

Abstract
Background and Objectives
Previous research has shown that elevated blood C-reactive protein (CRP) is associated with increased Alzheimer disease (AD) risk only in APOE ε4 allele carriers; the objective of this study was to examine the interactive effects of plasma CRP and APOE genotype on cognition and AD biomarkers.
Methods
Data from the Alzheimer's Disease Neuroimaging Initiative (ADNI) study were analyzed, including APOE genotype; plasma CRP concentrations; diagnostic status (i.e., mild cognitive impairment and dementia due to AD); Mini-Mental State Examination (MMSE) and Clinical Dementia Rating Dementia Staging Instrument scores; CSF concentrations of β-amyloid peptide (Aβ42), total tau (t-Tau) and phosphorylated tau (p-Tau); and amyloid (AV45) PET imaging. Multivariable regression analyses tested the associations between plasma CRP and APOE on cognitive and biomarker outcomes.
Results
Among 566 ADNI participants, 274 (48.4%) had no, 222 (39.2%) had 1, and 70 (12.4%) had 2 APOE ε4 alleles. Among only participants who had 2 APOE ε4 alleles, elevated CRP was associated with lower MMSE score at baseline (β [95% confidence interval] −0.52 [−1.01, −0.12]) and 12-month follow-up (β −1.09 [−1.88, −0.17]) after adjustment for sex, age, and education. The interaction of 2 APOE ε4 alleles and elevated plasma CRP was associated with increased CSF levels of t-Tau (β = 11.21, SE 3.37, p < 0.001) and p-Tau (β = +2.74, SE 1.14, p < 0.01). Among those who had no APOE ε4 alleles, elevated CRP was associated with decreased CSF t-Tau and p-Tau. These effects were stronger at the 12-month follow-up.
Discussion
CRP released during peripheral inflammation could be a mediator in APOE ε4–related AD neurodegeneration and serve as a drug target for AD.
APOE ε4 allele is a major genetic risk factor for late-onset Alzheimer disease (AD).1 However, even among APOE ε4 carriers who are >90 years of age,2 not all APOE ε4 carriers develop dementia. One potential mediator may be C-reactive protein (CRP). Elevated blood CRP has been linked to increased AD risk only in APOE ε4 allele carriers, suggesting that CRP might modify APOE ε4 effects leading to AD-related neurodegeneration.3 Still, the interactions between APOE ε4 and CRP on cognitive decline and AD pathology are unknown.
CRP is a protein involved in immune response to toxins or injuries in systemic inflammation,4 and CRP levels increase with age.4 There are 2 types of CRP: native CRP (pCRP) is a pentameric oligoprotein and acute-phase reactant that is produced during active inflammatory reaction,5 and monomeric CRP (mCRP), or free subunits of pCRP, is produced during and after the acute phase by the irreversible dissociation of pCRP, which has much lower aqueous solubility and damaged tissues.6 CRP has been implicated in the pathogenesis of chronic diseases associated with aging, including cardiovascular disease,7 age-related macular degeneration,8 and poststroke inflammation.9
The objective of this study was to investigate the independent and interactive effects of APOE ε4 status (0, 1, and 2 APOE ε4 alleles) and peripheral CRP on cognition and in vivo AD biomarkers, including CSF β-amyloid peptide 42 (Aβ42), total tau (t-Tau), and phosphorylated tau (p-Tau), in participants from the Alzheimer's Disease Neuroimaging Initiative (ADNI) study.10
Methods
ADNI Study
Data were obtained from the ADNI database (adni.loni.usc.edu). The ADNI was launched in 2003 as a public-private partnership led by principal investigator Michael W. Weiner, MD. The overall objective of ADNI is to develop and validate in vivo biomarkers for AD. Detailed methodology and description of ADNI can been found on the ADNI website. There are 4 phases of the ADNI study: ADNI1, ADNIGO, ADNI2, and ADNI3. Only the ADNI1 dataset has plasma CRP data. Therefore, this study included only participants from the ADNI1 database.
Standard Protocol Approvals, Registrations, and Patient Consents
The ADNI study has 63 sites (including Boston University) across the United States and Canada. The protocol was approved by the Institutional Review Board from each institute/site for the experiments using human participants described in this study. This study obtained the deidentified data for data analyses.
Participants
This study included 566 ADNI1 participants who had APOE genotype data, baseline plasma CRP measurements, a research diagnosis of cognitively normal (CN), mild cognitive impairment (MCI), or AD dementia, as well as 2-year follow-up measurements of the Mini-Mental State Examination (MMSE) and Clinical Dementia Rating Scale (CDR). We also obtained data for participants who had PET AV45 data; CSF Aβ42, t-Tau and p-Tau; CSF neurogranin; plasma and CSF neurofilament light chain (NfL); and plasma APOE protein.
