasco J, Tessarollo L, Swing DA, Court DL, Jenkins NA, and Copeland NG (2001). A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics 73, 56–65. [DOI] [PubMed] [Google Scholar]
- Li MZ, and Elledge SJ (2007). Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat. Methods 4, 251–256. [DOI] [PubMed] [Google Scholar]
- Li-Kroeger D, Kanca O, Lee PT, Cowan S, Lee MT, Jaiswal M, Salazar JL, He Y, Zuo Z, and Bellen HJ (2018). An expanded toolkit for gene tagging based on MiMIC and scarless CRISPR tagging in Drosophila. eLife 7, 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link N, and Bellen HJ (2020). Using Drosophila to drive the diagnosis and understand the mechanisms of rare human diseases. Development 147, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan H, Peabody NC, Vinson CR, and White BH (2006). Refined spatial manipulation of neuronal function by combinatorial restriction of transgene expression. Neuron 52, 425–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzoni R, Urrios A, Velazquez-Garcia S, de Nadal E, and Posas F (2016). Synthetic biology: insights into biological computation. Integr. Biol 8, 518–532. [DOI] [PubMed] [Google Scholar]
- Markaki M, Craig RK, and Savakis C (2004). Insect population control using female specific pro-drug activation. Insect Biochem. Mol. Biol 34, 131–137. [DOI] [PubMed] [Google Scholar]
- Matinyan N, Gonzalez Y, Dierick HA, and Venken KJT (2021). Determining effective drug concentrations for selection & counterselection genetics in Drosophila melanogaster (STAR Protocols). 10.1016/j.xpro.2021.100783. [DOI] [PMC free article] [PubMed]
- Matthews BJ, and Vosshall LB (2020). How to turn an organism into a model organism in 10 ‘easy’ steps. J. Exp. Biol 223 (Pt, Suppl 1), jeb218198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahan S, Kohl KP, and Sekelsky J (2013). Variation in meiotic recombination frequencies between allelic transgenes inserted at different sites in the Drosophila melanogaster genome. G3 (Bethesda) 3, 1419–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith JM, Underhill A, McArthur CC, and Eggleston P (2013). Next-generation site-directed transgenesis in the malaria vector mosquito Anopheles gambiae: self-docking strains expressing germline-specific phiC31 integrase. PLoS ONE 8, e59264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DE, Cook KR, Arvanitakis AV, and Hawley RS (2016). Third chromosome balancer inversions disrupt protein-coding genes and influence distal recombination events in Drosophila melanogaster. G3 (Bethesda) 6, 1959–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DE, Cook KR, Hemenway EA, Fang V, Miller AL, Hales KG, and Hawley RS (2018). The molecular and genetic characterization of second chromosome balancers in Drosophila melanogaster. G3 (Bethesda) 8, 1161–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen RM, and Kingston RE (2009). Selection of transfected mammalian cells. Curr. Protoc. Mol. Biol Chapter 9, 5. [DOI] [PubMed] [Google Scholar]
- Mosimann C, Puller AC, Lawson KL, Tschopp P, Amsterdam A, and Zon LI (2013). Site-directed zebrafish transgenesis into single landing sites with the phiC31 integrase system. Dev. Dyn 242, 949–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosková L, Stránecký V, Hartmannová H, Přistoupilová A, Barešová V, Ivánek R, Hůlková H, Jahnová H, van der Zee J, Staropoli JF, et al. (2011). Mutations in DNAJC5, encoding cysteine-string protein alpha, cause autosomal-dominant adult-onset neuronal ceroid lipofuscinosis. Am. J. Hum. Genet 89, 241–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva-Trastoy M, Defais M, and Larminat F (2005). Resistance to the antibiotic Zeocin by stable expression of the Sh ble gene does not fully suppress Zeocin-induced DNA cleavage in human cells. Mutagenesis 20, 111–114. [DOI] [PubMed] [Google Scholar]
- Pédelacq JD, Cabantous S, Tran T, Terwilliger TC, and Waldo GS (2006). Engineering and characterization of a superfolder green fluorescent protein. Nat. Biotechnol 24, 79–88. [DOI] [PubMed] [Google Scholar]
- Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, et al. (2019). The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47 (D1), D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer BD, Jenett A, Hammonds AS, Ngo TT, Misra S, Murphy C, Scully A, Carlson JW, Wan KH, Laverty TR, et al. (2008). Tools for neuroanatomy and neurogenetics in Drosophila. Proc. Natl. Acad. Sci. USA 105, 9715–9720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer BD, Ngo TT, Hibbard KL, Murphy C, Jenett A, Truman JW, and Rubin GM (2010). Refinement of tools for targeted gene expression in Drosophila. Genetics 186, 735–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer BD, Truman JW, and Rubin GM (2012). Using translational enhancers to increase transgene expression in Drosophila. Proc. Natl. Acad. Sci. USA 109, 6626–6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirrotta V (1988). Vectors for P-mediated transformation in Drosophila. Biotechnology 10, 437–456. [DOI] [PubMed] [Google Scholar]