Plasma CRP
All participants in this study analysis had data on plasma CRP. A subset of the ADNI1 cohort had overnight fasting plasma samples and were analyzed with a 190-analyte multiplex immunoassay panel, which included CRP measures. Next to other proteins, plasma CRP was measured by using targeted multiplex proteomic strategies implemented in the Biomarkers Consortium Project of the ADNI study.11 The Luminex xMAP technology (Luminex Corp, Austin, TX) with a low-based laser apparatus was used.
Cognitive Diagnoses and MMSE and CDR Scores
For ADNI, diagnoses of CN, MCI, and AD dementia are based on established research diagnostic criteria.12 MMSE scores served as a measure for global cognitive status.13 The MMSE is a brief cognitive screening measure with scores ranging from 0 to 30 that is commonly used to detect and monitor dementia severity. The CDR Sum of Boxes (CDR-SB)14 was also used to assess overall severity of clinical impairment. MMSE and CDR-SB scores were measured at baseline and 12 and 24 months after the CRP measurements.
Fluid Biomarkers
Aliquots of CSF samples, which had never been thawed, were measured and analyzed by electrochemiluminescence immunoassays (lot for each analyte: P09 for Aβ1-42 and P02 for t-tau and p-tau181). The Roche Elecsys β-Amyloid (1–42) CSF, Elecsys Total-Tau CSF, and Elecsys Phospho-Tau (181P) CSF immunoassays (Elecsys Corp, Lenexa, KS) were used following the Roche Study Protocol at the University of Pennsylvania/ADNI Biomarker Laboratory and in accord with the preliminary kit manufacturer's instructions.15 We also obtained the data on plasma APOE protein, CSF neurogranin, and plasma and CSF NfL from the ADNI database.
18F-AV45 PET
Summary data (standardized uptake value ratio [SUVR]) of 18F-AV45 (florbetapir) PET, obtained 2 years after baseline CRP measurements, were used. The techniques, methods, and results related to amyloid imaging in ADNI have been reported elsewhere.16 The AV45 summary data were calculated from the cortical sum of regions of interest divided by the whole cerebellum reference region. In this study, we used the AV45 variable from the R ADNIMERGE package as the florbetapir mean of whole cerebellum (reference region) in regions defined by FreeSurfer. The AV45 studies were conducted after 3 to 10 years after plasma CRP was measured.
Statistical Analysis
Statistical analysis was performed with R (version 3.60; R Foundation for Statistical Computing) statistical software. The concentrations of CRP, APOE protein, NfL, and neurogranin were log-transformed due to skewed distributions. One-way analysis of variance was used to compare means when there were >2 groups, and χ2 tests were used for binary variables in Table 1. Multivariable linear regression models were implemented to investigate the interaction effects between CRP and APOE ε4 (CRP × APOE ε4) on MMSE scores, CDR-SB scores, AD fluid, and PET biomarkers. By using a forward selection procedure, we included different sets of variables in 3 models: model 1 was unadjusted; model 2 was adjusted for age, sex, education, and APOE genotype; and model 3 added the CRP × APOE ε4 interaction variable. Due to the skewed distribution of MMSE and CDR-SB scores, we generated a bootstrapped 95% confidence interval (CI) for the regression coefficients (β). The bootstrapped CI is based on 10,000 replications, and we reported the bias-corrected and accelerated bootstrap interval. Follow-up Pearson correlations and scatterplots were conducted to depict the cross-sectional and longitudinal relationships between plasma CRP and MMSE scores, CDR-SB scores, AV45 PET scan, and CSF AD biomarkers. These analyses were conducted with the whole sample and stratified by APOE genotypes status. All tests were 2 sided, and statistical significance was defined by a value of p < 0.05.
Table 1.
Demographic, Cognitive Diagnoses, and Tau AD Biomarkers Based on APOE Genotypes in ADNI Study

Data Availability
Supplement Tables and Figures are available from the Dryad Digital Repository: doi.org/10.5061/dryad.08kprr52f.
Results
Sample Characteristics
The sample included 566 ADNI participants who had complete APOE genotype data and baseline plasma CRP measurements. The average age of the sample was 74.8 ± 7.4 years (mean ± SD), and 215 (38%) participants were female. Participants were stratified into 3 APOE genotype groups: (1) no APOE ɛ4 allele (ε4 noncarriers, n = 274), (2) 1 APOE ɛ4 allele (n = 222), and (3) 2 APOE ɛ4 alleles (genotype ε4/4¸ n = 70) (Table 1). There were no statistically significant differences between the groups in sex or education.
The dataset included 58 individuals with CN (10.2%), 396 participants with MCI (70.0%), and 112 participants with AD dementia (19.8%). The group with 2 APOE ε4 alleles had the highest rate of AD dementia, the lowest average MMSE scores, and the highest CDR-SB scores (Table 1). The group with 2 APOE ɛ4 alleles had lower levels of CRP (mean ± SD −0.21 ± 1.20 mg/L vs 0.02 ± 1.21 mg/L vs 0.64 ± 1.15 mg/L, p < 0.001) and plasma APOE protein (p < 0.001) than those with 1 APOE ɛ4 allele, followed by those who had no APOE ɛ4 allele. Greater number of APOE ɛ4 alleles corresponded to higher levels of CSF t-Tau, p-Tau, and neurogranin, as well as lower CSF Aβ42 (Table 1). This finding remained significant after CN participants were excluded (eTable 1 [doi.org/10.5061/dryad.08kprr52f]). Among the 3 APOE allele groups, there were no differences in NfL in either CSF or plasma.
Negative Association Between Plasma CRP and Cognitive Scores in the Presence of 2 APOE ɛ4 Alleles
First, we used bootstrap regression analyses to study the association between plasma CRP and baseline MMSE and CDR-SB scores. Unadjusted and adjusted regressions were performed for age, sex, education, and APOE ɛ4 alleles (Table 2). Although plasma CRP as an independent variable was not associated with MMSE scores in unadjusted or adjusted models, the interaction of APOE ɛ4 and CRP was negatively associated with MMSE scores at baseline (β [95% CI] = −0.34 [−0.57, −0.11], p < 0.01) and at 12-month follow-up (β [95% CI] = −0.60 [−1.01, −0.15], p < 0.01) (model 3). There was no statistical significance at the 24-month follow-up. After stratification by APOE genotypes, the negative association patterns were observed in the group of participants who had 2 APOE ɛ4 alleles but not in any of the other APOE genotype groups (Table 3, MMSE score section). The interaction between CRP and APOE ɛ4 alleles on CDR-SB scores showed similar relationships in the bootstrap regression models (eTable 2 [doi.org/10.5061/dryad.08kprr52f]).
Table 2.
Bootstrap Regression Analyses of the Interaction Effect Between 2 APOE ε4 Alleles and CRP on MMSE Scores

Table 3.
Stratified Analysis of the CRP Association With MMSE, and CSF t-Tau and p-Tau Among Different APOE Genotype Groups

Next, we used Pearson correlation analyses to test the relationship between baseline plasma CRP concentrations and cognition in those with 0, 1, and 2 APOE ɛ4 alleles. Because of imbalanced numbers of individuals with CN in each APOE group in Table 1, we excluded those with CN to conduct scatterplots. Plasma CRP levels were negatively correlated with MMSE scores at baseline among those who had 2 APOE ɛ4 alleles (r = −0.29, p = 0.015); this effect remained statistically significant at the 12-month follow-up (r = −0.26, p = 0.025) but not at the 24-month follow-up (Figure 1A). Among participants with MCI, there was no statistically significant association between baseline plasma CRP and MMSE score. However, there was a statistically significant effect at the 12-month follow-up (r = −0.42, p = 0.0045) in the 2 APOE ɛ4 allele group (eFigure 2A [doi.org/10.5061/dryad.08kprr52f]).
Figure 1. Association Between Baseline Plasma CRP and MMSE and CDR-SB Scores and CSF Biomarkers (t-Tau and p-Tau) Among Participants With MCI and AD Dementia Across 3 APOE Genotypes.

Because of imbalanced numbers of cognitive normal (CN) controls in each APOE group in Table 1, we excluded CN from the scatterplots. Participants with mild cognitive impairment (MCI) (n = 396) or Alzheimer Disease (AD) dementia (n = 112) were divided into 3 groups based on APOE genotypes: 0 APOE ε4 alleles (APOE ε4 = 0), 1 APOE ε4 allele (APOE ε4 = 1), and 2 APOE ε4 alleles (APOE ε4 = 2). Relationships between baseline plasma C-reactive protein (CRP) and trajectory cognitive tests or CSF AD biomarkers at baseline, 12 months, and 24 months were examined. Associations between baseline CRP and Mini-Mental State Examination (MMSE) scores (A), Clinical Dementia Rating Sum of Boxes (CDR-SB) score (B), CFS total tau (t-Tau) (C), and CFS phosphorylated tau (p-Tau) (D) are shown by using scatterplots with a linear regression line with 95% confidence bands (shaded area), the Pearson coefficient of correlation, and its p value.
Among those who had 1 APOE ε4 allele, plasma CRP levels were slightly correlated with MMSE scores at baseline (r = −0.17, p = 0.015) but not at follow-ups. Among those who had no APOE ε4 allele, plasma CRP levels were not associated with MMSE score (Figure 1A).