- Pondeville E, Puchot N, Meredith JM, Lynd A, Vernick KD, Lycett GJ, Eggleston P, and Bourgouin C (2014). Efficient ΦC31 integrase-mediated site-specific germline transformation of Anopheles gambiae. Nat. Protoc 9, 1698–1712. [DOI] [PubMed] [Google Scholar]
- Poteete AR, Rosadini C, and St Pierre C (2006). Gentamicin and other cassettes for chromosomal gene replacement in Escherichia coli. Biotechniques 41, 261–262, 264. [DOI] [PubMed] [Google Scholar]
- Potter CJ, Tasic B, Russler EV, Liang L, and Luo L (2010). The Q system: a repressible binary system for transgene expression, lineage tracing, and mosaic analysis. Cell 141, 536–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radman I, Greiss S, and Chin JW (2013). Efficient and rapid C. elegans transgenesis by bombardment and hygromycin B selection. PLoS ONE 8, e76019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakowski SA, and Filutowicz M (2013). Plasmid R6K replication control. Plasmid 69, 231–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese MG, Eeckman FH, Kulp D, and Haussler D (1997). Improved splice site detection in Genie. J. Comput. Biol 4, 311–323. [DOI] [PubMed] [Google Scholar]
- Riabinina O, Luginbuhl D, Marr E, Liu S, Wu MN, Luo L, and Potter CJ (2015). Improved and expanded Q-system reagents for genetic manipulations. Nat. Methods 12, 219–222, 5, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts JA, Miguel-Escalada I, Slovik KJ, Walsh KT, Hadzhiev Y, Sanges R, Stupka E, Marsh EK, Balciuniene J, Balciunas D, and Müller F (2014). Targeted transgene integration overcomes variability of position effects in zebrafish. Development 141, 715–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson HM, Preston CR, Phillis RW, Johnson-Schlitz DM, Benz WK, and Engels WR (1988). A stable genomic source of P element transposase in Drosophila melanogaster. Genetics 118, 461–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossignol M, Basset A, Espéli O, and Boccard F (2001). NKBOR, a mini-Tn10-based transposon for random insertion in the chromosome of Gram-negative bacteria and the rapid recovery of sequences flanking the insertion sites in Escherichia coli. Res. Microbiol 152, 481–485. [DOI] [PubMed] [Google Scholar]
- Rubin GM, and Spradling AC (1982). Genetic transformation of Drosophila with transposable element vectors. Science 218, 348–353. [DOI] [PubMed] [Google Scholar]
- Ryder E, and Russell S (2003). Transposable elements as tools for genomics and genetics in Drosophila. Brief. Funct. Genomics Proteomics 2, 57–71. [DOI] [PubMed] [Google Scholar]
- Saltzman AB, Leng M, Bhatt B, Singh P, Chan DW, Dobrolecki L, Chandrasekaran H, Choi JM, Jain A, Jung SY, et al. (2018). GpGrouper: A peptide grouping algorithm for gene-centric inference and quantitation of bottom-up proteomics data. Mol. Cell. Proteomics 17, 2270–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano H, Nakamura A, and Kobayashi S (2002). Identification of a transcriptional regulatory region for germline-specific expression of vasa gene in Drosophila melanogaster. Mech. Dev 112, 129–139. [DOI] [PubMed] [Google Scholar]
- Sarrion-Perdigones A, Falconi EE, Zandalinas SI, Juárez P, Fernándezdel-Carmen A, Granell A, and Orzaez D (2011). GoldenBraid: an iterative cloning system for standardized assembly of reusable genetic modules. PLoS ONE 6, e21622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrion-Perdigones A, Vazquez-Vilar M, Palací J, Castelijns B, Forment J, Ziarsolo P, Blanca J, Granell A, and Orzaez D (2013). GoldenBraid 2.0: a comprehensive DNA assembly framework for plant synthetic biology. Plant Physiol. 162, 1618–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrion-Perdigones A, Chang L, Gonzalez Y, Gallego-Flores T, Young DW, and Venken KJT (2019). Examining multiple cellular pathways at once using multiplex hextuple luciferase assaying. Nat. Commun 10, 5710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple JI, Garcia-Verdugo R, and Lehner B (2010). Rapid selection of transgenic C. elegans using antibiotic resistance. Nat. Methods 7, 725–727. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, and Tsien RY (2004). Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol 22, 1567–1572. [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh V (2014). Recent advances and opportunities in synthetic logic gates engineering in living cells. Syst. Synth. Biol 8, 271–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steller H, and Pirrotta V (1985). A transposable P vector that confers selectable G418 resistance to Drosophila larvae. EMBO J. 4, 167–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steller H, and Pirrotta V (1986). P transposons controlled by the heat shock promoter. Mol. Cell. Biol 6, 1640–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun FF, Johnson JE, Zeidler MP, and Bateman JR (2012). Simplified insertion of transgenes onto balancer chromosomes via recombinase-mediated cassette exchange. G3 (Bethesda) 2, 551–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, et al. (2019). STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47 (D1), D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasic B, Hippenmeyer S, Wang C, Gamboa M, Zong H, Chen-Tsai Y, and Luo L (2011). Site-specific integrase-mediated transgenesis in mice via pronuclear injection. Proc. Natl. Acad. Sci. USA 108, 7902–7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DD, Donnelly CA, Wood RJ, and Alphey LS (2000). Insect population control using a dominant, repressible, lethal genetic system. Science 287, 2474–2476. [DOI] [PubMed] [Google Scholar]
- Tobaben S, Thakur P, Fernández-Chacón R, Südhof TC, Rettig J, and Stahl B (2001). A trimeric protein complex functions as a synaptic chaperone machine. Neuron 31, 987–999. [DOI] [PubMed] [Google Scholar]
- Vara J, Perez-Gonzalez JA, and Jimenez A (1985). Biosynthesis of puromycin by Streptomyces alboniger: characterization of puromycin N-acetyltransferase. Biochemistry 24, 8074–8081. [DOI] [PubMed] [Google Scholar]
- Venken KJT, and Bellen HJ (2007). Transgenesis upgrades for Drosophila melanogaster. Development 134, 3571–3584. [DOI] [PubMed] [Google Scholar]
- Venken KJT, and Bellen HJ (2014). Chemical mutagens, transposons, and transgenes to interrogate gene function in Drosophila melanogaster. Methods 68, 15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venken KJT, He Y, Hoskins RA, and Bellen HJ (2006). P[acman]: a BAC transgenic platform for targeted insertion of large DNA fragments in D. melanogaster. Science 314, 1747–1751. [DOI] [PubMed] [Google Scholar]
- Venken KJT, Kasprowicz J, Kuenen S, Yan J, Hassan BA, and Verstreken P (2008). Recombineering-mediated tagging of Drosophila genomic constructs for in vivo localization and acute protein inactivation. Nucleic Acids Res. 36, e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venken KJT, Carlson JW, Schulze KL, Pan H, He Y, Spokony R, Wan KH, Koriabine M, de Jong PJ, White KP, et al. (2009). Versatile P [acman] BAC libraries for transgenesis studies in Drosophila melanogaster. Nat. Methods 6, 431–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venken KJT, Popodi E, Holtzman SL, Schulze KL, Park S, Carlson JW, Hoskins RA, Bellen HJ, and Kaufman TC (2010). A molecularly defined duplication set for the X chromosome of Drosophila melanogaster. Genetics 186, 1111–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venken KJT, Schulze KL, Haelterman NA, Pan H, He Y, Evans-Holm M, Carlson JW, Levis RW, Spradling AC, Hoskins RA, and Bellen HJ (2011). MiMIC: a highly versatile transposon insertion resource for engineering Drosophila melanogaster genes. Nat. Methods 8, 737–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venken KJT, Sarrion-Perdigones A, Vandeventer PJ, Abel NS, Christiansen AE, and Hoffman KL (2016). Genome engineering: Drosophila melanogaster and beyond. Wiley Interdiscip. Rev. Dev. Biol 5, 233–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent JP, and Girdham C (1997). Promoters to express cloned genes uniformly in Drosophila. Methods Mol. Biol 62, 385–392. [DOI] [PubMed] [Google Scholar]
- Volohonsky G, Terenzi O, Soichot J, Naujoks DA, Nolan T, Windbichler N, Kapps D, Smidler AL, Vittu A, Costa G, et al. (2015). Tools for anopheles gambiae transgenesis. G3 (Bethesda) 5, 1151–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Buchholz F, Muyrers JP, and Stewart AF (1998). A new logic for DNA engineering using recombination in Escherichia coli. Nat. Genet 20, 123–128. [DOI] [PubMed] [Google Scholar]
- Zhang YQ, Henderson MX, Colangelo CM, Ginsberg SD, Bruce C, Wu T, and Chandra SS (2012). Identification of CSPa clients reveals a role in dynamin 1 regulation. Neuron 74, 136–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Arcia S, Perez B, Fernandez-Funez P, and Rincon-Limas DE (2013). pΔTubHA4C, a new versatile vector for constitutive expression in Drosophila. Mol. Biol. Rep 40, 5407–5415. [DOI] [PubMed] [Google Scholar]
- Zhang X, Koolhaas WH, and Schnorrer F (2014). A versatile two-step CRISPR- and RMCE-based strategy for efficient genome engineering in Drosophila. G3 (Bethesda) 4, 2409–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data for Figure 5F is available as Table S4. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD026579. This paper does not report original code. Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.