Plasma CRP levels also positively correlated with CDR-SB score only in participants with 2 APOE ɛ4 alleles at baseline (r = 0.34, p = 0.0045). Results for CDR-SB score at follow-ups were similar (Figure 1B). Among those with MCI, this association was observed only at the 12-month follow-up, not at baseline or the 24-month follow-up (eFigure 2 [doi.org/10.5061/dryad.08kprr52f]). There was no association between plasma CRP and CDR-SB score among those with 0 or 1 APOE ɛ4 allele.
Positive Correlation Between Plasma CRP and Brain Aβ in the Total Sample but Not Among APOE Genotype Groups
The association between plasma CRP and CSF levels of Aβ42 was tested. eFigure 1A (doi.org/10.5061/dryad.08kprr52f]) shows that plasma CRP measurements were positively associated with CSF Aβ42 levels in the entire sample (r = 0.12, p = 0.043). However, this relationship was due to collinearity because both plasma CRP and CSF Aβ42 from the highest to the lowest followed the pattern of no APOE ɛ4 >1 APOE ɛ4 >2 APOE ɛ4 (Table 1). In each APOE genotype group, there was no association between plasma CRP and CSF Aβ42 (eFigure 1C [doi.org/10.5061/dryad.08kprr52f]). In multivariable regression analyses across the total sample, neither plasma CRP alone nor the APOE ɛ4 × CRP interaction term was associated with CSF Aβ42, suggesting collinearity of APOE genotype and CRP (eTable 3 [doi.org/10.5061/dryad.08kprr52f]).
Plasma CRP was negatively associated with AV45 PET SUVR values (r = −0.23, p = 0.023) in the entire sample (eTable 1 and eFigure 1B [doi.org/10.5061/dryad.08kprr52f]). However, there was no association between plasma CRP and AV45 PET SUVR in multivariable regression analysis (data not shown). Again, there was no statistically significant relationship between CRP and AV45 PET SUVR in each APOE genotype group (eFigure 1D [doi.org/10.5061/dryad.08kprr52f]).
Positive Correlation Between Plasma CRP and CSF t-Tau Only in the Presence of 2 APOE ɛ4 Alleles
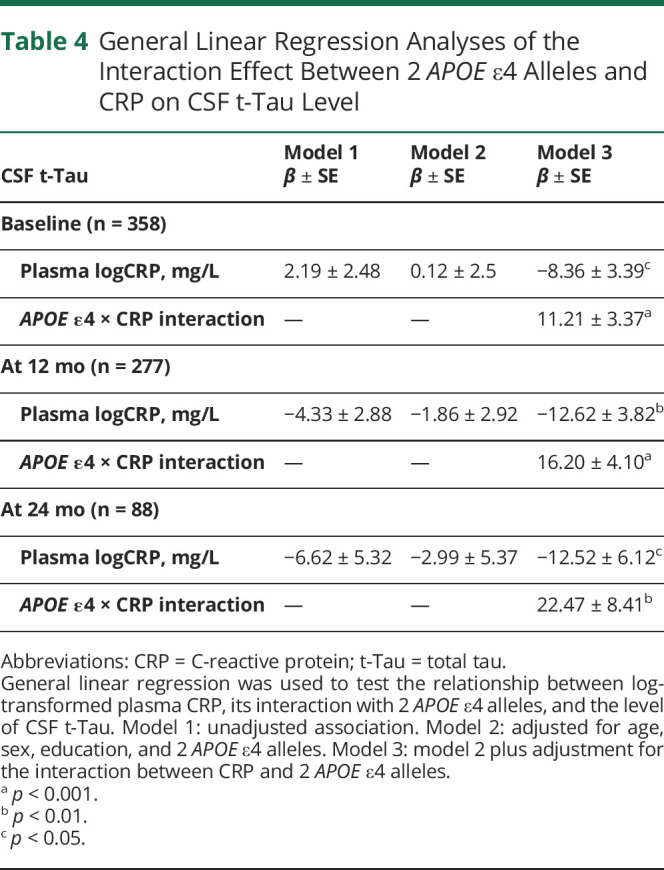
Using unadjusted and multivariable linear regression models, Table 4 shows that plasma CRP was not associated with CSF t-Tau levels (models 1 and 2). However, the interaction with 2 APOE ɛ4 alleles and plasma CRP on CSF t-Tau levels (model 3) was significant at baseline (β = 11.21, SE = 3.37, p < 0.001), 12 months (β = +16.20, SE = 4.10, p < 0.001), and 24 months (β = 22.47, SE 8.41, p < 0.01). After stratification by APOE ɛ4 group, there was a positive association between baseline plasma CRP and CSF t-Tau levels at baseline and the 12-month and 24-month follow-up in the group of participants who had 2 APOE ε4 alleles (Table 3). However, there were negative associations in the group with no APOE ɛ4 alleles and no association in the group of 1 APOE ɛ4 allele.
Table 4.
General Linear Regression Analyses of the Interaction Effect Between 2 APOE ε4 Alleles and CRP on CSF t-Tau Level
Due to the small sample sizes of participants with CN who had brain biomarkers in the APOE ɛ4 groups with 1 (n = 5) and 2 (n = 0) alleles (Table 1), we excluded those with CN and conducted Pearson correlation analyses to test the association between plasma CRP and CSF tau biomarkers. Figure 1C shows that baseline plasma CRP levels were positively correlated with baseline (r = 0.37, p = 0.013) and 12-month (r = 0.43, p = 0.011) and 24-month (r = 0.74, p = 0.035) CSF t-Tau levels in the 2 APOE ɛ4 allele group. In contrast, in the group without an APOE ɛ4 allele, baseline plasma CRP was negatively correlated with baseline (r = −0.16, p = 0.07) and 12-month (r = −0.25, p = 0.015) and 24-month (r = −0.34, p = 0.05) CSF t-Tau levels. Among those who had 1 APOE ɛ4 allele, there was no positive or negative relationship between plasma CRP and CSF t-Tau. Sensitivity analyses examined this relationship in participants with MCI only (eFigure 2C [doi.org/10.5061/dryad.08kprr52f]). Among only participants with MCI who had no APOE ɛ4 allele, there was an effect between baseline plasma CRP and 24-month CSF t-Tau levels (r = −0.46, p = 0.012).
Positive Correlation Between Plasma CRP and CSF p-Tau Only in the Presence of 2 APOE ɛ4 Alleles
Plasma CRP was not associated with CSF p-Tau (models 1 and 2) (Table 5). However, the interaction between 2 APOE ɛ4 alleles and plasma CRP (model 3) was positively associated with CSF p-Tau at baseline (β = 2.74, SE 1.14, p < 0.05) and at the 12-month follow-up (β = 3.54, SE 1.66, p < 0.05). There was no statistical significance at the 24-month follow-up (Table 5). When stratified by APOE ɛ4 allele group, similar trends of the relationship were observed but did not reach statistical significance at most time points (Table 3).
Table 5.
General Linear Regression Analyses of the Interaction Effect Between 2 APOE ε4 Alleles and CRP on CSF p-Tau Level
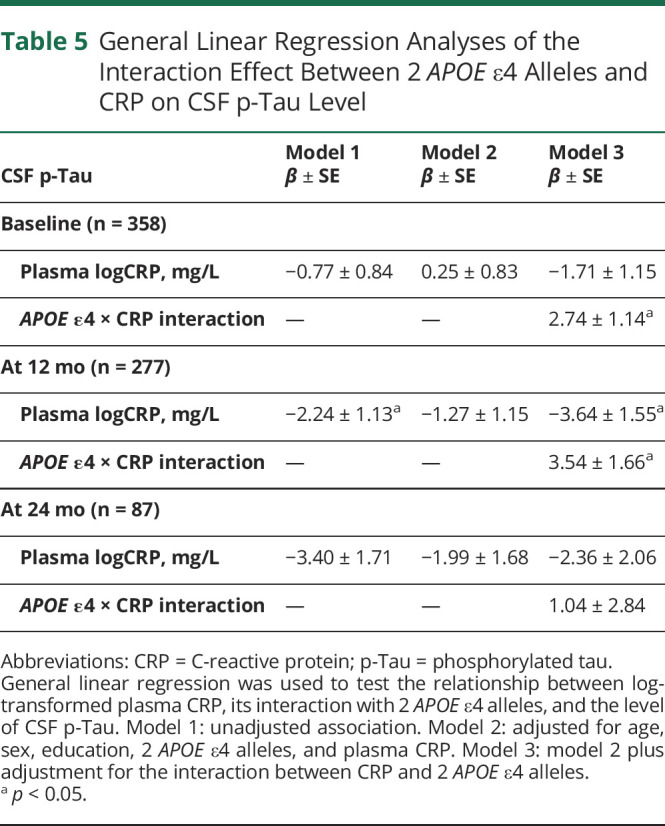
Figure 1D shows positive correlations between baseline plasma CRP levels and CSF p-Tau levels in the 2 APOE ɛ4 allele group, although this association did not reach statistical significance. In participants without the APOE ɛ4 allele, baseline plasma CRP negatively correlated with CSF p-Tau at the 24-month follow-up (r = −0.22, p = 0.036). Among participants with MCI, similar trends were observed (eFigure 2D [doi.org/10.5061/dryad.08kprr52f]).
Discussion
This study extends our previous research3 by investigating the interaction between APOE ɛ4 and CRP levels on cognitive decline and in vivo biomarkers of amyloid and tau using longitudinal data from the ADNI study. Results showed that higher baseline plasma CRP levels predicted accelerated global cognitive decline at a 1-year follow-up among individuals who had 2 APOE ɛ4 alleles. No such effect was observed among participants who had 1 or 0 APOE ɛ4 alleles. We additionally observed a positive association between baseline plasma CRP and trajectory levels of CSF t-Tau and p-Tau biomarkers among those who had 2 APOE ɛ4 alleles. This effect was reversed among those without an APOE ɛ4 allele. Our results suggest that participants with homozygous APOE ɛ4 alleles might be more susceptible to the effects of peripheral inflammatory factors on AD risk compared with those with 1 or 0 APOE ε4 allele. Thus, CRP may play an important mediator role in the APOE ɛ4–related pathway for AD risk. Treatment of chronic low-grade inflammation to reduce CRP levels, especially in 2 APOE ɛ4 allele carriers, may delay AD onset.
Baseline CRP was negatively associated with global cognitive function in the group of participants who had 2 APOE ɛ4 alleles, both cross-sectionally and longitudinally (Figure 1). CRP is involved in the immune response to toxins or injuries in systemic inflammation4 and is elevated in age-related diseases (e.g., cardiovascular diseases,7 age-related macular degeneration,8 and poststroke inflammation9). Infections of the respiratory, gastrointestinal, and urinary tract systems are common in older adults and can trigger chronic low-grade inflammation (i.e., high CRP levels).3 Such inflammatory response may increase susceptibility to AD, especially among those already at risk (i.e., APOE ɛ4 carriers).
Higher plasma CRP was also associated with higher CSF t-Tau and p-Tau levels only in those with 2 APOE ɛ4 alleles. Although peripheral CRP modestly correlated with fluid and PET biomarkers of amyloidosis, the CRP-Aβ relationships might be due to collinearity with APOE genotype. Compared with Aβ, tau biomarkers are more strongly associated with cognitive function.17 The amyloid cascade hypothesis is the most common proposed mechanism of AD pathogenesis whereby Aβ triggers the development of p-Tau, resulting in neurodegeneration.21 CRP may represent one causal pathway by which Aβ leads to tau phosphorylation, especially when it comes to APOE ɛ4–associated pathways to AD. However, the mechanisms by which APOE ɛ4 and CRP levels lead to tauopathy and cognitive decline are still unknown. One hypothesis is that APOE ɛ4 may make blood-facing endothelia in the brain more vulnerable to increased plasma CRP. Supporting this idea, a preclinical AD study found that mCRP exacerbates the severity of tauopathy in the brain of mice with transgenic genes of amyloid precursor protein and tau.22
Baseline plasma CRP had a consistent effect on tauopathy biomarkers and MMSE and CDR-SB scores at the 12-month, but not 24-month, follow-up in the 2 APOE ɛ4 alleles group. CSF AD biomarkers are shown to be elevated 10 to 20 years before the onset of clinical symptoms.23 Our findings suggest that CRP might enhance disease progression at an earlier rather than later stage of the long neurodegenerative process. Although genetic risk factors such as APOE ɛ4 are present across the lifespan, disease onset does not occur until later in life.2 APOE ɛ4 is the major genetic risk factor for AD,24 and delaying onset by 5 years can reduce AD risk by nearly 50%.25 Early detection and treatment of inflammation may be an important therapeutic target to prevent chronic inflammation and subsequent AD risk in APOE ε4 carriers.
Finally, plasma CRP was negatively associated with CSF biomarkers of tau at follow-up time points among those who were APOE ɛ4 noncarriers, as opposed to those who were 2 APOE ɛ4 allele carriers. APOE ɛ4 noncarriers had higher CRP levels compared with APOE ɛ4 carriers (Table 1). One possible explanation may be that APOE ɛ2 vs APOE ɛ4 brains are differentially affected by molecular pathways from chronic inflammation such as CRP and mCRP. APOE ɛ2 carriers may be more efficient at clearing t-Tau, p-Tau, and other brain AD pathologic proteins, so that APOE ɛ2 brains may be more resilient to chronic low-grade inflammation and AD risk. Whereas peripheral chronic low-grade inflammation may facilitate a clearance mechanism for tauopathy in those with APOE ɛ2 allele, inhibiting the inflammatory reaction in APOE ɛ2 carriers may not delay AD pathogenesis. Previous prevention trials of anti-inflammatory drugs for AD have failed,26 potentially because of differences in APOE status. APOE ɛ4 carriers may be a more appropriate population for future clinical trials of anti-inflammatory drugs.
This study has several limitations. The main limitation of this study is the lack of CN individuals in the group of 2 APOE ε4 alleles in the ADNI1 dataset (Table 1). Thus, we were limited to investigating the relationship between plasma CRP and AD biomarkers in normal cognition and at an earlier preclinical stage than MCI. In addition, the methods used to measure plasma CRP levels in the ADNI1 study are different from those used in clinical practice and other studies. Most studies such as the Framingham Heart Study (FHS) and clinical practice use enzymatic immunoassays (ELISAs) to measure serum CRP.27 ELISA specifically targets CRP. In contrast, the ADNI1 study used a targeted multiplex proteomic technique to measure CRP, along with 189 other proteins. The specificity of this approach is inferior to that of ELISA. Future research should examine the effect of CRP levels on cognitive decline using a more comprehensive neuropsychological test battery instead of assessing only MMSE scores. Nevertheless, based on 3 studies—the findings reported here, the results from our published FHS study on CRP and AD,3 and the preclinical result of CRP on AD5—it is likely that CRP, specifically mCRP, plays a meaningful role in APOE ε4–related pathways to AD.
Our findings suggest that CRP may represent an explanatory factor of the APOE ɛ4 vs ɛ2 risk pathway toward AD. Our results further suggest that APOE ɛ2 brains may be more resilient to peripheral inflammatory attacks. Because systemic infections are common among older adults, recovery of the immune system to normal CRP levels (<1 mg/L) could be critical for certain genotypes such as APOE ɛ4 in mitigating AD risk. Targeting APOE ɛ4 carriers may be an important consideration for future anti-inflammatory clinical trials.
Acknowledgment
Data collection and sharing for this project were funded by the ADN) (NIH grant U01 AG024904) and Department of Defense ADNI (Department of Defense award W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, by the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc; Biogen; Bristol-Myers Squibb Co; CereSpir, Inc; Cogstate; Eisai 32Inc; Elan Pharmaceuticals, Inc; Eli Lilly and Co; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc; Fujirebio; GE Healthcare; IXICO Ltd; Janssen Alzheimer Immunotherapy Research & Development, LLC; Johnson & Johnson Pharmaceutical Research & Development LLC; Lumosity; Lundbeck; Merck & Co, Inc; Meso Scale Diagnostics, LLC; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corp; Pfizer Inc; Piramal Imaging; Servier; Takeda Pharmaceutical Co; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Glossary
- Aβ
β-amyloid peptide
- AD
Alzheimer disease
- ADNI
Alzheimer's Disease Neuroimaging Initiative
- CDR
Clinical Dementia Rating Scale
- CDR-SB
CDR Sum of Boxes
- CI
confidence interval
- CN
cognitively normal
- CRP
C-reactive protein
- FHS
Framingham Heart Study
- MCI
mild cognitive impairment
- mCRP
monomeric CRP
- MMSE
Mini-Mental State Examination
- NfL
neurofilament light chain
- p-Tau
phosphorylated tau protein
- pCRP
native CRP
- SUVR
standardized uptake value ratio
- t-Tau
total tau protein
Appendix. Authors
Study Funding
This work was financially supported by National Institute on Aging grants R01AG049899, R01AG059424, and K24AG050842 (W.Q.Q) and a Boston University Alzheimer’s Disease Center pilot grant (Q.T).
Disclosure
Q. Tao, T.F.A. Ang, S.C. Akhter-Khan, I.S. Itchapurapu, R. Killiany, X. Zhang, A. Budson, K.W. Turk, L. Goldstein, J. Mez, M.L. Alosco, and W.W.Q. Qiu report no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
References
- 1.Strittmatter WJ, Saunders AM, Schmechel D, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA. 1993;90(5):1977-1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tanzi RE. A genetic dichotomy model for the inheritance of Alzheimer's disease and common age-related disorders. J Clin Invest. 1999;104(9):1175-1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tao Q, Ang TFA, DeCarli C, et al. Association of chronic low-grade inflammation with risk of Alzheimer disease in ApoE4 carriers. JAMA Netw Open. 2018;1(6):e183597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stephensen CB, Gildengorin G. Serum retinol, the acute phase response, and the apparent misclassification of vitamin A status in the third National Health and Nutrition Examination Survey. Am J Clin Nutr. 2000;72(5):1170-1178. [DOI] [PubMed] [Google Scholar]
- 5.Slevin M, Krupinski J. A role for monomeric C-reactive protein in regulation of angiogenesis, endothelial cell inflammation and thrombus formation in cardiovascular/cerebrovascular disease? Histol Histopathol. 2009;24(11):1473-1478. [DOI] [PubMed] [Google Scholar]
- 6.Caprio V, Badimon L, Di Napoli M, et al. pCRP-mCRP dissociation mechanisms as potential targets for the development of small-molecule anti-inflammatory chemotherapeutics. Front Immunol. 2018;9:1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang J, Tang B, Liu X, et al. Increased monomeric CRP levels in acute myocardial infarction: a possible new and specific biomarker for diagnosis and severity assessment of disease. Atherosclerosis. 2015;239(2):343-349. [DOI] [PubMed] [Google Scholar]
- 8.Chirco KR, Whitmore SS, Wang K, et al. Monomeric C-reactive protein and inflammation in age-related macular degeneration. J Pathol. 2016;240(2):173-183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Slevin M, Matou-Nasri S, Turu M, et al. Modified C-reactive protein is expressed by stroke neovessels and is a potent activator of angiogenesis in vitro. Brain Pathol. 2010;20(1):151-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnstone D, Milward EA, Berretta R, Moscato P. Multivariate protein signatures of pre-clinical Alzheimer's disease in the Alzheimer's Disease Neuroimaging Initiative (ADNI) plasma proteome dataset. PLoS One. 2012;7(4):e34341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ray S, Britschgi M, Herbert C, et al. Classification and prediction of clinical Alzheimer's diagnosis based on plasma signaling proteins. Nat Med. 2007;13(11):1359-1362. [DOI] [PubMed] [Google Scholar]
- 12.Aisen PS, Petersen RC, Donohue MC, et al. Clinical core of the Alzheimer's Disease Neuroimaging Initiative: progress and plans. Alzheimers Dement. 2010;6(3):239-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Folatein M, Liu T, Peter I, et al. The homocysteine hypothesis of depression. Am J Psychiatry. 2007;164(6):861-867. [DOI] [PubMed] [Google Scholar]
- 14.Hughes CP, Berg L, Danziger WL, Coben LA, Martin RL. A new clinical scale for the staging of dementia. Br J Psychiatry. 1982;140:566-572. [DOI] [PubMed] [Google Scholar]
- 15.Hansson O, Seibyl J, Stomrud E, et al. CSF biomarkers of Alzheimer's disease concord with amyloid-β PET and predict clinical progression: a study of fully automated immunoassays in BioFINDER and ADNI cohorts. Alzheimers Dement. 2018;14(11):1470-1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jagust WJ, Bandy D, Chen K, et al. The Alzheimer's Disease Neuroimaging Initiative positron emission tomography core. Alzheimers Dement. 2010;6(3):221-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM. Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol. 2007;64(3):343-349. [DOI] [PubMed] [Google Scholar]
- 18.Rowe CC, Ng S, Ackermann U, et al. Imaging beta-amyloid burden in aging and dementia. Neurology. 2007;68(20):1718-1725. [DOI] [PubMed] [Google Scholar]
- 19.Ong KT, Villemagne VL, Bahar-Fuchs A, et al. Aβ imaging with 18F-florbetaben in prodromal Alzheimer's disease: a prospective outcome study. J Neurol Neurosurg Psychiatry. 2015;86(4):431-436. [DOI] [PubMed] [Google Scholar]
- 20.Timmers T, Ossenkoppele R, Verfaillie SCJ, et al. Amyloid PET and cognitive decline in cognitively normal individuals: the SCIENCe project. Neurobiol Aging. 2019;79:50-58. [DOI] [PubMed] [Google Scholar]
- 21.Tsai LH, Delalle I, Caviness VS Jr., Chae T, Harlow E. p35 Is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature. 1994;371(6496):419-423. [DOI] [PubMed] [Google Scholar]
- 22.Slevin M, Matou S, Zeinolabediny Y, et al. Monomeric C-reactive protein: a key molecule driving development of Alzheimer's disease associated with brain ischaemia? Scientific Rep. 2015;5:13281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):280-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roses AD, Strittmatter WJ, Pericak-Vance MA, Corder EH, Saunders AM, Schmechel DE. Clinical application of apolipoprotein E genotyping to Alzheimer's disease. Lancet. 1994;343(8912):1564-1565. [DOI] [PubMed] [Google Scholar]
- 25.Wilson D, Peters R, Ritchie K, Ritchie CW. Latest Advances on interventions that may prevent, delay or ameliorate dementia. Ther Adv Chronic Dis. 2011;2(3):161-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Follow-up evaluation of cognitive function in the randomized Alzheimer's disease anti-inflammatory prevention trial and its follow-up study. Alzheimers Dement. 2015;11(2):216-225.e211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rost NS, Wolf PA, Kase CS, et al. Plasma concentration of C-reactive protein and risk of ischemic stroke and transient ischemic attack: the Framingham study. Stroke. 2001;32(11):2575-2579. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Supplement Tables and Figures are available from the Dryad Digital Repository: doi.org/10.5061/dryad.08kprr52f.